Chapter 21 Neurotransmitters and Drug Abuse
Monoamines
Dopamine
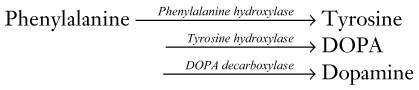
Anatomy
Three “long dopamine tracts” hold the greatest clinical importance in neurology:
1. The nigrostriatal tract, the major component of the extrapyramidal motor system, synthesizes most dopamine in the brain. This tract projects from the substantia nigra, the crescentic pigmented nuclei in the midbrain (Fig. 21-1), to the predominantly D2 receptors of the striatum (the caudate nucleus and putamen [see Chapter 18]).
2. The mesolimbic tract projects from the ventral tegmental area, situated in the inferior medial portion of the midbrain, to the amygdala and other portions of the limbic system. Its receptors are predominantly D4. The mesolimbic tract appears to propagate the positive symptoms of psychosis, whereas antipsychotic agents, by blocking dopamine transmission in this tract, reduce dopamine activity in the limbic system and thus suppress psychotic symptoms.
3. The mesocortical tract also projects from the ventral tegmental area, but it terminates primarily in the frontal cortex. It also terminates in the cingulate and prefrontal gyrus, creating an overlap with the mesolimbic system. The mesocortical tract likely propagates negative symptoms of psychosis. In addition, because illicit drugs affect both the mesolimbic and mesocortical tracts, these tracts serve as the neural substrates for drug addiction.
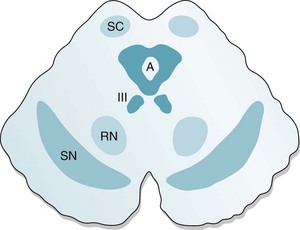
FIGURE 21-1 Coronal view of the midbrain, which gives rise to several dopamine-producing tracts – including the nigrostriatal, mesolimbic, and mesocortical. The nigrostriatal tract begins in the substantia nigra (SN), the large curved black structures in the base of the midbrain (see Fig. 18-2). The red nuclei (RN), which receive cerebellar outflow tracts, sit above the substantia nigra. The upper portion of the midbrain, the tectum (Latin, roof; tego, to cover), contains the aqueduct of Sylvius (A), which is surrounded by the periaqueductal gray matter, and the superior colliculi (SC). The oculomotor nuclei (III), which give rise to the third cranial nerves, lie midline, just below the aqueduct.
Receptors
The D1 and D2 receptors remain the most important in extrapyramidal system disorders (Table 21-1). Both types of these receptors are plentiful in the striatum, but D1 receptors are more abundant and more widely distributed.
| D1 | D2 | |
|---|---|---|
| Effect of stimulation on cyclic AMP production | Increased | Decreased |
| Greatest concentrations | Striatum, limbic system, and cerebral cortex | Striatum, substantia nigra |
| Effect of dopamine | Weak agonist | Strong agonist |
| Effects of dopamine agonists | Vary with agonist | Strong |
| Effect of phenothiazines | Strong antagonist | Strong antagonist |
| Effect of butyrophenones | Weak antagonists | Strong antagonists |
| Effect of clozapine | Weak antagonist | Weak antagonist |
When antipsychotics block D2 receptors, the reduced dopamine activity induces parkinsonism, raises prolactin production (inducing galactorrhea), and places patients at risk for tardive dyskinesia. Some atypical antipsychotic agents, particularly risperidone and its active metabolite paliperidone (Invega), raise prolactin serum concentration and induce galactorrhea. Even nonpsychiatric medications that decrease dopamine activity, such as metoclopramide (Reglan), which blocks D2 receptors, and tetrabenazine (see Chapter 18), which depletes dopamine from its presynaptic storage vesicles, increase prolactin serum concentration and induce parkinsonism. In contrast, clozapine, which shows little D2 receptor affinity, does not lead to either of these problems.
Physicians should bear in mind that finding an elevated serum prolactin level does not always indicate use of a dopamine-blocking medication or the presence of a pituitary tumor (see Chapter 19). The most common cause of serum prolactin elevation is pregnancy.
Conditions due to Reduced Dopamine Activity
Dopamine-Responsive Dystonia.
L-dopa, even in small amounts, inserts itself into the synthetic pathway. It bypasses the enzyme deficiencies and allows the nigrostriatal neurons to synthesize enough dopamine to reverse the symptoms (see Chapter 18). Small doses of L-dopa restore normal mobility throughout the entire day. The response to L-dopa is so reliable that it serves as a preliminary diagnostic test for DRD.
Parkinson Disease.
1. As a first-line treatment, they administer the dopamine precursor L-dopa. As long as enough nigrostriatal (presynaptic) neurons remain intact, DOPA decarboxylase converts L-dopa to sufficient quantities of dopamine to reverse the symptoms (see Fig. 18-3).
2. As the disease progresses, the presynaptic neurons degenerate beyond the stage where they can synthesize, store, and appropriately release dopamine. At this time, if not as a first-line treatment, neurologists add a dopamine receptor agonist to the medication regimen. As a substitute or supplement for L-dopa, dopamine agonists, such as pramipexole, ropinirole, and apomorphine, stimulate dopamine receptors. That stimulation replicates dopamine’s effect and alleviates symptoms.
3. As long as dopamine synthesis continues, neurologists prescribe medicines that slow L-dopa metabolism in the periphery but allow DOPA to continue forming dopamine centrally. Working in that way, two Parkinson disease medications – carbidopa and entacapone – inactivate enzymes that metabolize L-dopa and thereby enhance dopamine activity (see Fig. 18-11). Carbidopa inactivates DOPA decarboxylase. Entacapone inhibits COMT, which normally inactivates L-dopa by converting it to 3-O-methyldopa. Both enzyme inhibitors act almost entirely outside the CNS because they have little ability to penetrate the blood–brain barrier. Therefore, they do not interfere with basal ganglia dopamine synthesis. Giving these enzyme inhibitors along with L-dopa enables small doses of L-dopa to be effective. Moreover, the small doses of dopamine reduce its systemic side effects, such as nausea, vomiting, cardiac arrhythmias, and hypotension.
Neuroleptic-Malignant Syndrome.
An acute absence of dopamine activity causes the parkinson-hyperpyrexia or central dopaminergic syndrome, which neurologists still call the neuroleptic-malignant syndrome (NMS) (see Chapter 6). Administering dopamine agonists may compensate for the absence of dopamine activity and alleviate some of the symptoms, but treatment is generally supportive. Patients usually require parenteral treatment, such an intramuscular injections of the dopamine agonist apomorphine, because the severe muscle rigidity prevents their swallowing pills and the urgency of the situation demands treatment with rapid onset of action.
Conditions due to Excessive Dopamine Activity
As a less dramatic example of the effects of excessive dopamine, some Parkinson disease patients become overly involved with stimulating activities, such as sex and gambling. In these cases, neurologists diagnose the dopamine dysregulation syndrome or impulse control disorder (see Chapter 18). They usually ascribe the aberrant behavior to dopamine-induced novelty seeking and inattention.
Norepinephrine and Epinephrine
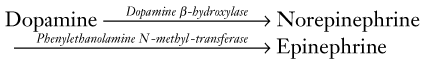
Anatomy
CNS norepinephrine synthesis takes place primarily in the locus ceruleus, which is located in the dorsal portion of the pons (Fig. 21-2). Neurons from the locus ceruleus project to the cerebral cortex, limbic system, and reticular activating system. In addition, whereas dopamine tracts remain confined to the brain, norepinephrine tracts project down into the spinal cord.
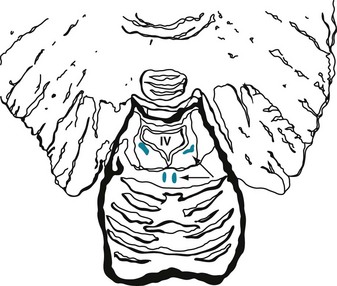
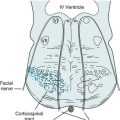
FIGURE 21-2 Coronal view of the pons, fourth ventricle (IV), and cerebellum. This portion of the brainstem contains paired locus ceruleus (diagonal arrow) and dorsal raphe nucleus (horizontal arrow). The locus ceruleus, which sits lateral and inferior to the fourth ventricle, gives rise to norepinephrine tracts. The dorsal raphe nucleus gives rise to serotonin tracts that spread cephalad, to the diencephalon and cerebrum. A caudal raphe nucleus, in the pons and medulla (not pictured), gives rise to serotonin tracts that spread to the spinal cord.
Receptors
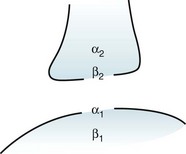
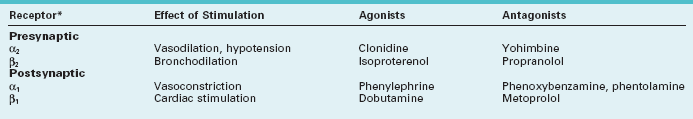
Norepinephrine receptors are located in the cerebral cortex, brainstem, and spinal cord. The α2 and β2 receptors, termed “autoreceptors,” are situated on presynaptic neurons. Through a feedback mechanism, these presynaptic receptors modulate norepinephrine synthesis and release (Fig. 21-3). Postsynaptic receptors are also of two varieties, α1 and β1, and they produce varied and sometimes almost opposite effects (Table 21-2).
Conditions due to Alterations in Norepinephrine Activity
In the opposite situation, excessive stimulation of β2-adrenergic sites leads to tremor and bronchodilatation. For example, isoproterenol and epinephrine, particularly when used for asthma, cause tremor as they alleviate bronchospasm. Conversely, β-blockers, which are contraindicated in asthma patients, suppress essential tremor (see Chapter 18).
Serotonin
Anatomy
Serotonin-producing CNS neurons reside predominantly in the dorsal raphe nuclei, which are located in the midline of the dorsal midbrain and pons (see Figs 21-2 and 18-2). Serotonin tracts project rostrally (upward) to innervate the cortex, limbic system, striatum, and cerebellum. They also innervate intracranial blood vessels, particularly those around the trigeminal nerve.
Other serotonin-producing nuclei, the caudal raphe nuclei, are located in the midline of the lower pons and medulla. They project caudally (downward) to the dorsal horn of the spinal cord to provide analgesia (see Chapter 14).
Conditions due to Alterations in Serotonin Activity
Serotonin plays a major role in the daily sleep–wake cycle. The activity of serotonin-producing cells reaches its highest level during arousal, drops to quiescent levels during slow-wave sleep, and disappears during rapid eye movement (REM) sleep (see Chapter 17).
Although increased serotonin activity is often therapeutic, excessive activity poses a danger. For example, combinations of medicines that simultaneously block serotonin reuptake and inhibit its metabolism lead to serotonin accumulation. These combinations may cause toxic levels and the serotonin syndrome (see Chapters 6 and 18).
Acetylcholine
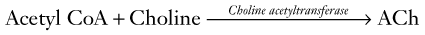
Anatomy
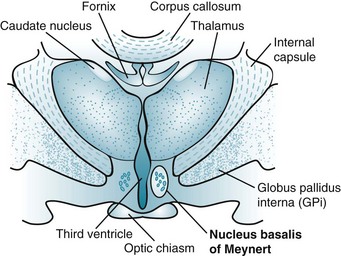
In the CNS, most ACh tracts originate in the nucleus basalis of Meynert (also known as the substantia innominata) and adjacent nuclei situated in the basal forebrain (a rostral portion of the brainstem) (Fig. 21-4). These nuclei project their cholinergic tracts throughout the cerebral cortex but particularly to the hippocampus, amygdala, and cortical association areas. In addition, ACh serves as the autonomic nervous system and neuromuscular junction neurotransmitter (see Fig. 6-1).
Receptors
ACh receptors fall into two categories, nicotinic and muscarinic. One or the other predominates in either the cerebral cortex or the neuromuscular junction, produces excitatory or inhibitory actions, and remains vulnerable to different blocking agents (Table 21-3).
TABLE 21-3 Cerebral Cortex and Neuromuscular Junction Acetylcholine (ACh) Receptors
| Cerebral Cortex | Neuromuscular Junction | |
|---|---|---|
| Predominant ACh receptors | Muscarinic | Nicotinic |
| Main action | Excitatory or inhibitory | Excitatory |
| Agents that block receptor | Atropine, scopolamine | Curare, α-bungarotoxin |
Conditions due to Reduced ACh Activity at the Neuromuscular Junction
In the PNS, decreased neuromuscular ACh activity – from either impaired presynaptic ACh release or blockade of postsynaptic ACh receptors – leads to muscle paralysis. Conditions that interfere with ACh activity have different etiologies and induce distinct patterns of weakness. For example, in Lambert–Eaton syndrome, the paraneoplastic disorder, antibodies impair the release of ACh from presynaptic neurons and cause weakness of limbs (see Chapters 6 and 19). Botulinum toxin also impairs ACh release from the presynaptic neuron; when ingested as a food poison (botulism), it primarily causes potentially fatal weakness of ocular, facial, limb, and respiratory muscles. When injected into affected muscles for treatment of focal dystonia, medicinal botulinum toxin inhibits forceful muscle contractions because it slows or prevents ACh release from the presynaptic neuron at the neuromuscular junction (see Chapter 18).
At the postsynaptic neuron of the neuromuscular junction, curare, other poisons, and antibodies, such as those associated with myasthenia gravis, each block ACh receptors. The pattern of weakness in myasthenia gravis is distinctive: Patients have asymmetric paresis of the extraocular and facial muscles, but not the pupils. To overcome the ACh receptor blockade in myasthenia gravis, neurologists administer anticholinesterase (the common contraction of antiacetylcholinesterase) medications, such as edrophonium (Tensilon), pyridostigmine, and physostigmine, to inhibit cholinesterase and thereby reduce the breakdown of ACh in the synaptic cleft (see Chapter 6 and Fig. 7-6). The reliability of edrophonium in temporarily reversing myasthenia-induced paralysis has led to the “Tensilon test” (see Fig. 6-4).
Conditions due to Reduced ACh Activity in the CNS
In Alzheimer disease, the cerebral cortex has markedly reduced cerebral ACh concentrations, ChAT activity, and muscarinic receptors (see Chapter 7). In addition, some nicotinic receptors are depleted.
To counteract the ACh deficiency in Alzheimer disease, neurologists have attempted several strategies to enhance its synthesis or slow its metabolism. In hopes of driving ACh synthesis, they have administered precursors, such as choline and lecithin (phosphatidylcholine). Although analogous to providing a dopamine precursor (L-dopa) in Parkinson disease treatment, this strategy failed in reversing the symptoms of Alzheimer disease. A complementary strategy has been to slow ACh metabolism by administering long-acting cholinesterase inhibitors that cross the blood–brain barrier. Several such cholinesterase inhibitors, such as donepezil, may slow the progression of dementia in Alzheimer disease and several other illnesses (see Chapter 7).
Anticholinergic Syndrome
Chlorpromazine, other typical antipsychotic agents, and tricyclic antidepressants may block muscarinic ACh receptors and cause physical anticholinergic side effects, including drowsiness, dry mouth, urinary hesitancy, constipation, and accommodation paresis (see Chapter 12). Anticholinergic activity may also rise in individuals taking nonpsychiatric medicines, such as atropine, scopolamine, benztropine, and trihexyphenidyl. With enough anticholinergic activity, individuals develop the anticholinergic syndrome: dilated pupils, elevated pulse and blood pressure, dry skin and hyperthermia, and delirium that may progress to coma. In this situation physicians may administer physostigmine, an anticholinesterase that crosses the blood–brain barrier, to restore ACh activity.
Neuropeptides
Gamma-Aminobutyric Acid – An Inhibitory Amino Acid Neurotransmitter

Conditions due to Alterations in GABA Activity
In a different situation, flumazenil, a benzodiazepine antagonist, blocks the actions of GABA at its receptor and reverses benzodiazepine-induced stupor and hepatic encephalopathy. In the case of hepatic encephalopathy, flumazenil presumably displaces false, benzodiazepine-like neurotransmitters from GABA receptors (see Chapter 7).
Glycine – Another Inhibitory Amino Acid Neurotransmitter

Conditions due to Alterations in Glycine Activity
Reduced glycine activity characterizes tetanus and strychnine poisoning. In both conditions, loss of inhibitory activity causes patients to suffer from unrelenting powerful muscle contractions. In tetanus (see Chapter 6), the toxin (tetanospasmin) prevents the presynaptic release of glycine and GABA. Due mostly to the loss of glycine-induced muscle inhibition, tetanus patients suffer “tetanic contractions” of limb, facial, and jaw muscles, typically manifest as “lockjaw.” In a different mechanism of action but with similar results, strychnine blocks the postsynaptic glycine receptor. Strychnine poisoning leads to fatal limb, larynx, and trunk muscle contractions.
Glutamate – An Excitatory Amino Acid Neurotransmitter

Conditions due to Alterations in NMDA Activity
Excessive NMDA activity floods the neuron with potentially lethal concentrations of calcium and sodium in several disorders. Through this process, excitotoxicity, glutamate–NMDA interactions lead to neuron death through apoptosis. Excitotoxicity may be intimately involved in the pathophysiology of epilepsy, stroke, neurodegenerative diseases (such as Parkinson and Huntington diseases), head trauma, and, conceivably, schizophrenia. In certain paraneoplastic syndromes, particularly one triggered by ovarian teratomas in young women, antibodies directed toward NMDA receptors create an autoimmune limbic encephalitis (see Chapter 19).
Other Neuropeptides
Endorphins, enkephalins, and substance P, which are situated in the spinal cord and brain, provide endogenous analgesia in response to painful stimuli (see Chapter 14). Substance P and, to a lesser degree, other neuropeptides are depleted in Alzheimer disease. The other neuropeptides that are reduced include somatostatin, cholecystokinin, and vasoactive intestinal peptide.
A pair of neuropeptides, hypocretin-1 and -2, also known as orexin A and B, are excitatory neurotransmitters that play a crucial – but an ill-defined – role in locomotion, metabolism, appetite, and the sleep–wake cycle (see Chapter 17). Synthesized in the hypothalamus, where they are cleaved from a large precursor protein, these neuropeptides increase during wakefulness and REM sleep, and decrease during non-REM sleep. Unlike normal individuals, narcolepsy patients have little or no hypocretin in their CSF and their hypothalamus is devoid of hypocretin-producing cells.
Nitric Oxide
Synthesis and Metabolism
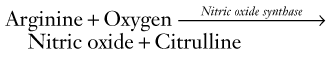
Conditions due to Alterations in NO Activity
The best-known role of NO is in generating erections (see Chapter 16). With sexual stimulation of the penis, parasympathetic neurons produce and release NO. NO then promotes the production of cyclic guanylate cyclase monophosphate (cGMP), which dilates the vascular system and creates an erection. Sildenafil slows the metabolism of cGMP and increases blood flow in the penis, which, in turn, produces, strengthens, and prolongs erections.
Neurologic Aspects of Drug Abuse
Cocaine
Clinical Effects
The immediate effects of cocaine typically consist of a short period of intense euphoria accompanied by a sense of increased sexual, physical, and mental power. Individuals using cocaine remain fully alert – even hypervigilant. Their heightened awareness is a prominent exception to the general rule that patients in delirium or a toxic-metabolic encephalopathy are lethargic or stuporous and have a fluctuating level of consciousness (see Chapter 7). In contrast to the miosis produced by opioids, particularly heroin, cocaine users have dilated pupils.
Cocaine, like other stimulants, not only reduces sleep time, it suppresses REM sleep, often to the point of eliminating it. Another characteristic effect follows individuals’ discontinuing cocaine: They experience a rebound in REM sleep as if to compensate for the loss of REM during intoxication (see Chapter 17).
Overdose
In contrast, use or even an overdose of barbiturates, benzodiazepines, or alcohol rarely causes strokes or seizures. Withdrawal from any of these substances, however, frequently precipitates not only seizures but also status epilepticus (see Chapter 10).
Probably because cocaine suddenly increases dopamine activity, its use also produces involuntary movements, such as tic-like facial muscle contractions, chorea, tremor, dystonia, and repetitive, purposeless behavior (stereotypies; see Chapter 18). Cocaine also exacerbates tics in individuals with Tourette or other tic disorder. When cocaine induces chorea, patients’ legs and feet move incessantly, and they cannot stand at attention. Neurologists label these involuntary movements “crack dancing,” and point out that they mimic chorea, restless legs syndrome, akathisia, and undertreated agitation.
Treatment of Intoxication and Overdose
Behavioral and cognitive aspects of cocaine overdose generally resolve spontaneously. Rest, seclusion, and, if necessary, benzodiazepines will moderate agitation and related symptoms. Dopamine-blocking antipsychotic agents will reduce or eliminate psychosis and violent behavior; however, because some neuroleptics, as well as cocaine itself, may lower the seizure threshold, physicians must choose and administer them judiciously. Potentially life-threatening hypertension, another hazard of cocaine, may require aggressive treatment with an α-blocker antihypertensive medication, such as phentolamine (see Table 21-2).
Opioids
Pharmacology
Opioid is a broad term encompassing medical narcotics, “street” narcotics, and endogenous opiate-like substances (see endorphins, Chapter 14). Although opioids may inhibit the reuptake of monamines and affect other neurotransmitters, their primary action is directly on a group of specific opioid receptors that are situated in the brain and spinal cord. Of the several opioid receptors, the mu receptors mediate opioid-induced euphoria.
Overdose
Opioid overdose causes a characteristic triad of coma, miosis, and potentially fatal respiratory depression. The respiratory depression takes the form of slowed rate rather than shallower depth of breathing, which often causes neurogenic pulmonary edema. The coma routinely leads to nerve compression from prolonged, static positioning of the body (see Chapter 5). In contrast to the complications of stimulants, opioid-induced strokes or seizures rarely occur.
Gamma-hydroxybutyrate
Nevertheless, sodium oxybate has legitimate use in the treatment of narcolepsy with cataplexy (see Chapter 17), where it has approved indications for treatment of cataplexy and excessive daytime sleepiness. Its benefits likely stem, in large part, from its ability to increase restful slow-wave sleep. Physicians and pharmacies must monitor prescriptions and distribution for sodium oxybate because of its potential nefarious use.
Benarroch EE. GABAB receptors. Neurology. 2012;78:578–584.
Hilker R, Thomas AV, Klein JC, et al. Dementia in Parkinson’s disease: Functional imaging of cholinergic and dopaminergic pathways. Neurology. 2005;65:1716–1722.
Hshieh TT, Fong TG, Marcantonio ER, et al. Cholinergic deficiency hypothesis in delirium: A synthesis of current evidence. J Gerontol. 2008;63A:764–772.
McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: Clinical course, treatments, and outcomes. Arch Neurol. 2012;69:230–238.
Stahl SM. Essential Psychopharmacology: Neuroscientific Basis and Practical Applications, 3rd ed. Cambridge: Cambridge University Press; 2008.
Baylen CA, Rosenberg H. A review of the acute subjective effects of MDMA/ecstasy. Addiction. 2006;101:933–947.
Benowitz NL. Nicotine addiction. N Engl J Med. 2010;362:2295–2303.
Derlet RW, Rice P, Horowitz BZ, et al. Amphetamine toxicity: Experience with 127 cases. J Emerg Med. 1989;7:157–161.
Dobbs MR. Clinical Neurotoxicology. Philadelphia: Saunders; 2009.
Hays JT, Ebbert JO. Varenicline for tobacco dependence. N Engl J Med. 2008;359:2018–2024.
Herning RI, Better W, Tate K, et al. Neuropsychiatric alterations in MDMA users. Ann NY Acad Sci. 2005;1053:20–27.
Honarmand K, Tierney MC, O’Connor, et al. Effects of cannabis on cognitive function in patients with multiple sclerosis. Neurology. 2011;76:1153–1160.
McCarron MM, Schulze BW, Thompson GA, et al. Acute phencyclidine intoxication: Incidence of clinical findings in 1,000 cases. Ann Emerg Med. 1981;10:237–242.
Penders TM, Gestring R. Hallucinatory delirium following use of MDPV: “Bath salts. Gen Hosp Psychiatry. 2011;33:525–526.
Schweinsburg AD, Brown SA, Tapert SF. The influence of marijuana use on neurocognitive functioning in adolescents. Curr Drug Abuse Rev. 2008;1:99–111.
Snead OC, Gibson KM. γ-hydroxybutyric acid. N Engl J Med. 2005;352:2721–2732.
Spivey WH, Euerle B. Neurologic complications of cocaine abuse. Ann Emerg Med. 1990;19:1422–1428.
Tait RJ, Mackinnon A, Christensen H. Cannabis use and cognitive function: 8-year trajectory in a young adult cohort. Addiction. 2011;106:2195–2203.
6. A former heroin addict has a history of epilepsy as a teenager. While attending his methadone maintenance program, he developed a flurry of seizures. Emergency room physicians responded and administered phenytoin (Dilantin). Although it controlled the seizures, the patient became agitated and combative. He also reported nausea and abdominal pain. A psychiatrist found piloerection and muscle cramps. Clinical chemistry, urine analysis, and hematology laboratory results were normal except for the presence of opiates or their metabolic products in the urine. Which is the best course of action?
25. Which two comparisons of Lambert–Eaton syndrome and myasthenia gravis are true?
57. Recalling that the neurologist explained that antiparkinson medicines cause nocturnal hallucinations, a patient’s family suddenly discontinued all his Parkinson disease medicines because he had developed severe nocturnal visual hallucinations. Rigidity returned on the first day. On the next day, when he became “stiff as a board,” febrile, and delirious, his family brought him to the emergency room. The medical staff found him stuporous, febrile, and rigid. While awaiting blood test results, which treatments should the staff administer in addition to providing supportive therapy, e.g., fluids, glucose, and electrolytes?
58. The police bring a young man to the psychiatric emergency room after he wandered through the streets saying that he took some medicine from a friend. He appears anxious, but he repeats that he feels “close to everyone.” His pupils have a normal diameter and reactivity to light. His eyes have full, conjugate movement without nystagmus. He grinds his teeth. Which is the most likely intoxicant?
a. GABAA receptors are more numerous and more widely distributed than GABAB receptors.
63. A 55-year-old bartender, who stated that he has smoked two packs of cigarettes each day but denied ever drinking alcohol, developed chronic obstructive lung disease and pneumonia for which his physician hospitalized him. After the pneumonia began to respond to treatment and his blood gas results started to return toward normal, he became cantankerous, then agitated, and sullen. When he demanded to be released, his intern called for neurology and psychiatry consultations. They determined that his physical neurologic examination, routine blood tests, and head CT were all normal. The patient insisted on a cigarette, but the medical staff would not allow him to smoke in his area of the hospital and they could not move him to a suitable location. Which would be the first, best treatment?
103. A 30-year-old man presented to a neurologist because of increasingly severe and prolonged low and middle back pain. When upright, he had hyperlordosis and limited range of movement about the low back and legs. He had begun to walk like a “tin soldier.” Extensive testing revealed antibodies to glutamic acid decarboxylase (GAD) in the serum and spinal fluid. What is the most likely diagnosis?