[level-membership-for-emergency-medicine-category]
Chapter 85 Non–North American Travel and Exotic Diseases
Travelers to tropical and subtropical areas of the world where hygienic conditions are poor and ecologic conditions are permissive may encounter infectious agents that are no longer endemic or have never existed in temperate regions of the world. Although economic development and industrialization of developing countries of the tropics have resulted in a decreased health burden of many tropical infectious diseases, it is important to realize that there is still a risk for exposure for the traveler who is unaware of appropriate measures to prevent or treat such conditions. The most important consideration in the management of this problem, which is increasing as international travel expands, is appropriate preventive measures through counsel with a travel medicine specialist and prophylaxis using safe drugs and vaccines. This topic has recently been reviewed in several excellent publications.*
This chapter is concerned with infectious diseases that are uncommon or do not exist in North America and with which most health professionals in North America have scant familiarity. Other chapters give specific details relevant to malaria (Chapter 49), tick-borne diseases (Chapter 51), infectious diarrheas (Chapter 68), and travel medicine (Chapter 84). The infectious diseases considered in this chapter should not be considered a complete listing. This is especially important to keep in mind in an era when diseases once thought to be eliminated or nonexistent in North America are emerging or reemerging coincidental with large-scale movements of human and vector populations.
Major Viral Infections
Yellow Fever
European physicians did not recognize until the late 1490s the clinical syndrome now known as yellow fever. Initially described by Columbus in the West Indies, large-scale epidemics were later observed throughout the Americas and tropical Africa in the 1700s and 1800s. After epidemic yellow fever in Texas, Louisiana, and Tennessee caused 20,000 deaths in the 1880s, the Yellow Fever Commission was organized to study the problem. Identification of the mosquito vector, Aedes aegypti, and definitive studies conducted by the U.S. military under the leadership of Walter Reed were followed by massive campaigns to eradicate mosquito breeding sites. This led to virtual elimination of urban yellow fever from the Americas. The last case of yellow fever acquired in the continental United States was reported in 1911. Because it is difficult if not impossible to eliminate jungle reservoirs, there continue to be cases reported annually from South America and tropical Africa. Larger outbreaks secondary to resurgent vector populations have occurred in recent years in tropical West Africa.14,35,38,70
Ecology and Epidemiology
Currently, both the Americas and Africa have a constant low level of jungle yellow fever because of inability to control either the monkey reservoir or the mosquito vector. Overall there are about 200,000 cases per year, resulting in approximately 30,000 deaths, occurring primarily in sub-Saharan Africa.79 Some suggest that these rates are underestimated by at least 10-fold. Persons at risk include workers or travelers in or near the tropical rainforest canopy. Urban yellow fever had been reduced in the western hemisphere through massive campaigns to control breeding and spread of the Aedes vector. However, the benefits of these campaigns have declined, and there is currently an increased threat of further outbreaks of disease. Introduction of Aedes albopictus, an aggressive anthropophilic dengue vector from Southeast Asia, and reemergence of A. aegypti into the Americas raise the specter of increased yellow fever transmission in the western hemisphere.65 Less-intense vector control measures and a more complex ecology have made elimination of urban yellow fever in Africa even more difficult.
Prevention
Avoidance of this potentially fatal infection is possible through use of yellow fever vaccine. The vaccine strain 17D is an attenuated live virus grown in chicken embryos. Greater than 95% of persons vaccinated achieve significant antibody 1evels. Repeat vaccinations are recommended every 10 years, although persistent antibody titers have been detected as long as 30 to 40 years after vaccination. Yellow fever vaccine is generally well tolerated, with headache or malaise occurring in less than 10% of those vaccinated. Rare allergic side effects occur primarily in persons with hypersensitivity to eggs. Other serious adverse events, including death, have been reported, with the greater risk being associated with age older than 60 years.5,49,66,81 Vaccination is not recommended in the first 6 months of life or in other situations where live virus vaccines are contraindicated. Although pregnant women have received the vaccine without adverse effect to themselves or their infants, it is not recommended for use in this group because of possible teratogenic effects. Other means of reducing the risk for yellow fever (and any mosquito-borne infectious disease) include liberal use of mosquito repellent and netting in endemic areas. Outbreak control in endemic countries is primarily through focused vaccination campaigns.
Dengue fever
Dengue fever has been reported since the late 1700s. Since World War II, increased attention has focused on the dengue virus, largely as a result of recognition of dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS). First noted in Southeast Asia, DHF and DSS have attained worldwide distribution in the last 30 years.34,53,67,86 Dengue is the most common insect-borne viral infection in the world. The infection has been reported in more than 100 countries, with 50 to 100 million dengue infections each year resulting in approximately 500,000 cases of life-threatening disease (DHF and DSS) annually.36,37
Virology and Pathophysiology
The etiologic agent is a single-stranded RNA flavivirus, which may be one of four serotypes, denoted DEN-1 through DEN-4. As with yellow fever, local viral replication is followed by dissemination to lymphocyte- and macrophage-rich areas, where most of the reproductive activity occurs. Infection with one virus serotype provides long-lasting immune protection against that type only. After infection with one serotype, a subsequent infection with a heterologous serotype may result in a more severe clinical course. Non-neutralizing antibodies produced in response to infection to the primary infection are thought to facilitate entrance of the heterologous virus into host macrophages. Although cases of DHF and DSS may result from this “immune enhancement,” severe DHF and DSS also occur with other serotypes in the absence of previous infection with a heterologous dengue virus serotype.60,61 Pathologic studies of DHF and DSS show hemorrhage, congestion, and perivascular edema of multiple organs. The liver may show areas of focal necrosis. As with yellow fever, the extent of pathologic findings does not correspond to severity of the clinical course.
Ecology and Epidemiology
A. aegypti is the principal vector for dengue viruses worldwide. In the Americas and Asia, viral transmission is maintained through a mosquito–human cycle without a major animal reservoir. Monkey carriers have been identified in Africa and Asia, but their importance in transmission is unclear. A. albopictus, an anthropophilic dengue vector from Southeast Asia, has also been recognized recently in the western hemisphere. Both of these mosquitoes are capable of large-scale transmission to humans in endemic areas. Currently, dengue is endemic in tropical and subtropical Asia, Africa, South America, and the Caribbean basin. In the early 1920s, large epidemics occurred in Texas, where dengue infections were reported in 500,000 inhabitants. In the last 30 years, endemic transmission on the mainland United States has been documented only in Texas. Travelers to Southeast Asia have highest risk for dengue, especially when travel takes place during periods of high transmission and epidemic dengue.98
Clinical Presentation
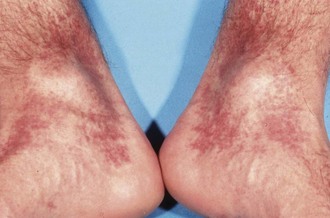
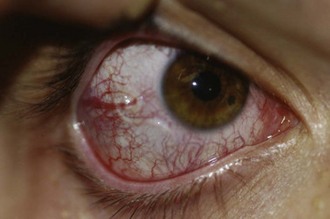
Most dengue infections appear after an incubation period of 2 to 14 days, either as an undifferentiated viral syndrome with fever and mild respiratory or gastrointestinal symptoms or as dengue (“break-bone”) fever with bone pain, generalized myalgia, severe headache, and retroorbital pain. Febrile illnesses that appear more than 2 weeks after putative exposure to dengue virus are unlikely to be due to this virus. After 1 to 3 days, a quiescent period may ensue. There may be a subsequent second episode of fever accompanied by a patchy maculopapular or morbilliform rash that spreads outward from the chest and that ultimately desquamates. Lymphadenopathy and leukopenia occur during this phase of the illness. The distinct severe forms of dengue disease referred to as either DHF or DSS may occur around the usual time of recovery. These are due to the development of capillary leak syndrome with associated hemorrhagic manifestations (Figure 85-1). The advanced forms have the unique feature that the platelet count decreases to less than 100,000 per mm3 and hematocrit increases by more than 20%. The severity is classified as grade I to IV, according to World Health Organization guidelines. In cases of grade I DHF, the only hemorrhagic manifestation is a positive tourniquet test, in which inflation of a tourniquet to midway between systolic and diastolic blood pressure for 5 minutes leads to development of 20 or more petechiae per square inch distal to the tourniquet. A complete blood cell count classically shows decreased platelet and leukocyte counts and increase in hematocrit value. Grade II DHF is defined as the above with hemorrhage from any site (e.g., gingiva, nares, conjunctivae). Grade III DSS includes clammy skin, hypotension, or a narrow pulse pressure (<20 mm Hg) in a patient with DHF. An undetectable blood pressure defines grade IV DHF and DSS. Most studies have noted DHF and DSS primarily in infants and young children, usually with a history or serologic evidence of previous heterologous dengue infection, but there is an increasing trend of cases in adults.
Lassa Fever
Epidemiology
The principal animal host for this virus is a rat, Mastomys natalensis, which prefers living in and around human dwellings. The rodents become chronically infected, secreting viral particles for long periods. Natural infection in humans occurs after rodent contamination of food and drink, inhalation of aerosolized rodent secretions, or contact with rodent material through skin abrasions. Lassa fever has been reported in several areas of sub-Saharan West Africa, and large outbreaks have been noted in Nigeria, Sierra Leone, Guinea, and Liberia.21,62 It affects up to 500,000 people with 5000 deaths annually.54 Complete seroprevalence data are lacking, making definition of an endemic area impossible at this time. Secondary human infection has been reported and may occur after contact with infected secretions.
Virology and Pathophysiology
Lassa virus is a single-stranded RNA arenavirus. Proliferation and dissemination presumably occur after initial replication at the inoculation site. As with the flaviviral diseases, the extent of end-organ involvement noted at autopsy does not account for the rapid death of infected patients. Recent work in an animal model provides evidence for platelet dysfunction and an endothelial cell defect in shock caused by Lassa fever virus.31 DIC, believed to be a major cause of bleeding and death in patients with other viral hemorrhagic fevers, appears to play a relatively minor role in arenavirus infections. The liver is most consistently the organ in which pathologic changes are observed at autopsy.
Clinical Presentation
Most seroconversions to Lassa virus are not accompanied by obvious symptoms.63,64,69 Only 5% to 14% of seroconverters experienced a febrile illness. The incubation period is between 3 and 21 days. Patients hospitalized with Lassa fever show a distinct clinical syndrome. Fever, malaise, and purulent pharyngitis often develop after the insidious onset of headache. Retrosternal chest pain, possibly a result of pharyngitis and esophagitis, suggests the diagnosis. The combined presence of retrosternal chest pain, fever, pharyngitis, and proteinuria is the best predictor of Lassa fever.62 Hemorrhagic complications (hematemesis, vaginal bleeding, hematuria, lower gastrointestinal bleeding, and epistaxis) were seen in fewer than 25% of patients with Lassa fever. Nonfatal disease usually begins to resolve in 8 to 10 days. The combined presence of fever, sore throat, and vomiting was associated with a poor prognosis (relative risk for death = 5.5). Terminal stages of fatal disease were accompanied by hypotension, encephalopathy, and respiratory distress caused by stridor (presumably secondary to laryngeal edema). The most common complication after recovery from Lassa fever is sensorineural hearing loss, presumably due to host immune response reactions against elements of the inner ear.
Management
The probability of transmission of Lassa fever virus to medical and nursing staff can be reduced by routine blood and body fluid precautions as well as strict barrier nursing. Barrier nursing includes wearing gloves, gown, mask, shoe covers, and, if there is risk for splashing fluids, goggles whenever entering the patient’s room. Decontamination of solid articles and rooms may be accomplished with 0.5% sodium hypochlorite solution. Recommendations for the management of patients with viral hemorrhagic fever have been published.17,11
Ebola and Marburg Viruses
Ebola and Marburg viruses are closely related large-RNA viruses known as filoviruses. They cause severe viral hemorrhagic fever syndromes with some of the highest case fatality rates (approximately 90%) of any known infectious disease. Both are endemic in focal areas of central and southern Africa.73 Ebola virus seropositivity has been noted in Sudan, Democratic Republic of the Congo, the Central African Republic, Côte d’Ivoire, and Kenya. A strain of Ebola known as Ebola Reston has been found in monkeys imported into the United States from the Philippines. More recently, there have been outbreaks with fatalities in Gabon, the Democratic Republic of Congo, and Angola. Marburg disease is found in South Africa, Zimbabwe, and Kenya. In 2005, there was an outbreak that caused over 300 deaths in Angola.18 Although there is not definitive evidence indicating the animal reservoir that maintains these filoviruses in nature, current evidence strongly suggests that bats are involved. Person-to-person transmission has been well documented, primarily through contaminated needles and contact with the secretions of infected individuals.72,74
Pathophysiology and Clinical Presentation
Marburg and Ebola viruses are presumed to act through similar pathophysiologic mechanisms that involve initial infection of monocytes, macrophages, and dendritic cells that are then distributed through the circulation to many organs and cell types. The viruses suppress both innate and adaptive host immune responses, leading to overwhelming infection and wide release of proinflammatory cytokines and chemokines causing fever, vascular instability, hypotension, and shock followed by multiorgan failure and death.7,22,41,57
Patients present after an incubation period of 4 to 10 days with fever, headache, and myalgias. Diarrhea and abdominal pain occur commonly. In many victims, rash, conjunctivitis, sore throat, and chest pain appear early in the disease. As in other hemorrhagic fevers, hemorrhage, hypotension, shock, and electrolyte abnormalities mark fatal courses. The high mortality reported in various outbreaks and transmission to health care workers taking care of patients emphasizes the importance of intensive supportive care and precautions that limit contact with body secretions of infected individuals.72
Diagnosis and Treatment
If these diseases are suspected, strict isolation procedures should be instituted and the local health authorities and the CDC notified immediately. Diagnosis may be made on a serologic basis or by polymerase chain reaction (PCR). There appears to be no serologic cross reactivity between the two viruses. Although anecdotal reports suggest the efficacy of immune sera in therapy, this has not been consistently observed in experimental studies. There are currently no specific antiviral therapies for Marburg or Ebola virus infection. Care is supportive. Vaccines are in development with one in Phase I testing.7,47,60,85
Crimean-Congo Hemorrhagic Fever
Virology and Epidemiology
The etiologic agent of Crimean-Congo hemorrhagic fever (CCHF) is a bunyavirus. Ixodid ticks serve as both reservoirs and vectors of the virus. Infection in humans results from tick bites or direct contact with infected secretions from crushed ticks, animals, or humans. Most cases occur in individuals with occupations or living conditions that bring them in contact with domestic goats, sheep, or cattle on which ticks feed. The disease has been observed in southeastern Europe, south central Asia, the Middle East, and much of Africa.95 Nosocomial transmission through contact with infected body fluids has been well documented.32,89,90,91
Pathophysiology and Clinical Presentation
Pathophysiologic mechanisms are presumably similar to those of other hemorrhagic fevers.28 One in five infections results in clinical disease with a case fatality rate ranging from 10% to 50%. The incubation period is approximately 1 week with initial symptoms of fever, severe headache, myalgias, vomiting, and diarrhea. Various forms of hemorrhage, including petechiae, large ecchymoses, melena, and hematemesis, are more pronounced in CCHF than in other hemorrhagic viral diseases. Severe cases progress rapidly to DIC, shock, and death.
Management
Initial management is similar to those for Lassa, Marburg, and Ebola virus infections, with strict patient isolation and notification of health authorities. Supportive therapy with attention to fluid balance and electrolytes in addition to oxygenation and hemodynamic support is the primary treatment. Although not confirmed in clinical trials, ribavirin has good activity in vitro against CCHF virus. The CDC recommends that patients believed to have CCHF receive intravenous (IV) ribavirin in the doses suggested for treatment of Lassa fever.27 Persons in contact with CCHF patients should receive prophylactic ribavirin as suggested for Lassa fever contacts. To date, almost all therapy has used the oral form of ribavirin.
Hemorrhagic Fever with Renal Syndrome and Hantavirus Pulmonary Syndrome
Hantaviruses, when transmitted from rodent reservoirs, cause two significant human diseases, hemorrhagic fever with renal syndrome (HFRS) in Asia and Europe, and hantavirus pulmonary syndrome (HPS) in the Americas. HFRS first came to the attention of Western medical science during the Korean conflict, when febrile illness accompanied by bleeding and renal failure developed in 3000 United Nations troops and was ultimately found to be caused by the hantavirus species Hantaan virus.40 Mortality ranged from 5% to 10%. A similar, less severe syndrome (nephropathia epidemica) had been recognized in Scandinavia since the 1930s. HPS was first recognized in a cluster of deaths in the southwestern United States in 1993. A nonspecific febrile illness is followed by shock and alveolar pulmonary edema caused by the hantavirus species Sin Nombre virus.23
Epidemiology
Hantaviruses cause chronic, nondebilitating infections of various rodent species. Human infection is initiated by contact with rodent secretions or inhalation of aerosolized rodent material. The disease occurs most commonly in rural areas, although occasional urban outbreaks occur, presumably with the common house rat as vector. Cases have been described most often from Asia, including China, Korea, Japan, and the Soviet Union, but the disease also occurs in Eastern Europe. A recent epidemiologic study from China found the highest rates of infection in men who engaged in heavy farm work and slept on the ground (rather than on raised wooden beds).99 The Sin Nombre virus appears to cause chronic infection of the deer mouse, Peromyscus maniculatus, which is the main reservoir of the virus in the United States. Since the initial outbreak, additional cases have been described across the United States and South America. The risk for infection is likely to be related to rodent exposure, but transmission is infrequent.
Management
Care of patients with HFRS is supportive. With HFRS, renal dysfunction occurs early and may require institution of dialysis soon after diagnosis to prevent fluid overload and to correct electrolyte disturbances. Patients’ secretions should be handled with care, and enteric precautions (but not strict isolation) are prudent. It is not clear whether person-to-person transmission of the virus through direct inoculation occurs. For the hantaviruses, viremia recedes and antibody levels rise as the clinical phase appears. Accordingly, nosocomial transmission or hematogenous transmission with hantavirus infections has not been frequently documented, although presumed nosocomial transmission has been reported.99 No vaccine is available.
Japanese B Encephalitis
Epidemiology
JE is the most common cause of encephalitis in Asia. Of the estimated 35,000 to 50,000 cases annually, 20% to 30% of infected individuals die and of those that recover, 30% to 50% have neurologic sequelae.30,46 Transmission correlates with monsoon rains in the tropics and in the summer and fall seasons in temperate regions. Rice field–breeding and other culicine mosquitoes serve as the vectors. In addition to humans, birds and pigs can be infected. Pigs play an important role as amplifying hosts because they develop high-grade viremia from which large numbers of mosquitoes may be infected. Most infections in endemic areas occur in children, whereas all age-groups of previously unexposed populations are at risk. Transmission of JE currently occurs in India, Southeast Asia, China, Korea, Indonesia, and the Western Pacific region.30 Routine use of JE vaccine in Japan has been eliminated because of low risk in this country. Recent outbreaks and case reports of JE in islands of the Torres Strait, which runs between Northern Australia and Papua New Guinea, indicate that the virus spread southward from Asia, presumably by migratory ardeid birds.39
Clinical Presentation
Incubation period is typically 2 to 15 days. Most infections do not cause clinical illness. Many patients recall a mild undifferentiated febrile illness, which probably coincides with the viremic phase of infection. Patients with encephalitis often report a similar prodrome. The encephalitis syndrome is not easily distinguished from other arboviral encephalitides. The patient usually complains of headache, lethargy, fever, and confusion and may display tremors or seizures. One clinical series suggested that the presence on admission of (1) unresponsiveness to pain, (2) low levels of anti–Japanese B encephalitis virus IgG or IgM antibodies (in serum or cerebrospinal fluid [CSF]), or (3) virus in CSF culture was associated with death. Of the 16 patients with fatal disease, all died within 7 days of hospitalization.15
Named Hepatitis Viruses
Hepatitis A
Epidemiology
Hepatitis A virus is transmitted primarily by the fecal–oral route by either person-to-person contact or ingestion of contaminated food or water. Food items commonly associated with outbreaks are raw or undercooked clams and shellfish. Risk factors include contact with a hepatitis A–infected person, international travel, household or personal contact with a child who attends a child care center, foodborne outbreaks, male homosexual activity, and use of illegal drugs.3,59 Occasional cases are associated with exposure to nonhuman primates. Transmission by blood transfusion has been reported, but this is an uncommon source of infection. Hepatitis A is endemic worldwide, but underdeveloped nations have a higher prevalence than those in North America. Most persons in these areas show serologic evidence of past infection with hepatitis A virus. Hepatitis A is a common viral infection occurring in travelers, but rates are declining with increased use of hepatitis A vaccine
Hepatitis B
Epidemiology
With the widespread use of serologic markers for hepatitis B disease, it became apparent that spread occurs through exchange of blood, semen, or, rarely, saliva of infected people. Although spread is possible from persons with acute disease, the primary sources of viral particles are chronic carriers. Persons are defined as carriers if blood samples obtained 6 months apart both contain hepatitis B surface antigen particles (HBsAg). The carrier state follows acute infection in up to 90% of infected infants and 10% of adults. Risk factors for acquisition of hepatitis B infection in the United States include IV drug use, homosexual activity, and working in health care. In the United States, most victims are adults, and the carrier rate in the general population is less than 0.5%. In many areas of the developing world, most infections occur in infancy or childhood, and chronic carriers may constitute as much as 10% to 20% of the total population; thus travelers are more likely to be exposed to carriers than is the nontraveling population. The risk is higher in persons regularly exposed to body fluids, including medical personnel and persons with many sexual partners.100
Virology and Pathophysiology
Hepatitis B virus is a deoxyribonucleic acid (DNA) virus unrelated to the agent responsible for hepatitis A. Infection occurs naturally in humans and can be induced easily in some nonhuman primates. Most hepatitis B infections are subclinical. In those resulting in clinical disease, entry of the virus into the liver is followed by viral replication and hepatocyte necrosis. HBsAg, a viral particle, appears in the bloodstream within 3 months of infection. In most cases, IgM antibody to the hepatitis B core antigen (HBcAg) appears first, followed by anti-HBsAg (surface) antibody. Antibody to a third hepatitis antigen, the e antigen (HBeAg), is present for variable periods. The course of the disease varies widely, depending on a number of factors that are not well defined. In brief, most cases are self-limited and resolve in 4 to 6 months. In these patients, anti-HBsAg or anti-HBcAg IgG antibodies can be detected for years after the episode of hepatitis. Chronic carriers do not develop anti-HBsAg antibody, but rather maintain measurable levels of HBsAg. Similarly, carriers with persistent HBeAg detectable in blood samples appear to be more infectious than are carriers without circulating HBeAg. The intricate network of antibody-antigen relationships in hepatitis B is believed to play a role not only in development of acute and chronic hepatitis but also in the many extrahepatic syndromes associated with hepatitis B. Immune complex formation has been suggested as etiologic in hepatitis B–associated arthritis, rash, arteritis, and renal disease.55
Clinical Presentation
The incubation period for hepatitis B ranges from 7 to 22 weeks; however, the patient may be antigenemic for a large portion of that time. Manifestations of hepatitis B infection are similar to those of hepatitis A and include fever, anorexia, nausea, vomiting, and abdominal pain. In addition, a prodrome of rash, arthralgia or arthritis, and fever is seen in up to 20% of hepatitis B patients, compared with its rarity in hepatitis A. Glomerulonephritis is occasionally seen. Jaundice usually appears a short time after the onset of gastrointestinal symptoms. In the self-limited form of disease, recovery is complete by 6 months. Some infections follow a fulminant course; case fatality rates are 2% or less in most series. In addition to complete resolution or death, three other sequelae are possible with acute hepatitis B. A person may become an asymptomatic chronic carrier and remain HBsAg positive but have no detectable active hepatitis. Another sequela is chronic persistent hepatitis, a term used to describe persistent but not progressive, hepatic inflammation (usually monitored by serum transaminase levels), often with HBsAg in the serum. Persons with chronic active hepatitis may be HBsAg positive and have progressive hepatitis, which may result in cirrhosis and death directly related to liver disease. Any of these three conditions results in the presence of hepatitis B viral particles in the blood.55
Management
Management is similar to that of hepatitis A. Prolonged viremia makes blood and body fluid precautions necessary until the absence of HBsAg antigen and presence of antibody to HBsAg are established. For patients with chronic infection, therapy with interferon-α and lamivudine is recommended.55,71 Even in individuals with good prognostic indicators, the response rate in terms of long-term clearance of virus and seroconversion only approaches 30%.
Prevention
Universal immunization of U.S. infants beginning at birth and catch-up immunization of children and adolescents are the current recommendations. Vaccination for at-risk adults is also advised. Travelers to highly endemic areas who stay for 6 or more months or have close contact with inhabitants should be vaccinated. Available vaccines are discussed in Chapter 84.
Delta Hepatitis (Hepatitis D)
Epidemiology
Delta virus infection is found only in patients concomitantly or previously infected with hepatitis B. Transmission follows a pattern similar to hepatitis B. In the United States, affected populations are IV drug abusers and multiply transfused hemophiliacs. Serologic evidence of delta virus disease has been documented in the Mediterranean basin, West Africa, and parts of South America.93
Virology and Pathophysiology
The delta agent has been termed a defective virus because it requires hepatitis B virus activity for its own replication.78 The agent is a single-strand of RNA enclosed in a protein coat of HBsAg. The delta agent infects cells at approximately the same time as does hepatitis B virus (coinfection), or it may be introduced later in the course of persistent hepatitis B infection (superinfection). In coinfected patients, the clinical picture may not differ from hepatitis B, but a higher percentage of such patients develop severe disease than do those with hepatitis B alone. Patients superinfected with the delta agent develop flare-ups of hepatitis, which may become fulminant. After the acute infection, the delta agent can cause progressive disease in previously stable hepatitis B patients. In general, infection with the delta agent worsens the prognosis of hepatitis B disease. The diagnosis can be made by detection of antibody to the delta antigen in the serum.78 All hepatitis B–infected individuals should be tested for anti–hepatitis D virus (HDV) IgG antibodies at least once.
Management and Prevention
Management of acute hepatitis consists of supportive care. Only interferon-α has proved antiviral activity against HDV and is associated with clearance in approximately 25% of infected patients.29,93 Precautions against transmission are the same as for hepatitis B. There is no specific vaccine or Ig for the delta agent. The best preventive measure is to be vaccinated for hepatitis B because delta agent infection cannot occur in the absence of the former virus.
Hepatitis C
As serologic methods for the diagnosis of hepatitis A, hepatitis B, and delta agent were developed, it became apparent that there was a group of persons with hepatitis for which no etiologic agent had been identified. This syndrome was previously termed non-A, non-B (NANB) hepatitis and thought to be caused by a heterogeneous group of etiologies. It is now clear that a majority of such cases were due to hepatitis C.45
Diagnosis
Two types of tests are available for diagnosis, including Ig enzyme immunoassays and RNA detection tests. Enzyme immunoassays detect only IgG antibodies, and assays for IgM or early/acute infection are not available. False negative results may occur early in the course of acute infection. Within 15 weeks of exposure, the majority of hepatitis C–infected patients will seroconvert. Viral RNA can be detected in blood 1 to 2 weeks after exposure and before IgG detection. Assays for detection of hepatitis C RNA are used to detect infection in infants born to hepatitis C–infected mothers, for monitoring patients on antiviral therapy, and to identify seropositive patients who have persistent hepatitis C infection.45 RNA tests can give false-positive and false-negative results. Viral RNA can also be shed intermittently, making a single negative assay inconclusive.
Management and Prevention
Prevention of hepatitis C largely depends on risk reduction, especially with respect to IV drug use. Pooled immunoglobulin has been used after exposure, but this should be procured from donors screened for hepatitis C. It is, however, not generally recommended. Unlike hepatitis B, protective antibody responses have not been demonstrated. Treatment of acute hepatitis C is supportive.58 People with chronic hepatitis C infection are at risk for developing cirrhosis and primary hepatocellular carcinoma. Treatments are expensive and have significant side effects. They are effective in only 50% of infected individuals. Interferon-α is the drug of choice and may be used in combination with ribavirin. Response depends on the hepatitis C infecting genotype and patient comorbidities.13,44,45
Hepatitides E, F, and G
Hepatitis E is an RNA virus provisionally placed in the Caliciviridae family. It is the second most common cause of viral hepatitis transmitted via the enteric route. The epidemiologic characteristics are similar to hepatitis A. However, hepatitis E has animals (pigs and deer) as its reservoir.87 This group of infections is especially important in the Indian subcontinent, the Middle East, and Africa. The incubation period is 2 to 6 weeks. The disease is usually self-limited but may be associated with severe illness in pregnant women. Diagnosis in travelers from endemic areas can be made on the basis of IgM antibody to hepatitis E in serum or testing of stool for viral antigen. PCR for hepatitis E may be available in some centers. In the United States, testing for hepatitis E is best undertaken in returned travelers with clinical hepatitis, although a more severe illness may occur in persons with underlying liver disease. Vaccines are not available. Prophylaxis is appropriate advice for travelers and involves counseling with respect to precautions regarding ingestion of food and water in endemic areas.
Major Bacterial Infections
This section reviews several bacterial diseases of relevance to the overseas traveler, including typhoid fever, meningococcal disease, pertussis, diphtheria, and tetanus. Other chapters deal with bacterial causes of gastroenteritis and diarrhea (Chapter 68), tick-borne diseases (Chapter 51), and zoonoses (Chapter 59).
Typhoid Fever
Epidemiology
Typhoid fever occurs worldwide, but its prevalence and attack rates are much higher in underdeveloped countries. Humans are the only host for S. typhi, the most common cause of the typhoid fever syndrome. Nearly all cases are contracted through ingestion of contaminated food or water. Transmission occurs through a variety of mechanisms, the most common of which are contact with a chronic carrier of the organism, especially food handlers, and ingestion of untreated waste material or sewage. Improved sewage and tracking of chronic carriers have markedly reduced the incidence in developed countries, although several hundred cases a year are reported to the CDC. Typhoid fever is estimated to cause 26 million illnesses and 200,000 deaths per year.20 This may be underestimated because diagnosis can be difficult (especially in children) and because typhoid fever is not a reportable disease in most countries. Travel to Asia and especially India is associated with the highest risk for contracting typhoid fever.26
Clinical Presentation
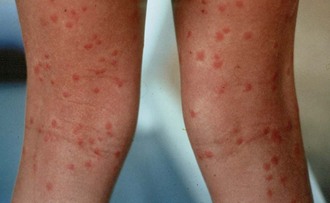
After exposure to the pathogen, 10 to 14 days usually pass before the onset of clinical illness. Some patients may experience gastroenteritis early in the course of disease, and abdominal pain or diarrhea may be present at the time the classic typhoid fever picture develops. Fever is usually the first sign of disease. Fever increases slowly over several days and may remain constant for 2 to 3 weeks, after which time defervescence begins. With antibiotic therapy, fever resolves more rapidly, often within 3 to 4 days. Relative bradycardia may accompany fever. Most victims also report headache, malaise, and anorexia. Rose spots (2- to 4-mm [0.08- to 0.16-inch] maculopapular blanching lesions) are classically described on the trunk, although they are not seen in the majority of patients. Hepatomegaly and splenomegaly have been reported in a large number of patients.43 Laboratory investigations early in the course may show a high white blood cell (WBC) count, anemia, and mild elevations of serum hepatic enzyme levels, including AST, lactate dehydrogenase, and alkaline phosphatase. Later in the course of the disease, leukopenia (WBC <3500/mm3) develops. Uncomplicated and untreated typhoid fever resolves in 3 to 4 weeks. Several complications may herald or contribute to death. Intestinal perforation, presumably secondary to necrosis of lymphoid areas of the bowel wall, may lead to peritonitis and death. Significant gastrointestinal hemorrhage may occur but rarely is fatal. Secondary pneumonia is common. A subgroup of patients has more severe disease, which may include myocardial involvement, mental status changes, hyperpyrexia, and multisystem failure. The overall case fatality rate has ranged from 12% to 32% in the developing world but is less than 2% in industrialized nations.25
Diagnosis
Culture of a bacterial species associated with the syndrome (most likely S. typhi) from a normally sterile fluid is the gold standard; however, it is expensive and requires expertise that many developing countries cannot afford. Multiple studies have evaluated the usefulness of various diagnostic tests. In general, bone marrow culture is the most sensitive method, detecting up to 90% of cases, whereas blood cultures are less sensitive. Both methods are most useful in the first week of disease. Stool cultures (and string test cultures) may be positive later in the course of disease but provide only circumstantial evidence of the causative agent. Other serodiagnostic methods are in development but have not yet proved useful.4
Management
Chloramphenicol had been the mainstay of treatment for typhoid fever since the late 1940s. Ampicillin and trimethoprim-sulfamethoxazole were the traditional alternatives. In the last decade, multiple drug resistance has increased, so ciprofloxacin is now the first-line antibiotic. Unfortunately, increasing resistance to quinolones has been observed, especially from the Indian subcontinent. In cases of quinolone resistance, either laboratory or clinical, ceftriaxone or other third-generation cephalosporins are indicated.6 Other treatment modalities include fluid support and adequate nutrition. Corticosteroids have been used empirically for many years. A single randomized double-blind study showed that administration of high-dose dexamethasone (3 mg/kg for the first dose, followed by 1 mg/kg every 6 hours for eight more doses) with chloramphenicol resulted in significantly lower mortality in patients with severe typhoid fever than in those treated with chloramphenicol alone.42 Severe typhoid fever was defined in this study by the presence of obtundation, delirium, stupor, coma, or shock (systolic blood pressure <90 mm Hg for those 12 years or older and <80 mm Hg in younger children). High-dose steroids are not recommended for those with less severe disease and should be used cautiously in those with severe disease.19
Prevention
Two vaccines exist for prophylactic use. An oral vaccine was licensed for use in the United States in 1989. The vaccine contains the Ty21 strain of S. typhi, and is associated with minimal side effects. The Ty21a vaccine is given as a four-dose series, with 1 day between each dose. Immunosuppression, antibiotic use, and gastroenteritis are contraindications to the use of this vaccine. The parenteral vaccine contains the Vi (virulence) polysaccharide of S. typhi purified from formalin-killed bacteria. It is administered as a single intramuscular injection and is therefore most useful when vaccination is required on short notice. A booster is required every 2 years.94 Even when the vaccine is given before travel, it is important to observe routine precautions for ingestion of food and water to prevent typhoid fever and acquisition of other pathogens by the fecal–oral route.
Meningococcal Disease
Epidemiology
Cases of meningococcal disease occur sporadically worldwide, with epidemic disease generally limited to developing nations. The five predominant strains of meningococcal infections are A, B, C, Y, and W-135. Serogroup A (and less so serogroup C) is most frequently associated with epidemics in sub-Saharan Africa. Increasingly, serogroup W-135 has emerged in Saudi Arabia (associated with the Hajj pilgrimage) and West Africa.97 Epidemic situations clearly pose the greater health problem to both travelers and resident populations. The “meningitis belt” in sub-Saharan Africa (a region that extends from Ethiopia in the east to Senegal in the west) experiences 100 to 800 cases/100,000/yr. In contrast, the U.S. population experiences approximately 1 case/100,000/yr, and the U.K. population experiences 2 to 3 cases/100,000/yr.76 Particularly in sub-Saharan Africa and China, the disease demonstrates yearly incidence peaks and periodic massive outbreaks, the exact determinants of which are unknown. Transmission of the organism occurs by exchange of respiratory secretions; contact is believed to be important in the spread of the disease. Asymptomatic transient nasopharyngeal carriage of the meningococcus, occurring with a baseline prevalence of 5% to 10%, may increase during epidemic periods and in close contacts of cases. The secondary attack rate among household contacts of patients with sporadic disease is 2 : 1000 to 4 : 1000, whereas that in epidemics ranges from 11 : 1000 to 45 : 1000 household contacts.
Clinical Presentation
Meningitis caused by N. meningitidis classically begins with fever, headache, and a stiff neck. It may also be accompanied by bacteremia and any of several skin manifestations, including petechiae, pustules, or maculopapular rash. In either meningitis or bacteremia, progression of petechiae to broad ecchymoses is a poor prognostic sign. As with septic meningococcemia, severe meningitis may progress with mental status deterioration, hypotension, congestive heart failure, DIC, and death. The case fatality rate of meningococcal meningitis with or without bacteremia is now estimated to be about 10%.51 In classic cases of meningitis or bacteremia with sepsis, the peripheral WBC count is elevated, with polymorphonuclear cell predominance. CSF typically is purulent, usually with more than 500 polymorphonuclear cells/mm3. There may be a more heterogeneous cell population and fewer cells if CSF is obtained early in the course or if the patient has been treated with antibiotics. The CSF glucose level is usually low and protein high, as in other bacterial meningitides. Gram stain of CSF may show the gram-negative diplococci. Meningococcal pneumonia is a well-known but less common clinical entity described in military recruit populations involving serogroup Y organisms.
Management
Treatment of meningococcal meningitis or sepsis is a medical emergency. Fortunately, the organism remains sensitive to a large number of antibiotics. Treatment of choice is penicillin G, 300,000 units/kg/day (up to 24 million units/day), given intravenously in divided doses every 2 hours. Between 7 and 10 days of therapy for serious disease is appropriate. Ceftriaxone is an alternative antimicrobial agent. If the patient is allergic to penicillin, chloramphenicol (100 mg/kg/day) may be given, although the emergence of chloramphenicol-resistant strains of N. meningitidis is of great concern.33 Antibiotic treatment before culture or hospital referral is recommended in clinically suspected cases. Supportive care should include close monitoring for hypotension and cardiac failure. Fluids and vasoactive and cardioselective agents may be important. This type of support necessitates invasive monitors and intensive care unit technology. Development of DIC is an ominous sign. Although focal bleeding and adrenal necrosis may lead to acute adrenal insufficiency, the role of replacement steroids in the treatment of Waterhouse-Friderichsen syndrome is unclear. Because the infectious agent has been found in household contacts and in those with exposure to oral secretions, contacts should receive prophylaxis to eradicate the organism. Rifampin, 600 mg by mouth every 12 hours for four doses, is standard adult prophylaxis. Children should receive 10 mg/kg of rifampin every 12 hours for four doses if they are older than 1 month and 5 mg/kg every 12 hours for four doses if they are younger than 1 month. More recently, alternate regimens using ceftriaxone and ciprofloxacin have also been proved to be efficacious, although rifampin remains the standard.
Prevention
Effective meningococcal vaccines are available that target serogroups A, C, Y, and W-135. MPSV4 is a tetravalent meningococcal polysaccharide vaccine containing serogroups A, C, Y and W-135 licensed for use in children greater than 2 years and adults. It can be given as one subcutaneous injection of 0.5 mL. Clinical efficacy of more than 65% is maintained for at least 3 years in persons immunized at 4 years of age or older, although protection wanes more rapidly in children vaccinated at younger than 4 years of age. The tetravalent meningococcal conjugate vaccine (MCV4) also contains serogroups A, C, Y and W-135 and is licensed for use in children older than 2 years and adults. With increased meningococcal disease rates in college dormitories and military barracks, it is recommended that all adolescents 11 to 18 years of age be immunized with a single dose of MCV4. In addition, asplenic individuals, those with specific complement deficiencies, and travelers to meningitis belt countries during the dry season or to regions with ongoing epidemics are advised to be vaccinated. Vaccination is a Hajj visa requirement for Hajj/Umrah pilgrims traveling to Saudi Arabia.96 Because the areas in which epidemic meningococcal disease occur change over time, it is prudent to check with appropriate authorities to determine current recommendations for meningococcal vaccination before travel.
Pertussis
Prevention
Universal immunization of children younger than 7 years, as well as boosters for adolescents (age 11 to 18 years) and adults, is recommended. In the United States, acellular pertussis vaccines are currently used. The pertussis vaccine is combined with diphtheria and tetanus toxoids (pediatric DTaP formulation and adolescent/adult Tdap formulations). The Tdap replaces Td to boost waning pertussis immunity in the adult population where the majority of pertussis infections are perpetuated. In cases of pertussis exposure, immunization of unimmunized or underimmunized individuals should be initiated. In addition, chemoprophylaxis (with azithromycin, erythromycin, or clarithromycin) should be started for all household contacts and other close contacts regardless of age and immunization status. If 21 days has elapsed since exposure, chemoprophylaxis is of limited value.2
Diphtheria
Management
Because the toxin and not the organism per se mediates life-threatening clinical manifestations of diphtheria, neutralization of absorbed toxin is crucial. A horse-derived antitoxin is available and should be administered as soon as the diagnosis is seriously considered. A 0.1-mL test dose of intradermal antitoxin diluted to a 1 : 1000 concentration (with a saline control) is observed for 20 minutes. If no reaction occurs, full doses can be given intravenously. Antitoxin should be diluted to 1 : 20 in saline and given no faster than 1 mL/min. For mild cases, 20,000 units may be adequate, 40,000 units for moderate cases, and as much as 80,000 to 120,000 units for severely ill patients.17 Erythromycin or penicillin G may be given to eradicate the carrier state, although their use has no effect on the clinical course of disease. Close observation is crucial to evaluate the need for respiratory support, especially in young children. Serial electrocardiograms and neurologic evaluation establish the onset of complications. If significant conduction abnormalities are present, continuous heart monitoring should be undertaken. Strict bed rest is recommended for all patients for 2 to 3 weeks. Immunization should be given during convalescence because disease survival does not necessarily confer immunity.
Tetanus
Clinical Presentation
A tetanus-prone wound precedes most adult disease, which may not be evident at the time of presentation. Localized tetanus, with spasm of a focal set of muscle groups, may occur and remain localized for weeks, then slowly resolve. This form of tetanus is much less common than is the generalized form, which often begins with trismus, or spasm of the masticator muscle group (Figure 85-2). Gradual onset of spasm of other muscle groups usually involves the trunk and extremities. Because the posterior muscles are stronger during spasms, the victim exhibits lumbar lordosis, with the neck and legs extended and arms flexed at the elbows (opisthotonos). Spasms seem to be exacerbated by external stimuli, such as sudden sound or light. The primary danger is loss of ability to breathe, especially during prolonged spasms. Respiratory failure is the main cause of death. The clinical picture in neonatal tetanus is similar but begins with restlessness and failure to nurse, with progression to tetany and sympathetic overactivity. There is no definitive laboratory test to confirm the diagnosis of tetanus, but the clinical picture is adequate in the majority of cases.

Management
Emergency medical treatment of tetanus patients should include (1) excision of the wound, (2) administration of human tetanus immunoglobulin (TIG; 3000 to 6000 units in a single dose), and (3) administration of an antibiotic effective against C. tetani, such as penicillin or metronidazole for 10 to 14 days.83 Depending on the severity of disease, different levels of supportive care and sedation may be appropriate. Diazepam may be given to mildly affected patients for sedation. Patients should be evaluated carefully for dysphagia. If dysphagia is present or other respiratory difficulties arise, endotracheal intubation or a tracheostomy should be performed. With prolonged spasms, hypoxia and cyanosis may occur; mechanical ventilation with pharmacologic paralysis is appropriate. At the same time, attention must be given to fluid balance and nutrition. Enteral feeding by a nasogastric tube is the least invasive way to supply both. β-Blockers have been suggested to relieve symptoms of autonomic overactivity, such as tachycardia and hypertension, but there is no proved benefit to their prophylactic use. Sources of sensory stimulation should be reduced when the spasms are uncontrolled.
Major Protozoan Infections Other Than Malaria
African Trypanosomiasis
Trypanosoma brucei rhodesiense (East Africa) and T. brucei gambiense (West Africa) are important infectious diseases in Africa and have provided remarkable insights into the importance of antigenic variation as a strategy used by parasites to avoid the immune response.68 T. brucei gambiense causes African sleeping sickness, and T. brucei rhodesiense causes an acute disease that may end in heart failure. The parasites are transmitted to humans by tsetse flies (Glossina spp.) in sub-Saharan Africa. Metacyclic promastigotes are injected into the bloodstream through the saliva of the biting tsetse fly and divide into long slender forms in the bloodstream. These eventually differentiate into short stumpy forms, which are taken up in the blood meal of the tsetse. Once in the fly, the parasite differentiates into procyclic forms. It takes approximately 3 weeks for the protozoa to develop into infective metacyclics within the tsetse fly. Approximately 15,000 human cases are reported each year. In East Africa, animals such as antelope, bushbuck, and hartebeest serve as reservoirs. In West and Central Africa, humans are the only reservoir.
South American Trypanosomiasis (Chagas’ Disease)
Clinical Manifestations
Affected individuals generally do not recall initial contact with the insects, during which time triatomid feces containing the protozoan organisms are deposited on broken skin or mucous membranes and then multiply within local macrophages. The macrophages rupture and elicit an inflammatory reaction that appears as a nodule with slightly painful satellite nodules or draining lymph nodes. A symptomatic phase, characterized by fever and diffuse lymph node enlargement, develops. Hepatosplenomegaly may also occur. In severe cases, acute myocarditis, pericarditis, or endocarditis is seen. After several months, the acute phase resolves, and chronic disease appears, characterized by cardiomyopathy, megaesophagus, or megacolon.77 It is rare for the traveler to develop these signs or symptoms.
Leishmaniasis
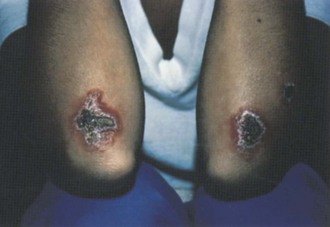
Humans may be infected by Leishmania species that cause three clinical syndromes: cutaneous, mucosal, or visceral leishmaniasis. These intracellular parasites are transmitted by phlebotomine sandflies. Various forms of the infection occur throughout Latin and Central America, Africa, the Middle East, and Asia (Figure 85-3). Cutaneous lesions are caused by Leishmania tropica, Leishmania major and Leishmania aethiopica (Old World species), and by Leishmania mexicana, Leishmania amazonensis, Leishmania braziliensis, Leishmania panamensis, Leishmania guyanensis, and Leishmania peruviana (New World species). Cutaneous disease begins as a small ulcer with raised borders. Ulcers can persist as nodules or papules. In the chronic phase, these nonhealing ulcers frequently become secondarily infected by bacteria.56 Mucosal leishmaniasis is typically caused by L. braziliensis, L panamensis, and L. guyanensis. It begins as a single nodule and months to years after the cutaneous lesion heals, progresses to the oropharyngeal or nasal mucosa, where it causes severe destruction. This disease occurs primarily in residents of the Amazon basin. Visceral leishmaniasis (kala-azar) is caused by Leishmania donovani, Leishmania infantum, and Leishmania chagasi. Affected individuals generally do not recall an initial skin lesion. Several months after inoculation, fever, abdominal discomfort, and weakness develop and become progressively more severe. Nausea and vomiting are protracted, the skin becomes dry and dark, and abdominal distention with hepatosplenomegaly eventually appears. Diagnosis is made by demonstration of the presence of the parasite in tissue biopsy or needle aspiration of affected tissue. Serologic testing is usually positive in mucosal or visceral leishmaniasis (available at the CDC). Treatment is indicated for mucosal and visceral leishmaniasis; liposomal amphotericin B is the only U.S. Food and Drug Administration (FDA)–approved treatment. Protection from sandfly bites prevents the disease. All forms of leishmaniasis are rare in travelers and nonresidents of endemic areas.
Major Helminthic Infections
Schistosomiasis
Clinical Manifestations
Signs and symptoms of infection vary among the three schistosome species. The initial presentation of acute S. mansoni infection may include fever, anorexia, weight loss, and abdominal pain. This unusual symptom complex, which occurs in individuals with heavy infection, has been referred to as Katayama fever and appears 18 to 60 days after exposure.80 Travelers with light or moderate exposure, however, usually have no specific signs or only mild local dermatitis (swimmer’s itch) associated with contact with cercariae, the infective stage of the parasite released by snails (Figure 85-4). In persons with established infections, the prevalence of clinical manifestations is low. Most individuals have no signs specifically attributable to S. mansoni infection. Hepatomegaly or splenomegaly, attributable to portal hypertension after granulomatous reactions to eggs deposited in the liver, occurs in 15% of subjects. Eggs may also embolize to the lungs and induce granulomatous lesions and cor pulmonale. Those at greatest risk are persons who have the heaviest intensity of infection as judged by fecal egg counts. These complications may ultimately result in esophageal and gastrointestinal varices, which cause acute blood loss. The manifestations of S. japonicum infections are similar to S. mansoni infections, except that Katayama fever appears to be more frequent in the former case. In addition, there is a unique manifestation of S. japonicum infection attributable to embolization of eggs to the brain. Generalized or jacksonian seizures are the major signs of cerebral schistosomiasis. Because S. haematobium adult worms inhabit the venous system of the genitourinary tract, the signs and symptoms of this helminth infection are primarily secondary to granulomatous reactions to eggs present in the ureters and bladder wall. Dysuria and hematuria have been reported in many individuals who reside in endemic areas. Treatment for all species is a one-time dose of praziquantel. Intense reactions may benefit from a course of oral corticosteroids.
Filariases
Onchocerciasis
O. volvulus is transmitted to humans by the bite of Simulium species of blackflies in Central America and West and Central Africa. Infective, or third-stage, larvae eventually develop into adult worms contained in deep subcutaneous nodules that are asymptomatic and may be palpable. Microfilariae are released from adult female worms and cause dermatitis as they migrate through the skin. The organisms have a propensity to invade the eye (especially the anterior chamber and cornea), where they cause blindness. Diagnosis is based on prolonged residence in an endemic area (e.g., as in Peace Corps volunteers) and parasitologic identification in skin snips or slit-lamp examination of the eye. Treatment is with ivermectin to clear microfilariae.88
Lymphatic Filariasis
B. malayi and W. bancrofti are transmitted by mosquitoes. Infective larvae eventually develop into lymphatic-dwelling adult worms, which release microfilariae into the bloodstream. Although chronic infection and recurrent exposure are associated with a wide variety of clinical manifestations, including tropical pulmonary eosinophilia, acute lymphangitis, and elephantiasis, these manifestations are rare in nonresidents of endemic areas. The only definitive diagnostic test is identification of parasites in the bloodstream. Because nonresidents and many residents who are infected may not have detectable parasitemia, other laboratory studies (eosinophilia, elevated serum IgE level) must be used as aids in diagnosis. Diethylcarbamazine (6 mg/kg/d in three doses for 12 days) is the treatment.10
Loiasis (Loaiasis)
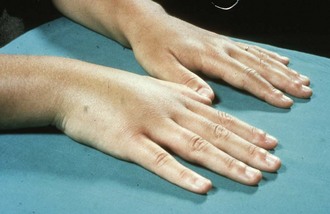
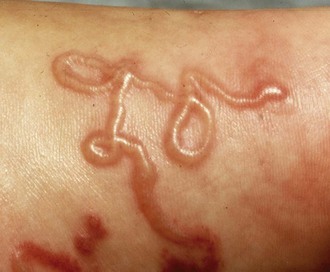
L. loa is transmitted to humans by the bites of tabanid flies that live along river edges in Central and West Africa. Microfilariae migrate in the bloodstream, whereas adult worms migrate in cutaneous tissues. The major disease manifestation is Calabar swellings, which are characterized as egg-sized or smaller raised lesions, predominantly over the extremities, that are tender and surrounded by edematous skin (Figure 85-5). They may migrate and last several days. Migration of the worm across the eye is known as loiasis, loa loa, or African eye worm (Figure 85-6). Their pathogenesis may be related to migration of adult worms or release of antigens that elicit immunologic hypersensitivity reactions. Treatment is with diethylcarbamazine at a dose of 9 mg/kg body weight/day in three doses for 12 days. Retreatment is occasionally required.12
Intestinal Helminth Infections
Ascariasis
Diagnosis of ascariasis may be made by identification of one of several parasite stages. Adult ascarids occasionally migrate from the mouth or anus. Ascaris larvae may rarely be observed in sputum or gastric washings. The most common means of diagnosis is identification of eggs in feces. Eggs are ovoid, 35 to 70 mm [1.4 to 2.8 inches] in diameter, and consist of an outer white shell and brownish ovum internally. The eggs are not produced until approximately 9 weeks after infection. Intestinal ascariasis is treated with albendazole or mebendazole. An alternative regimen that avoids the use of benzimidazoles (e.g., for treatment of pregnant women) is pyrantel pamoate (11 mg/kg body weight to a maximum of 1 g).9
Hookworm
Ancylostoma duodenale and Necator americanus infections occur most commonly in the tropics but also in temperate climates where sanitation is poor. Hookworm is second only to A. lumbricoides in terms of the number of people infected. Humans are infected percutaneously by third-stage larvae in the soil. The larvae enter the bloodstream, pass to the lungs, and rupture the alveolar lining to eventually ascend the trachea and descend the esophagus to differentiate into adult worms. These adult worms contain cutting plates on the anterior end and feed on host blood obtained through their attachment sites in the upper small intestine. It has been estimated that each N. americanus infection causes 0.03 mL of blood loss per day, whereas the A. duodenale hookworm consumes 0.26 mL per day. Iron deficiency anemia, especially in persons with low iron intake, is the major clinical manifestation of hookworm infection. The diagnosis may be made by identification of hookworm eggs in feces. The eggs are round, 40 to 60 mm [1.6 to 2.4 inches] in diameter, and have a “smoother” shell than do Ascaris eggs. Although multiple drugs are effective in treatment, albendazole is most readily available. Supplemental iron should be given to persons when necessary. Infection with hookworm is rare in the traveler from a developed country. Migrating animal hookworms, such as Ancylostoma braziliense and Ancylostoma caninum, may create serpiginous lesions in superficial tissues of humans (Figure 85-7).9
Strongyloidiasis
Strongyloides stercoralis infection occurs in tropical and temperate regions. The infection is initiated by contact with soil containing infective third-stage larvae. The helminth follows a route within the host similar to that described for hookworms. In addition, there is an autoinfection cycle in which larvae released in the intestine may penetrate the mucosa directly and then migrate through the liver and lungs. This occurs only in immunocompromised individuals. Many persons with S. stercoralis infection are asymptomatic. Some persons, however, have cutaneous or intestinal manifestations. The former are urticarial lesions around the buttocks and waist that last 1 to 2 days. These are secondary to penetration of larvae present in the feces. Other symptoms include indigestion, abdominal cramps, and diarrhea. Diagnosis is made by identification of larvae in fresh stools or gastrointestinal washings. Rhabditiform larvae with a length of 250 mm (9.8 inches) and a width of 10 to 20 mm (0.4 to 0.8 inches) are most commonly observed, although filariform larvae may also be present. Treatment is with ivermectin.84
Enterobiasis
Enterobiasis, or pinworm infection, exists in all parts of the world. Eggs are passed from female worms in the colon. Infection is transmitted by ingestion of Enterobius vermicularis eggs, which develop into gravid adult female worms in the large bowel. The infection is especially common in crowded settings where sanitation is poor. The diagnosis may be made by identification of adult worms migrating along the perianal area or by eggs deposited in the same area. Eggs are detected by applying a piece of sticky cellophane tape to the area and inspecting it microscopically. Treatment is with pyrantel pamoate or albendazole. Repeated treatments as a result of reinfection in crowded settings are commonly required.52
1 Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). Update: Prevention of hepatitis A after exposure to hepatitis A virus and in international travelers—updated recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep. 2007;56:1080.
2 American Academy of Pediatrics Committee on Infectious Diseases. Prevention of pertussis among adolescents: Recommendations for use of tetanus toxoid, reduced diphtheria toxoid, and acellular pertussis (Tdap) vaccine. Pediatrics. 2006;117:965.
3 Askling HH, Rombo L, Andersson Y, et al. Hepatitis A risk in travelers. J Travel Med. 2009;16:233.
4 Baker S, Favorov M, Dougan G. Searching for the elusive typhoid diagnostic. BMC Infect Dis. 2010;10:45.
5 Barrett AD, Teuwen DE. Yellow fever vaccine—how does it work and why do rare cases of serious adverse events take place? Curr Opin Immunol. 2009;21:308.
6 Basnyat B, Maskey AP, Zimmerman MD, et al. Enteric (typhoid) fever in travelers. Clin Infect Dis. 2005;41:1467.
7 Bausch DG, Sprecher AG, Jeffs B, et al. Treatment of Marburg and Ebola hemorrhagic fevers: A strategy for testing new drugs and vaccines under outbreak conditions. Antiviral Res. 2008;78:150.
8 Bazemore AW, Huntington M. The pretravel consultation. Am Fam Physician. 2009;80:583.
9 Bethony J, Brooker S, Albonico M, et al. Soil-transmitted helminth infections: Ascariasis, trichuriasis, and hookworm. Lancet. 2006;367:1521.
10 Bockarie MJ, Taylor MJ, Gyapong JO. Current practices in the management of lymphatic filariasis. Expert Rev Anti Infect Ther. 2009;7:595.
11 Borio L, Inglesby T, Peters CJ, et al. Hemorrhagic fever viruses as biological weapons: Medical and public health management. JAMA. 2002;287:2391.
12 Boussinesq M. Loiasis. Ann Trop Med Parasitol. 2006;100:715.
13 Brok J, Gluud LL, Gluud C: Ribavirin plus interferon versus interferon for chronic hepatitis C. Cochrane Database Syst Rev (1): CD005445.
14 Bryan CS, Moss SW, Kahn RJ. Yellow fever in the Americas. Infect Dis Clin North Am. 2004;18:275.
15 Burke DS, Lorsomrudee W, Leake CJ, et al. Fatal outcome in Japanese encephalitis. Am J Trop Med Hyg. 1985;34:1203.
16 Callahan MV, Hamer DH. On the medical edge: Preparation of expatriates, refugee and disaster relief workers, and Peace Corps volunteers. Infect Dis Clin North Am. 2005;19:85.
17 Centers for Disease Control and Prevention. Availability of diphtheria antitoxin through an investigational new drug protocol. MMWR Morb Mortal Wkly Rep. 1997;46:380.
18 Centers for Disease Control and Prevention. Outbreak of Marburg virus hemorrhagic fever—Angola, October 1, 2004-March 29, 2005. MMWR Morb Mortal Wkly Rep. 2005;54:308.
19 Cooles P. Adjuvant steroids and relapse of typhoid fever. J Trop Med Hyg. 1986;89:229.
20 Crump JA, Luby SP, Mintz ED. The global burden of typhoid fever. Bull World Health Organ. 2004;82:346.
21 Demby AH, Inapogui A, Kargbo K, et al. Lassa fever in Guinea: II. Distribution and prevalence of Lassa virus infection in small mammals. Vector Borne Zoonotic Dis. 2001;1:283.
22 Dolnik O, Kolesnikova L, Becker S. Filoviruses: Interactions with the host cell. Cell Mol Life Sci. 2008;65:756.
23 Duchin JS, Koster FT, Peters CJ, et al. Hantavirus pulmonary syndrome: A clinical description of 17 patients with a newly recognized disease—The Hantavirus Study Group. N Engl J Med. 1994;330:949.
24 Durbin AP, Whitehead SS. Dengue vaccine candidates in development. Curr Top Microbiol Immunol. 2010;338:129.
25 Edelman R, Levine MML. Summary of an international workshop on typhoid fever. Rev Infect Dis. 1986;8:329.
26 Ekdahl K, de Jong B, Andersson Y. Risk of travel-associated typhoid and paratyphoid fevers in various regions. J Travel Med. 2005;12:197.
27 Ergonul O. Treatment of Crimean-Congo hemorrhagic fever. Antiviral Res. 2008;78:125.
28 Ergonul O, Celikbas A, Dokuzoguz B, et al. Characteristics of patients with Crimean-Congo hemorrhagic fever in a recent outbreak in Turkey and impact of oral ribavirin therapy. Clin Infect Dis. 2004;39:284.
29 Farci P, Chessa L, Balestrieri C, et al. Treatment of chronic hepatitis D. J Viral Hepat. 2007;14:58.
30 Fischer M, Lindsey N, Staples JE, et al. Japanese encephalitis vaccines: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2010;59:1.
31 Fisher-Hoch S, McCormick JB, Sasso D, et al. Hematologic dysfunction in Lassa fever. J Med Virol. 1988;26:127.
32 Fisher-Hoch SP, Khan JA, Rehman S, et al. Crimean Congo-haemorrhagic fever treated with oral ribavirin. Lancet. 1995;346:472.
33 Galimand M, Gerbaud G, Guibourdenche M, et al. High-level chloramphenicol resistance in Neisseria meningitidis. N Engl J Med. 1998;339:868.
34 Gao X, Nasci R, Liang G. The neglected arboviral infections in mainland China. PLoS Negl Trop Dis. 2010;4:e624.
35 Gould LH, Osman MS, Farnon EC, et al. An outbreak of yellow fever with concurrent chikungunya virus transmission in South Kordofan, Sudan, 2005. Trans R Soc Trop Med Hyg. 2008;102:1247.
36 Gubler DJ. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 2002;10:100.
37 Gubler DJ. The global emergence/resurgence of arboviral diseases as public health problems. Arch Med Res. 2002;33:330.
38 Gubler DJ. The changing epidemiology of yellow fever and dengue, 1900 to 2003: Full circle? Comp Immunol Microbiol Infect Dis. 2004;27:319.
39 Hanna JN, Ritchie SA, Phillips DA, et al. An outbreak of Japanese encephalitis in the Torres Strait, Australia, 1995. Med J Aust. 1996;165:256.
40 Haemorrhagic fever with renal syndrome: Memorandum from a WHO meeting. Bull World Health Organ. 1983;61:269.
41 Hoenen T, Groseth A, Falzarano D, et al. Ebola virus: Unravelling pathogenesis to combat a deadly disease. Trends Mol Med. 2006;12:206.
42 Hoffman SL, Punjabi NH, Kumala S, et al. Reduction of mortality in chloramphenicol-treated severe typhoid fever by high-dose dexamethasone. N Engl J Med. 1984;310:82.
43 Hornick RB, Greisman SE, Woodward TE, et al. Typhoid fever: Pathogenesis and immunologic control. N Engl J Med. 1970;283:686.
44 Jain MK, Zoellner C. Role of ribavirin in HCV treatment response: Now and in the future. Expert Opin Pharmacother. 2010;11:673.
45 Jara P, Hierro L. Treatment of hepatitis C in children. Expert Rev Gastroenterol Hepatol. 2010;4:51.
46 Jelinek T. Ixiaro: A new vaccine against Japanese encephalitis. Expert Rev Vaccines. 2009;8:1501.
47 Jones SM, Feldmann H, Ströher U, et al. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat Med. 2005;11:786.
48 Kennedy PG. Human African trypanosomiasis of the CNS: Current issues and challenges. J Clin Invest. 2004;113:496.
49 Khromava AY, Eidex RB, Weld LH, et al. Yellow fever vaccine: An updated assessment of advanced age as a risk factor for serious adverse events. Vaccine. 2005;23:3256.
50 Kirkpatrick BD, Alston WK. Current immunizations for travel. Curr Opin Infect Dis. 2003;16:369.
51 Koch S, Steffen R. Meningococcal disease in travelers: Vaccination recommendations. J Travel Med. 1994;1:4.
52 Kucik CJ, Martin GL, Sortor BV. Common intestinal parasites. Am Fam Physician. 2004;69:1161.
53 Kukreti H, Mittal V, Chaudhary A, et al. Continued persistence of a single genotype of dengue virus type-3 (DENV-3) in Delhi, India since its re-emergence over the last decade. J Microbiol Immunol Infect. 2010;43:53.
54 Lassa fever. WHO Newsletter, Geneva, 2005.
55 Liaw YF, Chu CM. Hepatitis B virus infection. Lancet. 2009;373:582.
56 Magill AJ. Cutaneous leishmaniasis in the returning traveler. Infect Dis Clin North Am. 2005;19:241.
57 Mahanty S, Bray M. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect Dis. 2004;4:487.
58 Maheshwari A, Thuluvath PJ. Management of acute hepatitis C. Clin Liver Dis. 2010;14:169.
59 Marano C, Freedman DO. Global health surveillance and traveler’s health. Curr Opin Infect Dis. 2009;22:423.
60 Martin JE, Sullivan NJ, Enama ME, et al. A DNA vaccine for Ebola virus is safe and immunogenic in a phase I clinical trial. Clin Vaccine Immunol. 2006;13:1267.
61 Martina BE, Koraka P, Osterhaus AD. Dengue virus pathogenesis: An integrated view. Clin Microbiol Rev. 2009;22:564.
62 McCormick JB, Fisher-Hoch SP. Lassa fever. Curr Top Microbiol Immunol. 2002;262:75.
63 McCormick JB, King IJ, Webb PA, et al. A case-control study of the clinical diagnosis and course of Lassa fever. J Infect Dis. 1987;155:445.
64 McCormick JB, Webb PA, Krebs JW, et al. A prospective study of the epidemiology and ecology of Lassa fever. J Infect Dis. 1987;155:437.
65 Monath TP. Aedes albopictus, an exotic mosquito vector in the United States. Ann Intern Med. 1986;105:449.
66 Monath TP, Cetron MS, McCarthy K, Nichols R, et al. Yellow fever 17D vaccine safety and immunogenicity in the elderly. Hum Vaccin. 2005;1:207.
67 Morrison AC, Minnick SL, Rocha C, et al: Epidemiology of dengue virus in Iquitos, Peru 1999 to 2005: Interepidemic and epidemic patterns of transmission, PLoS Negl Trop Dis 4:e670
68 Morrison LJ, Marcello L, McCulloch R. Antigenic variation in the African trypanosome: Molecular mechanisms and phenotypic complexity. Cell Microbiol. 2009;11:1724.
69 Ogbu O, Ajuluchukwu E, Uneke CJ. Lassa fever in West African sub-region: An overview. J Vector Borne Dis. 2007;44:1.
70 Onyango CO, Ofula VO, Sang RC, et al. Yellow fever outbreak, Imatong, southern Sudan. Emerg Infect Dis. 2004;10:1063.
71 Papatheodoridis GV, Manolakopoulos S, Dusheiko G, et al. Therapeutic strategies in the management of patients with chronic hepatitis B virus infection. Lancet Infect Dis. 2008;8:167.
72 Peters CJ. Marburg and Ebola—arming ourselves against the deadly filoviruses. N Engl J Med. 2005;352:2571.
73 Peterson AT, Bauer JT, Mills JN. Ecologic and geographic distribution of filovirus disease. Emerg Infect Dis. 2004;10:40.
74 Peterson AT, Carroll DS, Mills JN, et al. Potential mammalian filovirus reservoirs. Emerg Infect Dis. 2004;10:2073.
75 Pickering LK, Baker CJ, Freed GL, et al. Immunization programs for infants, children, adolescents, and adults: Clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2009;49:817.
76 Pollard AJ. Global epidemiology of meningococcal disease and vaccine efficacy. Pediatr Infect Dis J. 2004;23:S274.
77 Rassi AJr, Rassi A, Marin-Neto JA. Chagas disease. Lancet. 2010;375:1388.
78 Rizzetto M, Hepatitis D. Thirty years after. J Hepatol. 2009;50:1043.
79 Robertson SE, Hull BP, Tomori O, et al. Yellow fever: A decade of reemergence. JAMA. 1996;276:1157.
80 Ross AG, Vickers D, Olds GR, et al. Katayama syndrome. Lancet Infect Dis. 2007;7:218.
81 Roukens AH, Visser LG. Yellow fever vaccine: Past, present and future. Expert Opin Biol Ther. 2008;8:1787.
82 Ryan CA, Hargrett-Bean NT, Blake PA. Salmonella typhi infections in the United States, 1975-1984: Increasing role of foreign travel. Rev Infect Dis. 1989;11:1.
83 Schofield F. Selective primary health care: Strategies for control of disease in the developing world: XXII. Tetanus: A preventable problem. Rev Infect Dis. 1986;8:144.
84 Segarra-Newnham M. Manifestations, diagnosis, and treatment of Strongyloides stercoralis infection. Ann Pharmacother. 2007;41:1992.
85 Sullivan NJ, Geisbert TW, Geisbert JB, et al. Accelerated vaccination for Ebola virus haemorrhagic fever in non-human primates. Nature. 2003;424:681.
86 Teixeira MG, Costa MC, Coelho G, et al. Recent shift in age pattern of dengue hemorrhagic fever, Brazil. Emerg Infect Dis. 2008;14:1663.
87 Teo CG. Much meat, much malady: Changing perceptions of the epidemiology of hepatitis E. Clin Microbiol Infect. 2010;16:24.
88 Udall DN. Recent updates on onchocerciasis: Diagnosis and treatment. Clin Infect Dis. 2007;44:53.
89 van de Wal BW, Joubert JR, van Eeden PJ, et al. A nosocomial outbreak of Crimean-Congo haemorrhagic fever at Tygerberg Hospitala: IV. Preventive and prophylactic measures. S Afr Med J. 1985;68:729.
90 van Eeden PJ, Joubert JR, van de Wal BW, et al. A nosocomial outbreak of Crimean-Congo haemorrhagic fever at Tygerberg Hospital: I. Clinical features. S Afr Med J. 1985;68:711.
91 van Eeden PJ, van Eeden SF, Joubert JR, et al. A nosocomial outbreak of Crimean-Congo haemorrhagic fever at Tygerberg Hospital: II. Management of patients. S Afr Med J. 1985;68:718.
92 Webster DP, Farrar J, Rowland-Jones S. Progress towards a dengue vaccine. Lancet Infect Dis. 2009;9:678.
93 Wedemeyer H, Manns MP. Epidemiology, pathogenesis and management of hepatitis D: Update and challenges ahead. Nat Rev Gastroenterol Hepatol. 2010;7:31.
94 Whitaker JA, Franco-Paredes C, del Rio C, et al. Rethinking typhoid fever vaccines: Implications for travelers and people living in highly endemic areas. J Travel Med. 2009;16:46.
95 Whitehouse CA. Crimean-Congo hemorrhagic fever. Antiviral Res. 2004;64:145.
96 Wilder-Smith A. Meningococcal disease: Risk for international travellers and vaccine strategies. Travel Med Infect Dis. 2008;6:182.
97 Wilder-Smith A, Goh KT, Barkham T, et al. Hajj-associated outbreak strain of Neisseria meningitidis serogroup W135: Estimates of the attack rate in a defined population and the risk of invasive disease developing in carriers. Clin Infect Dis. 2003;36:679.
98 Wilson ME, Freedman DO. Etiology of travel-related fever. Curr Opin Infect Dis. 2007;20:449.
99 Xu ZY, Guo CS, Wu YL, et al. Epidemiological studies of hemorrhagic fever with renal syndrome: Analysis of risk factors and mode of transmission. J Infect Dis. 1985;152:137.
100 Zuckerman JN, Hoet B. Hepatitis B immunisation in travellers: Poor risk perception and inadequate protection. Travel Med Infect Dis. 2008;6:315.
[/level-membership-for-emergency-medicine-category][not-level-membership-for-emergency-medicine-category]
Chapter 85 Non–North American Travel and Exotic Diseases
Travelers to tropical and subtropical areas of the world where hygienic conditions are poor and ecologic conditions are permissive may encounter infectious agents that are no longer endemic or have never existed in temperate regions of the world. Although economic development and industrialization of developing countries of the tropics have resulted in a decreased health burden of many tropical infectious diseases, it is important to realize that there is still a risk for exposure for the traveler who is unaware of appropriate measures to prevent or treat such conditions. The most important consideration in the management of this problem, which is increasing as international travel expands, is appropriate preventive measures through counsel with a travel medicine specialist and prophylaxis using safe drugs and vaccines. This topic has recently been reviewed in several excellent publications.*
This chapter is concerned with infectious diseases that are uncommon or do not exist in North America and with which most health professionals in North America have scant familiarity. Other chapters give specific details relevant to malaria (Chapter 49), tick-borne diseases (Chapter 51), infectious diarrheas (Chapter 68), and travel medicine (Chapter 84). The infectious diseases considered in this chapter should not be considered a complete listing. This is especially important to keep in mind in an era when diseases once thought to be eliminated or nonexistent in North America are emerging or reemerging coincidental with large-scale movements of human and vector populations.
Major Viral Infections
Yellow Fever
European physicians did not recognize until the late 1490s the clinical syndrome now known as yellow fever. Initially described by Columbus in the West Indies, large-scale epidemics were later observed throughout the Americas and tropical Africa in the 1700s and 1800s. After epidemic yellow fever in Texas, Louisiana, and Tennessee caused 20,000 deaths in the 1880s, the Yellow Fever Commission was organized to study the problem. Identification of the mosquito vector, Aedes aegypti, and definitive studies conducted by the U.S. military under the leadership of Walter Reed were followed by massive campaigns to eradicate mosquito breeding sites. This led to virtual elimination of urban yellow fever from the Americas. The last case of yellow fever acquired in the continental United States was reported in 1911. Because it is difficult if not impossible to eliminate jungle reservoirs, there continue to be cases reported annually from South America and tropical Africa. Larger outbreaks secondary to resurgent vector populations have occurred in recent years in tropical West Africa.14,35,38,70
Ecology and Epidemiology
Currently, both the Americas and Africa have a constant low level of jungle yellow fever because of inability to control either the monkey reservoir or the mosquito vector. Overall there are about 200,000 cases per year, resulting in approximately 30,000 deaths, occurring primarily in sub-Saharan Africa.79 Some suggest that these rates are underestimated by at least 10-fold. Persons at risk include workers or travelers in or near the tropical rainforest canopy. Urban yellow fever had been reduced in the western hemisphere through massive campaigns to control breeding and spread of the Aedes vector. However, the benefits of these campaigns have declined, and there is currently an increased threat of further outbreaks of disease. Introduction of Aedes albopictus, an aggressive anthropophilic dengue vector from Southeast Asia, and reemergence of A. aegypti into the Americas raise the specter of increased yellow fever transmission in the western hemisphere.65 Less-intense vector control measures and a more complex ecology have made elimination of urban yellow fever in Africa even more difficult.
Prevention
Avoidance of this potentially fatal infection is possible through use of yellow fever vaccine. The vaccine strain 17D is an attenuated live virus grown in chicken embryos. Greater than 95% of persons vaccinated achieve significant antibody 1evels. Repeat vaccinations are recommended every 10 years, although persistent antibody titers have been detected as long as 30 to 40 years after vaccination. Yellow fever vaccine is generally well tolerated, with headache or malaise occurring in less than 10% of those vaccinated. Rare allergic side effects occur primarily in persons with hypersensitivity to eggs. Other serious adverse events, including death, have been reported, with the greater risk being associated with age older than 60 years.5,49,66,81 Vaccination is not recommended in the first 6 months of life or in other situations where live virus vaccines are contraindicated. Although pregnant women have received the vaccine without adverse effect to themselves or their infants, it is not recommended for use in this group because of possible teratogenic effects. Other means of reducing the risk for yellow fever (and any mosquito-borne infectious disease) include liberal use of mosquito repellent and netting in endemic areas. Outbreak control in endemic countries is primarily through focused vaccination campaigns.
Dengue fever
Dengue fever has been reported since the late 1700s. Since World War II, increased attention has focused on the dengue virus, largely as a result of recognition of dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS). First noted in Southeast Asia, DHF and DSS have attained worldwide distribution in the last 30 years.34,53,67,86 Dengue is the most common insect-borne viral infection in the world. The infection has been reported in more than 100 countries, with 50 to 100 million dengue infections each year resulting in approximately 500,000 cases of life-threatening disease (DHF and DSS) annually.36,37
Virology and Pathophysiology
The etiologic agent is a single-stranded RNA flavivirus, which may be one of four serotypes, denoted DEN-1 through DEN-4. As with yellow fever, local viral replication is followed by dissemination to lymphocyte- and macrophage-rich areas, where most of the reproductive activity occurs. Infection with one virus serotype provides long-lasting immune protection against that type only. After infection with one serotype, a subsequent infection with a heterologous serotype may result in a more severe clinical course. Non-neutralizing antibodies produced in response to infection to the primary infection are thought to facilitate entrance of the heterologous virus into host macrophages. Although cases of DHF and DSS may result from this “immune enhancement,” severe DHF and DSS also occur with other serotypes in the absence of previous infection with a heterologous dengue virus serotype.60,61 Pathologic studies of DHF and DSS show hemorrhage, congestion, and perivascular edema of multiple organs. The liver may show areas of focal necrosis. As with yellow fever, the extent of pathologic findings does not correspond to severity of the clinical course.
Ecology and Epidemiology
A. aegypti is the principal vector for dengue viruses worldwide. In the Americas and Asia, viral transmission is maintained through a mosquito–human cycle without a major animal reservoir. Monkey carriers have been identified in Africa and Asia, but their importance in transmission is unclear. A. albopictus, an anthropophilic dengue vector from Southeast Asia, has also been recognized recently in the western hemisphere. Both of these mosquitoes are capable of large-scale transmission to humans in endemic areas. Currently, dengue is endemic in tropical and subtropical Asia, Africa, South America, and the Caribbean basin. In the early 1920s, large epidemics occurred in Texas, where dengue infections were reported in 500,000 inhabitants. In the last 30 years, endemic transmission on the mainland United States has been documented only in Texas. Travelers to Southeast Asia have highest risk for dengue, especially when travel takes place during periods of high transmission and epidemic dengue.98
Clinical Presentation
Most dengue infections appear after an incubation period of 2 to 14 days, either as an undifferentiated viral syndrome with fever and mild respiratory or gastrointestinal symptoms or as dengue (“break-bone”) fever with bone pain, generalized myalgia, severe headache, and retroorbital pain. Febrile illnesses that appear more than 2 weeks after putative exposure to dengue virus are unlikely to be due to this virus. After 1 to 3 days, a quiescent period may ensue. There may be a subsequent second episode of fever accompanied by a patchy maculopapular or morbilliform rash that spreads outward from the chest and that ultimately desquamates. Lymphadenopathy and leukopenia occur during this phase of the illness. The distinct severe forms of dengue disease referred to as either DHF or DSS may occur around the usual time of recovery. These are due to the development of capillary leak syndrome with associated hemorrhagic manifestations (Figure 85-1). The advanced forms have the unique feature that the platelet count decreases to less than 100,000 per mm3 and hematocrit increases by more than 20%. The severity is classified as grade I to IV, according to World Health Organization guidelines. In cases of grade I DHF, the only hemorrhagic manifestation is a positive tourniquet test, in which inflation of a tourniquet to midway between systolic and diastolic blood pressure for 5 minutes leads to development of 20 or more petechiae per square inch distal to the tourniquet. A complete blood cell count classically shows decreased platelet and leukocyte counts and increase in hematocrit value. Grade II DHF is defined as the above with hemorrhage from any site (e.g., gingiva, nares, conjunctivae). Grade III DSS includes clammy skin, hypotension, or a narrow pulse pressure (<20 mm Hg) in a patient with DHF. An undetectable blood pressure defines grade IV DHF and DSS. Most studies have noted DHF and DSS primarily in infants and young children, usually with a history or serologic evidence of previous heterologous dengue infection, but there is an increasing trend of cases in adults.
Lassa Fever
Epidemiology
The principal animal host for this virus is a rat, Mastomys natalensis, which prefers living in and around human dwellings. The rodents become chronically infected, secreting viral particles for long periods. Natural infection in humans occurs after rodent contamination of food and drink, inhalation of aerosolized rodent secretions, or contact with rodent material through skin abrasions. Lassa fever has been reported in several areas of sub-Saharan West Africa, and large outbreaks have been noted in Nigeria, Sierra Leone, Guinea, and Liberia.21,62 It affects up to 500,000 people with 5000 deaths annually.54 Complete seroprevalence data are lacking, making definition of an endemic area impossible at this time. Secondary human infection has been reported and may occur after contact with infected secretions.
Virology and Pathophysiology
Lassa virus is a single-stranded RNA arenavirus. Proliferation and dissemination presumably occur after initial replication at the inoculation site. As with the flaviviral diseases, the extent of end-organ involvement noted at autopsy does not account for the rapid death of infected patients. Recent work in an animal model provides evidence for platelet dysfunction and an endothelial cell defect in shock caused by Lassa fever virus.31 DIC, believed to be a major cause of bleeding and death in patients with other viral hemorrhagic fevers, appears to play a relatively minor role in arenavirus infections. The liver is most consistently the organ in which pathologic changes are observed at autopsy.
Clinical Presentation
Most seroconversions to Lassa virus are not accompanied by obvious symptoms.63,64,69 Only 5% to 14% of seroconverters experienced a febrile illness. The incubation period is between 3 and 21 days. Patients hospitalized with Lassa fever show a distinct clinical syndrome. Fever, malaise, and purulent pharyngitis often develop after the insidious onset of headache. Retrosternal chest pain, possibly a result of pharyngitis and esophagitis, suggests the diagnosis. The combined presence of retrosternal chest pain, fever, pharyngitis, and proteinuria is the best predictor of Lassa fever.62 Hemorrhagic complications (hematemesis, vaginal bleeding, hematuria, lower gastrointestinal bleeding, and epistaxis) were seen in fewer than 25% of patients with Lassa fever. Nonfatal disease usually begins to resolve in 8 to 10 days. The combined presence of fever, sore throat, and vomiting was associated with a poor prognosis (relative risk for death = 5.5). Terminal stages of fatal disease were accompanied by hypotension, encephalopathy, and respiratory distress caused by stridor (presumably secondary to laryngeal edema). The most common complication after recovery from Lassa fever is sensorineural hearing loss, presumably due to host immune response reactions against elements of the inner ear.
Management
The probability of transmission of Lassa fever virus to medical and nursing staff can be reduced by routine blood and body fluid precautions as well as strict barrier nursing. Barrier nursing includes wearing gloves, gown, mask, shoe covers, and, if there is risk for splashing fluids, goggles whenever entering the patient’s room. Decontamination of solid articles and rooms may be accomplished with 0.5% sodium hypochlorite solution. Recommendations for the management of patients with viral hemorrhagic fever have been published.17,11
Ebola and Marburg Viruses
Ebola and Marburg viruses are closely related large-RNA viruses known as filoviruses. They cause severe viral hemorrhagic fever syndromes with some of the highest case fatality rates (approximately 90%) of any known infectious disease. Both are endemic in focal areas of central and southern Africa.73 Ebola virus seropositivity has been noted in Sudan, Democratic Republic of the Congo, the Central African Republic, Côte d’Ivoire, and Kenya. A strain of Ebola known as Ebola Reston has been found in monkeys imported into the United States from the Philippines. More recently, there have been outbreaks with fatalities in Gabon, the Democratic Republic of Congo, and Angola. Marburg disease is found in South Africa, Zimbabwe, and Kenya. In 2005, there was an outbreak that caused over 300 deaths in Angola.18 Although there is not definitive evidence indicating the animal reservoir that maintains these filoviruses in nature, current evidence strongly suggests that bats are involved. Person-to-person transmission has been well documented, primarily through contaminated needles and contact with the secretions of infected individuals.72,74
Pathophysiology and Clinical Presentation
Marburg and Ebola viruses are presumed to act through similar pathophysiologic mechanisms that involve initial infection of monocytes, macrophages, and dendritic cells that are then distributed through the circulation to many organs and cell types. The viruses suppress both innate and adaptive host immune responses, leading to overwhelming infection and wide release of proinflammatory cytokines and chemokines causing fever, vascular instability, hypotension, and shock followed by multiorgan failure and death.7,22,41,57
Patients present after an incubation period of 4 to 10 days with fever, headache, and myalgias. Diarrhea and abdominal pain occur commonly. In many victims, rash, conjunctivitis, sore throat, and chest pain appear early in the disease. As in other hemorrhagic fevers, hemorrhage, hypotension, shock, and electrolyte abnormalities mark fatal courses. The high mortality reported in various outbreaks and transmission to health care workers taking care of patients emphasizes the importance of intensive supportive care and precautions that limit contact with body secretions of infected individuals.72
Diagnosis and Treatment
If these diseases are suspected, strict isolation procedures should be instituted and the local health authorities and the CDC notified immediately. Diagnosis may be made on a serologic basis or by polymerase chain reaction (PCR). There appears to be no serologic cross reactivity between the two viruses. Although anecdotal reports suggest the efficacy of immune sera in therapy, this has not been consistently observed in experimental studies. There are currently no specific antiviral therapies for Marburg or Ebola virus infection. Care is supportive. Vaccines are in development with one in Phase I testing.7,47,60,85
Crimean-Congo Hemorrhagic Fever
Virology and Epidemiology
The etiologic agent of Crimean-Congo hemorrhagic fever (CCHF) is a bunyavirus. Ixodid ticks serve as both reservoirs and vectors of the virus. Infection in humans results from tick bites or direct contact with infected secretions from crushed ticks, animals, or humans. Most cases occur in individuals with occupations or living conditions that bring them in contact with domestic goats, sheep, or cattle on which ticks feed. The disease has been observed in southeastern Europe, south central Asia, the Middle East, and much of Africa.95 Nosocomial transmission through contact with infected body fluids has been well documented.32,89,90,91
Pathophysiology and Clinical Presentation
Pathophysiologic mechanisms are presumably similar to those of other hemorrhagic fevers.28 One in five infections results in clinical disease with a case fatality rate ranging from 10% to 50%. The incubation period is approximately 1 week with initial symptoms of fever, severe headache, myalgias, vomiting, and diarrhea. Various forms of hemorrhage, including petechiae, large ecchymoses, melena, and hematemesis, are more pronounced in CCHF than in other hemorrhagic viral diseases. Severe cases progress rapidly to DIC, shock, and death.
Management
Initial management is similar to those for Lassa, Marburg, and Ebola virus infections, with strict patient isolation and notification of health authorities. Supportive therapy with attention to fluid balance and electrolytes in addition to oxygenation and hemodynamic support is the primary treatment. Although not confirmed in clinical trials, ribavirin has good activity in vitro against CCHF virus. The CDC recommends that patients believed to have CCHF receive intravenous (IV) ribavirin in the doses suggested for treatment of Lassa fever.27 Persons in contact with CCHF patients should receive prophylactic ribavirin as suggested for Lassa fever contacts. To date, almost all therapy has used the oral form of ribavirin.
Hemorrhagic Fever with Renal Syndrome and Hantavirus Pulmonary Syndrome
Hantaviruses, when transmitted from rodent reservoirs, cause two significant human diseases, hemorrhagic fever with renal syndrome (HFRS) in Asia and Europe, and hantavirus pulmonary syndrome (HPS) in the Americas. HFRS first came to the attention of Western medical science during the Korean conflict, when febrile illness accompanied by bleeding and renal failure developed in 3000 United Nations troops and was ultimately found to be caused by the hantavirus species Hantaan virus.40 Mortality ranged from 5% to 10%. A similar, less severe syndrome (nephropathia epidemica) had been recognized in Scandinavia since the 1930s. HPS was first recognized in a cluster of deaths in the southwestern United States in 1993. A nonspecific febrile illness is followed by shock and alveolar pulmonary edema caused by the hantavirus species Sin Nombre virus.23
Epidemiology
Hantaviruses cause chronic, nondebilitating infections of various rodent species. Human infection is initiated by contact with rodent secretions or inhalation of aerosolized rodent material. The disease occurs most commonly in rural areas, although occasional urban outbreaks occur, presumably with the common house rat as vector. Cases have been described most often from Asia, including China, Korea, Japan, and the Soviet Union, but the disease also occurs in Eastern Europe. A recent epidemiologic study from China found the highest rates of infection in men who engaged in heavy farm work and slept on the ground (rather than on raised wooden beds).99 The Sin Nombre virus appears to cause chronic infection of the deer mouse, Peromyscus maniculatus, which is the main reservoir of the virus in the United States. Since the initial outbreak, additional cases have been described across the United States and South America. The risk for infection is likely to be related to rodent exposure, but transmission is infrequent.
Management
Care of patients with HFRS is supportive. With HFRS, renal dysfunction occurs early and may require institution of dialysis soon after diagnosis to prevent fluid overload and to correct electrolyte disturbances. Patients’ secretions should be handled with care, and enteric precautions (but not strict isolation) are prudent. It is not clear whether person-to-person transmission of the virus through direct inoculation occurs. For the hantaviruses, viremia recedes and antibody levels rise as the clinical phase appears. Accordingly, nosocomial transmission or hematogenous transmission with hantavirus infections has not been frequently documented, although presumed nosocomial transmission has been reported.99 No vaccine is available.
Japanese B Encephalitis
Epidemiology
JE is the most common cause of encephalitis in Asia. Of the estimated 35,000 to 50,000 cases annually, 20% to 30% of infected individuals die and of those that recover, 30% to 50% have neurologic sequelae.30,46 Transmission correlates with monsoon rains in the tropics and in the summer and fall seasons in temperate regions. Rice field–breeding and other culicine mosquitoes serve as the vectors. In addition to humans, birds and pigs can be infected. Pigs play an important role as amplifying hosts because they develop high-grade viremia from which large numbers of mosquitoes may be infected. Most infections in endemic areas occur in children, whereas all age-groups of previously unexposed populations are at risk. Transmission of JE currently occurs in India, Southeast Asia, China, Korea, Indonesia, and the Western Pacific region.30 Routine use of JE vaccine in Japan has been eliminated because of low risk in this country. Recent outbreaks and case reports of JE in islands of the Torres Strait, which runs between Northern Australia and Papua New Guinea, indicate that the virus spread southward from Asia, presumably by migratory ardeid birds.39
Clinical Presentation
Incubation period is typically 2 to 15 days. Most infections do not cause clinical illness. Many patients recall a mild undifferentiated febrile illness, which probably coincides with the viremic phase of infection. Patients with encephalitis often report a similar prodrome. The encephalitis syndrome is not easily distinguished from other arboviral encephalitides. The patient usually complains of headache, lethargy, fever, and confusion and may display tremors or seizures. One clinical series suggested that the presence on admission of (1) unresponsiveness to pain, (2) low levels of anti–Japanese B encephalitis virus IgG or IgM antibodies (in serum or cerebrospinal fluid [CSF]), or (3) virus in CSF culture was associated with death. Of the 16 patients with fatal disease, all died within 7 days of hospitalization.15
Named Hepatitis Viruses
Hepatitis A
Epidemiology
Hepatitis A virus is transmitted primarily by the fecal–oral route by either person-to-person contact or ingestion of contaminated food or water. Food items commonly associated with outbreaks are raw or undercooked clams and shellfish. Risk factors include contact with a hepatitis A–infected person, international travel, household or personal contact with a child who attends a child care center, foodborne outbreaks, male homosexual activity, and use of illegal drugs.3,59 Occasional cases are associated with exposure to nonhuman primates. Transmission by blood transfusion has been reported, but this is an uncommon source of infection. Hepatitis A is endemic worldwide, but underdeveloped nations have a higher prevalence than those in North America. Most persons in these areas show serologic evidence of past infection with hepatitis A virus. Hepatitis A is a common viral infection occurring in travelers, but rates are declining with increased use of hepatitis A vaccine
