CHAPTER 85 Nonalcoholic Fatty Liver Disease
In 1980, Ludwig and colleagues from the Mayo Clinic coined the term nonalcoholic steatohepatitis (NASH) to describe a form of liver disease observed in middle-aged patients with abnormal liver biochemical test results and histologic evidence of alcoholic hepatitis but no history of alcohol abuse.1 Much has been learned about NASH since this initial description. NASH is part of the spectrum of nonalcoholic fatty liver disease (NAFLD), which encompasses simple fatty liver, NASH, and NAFLD-associated cirrhosis. NAFLD has emerged as a burgeoning clinical entity, now recognized as an important component of the metabolic syndrome, as well as an exciting area of basic and clinical investigation in the field of hepatology.
NONALCOHOLIC FATTY LIVER AND STEATOHEPATITIS
EPIDEMIOLOGY
The prevalence of NAFLD in the general population is undefined. Several studies have estimated the scope of this disorder in the United States. The Dallas Heart Study of more than 2200 adults documented hepatic triglyceride content by proton magnetic resonance spectroscopy (MRS) and found fatty liver in 31% of participants; the highest prevalence (45%) was among Hispanics.2 Most of the patients with fatty liver by MRS had normal liver biochemical test levels, although the normal range for serum aminotransferase levels in this study was wider than generally accepted. The National Health and Nutrition Examination Survey (NHANES) III, which included more than 15,700 adults, documented unexplained elevations of serum aminotransferase levels, presumably caused by NAFLD, in 2.8% to 5.5% of participants.3,4 Population-based estimates of NAFLD have been reported for other countries as well. These studies have documented NAFLD in 10% to 24% of the population, with the highest prevalence (up to 76%) among obese nondrinkers.5–7 Prevalence estimates vary widely depending on the information available in a given population and the diagnostic criteria that are used to establish the diagnosis (i.e., liver biochemical test levels, radiologic study results, or liver biopsy findings).
Most cases of NAFLD are discovered in the fourth to sixth decades of life, although NAFLD is also described, with increasing frequency, in obese children and adolescents, as well as in older adults. NAFLD may be present long before a diagnosis is established. In early clinical studies, the majority of patients with NAFLD were female; however, subsequent data have suggested that men may be affected as often as women and may be at greater risk for advanced forms of NAFLD, including NASH. The prevalence of NAFLD appears to vary by ethnicity. In the Dallas Heart Study, Hispanics demonstrated the highest prevalence (45%) of NAFLD, compared with 33% for whites and 24% for African Americans. The reasons for racial and ethnic disparities in the prevalence of NAFLD is not known but may be related, at least in part, to racial differences in body fat distribution8 and the prevalence of the metabolic syndrome, which is greatest in people of Hispanic descent.9 Other studies have also shown that African Americans and Mexican Americans have higher frequencies of unexplained serum aminotransferase elevations than do whites.3,4,9,10 Familial clustering of NAFLD may occur,11,12 which likely reflects both genetic and environmental predisposition to the metabolic conditions associated with NAFLD (see later).13
ETIOLOGY
Many agents and conditions have been associated with NAFLD. The causes may be divided into two broad categories (1) drugs and toxins and (2) metabolic abnormalities, either acquired or congenital. Potential causes of NAFLD are listed in Table 85-1.
| Acquired Metabolic Disorders |
* Tetracycline is cytotoxic by virtue of inhibiting mitochondrial β-oxidation.
Obesity is the condition most often reported in association with NAFLD. Since 1980, the proportion of Americans who are overweight (defined as a body mass index [BMI] > 25 kg/m2) or obese (BMI > 30 kg/m2) has increased markedly. In 2004, 66.2% of Americans adults were classified as overweight or obese, as were 17.4% of children ages 12 to 19 years.14 The health implications of the unremitting obesity epidemic are staggering, and NAFLD is a common byproduct in both adults and children. As noted earlier, most patients with NAFLD are obese. In morbidly obese patients (BMI > 35 kg/m2), including those referred for bariatric surgery, the frequency of NAFLD is as high as 90%, with advanced disease (i.e., NASH) seen in 9% to 40%.15–19 A correlation among BMI, degree of steatosis, and severity of liver injury has been demonstrated in several studies20–22; however, the distribution of body fat may be more important than the total adipose mass for the development of hepatic steatosis. Studies have shown a significant correlation between the risk of the metabolic syndrome, degree of hepatic steatosis, and waist-to-hip ratio, thus highlighting the importance of intra-abdominal or visceral fat as a predictor of NAFLD.23–25
NAFLD also is strongly associated with type 2 diabetes mellitus and glucose intolerance, with or without superimposed obesity.26 Type 2 diabetes mellitus, hyperglycemia, or glucose intolerance has been described in 20% to 75% of adult patients with NASH and may increase the risk of NASH more than twofold compared with that for nondiabetic persons. The presence of NAFLD in diabetic patients may also increase the risk of cardiovascular disease significantly.27 The association between type 2 diabetes mellitus and NAFLD appears strongest in morbidly obese patients.15 NAFLD has been associated with insulin resistance and hyperinsulinemia even in lean subjects with normal glucose tolerance.28 Diabetes mellitus may be an independent predictor of advanced NAFLD, including cirrhosis and hepatocellular carcinoma.29–32
Hyperlipidemia is found in a substantial proportion of patients with NAFLD. Data from the Dallas Heart Study revealed NAFLD in 60% of patients with mixed hyperlipidemia,33 and a study from Korea of potential living liver donors showed that hyperlipidemia was associated with a greater than twofold risk of significant (>30%) steatosis.34 Most patients with NAFLD have multiple risk factors, including central obesity, type 2 diabetes mellitus, and hyperlipidemia, although some affected persons lack all recognized risk factors. NAFLD has been associated with many drugs and toxins and metabolic, surgical, and genetic conditions (see Table 85-1) that have abnormal fat metabolism and mitochondrial injury or dysfunction in common. NAFLD is now recognized as the hepatic component of the metabolic syndrome, which includes hyperlipidemia, glucose intolerance, obesity, and systemic hypertension. The risk and severity of NAFLD increase with the number of components of the metabolic syndrome.28,35
PATHOGENESIS
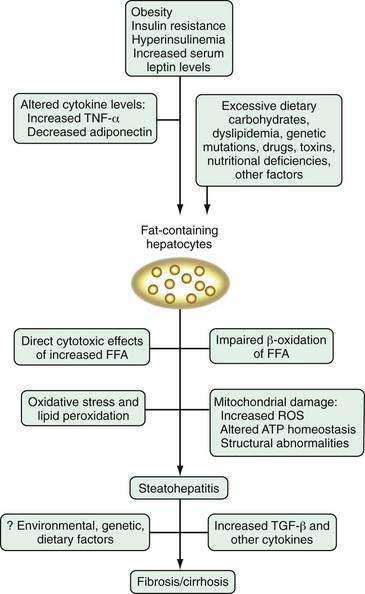
The pathogenesis of NAFLD is poorly understood, in part because of a lack of suitable animal models that mimic human NAFLD. In light of the variety of conditions that have been associated with NAFLD, it is not surprising that no single pathogenic mechanism has been identified. The prevailing theory is the “two-hit hypothesis,” first proposed by Day and James in 1998.36 This hypothesis states that dysregulation of fatty acid metabolism leads to steatosis, which is the first hepatic insult in NAFLD. Steatosis is associated with several cellular adaptations and altered signaling pathways, which render hepatocytes vulnerable to a “second hit.” The second insult may be one or more environmental or genetic perturbations, which cause hepatocyte necrosis and inflammation and activate the fibrogenic cascade, thereby leading to fibrosis and cirrhosis in a minority of patients with NAFLD.
Hepatic steatosis is the hallmark histologic feature of NAFLD. Normally, free fatty acid (FFA) is supplied to the liver through intestinal absorption (in the form of chylomicron remnants) or from lipolysis of adipose tissue, where FFA is stored as triglycerides. In the liver, FFA is oxidized by mitochondria, esterified into triglycerides, synthesized into phospholipids and cholesteryl esters, and secreted from the liver as very-low-density lipoprotein (VLDL). Under normal circumstances, fatty acid metabolism is under tight regulatory control by catecholamines, glucagon, growth hormone, and insulin. Hepatic triglyceride accumulation occurs when fatty acid metabolism shifts to favor net lipogenesis, rather than lipolysis. This shift occurs when the amount of FFA supplied to the liver from the intestine or adipose tissue exceeds the amount needed for mitochondrial oxidation, phospholipid synthesis, and synthesis of cholesteryl esters. Triglycerides also accumulate in the liver when synthesis of lipoprotein decreases or export of lipids from the liver is impeded (see also Chapter 72).
Current evidence points to insulin resistance and hyperinsulinemia as the primary pathogenic factors in steatosis in most patients with NAFLD. Strong laboratory and clinical evidence supports the association of peripheral insulin resistance and hyperinsulinemia with NAFLD, even in lean patients without obvious glucose intolerance.37–39 The molecular mechanism leading to insulin resistance is complex and not understood completely. In the setting of obesity and hyperinsulinemia, alterations in several molecules, including FFA, tumor necrosis factor-α (TNF-α), membrane glycoprotein PC-1, and leptin, interfere with the insulin signaling pathway. Diabetes mellitus and obesity are associated with increased amounts of FFA in plasma, caused in part by abnormal release of FFA by insulin-resistant adipocytes. Excess FFA contributes to hepatic insulin resistance by down-regulating insulin receptor substrate-1 (IRS-1) signaling.40,41 Insulin resistance and hyperinsulinemia lead to steatosis by means of a number of aberrant mechanisms of FFA disposal. In the liver, insulin stimulates fatty acid synthesis, down-regulates mitochondrial β-oxidation of FFA, blocks the secretion of triglycerides from hepatocytes by increasing intracellular degradation of VLDL and apolipoprotein B-100 (apoB-100), and blocks exocytosis of VLDL-containing vesicles.40,42,43 Also, patients with NASH have impaired hepatic synthesis of apoB-100, which also may contribute to hepatic triglyceride accumulation.44
Insulin resistance in NAFLD may be potentiated by aberrant levels or function of several important peptide mediators secreted by adipocytes, including TNF-α, leptin, and adiponectin. In noninflammatory states, TNF-α is derived from adipose tissue (including adipose tissue macrophages), and plasma levels of TNF-α correlate with body fat mass.45 TNF-α interferes with insulin signaling by down-regulating IRS-1 signaling via serine phosphorylation, likely through activation of stress-related protein kinases including Jun N-terminal kinase (JNK), which plays a key role in obesity-related insulin resistance. Activation of the inhibitor kappa-β kinase (IKK-β)/nuclear factor kappa β (NF-κβ) pathway by FFA may also play a role in reduced hepatic insulin sensitivity,46,47 and may increase production of additional inflammatory cytokines such as TNF-α and interleukin (IL)-6.48 Elevated TNF-α levels have been demonstrated in several studies of NAFLD49–52; however, the independent contribution of TNF-α to the pathogenesis and risk of progression of NAFLD is still unclear.
Adipocytokines are peptides produced by visceral adipose tissue. Adiponectin is secreted by adipocytes in inverse proportion to BMI and is a potent inhibitor of TNF-α. Serum adiponectin levels are reduced in obesity, insulin resistance, diabetes mellitus, and the metabolic syndrome.50 Delivery of recombinant adiponectin to mice fed a high-fat, alcohol-containing diet and to genetically obese (ob/ob) mice dramatically alleviates hepatomegaly, steatosis, inflammation, and elevated liver biochemical test levels in both murine populations.53 These therapeutic effects result in part from the ability of adiponectin to enhance hepatic fatty acid β-oxidation, decrease hepatic triglyceride content, and decrease hepatic insulin resistance. Furthermore, adiponectin suppresses hepatic and plasma concentrations of TNF-α. Studies have reported an inverse relationship between serum adiponectin levels and the degree of steatosis and hepatocyte injury in humans with NAFLD, and this inverse association may be independent of insulin resistance.51,54 Further studies are needed to determine whether an increase in the TNF-α/adiponectin ratio has a primary pathogenic role in the development of steatosis or is more directly correlated with progression from steatosis to steatohepatitis.
Leptin is a satiety hormone, derived from adipocytes, that controls food intake and energy regulation (see Chapter 1). Leptin is intimately involved with insulin signaling and regulation of glucose metabolism in peripheral tissues and may play an important role in regulating the partitioning of fat between mitochondrial β-oxidation and triglyceride synthesis in the liver.55 Severe steatosis and steatohepatitis develop in leptin-deficient (ob/ob) mice. Obesity in humans is associated with relative leptin resistance and high leptin levels, which may contribute to the genesis of steatosis by a negative impact on insulin signaling or may be a consequence of the chronic hyperinsulinemia associated with obesity. Several studies have examined the relationship between serum leptin levels and NAFLD, with conflicting results.56–59 One study has suggested that serum leptin levels in patients with NASH correlate with the severity of hepatic steatosis, independent of BMI, but not with the degree of hepatic inflammation or fibrosis.58 At present, the contribution of leptin to the pathogenesis of NAFLD is unclear.
Although insulin resistance and hyperinsulinemia are clearly pivotal to the development of steatosis, consensus is lacking on the subsequent insults that cause steatosis to progress to steatohepatitis and fibrosis in some patients. Similarities in the histologic features and natural history of alcoholic liver disease and NAFLD suggest that common mechanisms may be involved in the pathogenesis of these disorders. Chronic oxidative stress is believed to be central to the pathogenesis of alcohol-related liver damage. Processes that increase the production of oxidants in the liver during chronic alcohol exposure include the metabolism of ethanol to its reactive intermediate acetaldehyde; induction of microsomal ethanol-oxidizing enzymes, such as cytochrome P450 2E1 (CYP2E1), which generates reactive oxygen species (ROS) that can peroxidize cellular membranes, thereby causing cellular injury60; inhibition of mitochondrial electron transport chain activity; and depletion of mitochondrial glutathione.61 Activation of microsomal enzymes, including CYP2E1, in patients with NAFLD62–63 and mitochondrial production of ROS in murine models of NAFLD64,65 suggest that chronic oxidative stress and lipid peroxidation may also be central to the pathogenesis of NAFLD.
Increased levels of FFA can be directly toxic to hepatocytes through a number of mechanisms. An increased FFA concentration leads to lysosomal destabilization and stimulation of TNF-α.66 FFA also up-regulates cytochrome P450 isoenzymes, leading to enhanced generation of ROS and lipid peroxidation.67 An increased intracellular FFA concentration can lead to sustained up-regulation of peroxisomal proliferator-activated receptor-α (PPAR-α), which promotes fatty acid oxidation and disposal but also may increase oxidative stress through the production of dicarboxylic acid derivatives; PPAR-α also may predispose affected persons to carcinogenesis.44 FFA can be directly toxic to cellular membranes, lead to the formation of toxic fatty acid ethyl ethers, and cause overall disruption of mitochondrial function, thereby overwhelming the overlapping protective mechanisms designed to combat FFA hepatotoxicity.45
Endotoxin and endotoxin-mediated cytokine release are suspected in the pathogenesis of alcoholic steatohepatitis, in which increased serum levels of bacterial endotoxin and lipopolysaccharide (LPS) stimulate hepatic production of TNF-α, IL-6, and IL-8 and activate an inflammatory response that leads to hepatic necrosis (see Chapter 84).61 Bacterial endotoxin also may contribute to the development of NAFLD in some circumstances. Portal endotoxemia was believed to contribute to NASH and hepatic failure associated with surgical jejunoileal bypass (performed in the past to treat obesity), the risk of which was reduced with antibiotics. Yang and colleagues have demonstrated that ob/ob mice with steatosis are highly vulnerable to endotoxin-induced hepatocyte damage, and NASH rapidly develops in these animals after exposure to low doses of bacterial LPS.68 In addition, Zucker diabetic (fa/fa) rats and ob/ob mice demonstrate decreased Kupffer cell function, which may increase the vulnerability of steatotic hepatocytes to TNF-α–mediated liver damage.69 Small studies suggest a possible pathogenic role of bacterial endotoxins in human NAFLD as well,70,71 but these studies are far from conclusive.
A growing body of evidence suggests that mitochondrial changes and altered hepatic energy homeostasis may play roles in the pathogenesis of NAFLD. Studies have shown a decrease in the activity of mitochondrial respiratory chain complexes in steatotic livers, with a concomitant increase in mitochondrial ROS formation; these changes correlate with serum TNF-α levels, insulin resistance, and BMI.72,73 Ob/ob mice have increased levels of uncoupling protein, UCP-2, an inner mitochondrial membrane protein that mediates proton leak, uncouples adenosine triphosphate (ATP) synthesis, regulates ROS production, and may render fatty hepatocytes vulnerable to metabolic stressors65; however, the role of UCP-2 in NAFLD in humans is unknown. Studies have shown that mice and humans with NAFLD have diminished capacity for replenishing ATP stores after ATP depletion. Mitochondrial structural defects may be one cause of reduced ATP stores. Megamitochondria and crystalline mitochondrial inclusions have been identified in patients with NAFLD and may represent an adaptive process to oxidative stress or secondary injury.39,74 Limited data in patients with NASH suggest differential expression of several genes important for proper mitochondrial functioning, including genes involved in ROS scavenging, glucose metabolism, and fatty acid metabolism.75 In addition, mitochondrial DNA damage similar to that found in alcoholic liver disease and Wilson disease also may contribute to the development of NASH. Further animal and human studies are needed to determine whether mitochondrial dysfunction and ATP depletion are causes or consequences of NAFLD.
Fibrosis is a frequent histologic finding in advanced NAFLD but has not been well studied in this disease. Hepatic fibrosis results from activation and proliferation of hepatic stellate cells in the subendothelial space of Disse, with subsequent secretion of extracellular matrix components, including collagen types I and III (see Chapter 90). Factors proposed to initiate and perpetuate the fibrogenic process in stellate cells include inflammatory cytokines, angiotensin, alterations in the extracellular matrix, growth factors, and oxidative stress. In NAFLD, lipid peroxidation products may enhance hepatic production of transforming growth factor-β (TGF-β), which activates stellate cells. Endothelial cells, leukocytes, and Kupffer cells may stimulate the stellate cells to proliferate, possibly through the release of platelet-derived growth factor (PDGF), TGF-β, and other cytokines.76 In addition, hyperinsulinemia and hyperglycemia associated with NAFLD may stimulate release of connective tissue growth factor, an intermediate molecule involved in fibrogenesis.77 Finally, animal data suggest that leptin may perpetuate fibrogenesis in NAFLD by stimulating Kupffer cells and sinusoidal endothelial cells to produce TGF-β.78
Research into the pathogenesis of NAFLD is proliferating at a rapid pace, but the picture is far from clear (Fig. 85-1). Any one of the putative mechanisms discussed here is unlikely to explain the pathogenesis of NAFLD in all affected patients. More likely, NAFLD develops as a consequence of a “multi-hit” process. The first “hit” is steatosis induced primarily by insulin resistance and hyperinsulinemia. After steatosis develops, a number of factors, including lipid peroxidation, oxidative stress, cytokine alterations, mitochondrial dysfunction, and Kupffer cell activation, may incite an inflammatory response and fibrosis in some patients with genetic or environmental susceptibilities. The exact interplay among these and other factors remains to be elucidated, but understanding of the pathogenesis should be enhanced by the development and refinement of appropriate animal models, including the genetically obese, leptin-deficient ob/ob mouse and Zucker diabetic rats, in which steatosis develops; S-adenosylmethionine (SAM)-deficient mice, in which severe steatohepatitis develops79; the Otsuka-Long-Evans-Tokushima fatty (OLETF) rat model, in which a cholecystokinin-A receptor defect leads to hyperglycemia, obesity, insulin resistance, and hepatic steatosis80; and a high fat-fed rat model in which insulin resistance, elevated serum TNF-α levels, increased oxidative stress, mitochondrial lesions, and early fibrosis develop.81
CLINICAL, LABORATORY, AND IMAGING FEATURES
The clinical and laboratory features of NAFLD are summarized in Table 85-2. NAFLD usually is discovered incidentally because of elevated liver biochemical test levels or hepatomegaly noted during an evaluation for an unrelated medical condition. Most patients with NAFLD are asymptomatic, but some may describe vague right upper quadrant pain, fatigue, and malaise. Hepatomegaly has been described in up to 75% of patients with NAFLD but often is difficult to appreciate on physical examination because of obesity. Stigmata of chronic liver disease, such as splenomegaly, spider angiomata, and ascites, are rare, except in patients with NAFLD-associated cirrhosis.
Table 85-2 Clinical and Laboratory Features of Nonalcoholic Fatty Liver Disease
| SYMPTOMS | SIGNS | LABORATORY FEATURES |
|---|---|---|
| Common | ||
| None (48%-100% of patients) | Hepatomegaly | |
ANA, antinuclear antibodies; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Elevated liver biochemical test levels may be found in up to 50% of patients with simple steatosis and are present in approximately 80% of patients with advanced NAFLD. A mild-to-moderate (1.5- to 4-fold) elevation of the serum aspartate aminotransferase (AST) or alanine aminotransferase (ALT) level, or both, is usual, and levels seldom exceed 10 times the upper limit of normal. The serum ALT level usually is greater than the AST level, in contrast with the pattern of alcoholic hepatitis, in which the AST level is at least twofold higher than the ALT level (see Chapters 73 and 84). The alkaline phosphatase and gamma glutamyl transpeptidase (GGTP) levels may be elevated, but the serum bilirubin level, prothrombin time, and serum albumin level typically are normal, except in patients with NAFLD-associated cirrhosis. Up to one fourth of patients with NAFLD may have antinuclear antibodies (ANA) in low titers (less than 1 : 320).82 Antimitochondrial antibodies (AMA) and hepatitis B surface antigen are not detected. Antibody to hepatitis C virus (anti-HCV) must be absent to implicate NAFLD as the sole cause of abnormal liver biochemical test levels; however, steatosis, often in association with visceral obesity, frequently accompanies HCV infection and may be associated with a more aggressive course (see Chapter 79).83 Serum ceruloplasmin and α1-antitrypsin levels are within normal limits. Serum and hepatic iron levels may be elevated in patients with NAFLD. In particular, the serum ferritin level may be elevated in 20% to 50% of patients with NAFLD and may be a marker of more advanced disease.29,84 Nevertheless, the frequency of genetic hemochromatosis among patients with NAFLD is not increased. Clinical and laboratory findings do not correlate with the histologic severity of NAFLD. The entire histologic spectrum of NAFLD, including cirrhosis, can be seen in patients with normal or near-normal serum aminotransferase levels.31
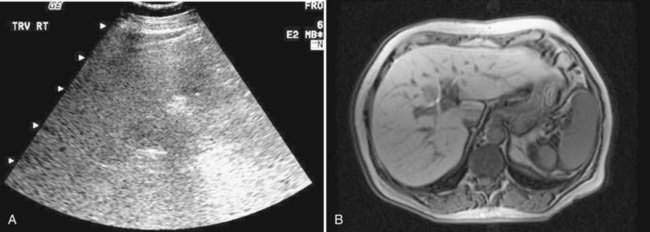
Imaging studies often are obtained during the evaluation of unexplained liver biochemical abnormalities or suspected NAFLD. Hepatic ultrasonography, the imaging modality employed most commonly, may reveal a “bright” liver of increased echogenicity, consistent with hepatic steatosis (Fig. 85-2). Fatty liver also can be documented by abdominal computed tomography (CT) scan (a fatty liver is lower in density than the spleen), and by magnetic resonance imaging (MRI), with which fat appears bright on T1-weighted imaging. A study that assessed the sensitivities of MRI, abdominal CT, and ultrasonography for distinguishing advanced NASH from simple steatosis showed that ultrasonography and CT had sensitivity rates of 100% and 93% for detecting hepatic fat involving greater than 33% of the liver, with positive predictive values of 62% and 76%, respectively.85 No radiologic modality, however, was able to distinguish simple steatosis from more advanced forms of NAFLD. Imaging studies such as ultrasonography may support the diagnosis of NAFLD but cannot predict the severity of disease and cannot replace liver biopsy for establishing the diagnosis with certainty.
HISTOPATHOLOGIC FEATURES
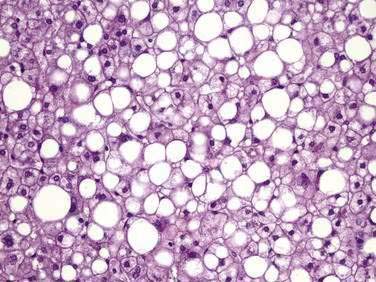
The major histologic features of NAFLD resemble those of alcohol-induced liver disease and include steatosis (fatty liver), steatohepatitis (fatty liver plus parenchymal inflammation with or without accompanying focal necrosis), and varying degrees of fibrosis, including cirrhosis. Steatosis is predominantly macrovesicular and usually is distributed diffusely throughout the liver lobule, although prominent microvesicular steatosis and zone 3 (perivenular) steatosis have been reported occasionally (Fig. 85-3). Mild lymphocytic, neutrophilic, or mixed inflammatory infiltrates also may be observed, and glycogenated nuclei are common.
NASH, which is an advanced form of NAFLD, is indistinguishable histologically from alcoholic hepatitis (Fig. 85-4 and Table 85-3). Steatosis is present in all cases and can affect the hepatic lobules either diffusely or primarily in the central zones. The degree of steatosis may correlate with the patient’s BMI and generally is more severe in NASH than in alcoholic hepatitis.86 Lobular inflammation is a hallmark feature of NASH and is characterized by infiltration of lymphocytes, other mononuclear cells, and polymorphonuclear neutrophils. The intensity of the inflammation varies with the severity of steatohepatitis and may be milder in NASH than in alcoholic hepatitis.87 Glycogenated nuclei may be present. Hepatocyte ballooning and hepatocyte necrosis of varying degrees often are present and may portend a worse prognosis.88,89 Mallory (or Mallory-Denk) bodies, which may be small, sparse, and inconspicuous, are seen frequently. Mild stainable iron may be present in up to 50% of the patients. Pericellular, perisinusoidal, and periportal fibrosis has been described in 37% to 84% of patients with NASH. The extent of fibrosis varies considerably, ranging from delicate strands surrounding small veins or groups of cells to densely fibrotic septa with distortion of the hepatic architecture. Perisinusoidal fibrosis is most common, especially in adults, is initially mild, and predominates in zone 3 around the terminal hepatic veins.41 Cirrhosis is found on initial biopsy in 7% to 16% of patients with NAFLD and abnormal liver biochemical test levels.15,29 The risk of cirrhosis in the setting of NAFLD may be greatest in morbidly obese patients. In NAFLD-associated cirrhosis, the typical histologic features of NAFLD may be minimal or absent, potentially leading to the misdiagnosis of cryptogenic cirrhosis.
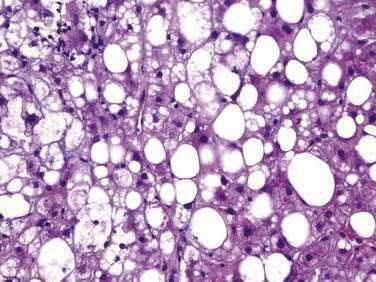
Table 85-3 Histologic Features of Nonalcoholic Fatty Liver Disease
| Present in All or Most Cases |
BMI, body mass index.
Strict histologic criteria for NASH have not yet been defined. At least two scoring systems have been proposed.90,91 To reach consensus on the pathologic classification of NASH, the Pathology Committee of the National Institutes of Health NASH Clinical Research Network has proposed a scoring system incorporating 14 histologic features.90 The unweighted sum of scores for steatosis, lobular inflammation, and hepatocellular ballooning is used to construct an activity score, which can be used to classify cases into categories designated “not NASH,” “borderline NASH,” or “NASH” with reasonable inter-rater agreement. Thus far, however, no scoring system for NAFLD or NASH has been widely adopted.
DIAGNOSIS
Establishing a definitive diagnosis of NAFLD requires both clinical and histologic data (Fig. 85-5). Most patients with NAFLD are evaluated because of chronically elevated liver biochemical test levels, with or without hepatomegaly. The combination of the patient’s history, physical examination, blood test results, and radiologic findings is useful for excluding other causes of liver disease. Laboratory testing should include liver biochemical tests, complete blood count, prothrombin time, anti-HCV, hepatitis B surface antigen, iron indices, ceruloplasmin in persons younger than 40 years of age, α1-antitrypsin, and AMA. Imaging studies may support the diagnosis (see earlier), but the absence of characteristic findings does not preclude a diagnosis of NAFLD. To establish a diagnosis of NAFLD, alcoholic liver disease must be excluded. Clinical and histologic data unreliably differentiate NAFLD from alcoholic liver disease in ambulatory patients. Therefore, the diagnosis of NAFLD should be entertained only in the absence of significant alcohol use (consumption of less than 20 to 40 g of alcohol per day in most clinical studies).
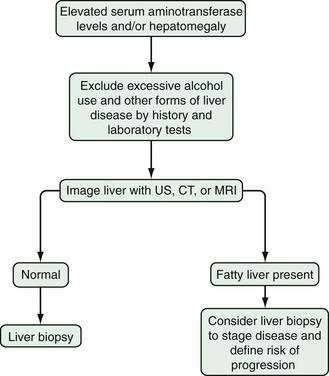
The Role of Liver Biopsy
The role of liver biopsy in establishing the diagnosis of NAFLD has been debated. Many practitioners consider NAFLD a diagnosis of exclusion, when clinical and laboratory examinations fail to reveal another cause of chronic liver disease. The diagnosis of NAFLD is also suggested when an imaging study provides evidence of fatty liver. Most patients with NAFLD do not undergo a liver biopsy, largely because the results usually will not affect management (therapeutic options are limited, see later). The argument for liver biopsy in all patients with presumed NAFLD is centered on several lines of evidence: The correlation among clinical, laboratory, and histologic findings in NAFLD is poor, and patients with normal liver biochemical test results can have significant liver injury on biopsy specimens31; liver biopsy is the only diagnostic test that can reliably quantify hepatic steatosis, necrosis, and fibrosis; and the histologic stage of NAFLD is the best prognostic indicator.92 In addition, understanding of the natural history and treatment of NAFLD have been hampered by the lack of histologic data in most clinical studies. The most prudent approach may be to select patients for whom liver biopsy might influence management. Several clinical and biochemical risk factors for progressive disease have been identified and may facilitate selection of patients for liver biopsy. Patients with obesity, longstanding or persistent liver biochemical test abnormalities (even with good glycemic control and after weight loss), older age, multiple components of the metabolic syndrome, an AST/ALT ratio greater than 1, markedly elevated liver biochemical test levels, symptoms and signs of portal hypertension, or evidence of fibrosis on an imaging study are more likely to have advanced disease (Table 85-4).29,89 The results of a liver biopsy in these patients might lead to a more aggressive treatment strategy, participation in clinical trials, or screening for hepatocellular carcinoma in the setting of cirrhosis.
Table 85-4 Risk Factors for Advanced* Nonalcoholic Fatty Liver Disease
| Clinical |
ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Noninvasive Markers of Fibrosis in NAFLD
Although percutaneous liver biopsy remains the standard for the diagnosis of NAFLD, it is costly, invasive, and associated with a small risk of complications. Sampling variability is common, and the large number of persons with NAFLD far outstrips the manpower available to perform liver biopsies. Significant progress has been made in developing simple, noninvasive, and quantitative tests to estimate the degree of hepatic fibrosis in a number of liver diseases, including NAFLD. The FibroTest (called FibroSure in the United States) is the best studied of these noninvasive tests (see Chapters 73 and 79). The panel of blood tests used to estimate hepatic fibrosis includes serum α2-macroglobulin, apolipoprotein A-1, haptoglobin, total bilirubin, and GGTP levels, and the necroinflammatory activity index combines the same five markers plus the serum ALT level. In a study of 167 patients with NAFLD, FibroTest was highly sensitive for detecting bridging fibrosis and cirrhosis.93 FibroTest cutoff value of .70 had a positive predictive value of 73% and a specificity of 98% for advanced fibrosis. A cutoff value of 0.30 had a negative predictive value of 90% for advanced fibrosis. Unfortunately, 33% of patients had a FibroTest score between 0.30 and 0.70, and in this range, the test is inaccurate for assessing the stage of fibrosis. Therefore, patients with a score in this range would need a liver biopsy for accurate staging.
Angulo and colleagues developed and validated another noninvasive fibrosis scoring system called the NAFLD Fibrosis Score, which is derived from clinical and laboratory information that is obtained easily in the context of any clinical encounter.94 Using this scoring algorithm, which incorporates age, BMI, hyperglycemia, AST/ALT ratio, platelet count, and serum albumin level, the authors defined a low cutoff value with a negative predictive value of 88% to 93% and a high cutoff value with a positive predictive value of 82% to 90%. Only 25% to 28% of cases were indeterminate and would therefore require liver biopsy for accurate staging. Additional noninvasive tests for fibrosis have been evaluated with variable success in small studies of NAFLD, including transient elastography (Fibroscan), which uses ultrasound to quantify liver stiffness and estimate fibrosis,95 serum dehydroepiandrosterone levels,96 and serum hyaluronic acid levels.97 One or more noninvasive indices of fibrosis is likely to be validated in the future and may supplant the need for liver biopsy in many, but not all, patients with NAFLD.
NATURAL HISTORY
The natural history of NAFLD is largely unknown because no prospective, longitudinal studies with histologic follow-up have been conducted in patients with NAFLD. In addition, the long-term complications of NAFLD are probably under-recognized and under-reported, in part because the characteristic feature of macrovesicular steatosis may diminish or disappear in late-stage NAFLD. The available data from retrospective studies suggest that NAFLD is a benign disease in most patients. The prognosis in patients with steatosis in the absence of hepatocyte necrosis and fibrosis clearly is favorable, with little potential for histologic or clinical progression.98 In some patients, however, NAFLD can lead to cirrhosis, liver failure, or hepatocellular carcinoma. Between 2003 and 2008, five retrospective studies were published that included a total of 215 patients with NAFLD who underwent paired liver biopsies.6,99–101 Follow-up periods ranged from 1 to 14 years, and histologic progression of fibrosis was documented in 32% to 41% of the patients. NAFLD was stable histologically in 34% to 50% and appeared to improve in 16% to 29%. Substantial variability in the rate of progression of fibrosis was observed among patients; histologic progression did not signal clinical deterioration in most cases, and no clinical or laboratory data reliably predicted the course of liver disease. These data must be interpreted with caution because variability in histologic staging may be the result of sampling error.102
A large population-based study from Olmsted County, Minnesota, included 420 patients with definite or presumed NAFLD based on imaging studies or, less frequently, liver biopsy findings.103 Cirrhosis developed in 3% of the patients over a mean follow-up of 7.6 years. Liver disease was the third leading cause of death in this cohort, behind malignancy and ischemic heart disease. These limited data suggest that NAFLD is an indolent condition, with few clinical sequelae in most patients, but can progress to irreversible, clinically important liver disease over a relatively short period of time in a minority of affected persons. This conclusion is supported by data from a classic study of 132 patients with NAFLD evaluated over an 18-year period. The study compared clinical outcomes based on the degree of injury on an index liver biopsy specimen.104 Each biopsy specimen was assigned to a histologic subgroup—types 1 to 4—that represented progressively severe disease as determined by the cumulative occurrence of steatosis, inflammation, hepatocyte necrosis, and fibrosis. Cirrhosis and liver-related deaths were more common (25% and 11%, respectively) in patients with NAFLD types 3 and 4 (steatosis, inflammation, and necrosis, with or without fibrosis) than in patients with NAFLD types 1 and 2 (steatosis without necrosis), although the mortality difference failed to reach statistical significance. In addition, the liver-related mortality rate of 11% among patients with NAFLD types 3 and 4 was higher than the age-adjusted death rate from chronic liver disease/cirrhosis in the general U.S. population (9.5 per 100,000 per year). These data suggest that the risk of liver-related complications in NAFLD correlates, at least to some extent, with the degree of hepatocellular injury and fibrosis found on an index liver biopsy specimen.
NAFLD and alcoholic hepatitis are similar histologically, but differ substantially in clinical outcomes (see Chapter 84). The five-year survival rate of patients with alcoholic hepatitis is only 50% to 75% because of the large proportion of patients (greater than 50%) in whom cirrhosis and its complications develop. A study has shown that the long-term survival of patients with NASH is significantly better than the long-term survival of patients with alcoholic hepatitis.86 In the minority of patients in whom NAFLD-associated cirrhosis develops, however, the outcome may be similar to that for other causes of cirrhosis. Strong circumstantial evidence suggests that NAFLD is the likely cause of many cases of cryptogenic cirrhosis105–107 and may be associated with the development of hepatocellular carcinoma.30,106,108 One study showed that the 5- to 10-year outcome of NAFLD-associated cirrhosis was similar to that for HCV-associated cirrhosis, although hepatocellular carcinoma was significantly less common in the patients with NAFLD.109
Large prospective studies are needed to define the natural history of NAFLD, but emerging evidence confirms that NAFLD can be progressive and associated with significant morbidity and mortality in some patients. The risks of liver-related morbidity and mortality are greatest in persons with evidence of advanced NAFLD (steatohepatitis with necrosis and fibrosis) on the initial liver biopsy specimen.104 If clinical and biochemical risk factors for progressive disease can be established, a subset of patients can be identified in whom a liver biopsy will have the greatest prognostic and therapeutic value (see earlier). Several potential risk factors have been identified in different populations (see Table 85-4). Women are over-represented in studies of NAFLD, but whether gender is an independent risk factor for advanced disease is unclear. Older age, obesity, diabetes mellitus, and an AST/ALT ratio greater than 1 were demonstrated in one study to be significant predictors of severe fibrosis (bridging/cirrhosis) in patients with NAFLD.29
In another study of overweight patients with abnormalities on liver biochemical tests, liver fibrosis was independently associated with hepatic necroinflammatory activity, BMI greater than 28 kg/m2, age older than 50 years, serum triglyceride level higher than 1.7 mmol/L, and serum ALT more than twice normal.43 In another study of morbidly obese patients referred for bariatric surgery, systemic hypertension, an elevated serum ALT level, and a high insulin resistance index were highly predictive of advanced NAFLD.15 As noted previously, however, the full spectrum of liver damage, including cirrhosis, has been documented in nonobese patients with near-normal liver biochemical test results.
TREATMENT
The optimal therapy for NAFLD has not been established. To date, no large, randomized treatment trials demonstrating resolution of steatosis, inflammation, and fibrosis have been conducted in patients with NAFLD. Small numbers of patients, varying inclusion criteria, and varying end points have limited the clinical impact of published studies. Historically, the treatment of NAFLD has consisted of weight loss, removal of offending drugs and toxins, and control of associated metabolic disorders, including diabetes mellitus and hyperlipidemia. Several case reports and small studies of diet and exercise have shown improvements in biochemical, ultrasonographic, and in some cases, histologic abnormalities in children and adults with NASH.52,110–112 Intensive nutritional counseling may lead to sustained weight loss and significant histologic improvement in some patients.113 Several small, largely uncontrolled studies also showed improvements in liver biochemical test results, steatosis, and fibrosis in a few patients who achieved modest weight loss with orlistat, a reversible inhibitor of gastric and pancreatic lipases.114,115 Bariatric surgery leads to massive weight loss and improves insulin sensitivity in most patients, normalizes some of the metabolic abnormalities involved in the pathogenesis of NAFLD, decreases the hepatic expression of mediators of liver inflammation and fibrosis, and improves hepatic histology in patients with NAFLD.116–120
No scientific data exist to support a particular commercial or medicinal diet plan for NAFLD, and no one correct dietary approach is likely to be suitable for all patients with NAFLD.121 Unfortunately, the therapeutic benefit of weight loss achieved with diet, medicinal aids, exercise, or surgery has not been examined in randomized, prospective studies with firm histologic end points. Until such studies are performed, a recommendation for moderate weight loss is reasonable in overweight patients with NAFLD, although sustained weight loss is seldom achieved. Rapid weight loss can exacerbate steatohepatitis in morbidly obese patients, especially after bariatric surgery122; therefore, the rate of weight loss and serial liver biochemical test results should be monitored carefully in patients on a weight reduction regimen. New therapeutic methods should capitalize on today’s improved understanding of the pathogenesis of NAFLD (Table 85-5).
Table 85-5 Potential Therapies for Nonalcoholic Fatty Liver Disease
ATP, adenosine triphosphate; TNF-α, tumor necrosis factor-α.
Antioxidants
Medications that minimize oxidative stress may prove useful. Vitamin E, an inexpensive yet potent antioxidant, has been examined as an agent for treatment of NAFLD in several small pediatric and adult studies, with varying results.52,123,124 In all studies, vitamin E was well tolerated, and most studies showed modest improvements in serum aminotransferase levels, ultrasonographic appearance of the liver, and, infrequently, histologic findings. Randomized controlled studies with histologic inclusion criteria and end points are needed, however, to determine if vitamin E, either alone or in combination with other medications, leads to histologic improvement in NAFLD. In light of the potentially negative effects of vitamin E on cardiovascular health,125 caution should be exercised in treating NAFLD with vitamin E until better studies are available.
Betaine, a metabolite of choline that raises SAM levels and decreases cellular oxidative damage, has shown promise in a small pilot study as a therapeutic agent for NASH.126 N-acetylcysteine, superoxide dismutase, and PPAR-α agonists such as ragaglitazar also may hold therapeutic promise,64,127,128 although clinical studies in humans are lacking.
Insulin-Sensitizing Agents
The association between hyperinsulinemic insulin resistance and NAFLD provides a logical target for treatment. Metformin, a biguanide that reduces hyperinsulinemia and improves hepatic insulin sensitivity, reduces hepatomegaly and hepatic steatosis in ob/ob mice,129 but results in human studies have been less impressive.130,131 Thiazolidinediones (TZDs), potent PPAR-α agonists, also are being investigated as possible agents for the treatment of NAFLD. PPAR-α is a nuclear receptor expressed in adipose tissue, muscle, and liver. In adipocytes, PPAR-α promotes cell differentiation and decreases lipolysis and FFA release. TZDs improve insulin sensitivity and hyperinsulinemia by increasing glucose disposal in muscle and decreasing hepatic glucose output. Treatment with troglitazone, a first-generation TZD, was associated with biochemical and histologic improvements in patients with NASH, but troglitazone subsequently was withdrawn from the market because of rare but serious hepatotoxicity. Rosiglitazone and pioglitazone, TZDs with low rates of hepatotoxicity, have been investigated in separate 48-week, single-arm treatment trials in patients with histologically proven NASH.132,133 In both studies, treatment was well tolerated and was associated with improved insulin sensitivity, normalization of liver biochemistries, and histologic improvement in most patients. A drawback of both TZDs, however, was substantial weight gain (4.0% to 7.3%) and increased total body adiposity. In addition, the durability of biochemical and histologic improvements after completion of therapy was not examined in either study. A follow-up study has suggested, however, that the beneficial effects of pioglitazone diminish when the drug is discontinued.134 A placebo-controlled European trial of rosiglitazone in 63 patients with NAFLD showed improvement in steatosis and serum aminotransferase levels but not in necroinflammation or fibrosis.135 Rosiglitazone was also associated with significant weight gain in the TZD-treated patients in this study. TZDs must be assessed in large, placebo-controlled trials before they can be recommended for routine use in patients with NAFLD.
Iron Reduction
High serum iron and ferritin levels have been identified in some patients with NAFLD, most of whom do not have genetic hemochromatosis or hepatic iron overload. Most investigators believe that increased serum iron indices are a by-product of hepatic inflammation, rather than a contributor to the pathogenesis of NAFLD, but a few small studies have suggested that iron depletion may have a therapeutic role in NAFLD by decreasing plasma insulin, glucose, and serum aminotransferase levels.136,137 The relationship between iron and insulin is complex, but the insulin-sparing effect of iron depletion may be the result of enhanced skeletal muscle glucose transport and metabolism and increased hepatic extraction and metabolism of insulin.138 The primary limitation of these studies is the lack of histologic inclusion criteria and end points, but the results are intriguing and merit further investigation.
Lipid-Lowering and Cytoprotective Agents
The usefulness of lipid-lowering and cytoprotective drugs for the treatment of NAFLD has been assessed in a few small trials, with varying results. Treatment with gemfibrozil was associated with biochemical improvement in 74% of patients in the treatment group, compared with 30% of untreated control subjects, but histologic features were not assessed.139 Treatment of NASH with atorvastatin, a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor, showed promise in a small pilot study,140 but statins have not been assessed in a large clinical trial. Ursodeoxycholic acid, a cytoprotective agent, showed promise in a pilot study of NASH but was not effective in a randomized placebo-controlled trial.141 The combination of ursodeoxycholic acid and vitamin E has shown some efficacy. Future therapies for NAFLD might include agents that increase adiponectin levels, neutralize TNF-α, improve mitochondrial ATP homeostasis, or alter leptin levels. When the pathogenesis of NAFLD is elucidated further, new therapeutic agents will likely be developed.
Liver Transplantation
Patients with NAFLD in whom end-stage liver disease develops should be evaluated for liver transplantation. The outcome of liver transplantation in these patients is good, although NAFLD can recur after liver transplantation.142,143 The risk factors for recurrent or de novo NAFLD after liver transplantation probably are multifactorial and include hypertriglyceridemia, obesity, diabetes mellitus, and glucocorticoid therapy.
FOCAL FATTY LIVER
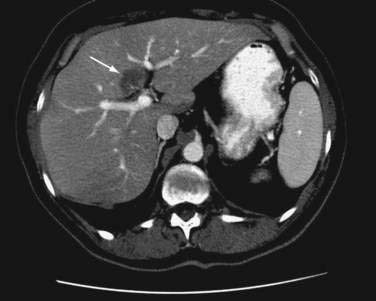
In contrast with NAFLD, which is a diffuse parenchymal process, focal fatty liver is a localized or patchy process that simulates a space-occupying lesion in the liver on imaging studies. This condition has been recognized increasingly in adults and children as a result of the improved sensitivity of abdominal imaging. Focal fatty liver has characteristic patterns on CT: usually a nonspherical shape, absence of mass effect, and CT attenuation values consistent with those of soft tissue.144 The density of focal fatty liver is close to that of water, unlike that of liver metastases, which have a density that is closer to that of hepatocytes. Ultrasonography and MRI can help confirm a diagnosis of focal fatty liver (Fig. 85-6). A presumptive diagnosis of focal fatty liver should not be made when a mass effect, areas of mixed hypo- and hyperechogenicity, an irregular shape, or a history of malignancy is present. In such cases, ultrasonographically guided fine-needle biopsy is recommended. No evidence exists to suggest that the pathogenesis of focal fatty liver is similar to that of NAFLD. In fact, the pathogenesis of focal fatty liver is uncertain and may involve altered venous blood flow to the liver, tissue hypoxia, and intestinal malabsorption of lipoproteins. Furthermore, in the absence of accompanying or background liver disease, the lesion often regresses. Therefore, no specific treatment is justified.
Adams LA, Lymp JF, St. Sauver J, et al. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology. 2005;129:113-21. (Ref 103.)
Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: A longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005;42:132-8. (Ref 99.)
Angulo P, Hui JM, Marchesini G, et al. The NAFLD fibrosis score: A noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology. 2007;45:846-54. (Ref 94.)
Day C, James O. Steatohepatitis: A tale of two “hits”? [editorial]. Gastroenterology. 1998;114:842-5. (Ref 36.)
Dixon J, Bhathal P, O’Brien P. Nonalcoholic fatty liver disease: Predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121:91-100. (Ref 15.)
El-Serag H, Tran T, Everhart J. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology. 2004;126:460-8. (Ref 30.)
Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology. 2006;43:S99-S112. (Ref 47.)
Hui J, Hodge A, Farrell G, et al. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40:46-54. (Ref 51.)
Lin H, Yang S, Chuckaree C, et al. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nature Med. 2000;6:998-1003. (Ref 129.)
Marchesini G, Bugianesi E, Forlani G, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917-23. (Ref 28.)
Matteoni C, Younossi Z, Gramlich T, et al. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413-19. (Ref 104.)
Promrat K, Lutchman G, Uwaifo G, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology. 2004;39:188-96. (Ref 133.)
Ratziu V, Giral P, Charlotte F, et al. Liver fibrosis in overweight patients. Gastroenterology. 2000;118:1117-23. (Ref 89.)
Ratziu V, Giral P, Jacqueminet S, et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135:100-10. (Ref 135.)
1. Ludwig J, Viggiano T, McGill D, Ott B. Nonalcoholic steatohepatitis. Mayo clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434-8.
2. Szczepaniak L, Nurenberg P, Leonard D, et al. Magnetic resonance spectroscopy to measure hepatic triglyceride content: Prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005;288:E462-8.
3. Clark J, Brancati F, Diehl A. The prevalence and etiology of elevated aminotransferase levels in the United States. Am J Gastroenterol. 2003;98:960-7.
4. Ruhl C, Everhart J. Determinants of the association of overweight with elevated serum alanine aminotransferase activity in the United States. Gastroenterology. 2003;124:71-9.
5. Amarapurkar D, Kamani P, Patel N, et al. Prevalence of non-alcoholic fatty liver disease: Population based study. Ann Hepatol. 2007;6:161-3.
6. Ekstedt M, Franzén L, Mathiesen U, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44:865-73.
7. Park S, Jeon W, Kim S, et al. Prevalence and risk factors of non-alcoholic fatty liver disease among Korean adults. J Gastroenterol Hepatol. 2006;21:138-43.
8. Perry A, Applegate E, Jackson M, et al. Racial differences in visceral adipose tissue but not anthropometric markers of health-related variables. J Appl Physiol. 2000;89:636-43.
9. Flores Y, Yee HJ, Leng M, et al. Risk factors for chronic liver disease in blacks, Mexican Americans, and whites in the United States: Results from NHANES IV, 1999-2004. Am J Gastroenterol. 2008;103:2231-8.
10. Schwimmer J, Deutsch R, Rauch JB, et al. Obesity, insulin resistance, and other clinicopathological correlates of pediatric nonalcoholic fatty liver disease. J Pediatr. 2003;143:500-5.
11. Abdelmalek M, Liu C, Shuster J, et al. Familial aggregation of insulin resistance in first-degree relatives of patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2006;4:1162-9.
12. Willner I, Waters B, Patil S, et al. Ninety patients with nonalcoholic steatohepatitis: Insulin resistance, familial tendency, and severity of disease. Am J Gastroenterol. 2001;96:2957-61.
13. Merriman R, Aouizerat B, Bass N. Genetic influences in nonalcoholic fatty liver disease. J Clin Gastroenterol. 2006;40:S30-S3.
14. Ogden C, Yanovski S, Carroll M, Flegal K. The epidemiology of obesity. Gastroenterology. 2007;132:2087-102.
15. Dixon J, Bhathal P, O’Brien P. Nonalcoholic fatty liver disease: Predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121:91-100.
16. Hsiao T, Chen J, Wang J. Insulin resistance and ferritin as major determinants of nonalcoholic fatty liver disease in apparently healthy obese patients. Int J Obes Relat Metab Disord. 2004;28:167-72.
17. Boza C, Riquelme A, Ibañez L, et al. Predictors of nonalcoholic steatohepatitis (NASH) in obese patients undergoing gastric bypass. Obes Surg. 2005;15:1148-53.
18. Luyckx F, Sheen A, Desaive C, et al. Effects of gastroplasty on body weight and related biological abnormalities in morbid obesity. Diab Metab. 1998;24:355-61.
19. Gholam PM, Flancbaum L, Machan JT, et al. Nonalcoholic fatty liver disease in severely obese subjects. Am J Gastroenterol. 2007;102:399-408.
20. Park J-W, Jeong G, Kim S, et al. Predictors reflecting the pathological severity of non-alcoholic fatty liver disease: Comprehensive study of clinical and immunohistochemical findings in younger Asian patients. J Gastroenterol Hepatol. 2007;22:491-7.
21. Moretto M, Kupski C, Mottin C, et al. Hepatic steatosis in patients undergoing bariatric surgery and its relationship to body mass index and co-morbidities. Obes Surg. 2003;13:622-4.
22. Hsiao PJ, Kuo KK, Shin SJ, et al. Significant correlations between severe fatty liver and risk factors for metabolic syndrome. J Gastroenterol Hepatol. 2007;22:2118-23.
23. Stranges S, Dorn J, Muti P, et al. Body fat distribution, relative weight, and liver enzyme levels: A population-based study. Hepatology. 2004;39:754-63.
24. Church T, Kuk J, Ross R, et al. Association of cardiorespiratory fitness, body mass index, and waist circumference to nonalcoholic fatty liver disease. Gastroenterology. 2006;130:2023-30.
25. Cheung O, Kapoor A, Puri P, et al. The impact of fat distribution on the severity of nonalcoholic fatty liver disease and metabolic syndrome. Hepatology. 2007;46:1091-100.
26. Targher G, Bertolini L, Padovani R, et al. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care. 2007;30:1212-18.
27. Targher G, Bertolini L, Rodella S, et al. Nonalcoholic fatty liver disease is independently associated with an increased incidence of cardiovascular events in type 2 diabetic patients. Diabetes Care. 2007;30:2119-21.
28. Marchesini G, Bugianesi E, Forlani G, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917-23.
29. Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology. 1999;30:1356-62.
30. El-Serag H, Tran T, Everhart J. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology. 2004;126:460-8.
31. Mofrad P, Contos MJ, Haque M, et al. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology. 2003;37:1286-92.
32. Davila JA, Morgan RO, Shaib Y, et al. Diabetes increases the risk of hepatocellular carcinoma in the United States: A population based case control study. Gut. 2005;54:533-9.
33. Browning JD. Statins and hepatic steatosis: Perspectives from the Dallas Heart Study. Hepatology. 2006;44:466-71.
34. Lee JY, Kim KM, Lee SG, et al. Prevalence and risk factors of non-alcoholic fatty liver disease in potential living liver donors in Korea: A review of 589 consecutive liver biopsies in a single center. J Hepatol. 2007;47:239-44.
35. Oh SY, Cho YK, Kang MS, et al. The association between increased alanine aminotransferase activity and metabolic factors in nonalcoholic fatty liver disease. Metabolism. 2006;55:1604-9.
36. Day C, James O. Steatohepatitis: A tale of two “hits”? [editorial]. Gastroenterology. 1998;114:842-5.
37. Chitturi S, Abeygunasekera S, Farrell G, et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35:373-9.
38. Pagano G, Pacini G, Musso G, et al. Nonalcoholic steatohepatitis, insulin resistance, and metabolic syndrome: Further evidence of an etiologic association. Hepatology. 2002;35:367-72.
39. Sanyal A, Campbell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183-92.
40. Angulo P, Lindor K. Insulin resistance and mitochondrial abnormalities in NASH: A cool look into a burning issue. Gastroenterology. 2001;120:1281-5.
41. Boden G. Fatty acid-induced inflammation and insulin resistance in skeletal muscle and liver. Curr Diab Rep. 2006;6:177-81.
42. Boden G. Interaction between free fatty acids and glucose metabolism. Curr Opin Clin Nutr Metab Care. 2002;5:545-9.
43. Neuschwander-Tetri B. A resistance movement in NASH. Am J Gastroenterol. 2001;96:2813-14.
44. Charlton M, Sreekumar R, Rasmussen D, et al. Apolipoprotein synthesis in nonalcoholic steatohepatitis. Hepatology. 2002;35:898-904.
45. Neuschwander-Tetri B, Caldwell S. Nonalcoholic steatohepatitis: Summary of an AASLD single topic conference. Hepatology. 2003;37:1202-19.
46. Cai D, Yuan M, Frantz DF, et al. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaβ. Nat Med. 2005;11:183-90.
47. Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology. 2006;43:S99-S112.
48. Carter-Kent C, Zein NN, Feldstein AE. Cytokines in the pathogenesis of fatty liver and disease progression to steatohepatitis: Implications for treatment. Am J Gastroenterol. 2008;103:1036-42.
49. Crespo J, Cayon A, Fernandez-Gil P, et al. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology. 2001;34:1158-63.
50. Czaja M. Liver injury in the setting of steatosis: Crosstalk between adipokine and cytokine. Hepatology. 2004;40:19-22.
51. Hui J, Hodge A, Farrell G, et al. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology. 2004;40:46-54.
52. Kugelmas M, Hill D, Vivian B, et al. Cytokines and NASH: A pilot study of the effects of lifestyle modification and vitamin E. Hepatology. 2003;38:413-19.
53. Xu A, Wang Y, Keshaw H, et al. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91-100.
54. Kaser S, Moschen A, Cayon A, et al. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut. 2005;54:117-21.
55. Harrison S, Di Bisceglie A. Advances in the understanding and treatment of nonalcoholic fatty liver disease. Drugs. 2003;63:2379-94.
56. Canbakan B, Tahan V, Balci H, et al. Leptin in nonalcoholic fatty liver disease. Ann Hepatol. 2008;7:249-54.
57. Chalasani N, Crabb DW, Cummings OW, et al. Does leptin play a role in the pathogenesis of human nonalcoholic steatohepatitis? Am J Gastroenterol. 2003;98:2771-6.
58. Chitturi S, Farrell G, Frost L, et al. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: A manifestation of lipotoxicity? Hepatology. 2002;36:403-9.
59. Uygun A, Kadayifci A, Yesilova Z, et al. Serum leptin levels in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2000;95:3584-9.
60. Kessova I, Cederbaum AI. CYP2E1: Biochemistry, toxicology, regulation and function in ethanol-induced liver injury. Curr Mol Med. 2003;3:509-18.
61. Hoek JB, Pastorino JG. Ethanol, oxidative stress, and cytokine-induced liver injury. Alcohol. 2002;27:63-8.
62. Chalasani N, Gorski C, Asghar M, et al. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology. 2003;37:544-50.
63. Weltman M, Farrell G, Hall P, et al. Hepatic cytochrome P450 2E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology. 1998;27:128-33.
64. Laurent A, Nicco C, Van Nhieu J, et al. Pivotal role of superoxide anion and beneficial effect of antioxidant molecules in murine steatohepatitis. Hepatology. 2004;39:1277-85.
65. Yang S, Zhu H, Li Y, et al. Mitochondrial adaptations to obesity-related oxidant stress. Arch Biochem Biophys. 2000;378:259-68.
66. Feldstein AE, Werneburg NW, Canbay A, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF expression via a lysosomal pathway. Hepatology. 2004;40:185-94.
67. Leclercq I, Farrell G, Field J, et al. CYP2E1 and CYP4A as microsomal catalysts of lipid peroxides in murine nonalcoholic steatohepatitis. J Clin Invest. 2000;105:1067-75.
68. Yang S, Lin H, Diehl AM. Fatty liver vulnerability to endotoxin-induced damage despite NFκB induction and inhibited caspase 3 activation. Am J Physiol Gastrointest Liver Physiol. 2001;281:G382-92.
69. Diehl A. Nonalcoholic steatosis and steatohepatitis IV. Nonalcoholic fatty liver disease abnormalities in macrophage function and cytokines. Am J Physiol. 2002;282:G1-G5.
70. Ruiz AG, Casafont F, Crespo J, et al. Lipopolysaccharide-binding protein plasma levels and liver TNF-alpha gene expression in obese patients: Evidence for the potential role of endotoxin in the pathogenesis of non-alcoholic steatohepatitis. Obes Surg. 2007;17:1374-80.
71. Thuy S, Ladurner R, Volynets V, et al. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J Nutr. 2008;138:1452-5.
72. Fromenty B, Robin M, Igoudjil A, et al. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 2004;30:121-38.
73. Pérez-Carreras M, Hoyo P, Martín M, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999-1007.
74. Le T, Caldwell S, Redick J, et al. The zonal distribution of megamitochondria with crystalline inclusions in nonalcoholic steatohepatitis. Hepatology. 2004;39:1423-9.
75. Sreekumar R, Rosado B, Rasmussen D, Charlton M. Hepatic gene expression in histologically progressive nonalcoholic steatohepatitis. Hepatology. 2003;38:244-51.
76. Pinzani M, Rombouts K. Liver fibrosis: From the bench to clinical targets. Dig Liver Dis. 2004;36:231-42.
77. Paradis V, Perlemuter G, Bonvoust F, et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: A potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology. 2001;34:738-44.
78. Saxena NK, Ikeda K, Rockey DC, et al. Leptin in hepatic fibrosis: Evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35:762-71.
79. Lu SC, Alvarez L, Huang Z-Z, et al. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. PNAS. 2001;98:5560-5.
80. Seo YS, Kim JH, Jo NY, et al. PPAR agonists treatment is effective in a nonalcoholic fatty liver disease animal model by modulating fatty-acid metabolic enzymes. J Gastroenterol Hepatol. 2008;23:102-9.
81. Lieber C, Leo M, Mak K, et al. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79:502-9.
82. Adams L, Lindor K, Angulo P. The prevalence of autoantibodies and autoimmune hepatitis in patients with nonalcoholic fatty liver disease. Am J Gastroenterol. 2004;99:1316-20.
83. Lonardo A, Adinolfi L, Loria P, et al. Steatosis and hepatitis C virus: Mechanisms and significance for hepatic and extrahepatic disease. Gastroenterology. 2004;126:586-97.
84. Bugianesi E, Manzini P, D’Antico S, et al. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology. 2004;39:179-87.
85. Saadeh S, Younossi Z, Remer E, et al. The utility of radiological imaging in nonalcoholic fatty liver disease. Gastroenterology. 2002;123:745-50.
86. Cortez-Pinto H, Baptista A, Camilo M, De Moura M. Nonalcoholic steatohepatitis—a long-term follow-up study: Comparison with alcoholic hepatitis in ambulatory and hospitalized patients. Dig Dis Sci. 2003;48:1909-13.
87. Zafrani E. Non-alcoholic fatty liver disease: An emerging pathological spectrum. Virchows Arch. 2004;444:3-12.
88. Gramlich T, Kleiner D, McCullough A, et al. Pathologic features associated with fibrosis in nonalcoholic fatty liver disease. Hum Pathol. 2004;35:196-9.
89. Ratziu V, Giral P, Charlotte F, et al. Liver fibrosis in overweight patients. Gastroenterology. 2000;118:1117-23.
90. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313-21.
91. Mendler MH, Kanel G, Govindarajan S. Proposal for a histological scoring and grading system for non-alcoholic fatty liver disease. Liver Int. 2005;25:294-304.
92. Scheuer P. Classification of chronic viral hepatitis: A need for reassessment. J Hepatol. 1991;13:372-4.
93. Ratziu V, Massard J, Charlotte F, et al. Diagnostic value of biochemical markers (FibroTest-FibroSURE) for the prediction of liver fibrosis in patients with non-alcoholic fatty liver disease. BMC Gastroenterology. 2006;6:6-19.
94. Angulo P, Hui JM, Marchesini G, et al. The NAFLD fibrosis score: A noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology. 2007;45:846-54.
95. Yoneda M, Yoneda M, Mawatari H, et al. Noninvasive assessment of liver fibrosis by measurement of stiffness in patients with nonalcoholic fatty liver disease (NAFLD). Dig Liver Dis. 2008;40:371-8.
96. Charlton M, Angulo P, Chalasani N, et al. Low circulating levels of dehydroepiandrosterone in histologically advanced nonalcoholic fatty liver disease. Hepatology. 2008;47:484-92.
97. Suzuki A, Angulo P, Lymp J, et al. Hyaluronic acid, an accurate serum marker for severe hepatic fibrosis in patients with non-alcoholic fatty liver disease. Liver Int. 2005;25:779-86.
98. Teli M, James O, Burt A, et al. The natural history of nonalcoholic fatty liver: A follow-up study. Hepatology. 1995;22:1714-19.
99. Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: A longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005;42:132-8.
100. Fassio E, Alvarez E, Dominguez N, et al. Natural history of nonalcoholic steatohepatitis: A longitudinal study of repeat liver biopsies. Hepatology. 2004;40:820-6.
101. Harrison S, Torgerson S, Hayashi P. The natural history of nonalcoholic fatty liver disease: A clinical histopathological study. Am J Gastroenterol. 2003;98:2042-7.
102. Ratziu V, Bugianesi E, Dixon J, et al. Histological progression of non-alcoholic fatty liver disease: A critical reassessment based on liver sampling variability. Aliment Pharmacol Ther. 2007;26:821-30.
103. Adams LA, Lymp JF, St. Sauver J, et al. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology. 2005;129:113-21.
104. Matteoni C, Younossi Z, Gramlich T, et al. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413-19.
105. Poonawala A, Nair S, Thuluvath P. Prevalence of obesity and diabetes in patients with cryptogenic cirrhosis: A case-control study. Hepatology. 2000;32:689-92.
106. Bugianesi E, Leone N, Vanni E, et al. Expanding the natural history of nonalcoholic steatohepatitis: From cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123:134-40.
107. Caldwell S, Oelsner D, Iezzoni J, et al. Cryptogenic cirrhosis: Clinical characterization and risk factors for underlying disease. Hepatology. 1999;29:664-9.
108. Marrero J, Fontana R, Su G, et al. NAFLD may be a common underlying liver disease in patients with hepatocellular carcinoma in the United States. Hepatology. 2002;36:1349-54.
109. Hui J, Kench J, Chitturi S, et al. Long-term outcomes of cirrhosis in nonalcoholic steatohepatitis compared with hepatitis C. Hepatology. 2003;38:420-7.
110. Hickman I, Jonsson J, Prins J, et al. Modest weight loss and physical activity in overweight patients with chronic liver disease results in sustained improvements in alanine aminotransferase, fasting insulin, and quality of life. Gut. 2004;53:413-19.
111. Knobler H, Schattner A, Zhornicki T, et al. Fatty liver—an additional and treatable feature of the insulin resistance syndrome. QJM. 1999;92:87-96.
112. Nobili V, Manco M, Devito R, et al. Lifestyle intervention and antioxidant therapy in children with nonalcoholic fatty liver disease: A randomized, controlled trial. Hepatology. 2008;48:119-28.
113. Huang MA, Greenson JK, Chao C, et al. One-year intense nutritional counseling results in histological improvement in patients with non-alcoholic steatohepatitis: A pilot study. Am J Gastroenterol. 2005;100:1072-81.
114. Harrison SA, Ramrakhiani S, Brunt EM, et al. Orlistat in the treatment of NASH: A case series. Am J. Gastroenterology. 2003;98:926-30.
115. Zelber-Sagi S, Kessler A, Brazowsky E, et al. A double-blind randomized placebo-controlled trial of orlistat for the treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2006;4:639-44.
116. Klein S, Mittendorfer B, Eagon JC, et al. Gastric bypass surgery improves metabolic and hepatic abnormalities associated with nonalcoholic fatty liver disease. Gastroenterology. 2006;130:1564-72.
117. Liu X, Lazenby AJ, Clements RH, et al. Resolution of nonalcoholic steatohepatitis after gastric bypass surgery. Obes Surg. 2007;17:486-92.
118. Dixon JB, Bhathal PS, O’Brien PE. Weight loss and non-alcoholic fatty liver disease: Falls in gamma-glutamyl transferase concentrations are associated with histologic improvement. Obes Surg. 2006;16:1278-86.
119. Dixon JB, Bhathal PS, Hughes NR, O’Brien PE. Nonalcoholic fatty liver disease: Improvement in liver histological analysis with weight loss. Hepatology. 2004;39:1647-54.
120. Clark JM. Weight loss as a treatment for nonalcoholic fatty liver disease. J Clin Gastroenterol. 2006;40(Suppl 1):S39-S43.
121. Zivkovic AM, German JB, Sanyal AJ. Comparative review of diets for the metabolic syndrome: Implications for nonalcoholic fatty liver disease. Am J Clin Nutr. 2007;86:285-300.
122. Luyckx F, Lefebvre P, Scheen A. Nonalcoholic steatohepatitis: Association with obesity and insulin resistance, and influence of weight loss. Diabetes Metab. 2000;26:98-106.
123. Harrison S, Torgerson S, Hayashi P, et al. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2003;98:2485-90.
124. Hasegawa T, Yoneda M, Nakamura K, et al. Plasma transforming growth factor-β1 and efficacy of alpha-tocopherol in patients with non-alcoholic steatohepatitis: A pilot study. Aliment Pharmacol Ther. 2001;15:1667-72.
125. Clarke MW, Burnett JR, Croft KD. Vitamin E in human health and disease. Crit Rev Clin Lab Sci. 2008;45:417-50.
126. Abdelmalek M, Angulo P, Jorgensen R, et al. Betaine, a promising new agent for patients with nonalcoholic steatohepatitis: Results of a pilot study. Am J Gastroenterol. 2001;96:2711-17.
127. Gulbahar O, Karasu Z, Ersoz G, et al. Treatment of nonalcoholic steatohepatitis with N-acetylcysteine [abstract]. Gastroenterology. 2000;118:A1444.
128. Ip E, Farrell G, Hall P, et al. Administration of the potent PPARalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39:1286-96.
129. Lin H, Yang S, Chuckaree C, et al. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nature Med. 2000;6:998-1003.
130. Marchesini G, Brizi M, Bianchi G, et al. Metformin in non-alcoholic steatohepatitis. Lancet. 2001;358:893-4.
131. Nair S, Diehl AM, Wiseman M, et al. Metformin in the treatment of non-alcoholic steatohepatitis: A pilot open label trial. Aliment Pharmacol Ther. 2004;20:23-8.
132. Neuschwander-Tetri B, Brunt E, Wehmeier K, et al. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with the PPAR-α ligand rosiglitazone. Hepatology. 2003;38:1008-17.
133. Promrat K, Lutchman G, Uwaifo G, et al. A pilot study of pioglitazone treatment for nonalcoholic steatohepatitis. Hepatology. 2004;39:188-96.
134. Lutchman G, Modi A, Kleiner DE, et al. The effects of discontinuing pioglitazone in patients with nonalcoholic steatohepatitis. Hepatology. 2007;46:424-9.
135. Ratziu V, Giral P, Jacqueminet S, et al. Rosiglitazone for nonalcoholic steatohepatitis: One-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135:100-10.
136. Facchini F, Hua N, Stoohs R. Effect of iron depletion in carbohydrate-intolerant patients with clinical evidence of nonalcoholic fatty liver disease. Gastroenterology. 2002;122:931-9.
137. Valenti L, Fracanzani AL, Dongiovanni P, et al. Iron depletion by phlebotomy improves insulin resistance in patients with nonalcoholic fatty liver disease and hyperferritinemia: Evidence from a case-control study. Am J Gastroenterol. 2007;102:1251-8.
138. Fernandez-Real J, Lopez-Bermejo A, Ricart W. Cross-talk between iron metabolism and diabetes. Diabetes. 2002;51:2348-54.
139. Basaranoglu M, Acbay O, Sonsuz A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis [correspondence]. J Hepatol. 1999;31:384.
140. Kiyici M, Gulten M, Gurel S, et al. Ursodeoxycholic acid and atorvastatin in the treatment of nonalcoholic steatohepatitis. Can J Gastroenterol. 2003;17:713-18.
141. Lindor K, Kowdley K, Heathcote E, et al. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: Results of a randomized trial. Hepatology. 2004;39:770-8.
142. Contos MJ, Cales W, Sterling RK, et al. Development of nonalcoholic fatty liver disease after orthotopic liver transplantation for cryptogenic cirrhosis. Liver Transpl. 2001;7:363-73.
143. Burke A, Lucey MR. Non-alcoholic fatty liver disease, non-alcoholic steatohepatitis and orthotopic liver transplantation. Am J Transplant. 2004;4:686-93.
144. Siegelman E, Rosen MA. Imaging of hepatic steatosis. Semin Liver Dis. 2001;21:71-80.