[level-membership-for-basic-science-category]
Chapter 22 Neuromuscular Junction
Skeletal Muscle
The most intensively studied effector endings are those that innervate muscle, particularly skeletal muscle. All neuromuscular (myoneural) junctions are axon terminals of somatic motor neurones. They are specialized for the release of neurotransmitter onto the sarcolemma of skeletal muscle fibres, causing a change in their electrical state that leads to contraction. Each axon branches near its terminal and subsequently innervates from several to hundreds of muscle fibres, depending on the precision of motor control required. The detailed structure of a motor terminal varies with the type of muscle innervated. Two major endings are recognized: those typical of extrafusal muscle fibres, and endings on the intrafusal fibres of neuromuscular spindles. In the former, each axon terminal usually ends midway along a muscle fibre in a discoidal motor end-plate (Figs 22.1–22.3). This type usually initiates action potentials, which are rapidly conducted to all parts of the muscle fibre. In the latter, the axon has numerous subsidiary branches that form a cluster of small expansions extending along the muscle fibre. In the absence of propagated muscle excitation, these excite the fibre at several points. Both types are associated with a specialized receptive region of the muscle fibre, the sole plate, where a number of muscle cell nuclei are grouped within the granular sarcoplasm.
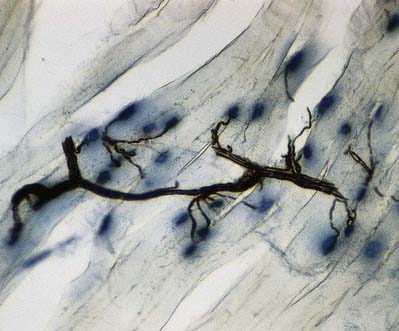
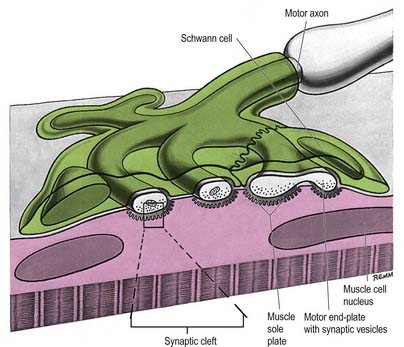
Fig. 22.2 Neuromuscular junction. Note the axonal motor end-plate and the deeply infolded sarcolemma.
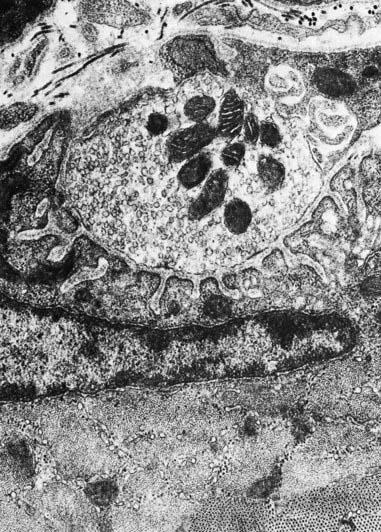
The sole plate contains numerous mitochondria, endoplasmic reticulum and Golgi complexes (see Figs 22.2, 22.3). The neuronal terminal branches are plugged into shallow grooves in the surface of the sole plate (primary clefts), from which numerous pleats extend for a short distance into the underlying sarcoplasm (secondary clefts). The axon terminal contains mitochondria and many clear 60-nm spherical vesicles, similar to those in presynaptic boutons, clustered over the zone of membrane apposition. The motor terminal is ensheathed by Schwann cells whose cytoplasmic projections extend into the synaptic cleft. The plasma membranes of the nerve terminal and the muscle cell are separated by a 30- to 50-nm gap, with a basal lamina interposed. The basal lamina follows the surface folding of the sole plate membrane into the secondary clefts. It contains specialized components, including specific isoforms of type IV collagen and laminin and agrin, a heparan sulphate proteoglycan. Endings of fast and slow twitch muscle fibres differ in detail: the sarcolemmal grooves are deeper, and the presynaptic vesicles more numerous, in the fast fibres.
Conduction of the Nervous Impulse
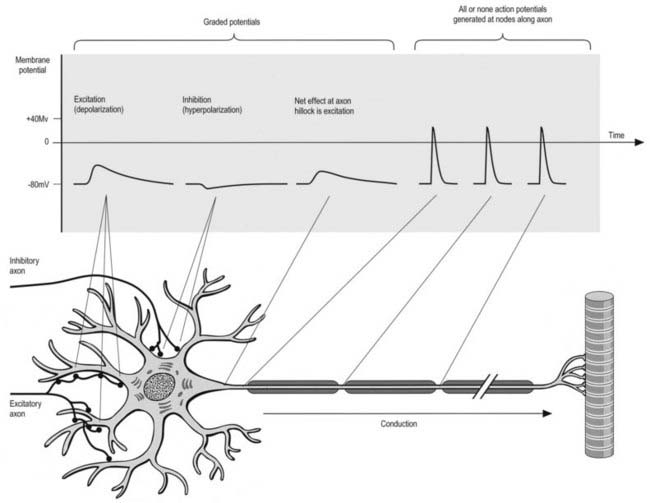
All cells generate a steady electrochemical potential across their plasma membranes (a membrane potential) because of the different ionic concentrations inside and outside the cell (Fig. 22.4). Neurones use minute fluctuations in this potential to receive, conduct and transmit information across their surfaces.
Autonomic Motor Terminations
Adrenergic sympathetic postganglionic terminals contain dense-core vesicles. Cholinergic terminals, which are typical of all parasympathetic and some sympathetic endings, contain clear spherical vesicles like those in motor end-plates of skeletal muscle. A third category of autonomic neurones has non-adrenergic, non-cholinergic endings that contain a wide variety of chemicals with transmitter properties. Conjugated purine (ATP, a nucleoside), is probably the neurotransmitter at these terminals, which are thus classed as purinergic. Typically, their axons contain large (80- to 200-nm), dense, opaque vesicles congregated in varicosities at intervals along axons. They are formed in many sites, including the external muscle layers and sphincters of the alimentary tract, lungs, vascular walls, urogenital tract and CNS. In the intestinal wall, neuronal somata lie in the myenteric plexus, and their axons spread caudally for a few millimetres, mainly to innervate circular muscle. Purinergic neurones are under cholinergic control from preganglionic sympathetic neurones. Their endings mainly hyperpolarize smooth muscle cells, causing relaxation (e.g. preceding peristaltic waves), opening sphincters and probably causing reflex distension in gastric filling.
Autonomic efferents also innervate glands, myoepithelial cells and adipose and lymphoid tissue.
Action Potential
The action potential is a brief complete reversal of polarity that propagates itself along membranes. It depends on an initial influx of sodium ions, which causes a reversal of polarity to about 40 mV (positive inside), followed by a rapid return to the resting potential as potassium ions flow out (the detailed mechanism differs somewhat between CNS and PNS). The whole process is completed in approximately 5 msec. For a particular neurone, the size and duration of action potentials are always the same (described as all or none), no matter how much a stimulus exceeds the threshold value.
Axonal conduction is naturally unidirectional, from dendrites and somata to axon terminals. When an action potential reaches the axon terminals, it causes depolarization of the presynaptic membrane, and as a result, quanta of neurotransmitter (corresponding to the content of individual vesicles) are released to change the degree of excitation of the next neurone, muscle fibre or glandular cell (Kandel and Schwartz 2000).
Kandel E.R., Schwartz J.H. Principles of Neural Science, fourth ed. New York: McGraw-Hill; 2000.
Lehmann-Horn F., Jurkat-Rott K., Rudel R. Periodic paralysis: understanding channelopathies. Neurology. 2006;66(Suppl. 1):62-69.
Miller A.E., editor. Myasthenic disorders and ALS: CONTINUUM Lifelong learning in neurology. Philadelphia: Lippincott Williams & Wilkins, 2009.
Ohno K., Engel A.G. Congenital myasthenic syndromes: genetic defects of the neuromuscular junction. Neurology. 2006;66(Suppl. 1):77-88.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 22 Neuromuscular Junction
Skeletal Muscle
The most intensively studied effector endings are those that innervate muscle, particularly skeletal muscle. All neuromuscular (myoneural) junctions are axon terminals of somatic motor neurones. They are specialized for the release of neurotransmitter onto the sarcolemma of skeletal muscle fibres, causing a change in their electrical state that leads to contraction. Each axon branches near its terminal and subsequently innervates from several to hundreds of muscle fibres, depending on the precision of motor control required. The detailed structure of a motor terminal varies with the type of muscle innervated. Two major endings are recognized: those typical of extrafusal muscle fibres, and endings on the intrafusal fibres of neuromuscular spindles. In the former, each axon terminal usually ends midway along a muscle fibre in a discoidal motor end-plate (Figs 22.1–22.3). This type usually initiates action potentials, which are rapidly conducted to all parts of the muscle fibre. In the latter, the axon has numerous subsidiary branches that form a cluster of small expansions extending along the muscle fibre. In the absence of propagated muscle excitation, these excite the fibre at several points. Both types are associated with a specialized receptive region of the muscle fibre, the sole plate, where a number of muscle cell nuclei are grouped within the granular sarcoplasm.
Fig. 22.2 Neuromuscular junction. Note the axonal motor end-plate and the deeply infolded sarcolemma.
The sole plate contains numerous mitochondria, endoplasmic reticulum and Golgi complexes (see Figs 22.2, 22.3). The neuronal terminal branches are plugged into shallow grooves in the surface of the sole plate (primary clefts), from which numerous pleats extend for a short distance into the underlying sarcoplasm (secondary clefts). The axon terminal contains mitochondria and many clear 60-nm spherical vesicles, similar to those in presynaptic boutons, clustered over the zone of membrane apposition. The motor terminal is ensheathed by Schwann cells whose cytoplasmic projections extend into the synaptic cleft. The plasma membranes of the nerve terminal and the muscle cell are separated by a 30- to 50-nm gap, with a basal lamina interposed. The basal lamina follows the surface folding of the sole plate membrane into the secondary clefts. It contains specialized components, including specific isoforms of type IV collagen and laminin and agrin, a heparan sulphate proteoglycan. Endings of fast and slow twitch muscle fibres differ in detail: the sarcolemmal grooves are deeper, and the presynaptic vesicles more numerous, in the fast fibres.
Conduction of the Nervous Impulse
All cells generate a steady electrochemical potential across their plasma membranes (a membrane potential) because of the different ionic concentrations inside and outside the cell (Fig. 22.4). Neurones use minute fluctuations in this potential to receive, conduct and transmit information across their surfaces.
[/not-level-membership-for-basic-science-category]
