22 Opioid intravenous anesthetics
Agonist: A drug with a specific cellular or receptor affinity that produces a predictable response.
Antagonist: A drug that exerts an opposite action to that of another or competes for the same receptor site or sites.
Breakthrough Pain: A transient increase in the intensity of pain from a baseline pain level that is no greater than moderate.
Dysphoria: A disorder of affect that is characterized by depression and anguish.
Endogenous: Originating inside the body.
Endorphins: These opioid peptides are produced in the body and are composed of many amino acid (protein) substances that attach to opioid receptors in the central nervous system and the peripheral nervous system for reduction of pain.
Exogenous: Originating outside the body.
Fixed Chest Syndrome: Rigidity of the diaphragmatic and intercostal muscles.
Mydriasis: Dilation of the pupil of the eye.
Opiate Receptor: Receptors that are transmembrane proteins that bind to endogenous opioid neuropeptides and exogenous morphine and similar compounds. They are designated mu, kappa, and delta subtypes of the opiate receptor.
Opioid: A drug that contains opium or a derivative of opium along with semisynthetic or synthetic drugs that have opium-like properties.
The immediate postanesthesia phase is when the patient is most vulnerable to complications (see Chapter 29).1 Many drugs that have residual anesthetic effects well into the postanesthesia period are used in modern anesthesia care. These agents include the potent inhaled agents, muscle relaxants, benzodiazepines, and opioids. Respiratory depression is the most common adverse event in the postanesthesia care unit (PACU); therefore the use and understanding of the various opioid agents optimize patient outcomes. In addition, the reduction of pain in the PACU is one of the primary focuses of care in the perianesthesia phase. In addition to assessing pain, the perianesthesia nurse must take into the evaluation of pain the preoperative, intraoperative, and postanesthesia phase of the surgical patient. Because all pain-reducing drugs must be taken into account during the pain assessment in the PACU, the mixed action or agonist-antagonist combination drugs are presented in this chapter. With a detailed description of the major opioids used in the perianesthesia period provided, the PACU nurse will be able to make excellent informed decisions in regard to the anticipated outcome of the patient of pain reduction and comfort.
Concept of opioids and opioid receptors
Opioids are the substances, either natural or synthetic, that are administered into the body (exogenous) and bind to specific receptors to produce a morphine-like or opioid agonist effect. The endogenous opioids are the endorphins. The endorphins, which are produced in the body, attach to the opioid receptors in the central nervous system (CNS) to activate the body’s pain modulating system. The term opioid is used because of the multitude of synthetic drugs with morphine-like actions; with the advent of receptor physiology, opioid has replaced the term narcotic, which is derived from the Greek word for “stupor” and usually refers to both the production of the morphine-like effects and the physical dependence.2
The naturally occurring alkaloids of opium are divided into two classes: phenanthrene and benzylisoquinoline. The principal phenanthrene series of drugs includes morphine, codeine, and thebaine. Papaverine and noscapine, which lack opioid activity, represent the benzylisoquinoline alkaloids of opium.3
The synthetic opioids have been produced with the modification of the chemical structure of the phenanthrene class of drugs. Drugs such as fentanyl (Sublimaze) and meperidine (Demerol) are examples of synthetic opioids.2,4
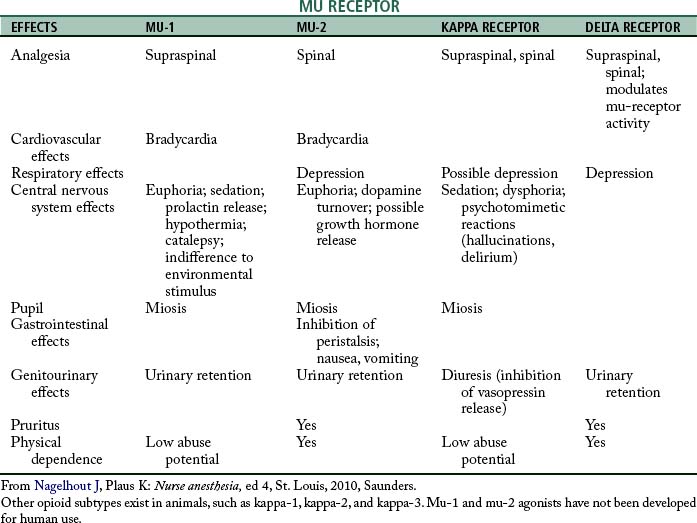
The identification of specific opioid receptors has enhanced the understanding of the agonist and antagonist actions of this category of drugs. The opioid receptors are located in the CNS, principally in the brain stem and spinal cord. These receptors have been determined by the pharmacologic effect they produce when stimulated by a specific agonist along with how the effect is blocked by a specific antagonist. The three major categories of opioid receptors are the mu (μ), delta (δ), and kappa (κ).4,5
The mu receptors are mainly responsible for the production of supraspinal analgesia effects with stimulation. These receptors are further divided into mu-1 and mu-2 types. Activation of the mu-1 receptors results in analgesia; when the mu-2 receptors are stimulated, hypoventilation, bradycardia, physical dependence, euphoria, and ileus can result. The mu receptors are activated by morphine, fentanyl, and meperidine. The drug that is specific to the mu-1 receptor is meptazinol, which is supraspinal in regard to analgesia; the mu-2 receptor analgesia occurs at the spinal level. Other characteristics of the mu-1 and mu-2 receptors are summarized in Table 22-1. Stimulation of the kappa receptors results in spinal analgesia, dysphoria, hallucinations, hypertonia, tachycardia, tachypnea, mydriasis, sedation, and miosis, with little effect on ventilation. The drugs that possess both opioid agonist and antagonist activities, such as nalbuphine (Nubain), have their principal action on the kappa opioid receptors. The delta opioid receptors, when stimulated, serve to modulate the activity of the mu receptors and cause depression and urinary retention. The drug naloxone (Narcan) attaches to all the opioid receptors and thus serves as an antagonist to all the opioid agonists (see Table 22-1).2,4
Opioids
The administration of opioids in the perioperative period is not without the concern of overdose. The major signs of overdose with opioids are miosis, hypoventilation, and coma. If the patient becomes severely hypoxemic, mydriasis can occur. Airway obstruction is a strong possibility because the skeletal muscles become flaccid. Hypotension and seizures can also occur. The treatment for an opioid overdose is mechanical ventilation and the slow titration of naloxone. Consideration must always be given to the fact that some patients who become overdosed with an opioid may indeed be already physically dependent. Naloxone can precipitate an acute withdrawal syndrome.2
Meperidine hydrochloride
Meperidine (Demerol) was discovered in 1939 by Eisleb and Schauman. Because it is chemically similar to atropine, it was originally introduced as an antispasmodic agent and was not used as an opioid anesthetic agent until 1947. The main action of this drug is similar to morphine; it stimulates the subcortical mu receptors, which results in an analgesic effect. Meperidine is approximately one tenth as potent as morphine and has a duration of action of 2 to 4 hours. The onset of analgesia is prompt (10 minutes) after subcutaneous or intramuscular administration. All pain, especially visceral, gastrointestinal, and urinary tract, is satisfactorily relieved. This drug causes less biliary tract spasm than morphine; however, in comparison with codeine, meperidine causes greater biliary tract spasm. It produces some sleepiness but causes little euphoria or amnesia. Meperidine increases the sensitivity of the labyrinthine apparatus of the ear, which explains the dizziness, nausea, and vomiting that sometimes occur in ambulatory patients.2,4
Meperidine is generally metabolized in the liver; less than 5% is excreted unchanged by the kidneys. However, because of a toxic metabolite of meperidine, patients who are administered this drug may have seizures.2 Meperidine is partially metabolized to normeperidine which has some analgesic effects, but more importantly, lowers the seizure threshold and can induce CNS excitability. Meperidine probably should not be administered to elderly patients because renal dysfunction may occur and less tolerance to normeperidine.
Because of its spasmolytic effect, meperidine is the drug of choice for biliary duct, distal colon, and rectal surgery. It offers the advantages of little interference with the physiologic compensatory mechanisms, low toxicity, smooth and rapid recovery, prolonged postoperative analgesia, excellent cardiac stability in patients at poor risk, and ease of detoxification and excretion.2,4,5 Meperidine is used most often with procedures now, and not typically for long-term pain management because of the effects of normeperidine over time.
Morphine
Morphine, one of the oldest known drugs, has only recently been used as an opioid intravenous anesthetic agent. Alkaloid morphine is from the phenanthrene class of opium. The exact mechanism of action of morphine is unknown. In humans, it produces analgesia, drowsiness, changes in mood, and mental clouding. The analgesic effect can become profound before the other effects are severe and can persist after many of the side effects have almost disappeared. With direct effect on the respiratory center, morphine depresses respiratory rate, tidal volume, and minute volume. Maximal respiratory depression occurs within 7 minutes after intravenous injection of the drug and 30 minutes after intramuscular administration. After therapeutic doses of morphine, the sensitivity of the respiratory center begins to return to normal in 2 or 3 hours, but the minute volume does not return to preinjection level until 4 or 5 hours have passed.
Morphine can cause nausea and vomiting, especially in ambulatory patients, because of direct stimulation of the chemoreceptor trigger zone. The emetic effect of morphine can be counteracted with opioid antagonists and phenothiazine derivatives such as prochlorperazine (Compazine), dexmedetomidine (Precedex), or the 5-HT3 receptor antagonist ondansetron (Zofran). Histamine release has been noted with morphine, and morphine also causes profound constriction of the pupils, stimulation of the visceral smooth muscles, and spasm of the sphincter of Oddi.5–7
Morphine is detoxified by conjugation with glucuronic acid. Ninety percent is excreted by the kidneys, and 7% to 10% is excreted in the feces via the bile.2
Methadone
The actions of this synthetic opioid agonist resemble morphine; side effects include depression of ventilation, miosis, constipation, and biliary tract spasm. Clinically, the sedative and euphoric actions of methadone appear to be less than those produced by morphine.5
Hydromorphone
Hydromorphone (Dilaudid), which is a derivative of morphine, was developed in Germany in the 1920s and released to the mass market in the late 1920s. The drug has a renewed popularity for the PACU and can be administered intravenously, intramuscularly, rectally, or orally. The drug profile in regard to its analgesia and side effects is similar to morphine. Hydromorphone is recommended for patients in renal failure because of its virtual lack of active metabolites after its breakdown in the liver. It has a high solubility and a rapid onset of action and appears to have less troublesome side effects and dependence liability profile as compared with morphine. Because of its high lipid solubility, hydromorphone can be administered via epidural or spinal for a wide area of anesthesia as compared with duramophine.2
Hydromorphone, like all opioids, is a CNS depressant and has actions and side effects similar to morphine. Its depressant effects can be enhanced with beta-blockers and alcohol. The duration of action of this drug is approximately 2 hours, with a peak action in approximately 30 minutes with intravenous administration.
Fentanyl
Janssen and colleagues8 introduced a series of highly potent meperidine derivatives that were found to render the patient free of pain without affecting certain areas in the CNS. Fentanyl (Sublimaze) appeared to be of special interest. In regard to analgesic properties, fentanyl is approximately 80- to 125-fold more potent as morphine and has a rapid onset of action of 5 to 6 minutes and a peak effect within 5 to 15 minutes. The analgesia lasts 20 to 40 minutes when administered intravenously. Via the intramuscular route, the onset of action is 7 to 15 minutes; the analgesia usually lasts 1 to 2 hours. When fentanyl is administered as a single bolus, 75% of the drug undergoes first-pass pulmonary uptake. That is, the lungs serve as a large storage site and this nonrespiratory function of the lung (see Chapter 12) limits the amount of fentanyl that actually reaches the systemic circulation. If the patient receives multiple doses of fentanyl via single injections or infusion, the first-pass pulmonary uptake mechanism becomes saturated and the patient has a prolonged emergence because of increased duration of the drug. Consequently, during the admission of the patient to the PACU, the postanesthesia nurse must determine the frequency and amount of intraoperative fentanyl administration. Patients who have received a significant amount of fentanyl via infusion or via titration should be continuously monitored for persistent or recurrent respiratory depression. In addition, fentanyl has been implicated in what is called a delayed-onset respiratory depression. In some patients, a secondary peak of the drug concentration in the plasma occurs approximately 45 minutes after the apparent recovery from the drug. This syndrome can occur when some of the fentanyl becomes sequestered in the gastric fluid and then can become recycled into the plasma in approximately 45 minutes. Therefore, in the PACU, all patients who have received fentanyl should be continuously monitored for respiratory depression for at least 1 hour from the time of admission to the unit.2
Fentanyl can be administered during surgery at three different dose ranges, depending on the type of surgery and the desired effect. For example, the low-dose range of 2 to 20 mcg/kg attenuates moderately stressful stimuli. The moderate dose range is 20 to 50 mcg/kg and strongly obtunds the stress response. The megadose range of as much as 150 mcg/kg blocks the stress response and is particularly valuable when protection of the myocardium is critical.4
Fentanyl can be used alone in a nitrous-opioid technique. It also is used in the PACU in the form of a low-dose intravenous drip for pain relief; however, fentanyl is usually given slowly intravenously in the PACU for breakthrough pain (see Table 22-2 for helpful calculation of milligram to microgram dosage information).
Table 22-2 Example of Conversion of Dosage Calculations from Milligrams to Micrograms
| MILLIGRAMS | MICROGRAMS | MILLILITERS OF FENTANYL |
|---|---|---|
| 0.025 | 25 | 0.5 |
| 0.05 | 50 | 1.0 |
| 0.10 | 100 | 2.0 |
| 0.15 | 150 | 3.0 |
| 0.20 | 200 | 4.0 |
| 0.25 | 250 | 4.5 |
| 0.50 | 500 | 10.0 |
| 1.00 | 1000 | 20.0 |
Sufentanil
Sufentanil is an analogue of fentanyl and is approximately fivefold to sevenfold more potent as fentanyl. Anesthesia with sufentanil can be induced more rapidly, with basically the same technique as that used for fentanyl, without an increase in the incidence rate of chest wall rigidity. However, sufentanil can produce chest wall rigidity; therefore if it is administered in the PACU, equipment for administration of oxygen with positive pressure and the skeletal muscle relaxant succinylcholine should be on hand. The incidence rate of hypertension with sufentanil is lower than with comparable doses of fentanyl. Bradycardia is infrequently seen in patients who receive sufentanil, and when high-dose sufentanil is used in combination with nitrous oxide-oxygen, the mean arterial pressure and cardiac output may be decreased. The recovery time from sufentanil from the time of injection is about the same as with fentanyl, because sufentanil is rapidly eliminated from tissue storage sites; consequently, the duration of action of sufentanil is about the same as with fentanyl. In addition, the incidence rates of postoperative hypertension, the need for vasoactive agents, and the requirements for postoperative analgesics are generally reduced in patients who are administered moderate or high doses of sufentanil in comparison with patients given inhalation agents. Of particular interest to the perianesthesia nurse is that sufentanil has an additive effect that is seen in patients who receive barbiturates, tranquilizers, other opioids, general anesthetics, or other CNS depressants. This effect is especially true of benzodiazepines because they can potentiate a profound hypotensive action. Therefore, when sufentanil is combined with any of these drugs, particular attention should be paid to any signs of decreased respiratory drive, increased airway resistance, or hypotension. Immediate countermeasures include maintenance of a patent airway with proper positioning of the patient, placement of an oral airway or endotracheal tube, and administration of oxygen. If indicated, naloxone should be used as a specific antidote for management of the respiratory depression. The duration of respiratory depression after overdose with sufentanil may be longer than the duration of action of the naloxone. Consequently, the patient should be constantly observed for the recurrence of respiratory depression, even after the initial successful treatment with naloxone. Hypotension can be treated with reversal with naloxone; however, fluids and vasopressors may be indicated (see Chapter 11).4
Alfentanil
Alfentanil is another analogue of fentanyl that is approximately one tenth as potent and has approximately one third the duration of action of fentanyl. The onset of action of this drug occurs in 1 or 2 minutes, and the duration of action is 20 to 30 minutes. Alfentanil appears to have significant advantages over currently available opioid anesthetics. For example, it has no cumulative drug effects, and once the infusion of alfentanil is terminated, the emergence time is predictable. Alfentanil, like fentanyl, produces minimal hemodynamic effects and offers a high therapeutic index. In fact, the therapeutic index for alfentanil is higher than those of fentanyl and other opioids. A therapeutic index is the ratio of the lethal dose to the effective dose; the higher the therapeutic index, the farther the lethal dose from the dose used for the desired effect. More specifically, the therapeutic index of fentanyl is 270, which means that it is approximately four times safer than morphine. The therapeutic index of alfentanil is approximately 2.5 times more favorable than that of fentanyl.9–11
Remifentanil
Remifentanil is a selective mu opioid agonist that has an analgesic potency almost equal to fentanyl and twentyfold more potent than alfentanil. This drug has some excellent pharmacologic properties in that it is brief in action, titratable, and noncumulative; it lacks histamine release; and it has a rapid recovery after the discontinuation of the drug. The onset of this drug is within 1 minute. When the drug is discontinued, it is metabolized quickly; its effects disappear within 4 minutes. The remifentanil anesthetic technique is excellent for suppression of the stress response and allows for excellent depression of neurologic responses. In regard to the immediate postanesthesia period, remifentanil is better than most intravenous opioid drugs in regard to residual effects because it has a rapid recovery and less risk of postoperative respiratory depression.
This drug should probably not be used in the PACU; however, if the drug is administered in the PACU, it should be administered only by an anesthesia provider who is experienced in the administration of the drug. Some of the adverse effects of the drug may accompany its administration.1,12,13
Partial agonist-antagonist drugs
Pentazocine
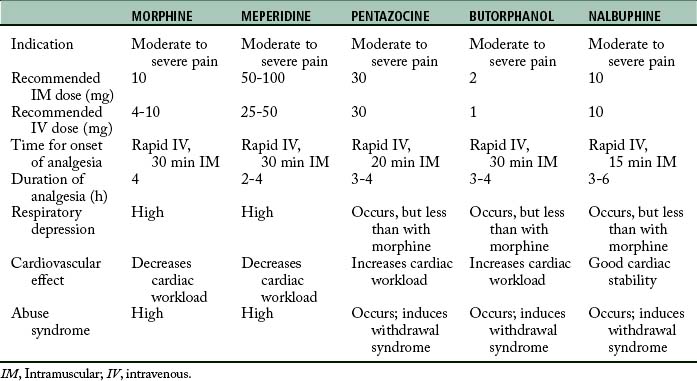
Studies of the relative potency of this drug indicate that 30 mg of pentazocine is analgesically equivalent to 10 mg of morphine and 75 mg of meperidine. Pentazocine has been established to relieve severe pain and is approximately twofold to fourfold less potent than morphine when administered parenterally.2,4,14–16
The onset of analgesic activity of pentazocine is approximately 2 or 3 minutes when it is given intravenously and 15 to 20 minutes when given intramuscularly. The duration of action is approximately 3 hours. When given orally, the drug is approximately one third as potent as when it is given intramuscularly.2,13
Butorphanol
Butorphanol is a synthetic analgesic that is chemically related to the nalorphine-cyclazocine series with both opioid and antagonist properties. More specifically, it serves as an agonist at the kappa and sigma opioid receptors. In regard to its analgesic potency, it is approximately fivefold more potent than morphine, thirtyfold more potent than meperidine, and twentyfold more potent than pentazocine. Butorphanol can produce sedation, nausea, and respiratory depression. The respiratory depression is plateaulike in that 2 mg of butorphanol depresses respiration to a degree equal to 10 mg of morphine. The magnitude of respiratory depression with butorphanol is not appreciably increased at doses of 4 mg. The duration of the respiratory depression is dose related and is reversible with naloxone. Intravenous administration of butorphanol can produce increased pulmonary artery pressure, pulmonary wedge pressure, left-ventricular end-diastolic pressure, systemic arterial pressure, and pulmonary vascular resistance. Consequently, this drug increases the workload of the heart, especially in the pulmonary circuit. Because of its antagonist properties, butorphanol is not recommended for patients who are physically dependent on opioids, because butorphanol can precipitate withdrawal symptoms in those patients. See Table 22-3 for an overview of the clinical pharmacology of butorphanol.4
Nalbuphine
Nalbuphine (Nubain) is a potent analgesic with opioid agonist and antagonist actions. It is chemically related to oxymorphone and naloxone. This drug is an antagonist at the mu receptors, a partial agonist at the kappa receptors, and an agonist at the sigma receptors. Nalbuphine is as potent as morphine and approximately threefold more potent as pentazocine on a milligram basis. At a dose of 10 mg/kg, nalbuphine causes the same degree of respiratory depression as does 10 mg of morphine. At higher doses, nalbuphine exhibits the same plateau effect as butorphanol (i.e., respiratory depression is not increased appreciably with higher doses). The respiratory depression produced by nalbuphine can be reversed with naloxone. Nalbuphine does not appear to increase the workload of the heart or to decrease cardiovascular stability. This drug has a lower abuse potential than does morphine; however, if it is given to a patient who is physically dependent on opioids, withdrawal symptoms may appear. Signs of withdrawal include abdominal cramps, nausea and vomiting, lacrimation, rhinorrhea, anxiety, restlessness, elevation of temperature, and piloerection. If these symptoms appear after the injection of nalbuphine, the administration of small amounts of morphine can relieve the objective effects of the syndrome. See Table 22-3 for an overview of the clinical pharmacology of nalbuphine.4
Opioid antagonists
Naloxone
An excessive dose of naloxone can increase blood pressure, a finding that may be seen as a response to pain. Too rapid reversal can induce nausea, vomiting, diaphoresis, and tachycardia. During the reversal procedure, the vital signs should be monitored; naloxone should be used with caution in patients with cardiac irritability.4,5
Selected methods of opioid administration
Intrathecal and epidural routes of administration
When 0.1 to 0.2 mg of preservative-free morphine (Duramorph) is administered into the subarachnoid space (intrathecal), the maximum concentration is reached in 5 to 10 minutes, with a duration of 80 to 200 minutes. When 5 mg of morphine is administered into the epidural space in the lumbar region, analgesia can last for as long as 24 hours. The patient should obtain pain relief in 30 to 60 minutes after injection. If appropriate pain relief is not achieved, incremental doses of 1 to 2 mg can be administered. The maximum dose in a 24-hour period is 10 mg.5
After spinal surgery, epidural morphine administered with the continuous epidural technique has advantages and disadvantages. Its advantage is a profound degree of pain relief, especially for the first 12 to 18 postoperative hours. However, disadvantages are related to displacement of the epidural catheter and the length of action of the epidural morphine. If the epidural catheter becomes displaced and the morphine has been injected, only partial pain relief ensues. Because of the possibility of profound respiratory depression, opioids must be administered cautiously. In fact, a nonopioid drug such as ketorolac may be especially useful in this circumstance. Depending on the anticipated amount and length of pain, a patient-controlled analgesia (PCA) device can be started in the patient for an immediate result of pain resolution. In addition, the postanesthesia nurse can have a profound effect on reducing the pain threshold by repositioning and reassuring the patient. The new technology of apnea monitors or end-tidal carbon dioxide monitors can be useful in detecting hypoventilation or apnea that can be created by the opioid in this technique. Therefore the use of either monitor on patients who are receiving epidural morphine certainly aids the postanesthesia nurse in monitoring for respiratory dysfunction.4
Other opioids that can be administered epidurally are fentanyl and sufentanil.14 These drugs offer some advantages over morphine; they are more suited for continuous infusion techniques because of their rapid onset and short duration of action. Because they have such a rapid clearance from the cerebrospinal fluid, less chance exists for these drugs to spread toward the head (rostral spread). Rostral spread, which is associated more with morphine, has been shown to produce side effects such as nausea, pruritus, and the previously discussed delayed respiratory depression syndrome. Naloxone reverses this side effect; however, the analgesic effect also is reversed. In this instance, nalbuphine administration should be considered to reverse the respiratory depression and preserve some of the analgesia.
Patient-controlled analgesia
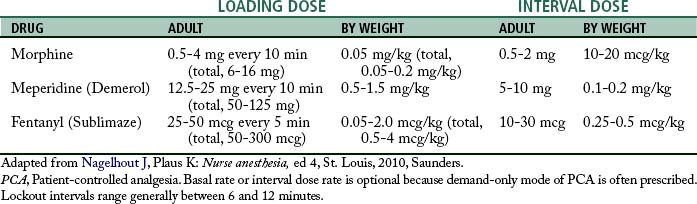
For an appropriate level of analgesia, a loading dose, followed by titration to effect, based on the pharmacokinetics of the opioid drug is used during surgery. The maintenance of an appropriate blood level of the opioid to achieve and maintain a level of analgesia is the goal of this technique. The blood level of the drug is called the minimum effective analgesic concentration (MEAC). Research has shown that the MEAC varies among individuals. Through the use of technology, the principles of this intraoperative technique have been continued into the immediate postoperative period. Intravenous PCA is the method of choice for patients who need continued analgesia. The peaks and valleys of analgesia can be avoided without the patient becoming totally dependent on the nurse’s response for pain relief. PCA allows the patient more control of the situation by allowing the patient to seek out a particular level of analgesia—the MEAC.4,5,14
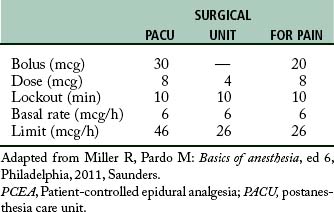
The amount of the self-dose bolus should be low to avoid an acute increase in blood levels of the drug above the MEAC because, along with the concern about overdose, blood levels above the MEAC have no analgesic value. The lockout interval, or delay, is the setting used to block the use of the self-dose bolus button for a period of time. During this time, the PCA pump does not deliver the drug, even when the patient pushes the button. The lockout interval is usually short, so that the patient can self-administer small incremental doses to maintain the MEAC, but the interval should be long enough to prevent overdosage. The basal infusion rate is usually set at a rate necessary to provide analgesia when the patient is resting. See Table 22-4 for a suggested protocol for drug administration in PCA.
Patient-controlled epidural analgesia
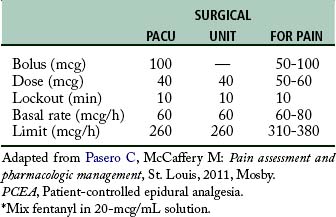
The concept of PCA has been adapted to epidural analgesia or PCEA. In this instance, the PCA infusion pump can be attached to the epidural catheter. The opioid drugs that can be used in this technique are fentanyl and sufentanil. They can be used with great success for analgesia after cesarean section. The basal infusion rate keeps the patient analgesic and comfortable. The patient can self-administer a bolus of the opioid if the analgesia provided with the basal infusion rate is not sufficient. In addition, the bolus opioid facilitates additional analgesia needed for turning and early ambulation. A suggested protocol for both fentanyl and sufentanil is provided in Tables 22-5 and 22-6. Because of the possibility of rostral spread, an apnea monitor is suggested for use on patients who are receiving patient-controlled epidural analgesia.4,5,14 (See Chapter 31.)
Monitoring the patient receiving opioids in the PACU
Along with monitoring of respiratory rate and depth, peripheral pulse oximetry and, if necessary, detection of expired carbon dioxide or arterial blood gas monitoring can be used. The nervous system clinical indicators of significant opioid action should also be monitored and include observation for excess sedation, lethargy, apathy, dysphoria, nausea, vomiting, pruritus (especially facial), miosis, and cough suppression.4,14
If the patient is determined to have opioid-induced respiratory depression, prophylactic supplemental oxygen should be administered and the modified stir-up regime should be instituted. If the patient is difficult to arouse, the airway should be supported and manually assisted ventilation with bag and mask may be needed. Should the patient have excessive secretions or vomitus, a person skilled in airway management should be summoned because tracheal intubation may be necessary. Pharmacologic treatment should include the use of naloxone. For the adult, treatment should start with lower doses such as 0.1 mg and titrate to effect to lessen the adverse cardiovascular effects that can occur when the opioid is completely reversed with high-dose naloxone.
Opiate detoxification in the PACU
Many methods of opioid detoxification have been developed since problems with addiction have occurred. The most common methods of opioid detoxification are methadone withdrawal, clonidine withdrawal, clonidine–naltrexone withdrawal, and anesthesia-assisted rapid opiate detoxification (AAROD).4
Summary
The pure opioid agonists were presented as were the mixed action agonist-antagonists. Appropriate reversal drugs were also presented with the methods of administration of these drugs in the PACU.
1. Atlee J. Complications in anesthesia, ed 2. Philadelphia: Saunders; 2007.
2. Brunton L, et al. Goodman and Gilman’s the pharmacological basis of therapeutics, ed 12. New York: McGraw-Hill Professional; 2010.
3. Hines R, Marschall K. Stoelting’s anesthesia and co-existing disease, ed 5. Philadelphia: Saunders; 2008.
4. Nagelhout J, Plaus K. Nurse anesthesia, ed 4. St. Louis: Saunders; 2010.
5. Stoelting R. Pharmacology and physiology in anesthetic practice, ed 4. Philadelphia: Lippincott Williams & Wilkins; 2005.
6. Shorten G, et al. Postoperative pain management: an evidence-based guide to practice. Philadelphia: Saunders; 2006.
7. Stancer-Smiley B, Paradise N. Does the duration of N2O administration affect postoperative nausea and vomiting? Nurse Anesth. 1991;2(1):13–20.
8. Stanley TH. The history and development of the fentanyl series. J Pain Symptom Manage. 1992;7(3):S3–7.
9. Aitkenhead A, et al. Textbook of anesthesia, ed 5. Philadelphia: Churchill Livingstone; 2007.
10. Barash P, et al. Clinical anesthesia, ed 6. Philadelphia: Lippincott Williams & Wilkins; 2009.
11. Fisher L. Anesthesia and uncommon diseases, ed 5. Philadelphia: Saunders; 2007.
12. Blouin R, Gross J. Ventilation and conscious sedation. Semin Anesth. 1996;15(4):335–342.
13. Miller R, et al. Miller’s anesthesia, ed 7. New York: Churchill Livingstone; 2010.
14. Pasero C, McCaffery M. Pain assessment and pharmacologic management. St. Louis: Mosby; 2011.
15. Pasero C, McCaffery M. Orthopaedic postoperative pain management. J Perianesth Nurs. 2007;22(3):160–174.
16. Shorten G, et al. Postoperative pain management: an evidence-based guide to practice. Philadelphia: Saunders; 2006.
American Association of Critical-Care Nurses: Core curriculum for progressive care nursing. Philadelphia: Saunders; 2010.
Alspach J. Core curriculum for critical care nursing, ed 6. Philadelphia: Saunders; 2005.
Babar M, et al. Effect of preoperative rectal indomethacin on postoperative pain reduction after open cholecystectomy. J Perianesth Nurs. 2010;25(1):7–10.
Barrett K, et al. Ganong’s review of medical physiology, ed 23. New York: McGraw-Hill Medical; 2009.
Borchardt M. Review of the clinical pharmacology and use of the benzodiazepines. J Perianesth Nurs. 1999;14(2):65–72.
Bready L, et al. Decision making in anesthesiology, ed 4. St. Louis: Mosby; 2007.
Conlay L, et al. Case files anesthesiology. New York: McGraw-Hill Medical; 2011.
Davis P, et al. Smith’s anesthesia for infants and children, ed 8. St. Louis: Mosby; 2011.
Deutschman C, Netigan P. Evidence-based practice of critical care. Philadelphia: Saunders; 2010.
Dorsch J, Dorsch S. Understanding anesthesia equipment, ed 5. Philadelphia: Lippincott Williams & Wilkins; 2007.
Drake R, et al. Gray’s anatomy for students, ed 2. Philadelphia: Churchill Livingstone; 2009.
Gallager C, Issenberg B. Simulation in anesthesia. Philadelphia: Saunders; 2007.
Hall J. Guyton and Hall textbook of medical physiology, ed 12. Philadelphia: Saunders; 2011.
Hines R, Marschall K. Handbook for Stoelting’s anesthesia and co-existing disease, ed 3. Philadelphia: Saunders; 2009.
Jones R. Desflurane and sevoflurane: inhalation anesthetics for this decade. Br J Anaesth.1990;65:527–536.
Kaplan J, et al. Cardiac anesthesia. New York: Churchill Livingstone; 2011.
Katoh T, et al. Blood concentration of sevoflurane and isoflurane on recovery from anesthesia. Br J Anaesth.1992;69:259–262.
Key K, et al. Use of propofol and emergence agitation in children: A literature review. AANA J. 2010;78(6):468–473.
Kier L, Dowd C. The chemistry of drugs for nurse anesthetists. Chicago: AANA Publishing, Inc; 2004.
Kulli J, Koch C. Does anesthesia cause loss of consciousness. Trends Neurosci. 1991;14(1):6–10.
Longnecker D, et al. Anesthesiology. New York: McGraw-Hill Medical; 2007.
Mason R. Murray and Nadel’s textbook of respiratory medicine, ed 5. Philadelphia: Saunders; 2011.
Miller R, Pardo M. Basics of anesthesia, ed 6. Philadelphia: Saunders; 2011.
Moos D, Cuddeford D. Implications of obstructive sleep apnea syndrome for the perianesthesia nurse. J Perianesth Nurs. 2006;21(2):103–118.
Prielipp R, Young C. Current drugs for sedation of critically ill patients. Semin Anesthesia Perioperative Med Pain. 2001;20(2):85–94.
Passannante A, et al. Anesthestic management of patients with obesity and sleep apnea. Anesthesiol Clin North Am. 2005;23:479–490.
Rakic A, Golembiewski J. Low-dose ketamine infusion for postoperative pain management. J Perianesth Nurs. 2009;24(4):254–257.
Redai I, et al. Are volatile anesthetics cardioprotective agents. Semin Anesthesia perioperative Med Pain. 2001;20(2):95–100.
Reves J, et al. Midazolam: Pharmacology and uses. Anesthesiology.1985;62:310–324.
Sebel P, Larson J. Propofol: a new intravenous anesthetic. Anesthesiology.1989;71:260–277.
Short T, Chui P. Propofol and midazolam act synergistically in combination. Br J Anaesth.1991;67:539–545.
Sandberg W, et al. The MGH textbook of anesthetic equipment. New York: Churchill Livingstone; 2011.
Schick L, Windle PE. Perianesthesia nursing core curriculum: preprocedure, phase I and phase II PACU nursing. ed 2. Philadelphia: Saunders; 2010.
Shepherd M. Multidisciplinary approach to developing guidelines for use of anesthetic agents in an intensive care unit. Anesthesia Today. 1995;6(1):15–18.
Sieber F. Geriatric anesthesia. New York: McGraw-Hill Medical; 2006.
Vincent J, et al. Textbook of critical care, ed 6. Philadelphia: Saunders; 2011.
White P. Perioperative drug manual, ed 2. Philadelphia: Saunders; 2005.