Neuromuscular Disorders
Principles of Disease
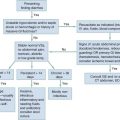
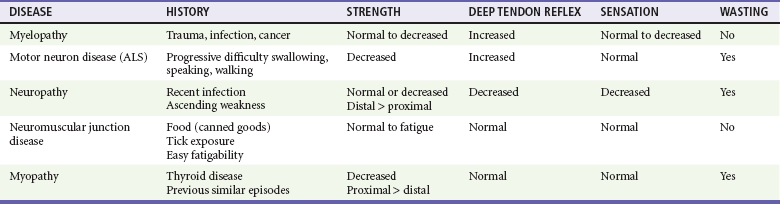
The neuromuscular unit has four components: the anterior horn cells of the spinal cord, the peripheral nerve, the neuromuscular junction, and the muscle innervated. The level of the pathologic process determines associated signs and symptoms (Table 108-1). Myelopathies involve the spinal cord; radiculopathies involve the nerve roots as they leave the spinal cord; neuropathies involve the peripheral nerves; and myopathies involve the muscle. The use of physical signs to differentiate these disorders is discussed in Chapter 13.
Clinical Findings
Physical Examination
The assessment of vital signs is important because some causes of weakness may result in dysregulation of the autonomic system. A systematic neurologic examination assesses the patient’s mental status, cranial nerves, motor function, sensory function, deep tendon reflexes, and coordination, including cerebellar function. The motor examination begins by determining whether the weakness is unilateral or bilateral and which muscle groups are involved. Key components of the examination include motor strength, muscle bulk, and presence of fasciculations. Box 108-1 provides the grading system used in motor strength assessment. Table 108-2 provides the findings used to distinguish upper motor neuron from lower motor neuron processes.

Differential Consideration
Myelopathies are spinal cord disorders that are manifested with signs of upper motor neuron dysfunction, such as muscle weakness with increased spinal reflexes, including an extensor plantar reflex (Babinski’s response). Muscle tone ranges from normal to slightly increased, eventually leading to spasticity. There may be bladder and bowel involvement. When sensory findings are present, they often define the level of the lesion. The presence of back pain suggests a compressive lesion, such as a herniated intravertebral disk, epidural hematoma, abscess, or tumor. Acute, painless spinal cord lesions include transverse myelitis and spinal cord infarction. Myelopathies are discussed in Chapter 106.
Neuropathies
Weakness from a neuropathy is often noted first in distal muscles and then ascends. Decreased grip strength and footdrop are common presentations. Muscle tone ranges from slightly diminished to flaccid, and deep tendon reflexes are diminished or absent. Patients exhibit varying degrees of altered sensation, muscle wasting, and fasciculations, depending on the duration of the symptoms. Disorders in the differential diagnosis include GBS, toxic neuropathies, diabetic neuropathy, and tick paralysis (which is caused by inhibition of both nerve conduction and function of the neuromuscular junction). Neuropathies are discussed in Chapter 107.
Specific Disorders
Disorders of the Neuromuscular Junction
Myasthenia Gravis
Perspective.: MG affects approximately 60,000 Americans.1 The age at onset is bimodal; women are most commonly affected between the ages of 20 and 40 years and men between 50 and 70 years. Whereas new cases of MG are occasionally diagnosed in the emergency department (ED), it is much more common for patients with established disease to present with exacerbations of their disorder, often caused by precipitating factors.
Principles of Disease.: MG results from autoantibodies directed against the nicotinic AChR at the neuromuscular junction or from antibodies against muscle-specific tyrosine kinase. This leads to destruction of AChRs with a decrease in the total number of available receptors. The autoantibodies further compete with ACh for binding at remaining receptors. With repeated stimulation, fewer and fewer receptor sites are available for ACh binding, and fatigue develops.
Clinical Features.: Patients with MG present with easy fatigability as the result of progressive weakness with repeated activity of affected muscle groups. Ocular symptoms are often the first manifestation of MG; typical symptoms are ptosis, diplopia, and blurred vision. Ocular muscle weakness is the first sign in up to 40% of patients, although 85% of patients with MG eventually have ocular involvement. When ptosis is present, it is often worse toward the end of the day. Respiratory failure is rarely the initial symptom of MG. Even so, up to 17% of patients may have weakness of the muscles of respiration.2 Bulbar muscles may be involved, producing dysarthria or dysphagia.
Lambert-Eaton myasthenic syndrome is a rare disorder. Almost 50% of cases are associated with small cell carcinoma of the lung. Autoantibodies cause inadequate release of ACh from nerve terminals, affecting both nicotinic and muscarinic receptors. With repeated stimulation, the amount of ACh in the synaptic cleft increases, leading to an increase in strength, the opposite of that seen with MG. The classic syndrome includes weakness that improves with use of muscles, particularly proximal hip and shoulder muscles; hyporeflexia; and autonomic dysfunction, most commonly seen as dry mouth.3 Management primarily focuses on treatment of the underlying neoplastic disorder, although intravenous immune globulin (IVIG) has been reported to be useful.4
New-Onset Myasthenia Gravis.: The diagnosis of MG is based on clinical findings and a combination of serologic testing, electromyographic testing, and the bedside edrophonium or ice bag test. Serum testing for AChR antibodies is positive in 80 to 90% of patients with MG, but it is not available in the ED setting.
The edrophonium test and ice bag test are similar to perform, and the results are based on their effect on the ptosis seen in patients with suspected MG. The production of edrophonium was discontinued in early 2008, and it will no longer be available once current stores are depleted. Edrophonium is a short-acting acetylcholinesterase-blocking agent that produces an increase of ACh in the synaptic cleft and a reduction in ptosis after intravenous administration. With the ice bag test, cooling decreases symptoms in MG, whereas heat exacerbates symptoms. In both tests, the change in the amount of ptosis is measured before and after administration of edrophonium or application of an ice bag. The distance from the upper to the lower eyelid in the most severely affected eye is measured first. If edrophonium is given, an intravenous test dose of 1 to 2 mg is given first as some patients have a severe reaction. If no adverse reaction is found and the patient does not dramatically improve in 30 to 90 seconds, a second dose of 3 mg is given. If there is still no response, a final dose of 5 mg is given for a total maximum dose of 10 mg.5 Atropine should be available at the bedside during the test. Because of the potential for cholinergic-induced increased airway secretions, this test should be used with caution in asthmatics and patients with chronic obstructive pulmonary disease. If an ice pack is used, it is applied to the affected eye for approximately 2 minutes, and the distance between the lids is measured again. A prospective evaluation of the ice bag approach found the test result to be positive (an improvement in distance of at least 2 mm) in 80% of patients with MG and no change in patients without MG.6
Myasthenic Crisis.: Myasthenic crisis is defined as respiratory failure leading to mechanical ventilation. It occurs in 15 to 20% of patients with MG,7 usually within the first 2 years of disease onset. Although it is potentially life-threatening, the mortality from this complication of MG has declined dramatically with aggressive care in the intensive care unit and the use of plasmapheresis or immunomodulatory therapy with high-dose IVIG and corticosteroids.
Crises are most often precipitated by underlying infection, aspiration, and medication changes, such as stopping anticholinergic medications or taking a new medication that precipitates weakness. Other precipitants can be surgery and pregnancy (Box 108-2). A precipitant may not be identified in up to 30% of cases.8
The initial step in managing the patient in crisis is stabilization of the airway. Noninvasive ventilation with biphasic positive airway pressure may be effective in managing patients who need ventilatory support.9
All patients with MG who present to the ED should be assessed for signs of myasthenic crisis even when they do not complain of weakness. Many commonly used drugs can adversely affect patients with MG (see Box 108-2). A patient with stable MG who has an acute medical or surgical condition requires a full neurologic examination. The decision to admit or to discharge a patient with MG from the ED should take into account the potential for neurologic deterioration.
Cholinesterase Inhibitors.: Pyridostigmine (60-120 mg PO every 4-6 hours) and neostigmine (15-30 mg PO every 4-6 hours) prolong the presence and activity of ACh in the synaptic cleft. They are the backbone of chronic outpatient therapy and provide symptomatic improvement. The most common side effects are those of excessive cholinergic stimulation, such as increased airway secretions and increased bowel motility. At extremes, there may be bradycardia or even worsening of weakness, simulating a myasthenic crisis. These drugs are often used as adjunctive therapy to control symptoms while other therapy is being instituted, after which they are usually discontinued.10 Cholinergic drug therapy, such as the intravenous administration of pyridostigmine, is not recommended for the treatment of myasthenic crises in the ED because plasmapheresis and IVIG are both safe and highly effective therapies.
Immunosuppressant Drugs.: Immunosuppressant drugs are often used for the chronic control of MG. Although they have no role in the acute management of a myasthenic crisis, they may be started before extubation of a patient recovering from a crisis. Cochrane reviews in 2005 and 2007 found support for the use of corticosteroids but only limited evidence that cyclosporine, cyclophosphamide, and azathioprine improve MG.11,12 Studies suggest that rituximab, a monoclonal antibody that decreases B-cell function, has an emerging role.13 Of note, the initiation of corticosteroids in patients with moderate to severe weakness may actually precipitate a worsening of weakness or even myasthenic crisis.
Thymectomy.: Whereas the association between thymoma and MG is not fully elaborated, it is well known that thymectomy for patients with thymoma can lead to remission of MG or enable a reduction in other medications. Thymectomy for patients with MG without thymoma has been shown to have similar benefits and is recommended by the American Academy of Neurology for patients younger than 60 years, with remission or improvement seen in up to 50% of cases.14 The onset of improvement after thymectomy is often delayed for 2 to 5 years.
Immunomodulatory Therapy.: Plasmapheresis and IVIG can be used for patients with exacerbations of MG or preoperatively in patients with stable MG.
Plasmapheresis removes the AChR antibodies and other immune complexes from the blood. The fall in AChR levels is associated with improvement in symptoms of MG. There is a risk of complications from hypotension or anticoagulation. Although there are no randomized controlled studies, a review yielded many case series with short-term benefit, especially in myasthenic crisis, and it is recommended by the American Academy of Neurology.15
A review of IVIG trials found one randomized controlled trial of IVIG versus placebo that demonstrated the benefit of IVIG. Another trial failed to show a difference between IVIG and plasmapheresis.16 The decision to institute either therapy is based on the input of the consulting neurologist and the resources available at the admitting hospital. If plasmapheresis is not readily available for a patient with myasthenic crisis, IVIG should begin with 1 to 2 g/kg.
Botulism
Principles of Disease.: Botulism is a toxin-mediated illness that can cause weakness leading to respiratory insufficiency. In 2009 the Centers for Disease Control and Prevention (CDC) reported 121 cases of botulism in the United States: 9% food-borne, 69% infant botulism, 19% wound botulism, and 3% unknown etiology. Clostridium botulinum is an anaerobic, spore-forming bacterium.17 Three of eight known toxins produced by C. botulinum (types A, B, and E) cause human disease. There has been an increase in reported cases of wound botulism in Washington State, California, England, and Germany associated with injection drug use.18 Botulism is also thought to be a potential agent for bioterrorism. The botulinum toxin works by binding irreversibly to the presynaptic membrane of peripheral and cranial nerves, inhibiting the release of ACh at the peripheral nerve synapse. As new receptors are generated, the patient improves.
Clinical Features.: The botulinum toxin blocks both voluntary motor and autonomic functions. Because the disorder is at the neuromuscular junction, there is no pain or sensory deficit. The onset of symptoms is 6 to 48 hours after the ingestion of tainted food. There may or may not be accompanying signs and symptoms of gastroenteritis, with nausea, vomiting, abdominal cramps, diarrhea, or constipation. The classic feature of botulism is a descending, symmetrical, flaccid paralysis. Cranial nerves and bulbar muscles are affected first, causing diplopia, dysarthria, and dysphagia, followed later by generalized weakness. Because the toxin decreases cholinergic output, anticholinergic signs may be seen in the form of constipation, urinary retention, dry skin and eyes, and increased temperature. Pupils are often dilated and not reactive to light. This can be a point of differentiation from MG. Deep tendon reflexes are normal or diminished.
Infantile botulism results from the ingestion of C. botulinum spores that are able to germinate and produce toxin in the high pH of the gastrointestinal tract of infants. The same spores are not active in the gut of adults because of the lower pH. The CDC reports approximately 100 cases per year.19 Botulism spores can survive in honey, so it is recommended that honey not be fed to infants. The clinical presentation includes constipation, poor feeding, lethargy, and weak cry; consequently, this diagnosis must be included in the differential diagnosis of the floppy infant.
Diagnostic Strategies.: The diagnosis is made by both clinical findings and exclusion of other processes. The toxin can be identified in serum and stool, but the assay is not commonly available in most hospitals and requires a prolonged turnaround time. If the suspected food source is available, it should also be tested for the toxin.
Management.: Treatment is initially focused on stabilization of the airway and supportive measures. In 2010 the CDC announced a new heptavalent botulinum antitoxin (HBAT) that is now the only antitoxin available in the United States for non-infant botulism. For suspected cases and to obtain HBAT, clinicians should contact their state health departments. The CDC also maintains a botulism duty officer at the CDC Emergency Operations Center (telephone, 770-488-7100).20 An intravenous human botulism immune globulin (BabyBIG) has been developed for treatment of infantile botulism and is available through the California Department of Public Health Infant Botulism Treatment and Prevention Program on-call physician at 510-231-7600.21
Tick Paralysis
Principles of Disease.: The pathogenesis of tick paralysis, also known as tick toxicosis, is not fully understood. It is known that a toxin, ixovotoxin, is injected while the tick feeds. The toxin appears to decrease the release of ACh at the neuromuscular junction and also reduces nerve conduction velocity. It may also have effects at autonomic ganglia, leading to pupillary signs. Colorado reports on average one case per year, although in 2006 four cases were reported during 1 week.22
Clinical Features.: Tick paralysis is an acute, ascending, flaccid motor paralysis that can be confused with GBS, botulism, and MG. Symptoms usually begin 1 to 2 days after the female tick has attached and begun to feed, although delays of up to 6 days have been reported.23 There may be associated ocular signs, such as fixed and dilated pupils, that can help distinguish it from GBS.
Management.: The management is supportive care and tick removal. A tick can be removed by use of forceps to grasp it as closely as possible to the point of attachment. Care should be taken not to leave mouth parts in the patient’s tissue. Although symptoms may resolve rapidly after removal of the tick, supportive measures such as intubation should not be withheld pending resolution of symptoms.
Disorders of the Muscles
Inflammatory Disorders
Principles of Disease.: The most common inflammatory myopathies are polymyositis (PM) and dermatomyositis (DM). PM may be idiopathic in nature, occur secondary to infections (viral or bacterial), or be seen in conjunction with other disorders, such as sarcoidosis and hypereosinophilic syndromes. Inflammatory myopathies cause weakness, pain, and tenderness of the muscles involved.
Clinical Features.: DM and PM can occur at any adult age, although DM may also affect children. There is a slightly increased incidence in women. They can be associated with various malignant neoplasms, such as of the breast, ovary, lung, and gastrointestinal tract, and lymphoproliferative disorders. Proximal muscle weakness predominates and leads to complaints of difficulty in rising from a seated position or climbing stairs and weakness in lifting the arms over the head. There is often pain and tenderness in these proximal muscles as well. There is a decrease in reflexes that is in proportion to the decrease in strength. Fasciculations are not seen, and atrophy is a very late finding.
Diagnostic Strategies.: Electrolyte abnormalities must be ruled out and the serum CK level checked. The CK level should be interpreted in light of the entire clinical picture; an elevated CK level does not establish the cause of weakness as a myopathy because some neuropathies can also produce an elevated CK level. Similarly, a normal CK level does not rule out a myopathy as the cause of weakness. Electromyography and muscle biopsy are used to confirm the diagnosis.24
Management.: PM and DM are usually managed with oral prednisone in a dose of 1 to 2 mg/kg/day. When steroids prove ineffective and during acute exacerbations, cytotoxic drugs such as azathioprine and methotrexate are added. Fortunately, the degree of rhabdomyolysis seen with the inflammatory myopathies is not sufficient to cause renal impairment.
Metabolic Disorders
Perspective.: Acute, generalized muscle weakness can be seen with a number of severe electrolyte abnormalities of any cause: hypokalemia, hyperkalemia, hypocalcemia, hypercalcemia, hypomagnesemia, and hypophosphatemia. Acute painless myopathies can also be seen with endocrine disorders involving the thyroid, parathyroid, or adrenal glands.
Principles of Disease.: These are autosomal dominant disorders of ion channels resulting in intermittent attacks of flaccid extremity weakness associated with either hyperkalemia or hypokalemia, although hypokalemia is more common. Periodic paralysis is most often associated with an inherited genetic mutation. Patients usually report a personal and family history of similar episodes.25
Clinical Features and Diagnostic Strategies.: Patients may suffer either isolated or recurrent episodes of flaccid paralysis. The lower limbs are involved more often than the upper, although both can be affected. Bulbar, ocular, and respiratory muscles are usually not involved. Onset is rapid; a prodrome of myalgias and muscle cramps may occur but is uncommon; mental status and sensory function are typically preserved, but reports of sensory nerve involvement have been documented.26 Males are more often affected than females, and there is a higher incidence in Asians, particularly Japanese, although it occurs in other ethnic groups.
Management.: Many cases resolve spontaneously with supportive care alone. The mainstay of management is the treatment of the underlying electrolyte imbalance. In the hypokalemic state, the total body potassium concentration is not depleted but has shifted intracellularly. Thus, in the repletion of potassium, caution is necessary to prevent overtreatment. For this reason, intravenous potassium should be used sparingly; one or two 10-mEq doses of potassium chloride, during 1 hour, is the maximum intravenous dose. This can be done in parallel with 40 mEq oral potassium repletion and retesting of serum potassium levels. Intravenous hydration helps redistribute the body’s potassium stores.
Thyrotoxic Periodic Paralysis.: The clinical picture of TPP is almost identical to that of hypokalemic FPP, and indeed a small number of patients with hypokalemic FPP have hyperthyroidism. In TPP, symptoms related to hyperthyroidism are often present at the same time the patient has weakness. The relation of the hyperthyroidism to hypokalemia is probably due to increased sodium-potassium–adenosine triphosphatase activity, which causes a rapid shift of potassium from the extracellular into the intracellular compartment. Treatment of the hyperthyroid symptoms, such as tachycardia, may help the paralysis as well. There are case reports of TPP in which the patient’s weakness did not respond to potassium replacement until propranolol was given to treat tachycardia.27,28 There is probably a genetic feature underlying this disorder because there is a higher incidence of repeated attacks of hypokalemic periodic paralysis among Japanese and Chinese patients with hyperthyroidism. It is important that all patients have thyroid function testing performed after a first episode of hypokalemic paralysis.




References
1. Phillips, LH. The epidemiology of myasthenia gravis. Semin Neurol. 2004;24:17.
1a. Thomas, CE, et al. Myasthenic crisis: Clinical features, mortality, complications and risk factors for prolonged intubation. Neurology. 1997;48:1253.
2. Massey, JM. Acquired myasthenia gravis. Neurol Clin. 1997;15:577.
3. O’Neill, JH, et al. The Lambert-Eaton myasthenic syndrome: A review of 50 cases. Brain. 1988;111:577.
4. Fergusson, D, et al. Use of intravenous immunoglobulin for treatment of neurologic conditions: A systematic review. Transfusion. 2005;45:1640.
5. Seybold, ME. The office Tensilon test for ocular myasthenia gravis. Arch Neurol. 1986;43:842.
6. Golnik, KC, et al. An ice test for the diagnosis of myasthenia gravis. Ophthalmology. 1999;106:1282.
7. Thomas, CE, et al. Myasthenic crisis: Clinical features, mortality, complications and risk factors for prolonged intubation. Neurology. 1997;48:1253.
8. Mayer, SA. Intensive care of the myasthenic patient. Neurology. 1997;48(Suppl 5):70.
9. Seneviratne, J, et al. Noninvasive ventilation in myasthenic crisis. Arch Neurol. 2008;65:54.
10. Berrouschot, J, et al. Therapy of myasthenic crisis. Crit Care Med. 1997;25:1228.
11. Schneider-Gold, C, et al. Corticosteroids for myasthenia gravis. Cochrane Database Syst Rev. (2):2005.
12. Hart, IK, et al. Immunosuppressive agents for myasthenia gravis. Cochrane Database Syst Rev. (4):2007.
13. Maddison, P, et al. The use of rituximab in myasthenia gravis and Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2011;82:671–673.
14. Gronseth, GS, Barohn, RJ. Practice parameter: Thymectomy for autoimmune myasthenia gravis (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55:7.
15. Gajdos, P, Chevret, S, Toyka, KV. Plasma exchange for generalised myasthenia gravis. Cochrane Database of Syst Rev. (4):2002.
16. Gajdos, P, Chevret, S, Toyka, KV. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev. (12):2012.
17. Centers for Disease Control and Prevention. Botulism Annual Summary, 2009. http://www.cdc.gov/nationalsurveillance/PDFs/Botulism_CSTE_2009.pdf.
18. Centers for Disease Control and Prevention. Wound botulism among black tar heroin users—Washington, DC, 2003. MMWR Morb Mortal Wkly Rep. 2003;52:885.
19. Centers for Disease Control and Prevention. Infant botulism—New York City, 2001-2002. MMWR Morb Mortal Wkly Rep. 2003;52:21.
20. Centers for Disease Control and Prevention. Investigational heptavalent botulinum antitoxin (HBAT) to replace licensed botulinum antitoxin AB and investigational botulinum antitoxin E. MMWR Morb Mortal Wkly Rep. 2010;59:299.
21. Arnon, SS, et al. Human botulism immune globulin for the treatment of infant botulism. N Engl J Med. 2006;354:462.
22. Centers for Disease Control and Prevention. Cluster of tick paralysis cases—Colorado, 2006. MMWR Morb Mortal Wkly Rep. 2006;55:933.
23. Felz, MW, et al. A six-year-old girl with tick paralysis. N Engl J Med. 2000;342:90.
24. Bartt, R. Autoimmune and inflammatory disorders. In: Goetz CG, ed. Goetz Textbook of Clinical Neurology. 3rd ed. Philadelphia: WB Saunders; 2007:1176.
25. Venance, SL, et al. The primary periodic paralyses: Diagnosis, pathogenesis and treatment. Brain. 2006;129:8.
26. Inshasi, J. Dysfunction of sensory nerves during attacks of hypokalemic periodic paralysis. Neuromusc Disord. 1999;9:227.
27. Shayne, P, Hart, A. Thyrotoxic periodic paralysis terminated with intravenous propranolol. Ann Emerg Med. 1994;24:734.
28. Yu, T, et al. Potassium chloride supplementation alone may not improve hypokalemia in thyrotoxic hypokalemic periodic paralysis. J Emerg Med. 2007;32:263.