[level-membership-for-pediatrics-category]
Neurosurgical Conditions
Brain Tumors
Brain tumors are rare in the first year of life, with an incidence of 1 per 100,000 infants.1 This incidence increases with age. By the age of 2 years, central nervous system (CNS) tumors occur in 2 to 5/100,000 children. Brain tumors represent the most common solid tumors in the pediatric population.2
In the group of brain tumors considered congenital, the most common are teratomas (37%), followed by primitive neuroectodermal tumors (PNETs) (12%), astrocytomas (10–15%), and choroid plexus tumors (8%).3 In a slightly older population of children, teratomas become less frequent and other neoplasms become relatively more common, including astrocytomas (34% of brain tumors in children younger than 24 months of age), PNETs (23%), and ependymomas (11%). There is also a group of tumors that are sometimes considered non-neoplastic. These include developmental tumors, which derive from aberrant proliferative growth during embryonic brain development and include craniopharyngiomas, lipomas, dermoids, and colloid cysts.4
Cerebellar Astrocytomas
Low-grade astrocytomas occur throughout the CNS and constitute a fourth of all pediatric brain tumors. Cerebellar astrocytomas are common (12–17% of pediatric CNS tumors), and their treatment has the most favorable outcome of all intra-axial neoplasms in the CNS.5 Most of these tumors are found in children younger than the age of 10 years. The symptoms of cerebellar astrocytomas include headache (80%), vomiting (80%), gait disturbance, and decreased level of consciousness. Signs include ataxia in 80%, papilledema, cranial nerve palsies (including blindness and diplopia), and dysmetria. The rapid progression of signs and symptoms is often a function of cerebrospinal fluid (CSF) obstruction because the tumor occupies the fourth ventricle or the aqueduct, and causes hydrocephalus. Treatment of the hydrocephalus is often required urgently in very sick children. Temporary diversion of CSF via external ventriculostomies or shunts often precedes removal of the tumor.
Operative excision of these tumors is often possible, but new neurologic deficits can occur postoperatively in 30% of patients, although at least half of these new deficits are transient.6 Postoperative deficits occur because of the proximity of vital brain stem and delicate cranial nerve structures near the tumor, which is often large and firm. Postoperative pseudomeningoceles (the bulging of skin at the occipital incision site) and hydrocephalus are not uncommon (10–25%) and require additional treatment. Temporary continuation of postoperative CSF diversion via external ventriculostomy is favored by many surgeons to reduce the likelihood of permanent ventriculoperitoneal shunts.
Ependymomas
Ependymomas represent about 10% of pediatric brain tumors and are slightly less common than PNETs and astrocytomas. More than two-thirds of ependymomas in children occur in the posterior fossa (Fig. 19-1) and the symptoms they cause are similar to posterior fossa or cerebellar astrocytomas: headache, vomiting, lethargy, and ataxia. These symptoms result from a combination of compression along the dorsal brain stem and hydrocephalus. Many extend from the obex (in the inferior aspect of the fourth ventricle) and then extrude through the lateral opening of this ventricle (foramen of Luschka) into the cerebellopontine angle. Here they invade and compress cranial nerves with resulting facial weakness, diplopia, swallowing dysfunction, and hearing loss. When large, they often extend beyond the foramen magnum and can compress the cervical cord. In regard to their cell of origin, they are not limited to the ependymal layer of the ventricle but can arise from cerebellar, cerebral, or spinal cord parenchyma.
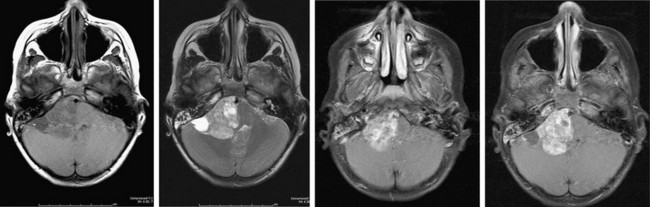
FIGURE 19-1 MR image of a posterior fossa ependymoma. The tumor involves the lateral aspect of the posterior fossa and contains calcifications typical of ependymomas. The brain stem is severely distorted by the mass effect of the tumor. The tumor also compresses cranial nerves on the right side, making complete tumor removal problematic. This patient had right facial weakness, diplopia, and mild left hemiparesis postoperatively.
Aggressive surgical resection has been the goal of treatment of these difficult tumors, but there is high probability of new or worsening neurologic deficits as a result of the operative dissection. There is a correlation between postoperative residual tumor and tumor recurrence and/or progression. In the 30% of children where the tumor is totally resected, there is still a 20% to 40% possibility of recurrence, even after conventional radiotherapy.7 In cases of near-total resection, the rate of progression-free survival falls to 30%.8 Most of the recurrences are local and radiation therapy has become the mainstay of treatment, even with complete resection. Very small children, usually younger than age 3 years, are faced with comparatively greater morbidity from conventional radiotherapy, and radiation therapy is often deferred. Chemotherapy has been used with some success in this group of patients. The role of chemotherapy without irradiation has generally not been favorable in older age groups. Retrospective studies have failed to prove substantial benefit in survival when chemotherapy is added to surgery and radiation therapy for newly diagnosed ependymomas.
Medulloblastomas
Medulloblastomas are the most common malignant solid tumor in children and constitute 20% of pediatric brain tumors.9 They are usually located in the posterior fossa and they are also referred to as PNETs because they are histologically identical to tumors (pineoblastomas, neuroblastomas, and retinoblastomas) located in other locations that are believed to have derived from progenitor subependymal neuroepithelial cells undergoing malignant transformation. Nearly half of medulloblastomas have chromosomal abnormalities, particularly the deletion of 17p chromosome that contains the tumor suppressor gene TP53.10
Hydrocephalus often occurs with medulloblastomas because of their location within the cerebellar vermis (a midline structure), often filling the fourth ventricle (Fig. 19-2). Children with this tumor often have symptoms of elevated intracranial pressure (ICP) (obtundation, headache, nausea/vomiting, irritability) and have signs suggestive of posterior fossa compression (dysmetria, ataxia, diplopia, head tilt, and papilledema). Lumbar puncture should not be done after computed tomography (CT) or magnetic resonance imaging (MRI) has established the presence of a posterior fossa tumor and obstructive hydrocephalus. CSF diversion (usually via an external drain) is usually done either before or in conjunction with craniotomy. Conversion of these temporary devices to permanent ventriculoperitoneal shunts is not uncommon in children with large tumors and marked preoperative ventriculomegaly.
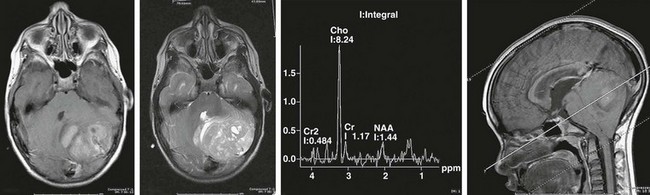
FIGURE 19-2 MRI and MRS (magnetic resonance spectroscopy) of a posterior fossa medulloblastoma, with compression of fourth ventricle. The MRS profile shows a choline peak with depression of N-acetyl aspartate (NAA) typical of aggressive tumors.
Complete resection of medulloblastomas is often possible, although permanent postoperative deficits can occur.6 The ‘posterior fossa syndrome’ can occur postoperatively in 10–15% of children and is characterized by mutism, drooling and swallowing difficulties, ocular palsies, and increasing ataxia.11 These problems resolve entirely in most patients after several months. Improvement is thought to occur with resolution of swelling within the inferior vermis.
Staging is important with medulloblastomas because patients have a predictable outcome depending on age, metastases, pathology, and extent of surgical resection. Poor survival is correlated with age younger than 4 years, residual tumor measuring more than 1.5 cm,2 and tumor dissemination, particularly ‘drop’ metastasis along the spinal column. After craniospinal radiation in eligible patients, survival occurs in 50–70% of patients with standard or low-risk medulloblastomas. Newer treatment protocols include chemotherapy first, which is followed by radiotherapy consisting of lowered cumulative craniospinal doses (24–36 Gy to the entire brain) with hyperfractionated delivery, usually 1 Gy twice daily. Survival in these ‘good risk’ patients is nearly 90% after five years. Survival in high-risk patients is 60–65% with current multimodality therapy, with the worst outcomes in affected infants. Children younger than 3 years of age are usually treated first with chemotherapy, with irradiation deferred for 1 to 2 years. Radiation is sometimes avoided altogether in the 40% with progression-free survival.12
Supratentorial Nonglial and Glial Neoplasms
Ependymomas that occur in the cerebral hemispheres remain problematic in terms of treatment, although hemispheric tumors generally have comparatively better outcomes. Incomplete resection (often because of diffuse involvement within critical brain regions) and leptomeningeal spread are significant adverse risk factors. Age is also a factor. Children younger than 3 years of age have significantly diminished progression-free survival (10–15%) compared with older children.13
As with medulloblastoma, glial and nonglial tumors often have genetic abnormalities, with chromosomal abnormalities and gene mutations. Many low-grade lesions progress to become more malignant. The pathway to this malignant progression is complex. Chromosomal translocations and mutations occur as initiating events before the amplification of deleterious genes that support tumor progression.14–16
With most benign tumors, there exists a strong association between extent of resection and outcome. From the neurosurgical viewpoint, maximal resection should be attempted without inordinate surgical morbidity. The advent of frameless stereotaxy for precise tumor localization, ‘functional’ localization with intraoperative monitoring (e.g., somatosensory evoked potential mapping to determine the location of the motor cortex), presurgical functional MRI to determine location of speech areas, and intraoperative scanning (via real-time ultrasonography [US], CT, or MRI) have each added considerably to the safety of the operation. Nonetheless, the operative approach can only achieve resection of targeted areas. Infiltrative tumors (which typically extend well beyond the target borders) cannot be ablated using current surgical technology. The roles of chemotherapy, molecular manipulation, and conformal radiation therapy remain essential to the goal of controlling high-grade brain neoplasms partially treated with operation. Unfortunately, high-grade brain lesions remain stubbornly resistant to the intensive multimodality treatments that follow surgical resection. Survival curves for highly malignant brain lesions have not changed substantially in the past several decades.
Radiotherapy for Pediatric CNS Tumors
The target for radiation therapy is cellular DNA. Ionizing radiation damages double-stranded DNA, leading to cell death. Unlike normal cells, which have a preserved ability to repair radiation damage, neoplastic cells often are replicating at abnormally high rates and radiation interferes with their mitotic or proliferative ability. With slowly growing tumors such as craniopharyngiomas, the response to radiation is subtle and such tumors may take many months to show a clinical response. The critical sublethal dose required to preserve normal tissue but damage brain tumors, the so-called therapeutic window, is quite well understood and depends on a number of factors, including vulnerability of affected tissue (which can depend on the age of the patient and locale of the target; optic nerve radiation, as an example, is poorly tolerated), tumor vulnerability, volume irradiated, total dose, fraction size, and interfraction interval. As total volume of irradiation increases, the cumulative radiation dose must necessarily decrease to reduce the morbidity of treatment. The conventional cumulative radiation dose for most pediatric CNS tumors is in the 50–60 Gy range, although some tumors (e.g., germinomas) are much more sensitive to radiation and can respond to treatment in the 30–50 Gy range.17 Tumor type is therefore an important determinant of the effectiveness of radiation therapy, and biopsy is often a prerequisite for treatment.
Treatment of Spasticity and Movement Disorders
Spasticity occurs because of an imbalance of excitatory Ia afferent nerves from muscle spindles into the spinal cord and inhibitory descending impulses from the basal ganglia and cerebellum. In most children, the inhibitory impulses are diminished because of early CNS injury or injury to the spinal cord, which conducts the descending inhibitory impulses. Hence, treatment is directed toward either increasing the inhibitory neurotransmitters (usually γ-aminobutyric acid [GABA]) or reducing the afferent excitatory transmission from muscle spindles. Baclofen achieves the former, and dorsal rhizotomy (via cutting afferent nerve roots) interrupts the reflex transmission from muscle spindles.18,19
Treatment of spasticity should aim to improve function and facilitate care. Multidisciplinary clinics usually assess the potential candidate for surgical treatment. The best candidates for lumbosacral rhizotomy are motivated, older children (age 5 to 6 years) with spastic diplegia (affecting the legs predominantly) who lack severe contractures and have relatively good leg strength. Children with weak legs and spasticity can lose function with rhizotomy because they depend on their increased tone to maintain marginal ambulatory function. Rhizotomy can produce, to their detriment, a nonadjustable and permanent decrease in spasticity. Oral baclofen can be most useful in these very young children. Baclofen pumps are advantageous in children with severe spasticity that interferes with their care and in children with quadriplegia, often with a greater reduction of spasticity in the legs than the arms. The treatment is not permanent and nonablative, and the dosing is amenable to adjustments. It is particularly useful in children with spinal cord insult from trauma or inflammatory processes (transverse myelitis), and in patients with familial spastic paraparesis.
Botulinum toxin produces neuromuscular blockade and thereby reduces muscle contractions and spasticity. It is typically injected into spastic muscles, and for a period of several months, will decrease spasticity and increase range of motion. These injections are often used to extend the period of time until a definitive procedure can be performed in very young children, often decreasing the risk of developing muscle contractures that become fixed deformities which are not amenable to treatment.20
Epilepsy Surgery
Epilepsy affects about 1% of the population and starts commonly in the first decade of life. Surgical management is a well-established therapy when medical treatment has not been successful. Temporal lobectomy has been a mainstay of operative care for more than 40 years, and temporal resections constitute more than half of the epilepsy operations performed in children. In a randomized, controlled trial of operation versus medical therapy, 64% of patients were free of disabling seizures after operation compared with only 8% of those in the medical group.21 In addition, surgery may curtail the cognitive and psychosocial disabilities that can occur with medically intractable seizures, particularly in remediable syndromes that begin in infancy before the acquisition of language and social skills. The quality of life for patients with epilepsy is unambiguously related to the recurrence of seizures, and uncontrolled seizures carry a substantial risk of disability and death.
The success of temporal lobe surgery has led to more aggressive approaches to control epilepsy originating elsewhere in the brain. The pathologic substrates of pediatric extratemporal epilepsies are quite diverse, including cortical dysplasia, developmental abnormalities of neuronal migration, gliosis, tumors, neurocutaneous disorders (e.g., Sturge–Weber syndrome, tuberous sclerosis), and inflammatory lesions. These entities are often intensely epileptogenic from an early age. In such lesions, the MRI abnormality does not strictly correlate with the source of seizures and the epileptogenic focus may, in fact, be relatively diffuse and involve eloquent cortex. Nonetheless, some series show excellent results in more than 50% of patients, particularly if there is complete resection of an epileptogenic focus, including the focal area of interictal abnormality.22 Intelligence quotient scores tend to be stable or improved in the majority of children selected for surgery.
In some patients, seizures can be lateralized to one hemisphere by preoperative studies but not precisely localized. These patients may be considered for hemispherectomy if there is significant unilateral dysfunction. As radical as such operative resection would appear, the improvement of hemisphere disconnection procedures is in the 70% range for seizure freedom, with likely hemiparesis and hemianopia found postoperatively (although these deficits are often present before surgery). Newer techniques involve a disconnection of the hemisphere without anatomic removal of the affected hemisphere, thereby reducing some of the complications associated with volumetric removal of large portions of the brain.23
Corpus callosum sectioning is a palliative approach in patients who have seizures without focal onset. In patients with drop attacks, division of all or part of the corpus callosum can result in improvement in about 60% of patients by reducing the severity of their seizures and decreasing the likelihood of severe injury from falling.24
Vagal nerve stimulation (VNS) is useful for the treatment of partial seizures, providing about a 50% reduction in seizure frequency in patients with intractable seizures who are not candidates for resection. The morbidity of implantation of the vagal nerve stimulator is extremely small, and the efficacy of the device rivals that of new antiepileptic medications. As opposed to medications, there is no toxicity and no issues of compliance.25
Hydrocephalus
Other than trauma, hydrocephalus is the single most common entity pediatric neurosurgeons are called on to manage. This disorder of CSF circulation and absorption accounts for 0.6% of all pediatric hospital admissions, 1.8% of all pediatric hospital days, and 3.1% of all pediatric hospital charges.26–29 The care of hydrocephalic patients is a major health care expenditure, hovering near $1 billion in the USA per year.30 Although hydrocephalus can afflict individuals at any age, the range of causes and manifestations are larger and often more complex in children.
CSF is a clear fluid, which is primarily secreted within the ventricles of the brain by the choroid plexus. A considerable volume may be formed by interstitial fluid from the intercellular clefts in the brain and spinal cord.31–33 The total production of CSF has been calculated at about 500 mL/day.34 The volume in the system turns over nearly four times per day.
The circulation of CSF is complex. CSF exits the brain through the fourth ventricle and has a pulsatile course through the subarachnoid space over the convexities of the brain, the basal cisterns, the spinal subarachnoid space, and ultimately back intracranially to the vertex of the brain. There, the CSF transits through midline arachnoid granulations into the venous system at the superior sagittal sinus. This transfer is passive from a high-pressure system into a low-pressure environment.
Escape of the CSF through alternative routes exists, although none is adequate to maintain normal nervous system function. Many mothers have observed that their children look puffy around the eyes when their shunt is malfunctioning. There is perhaps escape of CSF through the craniofacial lymphatics that might account for this observation. This transit of CSF has been confirmed in other mammals, and there is advancing evidence of this in humans.35–37
Congenital Hydrocephalus
The most common genetic hydrocephalus is X-linked hydrocephalus. It occurs in 1 : 30,000 male births and represents 2–5% of nonsyndromal cases of hydrocephalus. The aqueduct of Sylvius is narrowed, causing subsequent dilation of the third and lateral ventricles, sparing the fourth ventricle (Figs 19-3 to 19-5). In its fullest expression, other neurologic abnormalities can occur. Twenty-five percent of males with clear aqueduct stenosis will have X-linked hydrocephalus, which is very important in advising couples about future pregnancies.38 Other causes of aqueduct stenosis can be thickening of the tectum of the midbrain from hamartoma, glioma formation, or from intrauterine infections.39
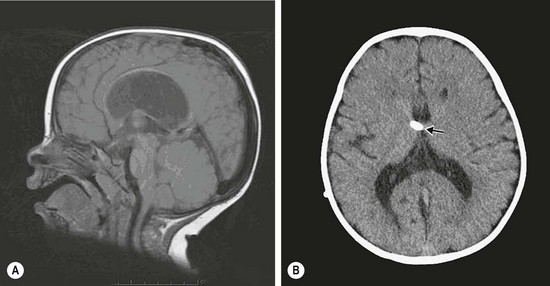
FIGURE 19-3 (A) Sagittal MR image showing aqueduct stenosis and dilated third and lateral ventricles. (B) Axial CT after ventriculoperitoneal shunt (arrow).
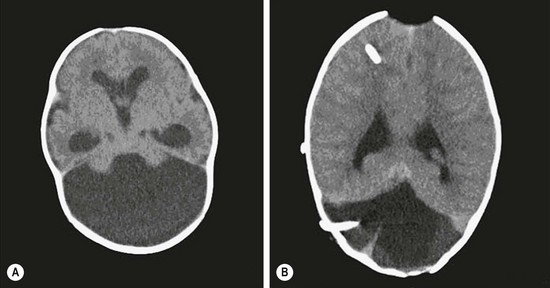
FIGURE 19-4 (A) Axial CT scan of Dandy–Walker malformation. (B) The axial CT scan shows supratentorial and posterior fossa shunt catheters. The posterior fossa shunt was required after the cyst continued to expand.
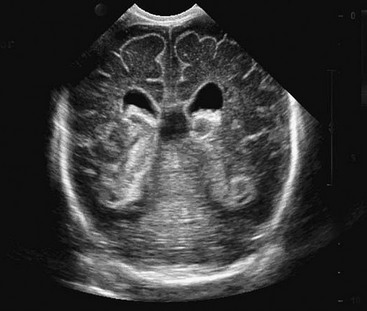
FIGURE 19-5 Ultrasound image shows a grade III bilateral intraventricular hemorrhage with ventricular dilation.
In Dandy–Walker malformations, the hydrocephalus is due to an alternation in CSF flow at the exit of the fourth ventricle. A Dandy–Walker malformation may be further complicated by a ‘double compartment’ hydrocephalus. The cyst formation may block escape of the CSF at the distal end of the aqueduct of Sylvius with resultant dilation of the third and lateral ventricles. In addition, the choroid plexus within the fourth ventricle will create CSF with no access to the subarachnoid space and subsequent enlargement of the cyst. In infants, this may lead to an unsightly distortion of the cranium and signs and symptoms of hindbrain compression. Not infrequently, additional surgical attention must be directed to the cyst, usually a CSF shunt catheter, either joined with a ventriculoperitoneal shunt or a separate shunt entirely (see Figs 19-3 and 19-4).40,41
Acquired Hydrocephalus
In the USA, there are more than 50,000 very low birth weight infants born each year. Almost 20% of these neonates suffer some degree of intraventricular hemorrhage, most related to bleeding within the germinal matrix adjacent to the ventricles of the brain (see Fig. 19-5). These hemorrhages are graded on scale of I to IV (Table 19-1). At least 25% of these neonates will develop posthemorrhagic hydrocephalus.42 Intraventricular blood is a powerful irritant to the ependymal lining of the ventricles as well as to the arachnoid membranes. The resultant inflammatory response and scarring can lead to obstruction of flow of CSF from the ventricle or, more commonly, an intense arachnoiditis with severe restriction of the CSF pathways at the base of the brain. Traumatic intracranial bleeding will manifest the same pathophysiology, leading to ‘post-traumatic’ hydrocephalus. In the worst cases, the resultant inflammation will be both intraventricular and in the subarachnoid space. It can lead to multiple scar septations within the ventricles and ‘multiple compartments’ hydrocephalus.43
TABLE 19-1
Grading System for Intraventricular Hemorrhage Based on Ultrasound Findings
| Grade | Cranial Ultrasound Findings |
| I | Hemorrhage in germinal matrix only |
| II | Ventricular hemorrhage without ventricular dilation |
| III | Ventricular hemorrhage with ventricular dilation |
| IV | Brain parenchymal hemorrhage |
It is extremely rare to see a child with congenital hydrocephalus become shunt independent. Thus, such children need to be approached with caution in making such a diagnosis.44,45
Diagnosis
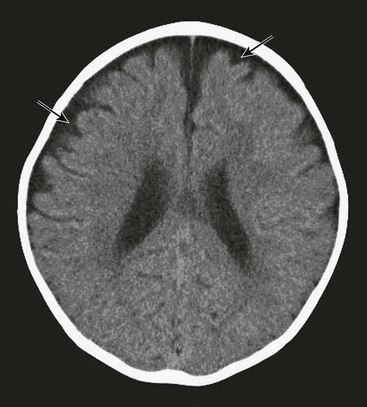
Suspicions are to be confirmed by brain imaging studies. Ultrasonography in infants with an open fontanelle is a good screening study, but even if hydrocephalus is diagnosed with that technique, MRI or CT is required to more fully understand the etiology (Fig. 19-6).
Treatment
The first reproducibly successful treatment for hydrocephalus of any type occurred in the early 1950s. Lumboureteral shunts were successfully employed in patients with communicating hydrocephalus.46 Success was then reported with ventricular to jugular vein shunts incorporating a miniaturized spring and ball valve.47 As silicone replaced the earlier stiffer plastics, the ventriculoperitoneal shunt became and remains the mainstay of hydrocephalus therapy.48
Development of shunt hardware has matured. The quality of the silicone has improved, making the tubing more pliable and less hazardous to abdominal organs. Valves can be adjusted to various opening pressures with external magnets. Tubing that incorporates antibiotics within or coating it is available. With these mechanical advances, life expectancy of the shunt itself has improved and the complications have diminished, although only slightly. Realistically, there is little difference in the performance of one brand of shunt over another, despite the claims of the multiple vendors.49
Shunt occlusions and infections are still vexing in our efforts to have these children lead a normal life. Nearly half of all shunt operations are revisions for occlusion or infection. Perioperative antibiotic usage, although not without controversy, has reduced the shunt infection rate to 8% or less in busy pediatric centers.50 There is no consensus as to which antimicrobial agents are most efficacious.51
Alternatives to ventriculoperitoneal shunts are still useful, if not required in some complex cases. Lumboperitoneal shunts have regained some popularity among pediatric neurosurgeons in selected patients with communication of CSF from the intracranial compartment to the spinal subarachnoid space. The development of a Chiari I malformation from chronic CSF drainage that draws the cerebellar tonsils downward has given many neurosurgeons pause before considering this shunt in young patients.52–54 When circumstances make the peritoneum inhospitable to CSF absorption, alternate termini for the tube include the venous system, pleural space, and even the gallbladder.55
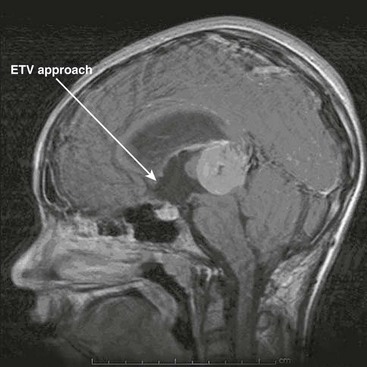
An alternative to CSF shunting has re-emerged with the miniaturization of endoscopic equipment. Creating an opening in the floor of the third ventricle to look around an obstruction at the aqueduct or the outlet of the fourth ventricle is very appealing. The concept dates from 1922 but was a failure. New technology for minimally invasive surgery has given a rebirth to the idea and, today, endoscopic third ventriculostomy (ETV) is selectively done in lieu of CSF shunts (Fig. 19-7). When successful, the hydrocephalus is managed without the need for foreign body implant and without the inherent risks of infection or valve or catheter failure. This approach has been found to be successful in 60% of properly selected children.56 The potential complications, such as uncontrollable hemorrhage, neuroendocrine dysfunction, and short-term memory loss, are severe, more so than with shunting. Nevertheless, there is great appeal in having hydrocephalus treated effectively without implanted hardware.
Shunt Malfunctions
Despite the progress in shunt design and materials, shunts will fail, and at a surprisingly high rate. Forty percent fail within the first year of implant. At 15 years, there is an 80% likelihood of failure. Again, the signs and symptoms will be age dependent. Infants may have minimal symptoms because of the expandable cranium, whereas older children may quickly become dreadfully ill as the ICP increases. Headache, vomiting, and altered mental status predominate as symptoms. The clinical assessment typically includes a brain imaging study, a shunt survey (plain films of the shunt) to look for a fracture of the tube, a shortened end, or a migration. Although a change in the ventricular size noted on imaging is a strong clue to the diagnosis, normal ventricular size can often be a falsely reassuring sign. It is not uncommon for symptomatic children to have no dilation of their ventricles, presumably related to loss of brain compliance.57,58 Often a percutaneous needle tap of the shunt reservoir or valve will be needed to measure pressure. Some surgeons use radioisotope or contrast flow studies (shuntograms). When in doubt, surgical exploration of the shunt is the best option.59
Spinal Dysraphism
Myelomeningocele
Myelomeningocele is the most frequently encountered spinal neural tube defect. In the USA, the incidence is 0.3 per 1,000 live births, a decrease of nearly 50% since the widespread use of folic acid in women planning pregnancy or begun in the first trimester.60 It is rarely a diagnostic problem and is now frequently discovered with fetal ultrasound or MRI.
Essentially, these are defects in the skin, fascia, posterior elements of the spine, and the conus medullaris of the cord, along with a failure in neurulation during the 26th to 28th weeks of gestation. Myelomeningocele is most frequent in the lumbar and lumbosacral area (Fig. 19-8). Often of considerable size, it might be confused with a sacral teratoma. The neonatal assessment may require MRI of the entire spinal canal to exclude concurrent anomalies. Cranial ultrasound is usually sufficient to evaluate for hydrocephalus. The surgical challenge is not so much in closing the exposed nervous system but in obtaining a tension-free cutaneous closure. At operation, the exposed end of the incompletely fused spinal cord is separated from the cutaneous attachments, and a pseudo-dural layer is closed over it. Not infrequently, the skin closure is the more complex segment of the operation, and plastic surgical techniques (relaxing flank incisions, skin grafts, rotational flaps) may be required to accomplish this closure.
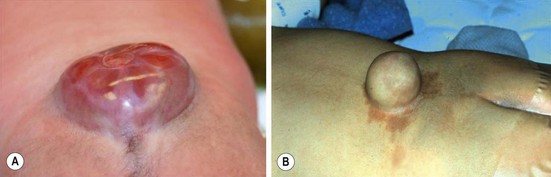
FIGURE 19-8 (A) A typical myelomeningocele shows a small neural placode at the dome with a large CSF-filled sack and little useful skin. (B) A meningocele with common surrounding port-wine stains is seen.
Prenatal closure of the myelomeningocele defect at three designated centers has confirmed the efficacy of this intervention in highly selected patients in a recently completed trial (Management of myelomeningocele study [MOMS]).61 The incidence of CSF shunts at one year was 40% in the prenatal surgery group vs 80% in those babies managed after birth (p < 0.001). Prenatal intervention also resulted in improvement in the composite score for mental development and motor function at 30 months (p = 0.007). Independent walking at 3 years was 42% in the prenatal group vs 21% in the postnatal surgery group (p = 0.01). However, prenatal operation was associated with higher rates of preterm birth, intraoperative complications, and uterine-scar defects apparent at delivery as well as a higher rate of maternal transfusion at delivery.
Lipomyelomeningocele
Lipomyelomeningocele is a complex anomaly that consists of a subcutaneous meningocele, fascia and bony defects, and a lipoma interfacing with the spinal cord, which is located dorsally, dorsolateral, or terminally. Externally, these lesions appear in the midline and can range from tiny subtle fatty lumps to large masses often accompanied by skin tags, port-wine stains, and an altered intragluteal fold (Fig. 19-9). Before the availability of CT, the connection with the spinal canal was often missed and cosmetic removal of the subcutaneous fat was performed, often with significant negative sequelae. These lesions often require tedious micro dissection with release of any tethering effect on the spinal cord to prevent neurologic (weakness, sensory loss), urologic (neurogenic bladder), or orthopedic (scoliosis, leg-length discrepancies) consequences. These children do not have Chiari malformation, sparing them the burden of hydrocephalus.
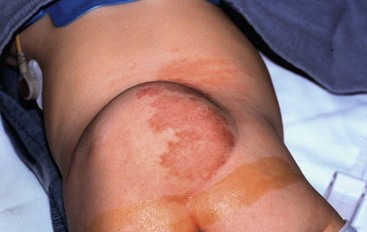
Whether to correct the asymptomatic infant with a lipomyelomeningocele is not without controversy. Unfortunately, the natural history of these anomalies is unknown. The rationale for early operation in these patients is to release any tethering on the spinal cord and prevent neurologic, urologic, or orthopedic consequences of spinal cord injury. However, a significant number of patients have suffered the same deficits postoperatively that the surgery was planned to prevent.62,63
References
1. Janisch, W, Haas, JF, Schreiber, D, et al. Primary central nervous system tumors in stillborns and infants: Epidemiological consideration. J Neurooncol. 1984; 2:113–116.
2. Grabb, PA, Albright, AL, Brain tumors of congenital and developmental origin in infants and children: Clinical features and natural history. The Practice of Neurosurgery. Tindall, GT, Cooper, PR, Barrow, DL, eds. The Practice of Neurosurgery; vol. 1. Williams & Wilkins, Baltimore, 1996:821–831.
3. Carmel, PW, Brain tumors of disordered embryogenesis. Neurological Surgery. Youmans, JR, eds. Neurological Surgery; vol. 4. WB Saunders, Philadelphia, 1996:2761–2781.
4. Lieberman, DM, Russo, CL, Berger, MS. Brain tumors during the first 2 years of life. In: Albright AL, Pollack IF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. New York: Thieme; 1999:463–490.
5. Steinbok, P, Mutat, A. Cerebellar astrocytomas. In: Albright AL, Pollack IF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. New York: Thieme; 1999:641–662.
6. Cochrane, DD, Gustavsson, B, Poskitt, KP, et al. The surgical and natural morbidity of aggressive resection for posterior fossa tumors in childhood. Pediatr Neurosurg. 1994; 20:19–29.
7. Pollack, I. Brain tumors in children. N Engl J Med. 1994; 331:1500–1507.
8. Sutton, L, Goldwein, J, Perilongo, G, et al. Prognostic factors in childhood ependymomas. Pediatr Neurosurg. 1990-1991; 16:57–65.
9. Roberts, RO, Lynch, CF, Jones, MP, et al. Medulloblastoma: A population-based study of 532 cases. J Neuro-pathol Exp Neurol. 1991; 50:134–144.
10. Raffel, C, Thomas, GA, Tishler, DM, et al. Absence of p53 mutations in childhood central nervous system primitive neuroectodermal tumors. Neurosurgery. 1993; 33:301–306.
11. Pollack, IF, Polinko, P, Albright, AL, et al. Mutism and pseudobulbar symptoms after resection of posterior fossa tumors in children: Incidence and pathophysiology. Neurosurgery. 1995; 37:885–893.
12. Duffner, PK, Horowitz, ME, Krischer, JP, et al. Postoperative chemotherapy and delayed radiation in children less than 3 years of age with malignant brain tumors. N Engl J Med. 1993; 328:1725–1731.
13. Pollack, IF, Gerszten, PC, Martinez, AJ, et al. Intracranial ependymomas of childhood: Long-term outcome and prognostic factors. Neurosurgery. 1995; 37:655–667.
14. Sanai, N, Alvarez-Buylia, A, Berger, M. Neural stem cells and the origin of gliomas. N Engl J Med. 2005; 353:811–822.
15. Croce, CM. Oncogenes and Cancer. N Engl J Med. 2008; 358:502–511.
16. Esteller, M. Epigenetics in cancer. N Engl J Med. 2008; 358:1148–1159.
17. Tarbell, N, Loeffler, JS. Radiotherapy for pediatric brain tumors. In: Albright AL, Pollack IF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. New York: Thieme; 1999:765–778.
18. Albright, AL, Barron, WB, Faick, MP, et al. Continuous intrathecal baclofen infusion for spasticity of cerebral origin. JAMA. 1993; 270:2475–2477.
19. Peacock, WJ, Arens, LJ, Berman, B. Cerebral palsy spasticity: Selective posterior rhizotomy. Pediatr Neurosci. 1987; 13:61–66.
20. Jankovic, J, Brin, MF. Therapeutic uses of botulinum toxin. N Engl J Med. 1991; 324:1186–1194.
21. Wiebe, S, Blume, WT, Girvin, JP, et al. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med. 2001; 345:311–318.
22. Morrison, G. Extratemporal epilepsy surgery in children. In: Albright AL, Pollack IF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. New York: Thieme; 1999:1127–1146.
23. Schramm, J, Kral, T, Clusmann, H. Transsylvian keyhole functional hemispherectomy. Neurosurgery. 2001; 49:891–901.
24. Black, PM, Holmes, G, Lombroso, C. Corpus callosum section for intractable epilepsy in children. Pediatr Neurosurg. 1992; 18:298–304.
25. Murphy, JV. Left vagal nerve stimulation in children with epilepsy. J Pediatr. 1999; 134:563–566.
26. Simon, TD, Riva-Cambrin Srivastaua, R, et al. or the hydrocephalus clinical network: Hospital care for children with hydrocephalus in the United States: Utilization, charges, comorbidities and deaths. J Neurosurg Pediatr. 2008; 1:131–138.
27. Cochrane, DD, Kestle, J. Ventricular shunting for hydrocephalus in children: Patients, procedures, surgeons, and institutions in English Canada, 1989-2001. Eur J Pediatr Surg. 2002; 12(1 Suppl):S6–S11.
28. Smith, ER, Butler, WE, Barker, FG, II. In-hospital mortality rates after ventricular peritoneal shunt procedures in the United States, 1998-2002: Relation to hospital and surgeon volume of care. J Neurosurg. 2004; 100(2 Suppl Pediatrics):90–97.
29. Berry, JG, Hall, MA, Sharma, V, et al. A multi-institutional five year analysis of initial and multiple shunt revisions in children. Neurosurgery. 2008; 62:445–453.
30. Patwardham, RV, Nanda, A. Implanted ventricular shunts in the United States: The billion-dollar-a-year cost of hydrocephalus treatment. Neurosurgery. 2005; 56:139–145.
31. Milhorat, TH. Cerebral Spinal Fluid and the Brain Edemas. Neuroscience Society of New York; 1987.
32. Milhorat, TH. Choroid plexus and CSF production. Science. 1969; 166:1514–1516.
33. Milhorat, TH, Hammock, MK, Fenstermacher, JD, et al. CSF production by the choroid plexus and brain. Science. 1971; 173:330–332.
34. Rekate, HL. The treatment of hydrocephalus. In: Albright AL, Pollack EF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. 2nd ed. New York: Thieme; 2008:103–105.
35. Boulton, M, Flessner, M, Armstrong, D, et al. Determination of volumetric CSF absorption into extracranial lymphatics in sheep. Am J Physiol. 1998; 274:88–96.
36. Erlich, SS, McComb, JG, Hyman, S, et al. Ultrastructure of the orbital pathways for CSF drainage in rabbits. J Neurosurg. 1989; 70:926–931.
37. Johnston, M, Zakharov, A, Papaiconomou, C, et al. Evidence of connections between CSF and nasal lymphatic vessels in humans, non-human primates and other mammalian species. CSF Res. 2004; 1:2.
38. Dirks, P. Genetics of hydrocephalus. In: Cinalli G, Maixner WJ, Sainte-Rose C, eds. Pediatric Hydrocephalus. New York: Springer-Verlag, 2005.
39. Barkovich, AJ, Newton, TH. MR of aqueductal stenosis: Evidence of a broad spectrum of tectal distortion. AJNR Am J Neuroradiol. 1989; 10:471–476.
40. Mohanty, A, Biswas, A, Satish, S, et al. Treatment options for Dandy-Walker malformation. J Neurosurg. 2006; 105(5 Suppl Pediatrics):348–356.
41. Yüceer, N, Mertol, T, Arda, N. Surgical treatment of 13 pediatric patients with Dandy-Walker syndrome. Pediatr Neurosurg. 2007; 43:358–363.
42. Boop, FA. Posthemorrhagic hydrocephalus of prematurity. In: Cinalli G, Maixner WJ, Sainte-Rose C, eds. Pediatric Hydrocephalus. New York: Springer-Verlag; 2005:121–126.
43. Fritsch, M, Mehdorn, M. Endoscopic intraventricular surgery for treatment of hydrocephalus and loculated CSF spaces in children less than one year of age. Pediatr Neurosurg. 2002; 36:183–188.
44. Rekate, HL. The treatment of hydrocephalus. In: Albright AL, Pollack EF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. 2nd ed. New York: Thieme; 2008:103–105.
45. Rekate, HL, Nulsen, FE, Mack, H, et al. Establishing the diagnosis of shunt independence. Monogr Neural Sci. 1982; 8:223–226.
46. Matson, DD. A new operation for treatment of communicating hydrocephalus. J Neurosurg. 1949; 6:238–247.
47. Nulsen, FE, Spitz, EB. Treatment of hydrocephalus by direct shunt from ventricle to jugular vein. Surg Forum (Am Coll Surg). 1952; 2:399–403.
48. Ames, RH. Ventricular peritoneal shunts in the management of hydrocephalus. J Neurosurg. 1967; 27:525–529.
49. Drake, JM, Kestle, JR, Milner, R, et al. Randomized trial of CSF shunt valve design in pediatric hydrocephalus. Neurosurgery. 1998; 43:294–305.
50. Piatt, JH, Carlson, CV. A search of determinates of cerebrospinal fluid shunt survival: Retrospective analysis of a 14-year institutional experience. Pediatr Neurosurg. 1993; 19:233–242.
51. Biyani, N, Grisaru-Soen, G, Steinbok, P, et al. Prophylactic antibiotics in pediatric shunt surgery. Childs Nerv Syst. 2006; 22:1465–1471.
52. Welch, K, Shillito, J, Strand, R, et al. Chiari I “malformation”: An acquired disorder? J Neurosurg. 1981; 55:604–609.
53. Wang, V, Barbaro, N, Lauton, M, et al. Complications of lumbar peritoneal shunts. Neurosurgery. 2007; 60:1045–1049.
54. Dagnew, E, van Loveren, H, Tew, J, Jr. Acute foramen magnum syndrome caused by an acquired Chiari malformation after lumbar drainage of cerebral spinal fluid: Report of three cases. Neurosurgery. 2002; 51:823–829.
55. Aldana, P, James, H, Postlethwait, R. Ventriculogallbladder shunts in pediatric patients. J Neurosurg Pediatr. 2008; 1:284–287.
56. Kadrian, D, van Gelder, J, Florida, D, et al. Long-term reliability of endoscopic third ventriculostomy. Neurosurgery. 2005; 56:1271–1278.
57. McNatt, SA, Kim, A, Hohuan, D, et al. Pediatric shunt malfunction without ventricular dilation. Pediatr Neurosurg. 2008; 44:128–132.
58. Winston, KR, Lopez, JA, Freeman, J. CSF shunt failure with stable normal ventricular size. Pediatr Neurosurg. 2006; 42:151–155.
59. Vinchon, M, Fichten, A, Delestret, I, et al. Shunt revision for asymptomatic failures: Surgical and clinical results. Neurosurgery. 2003; 52:347–356.
60. Diaz, M, McLone, D. Myelomeningocele. In: Albright AL, Pollack IF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. 2nd ed. New York: Thieme; 2008:338–366.
61. Adzick, NS, Tom, E, Spong, CY, et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N Engl J Med. 2011; 364:993–1004.
62. Cochrane, DD, Finley, C, Kestle, J, et al. The patterns of late deterioration in patients with transitional lipomyelomeningocele. Eur J Pediatr Surg. 2000; 10(Suppl 1):13–17.
63. Cochrane, DD. Occult spinal dysraphism. In: Albright AL, Pollack IF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. 2nd ed. New York: Thieme; 2008:367–393.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Neurosurgical Conditions
Brain Tumors
Brain tumors are rare in the first year of life, with an incidence of 1 per 100,000 infants.1 This incidence increases with age. By the age of 2 years, central nervous system (CNS) tumors occur in 2 to 5/100,000 children. Brain tumors represent the most common solid tumors in the pediatric population.2
In the group of brain tumors considered congenital, the most common are teratomas (37%), followed by primitive neuroectodermal tumors (PNETs) (12%), astrocytomas (10–15%), and choroid plexus tumors (8%).3 In a slightly older population of children, teratomas become less frequent and other neoplasms become relatively more common, including astrocytomas (34% of brain tumors in children younger than 24 months of age), PNETs (23%), and ependymomas (11%). There is also a group of tumors that are sometimes considered non-neoplastic. These include developmental tumors, which derive from aberrant proliferative growth during embryonic brain development and include craniopharyngiomas, lipomas, dermoids, and colloid cysts.4
Cerebellar Astrocytomas
Low-grade astrocytomas occur throughout the CNS and constitute a fourth of all pediatric brain tumors. Cerebellar astrocytomas are common (12–17% of pediatric CNS tumors), and their treatment has the most favorable outcome of all intra-axial neoplasms in the CNS.5 Most of these tumors are found in children younger than the age of 10 years. The symptoms of cerebellar astrocytomas include headache (80%), vomiting (80%), gait disturbance, and decreased level of consciousness. Signs include ataxia in 80%, papilledema, cranial nerve palsies (including blindness and diplopia), and dysmetria. The rapid progression of signs and symptoms is often a function of cerebrospinal fluid (CSF) obstruction because the tumor occupies the fourth ventricle or the aqueduct, and causes hydrocephalus. Treatment of the hydrocephalus is often required urgently in very sick children. Temporary diversion of CSF via external ventriculostomies or shunts often precedes removal of the tumor.
Operative excision of these tumors is often possible, but new neurologic deficits can occur postoperatively in 30% of patients, although at least half of these new deficits are transient.6 Postoperative deficits occur because of the proximity of vital brain stem and delicate cranial nerve structures near the tumor, which is often large and firm. Postoperative pseudomeningoceles (the bulging of skin at the occipital incision site) and hydrocephalus are not uncommon (10–25%) and require additional treatment. Temporary continuation of postoperative CSF diversion via external ventriculostomy is favored by many surgeons to reduce the likelihood of permanent ventriculoperitoneal shunts.
Ependymomas
Ependymomas represent about 10% of pediatric brain tumors and are slightly less common than PNETs and astrocytomas. More than two-thirds of ependymomas in children occur in the posterior fossa (Fig. 19-1) and the symptoms they cause are similar to posterior fossa or cerebellar astrocytomas: headache, vomiting, lethargy, and ataxia. These symptoms result from a combination of compression along the dorsal brain stem and hydrocephalus. Many extend from the obex (in the inferior aspect of the fourth ventricle) and then extrude through the lateral opening of this ventricle (foramen of Luschka) into the cerebellopontine angle. Here they invade and compress cranial nerves with resulting facial weakness, diplopia, swallowing dysfunction, and hearing loss. When large, they often extend beyond the foramen magnum and can compress the cervical cord. In regard to their cell of origin, they are not limited to the ependymal layer of the ventricle but can arise from cerebellar, cerebral, or spinal cord parenchyma.
FIGURE 19-1 MR image of a posterior fossa ependymoma. The tumor involves the lateral aspect of the posterior fossa and contains calcifications typical of ependymomas. The brain stem is severely distorted by the mass effect of the tumor. The tumor also compresses cranial nerves on the right side, making complete tumor removal problematic. This patient had right facial weakness, diplopia, and mild left hemiparesis postoperatively.
Aggressive surgical resection has been the goal of treatment of these difficult tumors, but there is high probability of new or worsening neurologic deficits as a result of the operative dissection. There is a correlation between postoperative residual tumor and tumor recurrence and/or progression. In the 30% of children where the tumor is totally resected, there is still a 20% to 40% possibility of recurrence, even after conventional radiotherapy.7 In cases of near-total resection, the rate of progression-free survival falls to 30%.8 Most of the recurrences are local and radiation therapy has become the mainstay of treatment, even with complete resection. Very small children, usually younger than age 3 years, are faced with comparatively greater morbidity from conventional radiotherapy, and radiation therapy is often deferred. Chemotherapy has been used with some success in this group of patients. The role of chemotherapy without irradiation has generally not been favorable in older age groups. Retrospective studies have failed to prove substantial benefit in survival when chemotherapy is added to surgery and radiation therapy for newly diagnosed ependymomas.
Medulloblastomas
Medulloblastomas are the most common malignant solid tumor in children and constitute 20% of pediatric brain tumors.9 They are usually located in the posterior fossa and they are also referred to as PNETs because they are histologically identical to tumors (pineoblastomas, neuroblastomas, and retinoblastomas) located in other locations that are believed to have derived from progenitor subependymal neuroepithelial cells undergoing malignant transformation. Nearly half of medulloblastomas have chromosomal abnormalities, particularly the deletion of 17p chromosome that contains the tumor suppressor gene TP53.10
Hydrocephalus often occurs with medulloblastomas because of their location within the cerebellar vermis (a midline structure), often filling the fourth ventricle (Fig. 19-2). Children with this tumor often have symptoms of elevated intracranial pressure (ICP) (obtundation, headache, nausea/vomiting, irritability) and have signs suggestive of posterior fossa compression (dysmetria, ataxia, diplopia, head tilt, and papilledema). Lumbar puncture should not be done after computed tomography (CT) or magnetic resonance imaging (MRI) has established the presence of a posterior fossa tumor and obstructive hydrocephalus. CSF diversion (usually via an external drain) is usually done either before or in conjunction with craniotomy. Conversion of these temporary devices to permanent ventriculoperitoneal shunts is not uncommon in children with large tumors and marked preoperative ventriculomegaly.
FIGURE 19-2 MRI and MRS (magnetic resonance spectroscopy) of a posterior fossa medulloblastoma, with compression of fourth ventricle. The MRS profile shows a choline peak with depression of N-acetyl aspartate (NAA) typical of aggressive tumors.
Complete resection of medulloblastomas is often possible, although permanent postoperative deficits can occur.6 The ‘posterior fossa syndrome’ can occur postoperatively in 10–15% of children and is characterized by mutism, drooling and swallowing difficulties, ocular palsies, and increasing ataxia.11 These problems resolve entirely in most patients after several months. Improvement is thought to occur with resolution of swelling within the inferior vermis.
Staging is important with medulloblastomas because patients have a predictable outcome depending on age, metastases, pathology, and extent of surgical resection. Poor survival is correlated with age younger than 4 years, residual tumor measuring more than 1.5 cm,2 and tumor dissemination, particularly ‘drop’ metastasis along the spinal column. After craniospinal radiation in eligible patients, survival occurs in 50–70% of patients with standard or low-risk medulloblastomas. Newer treatment protocols include chemotherapy first, which is followed by radiotherapy consisting of lowered cumulative craniospinal doses (24–36 Gy to the entire brain) with hyperfractionated delivery, usually 1 Gy twice daily. Survival in these ‘good risk’ patients is nearly 90% after five years. Survival in high-risk patients is 60–65% with current multimodality therapy, with the worst outcomes in affected infants. Children younger than 3 years of age are usually treated first with chemotherapy, with irradiation deferred for 1 to 2 years. Radiation is sometimes avoided altogether in the 40% with progression-free survival.12
Supratentorial Nonglial and Glial Neoplasms
Ependymomas that occur in the cerebral hemispheres remain problematic in terms of treatment, although hemispheric tumors generally have comparatively better outcomes. Incomplete resection (often because of diffuse involvement within critical brain regions) and leptomeningeal spread are significant adverse risk factors. Age is also a factor. Children younger than 3 years of age have significantly diminished progression-free survival (10–15%) compared with older children.13
As with medulloblastoma, glial and nonglial tumors often have genetic abnormalities, with chromosomal abnormalities and gene mutations. Many low-grade lesions progress to become more malignant. The pathway to this malignant progression is complex. Chromosomal translocations and mutations occur as initiating events before the amplification of deleterious genes that support tumor progression.14–16
With most benign tumors, there exists a strong association between extent of resection and outcome. From the neurosurgical viewpoint, maximal resection should be attempted without inordinate surgical morbidity. The advent of frameless stereotaxy for precise tumor localization, ‘functional’ localization with intraoperative monitoring (e.g., somatosensory evoked potential mapping to determine the location of the motor cortex), presurgical functional MRI to determine location of speech areas, and intraoperative scanning (via real-time ultrasonography [US], CT, or MRI) have each added considerably to the safety of the operation. Nonetheless, the operative approach can only achieve resection of targeted areas. Infiltrative tumors (which typically extend well beyond the target borders) cannot be ablated using current surgical technology. The roles of chemotherapy, molecular manipulation, and conformal radiation therapy remain essential to the goal of controlling high-grade brain neoplasms partially treated with operation. Unfortunately, high-grade brain lesions remain stubbornly resistant to the intensive multimodality treatments that follow surgical resection. Survival curves for highly malignant brain lesions have not changed substantially in the past several decades.
Radiotherapy for Pediatric CNS Tumors
The target for radiation therapy is cellular DNA. Ionizing radiation damages double-stranded DNA, leading to cell death. Unlike normal cells, which have a preserved ability to repair radiation damage, neoplastic cells often are replicating at abnormally high rates and radiation interferes with their mitotic or proliferative ability. With slowly growing tumors such as craniopharyngiomas, the response to radiation is subtle and such tumors may take many months to show a clinical response. The critical sublethal dose required to preserve normal tissue but damage brain tumors, the so-called therapeutic window, is quite well understood and depends on a number of factors, including vulnerability of affected tissue (which can depend on the age of the patient and locale of the target; optic nerve radiation, as an example, is poorly tolerated), tumor vulnerability, volume irradiated, total dose, fraction size, and interfraction interval. As total volume of irradiation increases, the cumulative radiation dose must necessarily decrease to reduce the morbidity of treatment. The conventional cumulative radiation dose for most pediatric CNS tumors is in the 50–60 Gy range, although some tumors (e.g., germinomas) are much more sensitive to radiation and can respond to treatment in the 30–50 Gy range.17 Tumor type is therefore an important determinant of the effectiveness of radiation therapy, and biopsy is often a prerequisite for treatment.
Treatment of Spasticity and Movement Disorders
Spasticity occurs because of an imbalance of excitatory Ia afferent nerves from muscle spindles into the spinal cord and inhibitory descending impulses from the basal ganglia and cerebellum. In most children, the inhibitory impulses are diminished because of early CNS injury or injury to the spinal cord, which conducts the descending inhibitory impulses. Hence, treatment is directed toward either increasing the inhibitory neurotransmitters (usually γ-aminobutyric acid [GABA]) or reducing the afferent excitatory transmission from muscle spindles. Baclofen achieves the former, and dorsal rhizotomy (via cutting afferent nerve roots) interrupts the reflex transmission from muscle spindles.18,19
[/not-level-membership-for-pediatrics-category]
