[level-membership-for-neurology-category]
34 Neuromuscular Junction Disorders
Disorders affecting the neuromuscular junction (NMJ) are among the most interesting and rewarding seen in the electromyography (EMG) laboratory. These disorders are generally pure motor syndromes that usually preferentially affect proximal, bulbar, or extraocular muscles. They are confused occasionally with myopathies. With knowledge of normal NMJ physiology (see Chapter 6), most of the abnormalities affecting the NMJ can be differentiated using a combination of nerve conduction studies, repetitive stimulation, exercise testing, and needle EMG.
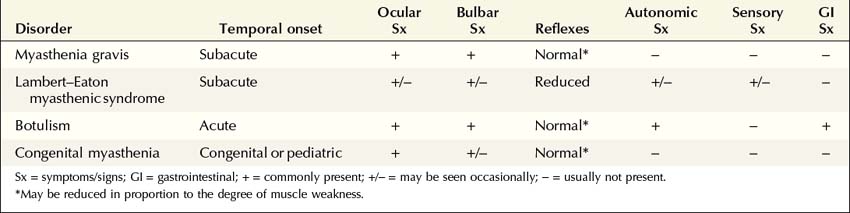
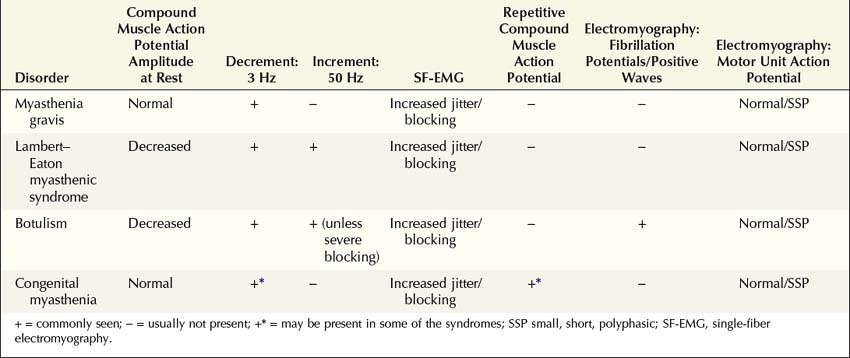
NMJ disorders can be classified into immune-mediated, toxic or metabolic, and congenital syndromes (Box 34–1). They usually are distinguished by their clinical and electrophysiologic findings (Tables 34–1 and 34–2). All are uncommon, but among them, myasthenia gravis (MG) and Lambert–Eaton myasthenic syndrome (LEMS) are the disorders most often encountered in the EMG laboratory. Both are immune-mediated disorders. In MG the autoimmune attack is postsynaptic; in LEMS the presynaptic membrane is the target of attack. Every electromyographer must understand the electrophysiology of these disorders so that appropriate electrodiagnostic tests can be applied and the correct diagnosis not overlooked.
Box 34–1
Disorders of the Neuromuscular Junction
Myasthenia Gravis
Electrophysiologic Evaluation
Like other disorders affecting the NMJ, the electrophysiologic evaluation of MG involves routine nerve conduction studies, repetitive nerve stimulation (RNS), exercise testing, routine EMG, and, in some cases, single-fiber EMG (SF-EMG) (Box 34–2).
Box 34–2
Electrophysiologic Evaluation of Myasthenia Gravis
1. Routine motor and sensory nerve conduction studies. Perform routine motor and sensory nerve conduction studies, preferably a motor and sensory nerve in one upper and one lower extremity. CMAP amplitudes should be normal. If CMAP amplitudes are low or borderline, repeat distal stimulation immediately after 10 seconds of exercise to exclude a presynaptic NMJ transmission disorder (e.g., Lambert–Eaton myasthenic syndrome).
2. Repetitive nerve stimulation (RNS) and exercise testing. Perform slow RNS (3 Hz) on at least one proximal and one distal motor nerve. Always try to study weak muscles. If any significant decrement (>10%) is present, repeat to ensure decrement is reproducible. If there is no significant decrement at baseline, exercise the muscle for 1 minute, and repeat RNS at 1, 2, 3, and 4 minutes looking for a decrement, secondary to post-exercise exhaustion. If at any time a significant decrement is present (at baseline or following post-exercise exhaustion), exercise the muscle for 10 seconds and immediately repeat RNS, looking for post-exercise facilitation (repair of the decrement).
3. Needle electromyography (EMG). Perform routine needle EMG of distal and proximal muscles, especially weak muscles. Patients with moderate to severe myasthenia gravis may display unstable or short, small, polyphasic motor unit action potentials. Recruitment is normal or early. Needle EMG must exclude severe denervating disorders or myotonic disorders, which may display an abnormal decrement on RNS.
4. Single-fiber EMG (SF-EMG). If the above are normal or equivocal in a patient strongly suspected of having myasthenia gravis, perform SF-EMG in the extensor digitorum communis and, if necessary, one other muscle, looking for jitter and blocking. It is always best to study a weak muscle. Normal SF-EMG in a clinically weak muscle excludes an NMJ disorder.
CMAP, compound muscle action potential; NMJ, neuromuscular junction.
Repetitive Nerve Stimulation
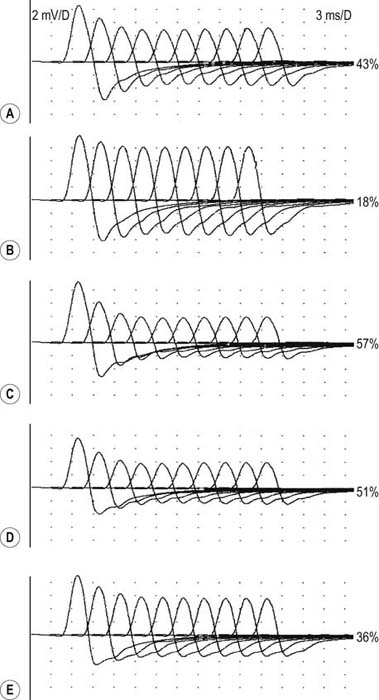
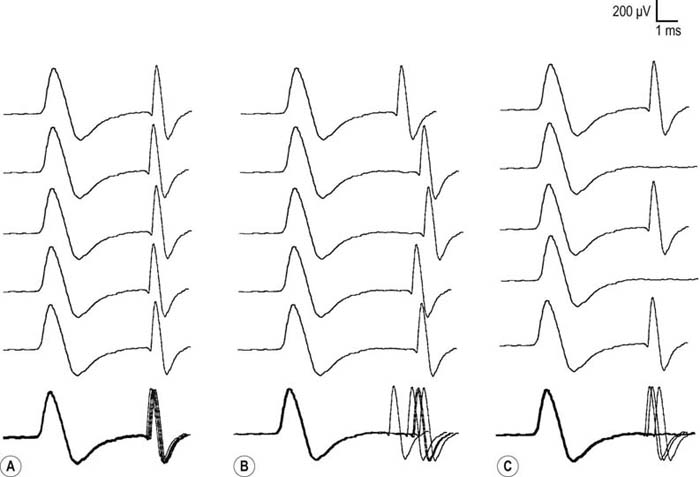
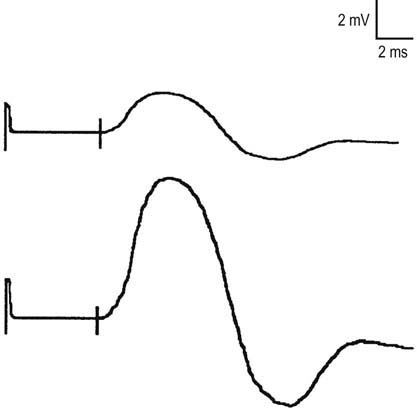
After the routine nerve conduction studies are completed, RNS studies are performed (see Chapter 6). These studies are abnormal in more than 50 to 70% of patients with generalized MG but often are normal in patients with the restricted ocular form of MG. A decremental response on RNS is the electrical correlate of clinical muscle fatigue and weakness. In normal subjects, slow RNS (3 Hz) results in little or no decrement of the CMAP, whereas in MG, a CMAP decrement of 10% or more is characteristically seen (Figure 34–1A). Both distal and proximal nerves should be tested. Although distal nerves are technically easier to study, the diagnostic yield increases with stimulation of proximal nerves (e.g., spinal accessory or facial nerves). This is not unexpected, because the proximal muscles usually are much more involved clinically than the distal ones. Facial RNS is especially important to perform in suspected anti-MuSK MG, where the yield of finding an abnormal decrement is much higher when examining a facial muscle than a limb muscle (probably reflecting the severe facial and bulbar involvement in some patients with anti-MuSK MG).
Exercise Testing
Exercise testing should be routinely used with all RNS studies (see Chapter 6). If there is no significant decrement on RNS studies at baseline (<10% decrement), the patient should perform 1 minute of exercise, followed by RNS at 1-minute intervals for the next 3 to 4 minutes, looking for a CMAP decrement secondary to post-exercise exhaustion. If at any time, either at baseline or following exercise, a significant decrement develops, the patient should perform a brief 10-second maximum isometric contraction, immediately followed by slow RNS, looking for an increment in the CMAP and “repair” of the decrement secondary to post-exercise facilitation (Figure 34–1).
Electromyography
Unstable MUAPs (see Chapter 15) occur when individual muscle fibers are either blocked or come to action potential at varying intervals, which leads to MUAPs that change in configuration from impulse to impulse. If some muscle fibers of a motor unit are blocked and never come to action potential, the motor unit effectively loses muscle fibers, becoming short, small, and polyphasic, similar to MUAPs seen in myopathy. Otherwise, the needle EMG findings in NMJ disorders usually are normal. In general, fibrillation potentials and other abnormal spontaneous activity are not seen in NMJ disorders, with the important exception of botulism (see section on Botulism).
Single-fiber Electromyography
When a motor axon is depolarized, the action potential normally travels distally and excites all the muscle fibers within that motor unit at more or less the same time (Figure 34–2). This variation in the time interval between the firing of adjacent single muscle fibers from the same motor unit is termed jitter and primarily reflects variation in NMJ transmission time. If the NMJ is compromised, the time it takes for the endplate potential to reach threshold is prolonged, which results in greater-than-normal variation between firing of adjacent muscle fibers. If the prolongation is severe enough, the muscle fiber may never reach action potential, resulting in blocking of the muscle fiber.
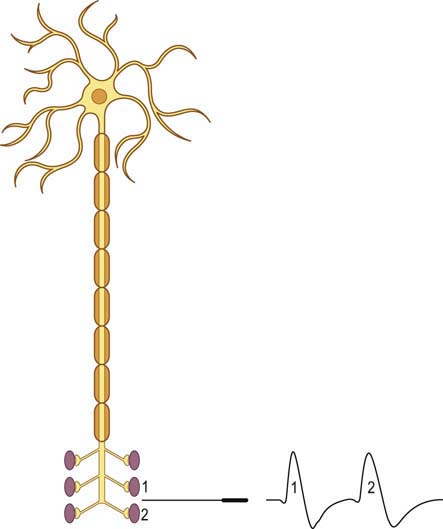
The goal of SF-EMG is to study two adjacent single muscle fibers, known as a pair, from the same motor unit. This is accomplished by changing the filters on the EMG machine and using a specialized SF-EMG needle. The low-frequency filter (high-pass) is increased to 500 Hz (normally 10 Hz in routine EMG). By using a high-pass filter of 500 Hz, the amplitudes of distant muscle fiber potentials are attenuated while those of the nearby fibers are preserved. The SF-EMG needle is a specially constructed needle with the active electrode (G1) located in a port along the posterior shaft of the needle and with a smaller leading surface area than the conventional concentric needle electrode (Figure 34–3). The reference electrode (G2) is the needle shaft. The result of these two modifications is that single-fiber muscle action potentials are recorded only if they are within 200 to 300 µm of the needle. The needle is placed in the muscle, and the patient is asked to activate the muscle in an even and constant fashion. The needle is moved until a single muscle fiber potential is located. With this single muscle fiber potential triggered on a delay line, the needle is slightly and carefully moved or rotated to look for a second potential that is time locked to the first potential (signifying that it is from the same motor unit).

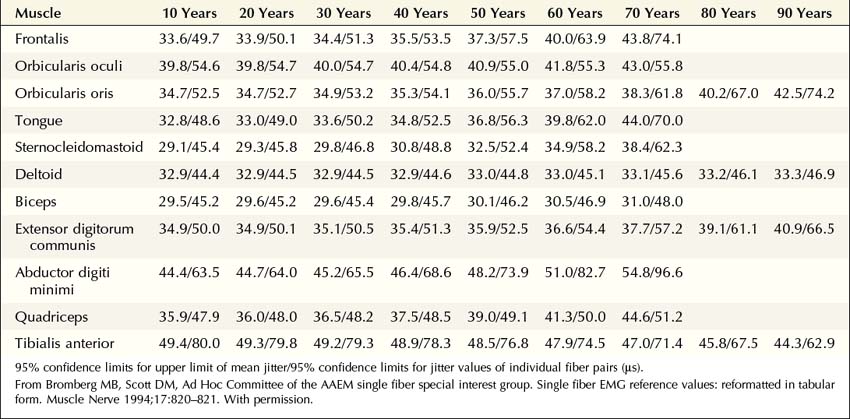
More recently, the regular disposable concentric EMG needle has been used for SF-EMG studies. The standard SF-EMG needle is expensive, and needs to be surgically sanitized between patients. Thus, the cost of the standard SF-EMG needle, along with the theoretical risk of transmitting infection (including prion diseases) despite sanitizing the needle, have prompted this change. In general, the values for jitter are comparable between the traditional SF and the concentric EMG needles. Single-fiber potentials should be accepted for analysis only if the potential is at least 200 µV in amplitude with a rise time of less than 300 µs. If a time-locked second potential is located, an interpotential interval between the two potentials (i.e., the pair) can be measured. By recording multiple consecutive firings of the muscle fiber action potential pairs, the difference between consecutive interpotential intervals can be calculated. This variation between consecutive interpotential intervals is the jitter. By recording 50 to 100 subsequent potentials, the mean consecutive difference (MCD), a measure of jitter, can be calculated between the triggered potential and the time-locked second single muscle fiber potential. Most modern EMG machines have programs that automatically perform the MCD calculation. This procedure is then repeated until 20 separate single-fiber pairs are collected, to calculate a mean MCD. This value is compared with the normal mean MCD for the muscle studied and the patient’s age (Table 34–3). There is also an upper limit of normal jitter for an individual pair, based on the muscle studied and the patient’s age. To call the latter abnormal, more than 10% of the pairs must exceed the limit (e.g., for 20 pairs, at least two must be abnormal). To make a diagnosis of an NMJ disorder, either the mean jitter must be abnormal or the upper limit of normal jitter must be abnormal in more than 10% of individual pairs. However, in most NMJ disorders, both will be abnormal. Increased jitter is consistent with an NMJ disorder (Figure 34–4). In addition to increased jitter, blocking may be seen on SF-EMG. Two time-locked, single-fiber muscle potentials from the same motor unit normally fire together. If the triggered potential fires steadily while the second potential fires only intermittently, blocking is occurring. Blocking, which is another marker of NMJ disease, usually occurs only when the jitter is markedly prolonged (e.g., MCD > 80–100 µs).
Lambert–eaton Myasthenic Syndrome
Electrophysiologic Evaluation
In the appropriate clinical setting, the electrophysiology of LEMS is diagnostic (Box 34–3). Single stimuli produce a reduced release of ACH quanta and a reduced endplate potential. At rest, many of the endplate potentials do not reach threshold, resulting in small-amplitude CMAPs on routine motor nerve conduction studies (Figure 34–5). Slow RNS (3 Hz) results in a decremental response similar to MG. However, rapid RNS (30–50 Hz) or brief (10 seconds) intense isometric exercise produces a marked increase in the CMAP amplitude (post-exercise facilitation) due to calcium accumulation in the presynaptic nerve terminal with subsequent enhancement of the release of ACH quanta (Figure 34–6). The CMAP commonly increments in amplitude by more than 100% (calculated by 100 × [(Highest amplitude − Initial amplitude)/Initial amplitude]. Brief, intense isometric exercise is preferable to rapid RNS, which can be quite painful. Brief exercise means 10 seconds of exercise. It has been definitely proven that the maximal increment occurs after 10 seconds. If longer exercise is used (e.g., 30 seconds), the increment may not reach the threshold criteria of a 100% increase in some patients. In the EMG laboratory, this marked post-exercise facilitation of the CMAP is the electrical correlate of the clinical facilitation of muscle strength and reflexes seen after brief exercise. Somewhat confusing in LEMS is the issue of slow RNS (3 Hz) before and after brief exercise. In both situations, there will be a decremental response. However, after brief exercise, the baseline CMAP is significantly larger (i.e., an incremental response) compared with the pre-exercise CMAP (Figure 34–7).
Box 34–3
Electrophysiologic Evaluation of Lambert–Eaton Myasthenic Syndrome
1. Routine motor and sensory nerve conduction studies. Perform routine motor and sensory nerve conduction studies in at least two nerves, preferably a motor and sensory nerve in one upper and one lower extremity. CMAP amplitudes usually are diffusely low or borderline, with normal latencies and conduction velocities.
2. Repetitive nerve stimulation (RNS) and exercise testing. To look for facilitation, either perform high-frequency (30–50 Hz) RNS or record a CMAP with distal stimulation before and after 10 seconds of maximal voluntary exercise. Exercise testing is better tolerated by patients and is always preferable to fast RNS unless the patient cannot cooperate (e.g., sedated patient, young child). Any increment greater than 40% is abnormal (calculated by [100 × (Highest amplitude − Initial amplitude)/Initial amplitude]). Most patients with LEMS have increments greater than 100%. Increments between 40 and 100% are equivocal for presynaptic disorders.
Perform slow RNS (3 Hz) on at least one proximal and one distal motor nerve as in MG (see Box 34–2). Decrements on slow RNS are common in LEMS but cannot differentiate this disorder from MG.
3. Needle electromyography (EMG). Perform routine needle EMG of distal and proximal muscles, especially weak muscles. Needle examination is usually normal. Similar to MG, motor unit action potentials may be unstable or short, small, and polyphasic with normal or early recruitment.
4. Single-fiber EMG (usually not required in LEMS). If performed, findings will be consistent with a neuromuscular junction disorder (increased jitter and blocking), but single-fiber EMG cannot routinely differentiate LEMS from other disorders of the neuromuscular junction.
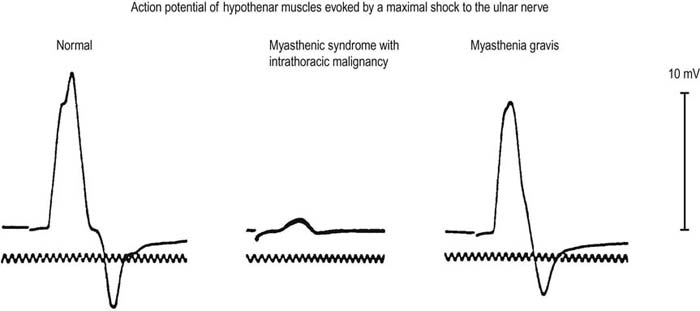
FIGURE 34–5 Compound muscle action potential amplitude in disorders of the neuromuscular junction.
(Reprinted from EH Lambert, et al. Myasthenic syndrome occasionally associated with bronchial neoplasm: neurophysiologic studies. In Viets HR, ed. Myasthenia gravis: the Second International Symposium. Springfield, IL: Thomas, 1961:363. With permission.)

FIGURE 34–6 Rapid repetitive nerve stimulation (50 Hz) in Lambert–Eaton myasthenic syndrome.
Note marked increment (>250%) in compound muscle action potential amplitude.
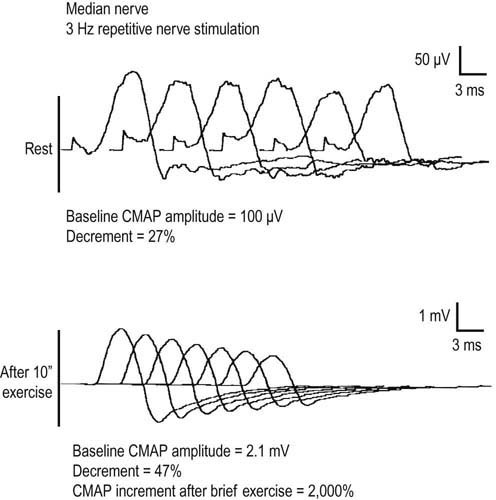
The diagnosis of LEMS is based on the clinical findings and a diagnostic study demonstrating marked post-exercise facilitation. Prior history of SCLC in a patient with proximal weakness should suggest the diagnosis. The diagnosis of LEMS must be suspected in any patient whose nerve conduction studies show low or borderline low CMAP amplitudes at rest with normal sensory responses. It is not unusual for these findings to be misinterpreted as neuropathy (low amplitudes, normal conduction velocities), even though the sensory potentials are normal. If a patient with LEMS also has a superimposed neuropathy, either from an unrelated cause or as a paraneoplastic process from underlying carcinoma, the diagnosis of LEMS is missed frequently. In any patient with low or borderline low CMAP amplitudes at rest, the distal motor stimulation should be repeated after 10 seconds of maximal exercise looking for post-exercise facilitation of the CMAP (Figure 34–8). Complicating the issue further is the fact that slow RNS in LEMS causes a decremental CMAP response similar to the decrement seen in MG. Many patients with LEMS initially are misdiagnosed with MG when their nerve conduction and RNS studies do not include exercise testing.
Botulism
Electrophysiologic Evaluation
The pathophysiology of botulism is presynaptic blocking of ACH, similar to LEMS. Likewise, the electrophysiologic evaluation and findings in botulism and LEMS are similar (Box 34–4). Sensory conduction studies are normal. CMAP amplitudes are decreased with normal latencies and conduction velocities. A decremental response may be seen with slow RNS. An incremental response characteristically occurs after brief exercise (10 seconds) or fast RNS (30–50 Hz). This finding usually is present in mild or early cases. However, the amount of increment is often not as dramatic as in LEMS, and many times it is lower than 100%. Note, in addition, that in severe botulism, if the amount of ACH release has dropped severely below threshold, even facilitation with rapid RNS or brief exercise may not result in a threshold response, and no increment occurs in the CMAP amplitude. Thus, the lack of an incremental response to rapid RNS or brief exercise cannot completely exclude the diagnosis of botulism.
Box 34–4
Electrophysiologic Evaluation of Botulism
1. Routine motor and sensory nerve conduction studies. Perform routine motor and sensory nerve conduction studies in at least two nerves, preferably a motor and sensory nerve in one upper and one lower extremity. CMAP amplitudes usually are diffusely low in amplitude or absent. Latencies and conduction velocities are normal.
2. Repetitive nerve stimulation (RNS) and exercise testing. To look for facilitation, either perform high-frequency (30–50 Hz) RNS or record a CMAP with distal stimulation before and after 10 seconds of maximal voluntary exercise. Exercise testing is better tolerated by patients and is always preferable to fast RNS unless the patient cannot cooperate (e.g., sedated patient, young child). Any increment greater than 40% is abnormal (calculated by [100 × (Highest amplitude − Initial amplitude)/Initial amplitude]). Most patients with botulism have increments greater than 100%. Increments between 40 and 100% are equivocal for presynaptic disorders. However, in severe botulism, the neuromuscular junction may be so blocked that facilitation following exercise or rapid RNS may not be seen, and there will be no CMAP increment. The absence of an increment does not exclude the diagnosis of botulism.
Perform slow RNS (3 Hz) on at least one proximal and one distal motor nerve as in myasthenia gravis (see Box 34–2). Decrements on slow RNS may be seen in botulism.
3. Needle electromyography (EMG). Perform routine needle EMG of distal and proximal muscles, especially weak muscles. Needle examination usually is markedly abnormal. After 4 to 5 days, denervating potentials (fibrillation potentials, positive sharp waves) are common. Motor unit action potentials usually are unstable or often short, small, and polyphasic, similar to myopathic motor unit action potentials. Recruitment may be normal, early, or reduced. If every muscle fiber of a motor unit is blocked by the toxin, there is an effective loss of motor units and decreased recruitment.
The needle EMG of botulism is quite interesting. Fibrillation potentials and positive sharp waves, signs of denervation, are common (Figure 34–9). Botulinum toxin is such a potent NMJ blocker that the muscle fibers are effectively chemo-denervated. Similar to other NMJ disorders, MUAPs may be normal or small, short and polyphasic, similar to myopathic MUAPs. Depending on the severity, recruitment may be normal, early, or reduced. The latter may occur if every muscle fiber of a motor unit is blocked by the botulinum toxin, effectively reducing the number of motor units. Likewise, SF-EMG shows increased jitter and blocking, signifying the underlying NMJ dysfunction.
Congenital Myasthenic Syndromes
 Example Cases
Example Cases
 Case 34–1
Case 34–1
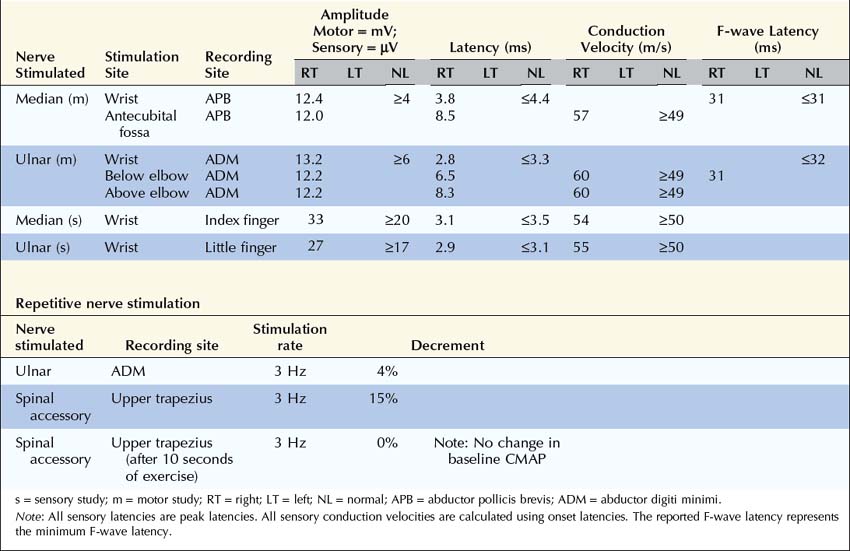
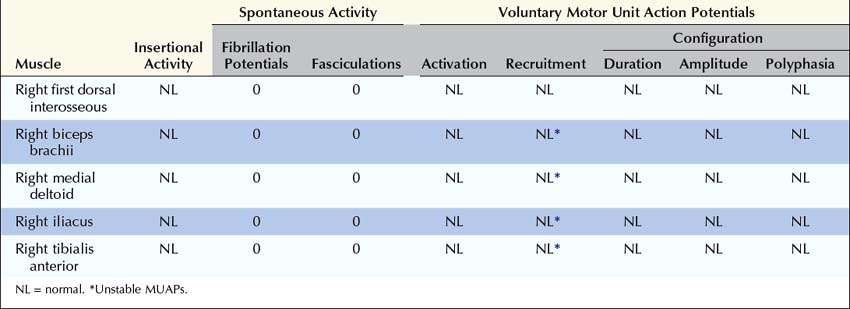
Summary
Why was no Decrement Present in the Ulnar Nerve While One was Present in the Spinal Accessory Nerve?
Are the Nerve Conduction Studies and Electromyography Consistent with the Diagnosis of Lambert–Eaton Myasthenic Syndrome?
 Case 34–2
Case 34–2
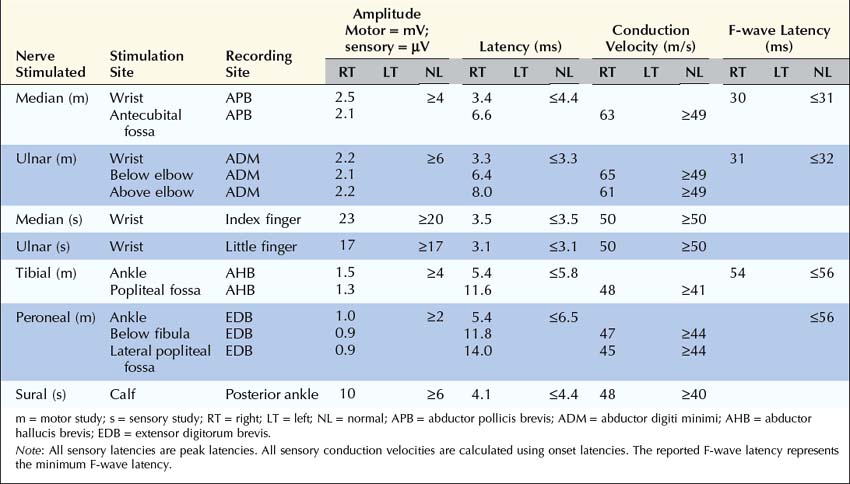
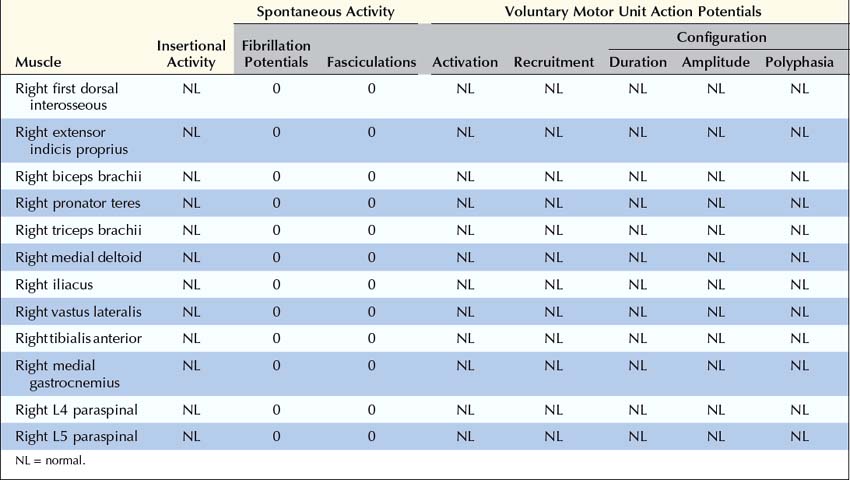

 Case 34–3
Case 34–3
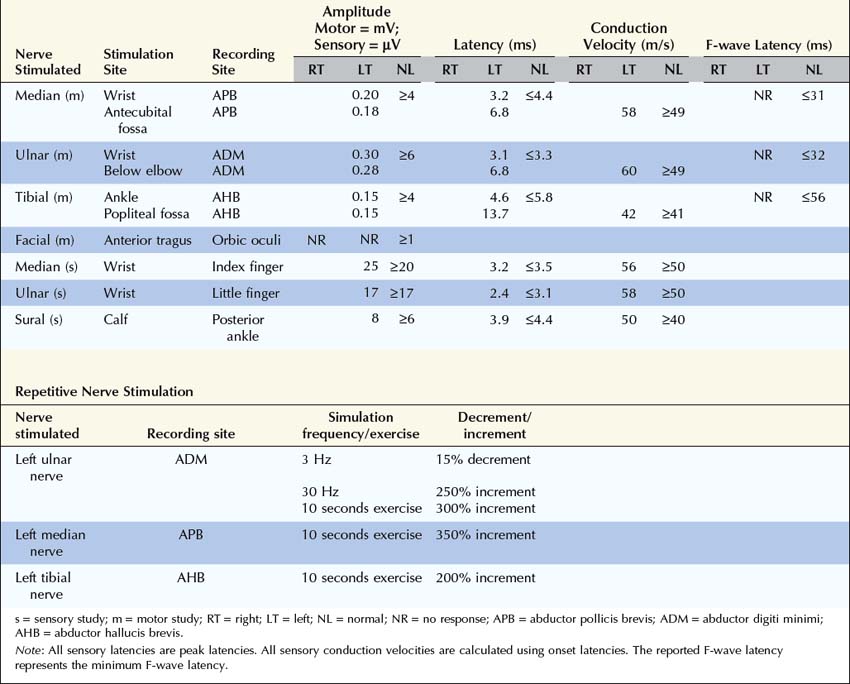
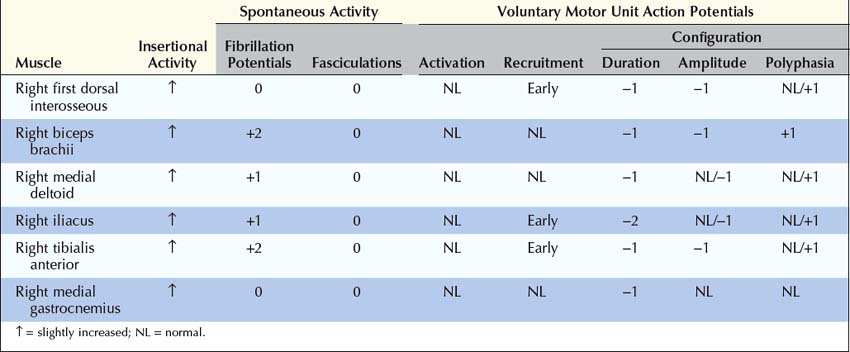
Summary
This case raises several important questions.
Are the Clinical and Electrophysiologic Findings Consistent with a Diagnosis of Myasthenia Gravis or Lambert–Eaton Myasthenic Syndrome?
Alseth E.H., Maniaol A.H., Elsais A., et al. Investigation for RAPSN and DOK-7 mutations in a cohort of seronegative myasthenia gravis patients. Muscle Nerve. 2011;43(4):574–577.
Engel A.G. Congenital myasthenic syndromes. Neurol Clin. 1994;2:401.
Engel A.G. Lambert–Eaton myasthenic syndrome. Ann Neurol. 1987;22:193.
Engel A.G. Congenital myasthenic syndromes. In: Katirji B., Kaminski H.J., Preston D.C., et al. Neuromuscular disorders in clinical practice. Boston: Butterworth-Heinemann, 2002.
Gilchrist J.M. Single fiber EMG. In: Katirji B., Kaminski H.J., Preston D.C., et al. Neuromuscular disorders in clinical practice. Boston: Butterworth-Heinemann, 2002.
Hantay D., Richard P., Koeniga J., et al. Congenital myasthenic syndromes. Curr Opin Neurol. 2004;17:539–551.
Hatanaka Y., Oh S.J. Ten-second exercise is superior to 30-second exercise for post-exercise facilitation in diagnosing Lambert–Eaton myasthenic syndrome. Muscle Nerve. 2008;37:572–575.
Hubbard J.I. Microphysiology of vertebrate neuromuscular transmission. Physiol Rev. 1973;53(Suppl):674.
Jablecki C. AAEM case report no. 3: myasthenia gravis. AAEM. 1991.
Jablecki C. Lambert–Eaton myasthenic syndrome. Muscle Nerve. 1984;7:250.
Keesey J.C. AAEM minimonograph, no. 33: electrodiagnostic approach to defects of neuromuscular transmission. AAEM. 1989.
Kimura J. Electrodiagnosis in diseases of nerve and muscle. Philadelphia: FA Davis; 1989.
Macdonell R.A., Rich J.M., Cros D., et al. The Lambert–Eaton myasthenic syndrome: a cause of delayed recovery from general anesthesia. Arch Phys Med Rehabil. 1992;73:98.
Milone M., Shen X.M., Selcen D., et al. Myasthenic syndrome due to defects in rapsyn: clinical and molecular findings in 39 patients. Neurology. 2009;73:228–235.
ONeill J.H., Murray N.M.F., Newsom-Davis J. The Lambert–Eaton myasthenic syndrome: a review of 50 cases. Brain. 1988;111:577.
Pasnoor M., Wolfe G.I., Nations S., et al. Clinical findings in MuSK-antibody positive myasthenia gravis: a U.S. experience. Muscle Nerve. 2010;41:370–374.
Shapiro B.E., Soto O., Shafquat S., et al. Adult botulism. Muscle Nerve. 1997;20:100.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
34 Neuromuscular Junction Disorders
Disorders affecting the neuromuscular junction (NMJ) are among the most interesting and rewarding seen in the electromyography (EMG) laboratory. These disorders are generally pure motor syndromes that usually preferentially affect proximal, bulbar, or extraocular muscles. They are confused occasionally with myopathies. With knowledge of normal NMJ physiology (see Chapter 6), most of the abnormalities affecting the NMJ can be differentiated using a combination of nerve conduction studies, repetitive stimulation, exercise testing, and needle EMG.
NMJ disorders can be classified into immune-mediated, toxic or metabolic, and congenital syndromes (Box 34–1). They usually are distinguished by their clinical and electrophysiologic findings (Tables 34–1 and 34–2). All are uncommon, but among them, myasthenia gravis (MG) and Lambert–Eaton myasthenic syndrome (LEMS) are the disorders most often encountered in the EMG laboratory. Both are immune-mediated disorders. In MG the autoimmune attack is postsynaptic; in LEMS the presynaptic membrane is the target of attack. Every electromyographer must understand the electrophysiology of these disorders so that appropriate electrodiagnostic tests can be applied and the correct diagnosis not overlooked.
Box 34–1
Disorders of the Neuromuscular Junction
Myasthenia Gravis
Electrophysiologic Evaluation
Like other disorders affecting the NMJ, the electrophysiologic evaluation of MG involves routine nerve conduction studies, repetitive nerve stimulation (RNS), exercise testing, routine EMG, and, in some cases, single-fiber EMG (SF-EMG) (Box 34–2).
Box 34–2
Electrophysiologic Evaluation of Myasthenia Gravis
1. Routine motor and sensory nerve conduction studies. Perform routine motor and sensory nerve conduction studies, preferably a motor and sensory nerve in one upper and one lower extremity. CMAP amplitudes should be normal. If CMAP amplitudes are low or borderline, repeat distal stimulation immediately after 10 seconds of exercise to exclude a presynaptic NMJ transmission disorder (e.g., Lambert–Eaton myasthenic syndrome).
2. Repetitive nerve stimulation (RNS) and exercise testing. Perform slow RNS (3 Hz) on at least one proximal and one distal motor nerve. Always try to study weak muscles. If any significant decrement (>10%) is present, repeat to ensure decrement is reproducible. If there is no significant decrement at baseline, exercise the muscle for 1 minute, and repeat RNS at 1, 2, 3, and 4 minutes looking for a decrement, secondary to post-exercise exhaustion. If at any time a significant decrement is present (at baseline or following post-exercise exhaustion), exercise the muscle for 10 seconds and immediately repeat RNS, looking for post-exercise facilitation (repair of the decrement).
3. Needle electromyography (EMG). Perform routine needle EMG of distal and proximal muscles, especially weak muscles. Patients with moderate to severe myasthenia gravis may display unstable or short, small, polyphasic motor unit action potentials. Recruitment is normal or early. Needle EMG must exclude severe denervating disorders or myotonic disorders, which may display an abnormal decrement on RNS.
4. Single-fiber EMG (SF-EMG). If the above are normal or equivocal in a patient strongly suspected of having myasthenia gravis, perform SF-EMG in the extensor digitorum communis and, if necessary, one other muscle, looking for jitter and blocking. It is always best to study a weak muscle. Normal SF-EMG in a clinically weak muscle excludes an NMJ disorder.
CMAP, compound muscle action potential; NMJ, neuromuscular junction.
Repetitive Nerve Stimulation
After the routine nerve conduction studies are completed, RNS studies are performed (see Chapter 6). These studies are abnormal in more than 50 to 70% of patients with generalized MG but often are normal in patients with the restricted ocular form of MG. A decremental response on RNS is the electrical correlate of clinical muscle fatigue and weakness. In normal subjects, slow RNS (3 Hz) results in little or no decrement of the CMAP, whereas in MG, a CMAP decrement of 10% or more is characteristically seen (Figure 34–1A). Both distal and proximal nerves should be tested. Although distal nerves are technically easier to study, the diagnostic yield increases with stimulation of proximal nerves (e.g., spinal accessory or facial nerves). This is not unexpected, because the proximal muscles usually are much more involved clinically than the distal ones. Facial RNS is especially important to perform in suspected anti-MuSK MG, where the yield of finding an abnormal decrement is much higher when examining a facial muscle than a limb muscle (probably reflecting the severe facial and bulbar involvement in some patients with anti-MuSK MG).
Exercise Testing
Exercise testing should be routinely used with all RNS studies (see Chapter 6). If there is no significant decrement on RNS studies at baseline (<10% decrement), the patient should perform 1 minute of exercise, followed by RNS at 1-minute intervals for the next 3 to 4 minutes, looking for a CMAP decrement secondary to post-exercise exhaustion. If at any time, either at baseline or following exercise, a significant decrement develops, the patient should perform a brief 10-second maximum isometric contraction, immediately followed by slow RNS, looking for an increment in the CMAP and “repair” of the decrement secondary to post-exercise facilitation (Figure 34–1).
Electromyography
Unstable MUAPs (see Chapter 15) occur when individual muscle fibers are either blocked or come to action potential at varying intervals, which leads to MUAPs that change in configuration from impulse to impulse. If some muscle fibers of a motor unit are blocked and never come to action potential, the motor unit effectively loses muscle fibers, becoming short, small, and polyphasic, similar to MUAPs seen in myopathy. Otherwise, the needle EMG findings in NMJ disorders usually are normal. In general, fibrillation potentials and other abnormal spontaneous activity are not seen in NMJ disorders, with the important exception of botulism (see section on Botulism).
Single-fiber Electromyography
When a motor axon is depolarized, the action potential normally travels distally and excites all the muscle fibers within that motor unit at more or less the same time (Figure 34–2). This variation in the time interval between the firing of adjacent single muscle fibers from the same motor unit is termed jitter and primarily reflects variation in NMJ transmission time. If the NMJ is compromised, the time it takes for the endplate potential to reach threshold is prolonged, which results in greater-than-normal variation between firing of adjacent muscle fibers. If the prolongation is severe enough, the muscle fiber may never reach action potential, resulting in blocking of the muscle fiber.
The goal of SF-EMG is to study two adjacent single muscle fibers, known as a pair, from the same motor unit. This is accomplished by changing the filters on the EMG machine and using a specialized SF-EMG needle. The low-frequency filter (high-pass) is increased to 500 Hz (normally 10 Hz in routine EMG). By using a high-pass filter of 500 Hz, the amplitudes of distant muscle fiber potentials are attenuated while those of the nearby fibers are preserved. The SF-EMG needle is a specially constructed needle with the active electrode (G1) located in a port along the posterior shaft of the needle and with a smaller leading surface area than the conventional concentric needle electrode (Figure 34–3). The reference electrode (G2) is the needle shaft. The result of these two modifications is that single-fiber muscle action potentials are recorded only if they are within 200 to 300 µm of the needle. The needle is placed in the muscle, and the patient is asked to activate the muscle in an even and constant fashion. The needle is moved until a single muscle fiber potential is located. With this single muscle fiber potential triggered on a delay line, the needle is slightly and carefully moved or rotated to look for a second potential that is time locked to the first potential (signifying that it is from the same motor unit).
More recently, the regular disposable concentric EMG needle has been used for SF-EMG studies. The standard SF-EMG needle is expensive, and needs to be surgically sanitized between patients. Thus, the cost of the standard SF-EMG needle, along with the theoretical risk of transmitting infection (including prion diseases) despite sanitizing the needle, have prompted this change. In general, the values for jitter are comparable between the traditional SF and the concentric EMG needles. Single-fiber potentials should be accepted for analysis only if the potential is at least 200 µV in amplitude with a rise time of less than 300 µs. If a time-locked second potential is located, an interpotential interval between the two potentials (i.e., the pair) can be measured. By recording multiple consecutive firings of the muscle fiber action potential pairs, the difference between consecutive interpotential intervals can be calculated. This variation between consecutive interpotential intervals is the jitter. By recording 50 to 100 subsequent potentials, the mean consecutive difference (MCD), a measure of jitter, can be calculated between the triggered potential and the time-locked second single muscle fiber potential. Most modern EMG machines have programs that automatically perform the MCD calculation. This procedure is then repeated until 20 separate single-fiber pairs are collected, to calculate a mean MCD. This value is compared with the normal mean MCD for the muscle studied and the patient’s age (Table 34–3). There is also an upper limit of normal jitter for an individual pair, based on the muscle studied and the patient’s age. To call the latter abnormal, more than 10% of the pairs must exceed the limit (e.g., for 20 pairs, at least two must be abnormal). To make a diagnosis of an NMJ disorder, either the mean jitter must be abnormal or the upper limit of normal jitter must be abnormal in more than 10% of individual pairs. However, in most NMJ disorders, both will be abnormal. Increased jitter is consistent with an NMJ disorder (Figure 34–4). In addition to increased jitter, blocking may be seen on SF-EMG. Two time-locked, single-fiber muscle potentials from the same motor unit normally fire together. If the triggered potential fires steadily while the second potential fires only intermittently, blocking is occurring. Blocking, which is another marker of NMJ disease, usually occurs only when the jitter is markedly prolonged (e.g., MCD > 80–100 µs).
