[level-membership-for-gastroenterology-and-hepatology-category]
Chapter 29 Nonepithelial Tumors of the Esophagus and Stomach
 Video related to this chapter’s topics
Video related to this chapter’s topicsEpidemiology
The clinical starting point for patients with lesions of nonepithelial origin is the discovery of a mass impinging on the GI tract mucosa from beneath—the so-called submucosal tumor (Fig. 29.1). In its classic form, a discrete tumorous appearance is present with overlying mucosa that, although most commonly bland, may be erythematous, pale, dimpled, or ulcerated. Lesions are often initially identified at esophagogastroduodenoscopy (EGD), but the patient may also be referred to an endoscopist for evaluation of an abnormal radiograph (e.g., barium-contrast examination).
Applied literally, the term submucosal would imply the presence of an intramural mass originating in the submucosal layer of the GI wall. However, the term has come to be used for a range of lesions that create a similar appearance, including intramural and extramural structures. Such submucosal tumors may include both neoplastic and nonneoplastic masses, and even mucosal neoplasms have been reported to exhibit a submucosal appearance.1 Examples of nonepithelial lesions in all four categories are listed in Table 29.1. This discussion centers on neoplasms that primarily originate from nonepithelial GI tract cell lines, but all types of pathology must be considered when developing a management plan.
Table 29.1 Types of Masses Causing Esophageal and Gastric Submucosal Tumors
| Neoplastic Masses | Nonneoplastic Masses | |
|---|---|---|
| Intramural masses | Stromal cell tumor | Varices |
| Lipoma | Duplication cyst | |
| Granular cell tumor | Inflammatory granuloma | |
| Lymphoma | Foreign body (e.g., surgical suture or clip) | |
| Fibrovascular polyp | Pancreatic rest | |
| Hemangioma/hemangiosarcoma | ||
| Lymphangioma/lymphangiosarcoma | ||
| Metastatic neoplasm | ||
| Extramural masses | Primary neoplasm of adjacent organs (benign and malignant) | Benign lymph node |
| Metastatic lymph node | Inflammatory mass of adjacent organs (e.g., pancreas, spleen) | |
| Organomegaly (e.g., spleen, liver) |
Depending on the clinical circumstances and type of tumor, the lesion may cause symptoms such as bleeding, obstruction, or pain. However, such lesions commonly are serendipitously found during evaluation for a different, unrelated problem. Because most such lesions are asymptomatic, epidemiologic data are skewed by the nature of their discovery incidental to a different, usually unrelated condition. In one study of 15,104 EGD reports, submucosal tumors were identified in 0.36%.2 Because most in this series were life-threatening tumors, the study database likely underreported less serious lesions. Many such lesions turn out to be normal extramural organs. Allgayer3 found that among 30 patients referred for submucosal tumors, normal extramural structures were present in 14 (47%). Motoo and coworkers4 also reported normal organs in 16 of 19 submucosal tumors, as did Caletti and colleagues5 in 10 of 25 tumors; organs identified include the spleen, liver, splenic vessels, and pancreas.
Because submucosal tumors are often left in situ, the pathologic distribution among tumors is unknown. It is reported that 1% to 3% of resected gastric tumors are stromal cell tumors6; it can be inferred that the actual incidence, when including tumors that were not resected, is considerably higher. In a small prospective study,7 among 45 submucosal tumors, most were found to have a benign appearance that required no follow-up. From these available data, it may be cautiously concluded that submucosal tumors are found in less than 1% of routine upper endoscopy examinations, half of such lesions are found to be normal extramural structures, most remaining lesions are benign, and stromal cell tumors constitute most such neoplasms.
Clinical Features
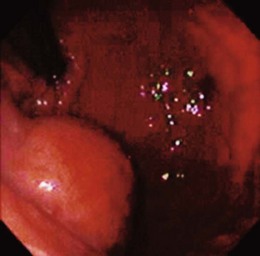
Lumps and bumps of all sorts are regularly encountered in endoscopic examinations; the decision regarding which to evaluate further depends on the endoscopic appearance, the clinical circumstances, and the inclination of the endoscopist. Because few standardized guidelines exist to direct such decisions, great variation in practice exists. Symptoms attributed to the mass nearly always drive further investigation, but our own endoscopic ultrasound (EUS) study of a subset of such lesions found that nearly 90% were asymptomatic.8 GI bleeding may be seen in many submucosal lesions, most commonly in the form of slow blood loss causing iron deficiency anemia. The surface of the tumor may be ulcerated in such cases (Fig. 29.2). Malignant tumors may be more prone to ulceration and bleeding9; this might be taken as a sign of a potentially malignant form that compels definitive treatment. However, benign lesions may also cause severe bleeding,10 and occasionally rapid hemorrhage may occur.11 Less often, GI tract obstruction may be caused by such masses,12 especially if the lesion is located in a narrow area such as the esophagogastric junction or pylorus; intussusception caused by such masses has been reported.13 Pain may be a presenting complaint, especially if the submucosal tumor is neoplastic or malignant.14
Because most lesions are incidentally found during endoscopic examination for another problem, the clinical features of submucosal masses are primarily those that, in the endoscopist’s opinion, compel further evaluation. Large size has been proposed as an ominous finding,15 and lesions with an ulcerated or irregular (lumpy) surface often undergo additional testing or treatment. Patients with submucosal tumors who have a prior history of malignancy should receive further evaluation to exclude metastatic disease. Finally, patients with submucosal lesions that change appearance on serial examination are usually directed by an alert clinician to further testing.
Pathology
Extramural masses compose half of suspected submucosal tumors and include normal organs, nonneoplastic masses, and extramural neoplasms. Normal liver, spleen, pancreas, gallbladder, colon, and kidney all have been reported to appear as suspected submucosal tumors.3–5 Vascular structures often produce the appearance of a discrete tumor, including normal vessels of the spleen16 and abnormal vessels such as varices and aneurysms.17 Neoplasms and nonneoplastic masses involving these same organs can also produce this appearance, as can such masses involving the peritoneum, mediastinum, and the lymph nodes adjacent to the upper GI tract. The various malignancies, cysts, and inflammatory masses of these structures need no further elaboration here because a large variety of such findings have been noted in the case report literature.
Masses that arise within the wall of the esophagus and stomach require further discussion, particularly because many are peculiar to the GI tract. Most neoplasms in this category are mesenchymal tumors, meaning that they arise from cells of mesodermal origin. Most such neoplasms are clinically benign, although, as shown subsequently, tumor histology may not provide reliable clues to malignant behavior. A large variety of such neoplasms have been described (Table 29.2), but most are exceedingly rare. The tumors most likely to be encountered in the esophagus and stomach in a routine clinical setting are discussed here.
Table 29.2 Classification of Gastrointestinal Mesenchymal Tumors
| Tumor Type | Examples |
|---|---|
| Stromal tumors | Smooth muscle tumors (leiomyoma, leiomyosarcoma), glomus tumors, leiomyomatosis, pleomorphic sarcoma |
| Neural tumors | Neuroma/neurofibroma, paraganglioma, ganglioneuromatosis |
| Endothelial and vascular tumors | Hemangioma, hemangiosarcoma, Kaposi’s sarcoma, lymphangioma |
| Lipocytic tumors | Lipoma, liposarcoma, lipohyperplasia (ileocecal valve), lipomatosis (colon) |
| Granular cell tumor | Granular cell tumor |
| Inflammatory fibroid polyp | Inflammatory fibroid polyp |
| Fibrohistiocytic tumors | Fibrovascular polyp, fibrous histiocytoma, desmoid tumors (mesentery), fibroepithelial polyp |
| Striated muscle tumors | Rhabdomyosarcoma |
Data from Lewin K, Riddel RH, Weinstein WM: Mesenchymal tumors. In: Gastrointestinal pathology and its clinical implications, New York, 1992, Igaku-Shoin, pp 284–341.
Gastrointestinal Stromal Tumor
Most mesenchymal GI tumors are pale, firm, spherical, or ovoid structures embedded in the wall of the affected organ. The microscopic appearance of musclelike eosinophilic, spindle-shaped cells in uniform sheets and the proximity of the tumors to the muscular wall layers led early observers to believe that these tumors were of myogenic origin18—hence the name leiomyoma and its variations (e.g., leiomyosarcoma, leiomyoblastoma). However, it later became clear that these neoplasms not only are not of obvious myogenic origin but also often lack any specific markers of differentiation whatsoever.19 Immunohistochemical analyses showed variable expression of smooth muscle features such as desmin and actin and neural proteins such as S-100.20 For the sake of clarity, these lesions came to be referred to as gastrointestinal stromal tumors (GISTs), an acknowledgment that they originate in mesenchymal stroma.
A significant breakthrough occurred with the discovery that most GI stromal tumors stain positive for a specific membrane protein, designated CD117,21 and subsequently identified as KIT, a tyrosine kinase receptor.22 Tyrosine kinases are a class of transmembrane receptors that mediate various cellular growth functions. The extracellular portion of KIT binds stem cell factor, causing a conformational change. The receptor subsequently forms a dimer with another activated KIT receptor, and the intracellular region of the resulting dimer triggers various cell signaling cascades.23 Among these are proliferation pathways such as mitogen-activated protein kinase (MAPK) and signal transducers and activators of transcription (STAT) proteins as well as antiapoptotic mediators such as AKT (protein kinase B).24 KIT and some other tyrosine kinases are important regulators of cell growth and proliferation.
Abnormal cell growth is a fundamental element of cancer physiology, and hyperfunction of the KIT receptor can lead to neoplasia. In this case, numerous gain-of-function mutations in the KIT gene have been described.25 These acquired (or, rarely, inherited) mutations produce KIT receptors that are capable of initiating the aforementioned intracellular growth cascades without activation by stem cell factor. The resulting dysregulated growth seems to be an important initial step to tumorigenesis. This concept is supported by the observation that nearly all GISTs express KIT. It has further been observed that the interstitial cells of Cajal (ICC) share some phenotypic and ultrastructural similarities with GIST and normally express the KIT receptor; this observation has led to the current hypothesis that GISTs arise from ICC26 or from ICC precursor cells. Finally, it has been noted that this gain-of-function mutation is not found in true leiomyomas.27 Some pathologists have suggested that CD117 positivity is required to confirm a diagnosis of GIST.28 It is now known, however, that a few otherwise obvious GISTs do not express KIT. Many of these tumors have been identified as expressing platelet-derived growth factor receptor α (PDGFRα), another distinct tyrosine kinase receptor that also mediates many of the same growth and antiapoptotic proliferation pathways.29 Several gain-of-function mutations have been reported in the PDGFRα gene, and the resulting tumors are otherwise indistinguishable from GISTs that are KIT-positive.
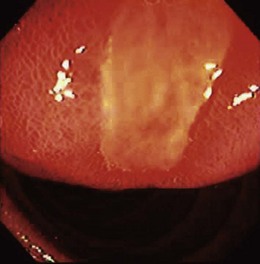
Nearly all upper GI tumors of this type occur in the stomach, but duodenal lesions have been described.30 Most esophageal stromal tumors lack the CD117 protein and may be true leiomyomas.28 The endoscopic appearance is a dome-shaped, firm submucosal mass; central umbilication or frank ulceration is common, and there may be a lobulated or irregular appearance. The tumors are most often solitary except in the case of specific disease entities such as Carney’s triad (GIST, pulmonary chondroma, and extraadrenal paraganglioma). Germline KIT mutation kindreds have been described,31 however, in which case multiple GISTs are seen. Giant sizes of greater than 10 cm have been noted, but most tumors are less than 3 cm.
Pathologically, the tumor usually consists of uniform pale tissue, although hemorrhagic and necrotic areas may be seen. Microscopically, the cells are spindle-shaped with uniform nuclei and general cytologic uniformity. Some cell groups may show epithelioid configurations (closely packed polygonal cells), and there may be nuclear pleomorphism. It has been observed more recently that the histologic pattern is sometimes related to the nature of the underlying genetic abnormality; KIT mutations at exon 13 or 17 more often show spindle-cell morphology,32 whereas PDGFRα GISTs often exhibit epithelioid histology.33 Ultrastructural cellular abnormalities have also been linked to specific gene mutations.34 However, malignant behavior has not been associated with specific mutation patterns.
It has long been known that malignant behavior in GISTs is difficult to predict given the relatively bland cytology and slow growth of these neoplasms. It has been reported that even small, benign-appearing stromal tumors have been known to metastasize.35 This discovery led to considerable confusion about the appropriate criteria to categorize these tumors as benign or malignant. Older pathologic scoring systems relied on numerous histologic features19 and were plagued with problems. More recent attention has focused on the size of the tumor and the number of mitoses observed (mitotic index), at least in part because these are easily quantifiable findings. In one study of 100 cases, tumors with more than 5 mitoses per 10 high-power fields (HPFs) were significantly more likely to metastasize, although 40% of malignant lesions in that study had fewer mitoses.36 In another study, multivariate analysis of various clinical and pathologic features in 122 specimens showed that more than 10 mitoses/50 HPFs correlated with poor outcome, whereas site, epithelioid histology, and tumor size were not independently predictive.37
Attempts to correlate tumor marker status such as CD117 positivity with malignant behavior have produced generally confusing or negative results,38 as have studies of specific KIT mutations27,39 and other tumor markers.40,41 Older GIST scoring systems have been abandoned following a National Institutes of Health (NIH) consensus conference, which defined malignancy risk based on size and mitotic index alone.42,43 The NIH criteria divide tumors into four categories of malignant risk (Table 29.3), an acknowledgment that even the most innocent lesion poses a slight but definite risk of malignant behavior.
Table 29.3 National Institutes of Health Criteria for Malignant Risk in Gastrointestinal Stromal Tumors
| Risk Level | Size (cm) | Mitoses/50 HPF |
|---|---|---|
| Very low risk | <2 | <5 |
| Low risk | 2–5 | <5 |
| Intermediate risk | <5 | 6–10 |
| 5–10 | <5 | |
| High risk | >5 | >5 |
| >10 | Any | |
| Any | >10 |
HPF, high-power field.
Data from Berman JJ, O’Leary TH: Gastrointestinal stromal tumor workshop. Hum Pathol 32:578–582, 2001; Toquet C, Le Neel JC, Guillou L, et al: Elevated (> or = 10%) MIB-1 proliferative index correlates with poor outcome in gastric stromal tumor patients: A study of 35 cases. Dig Dis Sci 47:2247–2253, 2002; Hedenbro JL, Ekelund M, Wetterberg P: Endoscopic diagnosis of submucosal gastric lesions: The results after routine endoscopy. Surg Endosc 5:20–30, 1991.
If all such neoplasms entail malignant risk, prudence might dictate that they should always be resected. However, more recent data suggest that GISTs are more common than previously thought. Up to 10% of resection and autopsy specimens contain such tumors,44 and microscopic GISTs (also called seedling GISTs, minimal GISTs, or GIST tumorlets) can be seen in 35% of some patient groups.45,46 These incidental and microscopic GISTs display the same KIT and PDGFRα mutations as their larger counterparts. The finding that synchronous GISTs from the same patient regularly show different gene mutations (and are independent sporadic GISTs) confuses the picture further.47 GISTs that were earlier classified as metastatic or recurrent may have been distinct neoplastic events. If it turns out that most small and asymptomatic GISTs stay that way, a conservative approach may be the most prudent.
A second breakthrough in GISTs has been the development of tyrosine kinase inhibitors such as imatinib mesylate that are effective in reducing the KIT enzyme activity and that are useful for tumor treatment. Imatinib targets the specific abnormal enzyme activity in the neoplasm and does not rely on generalized cytotoxicity for its effect. In an open label study of 147 patients with unresectable malignant GISTs, an overall response rate of 38% was seen.48 Among responders, results are often dramatic (Fig. 29.3). The recognition of the malignant potential of GIST, combined with the availability of effective treatment even for unresectable disease, has compelled new thinking in the accurate diagnosis of this neoplasm.
Glomus Tumors
Glomus tumors are paragangliomas that generally occur in the skin; a morphologically similar lesion has long been known in the stomach49 and is usually classified with GISTs. They are typically located in the antrum and are generally small, although they can range up to 5 cm.50 An immunohistochemical study of 32 cases showed that all examined glomus tumors were negative for desmin, S-100, and KIT,51 suggesting a different histogenesis from leiomyomas, neuromas, and GISTs. In this same study, one patient died of metastatic disease, suggesting a malignancy risk that is low but not zero.
Endothelial and Vascular Tumors
Cavernous hemangiomas are vascular neoplasms that rarely occur in the GI tract and even more rarely in the upper tract. They appear as sessile red or blue nodules and can be difficult to distinguish from vascular ectasias (which are not neoplastic). A malignant counterpart, hemangiosarcoma, is exceedingly rare but has been described in nearly all parts of the GI tract.52 Lymphatic tumors are also exceedingly rare in the GI tract, primarily being reported in the duodenum. They are described as having a smooth, sessile, polypoid translucent appearance endoscopically.53 Histologically, they are mucosal or submucosal and contain hamartomatous rather than true neoplastic elements52; they are always benign.
Lipocytic Tumors: Lipoma and Liposarcoma
Submucosal lipomas are usually harmless neoplasms arising from submucosal adipocytes. They are most common in the colon but may appear in any part of the GI tract, particularly the gastric antrum.54 The typical endoscopic appearance is a pale yellowish, soft submucosal tumor; usually a solitary lesion is seen. The overlying mucosa can sometimes be tented up with a biopsy forceps, and the lesion deforms easily when pushed with the forceps. Histologically, such tumors are encapsulated and consist of typical benign mature lipocytes. When they contain a large number of blood vessels, they are sometimes called angiolipomas. It is rarely necessary to evaluate them further or remove them unless they cause bleeding or obstruction. They are normally small, but giant lipomas have been described55 that can cause obstruction or intussusception.56 The clinically aggressive malignant form, liposarcoma, is exceedingly rare.57
Granular Cell Tumor
The esophagus is the most common site of granular cell tumors, but they may be found throughout the GI tract. They are most often located in the submucosa and often have a polypoid shape. Multiple tumors are common. They may be of Schwann cell origin and consist of masses of histiocytelike cells containing periodic acid–Schiff–positive cytoplasmic granules. A literature review of 117 cases found dysphagia as the most common symptom in roughly half of patients; three-fourths were smaller than 2 cm.58 It is uncertain that there is a malignant form of the neoplasm; the same review found that in four locally invasive cases distant metastases were not noted. They generally appear as yellowish, plaquelike, round or oval lesions less than 2 cm across.59 The overlying squamous epithelium of the esophagus may show pseudoepitheliomatous hyperplasia.60 If this is misinterpreted as metaplastic mucosal transformation, confusion may lead to additional investigations in search of an epithelial neoplasm.
Inflammatory Fibroid Polyp
Inflammatory fibroid polyps are uncommon tumors that can occur anywhere in the GI tract; the stomach is the most common site,19 but they have been described elsewhere.61 Pathologically, they consist of nonencapsulated myxoid stroma that includes blood vessels, inflammatory cells, and invariably eosinophils. The eosinophilic infiltrate had previously led to the (now discarded) designation of this tumor as localized eosinophilic gastroenteritis. They are often small, but giant tumors have been described that can cause (as usual) bleeding, obstruction, or intussusception.62 Current theories of their pathogenesis focus on myofibroblasts or fibroblasts,63 but the cell of origin is unknown. They tend to originate in the muscle layers of the GI tract wall, beginning as an intramural bulge but later assuming a polypoid shape; there may often be a significant extramural (subserosal) extension. There seems to be no malignant potential.
Fibrous (Fibrovascular) Polyp
Fibrous polyps are esophageal tumors that can grow to an enormous size. They occur predominantly in male patients, usually originate in the upper esophagus, and often assume a polypoid shape.64 Tumors greater than 15 cm have been described65 and can cause dysphagia, globus, bleeding, and asphyxiation from laryngeal impaction.
Although these tumors may have ulcerated overlying mucosa, the sheer size of the lesion may make it difficult to identify endoscopically.66 Histologically, a large variety of cytologic elements are seen, including spindle cells, lipocytes, mononuclear inflammatory cells, and vascular connective tissue. However, because some typical characteristics of inflammatory fibroid polyps are lacking and because of differing epidemiology and location, these lesions are classified separately from inflammatory fibroid polyps and should not be confused with them (despite the similar names). They are of uncertain histogenesis but do not seem to possess malignant potential. Nevertheless, because fatal outcomes have been described arising from the mechanical size of these tumors, removal is considered prudent.
Metastatic Tumors
Intramural metastases to the GI tract are less common than compression or invasion from extramural cancer. In a study of gastric mural metastases, the most common primary sites were lung, breast, and esophagus, but malignant melanoma was the most frequent tumor to metastasize to the stomach. Half appeared endoscopically as submucosal tumors, and the rest were ulcerated or fungating; one-third showed multiple lesions.67
Cystic Tumors
Various cystic intramural lesions may manifest as submucosal tumors. In an individual patient, it may be unclear until EUS is performed that these tumors are cystic. However, it is worth collectively listing lesions (some of which have already been discussed) that can assume such an appearance. Small lymphatic ectasias are a common endoscopic finding,52 particularly in the duodenum, and represent cystic structures less than 5 mm in size; they are not neoplasms. Duplication cysts are another nonneoplastic intramural cyst that may be seen as a submucosal tumor. These are very rarely encountered lesions in the upper GI tract68,69 and are due to embryonic epithelial nodules that fail to regress. Brunner’s gland hamartomas and heterotopic pancreas have also been described as having a cystic appearance.70 Among neoplastic cysts, lymphangiomas71 and hemangiomas72 (and hemangiosarcomas) assume a cystic appearance. In addition, any malignant neoplasm with central necrosis may appear as a cystic intramural lesion.
Differential Diagnosis
Conventional Endoscopy, Computed Tomography, and Transabdominal Ultrasound
Confronted with a submucosal tumor, the alert endoscopist should not permit the presence of mucosa between the endoscopic camera and the mass to preclude preliminary conclusions about the nature of the unseen tumor. Despite the multitude of possibilities already discussed, a short list of half a dozen lesions covers nearly everything likely to be found in all but the largest referral centers. Each has distinctive clinical and endoscopic features (Table 29.4) to provide an adequate preliminary assessment, which can be used to direct further management.
Table 29.4 Endoscopic Appearance of Typical Upper Gastrointestinal Submucosal Tumors
| Clinical Characteristics | Most Likely Tumor |
|---|---|
| Lower esophagus: <2 cm, plaquelike, firm, yellowish, sometimes multiple | Granular cell tumor |
| Upper esophagus: firm, large, polypoid | Fibrous (fibrovascular) polyp |
| Stomach: ovoid, firm, any size, single lesion | GIST |
| Stomach: <1 cm, firm, dimpled | Heterotopic pancreas |
| Any organ: translucent, soft | Lymphangioma |
| Any organ: soft, yellowish, soft, compressible, any size, single lesion | Lipoma |
GIST, gastrointestinal stromal tumor.
Routine endoscopic pinch biopsies are often performed but rarely yield diagnostic material, even when large size (jumbo) pinch forceps are used.73 Other efforts to obtain tissue during routine endoscopy have been described, including standard fine needle aspiration (FNA) needles,74 a special guillotine aspiration biopsy needle,75 and mucosal stripping after forceps biopsy76; these show variable success but have not yet gained widespread acceptance.
Conventional computed tomography (CT) has traditionally been unhelpful in evaluating intramural tumors because of their small size, but new CT methods show promise. Multidetector high-resolution CT scanners can identify most tumors larger than 1 cm, although further characterization may be difficult.77 Three-dimensional computerized reconstruction techniques are also able to detect intramural tumors reliably and may provide useful images.78 Ongoing improvements in CT resolution may yet result in clinically important images, but current conventional technology is not generally worthwhile.
Conventional transabdominal ultrasound has likewise been unhelpful in this setting, but similar to CT useful progress is being reported. High-resolution transabdominal ultrasound, using transducer frequencies of 5 MHz or 7.5 MHz and a water-filled stomach, provides remarkable imaging of tumors 10 mm in size and their relationship to the five-layer GI tract wall.79 Although unsuitable for obese patients or many anatomic tumor locations, this technique could potentially replace many EUS examinations of such tumors.
Endoscopic Ultrasound
High-resolution scanning from an intraluminal position makes EUS ideally suited for evaluation of submucosal tumors and for complete characterization of intramural masses that are identified. Extramural impression from normal and abnormal structures can be reliably shown, and intramural masses can be readily characterized. The ability to relate intramural tumors to the five-layer wall structure of the GI tract permits conclusions about the origin of the lesion (and its probable histology) and, where appropriate, the degree of local invasion (T stage). Table 29.5 lists some important features that can be derived from EUS and that are usually helpful diagnosing the lesion and directing further management.
Table 29.5 Important Endoscopic Ultrasound Features in Intramural Masses
| Attribute | Attribute Values |
|---|---|
| Location | Organ (e.g., stomach) and position (e.g., greater curve) |
| Size | Measured (in three dimensions if possible) |
| Background echogenicity | Hypoechoic, hyperechoic, or anechoic |
| Focal echogenicity | Hypoechoic foci, hyperechoic foci, both, or neither |
| Shape/margin shape | Round, oval; smooth margins, irregular margins |
| Margin definition | Well-defined margins, poorly defined margins |
| Position/origin relative to wall layers | Involves mucosa, submucosa, muscularis propria |
| Tumor extension or invasion | T stage relative to site organ |
Several studies have documented the ability of EUS to characterize such lesions accurately. Yasuda and coworkers80 reported their experience with 308 patients, including 210 submucosal tumors. Characteristic echo features of benign and malignant stromal tumors, varices, cysts, lipomas, lymphoma, and aberrant pancreas were described. Rösch and colleagues81 described the appearance of 102 submucosal lesions collected in a multicenter German study group. Characterizing the accuracy of EUS for this purpose is difficult and depends on the question being asked. The ability of EUS to measure tumor size accurately has been documented,82 as has reliability in identifying extramural structures. However, distinguishing among different classes of lesions (e.g., cystic, fatty, stromal) or pathologic entities (e.g., stromal tumor, carcinoid) is hampered by the fact that pathologic confirmation is not uniformly available in these research subject groups. Rösch and coworkers81 reported sensitivity and specificity figures of 64% to 92% and 80% to 100%, for various types of pathologic distinctions being examined.
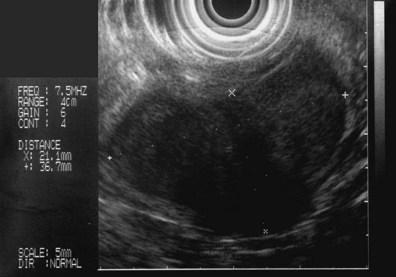
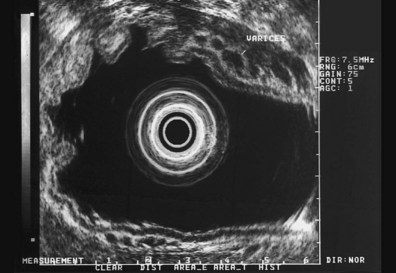
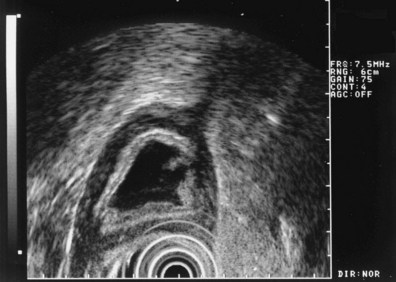
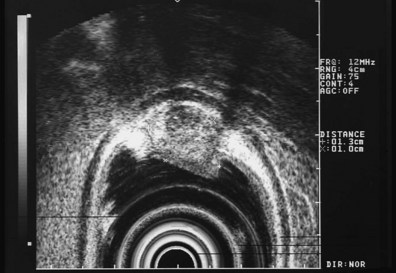
These and other authors have characterized the typical appearance of several intramural lesions. Lipomas (Fig. 29.4) are brightly echogenic structures with uniform echotexture and well-demarcated margins, and they are generally associated with the submucosal layer (layer three). They are easily deformed by compression from the transducer tip. Because they are virtually always benign, identifying continuity of the muscularis propria layer behind the tumor is helpful in confirming the diagnosis. Varices (Fig. 29.5) are also easy to recognize as anechoic vermiform structures. They are nearly always in groups, and extramural varices can usually be seen. When a variceal structure is seen in isolation, a hemangioma or lymphangioma should be considered because these may have a similar appearance. Cysts such as duplication cysts also have an anechoic internal structure (Fig. 29.6), although debris may be seen as hyperechoic foci within the cyst structure. Aberrant pancreas (Fig. 29.7) is usually suspected by the endoscopic appearance, and the EUS structure of a hypoechoic lesion associated with the submucosal layer is confirmatory. Internal echogenic spots are often observed with this lesion.
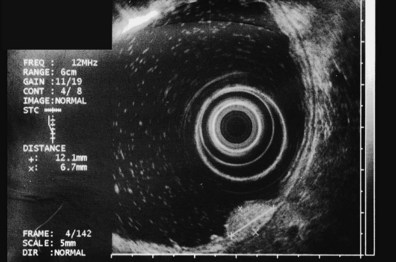
The most frequent finding is of a hypoechoic intramural structure (Figs. 29.8 and 29.9). Various tumors, mostly neoplasms, can have this appearance, including GISTs, carcinoids, granular cell tumors, lymphomas, and metastatic tumors. These lesions all show generally hypoechoic (ground-glass) background echotexture, often containing hyperechoic foci or hypoechoic and anechoic foci (or both hyperechoic and hypoechoic foci). The tumor margins are typically well defined and smooth, and the overall shape is round or oval. Despite the similarities, some EUS clues may help to distinguish among these lesions. Granular cell tumors, usually located in the esophagus, are typically seen within the third (submucosal) echo layer and are generally smaller than 2 cm (see Fig. 29.8A). Carcinoid tumors are most common in the mucosal and submucosal layers (see Fig. 29.8B), as are lymphomas (see Fig. 29.8C), whereas metastatic tumors (see Fig. 29.8D) may occupy any layer in any organ.
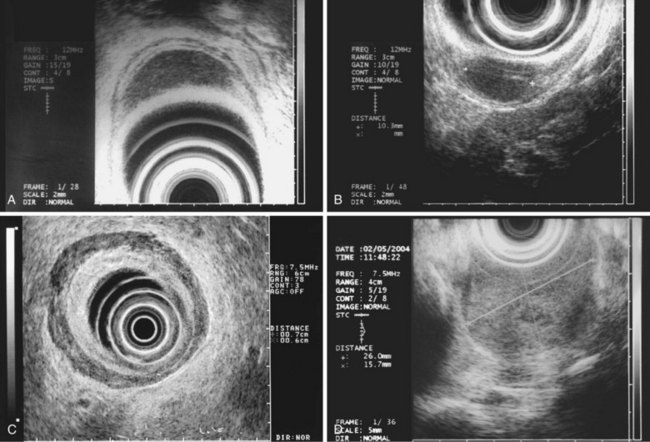
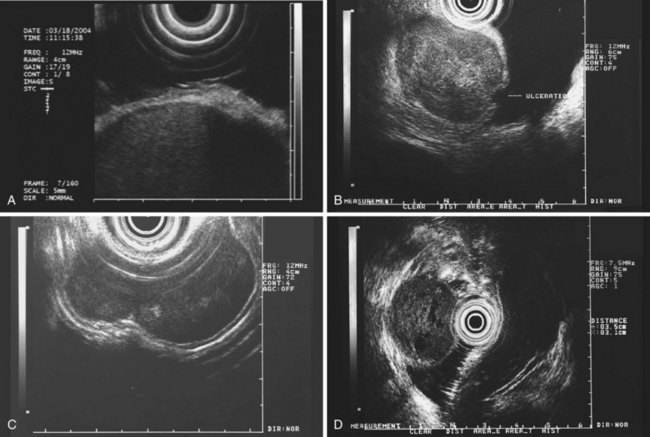
Stromal tumors (see Fig. 29.9) are typically located in the muscularis propria layer and indistinguishably blend into it. Their most innocent appearance is of rounded, well-circumscribed, smooth tumors of 1 to 2 cm with uniform echogenicity (see Fig. 29.9A). Other EUS features may be seen, however, and significant effort has been expended to determine whether those features can distinguish benign from malignant behavior. Giant size (see Fig. 29.9B) and irregular or knobby margins (see Fig. 29.9C) have been imputed to predict malignancy, as have internal foci that are hyperechoic or hypoechoic (see Fig. 29.9D).
Early proposals that all these features were ominous83 have been investigated further with mixed results. Tsai and coworkers84 showed a correlation of each of these features with malignant histology, but no single factor or combination of factors yielded satisfactory diagnostic accuracy. Chak and colleagues85 showed sensitivity figures of 80% to 100% for malignancy in a retrospective videotape study using these factors, but there was only fair to moderate intraobserver agreement for the factors themselves, especially for hyperechoic and hypoechoic foci; specificity was 80%. A multicenter prospective study, the largest prospective series to date (198 tumors), attempted to validate these criteria further and found that tumor size, surface ulceration, nonoval shape, and irregular or indistinct margins were associated with malignancy, whereas hyperechoic and hypoechoic internal foci were not correlated.8 The authors also reported that serial surveillance of initially innocent lesions by repeat EUS provided a very low yield of additional malignant neoplasms. Reviewing the literature on the subject collectively, most experts agree that size greater than 3 cm and irregular or indistinct margins are worrisome features.
Fine Needle Aspiration Biopsy
Given the ambiguous predictive value of EUS morphology in identifying tissue histology or predicting malignant behavior when considering hypoechoic intramural tumors, efforts to obtain diagnostic tissue assume greater importance. Transcutaneous sampling under CT or ultrasound guidance is possible for some lesions and can yield diagnostic material in up to three-fourths of lesions.86 Likewise, endoscopic FNA by direct puncture or under EUS guidance yields adequate diagnostic material in up to 90%74 and, when combined with immunohistochemical stains, can distinguish GIST from leiomyoma.87 Although such specimens are usually adequate to distinguish other neoplasms from GISTs, they are unable to distinguish reliably benign from malignant GISTs. The fact that the cellularity is insufficient to obtain a mitotic index is a major obstacle, but the addition of immunohistochemical stains in biopsy specimens, including c-KIT and Ki-67, improves the diagnostic accuracy of stromal tumors88 and can strongly suggest malignant risk. A more recent modeling study suggested that biopsy can improve clinical management of hypoechoic tumors,89 lending support to the routine performance of needle biopsy of hypoechoic tumors, with immunostaining when appropriate.
Treatment
Indications and Contraindications
Several clear-cut indications have emerged to direct resection of intramural tumors. Most obvious is the presence of symptoms that are caused by the lesion, such as bleeding, obstruction, or intussusception. Beyond this, lesions that are malignant or pose a significant risk of becoming malignant require resection, whereas clearly benign tumors, such as granular cell tumors and lipomas, pose no meaningful malignant risk and may be safely left in situ. Because GISTs compose most intramural tumors, it remains to identify which hypoechoic lesions are GISTs and how high the malignant risk is for a given GIST. For this reason, EUS combined with FNA biopsy and immunostaining is emerging as the diagnostic procedure of choice. In a more recent study, 71% of resected hypoechoic tumors were GISTs; 12% of these were malignant GISTs, and another 41% were GISTs of indeterminate malignant potential.8 Given what seems to be a significant malignant risk among such tumors, criteria to direct resection should have high sensitivity, even at the expense of low specificity. It seems reasonable to perform resection of hypoechoic tumors with size greater than 3 cm, irregular or indistinct margins, ulceration, or nonoval shape. Rapid growth on serial examination, although not a validated criterion, is nevertheless a sufficiently alarming finding also to direct removal of the tumor.
Description of Techniques
Some submucosal tumors are appropriate for endoscopic removal. Numerous reports document success in removal of tumors of the mucosa or submucosa, including lipomas, inflammatory fibroid polyps, carcinoids,90 and granular cell tumors.91 Stromal cell tumors that do not involve the muscularis propria can also be successfully removed endoscopically.92 Generally, a snare or inject/snare technique is described. Bleeding is the most common complication and may require transfusion, endoscopic therapy,90 or surgery.93 Despite the reported success of this technique, the number of tumors appropriate for endoscopic removal is limited. The most problematic intramural tumors are tumors that show EUS features of GIST; because most of these involve the muscularis propria, they are unsuitable for endoscopic treatment. Endoscopic removal is likely to remain of limited applicability for this condition.
Advances in minimally invasive surgery have made laparoscopic removal possible for many such tumors. Several small case series describe successful laparoscopic removal of GISTs in various gastric sites,94,95 including the posterior wall of the stomach.96 Tumors up to 7 cm in size can be removed.97 Surgical techniques described include tumor enucleation, wedge resection, and partial gastrectomy. A combined endoscopic and laparoscopic approach may be required when the tumor cannot be readily identified from the serosal surface.98 In these small series, few complications are reported,94 but conversion to open resection may be required.96 A more recent retrospective study comparing open and laparoscopic resection of GISTs found a shorter mean hospital stay for the latter (3.8 days vs. 6.2 days) but otherwise comparable technical and safety outcomes,99 and a smaller prospective study found a similar decrease in hospital stay but also noted hospital costs to be 31% less in the laparoscopy group.100 It has been emphasized that such resections require a high degree of technical skill in laparoscopic surgery.99 As experience widens, it is likely that laparoscopic resection will become the procedure of choice in the near future for GISTs requiring removal; the convenience and patient acceptance of this method may lead to decreased resistance to resection of GISTs of equivocal malignant potential. Natural orifice endoscopic surgery would be expected to offer a resection method well suited to mesenchymal tumors of all sorts including GISTs but has not been described for this purpose to date.
Future Trends
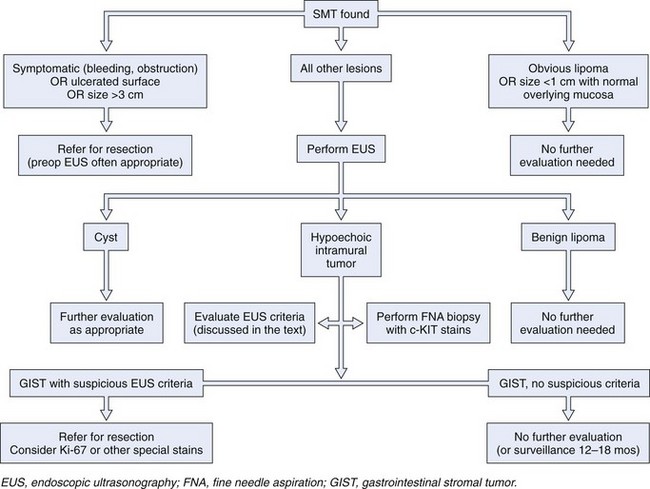
A more recent modeling suggests that the GI endoscopist who cannot establish definitive patient management in an average of 1.7 EUS examinations is wasting the patient’s time and money.8 This suggestion leads to what is, from the GI endoscopist’s perspective, a critical need. Clinical studies that reliably define submucosal tumors that require further investigation and that establish the performance characteristics of EUS and other diagnostic modalities would permit the endoscopist to direct further management of submucosal tumors safely and reliably. A provisional treatment algorithm (“fools rush in …”) is proposed here (Fig. 29.10), but substantial work remains to be done to validate the steps that would translate this provisional algorithm into a reliable management tool.
1 Kume K, Yoshikawa I, Yamazaki M, et al. A case of gastric cancer with features of submucosal tumor. Gastrointest Endosc. 2001;53:247-249.
2 Hedenbro JL, Ekelund M, Wetterberg P. Endoscopic diagnosis of submucosal gastric lesions: The results after routine endoscopy. Surg Endosc. 1991;5:20-30.
3 Allgayer H. Cost-effectiveness of endoscopic ultrasonography in submucosal tumors. Gastrointest Endosc Clin N Am. 1995;5:625-629.
4 Motoo Y, Okai T, Ohta H, et al. Endoscopic ultrasonography in the diagnosis of extraluminal compressions mimicking gastric submucosal tumors. Endoscopy. 1994;26:239-242.
5 Caletti G, Zani L, Bolondi L, et al. Endoscopic ultrasonography in the diagnosis of gastric submucosal tumor. Gastrointest Endosc. 1989;35:413-418.
6 Rohatgi A, Singh KK. Laparoendoscopic management of gastrointestinal stromal tumors. J Laparoendosc Adv Surg Tech. 2003;13:37-40.
7 Nickl N, Bhutani M, Catalano M, et al. Clinical implications of endoscopic ultrasound: The American Endosonography Club Study. Gastrointest Endosc. 1996;44:371-377.
8 Nickl N, Gress F, McClave S, et al. Hypoechoic intramural tumor study: Final report. Gastrointest Endosc. 2002;55:AB98.
9 De Waele B, Gillardin J, Creve U, et al. Upper gastrointestinal bleeding due to benign tumours of the stomach. Acta Chir Belg. 1987;87:322-325.
10 Hsu CC, Chen JJ, Changchien CS. Endoscopic features of metastatic tumors in the upper gastrointestinal tract. Endoscopy. 1996;28:249-253.
11 Johnson DC, GeGennaro VA, Pizzi WF, et al. Gastric lipomas: A rare cause of massive upper gastrointestinal bleeding. Am J Gastroenterol. 1981;75:299-301.
12 Treska V, Pesek M, Kreuzberg B, et al. Gastric lipoma presenting as upper gastrointestinal obstruction. J Gastroenterol. 1998;33:716-719.
13 Moues C, Steenvoorde P, Wiersma J, et al. Jejunal intussusception of a gastric lipoma: A review of the literature. Dig Surg. 2002;19:418-420.
14 Sanders L, Silberman M, Rossi R, et al. Gastric smooth muscle tumors: Diagnostic dilemmas and factors affecting outcome. World J Surg. 1996;20:992-995.
15 Rosch T, Lorenz R, Dancygier H, et al. Endosonographic diagnosis of submucosal upper gastrointestinal tract tumors. Scand J Gastroenterol. 1992;27:1-8.
16 Rosch T, Lorenz R, von Wichert A, et al. Gastric fundus impression caused by splenic vessels: Detection by endoscopic ultrasound. Endoscopy. 1991;23:85-87.
17 Sun MS, Wang HP, Lin JT. Gastroduodenal artery aneurysm mimicking a bleeding submucosal tumor. Gastrointest Endosc. 2001;54:621.
18 Golden T, Stout A. Smooth muscle tumors of the gastrointestinal tract and retroperitoneal tissues. Surg Gynecol Obstet. 1941;73:784-810.
19 Lewin K, Riddel RH, Weinstein WM. Mesenchymal tumors. In: Gastrointestinal pathology and its clinical implications. New York: Igaku-Shoin; 1992:284-341.
20 Ma CK, De Peralta MN, Amin MB, et al. Small intestinal stromal tumors: A clinicopathologic study of 20 cases with immunohistochemical assessment of cell differentiation and the prognostic role of proliferation antigens. Am J Clin Pathol. 1997;108:641-651.
21 Hirota S, Siozaki K, Moriyama Y. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-580.
22 Berman JJ, O’Leary TH. Gastrointestinal stromal tumor workshop. Hum Pathol. 2001;32:578-582.
23 Duffaud F, Blay JY. Gastrointestinal stromal tumors: Biology and treatment. Oncology. 2003;65:187-197.
24 Heinrich MD, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708-710.
25 Hirohita A, Osizaki K, Moriyama Y. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577-580.
26 Kindblom LG, Remotti HE, Aldenborg F, et al. Gastrointestinal pacemaker cell tumor (GIPACT): Gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152:2008-2011.
27 Lasota J, Jasinski M, Sarlomo-Rikala M, et al. Mutations in exon 11 of c-Kit occur preferentially in malignant versus benign gastrointestinal stromal tumors and do not occur in leiomyomas or leiomyosarcomas. Am J Pathol. 1999;154:53-60.
28 Miettinem M, Sobin LH, Sarlomo-Rikala M. Immunohistochemical spectrum of GISTs at different sites and their differential diagnosis with reference to CD117 (KIT). Mod Pathol. 2000;13:1134-1142.
29 Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708-710.
30 Miettinen M, Kopczynski J, Makhlouf HR, et al. Gastrointestinal stromal tumors, intramural leiomyomas, and leiomyosarcomas in the duodenum: A clinicopathologic, immunohistochemical, and molecular genetic study of 167 cases. Am J Surg Pathol. 2003;27:625-641.
31 Graham J, Debiec-Rychter M, Corless CL, et al. Imatinib in the management of multiple gastrointestinal stromal tumors associated with a germline KIT K642E mutation. Arch Pathol Lab Med. 2007;131:1393-1396.
32 Lasota J, Corless CL, Heinrich MC, et al. Clinicopathological profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: A multicenter study on 54 cases. Mod Pathol. 2008;21:476-484.
33 Sardelmann E, Pauls K, Merkelbah-Bruse S, et al. Gastrointestinal stromal tumors carrying PDGFRalpha mutations occur preferentially in the stomach and exhibit an epithelioid or mixed phenotype. Verh Dtsch Ges Pathol. 2004;88:174-183.
34 Agaram NP, Baren A, Arkun K, et al. Comparative ultrastructural analysis and KIT/PDGFRA genotype in 125 gastrointestinal stromal tumors. Ultrastruct Pathol. 2006;30:443-454.
35 Evans HL. Smooth muscle tumors of the gastrointestinal tract: A study of 56 cases followed for a minimum of 10 years. Cancer. 1985;56:2242-2250.
36 Ranchod M, Kempson R. Smooth muscle tumors of the gastrointestinal tract and retroperitoneum: A pathologic analysis of 100 cases. Cancer. 1977;34:255-262.
37 Cunningham RE, Federspiel BH, McCarthy WF, et al. Predicting prognosis of gastrointestinal smooth muscle tumors: Role of clinical and histologic evaluation, flow cytometry, and image cytometry. Am J Surg Pathol. 1993;17:588-594.
38 Li SQ, O’Leary TJ, Sobin LH, et al. Analysis of KIT mutation and protein expression in fine needle aspirates of gastrointestinal stromal/smooth muscle tumors. Acta Cytol. 2000;44:981-986.
39 Morey AL, Wanigesekera GD, Hawkins NJ, Ward RL. C-kit mutations in gastrointestinal stromal tumors. Pathology. 2002;34:315-319.
40 Takahashi R, Tanaka S, Kitadai Y. Expression of vascular endothelial growth factor and angiogenesis in gastrointestinal stromal tumor of the stomach. Oncology. 2003;64:266-274.
41 Toquet C, Le Neel JC, Guillou L, et al. Elevated (> or = 10%) MIB-1 proliferative index correlates with poor outcome in gastric stromal tumor patients: A study of 35 cases. Dig Dis Sci. 2002;47:2247-2253.
42 Berman JJ, O’Leary TJ. Gastrointestinal stromal tumor workshop. Hum Pathol. 2001;32:578-582.
43 Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33:459-465.
44 Abraham SC, Krasinskas AM, Hofstetter WL, et al. “Seedling” mesenchymal tumors (gastrointestinal stromal tumors and leiomyomas) are common incidental tumors of the esophagogastric junction. Am J Surg Pathol. 2007;31:1629-1635.
45 Agaimy A, Wünch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am J Surg Pathol. 2007;31:113-120.
46 Kawanowa K, Saluma Y, Sakuir S, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37:1527-1535.
47 Agaimy A, Dirnhofer S, Wunsch PH, et al. Multiple sporadic gastrointestinal stromal tumors (GISTs) of the proximal stomach are caused by different somatic KIT mutations suggesting a field effect. Am J Surg Pathol. 2008;32:1553-1559.
48 Dagher R, Cohen M, Williams G, et al. Approval summary: Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8:3034-3038.
49 Agawa H, Matsushita M, Nishio A, et al. Gastric glomus tumor. Gastrointest Endosc. 2002;56:903.
50 Appleman HD, Helwig EB. Glomus tumors of the stomach. Cancer. 1969;23:203-213.
51 Miettinen M, Paal E, Lasota J, et al. Gastrointestinal glomus tumors: A clinicopathologic, immunohistochemical, and molecular genetic study of 32 cases. Am J Surg Pathol. 2002;26:301-311.
52 Lewin K, Riddel RH, Weinstein WM. Vascular disorders. In: Gastrointestinal pathology and its clinical implications. New York: Igaku-Shoin; 1992:33-92.
53 Camilleri M, Satti M, Wood CB. Cystic lymphangioma of the colon: Endoscopic and histologic features. Dis Colon Rectum. 1982;25:813-816.
54 Johnson DC, DeGennaro VA, Pizzi WF, et al. Gastric lipomas: A rare cause of massive upper gastrointestinal bleeding. Am J Gastroenterol. 1981;75:299-301.
55 Hyun CB, Coyle WJ. Giant gastric lipoma. Gastrointest Endosc. 2002;56:905.
56 Moues CM, Steenvoorde P, Viersma JH, et al. Jejunal intussusception of a gastric lipoma: A review of literature. Dig Surg. 2002;19:418-420.
57 Seki K, Hasegawa T, Konegawa R, et al. Primary liposarcoma of the stomach: A case report and a review of the literature. Jpn J Clin Oncol. 1998;28:284-288.
58 Coutinho DS, Soga J, Yoshikawa T, et al. Granular cell tumors of the esophagus: A report of two cases and review of the literature. Am J Gastroenterol. 1985;80:758-762.
59 Fenoglio-Preiser CM, Lantz PE, Listrom MB, et al. Mesenchymal tumors. In: Gastrointestinal pathology: an atlas and text. New York: Raven Press; 1989:543-585.
60 Morson BC, Dawson IM, Day DW, et al. Polyps and tumors. In Morson & Dawson’s gastrointestinal pathology, ed 3, Oxford: Blackwell Scientific Publications; 1990:53-70.
61 Soon MS, Lin OS. Inflammatory fibroid polyp of the duodenum. Surg Endosc. 2000;14:86.
62 Shimer G, Helwig EB. Inflammatory fibroid polyp of the intestine. Am J Clin Pathol. 1984;81:708-714.
63 Makhlouf HR, Sobin LH. Inflammatory myofibroblastic tumors (inflammatory pseudotumors) of the gastrointestinal tract: How closely are they related to inflammatory fibroid polyps? Hum Pathol. 2002;33:307-315.
64 Lewin K, Riddel RH, Weinstein WM. Polyps and tumors. In: Gastrointestinal pathology and its clinical implications. New York: Igaku-Shoin; 1992:482.
65 Patel J, Kieffer RW, Martin M, et al. Giant fibrovascular polyp of the esophagus. Gastroenterology. 1984;87:953-956.
66 Burrell M, Toffler R. Fibrovascular polyp of the esophagus. Am J Dig Dis. 1973;18:714-718.
67 Oda, Kondo H, Yamao T, et al. Metastatic tumors to the stomach: Analysis of 54 patients diagnosed at endoscopy and 347 autopsy cases. Endoscopy. 2001;33:507-510.
68 Woolfolk GM, McClave, Jones WF, et al. Use of endoscopic ultrasound to guide the diagnosis and endoscopic management of a large gastric duplication cyst. Gastrointest Endosc. 1998;47:76-79.
69 Bhutani MS, Hoffman BJ, Reed C. Endosonographic diagnosis of an esophageal duplication cyst. Endoscopy. 1996;28:396-397.
70 Hizawa K, Matsumoto T, Kouzuki T, et al. Cystic submucosal tumors in the gastrointestinal tract: Endosonographic findings and endoscopic removal. Endoscopy. 2000;32:712-714.
71 Kim HS, Lee SY, Lee YD, et al. Gastric lymphangioma. J Korean Med Sci. 2001;16:229-232.
72 Araki K, Ohno S, Egashira A, et al. Esophageal hemangioma: A case report and review of the literature. Hepatogastroenterology. 1999;46:3148-3154.
73 Wegener M, Adamek R. Puncture of submucosal and extrinsic tumors: Is there a clinical need? Puncture techniques and their accuracy. Gastrointest Endosc Clin N Am. 1995;5:615-623.
74 Layfield LJ, Reichman A, Weinstein WM. Endoscopically directed fine needle aspiration biopsy of gastric and esophageal lesions. Acta Cytol. 1992;36:69-74.
75 Spandre M, Cavallero M, Pennazio M. Needle biopsy of submucosal lesions of the gastrointestinal tract. Surg Endosc. 1990;4:161-163.
76 Matsuoka J, Takai K, Kojima K, et al. Endoscopic submucosal tumor biopsy using Stiegmann-Goff endoscopic ligator. Acta Med Okayama. 2000;54:233-234.
77 Nishida T, Kumano S, Sugiura T, et al. Multidetector CT of high-risk patients with occult gastrointestinal stromal tumors. AJR Am J Roentgenol. 2003;180:185-189.
78 Ogata I, Komohara Y, Yamashita Y, et al. CT evaluation of gastric lesions with three-dimensional display and interactive virtual endoscopy: Comparison with conventional barium study and endoscopy. AJR Am J Roentgenol. 1999;172:1263-1270.
79 Tsai TL, Changchien CS, Hu TH, et al. Demonstration of gastric submucosal lesions by high-resolution transabdominal sonography. J Clin Ultrasound. 2000;28:125-132.
80 Yasuda K, Cho E, Nakamima M, et al. Diagnosis of submucosal lesions of the upper gastrointestinal tract by endoscopic ultrasonography. Gastrointest Endosc. 1990;36:S17-S20.
81 Rösch T, Kapfer B, Will U, et al. Accuracy of endoscopic ultrasonography in upper gastrointestinal submucosal lesions: A prospective multicenter study. Scand J Gastroenterol. 2002;37:856-862.
82 Murata Y, Yoshida M, Akimoto S, et al. Evaluation of endoscopic ultrasonography for the diagnosis of submucosal tumors of the esophagus. Surg Endosc. 1988;2:51-58.
83 Rösch T, Lorenz R, Dancygier H, et al. Endosonographic diagnosis of submucosal upper gastrointestinal tract tumors. Scand J Gastroenterol. 1992;27:1-8.
84 Tsai TL, Changchien CS, Hu TH, et al. Differentiation of benign and malignant gastric stromal tumors using endoscopic ultrasonography. Ghang Gung Med J. 2001;24:167-173.
85 Chak A, Canto MI, Rosch T, et al. Endosonographic differentiation of benign and malignant stromal tumors. Gastrointest Endosc. 1997;45:468-473.
86 Ballo MS, Guy CD. Percutaneous fine-needle aspiration of gastrointestinal wall lesions with image guidance. Diagn Cytopathol. 2001;24:16-20.
87 Stelow EB, Stanley MW, Mallery S, et al. Endoscopic ultrasound fine needle aspiration findings of gastrointestinal leiomyomas and gastrointestinal stromal tumors. Am J Clin Pathol. 2003;119:703-708.
88 Ando N, Goto H, Niwa Y, et al. The diagnosis of GI stromal tumors with EUS-guided fine needle aspiration with immunohistochemical analysis. Gastrointest Endosc. 2002;55:37-43.
89 Nickl N, Wackerbarth S, Gress F, et al. Management of hypoechoic intramural tumors: A decision tree analysis of EUS directed vs. surgical management. Gastrointest Endosc. 2000;51(4 Pt 2):AB176.
90 Wei SC, Wong JM, Shieh MJ, et al. Endoscopic resection of gastrointestinal submucosal tumors. Hepatogastroenterology. 1998;45:114-118.
91 Fujiwara Y, Watanabe T, Hamasaki N, et al. Endoscopic resection of two granular cell tumors of the oesophagus. Eur J Gastroenterol Hepatol. 1999;11:1413-1416.
92 Hyun JH, Jeen YT, Chun HJ, et al. Endoscopic resection of submucosal tumor of the esophagus: Results in 62 patients. Endoscopy. 1997;29:165-170.
93 Yu JP, Luo HS, Wang XZ. Endoscopic treatment of submucosal lesions of the gastrointestinal tract. Endoscopy. 1992;24:229-231.
94 Ludwig K, Wilhelm L, Scharlau U, et al. Laparoscopic-endoscopic rendezvous resection of gastric tumors. Surg Endosc. 2002;16:1561-1565.
95 Rohatgi A, Singh KK. Laparoendoscopic management of gastrointestinal stromal tumors. J Laparoendosc Adv Surg Tech. 2003;13:37-40.
96 Hepworth CC, Menzies D, Motson RW. Minimally invasive surgery for posterior gastric stromal tumors. Surg Endosc. 2000;14:349-353.
97 Walsh RM, Heniford BT. Laparoendoscopic treatment of gastric stromal tumors. Semin Laparosc Surg. 2001;8:189-194.
98 Agoi K, Hirai T, Mukiada H, et al. Laparoscopic resection of submucosal gastric tumors. Surg Today. 1999;29:102-106.
99 Matthews BD, Walsh RM, Kercher KW, et al. Laparoscopic vs. open resection of gastric stromal tumors. Surg Endosc. 2002;16:803-807.
100 Nickl N, Park A, Chak A, et al. A comparison of hospital costs and length of stay between laparoscopic and open resection of GI submucosal tumors. Gastroenterology. 1999;166(4 Pt 2):A1336.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
Chapter 29 Nonepithelial Tumors of the Esophagus and Stomach
Epidemiology
The clinical starting point for patients with lesions of nonepithelial origin is the discovery of a mass impinging on the GI tract mucosa from beneath—the so-called submucosal tumor (Fig. 29.1). In its classic form, a discrete tumorous appearance is present with overlying mucosa that, although most commonly bland, may be erythematous, pale, dimpled, or ulcerated. Lesions are often initially identified at esophagogastroduodenoscopy (EGD), but the patient may also be referred to an endoscopist for evaluation of an abnormal radiograph (e.g., barium-contrast examination).
Applied literally, the term submucosal would imply the presence of an intramural mass originating in the submucosal layer of the GI wall. However, the term has come to be used for a range of lesions that create a similar appearance, including intramural and extramural structures. Such submucosal tumors may include both neoplastic and nonneoplastic masses, and even mucosal neoplasms have been reported to exhibit a submucosal appearance.1 Examples of nonepithelial lesions in all four categories are listed in Table 29.1. This discussion centers on neoplasms that primarily originate from nonepithelial GI tract cell lines, but all types of pathology must be considered when developing a management plan.
Table 29.1 Types of Masses Causing Esophageal and Gastric Submucosal Tumors
| Neoplastic Masses | Nonneoplastic Masses | |
|---|---|---|
| Intramural masses | Stromal cell tumor | Varices |
| Lipoma | Duplication cyst | |
| Granular cell tumor | Inflammatory granuloma | |
| Lymphoma | Foreign body (e.g., surgical suture or clip) | |
| Fibrovascular polyp | Pancreatic rest | |
| Hemangioma/hemangiosarcoma | ||
| Lymphangioma/lymphangiosarcoma | ||
| Metastatic neoplasm | ||
| Extramural masses | Primary neoplasm of adjacent organs (benign and malignant) | Benign lymph node |
| Metastatic lymph node | Inflammatory mass of adjacent organs (e.g., pancreas, spleen) | |
| Organomegaly (e.g., spleen, liver) |
Depending on the clinical circumstances and type of tumor, the lesion may cause symptoms such as bleeding, obstruction, or pain. However, such lesions commonly are serendipitously found during evaluation for a different, unrelated problem. Because most such lesions are asymptomatic, epidemiologic data are skewed by the nature of their discovery incidental to a different, usually unrelated condition. In one study of 15,104 EGD reports, submucosal tumors were identified in 0.36%.2 Because most in this series were life-threatening tumors, the study database likely underreported less serious lesions. Many such lesions turn out to be normal extramural organs. Allgayer3 found that among 30 patients referred for submucosal tumors, normal extramural structures were present in 14 (47%). Motoo and coworkers4 also reported normal organs in 16 of 19 submucosal tumors, as did Caletti and colleagues5 in 10 of 25 tumors; organs identified include the spleen, liver, splenic vessels, and pancreas.
Because submucosal tumors are often left in situ, the pathologic distribution among tumors is unknown. It is reported that 1% to 3% of resected gastric tumors are stromal cell tumors6; it can be inferred that the actual incidence, when including tumors that were not resected, is considerably higher. In a small prospective study,7 among 45 submucosal tumors, most were found to have a benign appearance that required no follow-up. From these available data, it may be cautiously concluded that submucosal tumors are found in less than 1% of routine upper endoscopy examinations, half of such lesions are found to be normal extramural structures, most remaining lesions are benign, and stromal cell tumors constitute most such neoplasms.
Clinical Features
Lumps and bumps of all sorts are regularly encountered in endoscopic examinations; the decision regarding which to evaluate further depends on the endoscopic appearance, the clinical circumstances, and the inclination of the endoscopist. Because few standardized guidelines exist to direct such decisions, great variation in practice exists. Symptoms attributed to the mass nearly always drive further investigation, but our own endoscopic ultrasound (EUS) study of a subset of such lesions found that nearly 90% were asymptomatic.8 GI bleeding may be seen in many submucosal lesions, most commonly in the form of slow blood loss causing iron deficiency anemia. The surface of the tumor may be ulcerated in such cases (Fig. 29.2). Malignant tumors may be more prone to ulceration and bleeding9; this might be taken as a sign of a potentially malignant form that compels definitive treatment. However, benign lesions may also cause severe bleeding,10 and occasionally rapid hemorrhage may occur.11 Less often, GI tract obstruction may be caused by such masses,12 especially if the lesion is located in a narrow area such as the esophagogastric junction or pylorus; intussusception caused by such masses has been reported.13 Pain may be a presenting complaint, especially if the submucosal tumor is neoplastic or malignant.14
Because most lesions are incidentally found during endoscopic examination for another problem, the clinical features of submucosal masses are primarily those that, in the endoscopist’s opinion, compel further evaluation. Large size has been proposed as an ominous finding,15 and lesions with an ulcerated or irregular (lumpy) surface often undergo additional testing or treatment. Patients with submucosal tumors who have a prior history of malignancy should receive further evaluation to exclude metastatic disease. Finally, patients with submucosal lesions that change appearance on serial examination are usually directed by an alert clinician to further testing.
Pathology
Extramural masses compose half of suspected submucosal tumors and include normal organs, nonneoplastic masses, and extramural neoplasms. Normal liver, spleen, pancreas, gallbladder, colon, and kidney all have been reported to appear as suspected submucosal tumors.3–5 Vascular structures often produce the appearance of a discrete tumor, including normal vessels of the spleen16 and abnormal vessels such as varices and aneurysms.17 Neoplasms and nonneoplastic masses involving these same organs can also produce this appearance, as can such masses involving the peritoneum, mediastinum, and the lymph nodes adjacent to the upper GI tract. The various malignancies, cysts, and inflammatory masses of these structures need no further elaboration here because a large variety of such findings have been noted in the case report literature.
Masses that arise within the wall of the esophagus and stomach require further discussion, particularly because many are peculiar to the GI tract. Most neoplasms in this category are mesenchymal tumors, meaning that they arise from cells of mesodermal origin. Most such neoplasms are clinically benign, although, as shown subsequently, tumor histology may not provide reliable clues to malignant behavior. A large variety of such neoplasms have been described (Table 29.2), but most are exceedingly rare. The tumors most likely to be encountered in the esophagus and stomach in a routine clinical setting are discussed here.
Table 29.2 Classification of Gastrointestinal Mesenchymal Tumors
| Tumor Type | Examples |
|---|---|
| Stromal tumors | Smooth muscle tumors (leiomyoma, leiomyosarcoma), glomus tumors, leiomyomatosis, pleomorphic sarcoma |
| Neural tumors | Neuroma/neurofibroma, paraganglioma, ganglioneuromatosis |
| Endothelial and vascular tumors | Hemangioma, hemangiosarcoma, Kaposi’s sarcoma, lymphangioma |
| Lipocytic tumors | Lipoma, liposarcoma, lipohyperplasia (ileocecal valve), lipomatosis (colon) |
| Granular cell tumor | Granular cell tumor |
| Inflammatory fibroid polyp | Inflammatory fibroid polyp |
| Fibrohistiocytic tumors | Fibrovascular polyp, fibrous histiocytoma, desmoid tumors (mesentery), fibroepithelial polyp |
| Striated muscle tumors | Rhabdomyosarcoma |
Data from Lewin K, Riddel RH, Weinstein WM: Mesenchymal tumors. In: Gastrointestinal pathology and its clinical implications, New York, 1992, Igaku-Shoin, pp 284–341.
Gastrointestinal Stromal Tumor
Most mesenchymal GI tumors are pale, firm, spherical, or ovoid structures embedded in the wall of the affected organ. The microscopic appearance of musclelike eosinophilic, spindle-shaped cells in uniform sheets and the proximity of the tumors to the muscular wall layers led early observers to believe that these tumors were of myogenic origin18—hence the name leiomyoma and its variations (e.g., leiomyosarcoma, leiomyoblastoma). However, it later became clear that these neoplasms not only are not of obvious myogenic origin but also often lack any specific markers of differentiation whatsoever.19 Immunohistochemical analyses showed variable expression of smooth muscle features such as desmin and actin and neural proteins such as S-100.20 For the sake of clarity, these lesions came to be referred to as gastrointestinal stromal tumors (GISTs), an acknowledgment that they originate in mesenchymal stroma.
A significant breakthrough occurred with the discovery that most GI stromal tumors stain positive for a specific membrane protein, designated CD117,21 and subsequently identified as KIT, a tyrosine kinase receptor.22 Tyrosine kinases are a class of transmembrane receptors that mediate various cellular growth functions. The extracellular portion of KIT binds stem cell factor, causing a conformational change. The receptor subsequently forms a dimer with another activated KIT receptor, and the intracellular region of the resulting dimer triggers various cell signaling cascades.23 Among these are proliferation pathways such as mitogen-activated protein kinase (MAPK) and signal transducers and activators of transcription (STAT) proteins as well as antiapoptotic mediators such as AKT (protein kinase B).24 KIT and some other tyrosine kinases are important regulators of cell growth and proliferation.
Abnormal cell growth is a fundamental element of cancer physiology, and hyperfunction of the KIT receptor can lead to neoplasia. In this case, numerous gain-of-function mutations in the KIT gene have been described.25 These acquired (or, rarely, inherited) mutations produce KIT receptors that are capable of initiating the aforementioned intracellular growth cascades without activation by stem cell factor. The resulting dysregulated growth seems to be an important initial step to tumorigenesis. This concept is supported by the observation that nearly all GISTs express KIT. It has further been observed that the interstitial cells of Cajal (ICC) share some phenotypic and ultrastructural similarities with GIST and normally express the KIT receptor; this observation has led to the current hypothesis that GISTs arise from ICC26 or from ICC precursor cells. Finally, it has been noted that this gain-of-function mutation is not found in true leiomyomas.27 Some pathologists have suggested that CD117 positivity is required to confirm a diagnosis of GIST.28 It is now known, however, that a few otherwise obvious GISTs do not express KIT. Many of these tumors have been identified as expressing platelet-derived growth factor receptor α (PDGFRα), another distinct tyrosine kinase receptor that also mediates many of the same growth and antiapoptotic proliferation pathways.29 Several gain-of-function mutations have been reported in the PDGFRα gene, and the resulting tumors are otherwise indistinguishable from GISTs that are KIT-positive.
Nearly all upper GI tumors of this type occur in the stomach, but duodenal lesions have been described.30 Most esophageal stromal tumors lack the CD117 protein and may be true leiomyomas.28 The endoscopic appearance is a dome-shaped, firm submucosal mass; central umbilication or frank ulceration is common, and there may be a lobulated or irregular appearance. The tumors are most often solitary except in the case of specific disease entities such as Carney’s triad (GIST, pulmonary chondroma, and extraadrenal paraganglioma). Germline KIT mutation kindreds have been described,31 however, in which case multiple GISTs are seen. Giant sizes of greater than 10 cm have been noted, but most tumors are less than 3 cm.
Pathologically, the tumor usually consists of uniform pale tissue, although hemorrhagic and necrotic areas may be seen. Microscopically, the cells are spindle-shaped with uniform nuclei and general cytologic uniformity. Some cell groups may show epithelioid configurations (closely packed polygonal cells), and there may be nuclear pleomorphism. It has been observed more recently that the histologic pattern is sometimes related to the nature of the underlying genetic abnormality; KIT mutations at exon 13 or 17 more often show spindle-cell morphology,32 whereas PDGFRα GISTs often exhibit epithelioid histology.33 Ultrastructural cellular abnormalities have also been linked to specific gene mutations.34 However, malignant behavior has not been associated with specific mutation patterns.
It has long been known that malignant behavior in GISTs is difficult to predict given the relatively bland cytology and slow growth of these neoplasms. It has been reported that even small, benign-appearing stromal tumors have been known to metastasize.35 This discovery led to considerable confusion about the appropriate criteria to categorize these tumors as benign or malignant. Older pathologic scoring systems relied on numerous histologic features19 and were plagued with problems. More recent attention has focused on the size of the tumor and the number of mitoses observed (mitotic index), at least in part because these are easily quantifiable findings. In one study of 100 cases, tumors with more than 5 mitoses per 10 high-power fields (HPFs) were significantly more likely to metastasize, although 40% of malignant lesions in that study had fewer mitoses.36 In another study, multivariate analysis of various clinical and pathologic features in 122 specimens showed that more than 10 mitoses/50 HPFs correlated with poor outcome, whereas site, epithelioid histology, and tumor size were not independently predictive.37
Attempts to correlate tumor marker status such as CD117 positivity with malignant behavior have produced generally confusing or negative results,38 as have studies of specific KIT mutations27,39 and other tumor markers.40,41 Older GIST scoring systems have been abandoned following a National Institutes of Health (NIH) consensus conference, which defined malignancy risk based on size and mitotic index alone.42,43 The NIH criteria divide tumors into four categories of malignant risk (Table 29.3), an acknowledgment that even the most innocent lesion poses a slight but definite risk of malignant behavior.
Table 29.3 National Institutes of Health Criteria for Malignant Risk in Gastrointestinal Stromal Tumors
| Risk Level | Size (cm) | Mitoses/50 HPF |
|---|---|---|
| Very low risk | <2 | <5 |
| Low risk | 2–5 | <5 |
| Intermediate risk | <5 | 6–10 |
| 5–10 | <5 | |
| High risk | >5 | >5 |
| >10 | Any | |
| Any | >10 |
HPF, high-power field.
Data from Berman JJ, O’Leary TH: Gastrointestinal stromal tumor workshop. Hum Pathol 32:578–582, 2001; Toquet C, Le Neel JC, Guillou L, et al: Elevated (> or = 10%) MIB-1 proliferative index correlates with poor outcome in gastric stromal tumor patients: A study of 35 cases. Dig Dis Sci 47:2247–2253, 2002; Hedenbro JL, Ekelund M, Wetterberg P: Endoscopic diagnosis of submucosal gastric lesions: The results after routine endoscopy. Surg Endosc 5:20–30, 1991.
If all such neoplasms entail malignant risk, prudence might dictate that they should always be resected. However, more recent data suggest that GISTs are more common than previously thought. Up to 10% of resection and autopsy specimens contain such tumors,44 and microscopic GISTs (also called seedling GISTs, minimal GISTs, or GIST tumorlets) can be seen in 35% of some patient groups.45,46 These incidental and microscopic GISTs display the same KIT and PDGFRα mutations as their larger counterparts. The finding that synchronous GISTs from the same patient regularly show different gene mutations (and are independent sporadic GISTs) confuses the picture further.47 GISTs that were earlier classified as metastatic or recurrent may have been distinct neoplastic events. If it turns out that most small and asymptomatic GISTs stay that way, a conservative approach may be the most prudent.
A second breakthrough in GISTs has been the development of tyrosine kinase inhibitors such as imatinib mesylate that are effective in reducing the KIT enzyme activity and that are useful for tumor treatment. Imatinib targets the specific abnormal enzyme activity in the neoplasm and does not rely on generalized cytotoxicity for its effect. In an open label study of 147 patients with unresectable malignant GISTs, an overall response rate of 38% was seen.48 Among responders, results are often dramatic (Fig. 29.3). The recognition of the malignant potential of GIST, combined with the availability of effective treatment even for unresectable disease, has compelled new thinking in the accurate diagnosis of this neoplasm.
