Neuromuscular Disorders of the Gastrointestinal Tract
Dhanpat Jain
Introduction
Normal bowel motility depends on smooth muscle, the interstitial cells of Cajal (ICC), the intrinsic and extrinsic nerve supply and supporting cells, and various neuroendocrine peptides. Abnormalities in any of these components may result in bowel dysmotility. In addition, other inflammatory cells such as lymphocytes, eosinophils, and mast cells may act directly or indirectly on the neuromuscular apparatus of the bowel wall. The clinical manifestations of motility disorders depend on the extent and specific site of the abnormality. Some of these disorders manifest with distinct clinical features (e.g., idiopathic hypertrophic pyloric stenosis, Hirschsprung disease, achalasia), whereas others have nonspecific manifestations. Patients with idiopathic hypertrophic pyloric stenosis have projectile vomiting in the first month of life, often associated with an olive-sized abdominal mass. Patients with Hirschsprung disease have delayed passage of meconium. The pathogenesis of many of these conditions is poorly understood. In fact, many disorders have no specific pathologic features and lack standardized diagnostic criteria. This situation has led to marked variability in the approach to diagnostic workups among different laboratories.1
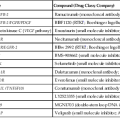
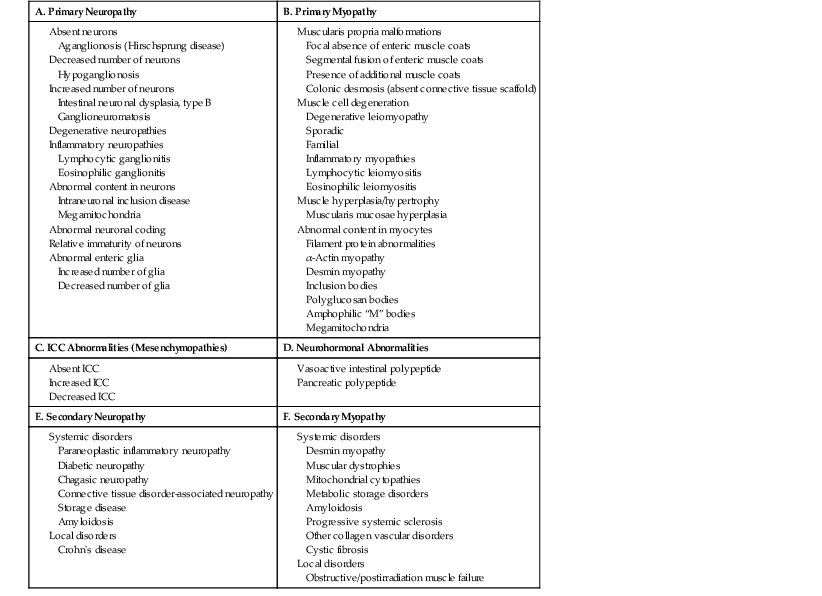
To address these issues, an international working group was formed in 2007. This group published a comprehensive guideline for handling of most specimen, including biopsies and resections, pertaining to gastrointestinal (GI) motility disorders. The classification proposed by this group is recognized as the London Classification of neuromuscular disorders of the GI tract. This classification has placed various primary neuropathies and myopathies into well-delineated categories. However, it includes only a short list of secondary disorders. A modified version of this classification is shown in Table 7.1. It is expected that this modification will lead to an improvement in the understanding of GI motility disorders.
Table 7.1
Classifications of Neuromuscular Disorders
| A. Primary Neuropathy | B. Primary Myopathy |
ICC, Interstitial cells of Cajal.
Muscle Coats of the Bowel Wall
Knowledge of the basic organization of the neuromuscular apparatus of the bowel is essential to understand and diagnose GI motility disorders. The neuromuscular framework of the bowel is similar throughout the tract. However, there are some minor variations.2 The bowel smooth muscle is composed of a thin superficial layer that separates mucosa from submucosa (muscularis mucosae) and a thick outer layer (muscularis propria), which has an inner circular and an outer longitudinal coat. The exception is the esophagus, in which the muscularis propria has only a single longitudinal muscle coat. The proximal part of the muscularis propria of the esophagus is formed entirely of skeletal muscle; the skeletal muscle merges with smooth muscle in the vicinity of the midesophagus (Fig. 7.1). Because of this anatomy, esophageal motility is susceptible to the effects of systemic disorders of both smooth and skeletal muscle. In the stomach, an additional inner oblique muscle layer is also present.
In contrast, the outer longitudinal layer in the colon forms thick localized bands of muscle termed tenia coli. The muscularis mucosae of the colon continues into the anal canal. The inner circular layer of muscularis propria of the rectum becomes thickened distally to form the internal anal sphincter. The external anal sphincter is formed of skeletal muscle and is connected to the skeletal muscle of the pelvic floor. The outer longitudinal muscle layer of the rectum continues between the inner and outer anal sphincters and then separates caudally into multiple septa, which diverge fanwise throughout the subcutaneous part of the external sphincter into the skin. These fibers are responsible for the characteristic corrugated appearance of the perianal skin. In addition, the muscle fibers from the outer longitudinal coat and the internal anal sphincter extend into the submucosa to form a meshwork of fibers surrounding the vascular plexuses (muscularis submucosa ani). The organization of the muscle layers in the appendix is similar to that of the colon, except that the appendix lacks tenia coli.
Neural Network of the Bowel
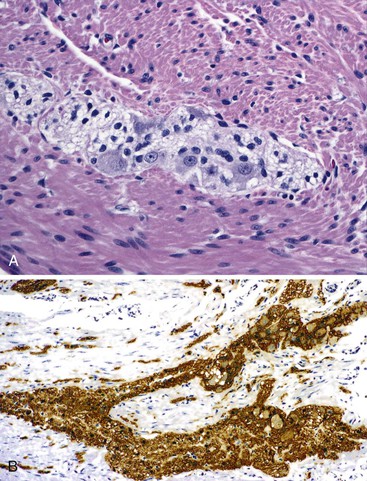
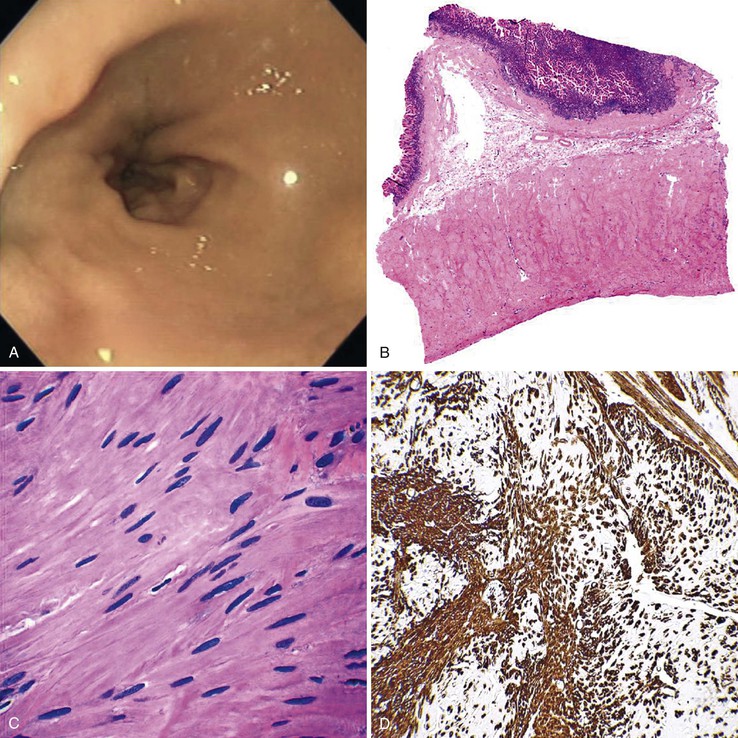
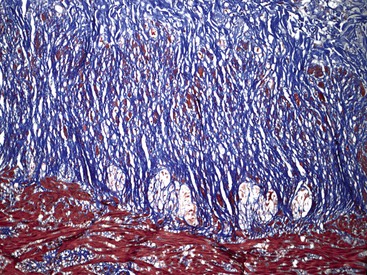
The organization of the neural network in the bowel is complex. The extrinsic nerve supply of the bowel wall consists of both sympathetic and parasympathetic nerve fibers that penetrate the wall and become the intrinsic neural plexus. The sympathetic fibers originate in the prevertebral ganglia and parallel the superior and inferior mesenteric arteries. The parasympathetic fibers are located alongside the posterior branch of the vagus nerve. The intrinsic neural system of the bowel wall is organized into three plexuses: the submucosal plexus (Meissner plexus), the deep submucosal plexus (Henle plexus), and the myenteric plexus (Auerbach plexus) (Fig. 7.2). The most easily identified and most prominent of these is the myenteric plexus, which is composed of clusters of ganglion cells connected by an intricate network of nerves located in the space between the inner circular and outer longitudinal muscle layers. Although the ganglion cells and nerve bundles are easily identified within these plexuses (Fig. 7.3, A), the intricacy of the neural meshwork of fibers is not easily detectable on hematoxylin and eosin (H&E)-stained tissue sections. Whole-mount specimens, silver stains, and/or immunohistochemical stains are usually needed to visualize the complexity of the neural network (see Fig. 7.3, B).3,4
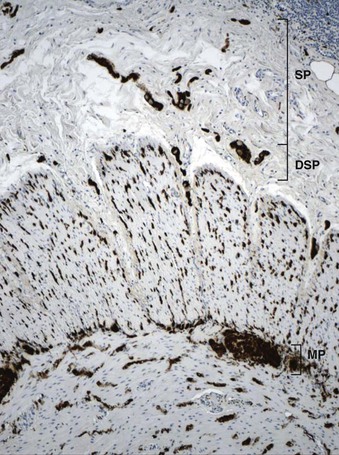
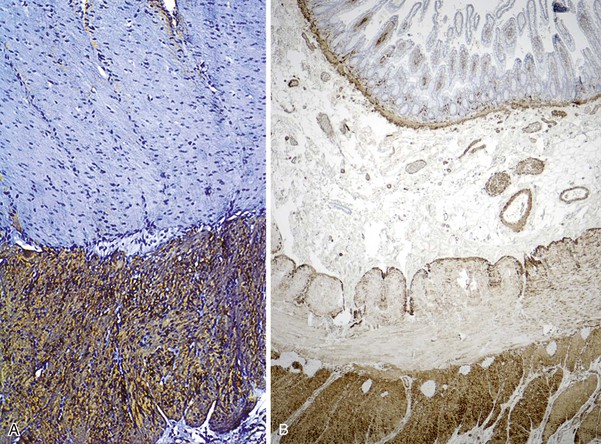
In addition to muscle fibers and the neural network, a third population of mesenchymal cells, the interstitial cells of Cajal (ICC), are critical for bowel motility. These cells generate a slow wave of depolarization and represent the “pacemaker cells” of bowel peristalsis.5,6 Their function is modulated by both intrinsic and extrinsic neural inputs. Because these cells are difficult to detect on routine tissue sections, most of our knowledge regarding their morphology and structural organization stems from ultrastructural studies.7–9 Ultrastructurally, these cells show a partial basal lamina, many intermediate filaments, darkly staining cytoplasm, abundant rough endoplasmic reticulum, sublamellar caveolae, oval indented nuclei, and lack of myosin filaments. Many of these features overlap with those of smooth muscle cells. ICC express CD117 (KIT), a tyrosine kinase receptor.10 Immunohistochemical stains with antibodies against KIT or ANO1 (formerly DOG1) can be used to visualize these cells.11–13 ICC are part of an intricate neural network and have a close association with smooth muscle cells and nerve endings. They are most easily identified surrounding the myenteric plexus, especially in the small bowel, where the network of cells extends into the inner and outer muscle coats (Fig. 7.4). In addition, there is a distinct ICC plexus in the submucosa. The distribution and organization of ICCs in the appendix is similar to that in the colon. The structural organization of ICC has been described in the various segments of the GI tract, from esophagus to anus. Minor regional differences within each bowel segment do exist, and the reader is referred to a review article by Venderwiden and Rumessen for more details.14
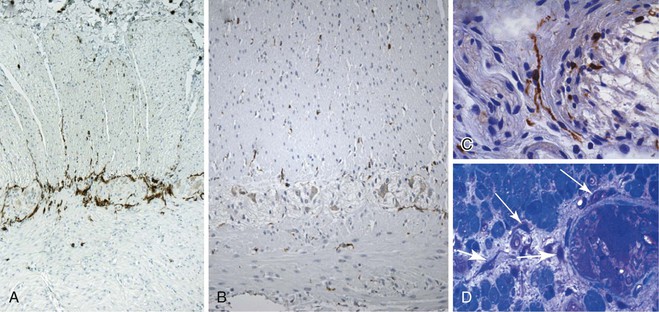
The neuromuscular organization of the appendix is similar to that of the colon and small bowel. However, the ganglia are sometimes embedded deeper into the circular or longitudinal layer (Fig. 7.5, A and B). The neural and ICC networks in the appendix are similar to those in the colon, but with less aggregation of ICC surrounding the myenteric plexus (see Fig. 7.5, B and C). Understanding of the neuromuscular organization of the appendix is helpful, because the appendix is sometimes examined intraoperatively to evaluate the extent of aganglionosis.15
Esophagus
Primary Achalasia
Achalasia is a motor disorder of the esophagus that is characterized by failure of the lower esophageal sphincter (LES) to relax in response to swallowing.16,17 Clinically, achalasia is divided into three subtypes: type I, classic type; type II, achalasia with compression; and type III, spastic achalasia.18 It is uncommon. The overall prevalence rate is less than 10 cases per 105 population.19 Its incidence has been fairly stable during the last 50 years. It is a disease of adults, mainly those older than 60 years of age, and affects both sexes equally. Achalasia is more frequent in North America, northwestern Europe, and Australia than in other regions, and it is more common in whites.
Clinical Features
The major clinical manifestations of achlasia differ between children and adults. A feeding aversion, failure to thrive, choking, recurrent pneumonia, nocturnal cough, aspiration, or nonspecific regurgitation typically develops in younger children (<5 years). Older children and adults often experience vomiting, chest pain, and dysphagia for solids and liquids. Heartburn is a common symptom (50%), even in untreated patients, although only a minority of patients have documented gastroesophageal reflux disease.20
The diagnosis is confirmed with imaging studies and manometry. Barium studies typically reveal reduced peristalsis, a characteristic beaklike deformity of the distal esophagus, and dilatation of the proximal esophagus. Manometry studies reveal abnormal peristalsis, increased intraluminal pressure, and incomplete and delayed relaxation of the LES. Endoscopy and endoscopic ultrasound studies are often performed to rule out coexisting mucosal pathology and to exclude secondary causes of achalasia (“pseudoachalasia”).
Pathogenesis
The most significant feature of achalasia is loss of myenteric ganglion cells. However, the cause of ganglion cell loss is unknown. Current data suggest that myenteric inflammation precedes loss of ganglion cells, but the initial inciting events that cause disease remain unknown.21 Environmental factors, viral infection, autoimmune mechanisms, and genetic predisposition have all been proposed. In addition, some data suggest familial aggregation. Rare familial forms associated with alacrima (absence of tears) and adrenocorticotropic hormone (ACTH) insensitivity have been described (Allgrove syndrome, or triple A syndrome).22,23 Concordance in monozygotic twins and an association with Down syndrome has also been reported. A significant association has been found with the class II human leukocyte antigen (HLA) DQw1 in white patients. The alleles identified, HLA DQB1*0602, DQA1*0101, and DRB1*15, are the same found to be associated with other autoimmune disorders, including multiple sclerosis, Goodpasture syndrome, Graves disease, myasthenia gravis, polymyositis, autoimmune polyglandular syndrome, and Sjögren and sicca syndromes. Antimyenteric neuronal antibodies have been identified in some patients.24–28 It has been demonstrated in an ex vivo model that on exposure to sera from patients with achalasia, gastric corpus mucosa undergoes phenotypic and functional changes that mimic achalasia.29 A factor in the serum, other than an antineuronal antibody, may be responsible for this phenomenon.29 Varicella-zoster viral DNA has been identified in the myenteric plexus in rare cases by in situ hybridization.30 Polypomorphisms in genes such as vasoactive intestinal polypeptide (VIP) receptor 1, KIT/CD117, and interleukin-23 receptor (IL23R) may increase susceptibility to achalasia.31–33 VIP is responsible for relaxation of esophageal smooth muscle, whereas KIT plays an important role in the function of ICC. The IL23 pathway is important in immune activation and plays a vital role in many chronic inflammatory disorders, including inflammatory bowel disease.
As noted previously, the pathogenesis of achalasia is poorly understood. However, progressive inflammatory destruction of myenteric ganglion cells is the most important underlying event. This condition results in failure of the LES to relax in response to swallowing.34 Esophageal peristalsis is decreased or completely absent. This results in esophageal dilatation, chronic stasis, and reactive hypertrophy of the muscularis propria. VIP, which was initially thought to be a major mediator of relaxation of the LES, shows a substantial decrease in VIP-containing neurons in the distal esophagus.35,36 It has been shown that nitric oxide is a primary esophageal inhibitory neurotransmitter. It colocalizes with VIP in ganglion cells. In addition, intrinsic nitrergic ganglion cells are lost, or markedly decreased, in achalasia. In fact, loss of VIP-positive ganglion cells is synonymous with loss of nitrergic ganglion cells.37,38 Although most early studies evaluated specimens only at the time of autopsy or esophagectomy, and therefore showed end-stage disease, study of esophagomyomectomy specimens has now given some insights into the early sequence of events in this condition.35 These studies have revealed that as the disease progresses, the inflammatory infiltrate decreases in intensity, but loss of ganglion cells and degeneration of the myenteric plexus become more prominent features.
Pathologic Features
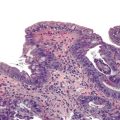
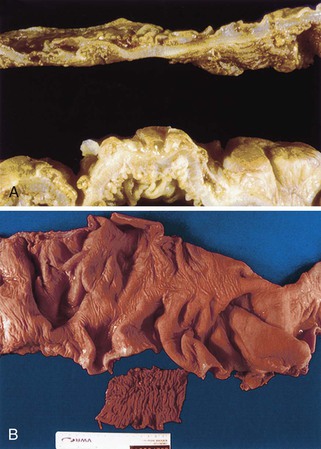
Grossly, the esophagus in achlasia shows dilation. The extent of dilatation depends on the severity and duration of disease (Fig. 7.6, A). The lumen often contains stagnant and foul-smelling, partially digested food. The distal end is typically narrowed and stenotic.
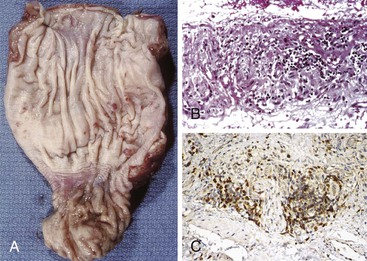
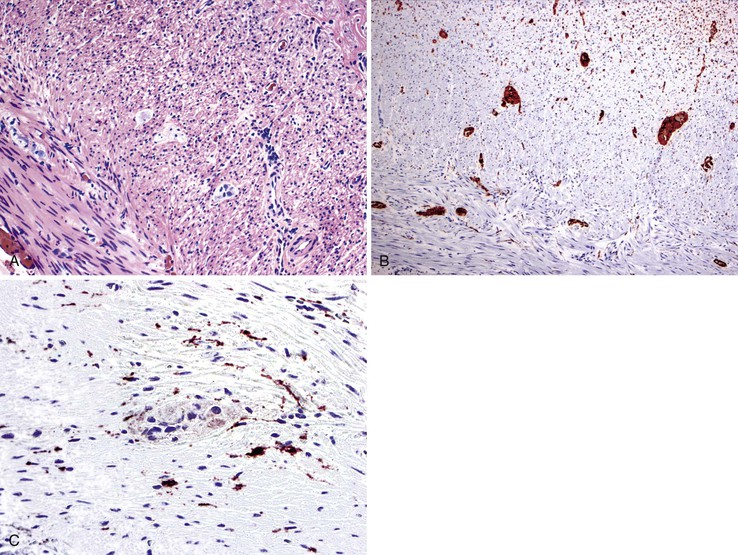
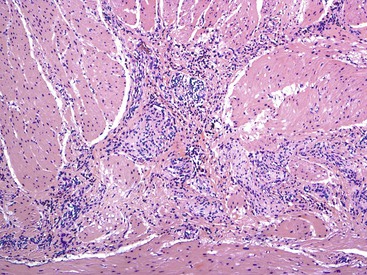
The main histologic abnormality in achalasia is related to the myenteric plexus, although numerous secondary changes are often present, presumably due to prolonged stasis and reflux. Widespread, often total, loss of myenteric ganglion cells is the cardinal feature of achalasia (Fig. 7.7; see Fig. 7.6, B and C). The ganglion cells may be better preserved in the more proximal portions of the esophagus.16 Some degree of neural hyperplasia may accompany neuronal loss (see Fig. 7.7). A variable amount of chronic inflammation is often admixed with eosinophils and plasma cells. Mast cells may be seen surrounding the myenteric nerves and residual ganglion cells39 (see Figs. 7.6, B and 7.7). In end-stage disease, the degree of inflammation may become minimal or disappear completely. One ultrastructural study showed that numerous mast cells are also present within the inflammatory infiltrate closely associated with the nerve fibers.40 Occasionally, lymphocytes may infiltrate the cytoplasm of ganglion cells (ganglionitis). The majority of chronic inflammatory cells are CD3-positive T cells, most of which are CD8-positive as well (Fig. 7.6, C), although the relative percentage of these cells decreases with progression of disease.34,41,42 A large subset of T cells represent either resting or activated cytotoxic cells.
Other changes frequently present are related to distal esophageal obstruction; they include muscularis propria hypertrophy, muscularis propria eosinophilia, and dystrophic calcification. Hypertrophied muscle may also show degenerative changes, including cytoplasmic vacuolation and liquefactive necrosis. The branches of the vagus nerve within the adventitia are unremarkable in most cases, although degenerative changes in the vagus nerve and in dorsal motor nuclei have been described.17 These changes may be caused by infection with a neurotrophic virus; however, no specific virus has been identified.42,43 The squamous mucosa also shows secondary changes, including diffuse hyperplasia, increased intraepithelial lymphocytes (“lymphocytic esophagitis”), papillomatosis, basal cell hyperplasia, and an increase in nonspecific lamina propria inflammation.44 Some of these changes mimic reflux esophagitis, although sustained lower esophageal pressure does not allow regurgitation of gastric contents in untreated cases.45 After esophagomyotomy, gastroesophageal reflux develops in as many as 50% of patients and can lead to Barrett’s esophagus in some cases.46,47
Differential Diagnosis
Biopsies are not performed to establish a diagnosis of achalasia; pathologists encounter this condition when a resection is performed or at autopsy. The role of biopsy in patients with achalasia is largely to exclude Barrett’s esophagus (after myotomy), dysplasia, and malignancy. The differential diagnosis of achalasia, both clinically and pathologically, is pseudoachalasia, which develops secondary to tumors or paraneoplastic syndrome. These are easily ruled out with esophageal manometry and imaging or with biopsies when a mass is present.
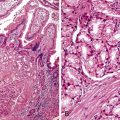
Occasionally, strictures at the gastroesophageal junction resulting from reflux, prior surgery, or trauma can mimic achalasia (Fig. 7.8, A and B). The esophagus may show muscular hypertrophy and neural hyperplasia similar to achalasia. However, on close examination, ganglion cells are easily identified in the neural plexus, and there is a lack of inflammatory or degenerative changes in the ganglion cells. In postinfectious or autoimmune-mediated ganglion cell loss, lymphocytic inflammation may be present in or around ganglion cells. Residual ganglion cells can often be identified (see Fig. 7.8, C).
In paraneoplastic achalasia, the diagnosis of malignancy is often already known, and despite histologic similarity with the idiopathic form, differentiation is seldom a problem clinically.
Chagas disease, which also causes massive dilation of the esophagus with ganglion cell loss, should be suspected in any patient from an endemic area. By the time achalasia-type features develop, the infectious organisms can no longer be demonstrated in the tissues, so one must rely on serologic evidence of infection. Patients with Chagas disease may also show dilatation of other hollow viscera, with ganglion cell loss.
Natural History and Treatment
Achalasia is a chronic disorder, and the treatment is largely palliative. Medications such as anticholinergics, nitrates, and calcium channel blockers are used in some circumstances but result in only partial benefit. Pneumatic dilatation and injection of botulinum toxin into the LES produce an initial response, but the results are usually short lasting. The best results are typically obtained with esophagomyotomy of the LES, with or without pneumatic dilatation. Patients with type II achalasia have the most favorable outcome and better response to treatment. Esophageal resection is usually reserved for end-stage cases. Patients have an increased long-term risk for squamous cell carcinoma of the esophagus.46,48 The risk is approximately 33-fold higher than in the general population.49 Studies show that the risk of adenocarcinoma is also higher, albeit to a lower degree.50
Secondary Achalasia
As discussed earlier, signs and symptoms indistinguishable from primary achalasia may be encountered with other conditions, such as Chagas disease, or in association with a neoplasm that directly invades the myenteric plexus. In some cases, a paraneoplastic phenomenon causes a secondary achalasia, as in paraneoplastic achalasia associated with small cell carcinoma. Rare associations have been described with other tumors, such as leiomyomatosis of the esophagus and sarcoidosis.51–53 In sarcoidosis, inflammation surrounding the myenteric plexus has been described, but without granulomas. One of the reported patients showed resolution of symptoms with steroid treatment.51
Chagas disease results from infection with the protozoan Trypanosoma cruzi.54,55 The infection is acquired from the bite of blood-sucking reduviid bugs. The geographic distribution of the disease is limited to certain parts of the world, including South and Central America and Africa. Chagas disease is uncommon in the United States, occurring almost exclusively among immigrants from endemic countries such as Brazil. Any part of the GI tract may be affected, but the esophagus and the sigmoid colon are the most frequent sites. Infection results in dysmotility and often massive dilatation (e.g., megaesophagus, megacolon) In the esophagus, the symptoms closely resemble idiopathic achalasia. Colonic involvement results in constipation and intestinal pseudo-obstruction. These features are seen in the chronic phase of disease, and by the time symptoms are observed, the organisms usually are no longer present in the myenteric plexus.
Idiopathic Muscular Hypertrophy
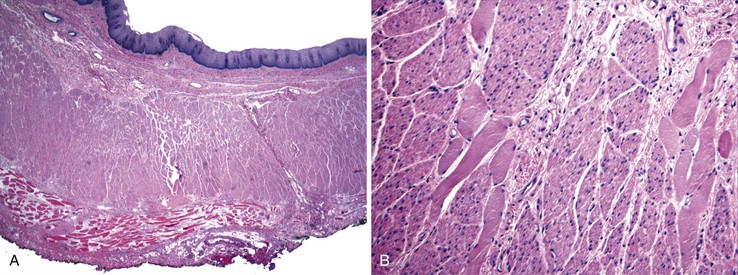
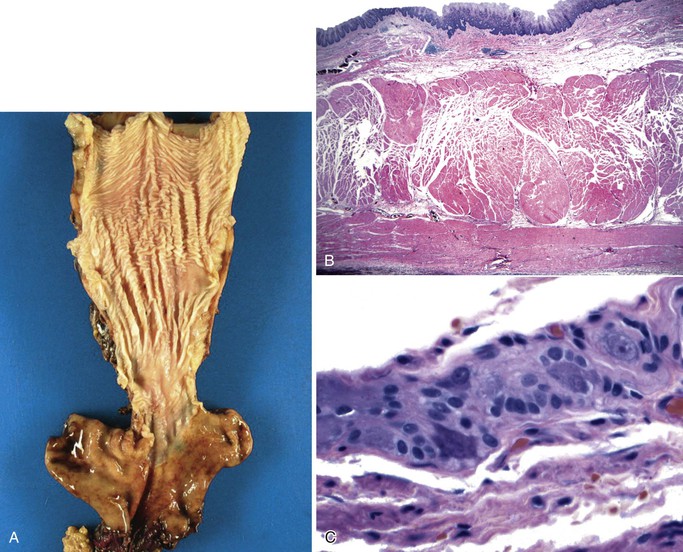
Idiopathic muscular hypertrophy of the esophagus is a poorly understood condition of uncertain etiology and clinical significance. Most of the cases reported with pathologic descriptions have been diagnosed at the time of autopsy. The condition can be diagnosed clinically with imaging techniques and esophageal motility studies. New clinical diagnostic criteria have been proposed.56,57 Some patients are symptomatic at presentation, with symptoms such as dysphagia, chest pain, vomiting, and weight loss, whereas others are entirely asymptomatic.58 Gastroesophageal reflux develops in some patients.57 Esophageal spasm and increased intraluminal pressure are believed to be the cause of symptoms. The disorder occurs in adults, with no gender or race predilection. Many patients also have diabetes. Some cases have shown to have autosomal dominant inheritance and association with bilateral cataracts and Alport-like nephropathy.59 Squamous cell carcinoma has also been described in some cases.60 Pathologically, the muscularis propria is markedly thickened, particularly toward the distal end of the esophagus58 (Fig. 7.9). In some cases, there is a mild degree of lymphocytic infiltration in the myenteric plexus. The vast majority lack evidence of muscle fiber degeneration, fibrosis, ganglion cell abnormalities, or neural plexus abnormalities.
Differential Diagnosis
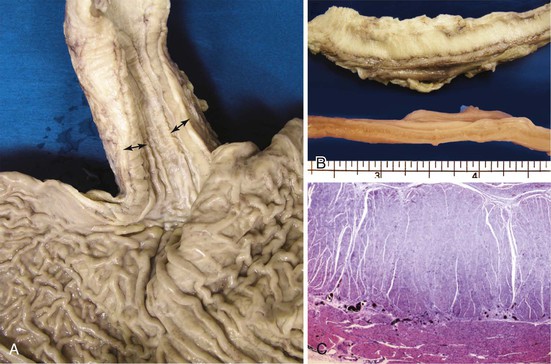
A variable degree of hypertrophy of the esophageal musculature may be seen in patients with distal obstruction of any cause, including achalasia. However, the cause of the obstruction is often obvious clinically (see Fig. 7.8, A). Histologically, the muscle fibers appears normal in idiopathic muscular hypertrophy, and ganglion cells and neural plexus are present. The key is to exclude distal obstruction in the presence of markedly hypertrophic muscularis propria.
Stomach
Idiopathic Hypertrophic Pyloric Stenosis
Idiopathic hypertrophic pyloric stenosis is a disorder characterized by thickened pyloric musculature and features of gastric outlet obstruction. Infantile, late-onset/adolescent, and adult forms of this disorder have been described. The presenting symptom in infants is projectile vomiting, usually within 2 to 4 weeks of birth.61–65 It occurs in approximately 1 in 1000 live births, has a high familial incidence and a strong male preponderance, and classically occurs in the first-born child. The incidence of this condition is rising in some countries (Britain and Ireland) but decreasing in others (United States, Germany, Canada, and Denmark).66–69 The overall incidence ranges from 0.18 to 8.8 per 1000 live births. Regional variation within countries is also known to occur.67
Clinical Features
Infants classically have progressive nonbilious vomiting, which gradually assumes a more characteristic projectile pattern. The hypertrophied pylorus may be palpated as an olive-sized epigastric mass in some individuals. Gastric peristalsis may be visible to the naked eye on examination of the abdomen. Some patients also have an associated congenital diaphragmatic hernia. More recent studies suggest that presentation with classic symptoms and signs is uncommon, however, so a higher degree of clinical suspicion is required to establish an early diagnosis.70 Idiopathic hypertrophic pyloric stenosis is uncommon in adults but has been reported.71 Most cases in adults are secondary to scarring caused by juxtapyloric peptic ulceration, inflammatory bowel disease, or tumors.72
Pathogenesis
The pathogenesis of the disease remains unclear. A genetic predisposition and other environmental precipitating factors have been implicated (e.g., bottle feeding, respiratory distress syndrome).73 Its incidence appears to be decreasing, parallel with an increasing trend towards breast feeding and young maternal age.69,74 Prenatal use of erythromycin and various other macrolide antibiotics has also been implicated as risk factors. However, the evidence is not conclusive.75,76 There is no association with Helicobacter pylori infection.77 Rare cases have been associated with esophageal atresia, as well as other types of congenital malformations and syndromes (e.g., Cornelia de Lange syndrome, Smith-Lemli-Opitz syndrome).78,79
The disease does not show any evidence of mechanical obstruction. In fact, the pylorus can be easily intubated in affected patients.80 Uncoordinated peristalsis of the stomach, including the pyloric musculature (“pylorospasm”), is one theory of pathogenesis. Other factors that may play a role include immaturity of the enteric nervous system, hormonal imbalance between gastrin and somatostatin, redundancy of the overlying mucosa, lack of KIT-positive ICC, and lack of nitric oxide synthase (NOS).81–86 Interestingly, homozygous transgenic mice that carry inactivating genes for NOS develop hypertrophy of the pylorus.87 In humans, a familial tendency and a high concordance rate in twins have been reported. The incidence is higher in monozygotic compared with dizygotic twins.88 At least five genetic loci (termed IHPS1 through IHPS5) have been associated with this disease. The strongest association is with IHPS1, which regulates the NOS1 gene.89 Neuronal NOS is a critical enzyme in the production of nitric oxide that mediates relaxation of pyloric smooth muscle. Several RET genomic variants associated with the disorder have also been identified. However, in contrast to Hirschsprung disease, these variants probably play a minor role in disease pathogenesis.90 Several linkage analysis studies have identified an association with loci on chromosomes 2, 3, 5, 6, 7, 11, 12, 16, and X.91–93 However, there is tremendous heterogeneity, and it is unlikely that a single gene is solely responsible for this disease. Within a family pedigree, the disease may be linked to a single locus or gene.94,95 Therefore, a commonly accepted theory is that environmental factors lead to this condition in a genetically predisposed host.79
Pathologic Features
The pathologic features are similar in children and adults (Fig. 7.10). Grossly, the pylorus is greatly thickened and fusiform in appearance. The proximal stomach may show dilation, depending on the severity and duration of obstruction. Histologically, cases of pyloric stenosis reveal a thick inner circular pyloric muscle (as much as four times normal thickness). The muscle fibers are disorganized, show increased intercellular collagen, and are sometimes associated with a mild lymphocytic infiltrate. The longitudinal muscle is frequently attenuated as well. The enteric nerve plexus is often hypertrophied and shows a relative increase in the number of Schwann cell nuclei. Glial cells show degeneration, characterized by pyknosis and vacuolation. ICC are markedly reduced or absent in the hypertrophied muscle layers, in the myenteric plexus, and in the outer longitudinal muscle.
Differential Diagnosis
Biopsies of the pyloric musculature are seldom performed in children; the diagnosis is made entirely on clinical grounds. When the disorder manifests in adults, biopsies are obtained to exclude a neoplasm or other lesions that can lead to pyloric stenosis.
Natural History and Treatment
Surgical myotomy is widely considered the definitive method of treatment. Pyloric hypertrophy typically disappears within a few months after the procedure.96 Histologic evaluation of the pylorus several months after myotomy often reveals normalization of the nerve fibers, glial cells, ICC, and neuronal NOS abnormalities.97 Therefore, the long-term outcome of affected patients after surgery is excellent.98
Gastroparesis
Gastroparesis is characterized by food retention in the stomach caused by delayed emptying. In most cases, it is associated with diabetes, largely related to autonomic dysfunction or after vagotomy, although some cases remain idiopathic.99 Most patients are asymptomatic, but intermittent vomiting that can become intractable develops in some patients. Such patients invariably have advanced complications of diabetes. The pathophysiologic basis for this disorder is poorly understood and disputed. Abnormalities of the vagus nerve are suspected, but findings have not been consistent.100,101
Pathologic examination of the gastric musculature was initially reported to be normal. However, more recent studies with newer techniques of examining the neuromuscular apparatus have shown a variety of subtle changes.102,103 Most cases appear to have normal musculature, but a few show mild atrophy with increased interstitial fibrosis.103 On H&E staining, amphophilic “M” bodies that are periodic acid–Schiff (PAS) negative are seen in many cases (Fig. 7.11, A and B).103 These are thought to represent degenerative changes in smooth muscle cells. They are different from PAS-positive polyglucosan inclusions that are seen in a variety of GI motility disorders (discussed later). In some cases, mild inflammation of the neural plexus and ganglia is present, and this is composed largely of T cells (see Fig. 7.11, C).102 Rare cases with marked inflammation of the neural plexus responsive to treatment with steroids have been reported.104 Some cases show a decreased number of ganglion cells, which includes both NOS-positive and NOS-negative neurons. Decreased numbers of ICC in the myenteric plexus and muscular layers have also been documented in some cases, whereas in some, the ICC lose their dendritic processes and acquire a more rounded phenotype (see Fig. 7.11, D).102 Animal models of gastroparesis also show loss of ICC.105
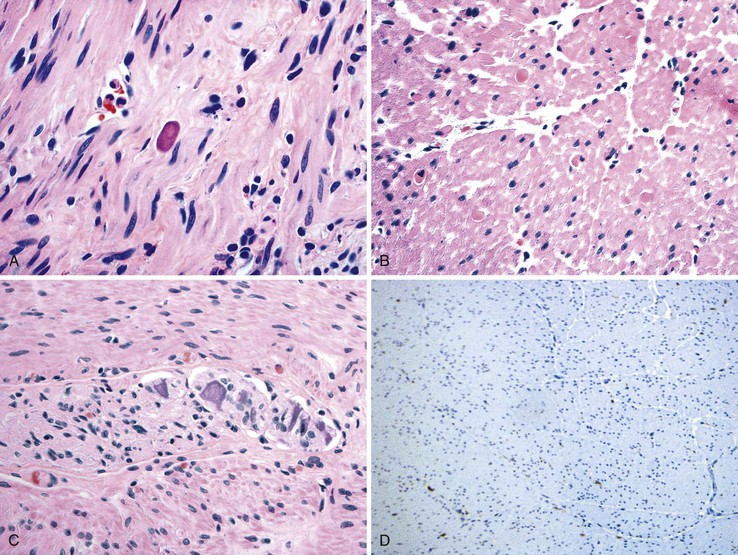
From a practical diagnostic perspective, recognition of muscle atrophy or cytoplasmic inclusion bodies is easy, whereas recognition of a subtle loss of ganglion cells or changes the in ICC network requires morphometry. About half of cases do not show any of these abnormalities, which has led to the speculation that all these changes may be secondary in nature. Because there are no therapeutic implications linked to specific histologic changes, there is no role for extensive morphometric workup in clinical practice.
Small and Large Intestine
Hirschsprung Disease
Hirschsprung disease is a heterogeneous group of disorders characterized by a lack of ganglion cells which results in bowel dysmotility.106,107 The most common form (75% to 80% of cases) involves the distal sigmoid colon and rectum; this is referred to as “short-segment” disease or “classic Hirschsprung” disease. In approximately 10% of cases, the disease (i.e., lack of ganglion cells) extends proximal to the splenic flexure; this is known as “long-segment” disease. Rarely (5%), the entire bowel is devoid of ganglion cells; this is termed “total bowel aganglionosis”. Zonal aganglionosis, in which the absence of ganglion cells is patchy, is extremely rare. Of course, this type may be responsible for surgery failures.108
Classic Hirschsprung disease is a congenital disorder. The estimated incidence is approximately 1 in every 5000 live births, with a striking male preponderance (3 to 4.5 : 1). Rare acquired forms, as well as adult cases, have also been described.109 Long-segment disease and total bowel aganglionosis show familial aggregation. However, classic Hirschsprung disease is usually sporadic in origin. A large number of associated conditions have been reported with Hirschsprung disease, including Down syndrome, cardiovascular malformations, neurofibromatosis, Waardenburg syndrome, Laurence-Monn-Bardet-Biedl syndrome, Ondine’s curse (Haddad syndrome), multiple endocrine neoplasia, neuroblastoma, total colonic agenesis, and imperforate anus, many of which belong to the category of neural crest disorders (“neurocristopathies”).107,110
Clinical Features
The earliest and most common form of clinical presentation is delayed (>48 hours) passage of meconium in the newborn. Infants and older children tend to have chronic constipation, often accompanied by abdominal distention and vomiting. Compensatory hypertrophy of the normally innervated proximal bowel develops in some patients and leads to a milder form of disease with diagnosis delayed until later in adulthood. However, the pathogenesis of adult-onset Hirschsprung disease remains poorly understood.111 The clinical diagnosis of Hirschsprung disease is facilitated with the use of imaging studies and rectal manometry, but it is ultimately established by suction mucosal biopsy. Enterocolitis, which is more common in Hirschsprung patients with Down syndrome, is a serious and occasionally life-threatening complication.112 The pathogenesis of enterocolitis is unknown. Defects in immunoglobulin A secretion and infection by toxigenic bacteria have both been implicated.
Pathogenesis
The pathogenesis of Hirschsprung disease involves failure of the neural crest–derived ganglion cell precursors to migrate appropriately, colonize, and survive in the bowel during embryogenesis. Mutations in at least eight different genes, each of which plays a role in the development and migration of enteric ganglion cells, have been recognized.106,107,113–118 Genes that have been associated or implicated in the pathogenesis of Hirschsprung disease include RET, EDNRB, EDN3, GDNF, SOX10, ECE1, NTN, ZEB2, PHOX2B, L1CAM, KIAA1279, TCF4, and NRG1.119 Approximately 50% of cases are associated with specific genetic abnormalities. In the rest, the underlying genetic alterations remain essentially unknown.119 Mutations of the RET protooncogene, the most frequent type of genetic abnormality, have been identified in 20% to 25% of short-segment cases and 40% to 70% of long-segment cases. Mutations in the other genes occur in fewer than 10% of cases.
Besides mutations, genetic polymorphisms in key genes, such as RET or NRG1, confer an increased risk of Hirschsprung disease.120,121 Recent gene expression array studies have also identified differential expression of a number of other genes (RELN, GAL, GAP43, NRSN1, and GABRG2) that were not previously implicated in Hirschsprung disease.122 These genes are involved in neuronal migration, nerve growth, nerve stimulation, neurotransmitter receptors, and smooth muscle relaxation.122 The mode of inheritance of Hirschsprung disease is variable. Familial forms of long- and short-segment disease are autosomal dominant with incomplete penetrance. However, variants associated with other congenital malformations are mostly autosomal recessive. Sporadic cases are believed to have a variable pattern of inheritance.
Pathologic Features
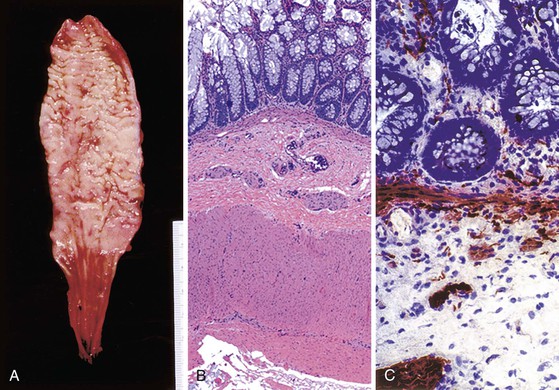
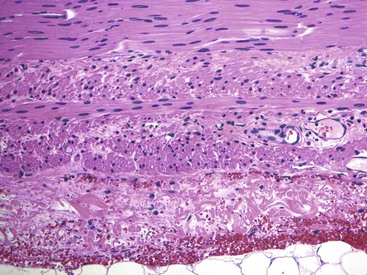
Classic cases of Hirschsprung disease reveal a distal narrow aperistaltic hypertonic segment, which is aganglionic, and a dilated proximal segment of colon caused by obstruction (Fig. 7.12, A). The distal hypertonic segment reveals a complete lack of ganglion cells in all neural plexuses and hypertrophy of Schwannian nerve fibers (see Fig. 7.12, B). In normal individuals, one to five ganglion cells are present, in clusters, for every 1 mm length of rectum. Mature ganglion cells are typically large cells with prominent nucleoli and amphophilic to basophilic cytoplasmic Nissl granules (see Fig. 7.3, A). In newborns, the cells are often smaller in size, and the nucleoli and cytoplasmic granules are not prominent, making their identification in tissue sections difficult (Fig. 7.13). The normal arrangement of ganglion cells in clusters and their association with nerve fibers facilitates their recognition. Immunostains to help identify ganglion cells have been recommended in difficult cases. A variety of antibodies may be useful, such as neuron-specific enolase (NSE), RET oncoprotein, Bcl-2, cathepsin D, protein gene product 9.5 (PGP9.5) HuC and HuC/D, or NeuN.107,123–126 Of these immunomarkers, HuC/D and NeuN are fairly sensitive and are the preferred markers (Fig. 7.14). However, in routine clinical practice, none of the neuronal markers offers a significant advantage over thorough histologic examination of H&E-stained sections by an experienced pathologist.127 When in doubt, some use neuronal markers in difficult cases. However, these antibodies have not been widely tested in clinical practice, and they should be used with caution despite claims of high specificity.127
A full-thickness transmural biopsy specimen offers better assessment of the neural plexuses because it allows visualization of the more prominent Auerbach plexus. However, such biopsies require general anesthesia and introduce risks of stricture and perforation. For these reasons, they are largely restricted to special cases. More commonly, blind rectal suction mucosal biopsies are obtained to establish a preoperative diagnosis, although one recent study showed that biopsies obtained with jumbo forceps are not only better but lead to fewer complications.128 The biopsy should be at least 3 mm in diameter, with submucosa representing at least one third of the entire thickness.129In practice, however, this is not always possible, and inadequacy rates for suction rectal mucosal biopsies range from 9% to 17%, mostly because of limited submucosa.129 This may necessitate full-thickness/seromuscular biopsies, especially in children older than 1 year of age. In addition, ganglion cells are scattered and fewer in number in the submucosa. Each cluster of ganglion cells contains approximately 2 to 7 cells, each cell being approximately 20 to 30 µm in diameter. Absence of ganglion cells in the submucosa in an adequate biopsy specimen of the rectum located more than 2 cm above the pectinate line is diagnostic of Hirschsprung disease.107Although hypertrophy of nerves (>40 µm thick), by itself, should not be considered sufficient evidence to establish the diagnosis, it can be a useful clue. Hypertrophic nerves may be absent in neonates and in patients with long-segment disease.
When ganglion cells are not seen in adequately studied serial sections of mucosal biopsies, supportive evidence can be obtained with the use of histochemical stains for acetylcholinesterase (AChE).130 In classic cases, AChE-positive nerve fibers are seen in the muscularis mucosae as well as in the lamina propria. These are lacking in biopsy specimens from normal individuals (see Fig. 7.12, C). Such fibers are few and difficult to demonstrate in newborns with Hirschsprung disease, but their number tends to increase with age. Some pathologists use a lactate dehydrogenase stain, which stains ganglion cells, in combination with AChE on frozen sections, for workup of suspected Hirschsprung disease.
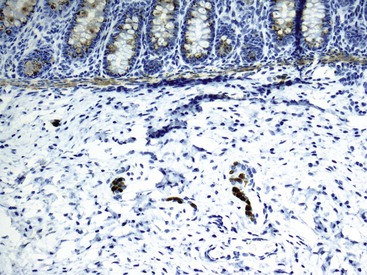
More recent studies have shown that calretinin immunostain is superior to AChE (which requires frozen sections) in the diagnostic workup of intestinal dysganglionoses.131–133 A few studies that compared these stains showed that calretinin stain is easier to interpret and superior to AChE.132,133 In these studies, there were no false-positive results with the calretinin stain, and only rare false-negatives, the latter mainly resulting from weak staining or inexperience of the pathologist. Normally, calretinin immunostain shows granular immunoreactivity in nerves fibers in the lamina propria, muscularis mucosae, and submucosa. It also stains ganglia (Fig. 7.15, A and B). Any positive staining in the nerve fibers, even when ganglia are not seen, excludes the possibility of Hirschsprung disease, whereas complete absence of staining supports this diagnosis (see Fig. 7.15, C and D). Calretinin immunoreactivity in mast cells is normal; it can be easily differentiated from neural positivity and seldom causes diagnostic problems (see Fig. 7.15, D).
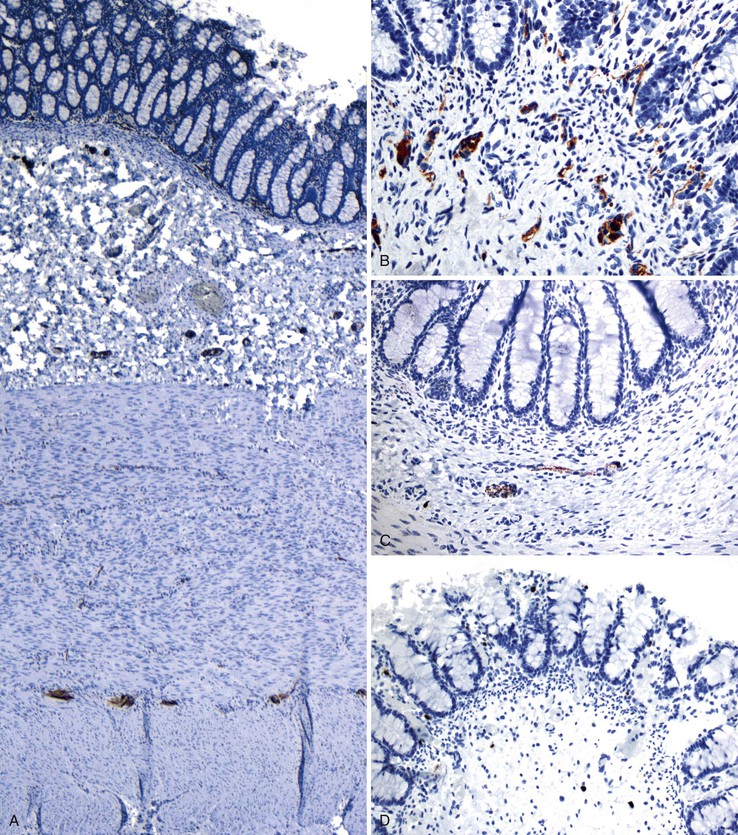
Equivocal AChE staining results in the presence of ganglion cells in biopsies. This can be seen in patients with Down syndrome and can be resolved by showing positive staining for calretinin. However, 2% to 10% of Down syndrome patients have associated Hirschsprung disease.134 Abnormalities of ICC have also been shown in some studies. However, it appears that this change is secondary to the absence of ganglion cells, and it is of little diagnostic value.135,136 A rare example of an extra layer of muscle in a patient with Hirschsprung disease associated with Mowat-Wilson syndrome has also been reported.137
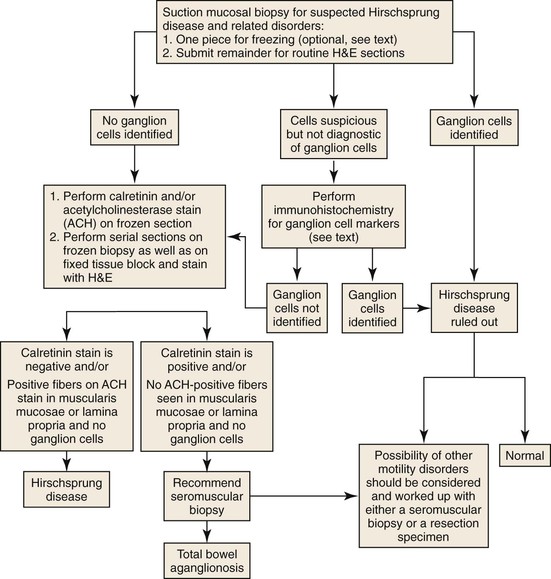
Preoperative biopsies are performed to establish a diagnosis before corrective surgery. Each laboratory should establish its own protocol for handling such cases (Fig. 7.16). The need for a piece of tissue to be kept for AChE staining is now considered redundant, because, as discussed earlier, calretinin staining is superior and can be performed on formalin-fixed tissue. However, a diagnosis of Hirschsprung disease rests on demonstration of an absence of ganglion cells or positive staining for calretinin. The latter can be difficult to determine due to sampling or technical reasons. Therefore, some pathologists use an AChE stain, because it may be the only positive finding. Multiple serial sections should be examined before a definitive diagnosis is rendered. Most laboratories obtain at least 50 to 100 serial sections stained with H&E before establishing a definite diagnosis of Hirschsprung disease.107 Unstained sections are used for calretinin staining. The presence of ganglion cells, or appropriate staining for calretinin in nerves, in a colonic biopsy rules out the possibility of conventional Hirschsprung disease.
The 10 to 25 mm of rectum located immediately above the pectinate line normally has a paucity of ganglion cells and prominent nerve fibers, which may lead to an erroneous diagnosis of Hirschsprung disease. This distance is shorter in neonates (approximately 10 mm) and increase to approximately 25 mm in children 3 years of age or older. To overcome this problem, some authorities advocate obtaining multiple biopsies at 1, 2, and 3 cm above the pectinate line. This is increased to 2, 3, and 5 cm in older children.129 A full-thickness biopsy can be obtained closer to the dentate line, because the hypoganglionosis zone extends for a shorter distance (approximately 5 mm) in the myenteric plexus. Biopsy specimens in this situation may reveal colonic to squamous transitional epithelium, indicating proximity to the pectinate line. The presence of this epithelium should be specifically mentioned in the pathology report.107 Presence of crush artifact, or small size of the biopsy specimen, are other factors that could lead to an incorrect diagnosis. One should not hesitate to request a repeat biopsy in this situation.
Some patients with Hirschsprung disease have zonal aganglionosis or hyperganglionosis with skip areas, involving the small intestine or colon.138,139 These cases are thought to be acquired in nature and of diverse etiology (viral enterocolitis, ischemia or other forms of injury). Other possibilities include inability of vagal and sacral crest cells to migrate appropriately. Occurrence of segmental aganglionosis, although rare, implies that absence of ganglion cells in the appendix cannot be used as absolute evidence of total bowel aganglionosis, particularly when making a decision regarding the extent of resection during a surgery.
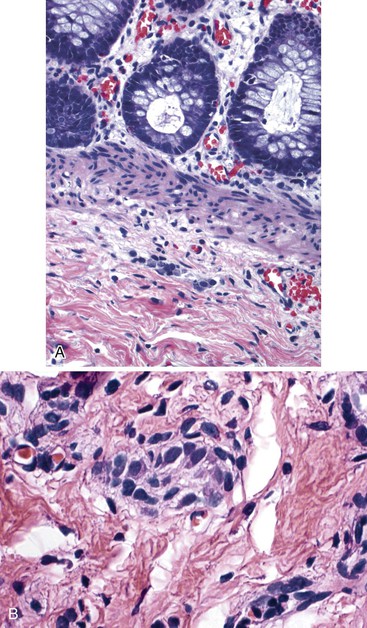
In addition, specific attention should be given in cases of Hirschsprung disease to the presence or absence of mucosal inflammation, which is not infrequently seen. Pathologic changes can resemble acute colitis, including the presence of cryptitis and crypt abscesses, neonatal necrotizing enterocolitis, ischemic colitis, or pseudomembranous colitis (Fig. 7.17).112,140,141 Severe cases with transmural necrotizing inflammation may progress to perforation.
Handling of resection specimens in Hirschsprung disease varies among different laboratories. One may follow the guidelines suggested by the International Working Group.129 The goals of examining a resection specimen are largely to document and define the extent of aganglionosis and the presence of normal ganglia at the proximal resection margin. One easy way to achieve this is to obtain transverse (en face) sections from the proximal and distal margins and then take a longitudinal strip along the entire length of the specimen. This may be submitted as multiple sequential sections or as a “Swiss roll” in a single block, keeping the orientation of the proximal and distal segments. Alternatively, one may submit multiple transverse sections from the entire specimen, spaced at 1cm intervals. Frozen samples may be obtained from the narrow involved segment, the transitional zone, and proximal normal-appearing colon for histochemical stains.
Differential Diagnosis
Establishing a diagnosis of Hirschsprung disease in suction mucosal biopsies remains a diagnostic challenge, especially in newborns and premature babies. The issue is largely related to reliable identification of ganglion cells. When ganglion cells are not identified, one must ensure that the biopsy specimen is adequate, that it was obtained from the correct anatomic area, and that special stains support the diagnosis. Patients with features suggestive of Hirschsprung disease who do not show absence of ganglion cells on biopsies remain a diagnostic challenge. The clinical differential diagnosis for these cases includes other types of dysganglionoses, intestinal pseudo-obstruction, constipation, ileus, toxic megacolon, and hypothyroidism. Once ganglion cells are identified in mucosal biopsy specimens, the possibilities include ultrashort-segment Hirschsprung disease and other dysganglionoses (hypoganglionosis and intestinal neuronal dysplasia). However, the diagnosis and even the existence of the latter entities remains controversial, so this possibility should be entertained only in resection specimens or full-thickness biopsies. In any case, most of these disorders are managed similarly. In some patients, an AChE staining pattern similar to Hirschsprung disease but with ganglion cells is believed to represent ultrashort-segment Hirschsprung disease. This remains a controversial entity and likely represents a heterogeneous group comprising aganglionosis, hypoganglionosis, and anal achalasia. Because the distal 2 to 3 cm of rectum normally contains only sparse ganglion cells, these diagnoses are difficult to establish.
Treatment and Follow-Up
Treatment involves surgical resection of the involved segment of bowel, followed by restoration of bowel continuity. This may be performed in two stages. A definitive anastomosis is performed after an initial colostomy. It may also be performed as a one-stage “endorectal pullthrough” procedure.107 Seromuscular biopsies, obtained laparoscopically, are usually submitted for intraoperative frozen section evaluation to ensure the presence of ganglion cells at the proximal margin of the resected segment before completion of the anastomosis and also to confirm a lack of ganglion cells in the affected segment of colon. However, use of frozen sections for evaluation of ganglion cells can be problematic and should not replace a preoperative suction mucosal biopsy.142
In premature babies or newborns, differentiation of immature ganglion cells from lymphocytes or plasma cells also can be problematic in frozen sections (see Fig. 7.13). Immature ganglion cells are smaller in size, and the nuclei lack prominent nucleoli. They are usually present in clusters, have discernible cytoplasm, and are often associated with nerve fibers. At the time of frozen section, toluidine blue, Geimsa, or Diff-Quick (Romanowski) stain, in addition to routine stains, may aid in identification of ganglion cells.
The prognosis in most surgically treated cases is excellent. Most patients are able to achieve continence. Some continue to have postoperative dysmotility despite anastomosis of normoganglionic segments. This phenomenon has been attributed to immaturity of ganglion cells, which improves with time.143 Long-term follow-up studies show that most patients achieve good quality of life into adulthood, with minimal or no problems. However, some patients continue to have incontinence, and others have problems with constipation and enterocolitis.144,145
Other Developmental Disorders of the Enteric Nervous System
This heterogeneous group of disorders includes hypoganglionosis, hyperganglionosis, and abnormal differentiation of ganglion cells. Enteric ganglia may also be involved secondarily by systemic metabolic defects such as lysosomal storage disorders. Neuropathic forms of familial intestinal pseudo-obstruction (discussed later) are considered another form of developmental abnormality.
Clinically, these disorders resemble Hirschsprung disease. However, ganglion cells are present. On AChE staining, they may mimic the pattern seen in Hirschsprung disease, with strong staining in some cases and little or no staining in others. Although diagnostic criteria for aganglionosis (Hirschsprung disease) are well established, objective criteria necessary to define and diagnose other enteric dysganglionoses are poorly established and often impractical to apply.146–149 This situation has resulted in the use of many different terms for these disorders, such as “variant Hirschsprung disease,” “pseudo-Hirschsprung disease,” or “Hirschsprung disease–allied disorders.” Subtle alterations in the number of ganglion cells are almost impossible to diagnose on routine suction mucosal biopsy specimens. Quantitation of the neural plexuses, neural and glial stroma, and ganglion cells is extremely difficult to perform, even on conventionally oriented transmural biopsies. Alterations in specific subtypes of ganglion cells can be resolved only by special stains and electrophysiologic studies, and protocols for clinical practice have not been established.4 It is likely that functional changes underlie the subtle morphologic alterations of ganglion cells but are not evident on routine studies, as was shown in a study revealing increased NOS-producing ganglion cells in cases of intestinal neuronal dysplasia.150 Study of whole-mount sections, along with application of special stains, has been advocated to better characterize these lesions.
Despite attempts to standardize the counting method for myenteric ganglia using neuronal markers and better guidelines for establishing the diagnosis, there is marked interobserver variability in this regard.125,151 The median number of ganglia in the myenteric plexus has been shown to be 8 per 10 millimeter (range, 2 to 12/mm) in routine stains and 15 per 10 millimeter (range, 2 to 23/mm) with use of special stains.152 The size of ganglion cells varies from 10 to 40 µm. The median number of neurons per ganglion has been reported to be 3 (range, 2 to 6) on H&E stain and 10 (range, 4 to 14) using special stains.152 The numbers are higher in whole-mount thick sections.152 Variability related to age, site, individual differences, and stretching of the tissues during processing makes it even more difficult to design criteria that can differentiate intestinal dysganglionoses based on mild to moderate alterations in ganglion cell numbers.125 Hence, the criteria suggested by the International Working Group are very conservative.
It is also unclear whether the histologic changes described are secondary in nature. This cannot be determined on the basis of evaluation of mucosal biopsy specimens but requires full-thickness biopsies or evaluation of resection specimens. Therefore, only cases (congenital or acquired) that show severe abnormalities are diagnosed reliably. The diagnostic utility of calretinin stain in mucosal biopsies in these conditions remains unknown.
Hypoganglionosis
The term hypoganglionosis refers to a reduction in the number of ganglia in neural plexuses, decreased number of ganglion cells per ganglion, and small size of ganglia.153 This condition may exist in some segments of the colon in patients with Hirschsprung disease or as an isolated (primary) condition leading to intestinal pseudo-obstruction. The genetic polymorphic variants or mutations associated with Hirschsprung disease are absent in this condition.
Due to methodologic variations, the International Working Group determined that there are no criteria that can be used uniformly among different laboratories or institutions. It has been suggested that the presence of one or fewer ganglia (clusters of neuronal cells) per millimeter, or two or fewer neurons per ganglion, constitutes hypoganglionosis (Fig. 7.18).129 The AChE stain shows very weak or absent staining in the lamina propria, and hypertrophic nerve twigs typically associated with Hirschsprung disease are usually lacking. A review of 92 cases reported in the literature showed that many features are similar to Hirschsprung disease, including male predominance (3 : 1 ratio) and a clinical presentation with severe chronic constipation, pseudo-obstruction, and/or enterocolitis. In contrast, only 32% of patients with hypoganglionosis were diagnosed in the neonatal period, compared with more than 90% of those with Hirschsprung disease, possibly because of the difficulty in establishing a definite diagnosis. Hypoganglionosis can be focal, segmental, or diffuse. The hypoganglionic segments are usually narrow, and the condition is associated with dilatation of the proximal bowel.111 However, occasionally one can see fewer numbers of ganglion cells in colonic resections performed for unrelated reasons and in patients without any motility issues. This puts into question the validity of evaluating ganglion cell numbers in motility disorders. The management, long-term outcome, and complications are also similar to those of Hirschsprung disease.
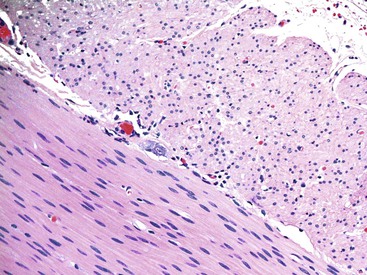
Hyperganglionosis
Hyperganglionosis is characterized by an increased number of ganglion cells and neural hyperplasia and has also been referred to as intestinal neuronal dysplasia (IND). Two forms, IND-A and IND-B, are recognized.154 IND-A is extremely rare, constituting fewer than 5% of all cases of IND. It is characterized by sympathetic aplasia, myenteric hyperplasia, and colonic inflammation.155,156 The myenteric plexus shows an increased number of ganglion cells in each ganglion. However, the submucosal ganglia are normal appearing, and ectopic ganglion cells are not present. The clinical presentation includes episodes of intestinal obstruction, bloody stools, and diarrhea. Surgical resection is believed to be curative.
IND-B is a controversial entity characterized by marked neural hypertrophy and an increased number of large ganglion cells in the submucosal neural plexuses. It constitutes approximately 95% of all IND cases.147,157,158 It is considered to represent an anomaly of the parasympathetic neural system of the bowel. The diagnostic criteria for IND-B have changed with time. Currently, diagnosis requires the presence of giant submucosal ganglia, defined as more than eight ganglion cell cross sections per submucosal ganglion. Such “giant” ganglia should constitute more than 20% of all ganglia. At least 25 submucosal ganglia should be evaluated (Fig. 7.19).149 The myenteric plexus show hypoganglionosis in some cases, and some are even associated with aganglionosis similar to Hirschsprung disease.158 Rare cases that are clinically similar but with increased numbers of ganglion cells only in the myenteric plexus have been described.159 The AChE stain reveals increased staining in the lamina propria, similar to Hirschsprung disease, early in the course of disease. Hyperplasia of the myenteric plexus may be evident in some cases. Involvement may be limited to the rectum, or it may be extensive. Small bowel involvement is rare. The clinical presentation is related to chronic constipation. Premature infants and those younger than 1 year of age normally show higher numbers of ganglion cells per submucosal ganglion (see Fig. 7.13, B). Therefore, it is recommended that a diagnosis of IND-B should be made only in children older than 1 and younger than 4 years of age.149
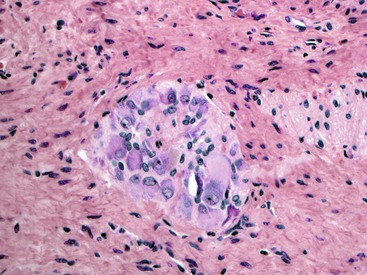
An increase or decrease in the number of ganglia may also be caused by mechanical obstruction. A rare case of mechanical obstruction caused by a Ladd band, associated with IND-B type changes, has been reported. Degeneration of ganglion cells (hypoganglionosis), enteric plexuses, and ICCs subsequently developed in this patient.160 Such an adaptive response combined with neuronal plasticity has also been shown in animal models of mechanical bowel obstruction.161,162
Homozygous mutant mice deficient in Ncx/Hox11L.1 show features similar to IND-B. However, these mutations have not been found in human patients.163,164 No specific genetic mutations are associated with this disorder, although a possible association with RET variants has been suggested.165
Management of IND-B depends on the extent of the involvement. Conservative treatment is possible in many patients.166–168 Surgical options are largely similar to those available for Hirschsprung disease.
Increased ganglion cells and ectopic ganglion cells in the lamina propria can also be seen in patients with intestinal ganglioneuromas. Sporadic lesions are usually single and localized, but they can also be diffuse (ganglioneuromatosis) (Fig. 7.20). Diffuse ganglioneuromatosis is almost always associated with multiple endocrine neoplasia (MEN IIb) and mutations in the RET protooncogene. It can manifest as bowel dysmotility. The presence of isolated ganglion cells in the deep lamina propria, by itself, is not considered abnormal.169
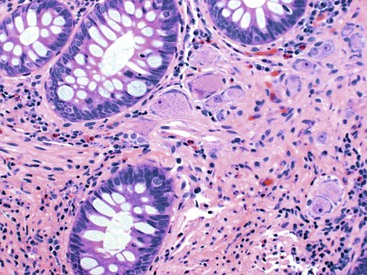
Despite several reports of IND-B and hypoganglionosis in the literature and many attempts to reach a consensus regarding diagnostic criteria,149,170,171 these entities remain controversial. Their clinical significance also remains unclear, and even their existence is still in question. IND-B is now considered a histologic pattern of disease, which does not require surgical treatment. The rarity of these conditions is highlighted by the fact that many experts in the field, at major institutions across the world, have spent their entire career without ever making a diagnosis of either hypoganglionosis or IND.
Intestinal Pseudo-obstruction
Intestinal pseudo-obstruction is defined as a clinical syndrome caused by inability of the bowel to propel its contents in patients without a mechanical obstruction. Both acute and chronic forms are recognized.
Acute Intestinal Pseudo-obstruction
Paralytic Ileus
Paralytic ileus is the most common cause of intestinal pseudo-obstruction. It occurs after abdominal surgery or abdominal trauma and in patients with peritonitis.172 The entire bowel becomes paralyzed and distended. The diagnosis is made on clinical grounds, and the treatment is supportive.172 No specific histopathologic changes are recognized in this condition.
Acute Idiopathic Intestinal Pseudo-obstruction (Ogilvie Syndrome)
Acute idiopathic pseudo-obstruction is a rare and potentially serious complication in patients who have recently undergone surgery or are ill from other causes.173 The pathogenesis is poorly understood, although temporary autonomic dysfunction is suspected. Patients demonstrate bowel dilatation, most often confined to the right colon, which can lead to transmural ischemia and perforation, the latter occurring most frequently in the cecum. Pathologic changes are nonspecific but mimic those of ischemia secondary to increased intramural pressure. In most instances, the condition resolves with supportive care.
Chronic Intestinal Pseudo-obstruction
Chronic intestinal pseudo-obstruction is caused by a variety of disorders that may affect various components of the bowel neuromuscular apparatus.174,175 It most commonly involves the small intestine or the colon, or both. It may involve the bowel (primary idiopathic types), or it may be part of a generalized or systemic disorder (secondary types). Dysmotility may be the result of abnormalities in any of the components of the neuromuscular apparatus of the bowel wall. Based on the component involved, four major categories have been recognized: conditions with abnormalities of the smooth muscle (myopathic form), those with abnormalities of the neural system (neuropathic form), those with ICC abnormalities (mesenchymopathic form), and those with abnormalities of neurohormonal peptides (see Table 7.1). Many disorders show involvement of more than one component, of which some may be primary in nature and others secondary. Idiopathic forms may be sporadic or familial.
The clinical features of chronic intestinal pseudo-obstruction are similar among the various subtypes. In most cases, particularly the familial ones, symptoms begin in childhood. Some patients remain asymptomatic until middle age, whereas others are entirely asymptomatic. The diagnosis is often delayed for many years (median age at diagnosis, 8 years). Many patients have had multiple exploratory laparotomies.176 Symptoms are typical of intestinal obstruction (e.g., abdominal distention, pain, vomiting). Distention may be gradual but it can become severe, especially if both the small intestine and the colon are involved. In general, these patients show alternating diarrhea and constipation rather than obstipation. Diarrhea is usually secondary to bacterial overgrowth because of stasis, and it may result in substantial weight loss. Perforation occurs rarely.
Primary Chronic Intestinal Pseudo-obstruction
Myopathic Forms.
Both sporadic and familial myopathic forms are well recognized (Table 7.2).174 Care must be exercised before considering a case to be sporadic because involved family members may be asymptomatic and a reliable family history may be difficult to obtain. Familial visceral myopathy may also involve other organs (e.g., urinary bladder, biliary tract) and has also been termed hollow visceral myopathy.177,178
Table 7.2
Primary Chronic Intestinal Pseudo-obstruction: Major Types of Familial Visceral Myopathies and Neuropathies
| Type | Mode of Transmission | Age at Onset | Symptoms | Gastrointestinal Lesions | Main Findings | Extraintestinal Involvement |
| Visceral Myopathy | ||||||
| I | AD | After first decade of life | Varied; dysphagia, constipation, and/or CIIP | Dilatation of esophagus, duodenum, colon | Involvement of both layers of muscularis propria | Dilation of bladder, uterine inertia, mydriasis |
| II | AR | Teenage years | Severe abdominal pain, CIIP | Dilation of stomach, mild dilatation of small intestine, small bowel diverticulosis | Involvement of both layers of muscularis propria | Ptosis and external ophthalmoplegia, mild degeneration of striated muscle |
| III | AR | Middle age | CIIP | Mild dilation of entire gastrointestinal tract | Involvement of both layers of muscularis propria | None |
| Visceral Neuropathy | ||||||
| I | AD | Any age | Abdominal pain diarrhea, abdominal distention, gastroparesis, CIIP | Segmental dilation of jejunum and ileum; proximal small intestine is always involved | Reduction and degeneration of myenteric plexus, loss and degenerative changes in neurons | None |
| II | AR | Infancy | Hypertrophic pyloric stenosis, short dilated small intestine, intestinal malrotation | None reported | Decreased neurons | CNS malformation with heterotopia and absence of operculum, patent ductus arteriosus |
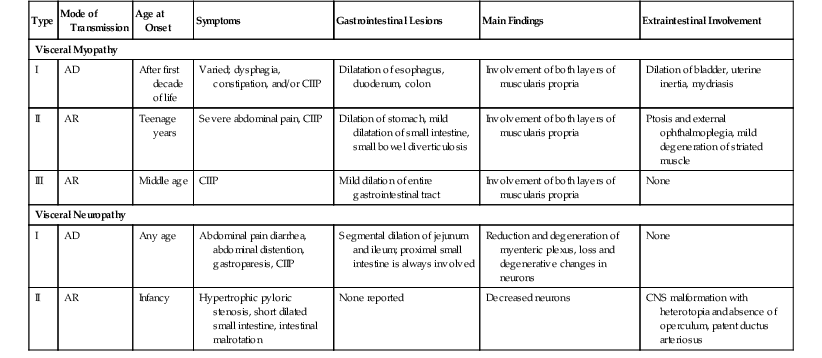
AD, Autosomal dominant, AR, autosomal recessive, CIIP, chronic idiopathic intestinal pseudo-obstruction, CNS, central nervous system; GI, gastrointestinal.
Type I is the most common. It is characterized by redundant colon, esophageal dilatation, megaduodenum, megacystis, and sometimes uterine inertia. Type II tends to show gastric dilatation, mild small-intestinal dilatation often with formation of diverticula, ptosis, and external ophthalmoplegia. In type III, the entire GI tract, from esophagus to rectum, may be involved and shows marked dilatation. Type IV is still poorly characterized and has been reported to be associated with gastroparesis, a tubular (narrow) small intestine, normal esophagus, and normal colon.179
In general, sporadic cases resemble autosomal recessive familial type III visceral myopathy. Other rare forms, partially resembling types I and III, with an autosomal recessive mode of transmission and esophageal and cardiac abnormalities, have also been described.180 Rare cases that have X-linked intestinal pseudo-obstruction associated with mutations in the FLNA gene show an extra layer of smooth muscle between the inner circular and outer longitudinal muscle coats.181 The FLNA gene encodes a protein (filamin A) that plays a role in cell shape and motility. These children have a variety of associated malformations, including intestinal malrotation and cardiac abnormalities. They are affected clinically early in life.
Although smooth muscle degeneration is believed to be responsible for bowel dysmotility, the pathogenesis in most cases remains obscure. Rare cases with actin or desmin abnormalities have been described.182,183 In cases of “desmin myopathy,” systemic skeletal and cardiac muscle involvement is also common. Rare cases show a T cell–rich inflammatory leiomyositis. These cases are possibly autoimmune in nature.184 A distinctive type of nonfamilial visceral myopathy has been described in young children from southern, central and eastern Africa (African visceral leiomyopathy).185 In some cases, absence of KIT-positive ICC has been thought to be the underlying mechanism (see later discussion).186 It is likely that this is a heterogeneous group of disorders, and many of the histologic changes probably represent end-stage disease. Many cases go unrecognized, so the spectrum of these disorders may be even wider than is currently believed.
Pathologic Features.
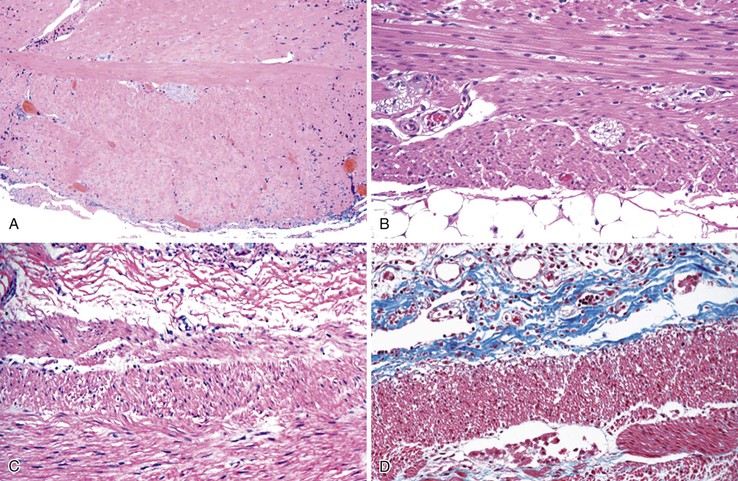
The involved segment of bowel is often dilated; it appears thick, normal, or thin depending on the degree of distention (Fig. 7.21). Initially, there is an absence of mucosal abnormalities, but inflammation, ulceration, and ischemia may develop secondary to stasis and extensive dilatation.174
Microscopically, the bowel reveals degeneration and fibrous replacement of the smooth muscle. Degenerative changes are most prominent in the muscularis propria, but they also affect the muscularis mucosae and therefore may occasionally be identified in mucosal biopsy specimens.187 Although the published literature suggests that the outer longitudinal layer tends to be more severely involved in this disorder, in our experience, either both layers are equally involved or the inner circular layer is involved in isolation (Fig. 7.22).174,178 These cases need to be differentiated from scleroderma based on other pathologic features and clinical findings. Muscle degeneration results in fibrosis, cytoplasmic vacuolation, variation in muscle fiber size, and thinning of the bowel wall (Fig. 7.23, A and C). Fibrosis may be subtle, requiring a trichrome stain to be fully appreciated (see Fig. 7.23, B and D).174 The relative thickness of the circular and longitudinal muscle layers varies among individuals and according to anatomic location and method of sectioning; measurement of their ratio has little practical significance. Other changes include variation in nuclear size and increased chromasia, increased mitotic activity, and variation in fiber size. Cytoplasmic inclusions of various types have been described. Amphophilic “M” bodies that are PAS negative can be seen in the muscularis propria, as in cases of slow transit constipation, and diabetic gastropathy (see Gastroparesis; see Fig. 7.11, A and B).188 These inclusions likely represent degenerating smooth muscle fibers. Similar PAS-positive intracytoplasmic eosinophilic inclusions (5 to 20 µm in size) are seen in polyglucosan body myopathy.189 These inclusions stain with polyglucosan antibodies, similar to Lafora bodies, and ultrastructurally they are composed of nonlysosomal filamentous material located beneath the sarcolemma and between myofibrils.190
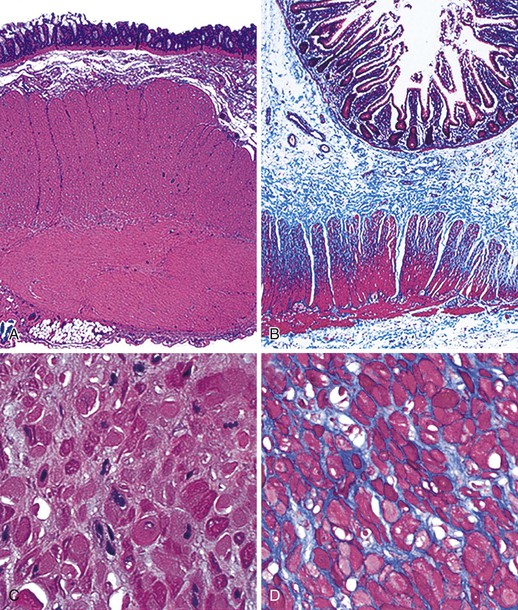
Rare familial forms of visceral myopathy associated with α-actin cytoplasmic inclusions have also been described. These inclusions may also be PAS positive, but they are best highlighted with an α-actin immnostain.191 Ultrastructurally, cytoplasmic inclusions represent aggregates of degenerated myofibrils.189 However, one must be aware of potential artifactual changes in the muscle that are commonly seen in surgical specimens (Fig. 7.24, A and B). Artifactual vacuolation, cytoplasmic pseudo-inclusions, and thinning of the muscle layers are not uncommon in colonic resections (see Fig. 7.23). Chronic ischemia and irradiation can also cause thinning of the muscularis propria and fibrosis. However, in these cases, the fibrosis is patchy, discontinuous and coarse, sometimes leading to complete extinction of the muscle layer in the involved region. Rare cases with deficient smooth muscle α-actin show an absence of staining with smooth muscle actin antibodies (αSMA), particularly in the inner circular muscle layer (Fig. 7.25, A).183,192 However, similar changes can be limited to the terminal ileum in cases without any evidence of dysmotility. Therefore, caution is warranted in interpreting this finding (see Fig. 7.25, B).193 Electron microscopy shows nonspecific degenerative changes, including mitochondrial vacuolation, and this may be the only evidence of myopathy in cases in which light microscopy results are normal.174,178,185,194 An extra layer of smooth muscle in the muscularis propria may be found in the large or small intestine in some cases. This phenomenon has also been reported in a case of X-linked intestinal pseudo-obstruction with FLNA gene mutation (Fig. 7.26).181,195 In that case, in addition to the extra layer of smooth muscle, multinucleated myocytes were seen in the innermost layer of muscularis porpria.181 Occasionally, one may see abnormal layering of muscle for a short distance, usually near the ileocolonic junction, without any motility issues (Fig. 7.27).
Neuropathic Forms.
This group of disorders includes abnormalities of the intrinsic or extrinsic neural network of the bowel. There are both sporadic and familial forms (see Table 7.2).174,196 The mode of inheritance may be autosomal dominant, autosomal recessive, or, rarely, X-linked.196–198 The autosomal dominant (type I) variant does not show extraintestinal manifestations. Autosomal recessive (type II) cases tend to show intranuclear inclusions in ganglion cells. Some cases are characterized by mental retardation and basal ganglia calcification. The X-linked form is associated with a short small intestine, malrotation, and pyloric hypertrophy.198
The pathogenesis of neurodegenerative changes remains obscure. Several mechanisms may be involved, including altered calcium signaling, mitochondrial dysfunction, autoimmunity, and free radical injury.199,200 Rare autosomal recessive cases involve a progressive, multisystem, neurodegenerative disorder and reveal abnormalities in mitochondrial DNA.201 Many genes have been identified that are responsible for the syndromic forms of intestinal pseudo-obstruction. These include the genes encoding thymidine phosphorylase (TYMP, also known as endothelial cell growth factor-1 or ECGF1), DNA polymerase-γ (POLG), and the transcription factor SOX10, some of which are associated with mitochondrial disorders.202 One study of 63 patients with idiopathic intestinal pseudo-obstruction detected mitochondrial myopathies that were not clinically suspected in 15 (19%).203 These included five cases with mutations in POLG, five with mutations in TYMP, and two with mutations in the mitochondrial tRNALeu(UUR) gene. A subsequent study of intestinal pseudo-obstruction in 12 children failed to reveal any evidence of a mitochondrial disorder. Mutations in TYMP have been shown to be responsible for familial cases of mitochondrial neurogastrointestinal encephalpomyopathy (MNGIE), a disorder characterized by intestinal pseudo-obstruction, progressive external ophthalmoplegia, ptosis, polyneuropathy, and leukoencephalopathy.204,205 Mutations in tRNALeu(UUR) are associated with MELAS (mitochondrial myopathy, epilepsy, lactic acidosis, and strokelike episodes) and with familial diabetes and recurrent pancreatitis.206
Because these mitochondrial disorders may not show specific changes on routine histopathology, the diagnostic workup should include serum levels of lactate and TYMP activity, brain magnetic resonance imaging (MRI), and muscle biopsies.203 The symptoms are typically severe, and total parenteral nutrition is often required. The presence of neurologic symptoms may lead to suspicion of an underlying mitochondrial disorder. Decreased ganglion cell survival may be a factor in some cases, as suggested by decreased BCL2 gene product in enteric ganglion cells.207 Some cases reveal inflammatory neuronal degeneration, which suggests an autoimmune or infectious etiology208; neuronal autoantibodies are detected in some patients (Table 7.3).209 One study suggested that antineuronal antibodies may contribute to neuronal dysfunction via activation of autophagy involving the Fas receptor complex.200 Some of these cases represent a paraneoplastic manifestation, whereas others remain idiopathic.210
Table 7.3
Antibodies Identified in Paraneoplastic CIIP
| Antibody | Target |
| Type I Anti-Hu | Neurons |
| Type II Anti-Ri | Neurons |
| Anti-Yo | Anti–Purkinje cell cytoplasmic antibodies |
| N-type voltage-gated calcium channel antibodies | N-type voltage-gated calcium channels |
| P/Q-type calcium channel antibodies | P/Q-type calcium channels |
| Ganglionic and muscle-type nicotinic acetylcholine receptor antibodies | Nicotinic acteylcholine receptors |
CIIP, Chronic idiopathic intestinal pseudo-obstruction.
Pathologic Features.
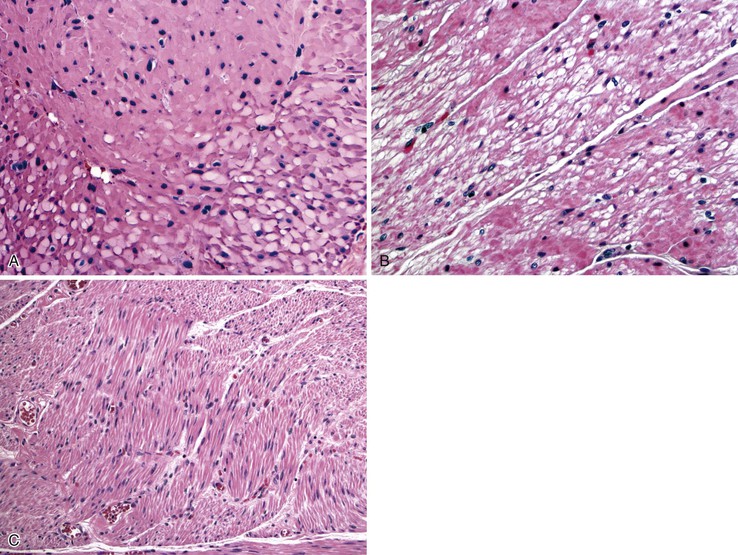
Gross findings are similar to other forms of intestinal pseudo-obstruction. Microscopic examination of routine sections may show a variety of qualitative and quantitative changes in ganglion cells, supporting glia, and nerve fibers. Determination of increased or decreased neuronal density remains controversial, despite attempts at standardization (see prior section on other developmental disorders of the enteric nervous system).152 Stretching of the tissues may result in a decrease in neuronal cell counts, which may also occur as a result of aging. In adults, the mean number of neurons per ganglion is estimated to be four in the myenteric plexus (three in the jejunum, six in colon) and two in the submucosal plexus.129,211 There are approximately eight ganglia per centimeter of bowel. Because of the lack of standardization, an International Working Group suggested criteria for patients with hypoganglionosis, which required one or fewer ganglia per millimeter length of bowel and two or fewer neurons per ganglion (see Hypoganglionosis). A variety of degenerative changes may be present as well, including cytoplasmic vacuolation, cell swelling, chromatolysis, and nuclear pyknosis and fragmentation (Fig. 7.28, A through C). In intranuclear inclusion neuropathy, cytomegalovirus (CMV)-like intranuclear inclusions may be identified in neurons (see Fig. 7.28, D).196,197,212,213 By electron microscopy, these inclusions represent non–membrane-bound, osmiophilic, proteinaceous material composed of 8-nm curved filaments, not viral particles.214 Megamitochondria may be seen in some cases, which is indicative of a mitochondrial disorder (see Fig. 7.28, B). Some cases show inflammation of the ganglia and myenteric plexus consisting of lymphocytes, eosinophils, or mast cells210 (Fig. 7.29). Although a few lymphocytes may be present surrounding a neural plexus, under normal circumstances, inflammatory cells are not present within ganglia or directly in contact with ganglion cells. Lymphocytes are mostly T cells. These can be highlighted and characterized with the use of immunohistochemical markers (CD45, CD3, CD20, CD25, CD4, CD8). However, their precise characterization adds little in clinical practice.
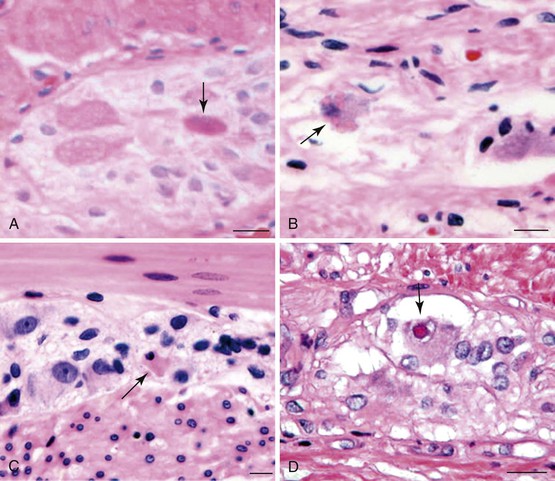
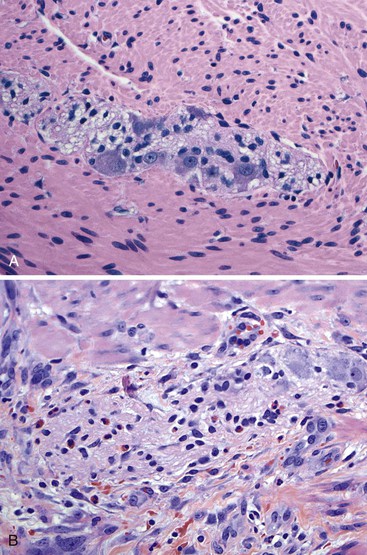
Eosinophilic myenteric ganglionitis has been described mainly in children, with only rare reports in adults (see Fig. 7.29, B).215,216 Increased mast cells have also been reported in bowel dysmotility and are believed to interact with the neural apparatus causing dysmotility. However, enumeration of mast cells or their identification around neural plexuses has unclear significance at present.
Subtle degenerative changes may also be present within glia and axons in these disorders.174 They are best appreciated with a special stain applied to thick en face or tangential embedded sections of the bowel or whole-mount preparations. The axons may show swelling and cytoplasmic vacuolation. However, such changes can also be seen in unrelated conditions and therefore are nonspecific (Fig. 7.30). In the past, silver stains were used to identify abnormalities of argyrophobic and argyrophilic ganglion cells. However, these stains are currently obsolete.