[level-membership-for-pulmolory-and-respiratory-category]
Chapter 62 Obesity Hypoventilation Syndrome
Pathophysiology
Obstructive Sleep Apnea
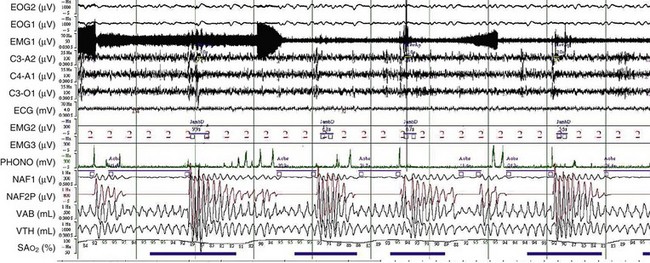
OSA may play a role in the pathogenesis of chronic hypercapnia in those patients, because in most cases, treatment of OSA corrects the hypercapnia. Indeed, to compensate for the effect of intermittent periodic breathing and resultant acute hypercapnia, normal subjects as well as patients with OSA will increase their tidal volume in the first breath after an apnea (hyperventilation). With physiologic differences in ventilatory responses between subjects, however, an overload in CO2 could result. The duration of the apneas also could contribute: When apneic episode duration becomes three times longer than the breathing interval, CO2 tends to accumulate despite maximal tidal volume, because there is insufficient time for adequate hyperventilation events (Figure 62-1). In addition, hypercapnia could blunt the ventilatory responses: The initial ventilation after an apneic episode is directly related to the volume of CO2 loaded during the preceding respiratory event and thus represents an index of CO2 ventilatory response. Hypercapnic patients demonstrated depression of this index of ventilatory compensation compared with that in eucapnic patients. It has been shown that the apnea-to-interapnea duration ratio is greater in hypercapnic patients than in eucapnic patients.
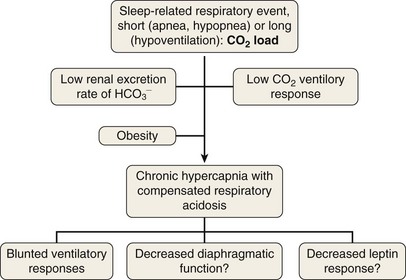
To elucidate the mechanisms that are involved in the development of hypercapnia in patients with OSA, Norman and co-workers have proposed, using a computer model, some hints for prediction of the transition from acute hypercapnia during sleep-disordered breathing (apnea, hypopnea, and hypoventilation) to chronic daytime hypercapnia. In their model, when the ventilatory CO2 response and renal HCO3− excretion were normal, increases in PaCO2 and HCO3− did not develop. The bicarbonate excretion during the day compensated for that retained during the night. When CO2 ventilatory response was very low, however, the model demonstrated a modest rise in PaCO2 and HCO3− measured during the awake state over multiple days. Similarly, when renal HCO3− excretion rate was lowered to simulate chloride deficiency, the model demonstrated a modest rise in daytime PaCO2 and HCO3−. The combination of low CO2 response and low renal HCO3− excretion rate produced a synergistic effect on the degree of elevation of daytime PaCO2. These workers suggested that hypercapnia results from an imbalance between the period of CO2 loading (short = apnea or hypopnea; long = hypoventilation) and inadequate compensation both during sleep and during the awake state. This pulmonary-renal interaction may contribute to the development and perpetuation of chronic daytime hypercapnia, which will lead to a blunted respiratory drive for the next sleep cycle (Figure 62-2).
Diagnosis
Sleep Laboratory Diagnosis
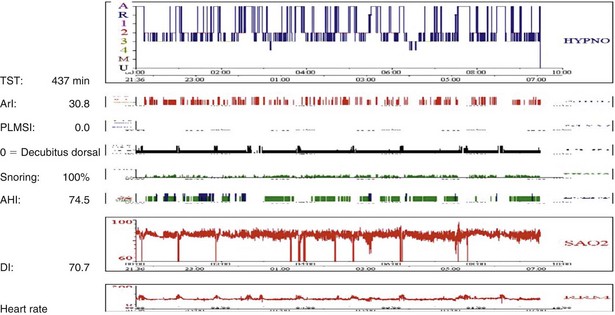
Sleep-disordered breathing can take either of two forms; the most common type is OSA, and the second one is central hypoventilation. Polysomnographic evaluation shows either a typical OSA pattern or periods of fall in SaO2 in REM stages alone (Figure 62-3) or throughout the entire sleep period (Figure 62-4). In many cases, sleep-disordered breathing is seen throughout sleep, with steep falls in saturation in non-REM sleep and much steeper falls in the REM stage (see Figure 62-4).
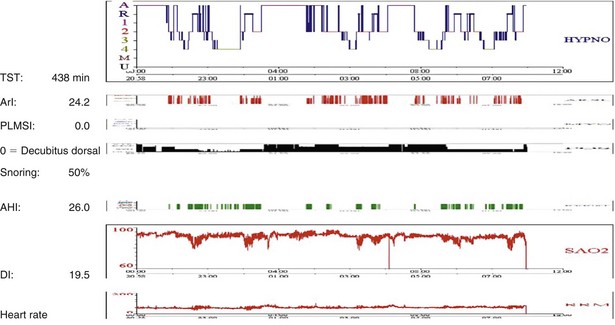
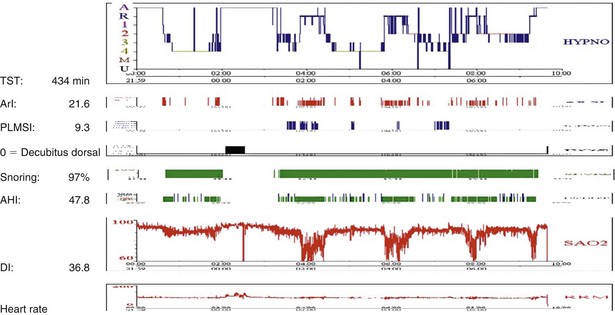
Figure 62-4 Note low SaO2 throughout the night and deeper falls in SaO2 related to the period of REM sleep. For abbreviations, see Figure 62-3.
In some patients, REM sleep may even be completely absent (Figure 62-5).
Treatment
Reversal of Sleep-Disordered Breathing
Controversies and Pitfalls
• Is the obesity hypoventilation syndrome just a particular form of obesity, so that losing weight would be enough to cure it?
• Does the obesity hypoventilation syndrome prevent weight loss via excessive daytime sleepiness, decrease in energy consumption, and exacerbated hunger sensation?
• What is the simpler and best screening method to identify patients at risk and to prompt an adequate diagnostic process?
• Is polysomnography necessary to manage patients with obesity hypoventilation syndrome?
• Is the respiratory treatment of the sleep-related causes of obesity hypoventilation syndrome (i.e., CPAP for the apnea-hypopnea syndrome or NIV for sleep-related hypoventilation) enough to protect the patient’s health, or is weight loss still a mandatory component of management?
• CPAP is an efficient therapy in many patients. A sizable number, however, do not respond to this intervention. What are the predictive factors that could rapidly identify such nonresponders?
• In patients needing NIV, how many can revert to a simpler CPAP treatment?
• In patients needing NIV, when is a shift to CPAP advisable?
• What is the cost-utility ratio of CPAP and NIV in OHS?
• CPAP or NIV treatment seems to decrease mortality, but this has been assessed in a limited number of patients. Long-term follow-up studies in larger cohorts are needed to confirm this major outcome and to determine the confounding effect of persistent obesity on survival.
Banerjee D, Yee BJ, Piper AJ, et al. Obesity hypoventilation syndrome: hypoxemia during continuous positive airway pressure. Chest. 2007;131:1678–1684.
Berg G, Delaive K, Manfreda J, et al. The use of health-care resources in obesity-hypoventilation syndrome. Chest. 2001;120:377–383.
Budweiser S, Riedl SG, Jorres RA, et al. Mortality and prognostic factors in patients with obesity-hypoventilation syndrome undergoing noninvasive ventilation. J Intern Med. 2007;261:375–383.
Chouri-Pontarollo N, Borel JC, Tamisier R, et al. Impaired objective daytime vigilance in obesity-hypoventilation syndrome: impact of noninvasive ventilation. Chest. 2007;131:148–155.
De Miguel Díez J, De Lucas Ramos P, Pérez Parra JJ, et al. [Analysis of withdrawal from noninvasive mechanical ventilation in patients with obesity-hypoventilation syndrome. Medium term results.]. Arch Bronconeumol. 2003;39:292–297.
Lumachi F, Marzano B, Fanti G, et al. Hypoxemia and hypoventilation syndrome improvement after laparoscopic bariatric surgery in patients with morbid obesity. In Vivo. 2010;24:329–331.
MacGregor MI, Block AJ, Ball WCJr. Topics in clinical medicine: serious complications and sudden death in the Pickwickian syndrome. Johns Hopkins Med J. 1970;126:279–295.
Marien H, Rodenstein D. Morbid obesity and sleep apnea. Is weight loss the answer? Clin Sleep Med. 2008;4:339–340.
Masa JF, Celli BR, Riesco JA, et al. The obesity hypoventilation syndrome can be treated with noninvasive mechanical ventilation. Chest. 2001;119:1102–1107.
Norman RG, Goldring RM, Clain JM, et al. Transition from acute to chronic hypercapnia in patients with periodic breathing: predictions from a computer model. J Appl Physiol. 2006;100:1733–1741.
Nowbar S, Burkart KM, Gonzales R, et al. Obesity-associated hypoventilation in hospitalized patients: prevalence, effects, and outcome. Am J Med. 2004;116:1–7.
Pérez de Llano LA, Golpe R, Ortiz Piquer M, et al. Short-term and long-term effects of nasal intermittent positive pressure ventilation in patients with obesity-hypoventilation syndrome. Chest. 2005;128:587–594.
Raurich JM, Rialp G, Ibáñez J, et al. Hypercapnic respiratory failure in obesity-hypoventilation syndrome: CO2 response and acetazolamide treatment effects. Respir Care. 2010;55:1442–1448.
Trakada GP, Steiropoulos P, Nena E, et al. Prevalence and clinical characteristics of obesity hypoventilation syndrome among individuals reporting sleep-related breathing symptoms in northern Greece. Sleep Breath. 2010;14:381–386.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]
Chapter 62 Obesity Hypoventilation Syndrome
Pathophysiology
Obstructive Sleep Apnea
OSA may play a role in the pathogenesis of chronic hypercapnia in those patients, because in most cases, treatment of OSA corrects the hypercapnia. Indeed, to compensate for the effect of intermittent periodic breathing and resultant acute hypercapnia, normal subjects as well as patients with OSA will increase their tidal volume in the first breath after an apnea (hyperventilation). With physiologic differences in ventilatory responses between subjects, however, an overload in CO2 could result. The duration of the apneas also could contribute: When apneic episode duration becomes three times longer than the breathing interval, CO2 tends to accumulate despite maximal tidal volume, because there is insufficient time for adequate hyperventilation events (Figure 62-1). In addition, hypercapnia could blunt the ventilatory responses: The initial ventilation after an apneic episode is directly related to the volume of CO2 loaded during the preceding respiratory event and thus represents an index of CO2 ventilatory response. Hypercapnic patients demonstrated depression of this index of ventilatory compensation compared with that in eucapnic patients. It has been shown that the apnea-to-interapnea duration ratio is greater in hypercapnic patients than in eucapnic patients.
To elucidate the mechanisms that are involved in the development of hypercapnia in patients with OSA, Norman and co-workers have proposed, using a computer model, some hints for prediction of the transition from acute hypercapnia during sleep-disordered breathing (apnea, hypopnea, and hypoventilation) to chronic daytime hypercapnia. In their model, when the ventilatory CO2 response and renal HCO3− excretion were normal, increases in PaCO2 and HCO3− did not develop. The bicarbonate excretion during the day compensated for that retained during the night. When CO2 ventilatory response was very low, however, the model demonstrated a modest rise in PaCO2 and HCO3− measured during the awake state over multiple days. Similarly, when renal HCO3− excretion rate was lowered to simulate chloride deficiency, the model demonstrated a modest rise in daytime PaCO2 and HCO3−. The combination of low CO2 response and low renal HCO3− excretion rate produced a synergistic effect on the degree of elevation of daytime PaCO2. These workers suggested that hypercapnia results from an imbalance between the period of CO2 loading (short = apnea or hypopnea; long = hypoventilation) and inadequate compensation both during sleep and during the awake state. This pulmonary-renal interaction may contribute to the development and perpetuation of chronic daytime hypercapnia, which will lead to a blunted respiratory drive for the next sleep cycle (Figure 62-2).
