11 Oliguria
 Definitions and Epidemiology
Definitions and Epidemiology
A number of definitions for oliguria can be found in the literature. Oliguria is often defined as urine output less than 200 to 500 mL per 24 hours. In order to standardize the use of the term across different studies and populations, the Acute Dialysis Quality Initiative (ADQI) recently adopted a definition of oliguria as urine output of less than 0.3 mL/kg/h for at least 24 hrs (www.ADQI.net). For all practical purposes, however, urine output under 0.5 mL/kg/h is usually considered inadequate for most critically ill patients.
Given the lack of consensus over definitions, it has been difficult to determine the incidence of oliguria. Some studies have estimated that up to 18% of medical and surgical intensive care unit (ICU) patients with intact renal function exhibit episodes of oliguria.1 Furthermore, 69% of ICU patients who develop acute kidney injury (AKI) are oliguric.2 Overall, AKI in the ICU has a poor prognosis (mortality rates range from 30%-70%), and oliguric renal failure is associated with worse outcome compared to nonoliguric renal failure, although this distinction is less clear for AKI. It is essential to understand the physiologic derangements leading to this exceedingly common problem.
 Pathophysiology
Pathophysiology
Urine output is a function of glomerular filtration, tubular secretion, and tubular reabsorption. Glomerular filtration is directly dependent on intravascular volume and renal perfusion. Renal perfusion in turn is a function of arterial pressure and renal vascular resistance. The intrarenal vasculature is capable of preserving glomerular filtration rate (GFR) in the face of varying systemic pressure through important neurohumoral autoregulating mechanisms that affect the afferent and efferent arterioles. The most important of these neurohumoral mechanisms is the renin-angiotensin-aldosterone system (Figure 11-1). Oliguria can be due to decreased GFR, increased tubular reabsorption of filtrate, or a combination of both. Oliguria also can be caused by mechanical obstruction to urine flow. In any case, oliguria is an insensitive clinical manifestation of AKI.
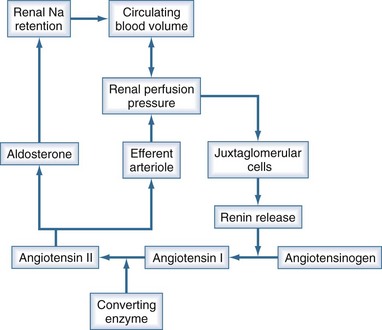
Reduction in glomerular filtration rate
Oliguria secondary to a decrease in GFR is usually related to one of the following conditions:
 Diagnostic Approach to Oliguria
Diagnostic Approach to Oliguria
Rule out Urinary Obstruction
The first step in diagnosis is to rule out urinary obstruction. A prior history of prostatic hypertrophy may provide some clues to the presence of distal obstruction. However, in the ICU setting, distal obstruction presenting as oliguria is commonly due to obstruction of the urinary catheter (especially in male patients). Hence, in patients with new-onset oliguria, the urinary catheter must be flushed or changed in order to rule out obstruction. Although uncommon in the acute setting, complete or severe partial bilateral ureteral obstruction may also lead to acute, “acute on chronic,” or chronic renal failure. Early diagnosis of urinary tract obstruction (UTO) is important, since many cases can be corrected, and a delay in therapy can lead to irreversible renal injury. Renal ultrasonography is usually the test of choice to exclude UTO.4 It is noninvasive, can be performed by the bedside, and also carries the advantage of avoiding the potential allergic and toxic complications of radiocontrast media. In the majority of affected patients, ultrasonography can establish the diagnosis of hydronephrosis and often establish its cause. Ultrasonography also can be useful for detecting other causes of renal disease such as polycystic kidney disease. However, under some circumstances, renal ultrasound may not yield good results. For example, in early obstruction or obstruction associated with severe dehydration, hydronephrosis may not be seen on the initial ultrasound examination but may appear later in the course of the disease. Computed tomography (CT) scanning should be performed if the ultrasound results are equivocal or if the kidneys are not well visualized. CT also is indicated if the cause of the obstruction cannot be identified by ultrasonography.
Laboratory Indices
Although most authorities advocate examining the urine sediment, the yield of urine microscopy in the ICU is very low. Urine sediment is typically bland or reveals hyaline and fine granular casts in a prerenal state. By contrast, ATN is often associated with coarse granular casts and tubular epithelial casts. However, the discrimination of these findings is limited, and AKI may be present in the absence of changes in urinary sediment, particularly with sepsis-induced AKI. The main utility of examining the urine sediment is in the detection of red cell casts, which indicate primary glomerular disease. The urine sediment in postrenal failure is often very bland; casts or sediment typically are absent. Occasionally a few red cells and white cells may be seen. Eosinophilia, eosinophiluria, and hypocomplementemia, if present (although insensitive and nonspecific), point to the diagnosis of atheroembolic etiology of acute oliguria.5
Table 11-1 lists laboratory values that can be useful for distinguishing prerenal from intrarenal causes of oliguria. The fractional excretion of filtered sodium (FENa) is calculated according to the following formula:
TABLE 11-1 Biochemical Indices Useful to Distinguish Prerenal from Intrarenal Acute Renal Failure
| Prerenal | Renal | |
|---|---|---|
| Osm u (mOsm/kg) | >500 | <400 |
| Na u (mmol/L or meq/L) | <20 | >40 |
| Urea/creatinine | >0.1 | <0.05 |
| U/S creatinine | >40 | <20 |
| U/S osmolality | >1.5 | >1 |
| FENa (%)* | <1 | >2 |
| FEurea (%) | <25 | >25 |
ARF, acute renal failure; S, serum; U, urine.
* ((u Na / s Na) / (u creat / s creat)) × 100
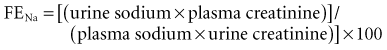
If the calculated FENa is less than 1%, a prerenal cause of oliguria should be suspected. Importantly, interpretation of the FENa is difficult or impossible if the patient has received diuretic or natriuretic agents (including dopamine and/or mannitol). Interpretation of the FENa also can be confounded by the presence of large amounts of endogenous osmotically active substances in the urine, such as glucose or urea. Drugs that interfere with the renin-angiotensin-aldosterone axis, such as ACE inhibitors or nonsteroidal antiinflammatory agents, also can confound the interpretation of FENa.
Several nephrotoxic factors, such as aminoglycosides, cyclosporine, and contrast media, are associated with FENa values below 1%, mimicking prerenal azotemia. Furthermore, sepsis may result in urine chemistries that resemble prerenal physiology even when renal blood flow is normal or increased.6
A low fractional excretion of urea (FEurea) (<35%) has been proposed to be more sensitive and specific than FENa in differentiating between prerenal and renal causes of AKI, especially when diuretics have been administered.7 The diagnostic accuracy of FENa versus FEurea was recently compared in 99 patients hospitalized at a tertiary care center; study subjects had developed a 30% increase in SCr concentration from baseline within 1 week.8 Patients were classified as having prerenal azotemia if the rise in SCr was transient and consistent with the clinical context. Each group also was subdivided according to exposure to diuretics. FEurea of 35% or less and FENa of 1% or less were then analyzed for their ability to predict prerenal azotemia. Sensitivity, specificity, and receiver operating characteristic (ROC) curves were generated for each index. Sensitivity and specificity of FEurea were 48% and 75%, respectively, in patients who did not receive diuretics, and 79% and 33%, respectively, in patients who received diuretics. Sensitivity and specificity of FENa were 78% and 75%, respectively, in patients not administered diuretics, and 58% and 81%, respectively, in those who received diuretics. ROC curves did not identify better diagnostic cutoff values for FEurea or FENa. Unfortunately, the study did not examine the combination of these indices, so neither test provides a level of diagnostic accuracy that can be relied on in clinical practice.
Clinical Parameters
It is common to assume that one can obtain a more accurate assessment of preload by measuring the central venous pressure (CVP) or pulmonary capillary occlusion pressure (PAOP). However, these parameters do not provide reliable estimates of fluid responsiveness.9 A cardiac index greater than 3.0 L/min/M2 generally suggests adequate preload, but it may not reflect optimal preload.10 The mixed venous oxygen saturation (SvO2) can serve as a surrogate for cardiac output, but again does not define optimal filling. In patients on mechanical ventilation and without spontaneous triggering of the ventilator, an arterial pulse-pressure variation of more than 13% is strongly predictive of adequate (or more than adequate) preload.11 In other cases, echocardiography may provide the only reliable evidence of fluid optimization (see Chapter 74).
Abdominal Compartment Syndrome
Another important and often overlooked reason for acute oliguria is abdominal compartment syndrome (ACS). ACS is defined as symptomatic organ dysfunction that results from an increase in intraabdominal pressure. Although this condition was initially described in trauma patients, ACS occurs in a wide variety of medical and surgical patients. ACS is sometimes seen after major abdominal surgeries requiring large-volume resuscitation, emergent laparotomies with tight abdominal wall closures, or abdominal wall burns with edema. ACS leads to AKI and acute oliguria mainly by directly increasing renal outflow pressure and thus reducing renal perfusion. Other mechanisms include direct parenchymal compression and arterial vasoconstriction mediated by stimulation of the sympathetic nervous and renin-angiotensin systems. Cardiac output also can be compromised by impaired venous return. These factors lead to decreased renal and glomerular perfusion and acute oliguria on this basis. Intraabdominal pressures over 15 mm Hg can lead to oliguria, and pressures over 30 mm Hg can cause anuria.12
 Treatment of Oliguria
Treatment of Oliguria
Ensuring Adequate Renal Perfusion
The mainstay of treatment of oliguria is identification and correction of the precipitating factors. Instituting appropriate supportive measures, such as avoidance of nephrotoxic agents and adjustment of doses of renally excreted drugs, is also important. Efforts should be made to optimize renal perfusion by correcting hypotension and supporting appropriate intravascular volume expansion. However, volume overload can also compromise renal perfusion (see abdominal compartment syndrome earlier), so fluid should be carefully prescribed in patients with oliguria. Correction of hypotension is especially crucial, since in sepsis and ischemic AKI, some of the important autoregulating mechanisms that help preserve GFR in the face of fluctuating BP are disrupted. Vasoactive drugs may be necessary in the ICU setting to increase mean arterial pressures to more than usual values to maintain adequate renal perfusion pressure and adequate urine output.13 In patients with chronic hypertension and renal vascular disease, the autoregulation curve can be shifted to the right, and higher than normal MAP may be required to ensure adequate renal perfusion. However, prior to initiation of treatment with vasoactive drugs, one must make sure the patient is adequately volume resuscitated. In many instances, the initial treatment consists of fluid challenges in the hope of correcting unrecognized volume depletion. Hemodynamic monitoring devices may provide important clues to the intravascular volume status that may enable a more streamlined, “goal-directed” approach to therapy.
Role of Diuretic Agents
The use of diuretic agents in oliguric renal failure is widespread despite the lack of convincing evidence supporting their efficacy. Traditionally, diuretics have been used in the early phases of oliguria to “jump start” the kidney and establish urine flow. Many clinicians believe that the absence of oliguria makes it easier to regulate intravascular volume status. Moreover, nonoliguric renal failure generally has a better prognosis than oliguric renal failure, and clinicians frequently use diuretics in an effort to avoid development of a low urine output state.14 A study by Anderson et al. in 1977 claimed a reduction in mortality from 50% to 26% by using high doses of a loop diuretic to convert oliguric to nonoliguric renal failure.15 This study excluded patients with shock and perioperative renal failure. More recent trials have failed to reproduce these results. A study in 1997 by Shilliday et al. examined the effect of loop diuretics on several outcomes in patients with AKI. While administration of loop diuretics increased average urine flow, there was no difference between the diuretic-treated and the placebo-treated groups with regard to the incidence of renal recovery, the need for renal replacement therapy, or death.16 Two other randomized controlled clinical trials by Brown et al. and Kleinknecht et al. have failed to find any evidence of benefit on survival with the use of loop diuretics in oliguric renal failure.17,18 The PICARD study group reported the results of a large cohort study of critically ill patients with AKI from 1989-1995.19 The study showed that diuretic use was associated with an increased risk of death or non-recovery of renal function. Recently a large observational study (BEST kidney study) showed that use of diuretics has no beneficial effect on clinical outcomes.20 Indeed, while not statistically significant, the odds ratio suggested that diuretic therapy might be harmful. Furthermore, high doses of loop diuretics can be associated with ototoxicity.
Vasoactive Agents
Other agents that have been used to treat oliguria include dopamine and related compounds. Because urine output often increases with the addition of low-dose dopamine, many intensivists assume that it has a beneficial effect. Indeed, low-dose dopamine has been advocated for nearly 30 years as therapy for oliguric renal failure on the basis of its action on DA1 receptors in doses of less than 5 µg/kg/min. However, there is abundant evidence that low-dose dopamine does not afford any renal protection in oliguria. Most evidence in favor of the treatment comes from uncontrolled trials or anecdotal studies. A comprehensive meta-analysis of dopamine in critically ill patients by Kellum et al. showed that dopamine did not prevent the onset of AKI, decrease mortality, or lessen the need for renal replacement therapy.21
Furthermore, there are important physiologic considerations that argue against a protective role for dopamine or any other dopamine receptor agonists (e.g., fenoldopam, dopexamine) in the oliguric state. First, the effect of dopamine agonists on urine output may be merely the natriuretic response mediated by inhibition of Na+/K+-ATPase at the tubular epithelial cell level.22 In other words, dopamine increases urine output because it is a diuretic. Second, administration of dopaminergic antagonists (e.g., metoclopramide) has not been associated with loss of renal function. Third, the effect of dopamine may be counteracted by increased plasma renin activity in critically ill patients. Fourth, a significant hysteresis effect has been shown for the action of dopamine on renal blood flow. Finally, although dopamine increases renal blood flow, it does not increase medullary oxygenation.23 Indeed, by increasing solute delivery to the distal tubule, dopamine agonists actually worsen medullary oxygen balance.24 Despite claims to the contrary, newer dopaminergic agonists (e.g., fenoldopam, dopexamine) not only suffer from these limitations but also can induce hypotension and thereby further increase the risk of renal injury.
 Conclusion
ConclusionBagshaw SM, Langenberg C, Bellomo R. Urinary biochemistry and microscopy in septic acute renal failure: a systematic review. Am J Kidney Dis. 2006;48(5):695-705.
Uchino S, Doig GS, Bellomo R, et al. Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators. Diuretics and mortality in acute renal failure. Crit Care Med. 2004;32(8):1669-1677.
1 Zaloga GP, Highes SS. Oliguria in patients with normal renal function. Anesthesiology. 1990;72(4):598-602.
2 Brivet FG, Kleinknecht DJ, Loirat P, Landais PJ. Acute renal failure in intensive care units–causes, outcome, and prognostic factors of hospital mortality; a prospective, multicenter study. French Study Group on Acute Renal Failure. Crit Care Med. 1996;24:192-198.
3 Thadhani RI, Camargo CAJr, Xavier RJ, Fang LS, Bazari H. Atheroembolic renal failure after invasive procedures. Natural history based on 52 histologically proven cases. Medicine (Baltimore). 1995;74:350-358.
4 Webb JA. The role of ultrasonography in the diagnosis of intrinsic renal disease. Clin Radiol. 1994;49:589-591.
5 Meyrier A, Buchet P, Simon P, Fernet M, Rainfray M, Callard P. Atheromatous renal disease. Am J Med. 1988;85:139-146.
6 Bagshaw SM, Langenberg C, Bellomo R. Urinary biochemistry and microscopy in septic acute renal failure: a systematic review. Am J Kidney Dis. 2006 Nov;48(5):695-705.
7 Carvounis CP, Nisar S, Guro-Razuman S. Significance of the fractional excretion of urea in the differential diagnosis of acute renal failure. Kidney Int. 2002;62:2223-2229.
8 Pepin MN, Bouchard J, Legault L, Ethier J. Diagnostic performance of fractional excretion of urea and fractional excretion of sodium in the evaluations of patients with acute kidney injury with or without diuretic treatment. Am J Kidney Dis. 2007;50:566-573.
9 Osman D, Ridel C, Ray P, et al. Cardiac filling pressures are not appropriate to predict hemodynamic response to volume challenge. Crit Care Med. 2007;35:64-68.
10 Kellum JA, Pinsky MR. Use of vasopressor agents in critically ill patients. Curr Opin Crit Care. 2002;8:236-241.
11 Michard F, Boussat S, Chemla D, et al. Relation between respiratory changes in arterial pulse pressure and fluid responsiveness in septic patients with acute circulatory failure. Am J Respir Crit Care Med. 2000;162:134-138.
12 Bailey J, Shapiro MJ. Abdominal compartment syndrome. Crit Care. 2000;4:23-29.
13 Berstyen AD, Holt AW. Vasoactive agents and the importance of renal perfusion pressure. New Horiz. 1995;3(4):650-651.
14 Majumdar S, Kjellstrand CM. Why do we use diuretics in acute renal failure. Semin Dial. 1996;9(6):454-459.
15 Anderson RJ, Linas SL, et al. Non oliguric acute renal failure. N Engl J Med. 1977;296:1134-1137.
16 Shilliday IR, Quinn KJ, Allison ME. Loop diuretics in the management of acute renal failure: a prospective, double-blind, placebo-controlled, randomized study. Nephrol Dial Transplant. 1998;12(12):2592-2596.
17 Brown CB, Ogg CS, Cameron JS. High dose furosemide in acute renal failure: a controlled trial. Clin Nephrol. 1981;15:90-96.
18 Kleinknecht D, Ganeval D, Gonzales-Duque LA, Fermanian J. Furosemide in acute oliguric renal failure. A controlled trial. Nephron. 1976;17:51-58.
19 Mehta RL, Pascual MT, Soroko S, et al. Diuretics, mortality, and nonrecovery of renal function in acute renal failure. JAMA. 2002;288(20):2547-2553.
20 Uchino S, Doig GS, Bellomo R, et al. Beginning and Ending Supportive Therapy for the Kidney (B.E.S.T. Kidney) Investigators. Diuretics and mortality in acute renal failure. Crit Care Med. 2004 Aug;32(8):1669-1677.
21 Kellum JA, Decker JM. Use of dopamine in ARF; a meta analysis. Crit Care Med. 2001;29:1526-1531.
22 Seri I, Kone BC, Gullans SR, Aperia A, Brenner BM, Ballerman BJ. Locally formed dopamine inhibits Na-K-ATPase activity in rat renal cortical tubule cells. Am J Physiol. 1988;255:F666-F673.
23 Heyman SN, Kaminski N, Brezis M. Dopamine increases renal medullary blood flow without improving regional hypoxia. Exp Nephrol. 1995;3(6):331-337.
24 Olsen NV, Hamsen JM, Ladefoged SD, et al. Renal tubular reabsorption of sodium and water during infusion of low-dose dopamine in normal man. Clin Sci. 1990;78:503-507.
