CHAPTER 10 Nonsteroidal Antiinflammatory Drugs
INTRODUCTION
The effective management of acute postoperative pain is important for both humanitarian and psychological reasons. Despite the advances in analgesic agents and delivery systems over recent years, postoperative pain is often undertreated.1,2
The undertreatment of acute postoperative pain has important implications for both patients and the healthcare system. Undertreated postoperative pain prevents sleep, which causes fatigue and delays mobilization.3 It prolongs patient suffering and is associated with a number of undesirable outcomes such as delayed healing, extended hospitalization, and the development of chronic pain conditions and depressive disorders.4–6 Its overall negative effect on patient function increases healthcare utilization and costs.7
Multimodal analgesia is currently recognized as the best postoperative method to reduce pain and analgesia requirements.8 Reduced pain improves recovery, reduces postoperative morbidity, and lowers costs.9–11 The multimodal approach uses a combination of drugs and delivery techniques. For example, the combination of drugs such as opioids, non-specific nonsteroidal antiinflammatory drugs (NSAIDs), cyclooxygenase-2 (COX-2)-specific inhibitors, and local anesthetics may decrease adverse effects and better optimize pain control. Delivery techniques such as patient-controlled analgesia, epidural blocks, and regional blocks are often better at optimizing pain control with fewer adverse effects than single-dose delivery.
Although opioids and non-specific NSAIDs are effective analgesic agents, their use is limited by their adverse effect profiles. Opioids can cause respiratory depression, nausea, vomiting, constipation, pruritis, urinary retention, hypotension, tolerance, and dependency.12 Non-specific NSAIDs can cause gastrointestinal toxicity, renal problems, platelet dysfunction, and bleeding complications, the latter of which may be particularly problematic in the surgical setting.13–15 The adverse effects of non-specific NSAIDs arise as a result of their inhibitory effect on COX-1.16,17 COX-2-specific inhibitors were developed to provide the beneficial antiinflammatory and analgesic effects of non-specific NSAIDs while sparing the protective functions mediated by COX-1.
EPIDEMIOLOGY AND UNDERTREATMENT OF POSTOPERATIVE PAIN
Postoperative pain is one of the most common types of acute pain. Based on data from the National Hospital Discharge Survey, 42.5 million procedures were performed on hospital inpatients during 2002 in the United States alone.18 The number of outpatient procedures is difficult to quantify, but probably exceeds this number.7 Up to 88% of patients may report postoperative pain, and of those, 34–62% experience moderate to severe postoperative pain.1,2
Although postoperative pain is treatable, over the last 50 years its undertreatment has been well documented in the literature.19 The reasons for this are not completely understood, but are probably fostered by the notion that pain is an inevitable consequence of surgery.2,20 Other reasons may include a lack of formal education in pain management, physicians’ concerns regarding potential opioid addiction and drug tolerance, and difficulties in assessing pain severity.21 Furthermore, patients themselves may fail to request or use analgesia because of fears about adverse effects or dependency.
PRACTICE GUIDELINES FOR PAIN MANAGEMENT IN THE PERIOPERATIVE SETTING
Several organizations have published treatment standards for the management of pain. The World Health Organization has produced guidelines for cancer pain relief,22 and the American Pain Society has developed guidelines for the management of acute and cancer pain (Table 10.1).23 The publication of the Joint Commission on Accreditation of Healthcare Organizations’ pain standards has been instrumental in promoting the increased acceptance of pain as the fifth vital sign.24,25
Table 10.1 American Pain Society Quality Improvement Guidelines for the Management of Acute and Cancer Pain23
| Recognize and treat pain promptly |
| Chart and display the patient’s self-report of pain |
| Commit to continue improvement of one or several outcome variables |
| Document outcomes based on data and provide prompt feedback |
| Make information about analgesic drugs readily available |
| Promise the patient attentive analgesic care |
| Define explicit policies for the use of advanced analgesic technologies |
| Examine the process and outcomes of pain management with the goal of continuous improvement |
Reprinted from JAMA, vol. 274, American Pain Society Quality of Care Committee, quality improvement guidelines for the treatment of acute pain and cancer pain, pp. 1874–1888, 1995, with permission from the American Medical Association.
PATHOPHYSIOLOGY OF POSTOPERATIVE PAIN
Nociception and inflammatory pain
Nociceptive input is initiated by tissue damage and prolonged by inflammation. In response to tissue injury, specialized peripheral nociceptors promote the release of inflammatory mediators, neurotransmitters, and growth factors from traumatized tissue and sequestrated intravascular cells.26,27 The resulting sensory input is conveyed by visceral and somatic neurons to the dorsal horns and ascending spinal pathways to the thalamic, limbic, and cortical structures involved in affective and sensory-discriminative responses. Although the sensory input is extensively modulated both peripherally and centrally, the end result is the subjective awareness of pain.28
This inflammatory pain is characterized by hypersensitivity to stimuli both at the site of injury (primary hyperalgesia) and at sites distant to the site of injury (secondary hyperalgesia).26,29 While primary hyperalgesia is the result of peripheral pain mechanisms, secondary hyperalgesia involves central modulatory systems, including central sensitization.29 Fortunately, the hypersensitivity caused by inflammation usually normalizes if the disease process is controlled.26
Peripheral and central sensitization
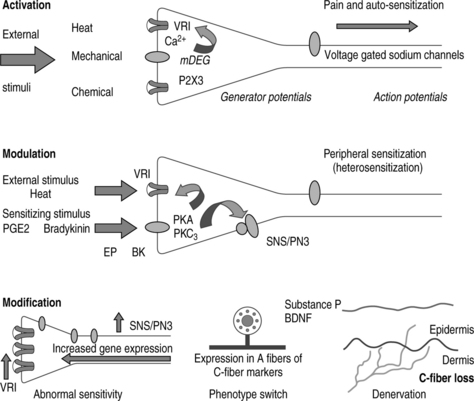
Peripheral sensitization or heterosensitization is caused by exposure of nociceptor terminals to sensitizing agents. Released by damaged tissue or by inflammatory cells, these sensitizing agents include the inflammatory mediators, prostaglandin E2, bradykinin, and serotonin as well as various nerve growth factors.26,30
By activating intracellular signaling cascades, increased peripheral input into the central nervous system increases neuronal synaptic responses and decreases neuronal inhibition. If this amplification of sensory input is prolonged, both nocuous and innocuous neuronal inputs cause activation of spinal cord cells. The result is central sensitization.26,31 Prolonged amplification results in anatomic and neurochemical changes leading to neural plasticity in primary sensory neurons and is categorized as activation, modulation, and modification (Fig. 10.1).26
Prostaglandin production and the expression of COX-1 and COX-2
The synthesis of prostanoids is essential for the generation of inflammatory pain.32 Arachidonic acid is released from phospholipids in the cell membrane by phospholipid A2 enzymes and is converted to prostaglandin G2 and then prostaglandin H2 in a 2-step reaction catalyzed by COX (Fig. 10.2).32 Prostaglandin H2 is converted into the prostaglandin isoforms prostaglandin E2, prostaglandin D2 or prostaglandin F2a, prostacyclin, or thromboxane A2 through the action of tissue-specific isomerases.32,33 Differential expression and induction patterns of these enzymes and prostanoid receptors have important roles in the cellular effects of the prostanoids.
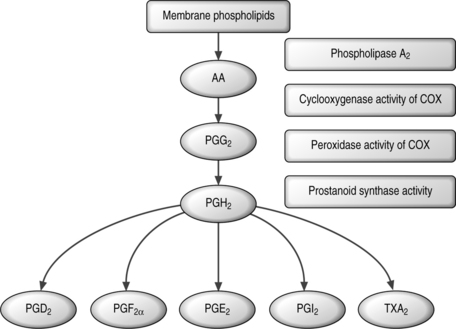
Fig. 10.2 Prostanoid biosynthetic pathway.32
Reprinted from Trends in Molecular Medicine, vol 8, Samad TA, Sapirstein A, and Woolf CJ, Prostanoids and pain: unraveling mechanisms and revealing therapeutic targets, pp. 390–396, 2002, with permission from Elsevier.
COX exists as two isoforms: COX-1, which is responsible for homeostatic prostanoid synthesis, and COX-2, which is responsible for proinflammatory prostanoid production.34 COX-1 is constitutive within platelets and is associated with the production of thromboxane, which strongly promotes platelet aggregation, and both COX-1 and COX-2 are constitutively expressed in the central nervous system, dorsal root ganglia, and kidney.35–38 In addition, COX-2 expression can be induced by several factors, including neurotransmitters and proinflammatory cytokines,39 and the role of inducible COX-2 in central nervous system responses to inflammation is well established. Its expression is upregulated within the central nervous system in response to interleukin-1β, leading to elevations in central prostanoid production.40
Several animal studies have assessed the relative contributions of COX-1 and COX-2 to prostaglandin release in the spinal cord. The selective COX-1 inhibitor SC560 significantly reduced nociceptive behavior and abolished spinal prostaglandin E2 release in a rat model, while celecoxib had no effect on either parameter.41 These findings indicated that nociception-evoked prostaglandin release was primarily caused by COX-1 and was independent of COX-2, and suggested that the efficacy of celecoxib in early injury-evoked pain may be lower than that of non-specific NSAIDs. In contrast, another study in a rat model suggested that COX-2, but not COX-1, mediated antihyperalgesic activity and release of spinal prostaglandin E2.42 In rats with chronic indwelling catheters, acute thermal hyperalgesia evoked by spinal delivery of substance P or N-methyl-D-aspartate and thermal hyperalgesia induced by carrageenan was suppressed by intrathecal and systemic COX-2 inhibitors, while systemic but not spinal administration of a COX-1 inhibitor reduced carrageenan-evoked thermal hyperalgesia, and neither systemic nor spinal COX-1 inhibition had an effect on spinal hyperalgesia evoked by substance P. Intrathecal substance P enhanced prostaglandin E2 release, and this effect was diminished by systemic delivery of non-specific NSAIDs and COX-2-specific inhibitors, but not by a COX-1-specific inhibitor. Therefore, constitutive spinal COX-2, but not COX-1, mediates acute antihyperalgesic effects of both spinal and systemic COX-2 inhibitors.
More recently, inhibition of constitutively expressed spinal COX-2 immediately following peripheral tissue injury has been found to reduce injury-induced activation of primary afferent neurons and spinal neurons and mechanical and thermal hyperalgesia prior to any measurable upregulation of COX-2 protein.36 This early research has suggested that constitutive spinal COX-2 may play a role in the development of hyperalgesia following peripheral tissue injury, and that blockade of this constitutive expression prior to injury may lessen the peripheral and central sensitization occurring after tissue injury. Considering the intrathecal potency of COX-2 inhibitors, the comparable efficacy of intrathecal and systemic COX-2 inhibitors in hyperalgesic states not associated with inflammation, and the onset of antihyperalgesic activity prior to COX-2 upregulation, it has been hypothesized that modulation of constitutive spinal COX-2 is a principal antihyperalgesic mechanism of COX-2 inhibitors.43
Role of COX-2 in peripheral sensitization
Inflammation induces the expression of COX-2, which in turn promotes the release of prostanoids. The resulting sensitization of peripheral nociceptor terminals causes primary hyperalgesia. Secondary hyperalgesia due to increased neuronal excitability in the spinal cord causes central sensitization.26,29
Role of COX-2 in central sensitization
Inflammation induces COX-2 expression in spinal cord neurons and other regions of the central nervous system. This widespread induction causes an 80-fold increase in the levels of prostaglandin E2 in the cerebrospinal fluid.40 The major inducer of COX-2 upregulation is interleukin-1β,44,45 and its role in the regulation of central prostanoid production was confirmed by intraspinal administration of an interleukin-converting enzyme or COX-2-specific inhibitor. Intraspinal injection resulted in decreased inflammation-induced central prostaglandin E2 levels and reduced centrally generated inflammatory pain hypersensitivity.40
These experimental findings were confirmed in a study in 30 patients undergoing thoracotomy for lobectomy who were randomized to receive nimesulide 100 mg b.i.d. or ibuprofen 400 mg t.i.d.46 Cerebrospinal fluid samples were analyzed for 6-keto-prostaglandin F1 α, the principle metabolite of prostacyclin. COX-1 and COX-2 activity was determined by measuring serum thromboxane B2 and endotoxin-induced prostaglandin E2 generation in whole blood. Nimesulide was found to be selective for COX-2, while ibuprofen was COX nonselective. Mean levels of 6-keto-prostaglandin F1 α in the cerebrospinal fluid increased following surgery and were significantly suppressed by nimesulide, but not by ibuprofen. Patients receiving nimesulide also had significantly lower pain scores (p<;0.001), morphine requirement (p=0.0175), and falls in peak expiratory flow rate (p<;0.001). This study demonstrated that increases in spinal prostaglandin synthesis after thoracotomy were repressed by a COX-2-selective agent.
PHARMACOLOGIC AGENTS FOR MANAGEMENT OF POSTOPERATIVE PAIN
Simple analgesics
Simple analgesics such as acetaminophen are a common monotherapy for treating mild to moderate pain. While acetaminophen can effectively treat pain of relatively low intensity, it does not provide any therapeutic benefit to an underlying inflammatory process. Acetaminophen is useful for aspirin-sensitive asthmatics and individuals at risk of gastrointestinal complications in whom non-specific NSAIDs are contraindicated.47 Although acetaminophen is routinely used in combination with opioids such as codeine, tramadol, hydrocodone, and oxycodone to reduce opioid use,48 this combination can result in more adverse effects than acetaminophen alone.
In adults, acetaminophen is administered to a maximum daily dose of 500 mg, although this should be reduced in patients with a history of heavy alcohol intake. Long-term use in combination with alcohol can be complicated by severe and sometimes fatal liver damage.47
Opioids
Opioids are highly effective in the management of severe, acute postoperative pain. The agonist or ‘pure’ morphine-like drugs range in potency from ‘less potent’ agents such as oxycodone, which is suitable for moderate pain, to potent drugs such as fentanyl, which is often used intraoperatively. There are also drugs such as buprenorphine, which possess both agonist and antagonist actions. Although the agonist–antagonist opioids are the least effective opioid type, they have the lowest risk of adverse events and typically show a ceiling effect in both analgesic activity and effect on respiratory depression.6,12 Finally, there are opioid antagonists such a naloxone that are pure antagonists without any analgesic effect.6
Although effective, opioid use is associated with multiple adverse effects, including sedation, hypotension, respiratory depression, emesis, paralytic ileus, urinary retention, pruritis, and dependence.49–51 These effects can complicate and lengthen the postoperative recovery period, and concerns about poor tolerability and perceived dependency can result in inadequate dosing. Controlling the variability in plasma concentration by the use of patient-assisted devices may reduce these adverse effects.
Even though opioids can provide a sustained analgesic effect and improved function in selected, well-monitored patients without the development of tolerance or significant toxicity,12 the use of adjunctive agents in combination with opioids is still often desirable. The synergistic coadministration of other analgesics reduces opioid doses and decreases adverse effects. In addition, opioids are ineffective when used alone for the management of movement-related pain, and supplementation with other types of medication is needed during postoperative mobilization.
Non-specific NSAIDs
Non-specific NSAIDs are often effective for mild to moderate pain following outpatient surgery,52 although their analgesic efficacy and adverse effects show marked variation among individuals. Non-specific NSAIDs may also be used as adjuncts to opioids for the management of more severe postoperative pain.
Monotherapy
Ketorolac tromethamine is the only parenteral non-specific NSAID approved for use in the United States and is the current standard for comparison. Although ketorolac is useful in the management of postoperative pain, it is a reversible inhibitor of platelet aggregation.53,54 Theoretically, platelet inhibitors should increase the risk of postoperative bleeding. However, a postmarketing surveillance study of patients receiving 10 272 courses of parenteral ketorolac therapy reported only a small increase in operative-site bleeding, which was limited to elderly patients and those receiving high-dose ketorolac or more than 5 days of treatment.55 Other studies, however, have reported an increase in operative and postoperative bleeding,56–59 including cases of severe and near-fatal postoperative bleeding.59 Studies in healthy volunteers have found single-dose ketorolac to prolong bleeding time significantly,60 with one study noting a 50% increase in bleeding time 4 hours after a single intramuscular dose.61 Consequently, ketorolac is not routinely used before surgery in which operative and postoperative bleeding may be problematic.
Ketoprofen is a commonly used non-specific NSAID that provides effective analgesia for mild to moderate pain. It is also used concurrently with opioids in the treatment of severe pain, and several studies have investigated its efficacy as a monotherapy for postoperative pain relief. A study in patients with moderate or severe pain following dental surgery reported that ketoprofen provided a superior level of meaningful pain relief at 6 hours compared with liquigel ibuprofen and acetaminophen. Ibuprofen, however, provided faster relief and superior overall efficacy.62 In a further study following dental surgery, ketoprofen demonstrated lower pain intensity from 2 to 6 hours after first drug intake and decreased swelling compared with acetaminophen.63
Following minor pediatric surgery, ketoprofen had an earlier onset and longer duration of analgesia than acetaminophen.64 Although the data were from too small a study to assess the effects of ketoprofen on hemorrhage rate, an anesthetic induction dose of ketoprofen in patients undergoing adenoidectomy did not increase the rate of blood loss.65
A meta-analysis of 7 randomized, double-blind tonsillectomy trials (n=505) showed that postoperative use of non-specific NSAIDs, such as ketorolac, ibuprofen, or ketoprofen, increased both the risk of postoperative bleeding requiring treatment and the risk of reoperation for hemostasis.66 The authors of the meta-analaysis searched PubMed® and the Cochrane Controlled Trials Register for randomized, double-blind studies published between January 1966 and May 2001. Trials were required to meet quality criteria for inclusion and were selected if they reported data on postoperative bleeding in patients treated with non-specific NSAIDs after tonsillectomy with or without adenoidectomy. In the 7 trials combined in the meta-analysis, the incidence of postoperative bleeding treated medically or surgically was 7.3%. The most common consequence of this bleeding was admission to the emergency department.
Adjunctive therapy
In the early postoperative period, a combination of opioids and non-specific NSAIDs can improve analgesic efficacy compared with either agent alone.12 The combined use of ketoprofen and opioids in patients with severe pain lowers opioid doses and reduces the incidence of opioid-related adverse events.67 Other studies have also shown that combination therapy lowers opioid requirements and decreases opioid-induced nausea, constipation, somnolence, and respiratory depression.52 The resulting increase in gastrointestinal motility may permit an earlier return to oral nutrition.68 Although the use of ketorolac and diclofenac has been reported to reduce morphine consumption by 30–40% following abdominal and orthopedic surgery,69,70 non-specific NSAIDs increase the risk of postoperative bleeding.13 For this reason, non-specific NSAIDs should probably not be used following hip arthroplasty. In addition, the common use of anticoagulation therapy with heparin following total knee surgery to prevent deep vein thrombosis also increases the risk of postoperative bleeding.
A study in patients receiving short-term ketorolac infusion following laparoscopic day surgery reported that, although ketorolac had an opioid-sparing effect over 36 hours of postoperative administration, it was not associated with continued benefits after this time.71 Although patients required significantly less fentanyl in the recovery ward and significantly less codeine prior to and following discharge, patients’ description of discomfort on performing common activities did not indicate any beneficial effect of ketorolac.
Adverse effects of non-specific NSAIDs
Although non-specific NSAIDs do not share many of the adverse effects associated with opioid use, non-specific COX inhibition can cause gastrointestinal ulceration and bleeding, platelet dysfunction, prolonged bleeding time, elevated blood pressure, renal toxicity, and hepatic dysfunction.13–15,55 These events may range from mild to potentially life-threatening complications.
The gastrointestinal adverse effects of non-specific NSAIDs are caused by inhibition of COX-1. In the gastrointestinal tract, COX-1 produces prostaglandin E2, which has a protective effect on the gastrointestinal mucosa by limiting acid damage and promoting mucus secretion, bicarbonate release, and adequate blood flow.16 The resulting gastrointestinal damage caused by COX-1 inhibition increases the incidence of ulceration, perforation, and potentially fatal gastrointestinal bleeding.15
Non-specific NSAIDs also inhibit normal platelet function and aggregation. Inhibition of COX-1 reduces platelet aggregation by blocking the production of thromboxane A2. Because COX-2 normally inhibits prostacyclin, a protein that increases platelet aggregation, normal platelet aggregation is further reduced.60
Although prostaglandins do not play a major role in the maintenance of basal renal function under normal physiologic conditions, prostaglandins produced by constitutive renal COX-1 and COX-2 may be vital to the maintenance of renal perfusion and glomerular filtration rate.72,73 In patients with compromised renal hemodynamics, such as the elderly or those with preexisting renal dysfunction or congestive heart failure, NSAID-mediated COX-1 inhibition can result in acute renal failure.74
COX-2-specific inhibitors
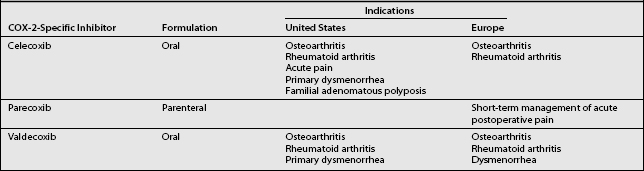
Because of the many adverse effects associated with opioids and non-specific NSAIDs, alternative analgesic agents are needed. The antiinflammatory and analgesic benefits of NSAIDs are due to the inhibition of COX-2 at the site of inflammation.34,39,75 The belief that selective inhibition of COX-2 would produce the beneficial effects of NSAIDs, but avoid disruption of COX-1-mediated protective functions, led to the development of COX-2-specific inhibitors. The indications for the currently available COX-2-specific inhibitors in the United States and Europe are presented in Table 10.2.
Table 10.2 Indications for Currently Available COX-2-Specific Inhibitors in the United States and Europe
Analgesic efficacy of COX-2-specific inhibitors
Studies of COX-2-specific inhibitors in the perioperative setting demonstrate that they provide effective analgesia while reducing opioid consumption in the range of 20–70%.7
Parenteral COX-2-specific inhibitors
The analgesic efficacy and opioid-sparing effects of parecoxib have been demonstrated in a number of studies of postoperative pain (Table 10.3). Following total hip arthroplasty, the use of parecoxib 20 mg or 40 mg significantly reduced the total amount of morphine used over 36 hours compared with placebo (p<0.01).76 Patients treated with parecoxib also reported significantly greater maximum pain relief and earlier discontinuation of morphine therapy. Parecoxib 20 mg or 40 mg has also demonstrated opioid-sparing effects and significant analgesic efficacy compared with morphine alone (p<0.05) following total knee replacement surgery.77 Another study compared parecoxib 20 mg and 40 mg, morphine 4 mg, and ketorolac 30 mg following unilateral total knee replacement surgery. Although the onset of analgesia in all groups was similar, the level and duration of analgesia with parecoxib 40 mg was significantly superior to morphine 4 mg.78
Table 10.3 Studies Investigating the Use of Parecoxib in the Management of Preoperative and Postoperative Pain
| Study | Treatment and Model | Results |
|---|---|---|
| Barton et al.81 |
Single-dose parecoxib 20 mg and 40 mg i.m. and i.v. have been compared with ketorolac 60 mg following oral surgery.79 The analgesic efficacy of both parecoxib dosing regimens was comparable, but time to use of rescue medication was longer following intramuscular administration. Parecoxib 40 mg was found to be comparable to ketorolac in most measures of analgesia, but had a longer duration of action. In another oral surgery study, single-dose parecoxib 20–100 mg and ketorolac 30 mg demonstrated a similarly rapid onset of analgesia (within 11 minutes) and comparable analgesic efficacy.80 Dose-proportional increases in the duration of analgesia were observed with parecoxib 50 mg and 100 mg, showing a significantly longer duration of analgesia than ketorolac.
Following gynecologic laparotomy surgery, parecoxib 20 mg or 40 mg demonstrated analgesic efficacy similar to that of intravenous ketorolac 30 mg and a similarly rapid time to onset of analgesia (10–23 min).81 Parecoxib also demonstrated similar analgesic efficacy and a longer duration of action than morphine 12 mg i.m. in this model.82 The use of preoperative single-dose parecoxib 40 mg followed by a single oral dose of valdecoxib 40 mg following laparoscopic cholecystectomy confirmed the opioid-sparing effects and analgesic efficacy of parecoxib compared with placebo.83
Parecoxib was well tolerated in all of these trials,77,78,80,82 and the adverse events reported in these studies (such as nausea and dizziness) are typical of those seen after surgery.84 Importantly, parecoxib showed no evidence of treatment-related serious adverse events such as wound bleeding, renal dysfunction, or upper gastrointestinal bleeding.78
Oral COX-2-specific inhibitors
The efficacy of the oral COX-2-specific inhibitors celecoxib and valdecoxib for the treatment of preoperative and postoperative pain is summarized in Table 10.4.
Table 10.4 Studies Investigating the use of Oral COX-2-Specific Inhibitors in the Management of Preoperative and Postoperative Pain
| Study | Treatment and Model | Results |
|---|---|---|
| Celecoxib | ||
| Gimbel et al.85 | ||
For treating moderate to severe pain following outpatient orthopedic surgery, multiple dosing with celecoxib 200 mg is significantly more effective and better tolerated than hydrocodone 10 mg/acetaminophen 100 mg.85 The use of celecoxib preoperatively in combination with other analgesics has also been reported to provide improved analgesic efficacy compared with single-agent therapy. Premedication with celecoxib 200 mg and acetaminophen 2000 mg significantly reduced the incidence of severe pain following otolaryngologic surgery compared with either agent alone.86 Patient satisfaction with treatment was also significantly higher with this combination than with acetaminophen alone.
A study in 60 patients undergoing spinal stabilization demonstrated significant opioid-sparing effects following preoperative administration of celecoxib 200 mg or rofecoxib 50 mg compared with placebo.87 Although analgesic efficacy was similar in both groups, rofecoxib had an extended analgesic effect.
Valdecoxib has demonstrated rapid analgesic efficacy with a long duration of action in various models of surgical pain. In addition, valdecoxib does not affect platelet aggregation and has fewer undesirable effects on the gastrointestinal mucosa compared with non-specific NSAIDs.88–91 In a comparative study in patients following oral surgery, valdecoxib 40 mg showed a significantly shorter time to perceptible pain relief and a faster time to onset of analgesia compared with rofecoxib 50 mg.92 Valdecoxib also provided significant pain relief 45 minutes after dosing, lasting up to 24 hours. A separate study also found that single-dose valdecoxib 40 mg had a significantly faster onset of action compared with rofecoxib 50 mg following oral surgery (p=0.05), although the magnitude and duration of analgesia were similar.93 In two other studies following oral surgery, the analgesic efficacy and tolerability of single-dose valdecoxib 20 mg or 40 mg showed a similar time to onset of analgesia and peak analgesic effect compared with oxycodone 10 mg/acetaminophen 1000 mg.94 However, both valdecoxib doses had a significantly longer duration of analgesia and superior tolerability compared with oxycodone/acetaminophen.
Valdecoxib has demonstrated opioid-sparing efficacy as part of multimodal treatment following hip arthroplasty.95 Patients receiving valdecoxib 20 mg or 40 mg b.i.d. preoperatively and postoperatively required less morphine than those receiving placebo, and experienced greater analgesic efficacy compared with morphine alone. Confirming previous findings of its lack of effect on platelet function and bleeding time,90,91 both valdecoxib doses were well tolerated and were not associated with an increase in bleeding episodes. Similar findings were reported in a study following total knee arthroplasty, in which morphine consumption was lower in patients receiving valdecoxib 40 mg or 80 mg daily compared with morphine alone.96 Valdecoxib-treated patients also experienced significantly lower maximum pain intensity and rated their study medication significantly higher than did the morphine group.
Although the opioid-sparing efficacy of COX-2-specific inhibitors should reduce the incidence of opioid-related adverse events in postoperative patients, studies of the postoperative use of COX-2-specific inhibitors in addition to standard-of-care opioids have not shown a consistent reduction in opioid-related adverse effects. These trials may have had a low sensitivity for assessing adverse events because they relied on investigator reports. A recent patient-reported health outcome analysis, based on a randomized, controlled, double-blind trial of patients after ambulatory laparoscopic cholecystectomy, allowed patients to self-assess their adverse events using the Symptom Distress Scale (SDS) questionnaire.97 Using the questionnaire, patients assessed 12 opioid-related symptoms by 3 ordinal measures: frequency, severity, and bothersomeness. The outcome analysis showed a significant dose–response relationship between postoperative opioid dose and opioid-related adverse effects.97 Furthermore, once postoperative opioid dose had reached a threshold, approximately every 3–4 mg increase in the morphine equivalent dose resulted in an additional opioid-related clinically meaningful event. These outcome results highlight the potential benefits of reducing postoperative opioid consumption using multimodal treatment with COX-2-specific inhibitors.
Potential drug–drug interactions in the surgical setting
Valdecoxib is primarily metabolized by hepatic cytochrome P450 (CYP) 3A4 and to a lesser degree by CYP2C9.98 Therefore, parecoxib (the prodrug of valdecoxib) may interact with other CYP3A4 substrates, so the potential for parecoxib to interact with medications commonly used in surgery has been investigated.
Frequently used in the perioperative setting, midazolam is a CYP3A4 substrate whose systemic clearance, magnitude, and duration of effect are highly susceptible to CYP3A4 drug interactions.99 Single-bolus parecoxib has not shown any effects on the plasma concentration, pharmacokinetics, or pharmacodynamics of midazolam when administered at doses expected to be used perioperatively.100 Similarly, bolus parecoxib has not altered the disposition or clinical effects of the opioid analgesics fentanyl or alfentanil, whose systemic clearance is affected by alterations in CYP3A4 activity.101 Parecoxib has also been found to have no effects on the plasma concentration, pharmacokinetics, or pharmacodynamics of propofol, which is the only commonly used anesthetic that undergoes significant metabolism by CYP2C9.102
COX-2-specific inhibitors and platelet function
Ketorolac has been associated with an increased risk of gastrointestinal and operative-site bleeding in patients aged 75 years or older and in patients receiving either higher doses of ketorolac or treatment lasting for more than 5 days.55,103 The effects of parecoxib and ketorolac on platelet function and bleeding time have been compared in healthy elderly (aged 65–95 years) and nonelderly subjects (aged 18–55 years).104 Parecoxib had little or no effect on arachidonate-induced platelet aggregation in either study population, while ketorolac demonstrated significant and sustained decreases in platelet aggregation throughout the drug administration period compared with parecoxib and placebo. A high degree of variability was noted in bleeding times in all treatment groups, but significant prolongation was recorded with ketorolac in both study groups. Parecoxib had no effect on serum thromboxane B2 levels in nonelderly subjects, but ketorolac resulted in marked reductions that were significantly greater than parecoxib and placebo at all time points (p<0.001). In elderly subjects, ketorolac significantly reduced serum thromboxane B2 levels (p=0.017 vs. placebo) versus parecoxib. The lack of effects on platelet aggregation and bleeding time recorded with parecoxib in this study suggest that, irrespective of age, parecoxib is less likely to be associated with increased bleeding during surgery than ketorolac and should be a better choice for treating postsurgical pain.
Similar findings have been noted with valdecoxib, which has demonstrated a lack of effect on platelet aggregation and bleeding time even when used at supratherapeutic doses.90,91 A study in healthy volunteers also found supratherapeutic doses of celecoxib (600 mg b.i.d.) had no effect on the normal mechanisms of platelet aggregation and hemostasis.105
Gastrointestinal tolerability of COX-2-specific inhibitors
Endoscopic studies have reported a 70–75% reduced risk of ulceration at 6 months for COX-2-specific inhibitors compared with non-specific NSAIDs, and an incidence of ulceration similar to placebo.106,107 Two large, randomized, double-blind outcomes studies investigated the risk of clinically important upper gastrointestinal events associated with the COX-2-specific inhibitor celecoxib. The CLASS study (Celecoxib Long-term Arthritis Safety Study) compared the gastrointestinal toxicity of celecoxib, ibuprofen, and diclofenac in patients with osteoarthritis and rheumatoid arthritis.108 The CLASS trial failed to show a statistically significant difference in the primary endpoint, the annualized incidence of upper gastrointestinal ulcers complications, between patients treated with celecoxib 400 mg b.i.d. and those treated with ibuprofen or diclofenac. The failure to show a difference was probably influenced by study design and methodology for evaluating gastrointestinal outcomes, unexpectedly high dropout rates, and concurrent use of low-dose aspirin. The second trial, SUCCESS-1 (SUCcessive Celecoxib Efficacy and Safety Study-1), was the largest, multinational, ‘real-world,’ randomized, controlled clinical trial of a COX-2-specific inhibitor.109 The SUCCESS-1 study showed that patients treated with celecoxib statistically had a significantly lower incidence of upper gastrointestinal complications compared with patients treated with naproxen or diclofenac (p=0.008). Several other observational studies and pooled analyses have also demonstrated the gastrointestinal safety profile of celecoxib.110,111
Studies with the newer COX-2-specific inhibitor valdecoxib have also noted a superior upper gastrointestinal safety profile compared with non-specific NSAIDs in healthy subjects.89 In addition, in patients with osteoarthritis, valdecoxib was associated with a significantly lower incidence of endoscopically detectable gastroduodenal ulcers compared with ibuprofen and diclofenac.112
The development of parecoxib has provided a COX-2-specific inhibitor that combines the analgesic efficacy of NSAIDs with an improved tolerability profile in a parenteral formulation. The gastrointestinal safety and tolerability of parecoxib, ketorolac, and naproxen over 7 days were compared in a placebo-controlled endoscopic study in 17 healthy elderly subjects who were at increased risk of upper gastrointestinal ulcer complications.113 The rate of ulcers was lower with parecoxib (0/4 subjects) and placebo (2/5 subjects) compared with ketorolac (4/4 subjects) and naproxen (2/4 subjects). Because of the unexpectedly high incidence of gastroduodenal ulceration in patients taking the non-specific NSAIDs, even after short-term treatment, the study was terminated early and the randomization blind broken.
A larger study compared the upper gastrointestinal effects of parecoxib, ketorolac, and placebo in healthy elderly subjects aged 65–75 years.114 The use of parecoxib or placebo was not associated with the development of gastric or duodenal lesions, while 22.6% of ketorolac-treated patients developed at least one ulcer within 5 days of administration (16.1% had gastric ulcers and 6.4% had duodenal ulcers; p<0.05 vs. parecoxib). The incidence of an ulcer or at least one erosion in the stomach was 90.3% with ketorolac, 13.8% with parecoxib, and 6.3% with placebo, while the incidence of duodenal ulcers/erosions was 45.2%, 10.3%, and 0%, respectively. This study thus confirms that parecoxib has a significantly reduced incidence of gastroduodenal mucosal injury compared with ketorolac.
Lastly, in a multicenter, randomized, double-blind, controlled study of 123 adults with endoscopically confirmed normal upper gastrointestinal mucosae at baseline, treatment for 7 days with parecoxib 40 mg b.i.d. had a gastrointestinal ulceration rate that was comparable to placebo and superior to ketorolac 30 mg 4 times daily for 5 days.115 During the 7-day study, no subjects treated with parecoxib or placebo developed gastroduodenal ulcers or ≥11 erosions/ulcers. Parecoxib was no more likely than placebo to increase the incidence of erosions or ulcers (12% vs. 7%, p=0.419). In contrast, in the ketorolac group, 11 (28%) subjects developed ulcers, 19 (48%) subjects developed ≥11 gastroduodenal erosions/ulcers, and the rate of combined ulcers/erosions was 85% (p<0.001 vs. placebo and parecoxib).
Renal effects of COX-2-specific inhibitors
Inhibition of renal COX-1 and COX-2 by non-specific NSAIDs inhibits prostaglandin-mediated renal function, including renal vascular tone and electrolyte and water excretion.116–118 These effects frequently cause edema and reduced blood pressure control in treated patients with hypertension. Therefore, since COX-2-specific inhibitors also spare inhibition of renal COX-1, it was hoped that they may have decreased rates of renovascular events compared with non-specific NSAIDs. However, subsequent recognition of the constitutive expression of COX-2 in the human kidney,37 coupled with the profound effects of prostaglandins on renal homoeostasis,72 indicate that COX-2-specific inhibitors are likely to share the nephrotoxic potential of non-specific NSAIDs.
In a historical cohort observational analysis of patients with stabilized hypertension, celecoxib was associated with a significantly lower incidence of outpatient blood pressure destabilization (2.27 per 1000 patient-days) compared with non-specific NSAIDs (ibuprofen, diclofenac, naproxen: 2.65 per 1000 patient-days; p<0.001) or rofecoxib (2.66 per 1000 patient-days; p<0.001).119 In two randomized trials, renal adverse events associated with 6 weeks’ treatment with celecoxib 200 mg q.d. or rofecoxib 25 mg q.d. were compared in 1900 elderly osteoarthritis patients taking antihypertensive agents.120,121 Celecoxib treatment was associated with less edema and blood pressure destabilization compared with rofecoxib.
In a long-term outcome study, rates of hypertension and edema in 8538 patients taking a non-specific NSAID, rofecoxib, or celecoxib during the previous 6 months were compared to patients not taking these agents.122 Compared with nonusers, patients treated with rofecoxib had increased rates of edema and hypertension. In contrast, patients taking celecoxib or non-specific NSAIDs had similar rates of edema and hypertension to the nonusers. Taken together, these studies suggest that the renal effects of COX-2-specific inhibitors may be molecule-specific rather than class-specific.
Furthermore, in a pooled analysis of 9 arthritis trials of 6–26 weeks duration (including 7 placebo controlled), chronic daily treatment with therapeutic doses of valdecoxib was associated with similar rates of hypertension and edema compared with naproxen, diclofenac, or ibuprofen.123
In terms of their effects of renal function, both non-specific NSAIDs and COX-2-specific inhibitors are generally safe when used in healthy subjects. However, in situations where renal hemodynamics are compromised, such as in the elderly or those with preexisting renal dysfunction or congestive heart failure, it is recommended that non-specific NSAIDs and COX-2-specific inhibitors should be used with caution.124
Potential cardiovascular complications of COX-2-specific inhibitors
In September 2004, rofecoxib was withdrawn from the market following an interim analysis of cardiovascular safety data from a prospective, randomized trial that showed an increased relative risk for confirmed cardiovascular events with rofecoxib versus placebo. The Adenomatous Polyp Prevention On Vioxx (APPROVe) trial was designed to evaluate the efficacy of rofecoxib 25 mg in preventing the recurrence of colorectal polyps in patients with a history of the disease.125 Only a minority of these patients had a known history of coronary artery disease. Preliminary data revealed that there was an approximate twofold increase in the rate of cardiovascular events with rofecoxib (46 events per 3059 patient years [1.5 events per 100 patient-years]) compared with placebo (26 events per 3327 patient years [0.78 events per 100 patient-years; p=0.008]). This difference did not become statistically significant until after 18 months of treatment.
Earlier studies had also suggested an increased risk of cardiovascular events in patients taking rofecoxib.126–128 The Vioxx Gastrointestinal Outcomes Research (VIGOR) trial showed that patients taking rofecoxib 50 mg had a fourfold increased risk of acute myocardial infarction compared with naproxen 500 mg twice daily.126 This was initially attributed to a potential cardioprotective effect of naproxen rather than a cardiotoxic effect of rofecoxib. However, a meta-analysis of 18 randomized controlled trials and 11 observational studies demonstrated that the cardioprotective effect of naproxen is small and could not have explained the findings of the VIGOR trial.129 The putative cardioprotective effect of naproxen has been further called into question since preliminary data from the Alzheimer Disease Antiinflammatory Prevention Trial (ADAPT; n=2400) demonstrated an increased risk of cardiovascular events in patients taking naproxen versus placebo.130 Because of this finding, the ADAPT study was prematurely halted; however, the events have not been adjudicated, and further evaluation is needed before conclusions about naproxen can be reached.
In contrast to the data for rofecoxib, numerous randomized, controlled trials and observational studies have found no elevated risk of cardiovascular events with celecoxib. Data from both the SUCCESS-1 and CLASS trials showed that there was no increased risk of acute myocardial infarction with celecoxib treatment,108,109 and the ADAPT trial described above also failed to find an increased risk for cardiovascular events for celecoxib.130 Furthermore, several other observational studies have shown a greater risk for cardiovascular events with rofecoxib than celecoxib. For example, Solomon et al. showed a statistically significantly higher rate of myocardial infarction in patients treated with rofecoxib compared with those treated with celecoxib.128 A dose–response relationship was also observed with rofecoxib, such that higher doses of rofecoxib were associated with a greater rate of myocardial infarction; no such relationship was seen with celecoxib.128 Similarly, Ray et al. demonstrated an association of myocardial infarction with high-dose rofecoxib, but not celecoxib treatment; new users of high-dose rofecoxib had a 2.2-fold greater risk of developing serious coronary heart disease compared with patients taking celecoxib.127 Two recent case-control studies have also shown a significantly higher risk of cardiovascular events with rofecoxib than with celecoxib.131,132
The only instance of a potential increase in cardiovascular events with celecoxib was initially reported in December 2004. Following the observations of an increased incidence of cardiovascular events with rofecoxib from the APPROVe trial, the data and safety monitoring board of a similar trial for celecoxib, the Adenoma Prevention with Celecoxib (APC) trial (n=2035), requested a similar assessment of cardiovascular data. This analysis found that celecoxib was associated with a dose-related increase in the risk of serious cardiovascular events, including death from cardiovascular causes, myocardial infarction, stroke, and heart failure.133 However, preliminary analyses from the Prevention of Spontaneous Adenomatous Polyps (PreSAP) trial, conducted in parallel to APC for the same indication, has not shown any increased cardiovascular risk for celecoxib.133
To date, there is no evidence of an increased cardiovascular risk with valdecoxib in patients with osteoarthritis or rheumatoid arthritis. A retrospective pooled analysis of four randomized, placebo-controlled trials of up to 3 months’ duration in patients with rheumatoid arthritis demonstrated that valdecoxib 10–80 mg daily (n=1945) was associated with a similar incidence of serious thrombotic events compared with naproxen 500 mg twice daily (n=744) or placebo (n=529).123 Similarly, a pooled analysis of 10 randomized, placebo-controlled trials in patients with osteoarthritis and rheumatoid arthritis (n=7934) also showed similar incidences of thrombotic events in patients taking valdecoxib, non-specific NSAIDs (e.g. naproxen, diclofenac, and ibuprofen) or placebo.134 In two studies of the investigational use of parecoxib/valdecoxib for postoperative pain relief following coronary artery bypass graft (CABG) surgery, a significantly greater incidence of cardiovascular/thromboembolic events was detected in the parecoxib/valdecoxib treatment group compared with the placebo treatment group, prompting the recent contraindication of valdecoxib in the treatment of postoperative pain immediately following CABG surgery,135,136 but this risk has not been seen in any other population.
Taken together, these data suggest that there may be a true difference in the cardiovascular risk profiles of different COX-2-specific inhibitors, with rofecoxib posing a higher risk than either celecoxib or valdecoxib. Possible mechanisms by which rofecoxib may increase the risk for cardiovascular events are not completely understood, but may include an increase in hypertension, thrombosis, and reduced vascular reactivity.132,137 Clearly, further studies are needed to explain the precise mechanism(s) involved in the cardiovascular safety profiles of COX-2-specific inhibitors and to evaluate whether true differences exist.
NON-SPECIFIC NSAIDs AND COX-2-SPECIFIC INHIBITORS: EFFECTS ON BONE HEALING AND SPINAL FUSION
Prostaglandins play an important role in bone healing, which is initiated by an inflammatory phase that involves a number of cytokines, growth factors, and arachidonic acid metabolites.138 Bone repair is mediated by two cell types: osteoblasts that are responsible for bone renewal, and osteoclasts that are involved in bone resorption. Both of these cell types are influenced by a range of mediators, which also influence the production of prostaglandins by bone. Osteoblasts produce prostaglandins that act locally to modulate bone metabolism, while prostaglandins produced by local tissues and inflammatory cells may influence skeletal tissues.139 The production of prostaglandins is also stimulated by weight bearing.140
Because of their effects on prostaglandin formation, non-specific NSAIDs and COX-2-specific inhibitors might be expected to affect bone healing. Although non-specific NSAIDs do not appear to alter normal bone homeostasis, they do affect osteogenesis during bone repair. COX-2 may potentially affect osteoclast and osteoblast function as well as angiogenesis, which may affect the delivery of osteoclasts and osteoblasts.139
Effect of non-specific NSAIDs on bone healing
Non-specific NSAIDs have been shown to inhibit fracture healing in human and animal models (Table 10.5). A variety of factors have been implicated in this phenomenon, including inhibition of bone-forming cells at endosteal bone surfaces, reductions in immune and inflammatory responses, and inhibition of prostaglandin synthesis.141,142
Table 10.5 Studies of Non-Specific NSAIDs and COX-2-Specific Inhibitors on Bone Healing
| Study | Treatment and Model | Results |
|---|---|---|
| NSAIDs | ||
| Dimar et al.144 | ||
In a rat model of spinal fusion, a statistically lower level of fusion was achieved with administration of indometacin than with saline (p<0.001).142 The results of this study led the authors to question the widespread use of non-specific NSAIDs in the postoperative period after spinal fusion.
A retrospective review of 288 patients who underwent an instrumented spinal fusion found that ketorolac had a significant adverse effect on fusion (p>0.001), with ketorolac-treated patients being five times more likely to experience nonunion.143 Smoking also significantly decreased the fusion rate (p>0.01), with smokers being 2.8 times more likely to develop nonunion. This study reported that non-specific NSAIDs significantly inhibited spinal fusion at doses typically used for postoperative pain control, and recommended that they should be avoided in the early postoperative period. In a similar retrospective study, 83 patients with isthmic spondylolisthesis who underwent decompressive surgery combined with posterolateral spine fusion and who took non-specific NSAIDs for more than 3 months postoperatively experienced a significantly lower spinal fusion rate (44%) compared with overall single-level (82%) and two-level fusion rates (74%).144 A further retrospective study of 32 patients with nonunion of a fracture of the diaphysis of the femur and 67 comparable patients with united fractures found a marked association between nonunion and the use of non-specific NSAIDs after injury (p=0.000001).145 Delayed healing was also noted in those patients who took non-specific NSAIDs and whose fractures had subsequently united.
Effect of COX-2-specific inhibitors on bone healing
Several animal studies have also examined the effects of COX-2-specific inhibitors on bone healing (see Table 10.5). In a rat model of femoral fracture, a significantly higher incidence of nonunions was observed with rofecoxib 8 mg/kg compared with ibuprofen 30 mg/kg (p<0.007) and controls (p<0.0001).146 Ibuprofen-treated animals also showed reduced healing. A study using a model of osteointegration in rabbits reported that bone formation was significantly suppressed by naproxen 110 mg/kg/d (p=0.031) and rofecoxib 12.5 mg/d (p=0.035) compared with controls.147 A rat model of femoral fracture investigating the effects of celecoxib 4 mg/kg, rofecoxib 3 mg/kg, and indometacin 1 mg/kg for 8 weeks reported similar findings.148 In this study, fracture healing failed in rats treated with celecoxib, rofecoxib, and indometacin, and in COX-2-deficient mice, suggesting that COX-2 function is essential for fracture healing. An analysis of bone healing in wild-type, COX-1 knockout, and COX-2 knockout mouse models of tibial fracture found that fracture healing was significantly delayed in COX-2 knockout mice.149 These investigators concluded that COX-2 may be involved in multiple processes leading to increased bone formation, although it is recognized that there are concerns about the validity of knockout models.139
In contrast, other animal studies have demonstrated that the effects of COX-2-specific inhibitors on bone healing are not significantly deleterious. In a rabbit model of spinal fusion, the rate of fusion was not significantly different in control animals and animals treated with celecoxib 10 mg/kg, but was significantly lower in animals receiving indometacin 10 mg/kg after 8 weeks (p<0.002 vs. placebo).150 These findings suggested that the effects of NSAIDs on bone healing were likely to be mediated by COX-1 inhibition, and indicated that a COX-2-specific inhibitor was the better choice if NSAID treatment was necessary following spinal arthrodesis.
PCR analysis of COX expression in a rat model of femoral fracture found that although the relative levels of COX-1 mRNA remained constant over a 21-day period, COX-2 mRNA levels showed peak expression during the first 14 days of healing and returned to basal levels by day 21.151 In this model, animals treated with ketorolac 4 mg/kg had a significant reduction in mechanical strength and stiffness at 21 days compared with controls (p<0.05), while mechanical strength in animals treated with parecoxib 0.3 and 1.5 mg/kg was not significantly different from that in controls. Although the rate of nonunion was higher in ketorolac-treated animals at 21 days, all fractures in the ketorolac and parecoxib groups had reunited at 5 weeks. Ketorolac also had a more deleterious effect on bone healing than parecoxib, which only slightly delayed bone healing, even at doses known to fully inhibit prostaglandin production. Similar findings have been reported in other animal studies of fracture healing. In a 12-week study of indometacin 1 mg/kg/d and celecoxib 3 mg/kg/d versus no drug in a rat model of femoral fracture, only indometacin-treated animals showed evidence of delayed healing at 4 weeks.152 By 12 weeks there were no significant differences among the three groups. A 12-week comparative study of ibuprofen 30 mg/kg, ketorolac 2 mg/kg, celecoxib 10 mg/kg, rofecoxib 1 and 5 mg/kg, and indometacin 21 mg/kg in a mouse fracture model found that only ketorolac-treated animals had a significant difference from placebo in bone healing at 4 weeks, although both NSAIDs and COX-2-specific inhibitors showed no effect on fracture healing by the end of the study.153
Few studies have been performed to assess the effects of COX-2-specific inhibitors on fracture healing in humans. No prospective analyses have been published and both animal and human data are conflicting. A retrospective analysis of 342 patients who underwent spinal fusion and who received rofecoxib 50 mg, celecoxib 200 mg, ketorolac 15–240 mg, or no NSAIDs in the 5 days following surgery found no statistically significant difference in nonunion rates among celecoxib, rofecoxib, and placebo.154 However, the nonunion rate in the ketorolac group was significantly higher than that in the other three groups (p=0.01) and the effects of ketorolac on nonunion appeared to be dose related.
CONCLUSIONS
Despite the availability of effective analgesic drugs and techniques, the treatment of postoperative pain is often suboptimal. The use of multimodal analgesia represents an effective strategy to optimize the management of pain, but many currently available analgesic drugs are associated with adverse event profiles that limit their use in the postoperative setting. COX-2-specific inhibitors are effective in the treatment of postoperative pain following a range of surgical procedures and have been shown to reduce the postsurgical requirement for opioids.155 Clinical studies demonstrate that COX-2 inhibition, administered prior to or soon after the initiation of surgical pain, may prevent peripheral and central sensitization.
The findings of preclinical studies on the effects of non-specific NSAIDs and COX-2-specific inhibitors in bone healing are often conflicting or inconclusive, and may not be predictive of the clinical situation in humans.156 Analysis of the results of animal and human fracture studies is also complicated by inconsistencies, including study design and dosages, and by confounding factors such as smoking.143 Some studies also lack sufficient power to detect differences in fusion rates.139 Although a recent comprehensive review of the literature reported that there was no evidence of a clinical effect of NSAIDs on bone healing, there were few large, quality studies investigating this subject. Further studies are needed to characterize the effects of non-specific NSAIDs and COX-2-specific inhibitors on bone healing in humans.
NEW DEVELOPMENTS
1 Oates JD, Snowdon SL, Jayson DW. Failure of pain relief after surgery: attitudes of ward staff and patients and patients to post-operative analgesia. Anesthesia. 1994;49:755-758.
2 Warfield CA, Kahn CH. Acute pain management: programs in US hospitals and experiences and attitudes among US adults. Anesthesiology. 1995;83:1090-1094.
3 Chauvin M. State of the art of pain treatment following ambulatory surgery. Eur J Anaesthesiol. 2003;20(Suppl 28):3-6.
4 Gureje O, Von Korff M, Simon GE, et al. Persistent pain and well-being: a World Health Organization Study in Primary Care. JAMA. 1998;280:147-151.
5 Perkins FM, Kehlet H. Chronic pain as an outcome of surgery. A review of predictive factors. Anesthesiology. 2000;93:1123-1135.
6 Shang AB, Gan TJ. Optimising postoperative pain management in the ambulatory patient. Drugs. 2003;63:855-867.
7 Stephens J, Laskin B, Pashos C, et al. The burden of acute postoperative pain and the potential role of the COX-2-specific inhibitors. Rheumatology. 2003;42(Suppl 3):iii40-iii52.
8 Tong D, Chung F. Postoperative pain control in ambulatory surgery. Surg Clin N Am. 1999;79:401-431.
9 Anon. Practice guidelines for acute pain management in the perioperative setting. A report by the American Society of Anesthesiologists Task Force on Pain Management, Acute Pain Section. Anesthesiology. 1995;82:1071-1081.
10 Kehlet H. Multimodal approach to control postoperative pathophysiology and rehabilitation. Br J Anaesth. 1997;78:606-617.
11 Kehlet H, Werner M, Perkins F. Balanced analgesia: what is it and what are its advantages in postoperative pain? Drugs. 1999;58:793-797.
12 Aronson MD. Nonsteroidal anti-inflammatory drugs, traditional opioids, and tramadol: contrasting therapies for the treatment of chronic pain. Clin Ther. 1997;19:420-432.
13 Brooks PM, Day RO. Nonsteroidal anti-inflammatory drugs – differences and similarities. N Engl J Med. 1991;324:1716-1725.
14 Vane JR. Towards a better aspirin. Nature. 1994;367:215-216.
15 Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340:1888-1899.
16 Schoen RT, Vender RJ. Mechanisms of nonsteroidal anti-inflammatory drug-induced gastric damage. Am J Med. 1989;86:449-458.
17 Cryer B. Nonsteroidal anti-inflammatory drugs and gastrointestinal disease. In: Feldman M, Scharschmidt BF, Sleisenger MH, editors. Sleisenger & Fordtran’s gastrointestinal and liver diseases. 6th edn. Philadelphia: WB Saunders; 1998:343-357.
18 DeFrances CJ, Hall MJ. 2002 National Hospital Discharge Survey. Advance data from Vital and Health Statistics; no. 342. US Department of Health and Human Services, Centers for Disease Control and Prevention Web site. Available: http://www.cdc.gov/nchs/data/ad/ad342.pdf, 24 Jun 2004.
19 Marks RM, Sachar EJ. Undertreatment of medical inpatient pain with narcotic analgesics. Ann Intern Med. 1973;78:173-181.
20 Spoeri RK, Ullman R. Measuring and reporting managed care performance: lessons learned and new initiatives. Ann Intern Med. 1997;127:726-732.
21 Hitchcock LS, Ferrell BR, McCaffery M. The experience of chronic nonmalignant pain. J Pain Symptom Manage. 1994;9:312-318.
22 World Health Organization. Cancer Pain Relief and Palliative Care. Report of a WHO Expert Committee. Geneva: World Health Organization; 1990. World Health Organization Technical Report Series, 804. pp. 1–75.
23 Anon. Quality improvement guidelines for the treatment of acute pain and cancer pain. American Pain Society Quality of Care Committee. JAMA. 1995;274:1874-1888.
24 Joint Commission on Accreditation of Healthcare Organizations. Pain Management and Assessment: An Organizational Approach. Oakbrook Terrace, Ill: Joint Commission on Accreditation of Healthcare Organizations, 2000. JCAHO Publication PAM-100
25 Lynch M. Pain: the fifth vital sign. Comprehensive assessment leads to proper treatment. Adv Nurse Pract. 2001;9:28-36.
26 Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;9:1765-1769.
27 Carlton SM, Coggeshall RE. Nociceptive integration: does it have a peripheral component? Pain Forum. 1998;7:71-78.
28 Carr DB, Goudas LC. Acute pain. Lancet. 1999;353:2051-2058.
29 Urban MO, Gebhart GF. Central mechanisms in pain. Med Clin N Am. 1999;83:585-596.
30 Shu X, Mendell LM. Nerve growth factor acutely sensitizes the response of adult rat sensory neurons to capsaicin. Neurosci Lett. 1999;274:159-162.
31 Ali Z, Meyer RA, Campbell JN. Secondary hyperalgesia to mechanical but not heat stimuli following a capsaicin injection in hairy skin. Pain. 1996;68:401-411.
32 Samad TA, Sapirstein A, Woolf CJ. Prostanoids and pain: unraveling mechanisms and revealing therapeutic targets. Trends Mol Med. 2002;8:390-396.
33 Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Ann Rev Biochem. 2000;69:145-182.
34 O’Banion MK. Cyclooxygenase-2: molecular biology, pharmacology, and neurobiology. Crit Rev Neurobiol. 1999;13:45-82.
35 Vanegas H. Bases for a spinal analgesic action of cyclooxygenase inhibitors. Proc West Pharmacol Soc. 2002;45:225-227.
36 Ghilardi JR, Svensson CI, Rogers SD, et al. Constitutive spinal cyclooxygenase-2 participates in the initiation of tissue injury-induced hyperalgesia. J Neurosci. 2004;24:2727-2732.
37 Kömhoff M, Grone HJ, Klein T, et al. Localization of cyclooxygenase-1 and -2 in adult and fetal human kidney: implication for renal function. Am J Physiol. 1997;272:F460-F468.
38 Nantel F, Meadows E, Denis D, et al. Immunolocalization of cyclooxygenase-2 in the macula densa of human elderly. FEBS Lett. 1999;457:475-477.
39 Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97-120.
40 Samad TA, Moore KA, Sapirstein A, et al. Interleukin-1beta-mediated induction of COX-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471-475.
41 Tegeder I, Niederberger E, Vetter G, et al. Effects of selective COX-1 and -2 inhibition on formalin-evoked nociceptive behaviour and prostaglandin E(2) release in the spinal cord. J Neurochem. 2001;79:777-786.
42 Yaksh TL, Dirig DM, Conway CM. The acute antihyperalgesic action of nonsteroidal, anti-inflammatory drugs and release of spinal prostaglandin E2 is mediated by the inhibition of constitutive spinal cyclooxygenase-2 (COX-2) but not COX-1. J Neurosci. 2001;21:5847-5853.
43 Svensson CI, Yaksh TL. The spinal phospholipase–cyclooxygenase–prostanoid cascade in nociceptive processing. Annu Rev Pharmacol Toxicol. 2002;42:553-583.
44 Maier JA, Hla T, Maciag T. Cyclooxygenase is an immediate-early gene induced by interleukin-1 in human endothelial cells. J Biol Chem. 1990;265:10805-10808.
45 Nakao S, Ogata Y, Shimizu-Sasaki E, et al. Activation of NFκB is necessary for IL-1β-induced cyclooxygenase-2 (COX-2) expression in human gingival fibroblasta. Mol Cell Biochem. 2000;209:113-118.
46 McCrory C, Fitzgerald D. Spinal prostaglandin formation and pain perception following thoracotomy: a role for cyclooxygenase-2. Chest. 2004;125:1321-1327.
47 Prescott LF. Paracetamol: past, present, and future. Am J Ther. 2000;7:143-147.
48 Anon. Weak opiate analgesics: modest practical merits. Prescrire Int. 2004;13:22-25.
49 Kehlet H, Rung GW, Callesen T. Postoperative opioid analgesia: time for a reconsideration? J Clin Anesth. 1996;8:441-445.
50 Mulroy MF. Monitoring opioids. Reg Anesth. 1996;21(Suppl 6):89-93.
51 Bowdle TA. Adverse effects of opioid agonists and agonist–antagonists in anaesthesia. Drug Saf. 1998;19:173-189.
52 Carpenter RL. Optimizing postoperative pain management. Am Fam Physician. 1997;56:835-844.
53 Niemi TT, Taxell C, Rosenberg PH. Comparison of the effect of intravenous ketoprofen, ketorolac and diclofenac on platelet function in volunteers. Acta Anaesthesiol Scand. 1997;41:1353-1358.
54 Niemi TT, Backman JT, Syrjala MT, et al. Platelet dysfunction after intravenous ketorolac or propacetamol. Acta Anaesthesiol Scand. 2000;44:69-74.
55 Strom BL, Berlin JA, Kinman JL, et al. Parenteral ketorolac and risk of gastrointestinal and operative site bleeding. A postmarketing surveillance study. JAMA. 1996;275:376-382.
56 Gallagher JE, Blauth J, Fornadley JA. Perioperative ketorolac tromethamine and postoperative hemorrhage in cases of tonsillectomy and adenoidectomy. Laryngoscope. 1995;105:606-609.
57 Gunter JB, Varughese AM, Harrington JF, et al. Recovery and complications after tonsillectomy in children: a comparison of ketorolac and morphine. Anesth Analg. 1995;81:1136-1141.
58 Reinhart DI. Minimising the adverse effects of ketorolac. Drug Saf. 2000;22:487-497.
59 Rudusky BM. Severe postoperative hemorrhage attributed to single-dose parenteral ketorolac-induced coagulopathy. Angiology. 2000;51:999-1002.
60 Dordoni PL, Della Ventura M, Stefanelli A, et al. Effect of ketorolac, ketoprofen and nefopam on platelet function. Anaesthesia. 1994;49:1046-1049.
61 Singer AJ, Mynster CJ, McMahon BJ. The effect of IM ketorolac tromethamine on bleeding time: a prospective, interventional, controlled study. Am J Emerg Med. 2003;21:441-443.
62 Olson NZ, Otero AM, Marrero I, et al. Onset of analgesia for liquigel ibuprofen 400 mg, acetaminophen 1000 mg, ketoprofen 25 mg, and placebo in the treatment of postoperative dental pain. J Clin Pharmacol. 2001;41:1238-1247.
63 Bjornsson GA, Haanaes HR, Skoglund LA. Ketoprofen 75 mg qid versus acetaminophen 1000 mg qid for 3 days on swelling, pain, and other postoperative events after third-molar surgery. J Clin Pharmacol. 2003;43:305-314.
64 Messeri A, Busoni P, Noccioli B, et al. Analgesic efficacy and tolerability of ketoprofen lysine salt vs paracetamol in common paediatric surgery. A randomized, single-blind, parallel, multicentre trial. Paediatr Anaesth. 2003;13:574-578.
65 Tuomilehto H, Kokki H. Parenteral ketoprofen for pain management after adenoidectomy: comparison of intravenous and intramuscular routes of administration. Acta Anaesthesiol Scand. 2002;46:184-189.
66 Marret E, Flahault A, Samama CM, et al. Effects of postoperative, nonsteroidal, antiinflammatory drugs on bleeding risk after tonsillectomy: meta-analysis of randomized, controlled trials. Anesthesiology. 2003;98:1497-1502.
67 Kokki H, Homan E, Tuovinen K, et al. Perioperative treatment with i.v. ketoprofen reduces pain and vomiting in children after strabismus surgery. Acta Anaesthesiol Scand. 1999;43:13-18.
68 Jain S, Datta S. Postoperative pain management. Chest Surg Clin N Am. 1997;7:773-799.
69 Hodsman NB, Burns J, Blyth A, et al. The morphine sparing effects of diclofenac sodium following abdominal surgery. Anaesthesia. 1987;42:1005-1008.
70 Fogarty DJ, O’Hanlon JJ, Milligan KR. Intramuscular ketorolac following total hip replacement with spinal anaesthesia and intrathecal morphine. Acta Anaesthesiol Scand. 1995;39:191-194.
71 Campbell L, Plummer J, Owen H, et al. Effect of short-term ketorolac infusion on recovery following laparoscopic day surgery. Anaesth Intensive Care. 2000;28:654-659.
72 Carmichael J, Shankel SW. Effects of nonsteroidal anti-inflammatory drugs on prostaglandins and renal function. Am J Med. 1985;78:992-1000.
73 Toto RD. The role of prostaglandins in NSAID-induced renal dysfunction. J Rheumatol. 1991;28:22-25.
74 Brater DC. Effects of nonsteroidal anti-inflammatory drugs on renal function: focus on cyclooxygenase-2-selective inhibition. Am J Med. 1999;107(Suppl 6A):65S-71S.
75 Wallace JL. Mechanism of non-steroidal anti-inflammatory drug (NSAID) induced gastrointestinal damage – potential for development of gastrointestinal tract safe NSAIDs. Can J Physiol Pharmacol. 1994;72:1493-1498.
76 Malan TPJr, Marsh G, Hakki SI, et al. Parecoxib sodium, a parenteral cyclooxygenase 2 selective inhibitor, improves morphine analgesia and is opioid-sparing following total hip arthroplasty. Anesthesiology. 2003;98:950-956.
77 Hubbard RC, Naumann TM, Traylor L, et al. Parecoxib sodium has opioid-sparing effects in patients undergoing total knee arthroplasty under spinal anaesthesia. Br J Anaesth. 2003;90:166-172.
78 Rasmussen GL, Steckner K, Hogue C, et al. Intravenous parecoxib sodium for acute pain after orthopedic knee surgery. Am J Orthop. 2002;31:336-343.
79 Daniels SE, Grossman EH, Kuss ME, et al. A double-blind, randomized comparison of intramuscularly and intravenously administered parecoxib sodium versus ketorolac and placebo in a post-oral surgery pain model. Clin Ther. 2001;23:1018-1031.
80 Mehlisch DR, Desjardins PJ, Daniels S, et al. Single doses of parecoxib sodium intravenously are as effective as ketorolac in reducing pain after oral surgery. J Oral Maxillofacial Surg. 2003;61:1030-1037.
81 Barton SF, Langeland FF, Snabes MC, et al. Efficacy and safety of intravenous parecoxib sodium in relieving acute postoperative pain following gynecologic laparotomy surgery. Anesthesiology. 2002;97:306-314.
82 Malan TPJr, Gordon S, Hubbard R, et al. The cyclooxygenase-2-specific inhibitor parecoxib sodium is as effective as 12 mg of morphine administered intramuscularly for treating pain after gynecologic laparotomy surgery. Anesth Analg. 2005;100:454-460.
83 Joshi GP, Viscusi ER, Gan TJ, et al. Effective treatment of laparoscopic cholecystectomy pain with intravenous followed by oral COX-2 specific inhibitor. Anesth Analg. 2004;98:336-342.
84 Cheer SM, Goa KL. Parecoxib (parecoxib sodium). Drugs. 2001;61:1133-1141.
85 Gimbel JS, Brugger A, Zhao W, et al. Efficacy and tolerability of celecoxib versus hydrocodone/acetaminophen in the treatment of pain after ambulatory orthopedic surgery in adults. Clin Ther. 2001;23:228-241.
86 Issioui T, Klein KW, White PF, et al. The efficacy of premedication with celecoxib and acetaminophen in preventing pain after otolaryngologic surgery. Anesth Analg. 2002;94:1188-1193.
87 Reuben SS, Connelly NR. Postoperative analgesic effects of celecoxib or rofecoxib after spinal fusion surgery. Anesth Analg. 2000;91:1221-1225.
88 Desjardins PJ, Shu VS, Recker DP, et al. A single preoperative oral dose of valdecoxib, a new cyclooxygenase-2 specific inhibitor, relieves post-oral surgery or bunionectomy pain. Anesthesiology. 2002;97:565-573.
89 Goldstein JL, Kivitz AJ, Verburg KM, et al. A comparison of the upper gastrointestinal mucosal effects of valdecoxib, naproxen and placebo in healthy elderly subjects. Aliment Pharmacol Ther. 2003;18:125-132.
90 Leese PT, Recker DP, Kent JD. The COX-2 selective inhibitor, valdecoxib, does not impair platelet function in the elderly: results of a randomized controlled trial. J Clin Pharmacol. 2003;43:504-513.
91 Leese PT, Talwalker S, Kent JD, et al. Valdecoxib does not impair platelet function. Am J Emerg Med. 2002;20:275-281.
92 Fricke J, Varkalis J, Swillich S, et al. Valdecoxibif more efficacious than rofecoxib in relieving pain associated with oral surgery. Am J Ther. 2002;9:89-97.
93 Christensen KS, Cawkwell GD. Valdecoxib versus rofecoxib in acute postsurgical pain: results of a randomized controlled trial. J Pain Symptom Manage. 2001;27:460-470.
94 Daniels SE, Desjardins PJ, Talwalker S, et al. The analgesic efficacy of valdecoxib vs. oxycodone/acetaminophen after oral surgery. J Am Dent Assoc. 2002;133:611-621.
95 Camu F, Beecher T, Recker DP, et al. Valdecoxib, a COX-2-specific inhibitor, is an efficacious, opioid-sparing analgesic in patients undergoing hip arthroplasty. Am J Ther. 2002;9:43-51.
96 Reynolds LW, Hoo RK, Brill RJ, et al. The COX-2 specific inhibitor, valdecoxib, is an effective, opioid-sparing analgesic in patients undergoing total knee arthroplasty. J Pain Symptom Manage. 2003;25:133-141.
97 Zhao SZ, Chung F, Hanna DB, et al. Dose-response relationship between opioid use and adverse effects after ambulatory surgery. J Pain Symptom Manage. 2004;28:35-46.
98 Karim A, Laurent A, Slater ME, et al. A pharmacokinetic study of intramuscular (IM) parecoxib sodium in normal subjects. J Clin Pharmacol. 2001;41:1-9.
99 Olkkola KT, Backman JT, Neuvonen PJ. Midazolam should be avoided in patients receiving the systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994;55:481-485.
100 Ibrahim A, Karim A, Feldman J, et al. The influence of parecoxib, a parenteral cyclooxygenase-2 specific inhibitor, on the pharmacokinetics and clinical effects of midazolam. Anesth Analg. 2002;95:667-673.
101 Ibrahim AE, Feldman J, Karim A, et al. Simultaneous assessment of drug interactions with low- and high-extraction opioids: application to parecoxib effects on the pharmacokinetics and pharmacodynamics of fentanyl and alfentanil. Anesthesiology. 2003;98:853-861.
102 Ibrahim A, Park S, Feldman J, et al. Effects of parecoxib, a parenteral COX-2-specific inhibitor, on the pharmacokinetics and pharmacodynamics of propofol. Anesthesiology. 2002;96:88-95.
103 Feldman HI, Kinman JL, Berlin JA, et al. Parenteral ketorolac: the risk for acute renal failure. Ann Intern Med. 1997;126:193-199.
104 Noveck RJ, Laurent A, Kuss M, et al. Parecoxib sodium does not impair platelet function in healthy elderly and non-elderly individuals. Clin Drug Investig. 2001;21:465-476.
105 Leese PT, Hubbard RC, Karim A, et al. Effects of celecoxib, a novel cyclooxygenase-2 inhibitor, on platelet function in healthy adults: a randomized, controlled trial. J Clin Pharmacol. 2000;40:124-132.
106 Laine L, Harper S, Simon T, et al. A randomized trial comparing the effect of rofecoxib, a cyclooxygenase 2-specific inhibitor, with that of ibuprofen on the gastroduodenal mucosa of patients with osteoarthritis. Gastroenterology. 1999;117:776-783.
107 Simon LS, Weaver AL, Graham DY, et al. Anti-inflammatory and upper gastrointestinal effects of celecoxib in rheumatoid arthritis. A randomized controlled trial. JAMA. 1999;282:1921-1928.
108 Silverstein FE, Faich G, Goldstein JL, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis. The CLASS study: a randomized, controlled trial. JAMA. 2000;284:1247-1255.
109 Singh G, Fort JG, Triadafilopoulos G, et al. SUCCESS-1: A global osteoarthritis (OA) trial in 13,274 randomized patients: Celecoxib provides similar efficacy to diclofenac and naproxen while providing significantly improved UGI safety. Arthritis Rheum. 2001;44(Suppl 9):S135.
110 Deeks JJ, Smith LA, Bradley MD. Efficacy, tolerability, and upper gastrointestinal safety of celecoxib for treatment of osteoarthritis and rheumatoid arthritis: systematic review of randomised controlled trials. BMJ. 2002;325:619-626.
111 Mamdani M, Rochon PA, Juurlink DN, et al. Observational study of upper gastrointestinal haemorrhage in elderly patients given selective cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs. BMJ. 2002;325:624.
112 Sikes DH, Agrawal NM, Zhao WW, et al. Incidence of gastroduodenal ulcers associated with valdecoxib compared with that of ibuprofen and diclofenac in patients with osteoarthritis. Eur J Gastroenterol Hepatol. 2002;14:1101-1111.
113 Harris SI, Kuss M, Hubbard RC, et al. Upper gastrointestinal safety evaluation of parecoxib sodium, a new parenteral cyclooxygenase-2-specific inhibitor, compared with ketorolac, naproxen, and placebo. Clin Ther. 2001;23:1422-1428.
114 Stoltz RR, Harris SI, Kuss ME, et al. Upper GI mucosal effects of parecoxib sodium in healthy elderly subjects. Am J Gastroenterol. 2002;97:65-71.
115 Harris SI, Stoltz RR, LeComte D, et al. Parecoxib sodium demonstrates gastrointestinal safety comparable to placebo in healthy subjects. J Clin Gastroenterol. 2004;38:575-580.
116 Whelton A, Maurath CJ, Verburg KM, et al. Renal safety and tolerability of celecoxib, a novel cyclooxygenase-2 inhibitor. Am J Ther. 2000;7(3):159-175.
117 Pope JE, Anderson JJ, Felson DT. A meta-analysis of the effects of nonsteroidal anti-inflammatory drugs on blood pressure. Arch Intern Med. 1993;153(4):477-484.
118 Whelton A, Hamilton CW. Nonsteroidal anti-inflammatory drugs: effects on kidney function. J Clin Pharmacol. 1991;31(7):588-598.
119 Zhao SZ, Burke TA, Whelton A, et al. Blood pressure destabilization and related healthcare utilization among hypertensive patients using nonspecific NSAIDs and COX-2-specific inhibitors. Am J Manag Care. 2002;8:S401-S413.
120 Whelton A, Fort JG, Puma JA, et al. Cyclooxygenase-2-specific inhibitors and cardiorenal function: a randomized, controlled trial of celecoxib and rofecoxib in older hypertensive osteoarthritis patients. Am J Ther. 2001;8:85-95.
121 Whelton A, White WB, Bello AE, et al. Effects of celecoxib and rofecoxib on blood pressure and edema in patients > or =65 years of age with systemic hypertension and osteoarthritis. Am J Cardiol. 2002;90:959-963.
122 Wolfe F, Zhao S, Pettitt D. Blood pressure destabilization and edema among 8538 users of celecoxib, rofecoxib, and nonselective nonsteroidal antiinflammatory drugs (NSAID) and nonusers of NSAID receiving ordinary clinical care. J Rheumatol. 2004;31(6):1143-1151.
123 Whelton A, White W, Kent J, et al. Hypertension and edema rates in 6,717 patients for the new COX-2 inhibitor, valdecoxib versus the conventional nonselective NSAIDs and placebo [abstract]. Am J Hypertens. 2003;16:P445.
124 LeLorier J, Bombardier C, Burgess E, et al. Practical considerations for the use of nonsteroidal anti-inflammatory drugs and cyclo-oxygenase-2 inhibitors in hypertension and kidney disease. Can J Cardiol. 2002;18:1301-1308.
125 Bresalier RS, Sandler RS, Quan H, et alfor the Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial Investigators. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. Published February 15, 2005 [epub ahead of print. 10.1056/NEJMoa050493]. Available at: http://content.nejm.org/cgi/content/abstract/NEJMoa050493v1. Accessed February 16, 2005.
126 Bombardier C, Laine L, Reicin A, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N Engl J Med. 2000;343:1520-1528.
127 Ray WA, Stein CM, Daugherty JR, et al. COX-2 selective non-steroidal anti-inflammatory drugs and risk of serious coronary heart disease. Lancet. 2002;360:1071-1073.
128 Solomon DH, Schneeweiss S, Glynn RJ, et al. Relationship between cyclooxygenase-2 inhibitors and acute myocardial infarction in older adults. Circulation. 2004;109:2068-2073.
129 Juni P, Nartey L, Reichenbach S, et al. Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet. 2004;364:2021-2029.
130 US Food and Drug Administration. FDA statement on naproxen. Available at: http://www.fda.gov/bbs/topics/news/2004/NEW01148.html. Accessed February 16, 2005
131 Graham DJ, Campen D, Hui R, et al. Risk of acute myocardial infarction and sudden cardiac death in patients treated with cyclo-oxygenase 2 selective and non-selective non-steroidal anti-inflammatory drugs: nested case-control study. Lancet. 2005;365:475-481.
132 Kimmel SE, Berlin JA, Reilly M, et al. Patients exposed to rofecoxib and celecoxib have different odds of nonfatal myocardial infarction. Ann Intern Med. 2005;142:157-164.
133 Solomon SD, McMurray JJV, Pfeffer MA, et alfor the Adenoma Prevention with Celecoxib (APC) Study Investigators. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. Published February 15, 2005 [epub ahead of print. 10.1056/NEJMoa050405]. Available at: http://content.nejm.org/cgi/content/abstract/NEJMoa050405v1. Accessed February 16, 2005
134 White WB, Strand V, Roberts R, et al. Effects of the cyclooxygenase-2 specific inhibitor valdecoxib versus nonsteroidal antiinflammatory agents and placebo on cardiovascular thrombotic events in patients with arthritis. Am J Ther. 2004;11:244-250.
135 Ott E, Nussmeier NA, Duke PC, et althe Multicenter Study of Perioperative Ischemia (McSPI) Research Group, Ischemia Research and Education Foundation (IREF) Investigators. Efficacy and safety of the cyclooxygenase 2 inhibitors parecoxib and valdecoxib in patients undergoing coronary artery bypass surgery. J Thorac Cardiovasc Surg. 2003;125:1481-1492.
136 Nussmeier NA, Whelton AA, Brown MT. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. Published February 15, 2005 [epub ahead of print. 10.1056/NEJMoa050330]. Available at: http://content.nejm.org/cgi/content/abstract/NEJMoa050330v1. Accessed February 16, 2005
137 Sowers JR, White WB, Pitt B, et althe Celecoxib Rofecoxib Efficacy and Safety in Comorbidities Evaluation Trial (CRESCENT) Investigators. The effects of cyclooxygenase-2 inhibitors and nonsteroidal anti-inflammatory therapy on 24-hour blood pressure in patients with hypertension, osteoarthritis, and type 2 diabetes mellitus. Arch Intern Med. 2005;165:161-168.
138 Dunstan C, Boyce R, Boyce B. Systemic administration of acidic fibroblast growth factor (FGF-1) prevents bone loss and increases new bone formation in ovariectomized rats. J Bone Miner Res. 1999;14:953-959.
139 Gajraj NM. The effect of cyclooxygenase-2 inhibitors on bone healing. Reg Anesth Pain Med. 2003;28:456-465.
140 Thorsen K, Kristoffersson AO, Lerner UH, et al. In situ microdialysis in bone tissue: stimulation of prostaglandin E2 release by weight-bearing mechanical loading. J Clin Invest. 1996;98:2446-2449.
141 Keller J, Bunger C, Andreassen TT, et al. Bone repair inhibited by indomethacin. Effects on bone metabolism and strength of rabbit osteotomies. Acta Orthop Scand. 1987;58:379-383.
142 Dimar JR2nd, Ante WA, Zhang YP, et al. The effects of nonsteroidal anti-inflammatory drugs on posterior spinal fusions in the rat. Spine. 1996;21:1870-1876.
143 Glassman SD, Rose SM, Dimar JR, et al. The effect of postoperative nonsteroidal anti-inflammatory drug administration on spinal fusion. Spine. 1998;23:834-838.
144 Deguchi M, Rapoff AJ, Zdeblick TA. Posterolateral fusion for isthmic spondylolisthesis in adults: analysis of fusion rate and clinical results. J Spinal Disord. 1998;11(6):459-464.
145 Giannoudis PV, MacDonald DA, Matthews SJ, et al. Non-union of the femoral diaphysis. The influence of reaming and non-steroidal anti-inflammatory drugs. J Bone Joint Surg Br. 2000;82:655-658.
146 Leonelli S, Goldberg B, Safanda J, et al. The effect of cyclooxygenase 2 (COX-2) inhibitors on bone healing. Presented at: 48th Annual Meeting of the Orthopaedic Research Society; February 9–12, 2002; Dallas, Tex.
147 Goodman S, Ma T, Trindade M. COX-2 selective NSAID decreases bone ingrowth in vivo. J Orthop Res. 2002;20:1164-1169.
148 Simon AM, Manigrasso MB, O’Connor JP. Cyclo-oxygenase 2 function is essential for bone fracture healing. J Bone Miner Res. 2002;17:963-976.
149 Zhang X, Schwarz EM, Young DA, et al. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J Clin Invest. 2002;109:1405-1415.
150 Long J, Lewis S, Kuklo T, et al. The effect of cyclooxygenase-2 inhibitors on spinal fusion. J Bone Joint Surg Am. 2002;84-A:1763-1768.
151 Gerstenfeld LC, Thiede M, Seibert K, et al. Differential inhibition of fracture healing by non-selective and cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs. J Orthop Res. 2003;21:670-675.
152 Brown KM, Saunders MM, Kirsch T, et al. Effect of COX-2-specific inhibition on fracture-healing in the rat femur. J Bone Joint Surg Am. 2004;86-A:116-123.
153 Mullis B, Copeland S, Weinhold P, et al. Effect of COX-2 inhibitors and NSAIDs on fracture healing in a mouse model. Presented at: 48th Annual Meeting of the Orthopaedic Research Society; February 9–12, 2002; Dallas, Tex.
154 Reuben S, Rizvi A, Steinberg R, et al. The effects of NSAIDs on spinal fusion. Presented at: American Society of Regional Anesthesia and Pain Medicine 27th Annual Spring Meeting & Workshops; April 25–28, 2002; Chicago, Ill. ASRA PD-16.
155 Katz WA. Cyclooxygenase-2-selective inhibitors in the management of acute and perioperative pain. Cleve Clin J Med. 2002;69(Suppl 1):S165-S175.
156 Arnoczky SP, Wilson JW. Experimental surgery of the skeletal system. In: Gay WI, Heavner JE, editors. Methods of animal experimentation. Vol. II: Research surgery and care of the research animal. London: Academic Press; 1996:67-108.