183 Nonsteroidal Antiinflammatory Agents
Nonsteroidal antiinflammatory drugs (NSAIDs) have important clinical uses in selected critically ill patients for treatment of pain and inflammatory states or reduction of fever.1–3 However, drugs in this class can cause serious side effects and/or affect other medications used concomitantly. The pharmacologic characteristics of NSAIDs related to cyclooxygenase-1 (COX-1) and COX-2 enzyme inhibition may result in severe gastrointestinal, cardiovascular, and renal side effects.4–8 Two highly COX-2-selective agents have been withdrawn from the market as a result of toxicity.9 Patient characteristics and differences in NSAID toxicity profiles are important for the judicious use of NSAIDs in the critically ill patient.
 NSAID Pharmacodynamics
NSAID Pharmacodynamics
NSAIDs include aspirin, indomethacin, ibuprofen, naproxen, diclofenac, and a product which is relatively more selective for the COX-2 isoform of cyclooxygenase (i.e., celecoxib). All NSAIDs have analgesic, antiinflammatory, and antipyretic properties. NSAIDs belong to a number of chemical families including acetic acids, oxicams, propionic acids, salicylates, fenamates, furanones and coxibs (Table 183-1). All NSAIDs are weakly acidic chemical compounds and share similarities in pharmacokinetic properties.10 Their absorption is primarily in the large surface area of the small intestine as well as in the stomach.10 Gastrointestinal absorption of NSAIDs occurs rapidly, usually within 15 to 30 minutes. Different product formulations, including enteric-coated and delayed-release preparations, decrease gastric emptying, and altered gastric transit time can delay drug absorption and time to peak effect.11 For example, 4 to 6 hours is required for peak absorption of enteric-coated products.11 After absorption, NSAIDs are more than 90% bound to albumin, which influences their distribution and drug-drug interaction potential. Hypoalbuminemia (e.g., due to alcoholic liver disease) can result in greater unbound drug and increased risk for NSAID-related adverse events.11
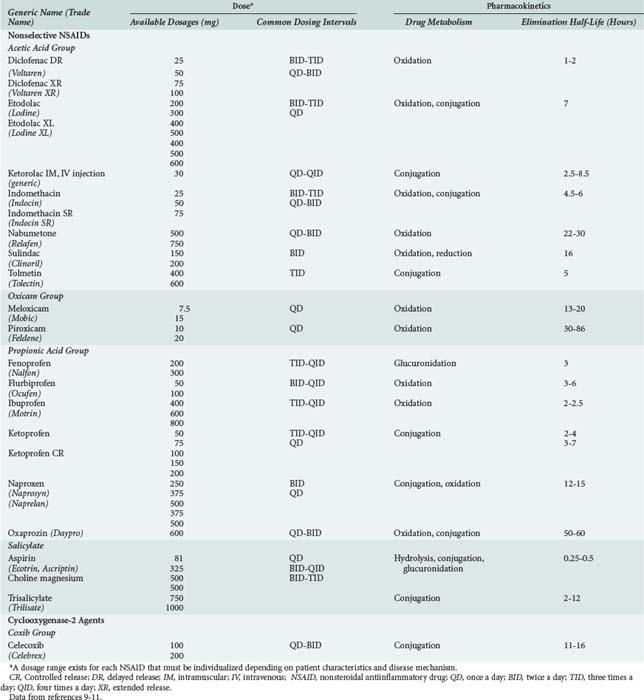
NSAIDs are primarily eliminated by renal and biliary excretion.12 The elimination half-lives of NSAIDs vary from 0.25 to 86 hours, which accounts for differences in dosing schedules (see Table 183-1).11 Factors that delay NSAID clearance increase their potential for adverse reactions. Reduced renal function prolongs NSAID half-life, and the dose should be lowered proportionally in patients with impaired kidney function.10,12 Some NSAIDs are hepatically metabolized to both active and inactive metabolites, primarily through the cytochrome P450 enzymes, glucuronidase enzymes, or both.10,13 Nabumetone and sulindac are prodrugs and require metabolism by the liver to generate pharmacologically active metabolites.10 Moderate to severe liver disease impairs NSAID metabolism, increasing the potential for NSAID toxicity. With advanced age, the hepatic clearance of diclofenac, etodolac, flurbiprofen, ibuprofen, indomethacin, meloxicam, nabumetone, naproxen, oxaprozin, piroxicam, and sulindac is slower because of decreased hepatic phase I oxidative, reductive, and hydrolytic catalytic reactions.10
NSAIDs have a number of physiologic effects, although their principal action is inhibition of the cyclooxygenase enzyme.1,2 COX is responsible for the production of prostaglandins (PG) and thromboxanes (TX), which are derived from arachidonic acid, an unsaturated fatty acid present in all body cell membranes. PGs and TXs mediate normal homeostatic functions of the upper gastrointestinal tract, kidneys, endothelium, vascular smooth muscle, and platelets, among other tissues and organs. PG and TX are critical in the inflammatory response because of their influences on vascular permeability, platelet function, and immune reactions. These autocoids are involved in both peripheral and central pain processing and have a role in fever production.14,15
As noted earlier, there are two isoforms of the COX enzyme: COX-1 and COX-2. Although the COX enzymes are coded on two separate genes, they share 63% structural homogeneity, have similar mechanisms of action, and produce identical compounds from arachidonic acid.14–17 Expression and regulation of COX-1 and COX-2 differ in various organs and tissues; however, their physiologic effects are overlapping.2,14,15,18 Differences in the structural configurations of COX-1 and COX-2 enzyme side chains determine whether a particular NSAID will inhibit the enzyme. Isoform nonselective NSAIDs (e.g., naproxen, ibuprofen) inhibit both COX-1 and COX-2, thereby decreasing production of PGs involved in both homeostatic and inflammatory actions. The COX-2-selective agent, celecoxib, is 50-fold more active against COX-2 than COX-1 and therefore exerts actions primarily in inflammatory processes.18,19 Both COX-1 and COX-2 are involved in pain processing.
Differences in COX-1 and COX-2 inhibition allow for comparisons of drug effect.6 Nonselective NSAIDs exert one of three kinetic models for inhibiting COX-1 and COX-2: (1) rapid, reversible binding (e.g., ibuprofen); (2) rapid, lower-affinity reversible binding followed by time-dependent, higher-affinity, slowly reversible binding (e.g., indomethacin); or (3) rapid, irreversible binding followed by covalent modification (e.g., aspirin). Aspirin is the only NSAID that covalently modifies both COX enzymes, thereby resulting in permanent inhibition of both isoforms.20 COX-2-selective inhibitors act on COX-2 by a time-dependent, slowly reversible mechanism. They also can affect COX-1 by a freely reversible and competitive mechanism. The result of this two-stage process by COX-2-selective agents is maximal inhibition of COX-2 with minimal inhibition of COX-1.16,17,21
The biochemical selectivity of NSAIDs is related to the in vitro drug concentration necessary to inhibit COX-2 activity completely and COX-1 activity by 50%.2,15 Data from clinical trials demonstrating a decreased incidence of gastrointestinal toxicity and an absence of platelet inhibition with COX-2-selective versus nonselective agents have been the clinical parameters used to distinguish among the various agents.2
COX converts arachidonic acid to the inactive precursor, PGG2, and then PGH2. PGH2 is metabolized in various tissues to physiologically active products including PGI2, PGE2, and TXA2. The concentration of different PGs determine their biological effects on tissues. PGs exert their effects by activating cell-membrane receptors of the superfamily of G protein–coupled receptors.1 PGI2 has important regulatory effects on renal blood flow, gastric mucosa, uterine smooth muscle, and bronchial smooth muscle. PGI2 also inhibits platelet aggregation. PGE2 is an abundant PG with important regulatory effects on fever and on the reproductive, gastrointestinal, neuroendocrine, and immune systems.22 PGE2 is present at sites of inflammation as a potent vasodilator in acute and chronic inflammatory diseases and in tissue injury. PGE2 also can promote labor and dysmenorrhea.22 TXA2 promotes platelet aggregation and vasoconstriction. TXA2 is released with tissue injury and plays a role in cellular responses to inflammation.22
PGs produced by COX-1 are primarily involved in maintaining the protective gastrointestinal mucosal barrier in the stomach and intestines, modulating intrarenal hemodynamics, influencing platelet function (especially aggregation), and regulating vascular homeostasis. For example, PGs produced by COX-1 provide gastric protection by reducing gastric acid secretion, stimulating mucus secretion, and promoting gastric mucosa vasodilation. Kidney function is affected by the localization of COX-1 in the collecting ducts and renal vasculature. COX-1 converts PGH2 to TXA2, which promotes platelet aggregation. Although the major role of COX-1 is homeostasis, COX-1 may contribute to PG production in certain inflammatory reactions, including those in the synovia of inflamed joints and atherosclerotic plaques.14,15,18
The primary role of COX-2 is in inflammatory reactions that result in PG production by fibroblasts, macrophages, endothelial cells, and synoviocytes. This enzyme is also important in pain and fever mechanisms.16 Certain other tissues express COX-2, especially the cortical macula densa, medullary interstitial cells, and the kidney vasculature.15 Small amounts of COX-2 are also found in the small intestine, ovary, uterus, bone, and brain.14,16 Because COX-2 expression is regulated by growth factors, its role in wound repair is under investigation.17
 Clinical Implications and Uses of NSAIDs in Critically Ill Patients
Clinical Implications and Uses of NSAIDs in Critically Ill Patients
Although NSAIDs have important analgesic, antiinflammatory, and antipyretic activity, their role in critically ill patients should be limited, owing to their potential for toxic side effects.23 In clinical trials, all nonselective and COX-2-selective NSAIDs in equipotent doses have demonstrated similar efficacy in relieving pain, inflammation, and fever.24–26 However, there is significant variability in the clinical effects of NSAIDs within and among patients, with approximately 70% to 80% of individuals responding to any particular agent.25 Lack of response to one NSAID does not preclude benefit from another.25 Differences in NSAIDs may be related to COX-1 and COX-2 inhibitory pharmacodynamics, because no definite clinical characteristics have been identified in nonresponders compared to responders.10,20 In contrast, toxicity profiles of nonselective and COX-2-selective agents have a defined relationship to the degree of COX-1 and COX-2 inhibition, especially for the development of gastrointestinal adverse events or antiplatelet effects.
Management of Pain and Inflammation
Pain management is a key issue for critical care clinicians. Although morphine is the primary analgesic of choice among critically ill patients with pain, NSAIDS may have a role in selected patients.26 Pain is initiated by activation of tissue nociceptors in various disease states by mechanical, thermal, and chemical stimuli. Surgical trauma and other forms of tissue injury induce expression of COX-2, and to a lesser extent COX-1, resulting in generation of PGs, especially PGE2.27–29 PGs sensitize A-δ and C primary afferent sensory nerve fibers that carry impulses to the dorsal horn of the spinal cord. Glutamate, substance P, and other mediators along with PGs are involved in dorsal horn pain processing.30 By inhibiting PGs at these different levels of the pain matrix, NSAIDs can have important effects on pain processing. For example, NSAIDs reduce PG-mediated protein kinase A phosphorylation of sodium channels in nociceptor terminals.
Surgical Pain
Inadequate postoperative pain control has been associated with increased morbidity, increased length of stay, and increased costs for intensive care unit (ICU) patients.31,32 Opioids are commonly prescribed for surgical and trauma pain and other acute pain states, but adverse events such as nausea and vomiting, drowsiness, and respiratory depression may limit their use in some patients and prolong postoperative recovery and increase costs.31 NSAIDs are effective in combination with other analgesics in managing the acute pain of tissue injury.33 For example, the use of NSAIDs in orthopedic and other types of surgeries including knee arthroscopy, hip replacement, spinal surgery, and gynecologic laparoscopy decreases postoperative opioid requirements.24,26,34–38 In addition, NSAID therapy alone can provide effective postoperative pain relief.31,38,39 However, because of the potential for adverse reactions (e.g., gastrointestinal toxicity, platelet inhibition with increased bleeding risk, renal dysfunction), especially with hypovolemia, NSAIDs should be used cautiously in the management of postoperative pain.24,31,39 Although COX-2-selective inhibitors are equally effective in pain relief as nonselective NSAIDs, they should be used cautiously in the ICU owing to their potential for cardiac and gastrointestinal toxicity.39 Recent studies of the use of COX-2-selective inhibitors in orthopedic and gynecologic surgeries demonstrated significant reductions in opioid consumption, decreased postoperative opioid side effects including nausea and vomiting, and improved subjectively.24,28,40,41 In addition, preoperative use of COX-2-selective agents in knee arthroscopy, compared with postoperative use, can delay the time to first analgesic request and decrease total opioid consumption.35 The clinical impact of NSAID therapy on wound and bone healing after surgery is unclear.42,43 NSAIDs, especially indomethacin, have been used perioperatively to reduce heterotopic bone formation after acetabular fracture surgery.44 Recent data from animal and human trials suggest that NSAIDs may impair bone healing after fractures because of the role of PGs in osteogenesis.42,43,45
Ketorolac is currently the only injectable NSAID available in the United States. Clinical studies comparing injectable ketorolac with morphine in managing postoperative pain after orthopedic, gynecologic, and major abdominal procedures have shown similar efficacy but slower onset of action with ketorolac.46 Combination therapy with ketorolac and morphine may provide analgesic benefit compared with morphine alone and reduce total morphine consumption.47 However, use of ketorolac in acute pain states is now limited because of postmarketing reports of toxicity including peptic ulcers, gastrointestinal bleeding, and renal insufficiency.46 In addition, dosing adjustments are necessary for patients with renal dysfunction and elderly patients, and duration of therapy never should exceed 5 days.48
Regional Inflammatory States
The antiinflammatory properties of NSAIDs are beneficial in specific acute disease states affecting critically ill patients, including systemic and regional rheumatic disorders and localized inflammatory conditions such as pleuropericarditis. PGs, especially PGI2 and PGE2, are induced by interleukin (IL)-1, IL-6, and IL-8. These PGs are important mediators of inflammatory reactions; they influence vascular reactivity and increase vascular permeability.22
Rheumatic diseases such as rheumatoid arthritis and systemic lupus erythematosus (SLE) present therapeutic challenges to the critical care clinician. Concerns about the role of NSAIDs in causation of certain acute problems, especially gastrointestinal bleeding and fluid retention, have arisen. NSAIDs are an important component of the therapeutic regimen for the synovitis of inflammatory arthritis and for serositis involving pleural or pericardial membranes.48,49 Discontinuation of NSAID therapy can result in a significant increase in synovitis. The antiinflammatory effects of NSAIDs often require higher doses than those needed for a chronic analgesic response. No significant differences in effectiveness in suppressing inflammation have been demonstrated among the various nonselective and COX-2-selective agents.24,25,50 Acute crystal-induced arthritis such as gout and pseudogout, although uncommon, still occasionally present in critically ill patients. Inflammation in response to uric acid and calcium pyrophosphate dihydrate crystals, respectively, is induced by immune mediators such as PGs, cytokines, bradykinin, and leukotrienes, which produce capillary dilation, neutrophil migration, and pain stimulation. NSAIDs, especially indomethacin, have been shown to be beneficial in acute crystal-induced arthritis.51 Aspirin should not be used for gout because it influences renal tubular uric acid excretion, thereby causing fluctuations in serum uric acid levels, potentially aggravating acute gouty arthritis. Because all NSAIDs except ketorolac are administered only orally and can be toxic, their use is limited in the critical care setting. Alternative treatments for crystal-induced arthritis include corticosteroids and colchicine for gout.
Occasionally, acutely ill patients require NSAID therapy for their antiinflammatory and analgesic properties because of concurrent regional musculoskeletal disorders such as shoulder or elbow tendonitis or back problems.52 Finally, NSAIDs have been used to treat pleuropericarditis of nonrheumatic origin, including viral serositis and postmyocardial infarction syndrome.
Cardiovascular Protection
Abnormalities within the cardiovascular system are frequently present in critically ill patients. Because PGs play pathophysiologic roles in coagulation and inflammatory mechanisms involved in cardiovascular diseases, NSAIDs, especially aspirin, have an important therapeutic role. TXA2, synthesized by platelets, is produced by COX-1 (together with another enzyme, thromboxane synthase); it promotes platelet aggregation on abnormal vascular endothelium and in areas of vascular stasis, leading to thrombosis. All nonselective NSAIDs inhibit COX-1; however, only aspirin does so irreversibly through acetylation.53 The U.S. Preventive Services Taskforce Report strongly recommended the use of aspirin in adult patients who are at risk for coronary heart disease.54 Practice guidelines recommend the use of low-dose aspirin (81-325 mg/d) for high-risk patients (i.e., 5% risk within 5 years) to reduce cardiovascular events including nonfatal myocardial infarction, fatal coronary heart disease, and nonhemorrhagic stroke.53,55 For those individuals with a lower risk of having a coronary heart disease event, the benefit was negated by the toxic effects of aspirin, including gastrointestinal events and hemorrhagic stroke. An update of the evidence from the U.S. Preventive Task Force was published in 2009.55 Following a systematic review of randomized clinical trials, the authors concluded that aspirin reduces the risk of coronary vascular disease (CVD) in adults without a history of CVD.55 Critical care clinicians should consider using aspirin for patients at risk for CVD; however, the risks must be weighed against the benefits because of the increased risk for gastrointestinal bleeding events.55
Although other nonselective NSAIDs reversibly inhibit the COX-1 enzyme in platelets, they have not been demonstrated to reduce cardiac events and currently should not be used for primary or secondary prevention of vascular disease. COX-2-selective agents do not have demonstrated cardiovascular benefits.56
Aspirin has been compared with adjusted-dose warfarin for the prevention of stroke in atrial fibrillation. The 8th American College of Chest Physicians (ACCP) Consensus Conference on Antithrombotic Therapy recommended that drug selection in atrial fibrillation be based on risk likelihood: adjusted-dose warfarin for patients with a high risk of stroke, aspirin or adjusted-dose warfarin for those with a moderate risk; and aspirin for those with a low risk.53 The risk for stroke is increased by a history of prior stroke, systemic embolus, hypertension, poor left ventricular systolic function, age older than 75 years, rheumatic mitral valvular disease, or a prosthetic heart valve.53 Although both adjusted-dose warfarin and aspirin confer significant stroke reduction in the case of atrial fibrillation, their concomitant use does not impart greater benefit, and increased side effects do occur.53
Aspirin therapy, alone or in combination with dipyridamole, can delay progression of established arterial occlusive disease in patients with chronic lower-extremity arterial insufficiency.53 Low-dose aspirin is beneficial in preventing morbidity and mortality from stroke and myocardial infarction in patients with peripheral arterial disease.53,57,58 In addition, low-dose aspirin is used as prophylaxis and treatment for ischemic cerebrovascular disease, alone and in combination with dipyridamole, and has been demonstrated to reduce the risk of stroke in individuals with transient ischemic attacks or completed ischemic strokes due to thrombosis.53
Fever
The mechanisms of fever involve either peripheral release of pyrogenic cytokines (IL-1, IL-6, tumor necrosis factor [TNF], and interferon [IFN]-α) from monocytes and macrophages or the presence of circulating endotoxins which stimulate central production of PGE2 via COX-2 in vascular endothelial cells. PGE2 targets hypothalamic thermoregulatory neurons.59–61 Knockout mice lacking the COX-2 enzyme are unable to mount a fever when exposed to exogenous pyrogens.59,60 The COX-1 enzyme is not involved in thermal regulatory control.59
Aspirin has long been recognized as an effective antipyretic agent, and it is the gold standard with which acetaminophen and other NSAIDs are compared. Studies evaluating the antipyretic properties of NSAIDs are primarily from the pediatric literature. A meta-analysis of adult fever trials has not been possible because of differences in patient populations, NSAID dosing schedules, and outcome measures. However, studies in adults have shown equal or superior antipyretic efficacy of NSAIDs compared with acetaminophen.60 NSAIDs are more effective in reducing fevers associated with cancer than those caused by infection, although the mechanism is unclear.60 Duration of action of various NSAIDs in fever is related to drug half-life and drug concentration in the hypothalamus, a parameter determined by drug transport across the blood-brain barrier.59 There are limited data evaluating clinical use of COX-2-selective agents for fever, but a beneficial effect has been demonstrated.62,63,64 Toxicities of the various nonselective NSAIDs limit their clinical utility compared with acetaminophen, especially in critically ill patients.60,65
Septic Shock
Ibuprofen and other NSAIDs have some physiologic effects in sepsis, although they do not reduce morbidity and mortality in septic shock, except possibly in patients with hypothermia.66,67 A prospective randomized trial evaluated the impact of ibuprofen on organ failure and mortality in 455 patients with sepsis syndrome.67 Significant improvements in temperature, heart rate, oxygen consumption, and lactic acidosis were noted, but there was no reduction in organ failure or 30-day mortality compared with standard care. The Surviving Sepsis Campaign guidelines do not list NSAIDs as a treatment option for septic shock.68
 Toxicity of NSAIDs
Toxicity of NSAIDs
Gastrointestinal Toxicity
PGs play a critical role in maintenance of the gastrointestinal mucosal barrier. Beneficial effects include maintenance of epithelial mucus secretion, mucosal blood flow, bicarbonate secretion, and epithelial proliferation. These protective mechanisms may be altered by NSAID-mediated inhibition of PG synthesis and local mucosal damage by the acidic NSAID compounds.69 The spectrum of gastrointestinal injury includes mucosal irritation and ulceration in the stomach and small intestine, with bleeding and perforation.
There are limited data available regarding the gastrointestinal toxicity of NSAIDs in critically ill patients. However, serious gastrointestinal complications including ulceration, perforation, and bleeding occur in 1% to 2% of short-term NSAID users and in 2% to 5% of patients on chronic NSAID therapy.70 A prospective observational trial conducted over a 19-week period identified admissions to a medical ICU related to adverse drug reactions (ADR). Bleeding secondary to both nonselective and selective NSAIDS, aspirin, and clopidogrel were the most common ADR admission diagnoses.71 Gastrointestinal lesions related to NSAIDs frequently are asymptomatic. For example, approximately 40% of patients with endoscopically proven gastritis do not have symptoms.70,72 Other studies confirm the occurrence of NSAID-induced gastrointestinal toxicities, although reported incidences vary depending on the characteristics of the study population and the evaluation tools used.73–75
The Arthritis, Rheumatism, and Aging Medical Information System (ARAMIS) prospectively evaluated outcomes including adverse effects from NSAID therapy in more than 36,000 patients with osteoarthritis or rheumatoid arthritis from 17 centers in the United States and Canada, accounting for more than 300,000 patient-years.70 Based on this study population, it was calculated that more than 16,000 NSAID-related deaths could occur yearly in the United States, a number similar to the death rate for leukemia and exceeding the rates for malignant melanoma and asthma.70 The CADEUS (COX-2 inhibitors and NSAIDs: description of users) study of 23,535 coxib and 22,919 traditional NSAIDs (tNSAIDs) identified hospitalizations for gastrointestinal and cardiovascular events.76 There were only 21 hospitalizations for gastrointestinal events, 12 in the coxib cohort and nine in the tNSAID cohort. The rates of gastrointestinal events of 0.39 per 100 patients were considerably lower than observed in older randomized clinical trials and may reflect the increased use of prophylactic proton pump inhibitors in at-risk patients.
When a critically ill patient is prescribed either nonselective or COX-2-selective therapy, evaluation of their gastrointestinal toxicity profile is essential. In non-ICU patients, evidence-based risk factors for gastrointestinal toxicity with chronic use of nonselective NSAIDs include high doses, age older than 65 years, history of previous upper-gastrointestinal ulcers or upper-gastrointestinal bleeding, and use of corticosteroids and/or oral anticoagulants.77–81 Data have also implicated cigarette smoking, alcohol use, and Helicobacter pylori infection as possible risk factors for nonselective NSAID-related gastrointestinal toxicity.62,82 Low-dose aspirin increases the occurrence of serious gastrointestinal complications if it is given concomitantly with either nonselective or COX-2-selective NSAIDs because of the COX-1-inhibiting and local effects of the aspirin.
Some individuals on chronic therapy who have an increased risk of developing nonselective NSAID-related gastrointestinal toxicities may benefit from COX-2-selective agents.76-62 It is unclear whether the risk of gastrointestinal toxicity differs between COX-2 selective and nonselective NSAIDs in critically ill patients. Gastrointestinal toxicity with both nonselective and COX-2-selective agents increases in patients older than 65 years of age; however, COX-2-selective agents pose a lower risk.62 The Celecoxib Long-Term Arthritis Safety Study (CLASS)63 and the Vioxx Gastrointestinal Outcomes Research (VIGOR)64 studies are NSAID safety trials that compared the gastrointestinal side effects of the COX-2-selective agents, celecoxib and rofecoxib, respectively, with those of nonselective NSAIDs.83,84 The primary outcome endpoints of the CLASS study (ulceration, perforation, and bleeding) were similar for celecoxib compared with ibuprofen or diclofenac, whereas the VIGOR trial showed a statistically significant decrease in primary endpoints (ulceration, perforation, bleeding, and symptomatic ulcers) for rofecoxib compared with naproxen. However, after consideration of symptomatic ulcers was added to the primary analysis of CLASS data, a significantly lower incidence of gastrointestinal toxicity was found with celecoxib. In addition, a subanalysis of CLASS that excluded patients receiving concomitant low-dose aspirin therapy revealed a significantly lower rate of gastrointestinal complications with or without inclusion of symptomatic ulcer in the celecoxib group. However, a U.S. Food and Drug Administration (FDA) report concluded that the CLASS study demonstrated no gastrointestinal advantage with celecoxib and requires gastrointestinal warning labeling similar to that required for nonselective NSAIDs.85 Differences in study methodology (study duration, subject selection, primary and secondary outcome endpoints), patient population (osteoarthritis and rheumatoid arthritis in CLASS, rheumatoid arthritis in VIGOR), and concomitant therapy (no aspirin in VIGOR) precluded making valid comparisons between the CLASS and VIGOR data. For critically ill patients who are experiencing gastrointestinal problems and taking nonselective or COX-2-selective NSAIDs, proper evaluation of the gastrointestinal tract is imperative and usually requires upper-gastrointestinal endoscopy. Furthermore, additive bleeding risks (e.g., heparin, other anticoagulants) and underlying disease-related factors are often introduced in the critical care setting. The mortality rate for NSAID-induced ulceration and bleeding is approximately 10%.72 The first step in the management of NSAID-related gastrointestinal ulcers is discontinuation of NSAID therapy.
Pharmacologic treatment options have focused on inhibition of acid secretion and replacement of PG deficiency; these options include proton pump inhibitors (PPIs), histamine-2 receptor antagonists (H2RA), misoprostol, and protective barrier agents.86–88 Suppression of acid by H2RAs can effectively heal gastric and duodenal ulcers on discontinuation of the NSAID.88 PPIs markedly suppress acid secretion and are very effective at healing gastric and duodenal ulcers, even if the NSAID is continued.62,88 PPIs and misoprostol have been shown to be superior to H2RAs for gastric ulcer healing.62,86,88 However, because misoprostol often is not well tolerated, its role in clinical practice is limited. Comparative studies in non-ICU patients of omeprazole, ranitidine, misoprostol, and sucralfate demonstrated a therapeutic advantage of the PPIs that range from 10% to 40%.62,88 For NSAID prophylaxis, PPIs are superior to H2RAs in reducing the risk of both gastric and duodenal ulceration.62,86,88 Although high doses of misoprostol and proton pump inhibitors are effective for preventing NSAID-induced gastric ulcers, misoprostol compliance is poor as a result of gastrointestinal side effects such as nausea and diarrhea.87,88–90
Monitoring for bleeding is another important factor in the critical care setting. The effects of aspirin and NSAIDs on fecal occult blood tests have been evaluated. Aspirin in doses less than 325 mg/d does not interfere with fecal occult blood testing and does not have to be discontinued during stool collection.62,90 Typically, NSAID-induced fecal blood loss is dose dependent and may correlate with the severity of endoscopically detected upper-gastrointestinal lesions.62,90 Fecal occult blood loss associated with COX-2 inhibitor therapy is reported to be similar to that observed with placebo.62,90
Cardiovascular and Renal Toxicity
All NSAIDs have the potential to aggravate hypertension, congestive heart failure, and edema.85 The COX-2-selective agents have been linked to serious cardiovascular side effects when compared with nonselective NSAIDs. This has led to withdrawal of rofecoxib and valdecoxib from the U.S. market, and only celecoxib remains.91 Thromboxane A-2 and prostacyclin (PGI2) are produced from prostaglandins by COX enzymes. TXA2 is expressed most prominently in platelets and leads to enhanced platelet aggregation and vasoconstriction. Conversely, PGI2 is a vasodilator and inhibitor of platelet aggregation. Although the mechanism is not completely understood, the COX-2-selective inhibitors create an imbalance between PGI2 and TXA2 characterized by excessive TXA2-mediated actions. The result is increased vascular tone, platelet aggregation, and vascular smooth muscle proliferation which lead to increased risk of thrombotic events. Not all agents have equal specificity for COX-2 enzymes. Thus, celecoxib is about 10-fold less active than rofecoxib against COX-2 in an in vitro model.9
The VIGOR trial64 reported a significant increase in the incidence of myocardial infarction for rofecoxib compared with naproxen, whereas the CLASS study63 showed no difference in cardiovascular endpoints between celecoxib and ibuprofen or diclofenac. Differences in methodology between the studies, especially the fact that low-dose aspirin was not permitted in the VIGOR trial, make them difficult to compare. In addition, the antiplatelet effects of the nonselective NSAIDs may have influenced the results. The results of the VIGOR trial were confirmed in the Adenomatous Polyp Prevention on Vioxx (APPROVe) trial.92 Compared to placebo, rofecoxib was associated with increased cardiovascular events that included myocardial infarction, cerebrovascular events, and other cardiac events (congestive heart failure, pulmonary edema, or cardiac failure). The subsequent events surrounding the withdrawal of rofecoxib from the market have been thoroughly summarized.93
Since the VIGOR, CLASS, and APPROVe studies, at least two large randomized trials have demonstrated increased risk of cardiovascular events with celecoxib.94,95 The Adenoma Prevention with Celecoxib (APC) trial identified an increased risk of myocardial infarction, stroke, and heart failure compared to placebo. Among the 2000 patients enrolled in the trial, cardiovascular events were observed in 2.5% of patients receiving celecoxib compared to 1.9% of those taking a placebo (relative risk [RR], 1.30; 95% confidence interval [CI], 0.65-2.62).94 A second study evaluating celecoxib, naproxen, or placebo in the Alzheimer Disease Anti-inflammatory Prevention Trial (ADAPT) was stopped early due to increased cardiovascular events.95 Both celecoxib and naproxen demonstrated a higher incidence of cardiovascular and cerebrovascular events (RR 1.10 and 1.63, respectively). Although none of these trials involved critically ill patients, selective COX-2 inhibitors should be used very cautiously in the ICU. Patients with any risk of cardiovascular disease, or patients taking aspirin or other medications for prevention or treatment of cardiovascular diseases, should not receive a COX-2-selective agent.92
Blood pressure elevation has been associated with both nonselective and COX-2-selective agents. Mean blood pressure increases of 3 to 6 mm Hg have been reported with short-term NSAID therapy, especially in patients with preexisting hypertension. Patients with cardiovascular disease, hypertension, renal or hepatic insufficiency, or advanced age should be monitored for fluid retention, which can affect blood pressure during therapy with NSAIDs (including COX-2-selective drugs).96,97 Data from studies of COX-2-selective agents indicate that there is a similar risk of hypertension, leading to labeling precautions for both COX-2-selective and nonselective NSAIDs.97,98 In addition, drug-drug interactions of nonselective NSAIDs or COX-2-selective inhibitors with antihypertensive agents including angiotensin-converting enzyme (ACE) inhibitors, β-adrenergic blockers, and diuretics may accentuate NSAID-mediated inhibition of renal PG production, thereby lessening antihypertensive efficacy.12 Monitoring for hypertension, renal function, and edema is recommended for all patients on NSAID therapy, especially those with the risk factors noted previously.96
Both COX-1 and COX-2 are constitutively expressed in the kidney, predisposing to renal problems with either NSAID class. The adverse effects of nonselective NSAIDs and those of COX-2-selective agents on renal function appear to be similar.98 NSAID-related renal toxicities most commonly include decreased glomerular filtration rate and decreased sodium excretion.95 The deleterious effects of NSAIDs on renal homeostasis are most pronounced in patients with renal insufficiency and volume depletion. The risk of deleterious effects also is increased in patients with hypertension, congestive heart failure, edema, chronic renal failure, advanced age, or concomitant diuretic therapy.96,98,83 Less commonly, patients present with NSAID-induced acute interstitial nephritis, which accounts for fewer than 2% of hospital admissions related to drug-induced renal failure.98 Nephrotic syndrome and papillary necrosis rarely have been associated with chronic NSAID use. Although nephrotic syndrome is generally reversible, papillary necrosis, which occurs most often with NSAID overdose, can result in permanent renal impairment.96 As with nonselective NSAIDs, the renal adverse effects of COX-2-selective agents tend to occur early in therapy and usually are reversible on discontinuation of the drug.98,83
Hypersensitivity Reactions
Hypersensitivity reactions to NSAIDs occur rarely, and they are more common in individuals with nasal polyps or asthma. Allergic reactions including bronchoconstriction, rhinitis, and urticaria have been associated with all nonselective NSAIDs and COX-2-selective inhibitors. Recent data suggest a role of altered COX-2 regulation associated with the aspirin-intolerant asthma/rhinitis syndrome.84 Because of the potential for cross-reactivity, avoidance of all NSAIDs is recommended in patients with a history of bronchoconstriction or allergic reactions to any NSAID.
Various cutaneous reactions have been described with all NSAIDs, especially skin eruptions of a pustular, acneiform nature that are often pruritic. Cessation of NSAID therapy is usually required. Because of its sulfonamide-like chemical structure, celecoxib should not be used in patients with a history of allergic cutaneous and other hypersensitivity reactions to sulfa drugs.13,99 Other less common adverse events associated with NSAIDs include hepatic abnormalities with elevated liver function tests, headache, confusion (especially in older individuals), sleep disturbances, and tinnitus. In rare cases, NSAIDs have been implicated in causing aseptic meningitis and, in children, Reye syndrome.13
Drug-Drug Interactions
Nonselective NSAIDs affect other antiplatelet agents via additive inhibition of platelet aggregation. The result is an increased bleeding risk with the concomitant use of NSAIDs and other antiplatelet agents such as heparin, low-molecular-weight heparin, warfarin, clopidogrel, ticlopidine, lepirudin, and argatroban.13,100 Similarly, concurrent therapy with drotrecogin-α (activated) and NSAIDs must be undertaken cautiously, especially with aspirin at a dose greater than 650 mg or with other NSAID agents.101 A COX-2-selective NSAID may be administered concurrently with warfarin with caution and appropriate monitoring.
Significant drug-drug interactions have been documented with use of NSAIDs and lithium. Both nonselective and COX-2-selective NSAIDs decrease lithium clearance and increase serum lithium concentrations by inhibiting renal PG production and altering intrarenal blood flow. Serum lithium levels and the clinical signs and symptoms of bipolar disorder should be evaluated in patients with concomitant NSAID use.13,100
Data are conflicting regarding the drug-drug interaction potential of ACE inhibitors and NSAIDs. Mixed results of significant drug interactions have been noted in hypertension, coronary artery disease, and congestive heart failure trials, particularly with doses of aspirin greater than 325 mg/day.102 In patients with congestive heart failure, ACE inhibitors increase bradykinin production, resulting in increased synthesis of the vasodilating PGs, prostacyclin and PGE2, which reduces cardiac afterload. By blocking COX and inhibiting the production of PGs, NSAIDs may antagonize the beneficial vasodilatory effects of ACE inhibitors, leading to decreased cardiac output and worsening of heart failure. In addition, concurrent use of ACE inhibitors and NSAIDs may reduce the beneficial effects of renal PGE2 on sodium excretion produced by ACE inhibitors.
Concurrent administration of digoxin and NSAIDs can decrease renal clearance of digoxin, increase plasma drug concentration, and potentiate digoxin toxicity.100
NSAIDs interact with anticonvulsant agents such as phenytoin and valproic acid by displacing the anticonvulsants from their protein-binding sites, which increases the free drug concentration and the potential for anticonvulsant toxicity.100
Combination use of corticosteroids and aspirin can increase renal clearance of salicylate and significantly decrease plasma salicylate concentrations. Therefore, a potential for aspirin toxicity exists when tapering high-dose steroids. Appropriate monitoring is warranted, especially for gastrointestinal bleeding, because of increased salicylate concentrations.100
 Overdose
Overdose
Despite the common use of NSAIDs in clinical practice, serious acute overdose and adverse sequelae have been reported. The National Poison Data System (NPDS) annual report from the American Association of Poison Control Centers documented 25 deaths due to aspirin and 5 from NSAID overdose in the year 2008.103 The mechanism of NSAID toxicity in overdose is related to both their acidic nature and their inhibition of PG production.
Prompt recognition and management of NSAID overdose is important. With salicylate overdose, the severity typically depends on the dose ingested and the salicylate concentration that correlates with the degree of acid-base disturbance. Measurement of salicylate serum levels is important in all cases of aspirin overdose to guide management.103–105 Serum salicylate levels should be measured 4 hours after ingestion and repeated in 2 to 4 hours to determine the peak concentration.102,103 If the acute ingestion was with an enteric-coated product, salicylate levels should be monitored for 12 hours because of the delay in absorption and time to peak concentration.104 Generally, salicylate levels of 300 to 600 mg/L are associated with mild toxicity, 600 to 800 mg/L with moderate toxicity, and greater than 800 mg/L with severe toxicity.103 For nonselective NSAIDs, plasma concentrations are not commonly measured and are less helpful because the half-life of many of these agents is relatively short.103
Although patients with overdoses of aspirin and other NSAIDs may be asymptomatic, depending on the amount ingested, common symptoms include nausea, vomiting, abdominal pain, tinnitus, hearing impairment, and central nervous system (CNS) depression. With higher-dose aspirin ingestion, metabolic acidosis, renal failure, greater CNS changes (e.g., agitation, confusion, coma), and hyperventilation with respiratory alkalosis occur. The presence of acidemia permits more salicylic acid to cross the blood-brain barrier, leading to more severe CNS toxicity.104 In addition, salicylate toxicity can stimulate the respiratory center, leading to hyperventilation and respiratory alkalosis.99 With other nonselective NSAID ingestions, symptoms are similar to those occurring with aspirin overdose.103,105
There is no antidote for salicylate or NSAID poisoning. Management varies depending on the amount of NSAID ingested and is directed at symptomatic support, prevention of further absorption, and correction of acid-base imbalance.103,104 Appropriate hydration should be administered in all overdose situations. Although evidence is limited for the benefit of absorption therapy in aspirin overdose, activated charcoal is often administered within 1 hour after aspirin ingestion and repeated hourly for 4 doses until the salicylate level peaks.78,103,104 Urine alkalinization increases salicylate elimination, especially in adult patients with salicylate levels of 600 to 800 mg/L and in the elderly.103 Because of the relatively neutral pKa of salicylic acid, increasing the urine pH from 5 to 8 is associated with a 10- to 20-fold increase in renal salicylate clearance. Infusion of 1 L of sodium bicarbonate (132 mEq/L) over 3 hours is the recommended regimen for urine alkalinization. The circulating potassium level should also be evaluated because of the acidosis. Consideration of potassium replacement is recommended; however, administration of sodium bicarbonate should not be delayed until potassium levels are stabilized.103,104 In severe cases of aspirin overdose, hemodialysis is effective at removing salicylate and correcting acid-base imbalances and has been shown to reduce morbidity and mortality. Hemodialysis should be considered in patients with salicylate levels greater than 800 mg/L and in the elderly. Hemodialysis also should be considered in patients with metabolic acidosis refractory to treatment, severe and symptomatic CNS toxicity (e.g., coma, convulsions), acute pulmonary edema, or acute renal failure.103,104 Urinary alkalinization should be continued while hemodialysis is administered. Figure 183-1 provides a treatment algorithm for management of salicylate toxicity.
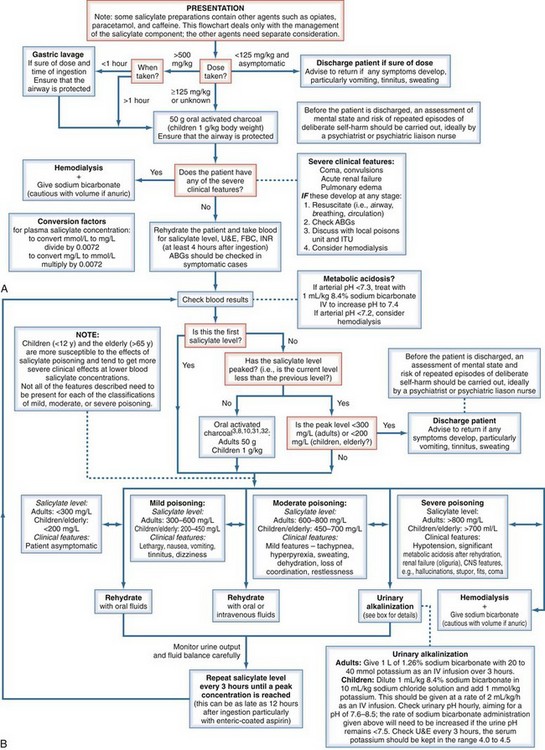
Figure 183-1 Evidence-based flowchart for management of salicylate poisoning.
(From Dargan PI, Wallace CI, Jones AL. An evidence-based flowchart to guide the management of acute salicylate [aspirin] overdose. Emerg Med J 2002;19:206–9, with permission of the BMJ Publishing Group.)
In non-aspirin NSAID overdose, management is also directed toward supportive care. In addition, activated charcoal is useful if administered within 1 hour after ingestion to patients who took more than 100 mg/kg body weight of ibuprofen or more than 10 tablets of other NSAIDs.103 If renal insufficiency occurs with the overdose, NSAID accumulation is more pronounced.105,106 Hepatotoxicity is more commonly seen with diclofenac overdoses, in female patients, in patients older than 50 years of age, and in patients with preexisting autoimmune disease.105
 Summary
SummaryKey Points
Rodriguez LAG, Tolosa LB. Risk of upper gastrointestinal complications among users of traditional NSAIDs and COXIBs in the general population. Gastroenterology. 2007;132:498-506.
Amer M, Bead VR, Bathon J, Blumenthal RS, Edwards DN. Use of nonsteroidal anti-inflammatory drugs in patients with cardiovascular disease. A cautionary tale. Cardiol Rev. 2010:18204-18212.
Silverstein FE, Faich G, Goldstein JL, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis the CLASS study: a randomized controlled trial. JAMA. 2000;284:1247-1255.
1 Vane JR, Botting RM. Mechanism of action of non-steroidal anti-inflammatory drugs. Am J Med. 1998;104:2S-8S.
2 Verburg KM, Maziasz TJ, Weiner E, et al. COX-2 specific inhibitors: Definition of a new therapeutic concept. Am J Ther. 2001;8:49-64.
3 Kean WF, Buchanan WW. The use of NSAIDS in rheumatic disorders 2005;a global perspective. Immunopharmacology. 2005;13:343-370.
4 Mamdani M, Rochon PA, Juurlink DN, et al. Observational study of upper gastrointestinal haemorrhage in elderly patients given selective cyclooxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs. BMJ. 2002;325:624-627.
5 Oviedo JA, Wolfe MM. Gastroprotection by coxibs: What do the Celecoxib Long-term Arthritis Safety Study and the Vioxx Gastrointestinal Outcomes Research Trial tell us? Rheum Dis Clin North Am. 2003;29:769-788.
6 Ofman JJ, Maclean CH, Straus WL, et al. A metaanlaysis of severe upper gastrointestinal complications of nonsteroidal anti-inflammatory drugs. J Rheumatol. 2002;29:804-812.
7 Laine L. Approaches to nonsteroidal anti-inflammatory drug use in the high-risk patient. Gastroenterology. 2001;120:594-606.
8 Hippisley-Cox J, Coupland C, Logan R. Risk of adverse gastrointestinal outcomes in patients taking cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs; population based nested case-control analysis. BMJ. 2005;331:1310-1316.
9 Sooriakumaran P. COX-2 inhibitors and the heart: are all coxibs the same? Post Grad Med J. 2006;82:242-245.
10 Davies NM, Skjodt NM. Choosing the right nonsteroidal anti-inflammatory drug for the right patient: a pharmacokinetic approach. Clin Pharmacokinet. 2000;38:377-392.
11 Needs CJ, Brooks PM. Clinical pharmacokinetics of the salicylates. Clin Pharmacokinet. 1985;10:164-171.
12 Brune K, Neubert A. Pharmacokinetic and pharmacodynamic aspects of the ideal COX-2 inhibitor: A pharmacologist’s perspective. Clin Exp Rheumatol. 2001;19:S51-S57.
13 Dabu-Bondoc S, Franco S. Risk-benefit perspectives in COX-2 blockade. Curr Drug Saf. 2008;3:14-23.
14 Buttar NS, Wang KK. The “aspirin” of the new millennium: Cyclooxygenase-2 inhibitors. Mayo Clin Proc. 2000;75:1027-1038.
15 Fitzgerald GA, Patrono C. Drug therapy: The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001;345:433-442.
16 Smith WL, DeWitt DL, Baravito RM. Cyclooxygenases: Structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145-182.
17 Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97-120.
18 Fiorucci S, Meli R, Bucci M, et al. Dual inhibitors of cyclooxygenase and 5-lipoxygenase: A new avenue in anti-inflammatory therapy. Biochem Pharmacol. 2001;62:1433-1438.
19 Vane JR, Botting RM. Anti-inflammatory drugs and their mechanism of action. Inflamm Res. 1998;47:S78-S87.
20 Marnett LJ, Rowlinson SW, Goodwin DC. Arachidonic acid oxygenation by COX-1 and COX-2. J Biol Chem. 1999;33:22903-22906.
21 Dewitt DL. COX-2-selective inhibitors: The new super aspirins. Mol Pharmacol. 1999;55:526-531.
22 Serhan CN, Bevy B. Success of prostaglandin E2 in structure-function is a challenge for structure-based therapeutics. Proc Natl Acad Sci U S A. 2003;100:8609-8611.
23 Cannon GW, Breedveld FC. Efficacy of cyclooxygenase-2-specific inhibitors. Am J Med. 2001;110:6S-12S.
24 Reuben SS. Update on the role of nonsteroidal anti-inflammatory drugs and coxibs in the management of acute pain. Curr Opin Anaesthesiol. 2007;20:440-450.
25 Ong CKS, Lirk P, Tan CH, Seymour RA. An evidence based update on nonsteroidal anti-inflammatory drugs. Clin Med Res. 2007;5:19-34.
26 Sessler CN, Varney K. Patient-focused sedation and analgesia in the ICU. Chest. 2008;133:552-565.
27 Ito S, Okuda-Ashitak E, Minami T. Central and peripheral roles of prostaglandins in pain and their interactions with novel neuropeptides nociceptin and nocistatin. Neurosci Res. 2001;41:299-332.
28 Buvanendran A, Kroin JS, Tuman KJ, et al. Effects of perioperative administration of a selective cyclooxygenase 2 inhibitor on pain management and recovery of function after knee replacement a randomized controlled trial. JAMA. 2003;290:2411-2418.
29 Dahl V, Raeder JC. Non-opioid postoperative analgesia. Acta Anaesthesiol Scand. 2000;44:1191-1203.
30 Samad TA, Moore KA, Sapirstein A, et al. Interleukin-1b-mediated induction of COX-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471-475.
31 Malchow RJ. The evolution of pain management in the critically ill trauma patient: Emerging concepts from the global war on terrorism. Crit Care Med. 2008;36(Suppl):S346-S352.
32 Bertolini G, Minelli C, Latronico N, et al. The use of analgesic drugs in postoperative patients: The neglected problem of pain control in intensive care units. An observational, prospective, multicenter study in 128 Italian intensive care units. Eur J Clin Pharmacol. 2002;58:73-77.
33 Dionne RA, Khan AA, Gordon AM. Analgesia and COX-2 inhibition. Clin Exp Rheumatol. 2001;19:S63-S70.
34 Aubrun F, Langerdon O, Heitz D, et al. Randomised, placebo-controlled study of the postoperative analgesic effects of ketoprofen after spinal fusion surgery. Acta Anaesthesiol Scand. 2000;44:934-939.
35 Reuben SS, Bhopatkar S, Maciolek H, et al. The preemptive analgesic effect of rofecoxib after ambulatory arthroscopic knee surgery. Anesth Analg. 2002;94:55-59.
36 Reuben SS, Connelly NR. Postoperative analgesic effects of celecoxib or rofecoxib after spinal fusion surgery. Anesth Analg. 2000;91:1221-1225.
37 White PF. The role of non-opioid analgesic techniques in the management of pain after ambulatory surgery. Anesth Analg. 2002;94:577-585.
38 Raeder JC, Seinei S, Vatsgar TT. Oral ibuprofen versus paracetamol plus codeine for analgesia after ambulatory surgery. Anesth Analg. 2001;92:1470-1472.
39 Recart A, Issioui T, White PF, et al. The efficacy of celecoxib premedication on postoperative pain and recovery times after ambulatory surgery: A dose-ranging study. Anesth Analg. 2003;96:1631-1635.
40 Joshi W, Connelly NR, Reuben SS. An evaluation of the safety and efficacy of administering rofecoxib for postoperative pain management. Anesth Analg. 2003;97:35-38.
41 Camu F, Beecher T, Recker DP, et al. Valdecoxib, a COX-2-specific inhibitor, is an efficacious, opioid-sparing analgesic in patients undergoing hip arthroplasty. Am J Ther. 2002;9:43-51.
42 Salari P, Abdollahi M. Controversial effects non-steroidal anti-inflammatory drugs on bone: a review. Inflamm Allergy Drug Targets. 2009;8:169-175.
43 Kwong FN, Harris MB. Recent developments in the biology of fracture repair. J Am Acad Orthop Surg. 2008;16:619-625.
44 Burd TA, Hughes MS, Anglen JO. Heterotopic ossification prophylaxis with indomethacin increases the risk of long-bone nonunion. J Bone Joint Surg Br. 2003;85:700-705.
45 Allami MK, Giannoudis PV. Comment on “Bone formation following intramedullary femoral reaming is decreased by Indomethacin and antibodies to insulin-like growth factors” (Letter to the editor). J Orthop Trauma. 2003;17:390-391.
46 Wheatley RG. Analgesic efficacy of ketorolac. Acta Anaesth Belg. 1996;47:135-142.
47 Gillis JC, Borgden RN. Ketorolac a reappraisal of its pharmacodynamic and pharmacokinetic properties and therapeutic use in pain management. Drugs. 1997;53:139-188.
48 Spodick DH. Acute pericarditis: Current concepts and practice. JAMA. 2003;289:1150-1153.
49 Maisch B, Ristic AD. Practical aspects of the management of pericardial disease. Heart. 2003;89:1096-1103.
50 Ioannou Y, Isenberg DA. Current concepts for the management of systemic lupus erythematosus in adults: A therapeutic challenge. Postgrad Med J. 2002;78:599-606.
51 Fam AG. Treating acute gouty arthritis with selective COX-2 inhibitors: preliminary evidence supports their relative efficacy and safety. BMJ. 2002;325:180-181.
52 Almekinders LC, Temple JD. Etiology, diagnosis, and treatment of tendonitis: An analysis of the literature. Med Sci Sports Exerc. 1998;30:1183-1190.
53 Patrono C, Baigent C, Hirsh J, Roth G. Antiplatelet drugs: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines (8th Edition). Chest. 2008;133:199S-233S.
54 Hayden M, Pignone M, Phillips C, et al. Aspirin for the primary prevention of cardiovascular events: A summary of the evidence for the U.S. preventive services task force. Ann Intern Med. 2002;136:161-172.
55 Wolff TW, Miller T, Ko S. Aspirin for the primary prevention of cardiovascular events: An update of the evidence for the U.S. preventive services task force. Ann Intern Med. 2009;150:405-410. 67
56 Solomon DH, Glynn RJ, Levin R, et al. Nonsteroidal anti-inflammatory drug use and acute myocardial infarction. Arch Intern Med. 2002;162:1099-1104.
57 Patrono C, Garcia Rodriquez L, Landolfie R, et al. Low-dose aspirin for the prevention of atherothrombosis. N Engl J Med. 2005;353:2373-2383.
58 Albers GW, Amarenco P, Easton JD, et al. Antithrombotic and thrombolytic therapy for ischemic stroke. Chest. 2001;119:300S-320S.
59 Simmons DL, Wagner D, Westover K. Nonsteroidal anti-inflammatory drugs, acetaminophen, cyclooxygenase 2, and fever. Clin Infect Dis. 2000;31:S211-S218.
60 Plaisance KI, Mackowiak PA. Antipyretic therapy: Physiologic rationale, diagnostic implications, and clinical consequences. Arch Intern Med. 2000;160:449-456.
61 Mackowiak PA. Concepts of fever. Arch Intern Med. 1998;158:1870-1881.
62 Lanas A, Soena F. Nonsteroidal anti-inflammatory drugs and lower gastrointestinal complications. Gastroenterol Clin North Am. 2009;38:333-352.
63 Silverstein FE, Faich G, Goldstein JL, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis the CLASS study: A randomized controlled trial. JAMA. 2000;284:1247-1255.
64 Bombardier C, Laine L, Reicin A, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N Engl J Med. 2000;343:1520-1528.
65 Plaisance KI. Toxicities of drugs used in the management of fever. Clin Infect Dis. 2000;31:S219-S223.
66 Minneci P, Deans K, Natanson C, Eichacker PQ. Increasing the efficacy of anti-inflammatory agents used in the treatment of sepsis. Eur J Clin Microbiol Infect Dis. 2003;22:1-9.
67 Bernard GR, Wheeler AP, Russell JA, et al. The effects of ibuprofen on the physiology and survival of patients with sepsis. N Engl J Med. 1997;336:912-918.
68 Dellinger RP, Levy MM, Carlet JM, et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36:296-327.
69 Meurer LN. Treatment of peptic ulcer disease and nonulcer dyspepsia. J Fam Pract. 2001;50:614-619.
70 Singh G, Ramey DR. NSAID-induced gastrointestinal complications: The ARAMIS perspective—1997. J Rheumatol. 1998;25(Suppl 51):8-16.
71 Rivkin A. Admissions to a medical intensive care unit related to adverse drug reactions. Am J Health-Syst Pharm. 2007;64:1840-1843.
72 Wolfe MM, Lichtenstein DV, Singh G. Medical progress: Gastrointestinal toxicity of nonsteroidal anti-inflammatory drugs. N Engl J Med. 1999;340:1888-1899.
73 Scheiman JM, Hindley CE. Strategies to optimized treatment with NSAIDS in patients at risk for gastrointestinal and cardiovascular adverse events. Clin Ther. 2010;32:667-677.
74 Hsiang KW, Chen TS, Lin HY, et al. Incidence and possible risk factors for clinical upper gastrointestinal events in patients taking selective cyclooxygenase-2 inhibitors: A prospective, observational cohort study in Taiwan. Clin Ther. 2010;32:1294-1303.
75 Rodriguez LAG, Tolosa LB. Risk of upper gastrointestinal complications among users of traditional NSAIDs and COXIBs in the general population. Gastroenterology. 2007;132:498-506.
76 Laharie D, Droz-Perroteau C, Benichou J, et al. Hospitalizations for gastrointestinal and cardiovascular events in the CADEUS cohort of traditional or coxib NSAID users. Br J Clin Pharmacol. 2010;69:295-302.
77 Schoenfeld P. An evidence-based approach to the gastrointestinal safety profile of COX-2-selective anti-inflammatories. Gastroenterol Clin North Am. 2001;30:1027-1045.
78 Wolfe F, Anderson J, Burke TA, et al. Gastroprotective therapy and risk of gastrointestinal ulcers: Risk reduction by COX-2 therapy. J Rheumatol. 2002;29:467-473.
79 Hawkey CJ. Gastrointestinal safety of COX-2 specific inhibitors. Gastroenterol Clin North Am. 2001;30:921-937.
80 Simon LS. COX-2 inhibitors: Are they nonsteroidal anti-inflammatory drugs with a better safety profile? Gastroenterol Clin North Am. 2001;30:1011-1025.
81 Lane JM. Anti-inflammatory medications: Selective COX-2 inhibitors. J Am Acad Orthop Surg. 2002;10:75-78.
82 Calam J. Clinical science of Helicobacter pylori infection: Ulcers and NSAIDs. Br Med Bull. 1998;54:55-62.
83 Harris CJ, Brater DC. Renal effects of cyclooxygenase-2 selective inhibitors. Curr Opin Nephrol Hypertens. 2001;10:603-610.
84 Picado C, Fernandez-Morata JC, Juan M, et al. Cyclooxygenase-2 mRNA is down expressed in nasal polyps from aspirin-sensitive asthmatics. Am J Respir Crit Care Med. 1999;160:291-296.
85 Risser A, Donovan D, Heintzman J, Page T. NSAID prescribing precautions. Am Fam Physician. 2009;80:1371-1378.
86 Agrawal NM, Campbell DR, Safdi MA, et al. Superiority of lansoprazole vs. ranitidine in healing nonsteroidal anti-inflammatory drug-associated gastric ulcers: Results of a double-blind, randomized, multicenter study. Arch Intern Med. 2000;160:1455-1461.
87 Hawkey CJ, Karrasch JA, Szczepanski L, et al. Omeprazole compared with misoprostol for ulcers associated with nonsteroidal anti-inflammatory drugs. N Engl J Med. 1998;338:727-734.
88 Lanza FL, Chan FKL, Quigley EMM. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol. 2009;104:728-738.
89 Greenberg PD, Cello JP, Rockey DC. Relationship of low-dose aspirin to GI injury and occult bleeding: A pilot study. Gastrointest Endosc. 1999;50:618-622.
90 Hunt RH, Bowen B, Mortensen ER. A randomized trial measuring fecal blood loss after treatment with rofecoxib, ibuprofen, or placebo in healthy subjects. Am J Med. 2000;109:201-206.
91 Bresalier RS, Sandler RS, Quan H, et al. Adenomatous polyp prevention on Vioxx (APPROVe) trial investigators. Cardiovascular events associated with rofecoxib in colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092-1102.
92 Amer M, Bead VR, Bathon J, Blumenthal RS, Edwards DN. Use of nonsteroidal anti-inflammatory drugs in patients with cardiovascular disease. A cautionary tale. Cardiol Rev. 2010:18204-18212.
93 Bennett JS, Daugherty A, Herrington D, et al. The use of non-steroidal anti-inflammatory (NSAIDs): a science advisory from the American Heart Association. Circulation. 2005;111:1713-1716.
94 Solomon SD, McMurray JJ, Pfeffer MA, et al. Adenoma Prevention with Celecoxib (APC) Study Investigators. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071-1080.
95 ADAPT Research Group. Cardiovascular and cerebrovascular events in the randomized, controlled Alzheimer’s Disease Anti-Inflammatory Prevention Trial (ADAPT). PLoS Clin Trials. 2006;1:e33.
96 LeLorier J, Bombardier C, Burgess E, et al. Practical considerations for the use of nonsteroidal anti-inflammatory drugs and cyclo-oxygenase-2 inhibitors in hypertension and kidney disease. Can J Cardiol. 2002;18:1301-1308.
97 Frishman WH. Effects of nonsteroidal anti-inflammatory drug therapy on blood pressure and peripheral edema. Am J Cardiol. 2002;89:18D-25D.
98 Komers R, Anderson S, Epstein M. Renal and cardiovascular effects of selective cyclooxygenase-2 inhibitors. Am J Kidney Dis. 2001;38:1145-1157.
99 Celebrex (celecoxib) [Manufacturer product information]. New York: Pfizer, Inc., June 2009.
100 Verbeeck RK. Pharmacokinetic drug interactions with nonsteroidal anti-inflammatory drugs. Clin Pharmacokinet. 1990;19:44-66.
101 Xigris (drotrecogin alpha) [Manufacturer product information]. Indianapolis, IN, Eli Lilly and Company, October 2008.
102 Takkouche B, Etminan M, Caamano F, et al. Interaction between aspirin and ACE inhibitors resolving discrepancies using a meta-analysis. Drug Saf. 2002;25:373-378.
103 Bronstein AC, Spyker DA, Cantilena LR, Green JL, Rumack BH, Giffin SL. 2008 annual report of the American Association of Poison Control Centers national poison data system (NPDS): 26th annual report. Clin Toxicol. 2009;47:911-1084.
104 Dargan PI, Wallace CI, Lones AL. An evidence-based flowchart to guide the management of acute salicylate (aspirin) overdose. Emerg Med J. 2002;19:206-209.
105 Volans G, Hartley V, McCrea S, et al. Non-opioid analgesic poisoning. Clin Med. 2003;3:119-123.
106 Al-Harbi NN, Domrongkitchaiporn S, Lirenman DS. Hypocalcemia and hypomagnesemia after ibuprofen overdose. Ann Pharmacother. 1997;31:432-434.
