37 Neuromuscular Disorders in the ICU
 The Motor Unit and Its Physiology
The Motor Unit and Its Physiology Muscles of Respiration
Muscles of Respiration
Three muscle groups may be defined based on their importance for respiration (Figure 37-1):1
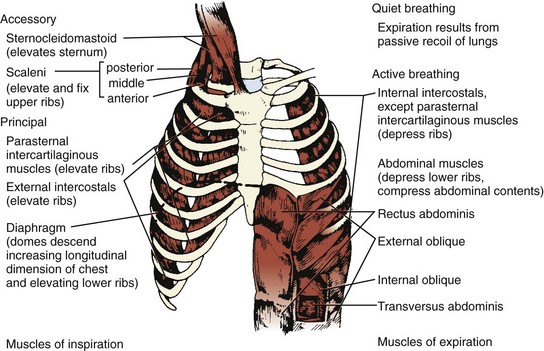
Figure 37-1 Major respiratory muscles.
Inspiratory muscles are indicated on the left and expiratory muscles are indicated on the right.
(From Garrity ER. Respiratory failure due to disorders of the chest wall and respiratory muscles. In: MacDonnell KF, Fahey PJ, Segal MS, editors. Respiratory Intensive Care. Boston: Little, Brown; 1987, p. 313.)
Clinical Presentation of Neuromuscular Respiratory Failure
Patients experiencing respiratory dysfunction due to neuromuscular disease typically present with a combination of upper airway dysfunction and diminished tidal volume (VT). Difficulty with swallowing liquids, including respiratory secretions, is the most typical presentation of pharyngeal weakness, although some patients have an equal or greater degree of difficulty with solid food. A hoarse or nasal voice may also signal problems with the upper airway. These conditions are noted in patients who are at risk for aspiration and present with difficulty with attempts at negative-pressure ventilation (cuirass or iron lung), because the weakened muscles may not be able to keep the airway open as the pressure falls.2 Paradoxical abdominal movement (inward movement of the abdomen during inspiration) is an important sign of diaphragmatic weakness.3
Regardless of the vital capacity, however, indications for intubation and mechanical ventilation include evidence of fatigue, hypoxemia despite supplemental oxygen administration, difficulty with secretions, and a rising PaCO2. In the absence of hypercapnia, occasional patients (e.g., those with myasthenia gravis) can be managed under very close observation in an ICU with less invasive techniques (e.g., bilevel positive airway pressure [BiPAP]).4
In addition to vital capacity, trended measurements of the maximum inspiratory pressure (PImax, more typically recorded as negative inspiratory force [NIF]), are useful indicators of ventilatory capacity. Inability to maintain a PImax greater than 20 to 25 cm H2O usually indicates a need for mechanical ventilation. Although the maximum expiratory pressure (PEmax) is a more sensitive indicator of weakness,5 it has not proved to be as useful as an indicator of the need for mechanical ventilation. A more detailed discussion of these variables and their use may be found elsewhere.6,7
Because a patient with neuromuscular respiratory failure has intact ventilatory drive,8 the fall in VT is initially matched by an increase in respiratory rate, keeping the PaCO2 normal or low until the vital capacity becomes dangerously reduced. Many patients initially maintain their PaCO2 in the range of 35 mm Hg because of either (1) a subjective sense of dyspnea at low VT or (2) hypoxia from atelectasis and increasing dead space. When the PaCO2 begins to rise in this circumstance, abrupt respiratory failure may be imminent.
The modest degree of hypoxia in most of these patients worsens when the PaCO2 begins to rise, displacing more oxygen from the alveolar gas. However, aspiration pneumonia and pulmonary embolism are also frequent causes of hypoxemia in these patients. To determine the relative contributions of these conditions to a patient’s hypoxemia, one can use a simplified version of the alveolar gas equation as follows (derived elsewhere)6,7:
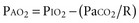
where PAO2 is the alveolar partial pressure of oxygen, PIO2 is the partial pressure of inspired oxygen (in room air, 150 mm Hg), and R is the respiratory quotient (on most diets, about 0.8). This allows estimation of the alveolar-arterial oxygen difference (PAO2 − PaO2). Under ideal circumstances in young people breathing room air, this value is about 10 mm Hg, but it rises to about 100 mm Hg when the fraction of inspired oxygen (FIO2) is 1.0. The alveolar air equation allows one to factor out the contribution of hypercarbia to the decrease in arterial partial pressure of oxygen (PaO2); it should be used to determine whether there is a cause of significant hypoxemia in addition to the displacement of oxygen by carbon dioxide.
Patients with orbicularis oris weakness may have artifactually low vital capacity and NIF measurements because they cannot form a tight seal around the spirometer mouthpiece. The need for nursing and respiratory therapy personnel who are experienced in the care of these patients is thus underscored. It is also important for physicians to observe these patients directly rather than relying solely on reported measurements. The physical findings associated with neuromuscular respiratory failure are reviewed elsewhere.6,7 Among the most important findings are rapid, shallow breathing,9 the recruitment of accessory muscles, and paradoxical movement of the abdomen during the respiratory cycle. Fluoroscopy of the diaphragm is occasionally valuable for the diagnosis of diaphragmatic dysfunction.10
Autonomic dysfunction commonly accompanies some of the neuromuscular disorders requiring critical care, such as Guillain-Barré syndrome, botulism, and porphyria (Table 37-1). In Guillain-Barré syndrome (discussed later) dysautonomia is common and may arise in parallel with weakness or may follow the onset of the motor disorder after one week or more.
| Location | Disorder | Associated Autonomic Dysfunction? |
|---|---|---|
| Spinal cord | Tetanus112 | Frequent |
| Anterior horn cell | Amyotrophic lateral sclerosis113 | No |
| Poliomyelitis | No | |
| Rabies | Frequent | |
| West Nile virus flaccid paralysis | No | |
| Peripheral nerve | Guillain-Barré syndrome | Frequent |
| Critical illness polyneuropathy | No | |
| Diphtheria | No, but cardiomyopathy and arrhythmias may occur | |
| Porphyria | Occasional | |
| Ciguatoxin (ciguatera poisoning) | Occasional | |
| Saxitoxin (paralytic shellfish poisoning) | No | |
| Tetrodotoxin (pufferfish poisoning) | No | |
| Thallium intoxication | No | |
| Arsenic intoxication114,115 | No | |
| Lead intoxication | No | |
| Buckthorn neuropathy | No | |
| Neuromuscular junction | Myasthenia gravis | No |
| Botulism116 | Frequent | |
| Lambert-Eaton myasthenic syndrome117 | Yes, frequent dry mouth and postural hypotension | |
| Hypermagnesemia118 | No | |
| Organophosphate poisoning | No | |
| Tick paralysis | No | |
| Snake bite | No | |
| Muscle | Polymyositis/dermatomyositis | No |
| Acute quadriplegic myopathy | No | |
| Eosinophilia-myalgia syndrome119 | No | |
| Muscular dystrophies120 | No, but cardiac rhythm disturbances may occur | |
| Carnitine palmitoyl transferase deficiency | No | |
| Nemaline myopathy121 | No | |
| Acid maltase deficiency122 | No | |
| Mitochondrial myopathy123 | No | |
| Acute hypokalemic paralysis | No | |
| Stonefish myotoxin poisoning | No | |
| Rhabdomyolysis | No | |
| Hypophosphatemia124 | No |
 Neuromuscular Disorders
Neuromuscular Disorders
Many chronic neuromuscular disorders and other central nervous system conditions affecting the suprasegmental innervation and control of respiratory muscles eventually compromise ventilation. In this chapter, however, we emphasize the more common acute and subacute neuromuscular disorders that precipitate or prolong critical illness due to ventilatory failure and autonomic dysfunction. A more complete listing of neuromuscular diseases appears in Table 37-1; reviews of this subject11,12 or the references listed in Table 37-1 may be consulted for details of the more rare disorders. Some of the diseases listed (e.g., Lambert-Eaton myasthenic syndrome) rarely cause respiratory failure in isolation but may be contributing causes in the presence of other conditions13 such as neuromuscular junction blockade intended only for the duration of a surgical procedure.14
Neuromuscular Diseases Precipitating Critical Illness
Guillain-Barré Syndrome
Guillain-Barré syndrome, or acute inflammatory demyelinating polyradiculoneuropathy, is typically a motor greater than sensory peripheral neuropathy with subacute onset, monophasic course, and nadir within 4 weeks. Although the precise etiology is unknown, Guillain-Barré syndrome is immune mediated and related to antibodies directed against peripheral nerve components. Approximately 1.7 cases occur per 100,000 population per year.15 Most patients suffer a demyelinating neuropathy, but in about 5% of cases the condition is a primary axonopathy.16 Numerous antecedents have been implicated17; the more frequent ones are listed in Box 37-1. The association with antecedent infections suggests that certain agents may elicit immune responses involving antibodies that cross-react with peripheral nerve gangliosides. In particular, the development of ganglioside antibodies has been observed in Guillain-Barré syndrome after Campylobacter jejuni infections, such as GM1 antibodies in axonal forms of Guillain-Barré syndrome18 and GQ1b antibodies in the Miller-Fisher variant of Guillain-Barré syndrome.19
The initial findings of patients with Guillain-Barré syndrome are subacute and progressive weakness, usually most marked in the legs, associated with sensory complaints but without objective signs of sensory dysfunction.20 Deep tendon reflexes are often significantly reduced or absent at presentation, though this finding may take several days to develop. The cerebrospinal fluid (CSF) typically reveals an albuminocytologic dissociation or elevated protein content without pleocytosis; this may not evolve until the second week of illness. The major reason to examine the CSF is to preclude other diagnoses. Although mild CSF lymphocytic pleocytosis (10-20 cells/mm3) may suggest the possibility of associated human immunodeficiency virus (HIV) infection, in most patients, the nucleated cell count is less than 10 cells/mm3.21 Although they may be normal initially, results of electrodiagnostic studies (motor and sensory nerve conduction studies and needle electromyography) often reflect segmental nerve demyelination with multifocal conduction blocks, temporally dispersed compound muscle action potentials, slowed conduction velocity, and prolonged or absent F waves.22 Differential diagnostic considerations for patients with suspected Guillain-Barré syndrome are primarily those listed in the “Peripheral Nerve” section of Table 37-1.
The components of treatment for patients with Guillain-Barré syndrome are as follows:
Many patients are too weak to trigger the ventilator; in such cases, the assist/control or intermittent mandatory ventilation mode is initiated. Weaning patients with Guillain-Barré syndrome from mechanical ventilation must wait for adequate improvement in strength. We usually shift to pressure support ventilation for weaning, although evidence of its superiority over intermittent mandatory ventilation or synchronized intermittent mandatory ventilation modes is only anecdotal. Although the majority of patients require mechanical ventilation for less than 4 weeks, as many as one-fifth need 2 or more months of support before they can breathe without assistance. Improvement in vital capacity to greater than 15 mL/kg and in NIF to greater than 25 cm H2O suggests that a patient has improved enough to begin weaning from the ventilator. A formula using a combination of ventilatory and gas exchange variables may allow more accurate determination of a patient’s ability to be weaned.23
Nutritional support should begin as soon as a patient is admitted, with appropriate concern for the risk of aspiration.24 Most mechanically ventilated patients with Guillain-Barré syndrome can be fed via soft, small-caliber feeding tubes; autonomic dysfunction affecting the gut occasionally requires total parenteral nutrition.
Immunotherapy for Guillain-Barré syndrome includes removal of autoantibodies with plasma exchange or immune modulation with high-dose intravenous immunoglobulin (IVIg). The efficacy of plasma exchange has been evaluated in a Cochrane systematic review of six class II trials comparing plasma exchange alone with supportive care.25 Most of the trials employed up to 5 plasma exchanges of 50 mL/kg over 2 weeks. In a large North American trial,25 the time needed to improve one clinical grade (being weaned from the ventilator or being able to walk) was reduced by 50% in the plasma exchange group by comparison with the control group. There was no significant benefit when plasma exchange was begun later than 2 weeks after symptom onset. A meta-analysis demonstrated more rapid recovery in ventilated patients treated with plasma exchange within 4 weeks of onset.26 The optimal number of plasma exchanges has been assessed in patients with mild (unable to run), moderate (unable to stand without assistance), and severe (requiring mechanical ventilation) Guillain-Barré syndrome by the French Cooperative Group.27 On the basis of this trial, two exchanges are better than none in mild Guillain-Barré syndrome; four are better than two in moderate Guillain-Barré syndrome; and six are no better than four in severe Guillain-Barré syndrome. Albumin is the preferred replacement solution.28 Treatment with IVIg for Guillain-Barré syndrome has also been examined in a Cochrane systematic review. Three randomized controlled trials demonstrated class I evidence that IVIg (2 g/kg over 2-5 days) is as effective as plasma exchange in Guillain-Barré syndrome patients with impaired walking.29 Complication rates were somewhat higher in the plasma exchange groups. A large international multicenter randomized trial compared plasma exchange (50 mL/kg × 5 exchanges over 8-13 days), IVIg (0.4 g/kg × 5 days), and plasma exchange followed by IVIg.30 No significant outcome differences between these therapies were found with respect to functional improvement at 4 weeks or at 48 weeks.
Evidence-based guidelines for Guillain-Barré syndrome immunotherapy have been published by the Quality Standards Subcommittee of the American Academy of Neurology.31 Plasma exchange is recommended for adult patients who cannot walk within 4 weeks of symptom onset. IVIg is recommended in these patients within 2 or possibly 4 weeks of symptom onset. Both treatments are deemed equivalent in efficacy, and combining treatment with plasma exchange and IVIg confers no additional benefit. In light of their therapeutic equivalence, the decision whether to employ plasma exchange or IVIg in treating acute Guillain-Barré syndrome may be determined by resource availability and by avoiding potential side effects related to a patient’s medical comorbidities. Patients with heart disease, renal insufficiency or failure, hyperviscosity, or IgA deficiency may be more susceptible to complications of treatment with IVIg, whereas plasma exchange may be complicated in patients with labile blood pressure, septicemia, and significant venous access problems.
Despite the autoimmune pathophysiology of Guillain-Barré syndrome and the efficacy of corticosteroids in more chronic forms of inflammatory neuropathy, corticosteroids have not demonstrated effectiveness in Guillain-Barré syndrome and are therefore not recommended for Guillain-Barré syndrome treatment.31 A large multicenter trial failed to demonstrate efficacy of high-dose intravenous methylprednisolone,32 and another large multicenter trial demonstrated no added clinical benefit in combined treatment with IVIg and methylprednisolone.33
West Nile Virus Acute Flaccid Paralysis Syndrome
The large outbreak of West Nile virus encephalitis in the summer of 1999 in New York City marked the emergence of a relatively new cause for neuromuscular weakness with the potential for neuromuscular respiratory compromise. West Nile virus is a flavivirus transmitted between birds and mosquitoes. Humans may acquire West Nile virus from the bite of an infected Culex species mosquito, and a corresponding peak in human disease occurs in the late summer and fall. West Nile virus may also be transmitted to humans by organ transplantation,34 blood and blood product transfusion,35 transplacental exposure,36 breast feeding,37 and percutaneous laboratory injuries.38 About 20% of humans experience a mild flulike illness lasting 3 to 6 days, and about 1 in 150 develop central nervous system disease, which usually presents as meningoencephalitis.39
In the initial North American outbreak of West Nile virus, about 10% of infected patients experienced flaccid weakness with clinical features resembling Guillain-Barré syndrome.40 In one report from the original outbreak, a patient developed electromyographic evidence for segmental demyelination compatible with Guillain-Barré syndrome.41 Although patients with West Nile virus infection exhibit a spectrum of clinical weakness,42 the most prominent and distinctive syndrome documented in several subsequent reports of West Nile virus infection is an acute “poliomyelitis-like” or acute flaccid paralysis syndrome with pathology localizing to the ventral horns of the spinal cord and/or ventral roots.43–49 These patients developed acute, asymmetrical, flaccid weakness in the absence of sensory abnormalities, diffuse areflexia, or bowel/bladder dysfunction. Some of the patients experienced concurrent meningoencephalitis, and a few required mechanical ventilation.44,45 West Nile virus acute flaccid paralysis syndrome may occur in the absence of overt encephalitic signs (e.g., fever, confusion) or meningismus. Although the risk for West Nile virus encephalitis is significantly increased with age,50 West Nile virus acute flaccid paralysis syndrome occurs in relatively younger patients.43–49
Electrodiagnostic studies in patients with West Nile virus acute flaccid paralysis syndrome demonstrate normal sensory potentials, the absence of findings suggesting segmental demyelination (e.g., motor conduction block, reduced conduction velocities, prolonged distal and F-wave latencies), low-amplitude compound muscle action potentials in affected regions, and marked denervation changes in affected limb and in corresponding paraspinal muscles on needle electromyography. Corresponding magnetic resonance imaging (MRI) findings are sometimes observed and include abnormal signal in the spinal cord on T2-weighted images47,48 and abnormal enhancement of the nerve roots and cauda equina.46,47 CSF analysis usually demonstrates mild pleocytosis with lymphocytic predominance, mild to moderate protein elevation, and normal glucose.51 Prognosis for recovery of strength in these patients appears poor.52
West Nile virus infection may be diagnosed by demonstrating West Nile virus RNA in serum, CSF, or other tissues by reverse-transcriptase polymerase chain reaction, although this is insensitive.53 More commonly, a diagnosis is made by demonstration of West Nile virus IgM in CSF or serum by antibody-capture enzyme-linked immunosorbent assay. When serum West Nile virus IgM is present, diagnosis is confirmed by a fourfold increase in West Nile virus IgG titers between acute and convalescent sera obtained 4 weeks apart. Positive IgM and IgG antibody titers should be confirmed by plaque-reduction viral neutralization assay to exclude false-positive results related to other flaviviral infections such as St. Louis encephalitis. Serology may not become positive until 8 days after symptom onset.39
Although there is currently no specific treatment for West Nile virus acute flaccid paralysis syndrome, a multicenter study to evaluate the efficacy of Israeli IVIg in patients with West Nile virus meningoencephalitis or weakness began in the summer of 2003. The IVIg for this study contains high levels of West Nile virus antibodies because it was prepared from sera obtained after an Israeli West Nile virus epidemic in 2000.54 Two candidate vaccines against West Nile virus are also being evaluated.51
Myasthenia Gravis
Myasthenia gravis is a consequence of autoimmune attack on the acetylcholine receptor complex at the postsynaptic membrane of the neuromuscular junction. This process results in clinical weakness with a fluctuating pattern that is most marked after prolonged muscle exertion. Myasthenia gravis occurs at a higher rate in early adulthood in women, but in later life the incidence rates for men and women become nearly equal. The reported prevalence is 14.2 cases per 100,000 population.55 Myasthenia gravis typically involves ocular muscle weakness producing ptosis and diplopia, as well as bulbar muscle weakness resulting in dysphagia and dysarthria. This diagnosis should be considered in patients who have acute respiratory failure with these cranial nerve findings. A clinical diagnosis of myasthenia gravis may be supported by edrophonium testing, by electrophysiologic studies including repetitive nerve stimulation studies and single-fiber electromyography, and by acetylcholine receptor and muscle-specific receptor tyrosine kinase (MuSK) antibody testing.
Approximately 20% of patients with myasthenia gravis develop myasthenic crisis with respiratory failure requiring mechanical ventilation.56 Intensivists may also encounter myasthenic patients for management of complications of immunomodulating treatment or for postoperative care after thymectomy The most common precipitating factors for myasthenic crisis include bronchopulmonary infections (29%) and aspiration (10%).57 Other precipitating factors include sepsis, surgical procedures, rapid tapering of immune modulation, beginning treatment with corticosteroids, pregnancy, and exposure to drugs that may increase myasthenic weakness (Box 37-2).58 Patients with myasthenia gravis are exceptionally sensitive to nondepolarizing neuromuscular blocking agents but are resistant to depolarizing agents.59 Thymomas are associated with more fulminant disease and have been identified in about one third of patients in myasthenic crisis.57
Although sometimes less appreciated than respiratory muscle weakness, upper airway muscle weakness is a common mechanism leading to myasthenic crisis.60 Oropharyngeal and laryngeal muscle weakness may result in upper airway collapse with obstruction, along with inability to swallow secretions that may also obstruct the airway and become aspirated. Because direct assessment of oropharyngeal muscle strength is impractical, a focused history and examination to assess surrogate muscles in the head and neck region is important. Findings of bulbar myasthenia associated with upper airway compromise include flaccid dysarthria with hypernasal, staccato, or hoarse speech, dysphagia (sometimes associated with nasal regurgitation), and chewing fatigue. Patients may exhibit facial weakness with difficulty holding air within the cheeks. Jaw closure is often weak and cannot be maintained against resistance. Patients with myasthenic tongue weakness may be unable to protrude the tongue into either cheek. Although neck flexors are often weaker, a dropped head syndrome due to neck extensor weakness may occur. Vocal cord abductor paralysis may produce laryngeal obstruction with associated stridor.61,62
Patients with features of impending myasthenic crisis including severe bulbar weakness, marginal vital capacity (less than 20 to 25 mL/kg), weak cough with difficulty clearing secretions from the airway, or paradoxical breathing while supine should be admitted to an ICU and made NPO to prevent aspiration.63 Serial vital capacity and NIF measurements may be used to monitor ventilatory function in impending myasthenic crisis. However, with significant bulbar weakness, these measurements are often inaccurate if the patient has difficulty sealing the lips around the spirometer mouthpiece or is unable to seal the nasopharynx. Vital capacity measurements may not reliably predict respiratory failure in myasthenia gravis, owing to the fluctuating nature of myasthenic weakness.64 The criteria for intubation and mechanical ventilation are similar to those discussed earlier for Guillain-Barré syndrome. If the upper airway is competent and there is no difficulty handling secretions or gross hypercapnia (PaCO2 > 50 mm Hg), intermittent nasal BiPAP may be a useful temporizing measure.4 The majority of patients who develop hypercapnia in myasthenic crisis require intubation, as do those who are becoming fatigued.
Plasma exchange is an effective short-term immunomodulating treatment for myasthenic crisis and for surgical preparation in symptomatic myasthenic patients. Significant strength improvement in myasthenic crisis is well documented in several series,65–69 although there have been no controlled clinical trials. We perform a series of five to six exchanges of 2 to 3 L every other day. Onset of improved strength is variable but generally occurs after two to three exchanges.
IVIg may represent an alternative short-term treatment for myasthenic exacerbations or crises in patients who are poor candidates for plasma exchange because of difficult vascular access or septicemia. Comparable efficacy for plasma exchange and IVIg was demonstrated in myasthenic exacerbations and crises in a relatively small randomized controlled trial of IVIg at 1.2 and 2 g/kg over 2 to 5 days.70 However, in a retrospective multicenter study of myasthenic crisis, plasma exchange proved more effective than IVIg in ability to extubate at 2 weeks and in 1-month functional outcome.69 Treatment failures to IVIg subsequently responding to plasma exchange have also been reported.71 Recent experience with preoperative IVIg for thymectomy in myasthenia gravis suggests that the time course of maximal response may be considerably delayed in some patients.72
In the context of myasthenic crisis, excessive dosing of cholinesterase inhibitors may superimpose a cholinergic crisis due to depolarization blockade and result in increased weakness. Other symptoms of cholinergic crisis include muscle fasciculations and prominent muscarinic symptoms including miosis, excessive lacrimation and salivation, abdominal cramping, nausea, vomiting, diarrhea, thick bronchial secretions, diaphoresis, and bradycardia. Cholinergic crisis is rare in contemporary series of myasthenic crisis,57 and it is now common practice to avoid repeated dose escalations of cholinesterase inhibitors in impending myasthenic crisis and to discontinue the use of cholinesterase inhibitors after intubation to reduce muscarinic complications. When there is a question of cholinergic excess contributing to respiratory insufficiency, it is most prudent to discontinue all cholinesterase inhibitors, protect the airway, and support respiration as necessary.
Thymectomy may result in long-term improvement in patients with a suspected thymoma or with a life expectancy of more than 10 years. However, a patient in acute respiratory failure is generally considered a poor operative risk, and thymectomy is generally delayed until the patient’s condition has improved.73 Post-thymectomy pain control and ventilatory function may be improved by postoperative administration of epidural morphine.74
Neuromuscular Diseases Secondary to Critical Illness and Its Treatment
Critical Illness Polyneuropathy
Critical illness polyneuropathy is a widespread axonal peripheral neuropathy that develops in the context of multiple organ failure and sepsis. This entity was recognized by several investigators in 198375–77 and has been further characterized in large part by Bolton and colleagues.78,79 In a prospective series of 43 consecutive patients with sepsis and multiorgan failure, 70% developed electrophysiologic evidence of a sensorimotor axonal neuropathy, and 15 patients developed difficulty weaning from mechanical ventilation as a consequence of the neuropathy.80 Critical illness polyneuropathy is possibly the most common neuromuscular cause of prolonged ventilator dependency in patients without prior known neuromuscular disease.81 Given the limitations to detailed clinical motor and sensory examinations in the setting of critical illness, the clinical features of critical illness polyneuropathy (extremity muscle weakness and wasting, distal sensory loss, and paresthesias) may not be recognized. Deep tendon reflexes are generally reduced or absent. In the setting of superimposed central nervous system insult with pyramidal tract dysfunction, however, deep tendon reflexes may be normal or increased.82
Electrodiagnostic studies are important in establishing a diagnosis of critical illness polyneuropathy, because the clinical findings may be unobtainable or indeterminate in this setting.82 Nerve conduction findings include normal or near-normal conduction velocity and latency values and significantly reduced compound muscle action potential and sensory nerve action potential amplitudes. Needle electrode examination reveals denervation changes that are most marked in distal muscles, including fibrillation potentials, positive sharp waves, and reduced recruitment of motor unit potentials.83 With recovery over time, the denervation potentials abate, and the motor unit potentials become polyphasic and enlarged. Peripheral nerve histopathology has revealed widespread primary axonal degeneration in distal motor and sensory fibers, and skeletal muscle has exhibited fiber-type grouping.79
Although the clinical history is usually adequate to distinguish between critical illness polyneuropathy and Guillain-Barré syndrome, the latter has developed in the context of recent surgery complicated by infection.84 In some such instances, it may be necessary to differentiate between these two peripheral neuropathic disorders in a patient with extremity weakness and inability to wean from mechanical ventilation. Although only a few severe cases of critical illness polyneuropathy have been associated with facial weakness,85 facial and oropharyngeal weakness are common in Guillain-Barré syndrome.84 Dysautonomia and occasionally external ophthalmoplegia are also observed in Guillain-Barré syndrome but have virtually never been attributed to critical illness polyneuropathy.85
Electrophysiologic findings are also helpful in distinguishing these two disorders. Features of segmental demyelination may be observed in Guillain-Barré syndrome on nerve conduction studies (e.g., reduced conduction velocity, prolonged distal and F-wave latencies, conduction block, and temporal dispersion of compound muscle action potentials); these findings are not observed in critical illness polyneuropathy. Needle electromyographic findings may differ in that relatively less spontaneous activity is observed in clinically weak muscles within the first few days in Guillain-Barré syndrome.83 Although electrophysiologic studies are quite helpful in demonstrating the classic demyelinating form of Guillain-Barré syndrome, an electrophysiologic distinction between axonal forms of Guillain-Barré syndrome and critical illness polyneuropathy may not be reliable. The mean CSF protein level in Guillain-Barré syndrome is significantly higher than in critical illness polyneuropathy, although there is overlap between these populations.83 Peripheral nerve histopathology may also distinguish between these two groups, because segmental demyelination and inflammatory changes may be observed in Guillain-Barré syndrome and are not seen in critical illness polyneuropathy.79
Although overall prognosis in critical illness polyneuropathy is dependent on recovery from the underlying critical illness, most patients who survive experience a functional recovery from the neuropathy within several months.79 Critical illness polyneuropathy may prolong ventilator dependence, but it does not worsen long-term prognosis.82 Proper positioning and padding are important to prevent compression neuropathies, because prognosis from superimposed compression neuropathies in the context of critical illness polyneuropathy is less favorable.82
The pathophysiology of critical illness polyneuropathy is unknown. No clear metabolic, drug, nutritional, or toxic factors have been identified,79 although the severity of critical illness polyneuropathy has been correlated with the amount of time in the ICU, the number of invasive procedures, an increased glucose level, a reduced albumin level,80 and the severity of multiple organ failure.86 Given the common antecedents of multiple organ failure and sepsis in which significant release of various cytokines occurs, increased microvascular permeability has been postulated to ultimately result in axonal hypoxia and degeneration as a consequence of endoneurial edema.87
Prolonged Effects of Neuromuscular Blocking Agents
Prolonged neuromuscular blockade may occur with most depolarizing and nondepolarizing agents, particularly when hepatic or renal function is impaired.88 In one study, administration of vecuronium for 2 or more consecutive days resulted in prolonged neuromuscular blockade and paralysis lasting from 6 hours to 7 days.89 Although vecuronium is hepatically metabolized, patients with renal failure were susceptible to prolonged effects due to delayed excretion of the active 3-desacetyl metabolite. Acidosis and elevated serum magnesium levels were also associated with prolonged paralytic effects of vecuronium. A peripheral nerve stimulator may be used to monitor muscle twitch responses to a train-of-four stimulus during use of neuromuscular blocking agents. Drug dosage should be titrated to preserve one or two twitches to avoid overdosing. Two- to 3-hertz repetitive nerve stimulation studies may also be used to confirm neuromuscular blockade when it is suspected. Since atracurium and cisatracurium do not require organ metabolism for clearance, they are rarely associated with this problem.
Acute Quadriplegic Myopathy
The syndrome known as acute quadriplegic myopathy90 or acute myopathy of intensive care91 was originally described in 1977 in a young woman who developed severe myopathy after treatment of status asthmaticus with high doses of corticosteroids and pancuronium.92 Subsequent to that report, there have been numerous citations of an acute myopathy developing in critically ill patients without preexisting neuromuscular disease. Acute quadriplegic myopathy has developed most frequently in the setting of severe pulmonary disorders in which neuromuscular blockade is used to facilitate mechanical ventilation, and high doses of corticosteroids are concurrently administered. In a majority of reported cases, myopathy developed when nondepolarizing neuromuscular blocking agents were used for more than 2 days.90–100 The development of acute, necrotizing myopathy with myosin loss also occurs in patients receiving high doses of corticosteroids and hypnotic doses of propofol and benzodiazepines to induce paralysis.101 This observation highlights the significance of high-dose corticosteroid exposure in the development of this syndrome and suggests that paralyzed muscles may be generally susceptible to the toxic effects of corticosteroids. The mechanism of this myosin abnormality appears to lie at the level of transcriptional regulation of protein synthesis.102 The occurrence of acute quadriplegic myopathy after organ transplantation may be caused by the use of high doses of corticosteroids to prevent graft rejection, along with perioperative exposure to neuromuscular blocking agents.103 Although most cases of acute quadriplegic myopathy have been associated with critical illness, high doses of corticosteroids, and paralytic agents, acute quadriplegic myopathy has developed after isolated corticosteroid exposure,90,104–107 isolated nondepolarizing neuromuscular blocking agent use,100,104,108 or neither.109 Factors that may impair neuromuscular transmission (e.g., hypermagnesemia, aminoglycoside exposure), factors that may slow the elimination of nondepolarizing neuromuscular blocking agents (e.g., hepatic or renal failure), and factors associated with critical illness (e.g., sepsis and acidosis) have also been associated with acute quadriplegic myopathy.93
In typical cases, a diffuse flaccid quadriparesis with involvement of respiratory muscles and muscle wasting evolves after several days of induced paralysis. External ophthalmoparesis has rarely been noted.110 Sensation remains intact, but deep tendon reflexes are reduced or absent. The creatine kinase level is commonly elevated, but this may not be observed if creatine kinase is measured well after the myopathy has developed. Although the paralysis may be quite severe and may necessitate or prolong mechanical ventilation, the prognosis from the myopathy itself is good, with functional recovery over several weeks to months.95 Electromyographic findings include reduced amplitude of compound motor action potentials with normal sensory nerve action potentials and normal nerve conduction velocities. M-wave amplitude improvement accompanies clinical recovery.100 Repetitive nerve stimulation studies may yield significant decremental responses while residual effects of nondepolarizing neuromuscular blocking agents or their active metabolites persist.93,100 Needle electromyography often reveals small, low-amplitude, polyphasic motor unit potentials exhibiting early recruitment, sometimes along with positive sharp waves and fibrillation potentials.
A spectrum of muscle histologic changes may be observed, ranging from type II fiber atrophy and loss of adenosine triphosphatase (ATPase) reactivity in atrophic fibers to fiber necrosis in severe cases. However, the distinctive finding in most cases of acute quadriplegic myopathy is an extensive loss of thick filaments corresponding to myosin loss.90,94,99,104,109 This finding may be demonstrated with immunohistochemical staining or electron microscopy. The increased expression of steroid receptors in denervated and immobilized muscle111 may render these muscles susceptible to toxic catabolic effects of steroids.90 Given the growing recognition of acute quadriplegic myopathy, the use of high doses of corticosteroids should be avoided if possible when neuromuscular blockade or induced paralysis is required.
Key Points
Hughes RA, Swan AV, Raphaël JC, Annane D, van Koningsveld R, van Doorn PA. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain. 130, 2007. 2245-2245
Chawla J, Gruener G. Management of critical illness polyneuropathy and myopathy. Neurol Clin. 2010;28:961-977.
An excellent comprehensive review of this difficult management problem.
Sejvar JJ, Haddad MB, Tierney BC, et al. Neurologic manifestations and outcome of West Nile virus infection. JAMA. 2003;290:511-515.
Thomas CE, Mayer SA, Gungor Y, et al. Myasthenic crisis: clinical features, mortality, complications, and risk factors for prolonged intubation. Neurology. 1997;48:1253-1260.
Witt NJ, Zochodne DW, Bolton CF, et al. Peripheral nerve function in sepsis and multiple organ failure. Chest. 1991;99:176-184.
1 Garrity ER. Respiratory failure due to disorders of the chest wall and respiratory muscles. In: MacDonnell KF, Fahey PI, Segal MS, editors. Respiratory Intensive Care. Boston: Little, Brown; 1987:312-320.
2 Jackson M, Kinnear W, King M, et al. The effect of five years of nocturnal cuirass-assisted ventilation in chest wall disease. Eur Respir J. 1993;6:630-635.
3 Mier-Jedrzejowicz AK, Brophy C, Moxham J, et al. Assessment of diaphragm weakness. Am Rev Respir Dis. 1988;137:877-883.
4 Rabinstein A, Wijdicks EFM. BiPAP in acute respiratory failure due to myasthenic crisis may prevent intubation. Neurology. 2002;59:1647-1649.
5 Black LF, Hyatt RE. Maximal static respiratory pressures in generalized neuromuscular disease. Am Rev Respir Dis. 1971;103:641-650.
6 Rochester DF, Truwit JD. Respiratory muscle failure in critical illness. In: Ayres SM, Grenvik A, Holbrook PR, et al, editors. Textbook of Critical Care. 3rd ed. Philadelphia: WB Saunders; 1995:637-643.
7 Alex CG, Tobin MJ. Assessment of pulmonary function in critically ill patients. In: Ayres SM, Grenvik A, Holbrook PR, et al, editors. Textbook of Critical Care. 3rd ed. Philadelphia: WB Saunders; 1995:649-659.
8 Borel CO, Teitelbaum IS, Hanley DF. Ventilatory failure and carbon dioxide response in ventilatory failure due to myasthenia gravis and Guillain-Barré syndrome. Crit Care Med. 1993;21:1717-1726.
9 Yang KL, Tobin MJ. A prospective study of indexes predicting the outcome of trials of weaning from mechanical ventilation. N Engl J Med. 1991;324:1445-1450.
10 Loh L, Goldman M, Newsom-Davis J. The assessment of diaphragm function. Medicine. 1977;56:165-169.
11 Bennett DA, Bleck TP. Diagnosis and treatment of neuromuscular causes of respiratory failure. Clin Neuropharmacol. 1988;11:303-347.
12 Kelly BJ, Luce JM. The diagnosis and management of neuromuscular diseases causing respiratory failure. Chest. 1991;99:1485-1494.
13 Bleck TP, Smith MC, Pierre-Louis JC, et al. Neurologic complications of critical medical illnesses. Crit Care Med. 1993;21:98-103.
14 Breucking E, Mortier W. Anesthesia in neuromuscular diseases. Acta Anaesthesiol Belg. 1990:127-132.
15 Kennedy RH, Danielson MA, Mulder DW, et al. Guillain-Barré syndrome: a 42-year epidemiologic and clinical study. Mayo Clin Proc. 1978;53:93-99.
16 Gupta SK, Taly AB, Surmh TG, et al. Acute idiopathic axonal neuropathy (AIAN): a clinical and electrophysiological observation. Acta Neurol Scand. 1994;89:220-224.
17 Ropper AH, Wijdicks EFM, Truax BT. Guillain-Barré Syndrome. Philadelphia: FA Davis; 1991.
18 Griffin JW, Li CY, Ho TW, et al. Pathology of the motor-sensory axonal Guillain-Barré syndrome. Ann Neurol. 1996;39:17-28.
19 Yuki N, Sato S, Tsuji S, et al. Frequent presence of anti-GQ1b antibody in Fisher’s syndrome. Neurology. 1993;43:414-417.
20 Hughes RA. The spectrum of acquired demyelinating polyradiculoneuropathy. Acta Neurol Belg. 1994;94:128-132.
21 Brannagan TH, Zhou Y. HIV-associated Guillain-Barré syndrome. J Neurol Sci. 2003;208:39-42.
22 Albers JW, Kelly JJ. Acquired inflammatory demyelinating polyneuropathies: clinical and electrodiagnostic features. Muscle Nerve. 1989;12:435-451.
23 Jabour ER, Rabil DM, Truwit JD, et al. Evaluation of a new weaning index based on ventilatory endurance and the efficiency of gas exchange. Am Rev Respir Dis. 1991;144:531-537.
24 Roubenoff RA, Borel CO, Hanley DF. Hypermetabolism and hypercatabolism in Guillain-Barré syndrome. J Parenter Enteral Nutr. 1992;16:464-472.
25 Raphael J-C, Chevret S, Hughes RAC, Annane D. Plasma exchange for Guillain-Barré syndrome (Cochrane review). Cochrane Database of Systematic Reviews 2 Oxford: Update Software. 2001.
26 Guillain-Barré Study Group. Plasmapheresis and acute Guillain-Barré syndrome. Neurology. 1985;35:1096-1104.
27 French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Appropriate number of plasma exchanges in Guillain-Barré syndrome. Ann Neurol. 1997;41:298-306.
28 French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Efficacy of plasma exchange in Guillain-Barré syndrome: Role of replacement fluids. Ann Neurol. 1987;22:753-761.
29 Hughes RAC, Raphael J-C, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome (Cochrane review). Cochrane Database of Systematic Reviews 3. Oxford: Update Software; 2001.
30 Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Randomized trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet. 1997;349:225-230.
31 Hughes RAC, Wijdicks EFM, Barohn R, et al. Practice parameter: immunotherapy for Guillain-Barré syndrome. Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003;61:736-740.
32 Guillain-Barré Syndrome Steroid Trial Group. Double-blind trial of intravenous methylprednisolone in Guillain-Barré syndrome. Lancet. 1993;341:586-590.
33 van Koningsveld R, Schmitz PIM, van der Meche FGA, et al. Effect of methylprednisolone when added to standard treatment with intravenous immunoglobulin for Guillain-Barré syndrome: randomized trial. Lancet. 2004;363:192-196.
34 Iwamoto M, Jernigan DB, Guasch A, et al. Transmission of West Nile virus from an organ donor to four transplant recipients. N Engl J Med. 2003;348:2196-2203.
35 Centers for Disease Control and Prevention. Investigations of West Nile virus infections in recipients of blood transfusions. MMWR Morb Mortal Wkly Rep. 2002;51:973-974.
36 Centers for Disease Control and Prevention. Intrauterine West Nile virus infection—New York, 2002. MMWR Morb Mortal Wkly Rep. 2002;51:1135-1136.
37 Centers for Disease Control and Prevention. Possible West Nile virus transmission to an infant through breast-feeding—Michigan, 2002. MMWR Morb Mortal Wkly Rep. 2002;51:877-878.
38 Centers for Disease Control and Prevention. Laboratory-acquired West Nile virus infections—United States, 2002. MMWR Morb Mortal Wkly Rep. 2002;51:1133-1135.
39 Petersen LR, Marfin AA. West Nile virus: A primer for the clinician. Ann Intern Med. 2002;137:173-179.
40 Nash D, Mostashari F, Fine A, et al. The outbreak of West Nile virus infection in the New York City area in 1999. N Engl J Med. 2001;344:1807-1814.
41 Ahmed S, Libman R, Wesson K, et al. Guillain-Barré syndrome: an unusual presentation of West Nile virus infection. Neurology. 2000;55:144-146.
42 Leis AA, Stokic DS, Webb RM, et al. Clinical spectrum of muscle weakness in human West Nile virus infection. Muscle Nerve. 2003;28:302-308.
43 Leis AA, Stokic DS, Polk JL, et al. A poliomyelitis-like syndrome from West Nile virus infection. N Engl J Med. 2002;347:1279-1280.
44 Glass JD, Samuels O, Rich MM. Poliomyelitis due to West Nile virus. N Engl J Med. 2002;347:1280-1281.
45 Centers for Disease Control and Prevention. Acute flaccid paralysis syndrome associated with West Nile virus infection—Mississippi and Louisiana, July-August 2002. MMWR Morbid Mortal Wkly Rep. 2002;51:708-709.
46 Sejvar JJ, Leis AA, Stokic DS, et al. Acute flaccid paralysis and West Nile virus infection. Emerg Infect Dis. 2003;9:788-793.
47 Jeha LE, Sila CA, Lederman RJ, et al. West Nile virus infection: a new acute paralytic illness. Neurology. 2003;61:55-59.
48 Li J, Loeb JA, Shy ME, et al. Asymmetric flaccid paralysis: a neuromuscular presentation of West Nile virus infection. Ann Neurol. 2003;53:703-710.
49 Flaherty ML, Wijdicks EFM, Stevens JC, et al. Clinical and electrophysiologic patterns of flaccid paralysis due to West Nile virus. Mayo Clin Proc. 2003;78:1245-1248.
50 Weiss D, Carr D, Kellachan J, et al. Clinical findings of West Nile virus infection in hospitalized patients, New York and New Jersey, 2000. Emerg Infect Dis. 2001;7:654-658.
51 Sampathkumar P. West Nile virus: Epidemiology, clinical presentation, diagnosis, and prevention. Mayo Clin Proc. 2003;78:1137-1144.
52 Sejvar JJ, Haddad MB, Tierney BC, et al. Neurologic manifestations and outcome of West Nile virus infection. JAMA. 2003;290:511-515.
53 Lanciotti RS, Kerst AJ, Nanci RS, et al. Rapid detection of West Nile virus from human clinical specimens, field-collected mosquitoes, and avian samples by a TaqMan reverse transcriptase-PCR assay. J Clin Microbiol. 2000;38:4066-4071.
54 Roos KL. Fever and asymmetrical weakness in the summer: evidence of a West Nile virus-associated poliomyelitis–like illness. Mayo Clin Proc. 2003;78:1205-1206.
55 Phillips LH, Torner JC, Anderson MS, et al. The epidemiology of myasthenia gravis in central and western Virginia. Neurology. 1992;42:1888-1893.
56 Fink ME. Treatment of the critically ill patient with myasthenia gravis. In: Ropper AH, editor. Neurological and Neurosurgical Intensive Care. New York: Raven Press; 1993:351-362.
57 Thomas CE, Mayer SA, Gungor Y, et al. Myasthenic crisis: clinical features, mortality, complications, and risk factors for prolonged intubation. Neurology. 1997;48:1253-1260.
58 Wright RB. Myasthenia. In: Klawans HL, Goetz CG, Tattler CM, editors. Textbook of Clinical Neuropharmacology and Therapeutics. New York: Raven Press; 1992:505-516.
59 Cullen DJ, Bigatello LM, DeMonaco HI. Anesthetic pharmacology and critical care. In: Chernow B, editor. The Pharmacologic Approach to the Critically Ill Patient. Baltimore: Williams & Wilkins; 1994:291-308.
60 Putman MT, Wise RA. Myasthenia gravis and upper airway obstruction. Chest. 1996;109:400-404.
61 Cridge PB, Allegra J, Gerhard H. Myasthenic crisis presenting as isolated vocal cord paralysis. Am J Emerg Med. 2000;18:232-233.
62 Schmidt-Nowara WW, Marder EJ, Feil PA. Respiratory failure in myasthenia gravis due to vocal cord paresis. Arch Neurol. 1984;41:567-568.
63 Rabinstein AA, Wijdicks EFM. Warning signs of imminent respiratory failure in neurological patients. Semin Neurol. 2003;23:97-104.
64 Rieder P, Louis M, Jolliet P, Chevrolet JC. The repeated measurement of vital capacity is a poor predictor of the need for mechanical ventilation in myasthenia gravis. Intensive Care Med. 1995;21:663-668.
65 Dau PC, Lindstrom JM, Cassel CK, et al. Plasmapheresis and immunosuppressive drug therapy in myasthenia gravis. N Engl J Med. 1977;297:1134-1140.
66 Kornfield P, Ambinder EP, Papatestas AE, et al. Plasmapheresis in myasthenia gravis: controlled study. Lancet. 1979;2:629.
67 Antozzi C, Gemma M, Regi B, et al. A short plasma exchange protocol is effective in severe myasthenia gravis. J Neurol. 1991;238:103-107.
68 Gracey DR, Howard FM, Divertie MB. Plasmapheresis in the treatment of ventilator-dependent myasthenia gravis patients: report of four cases. Chest. 1984;8:739-743.
69 Qureshi AI, Choundry MA, Akbar MS, et al. Plasma exchange versus intravenous immunoglobulin treatment in myasthenic crisis. Neurology. 1999;52:629-632.
70 Gajdos P, Chevret S, Clair B, et alMyasthenia Gravis Clinical Study Group. Clinical trial of plasma exchange and high-dose intravenous immunoglobulin in myasthenia gravis. Ann Neurol. 1997;41:789-796.
71 Stricker RB, Kwiatkowska BJ, Habis JA, Kiprov DD. Myasthenic crisis: response to plasmapheresis following failure of intravenous gamma-globulin. Arch Neurol. 1993;50:837-840.
72 Huang C-S, Hsu H-S, Kao K-P, et al. Intravenous immunoglobulin in the preparation of thymectomy for myasthenia gravis. Acta Neurol Scand. 2003;60:1805-1810.
73 Turani E, Szathmary I, Molnar J, et al. Myasthenia gravis: prognostic significance of clinical data in the prediction of post-thymectomy respiratory crises. Acta Chir Hung. 1992-1993;33:353-360.
74 Kirsch IR, Diringer MN, Borel CO, et al. Preoperative lumbar epidural morphine improves postoperative analgesia and ventilatory function after transsternal thymectomy in patients with myasthenia gravis. Crit Care Med. 1991;19:1474-1479.
75 Rivner MH, Kim S, Greenberg M, et al. Reversible generalized paresis following hypotension: a new neurological entity. Abstr Neurol. 1983;33(Suppl 2):164.
76 Bolton CF, Brown JD, Sibbald WJ. The electrophysiologic investigation of respiratory paralysis in critically ill patients. Abstr Neurol. 1983;33(Suppl 2):240.
77 Roelofs RJ, Cerra F, Bielka N, et al. Prolonged respiratory insufficiency due to acute motor neuropathy: A new syndrome? Abstr Neurol. 1983;33(Suppl 2):240.
78 Bolton CF, Gilbert JJ, Hahn AF, Sibbald WJ. Polyneuropathy in critically ill patients. J Neurol Neurosurg Psychiatry. 1984;47:1223-1231.
79 Zochodne DW, Bolton CF, Wells GA, et al. Critical illness polyneuropathy: a complication of sepsis and multiple organ failure. Brain. 1987;110:819-842.
80 Witt NJ, Zochodne DW, Bolton CF, et al. Peripheral nerve function in sepsis and multiple organ failure. Chest. 1991;99:176-184.
81 Spitzer AR, Giancarlo T, Maher L, et al. Neuromuscular causes of prolonged ventilator dependency. Muscle Nerve. 1992;15:682-686.
82 Hund EF, Fogel W, Krieger D, et al. Critical illness polyneuropathy: clinical findings and outcomes of a frequent cause of neuromuscular weaning failure. Crit Care Med. 1996;24:1328-1333.
83 Bolton CF, Laverty DA, Brown JD, et al. Critically ill polyneuropathy: electrophysiological studies and differentiation from Guillain-Barré syndrome. J Neurol Neurosurg Psychiatry. 1986;49:563-573.
84 Arnason BGW, Soliven B. Acute inflammatory demyelinating polyradiculoneuropathy. In: Dyck PJ, Thomas PK, editors. Peripheral Neuropathy. 3rd ed. Philadelphia: WB Saunders; 1993:1437-1497.
85 Leijten FS, de Weerd AW. Critical illness polyneuropathy: a review of the literature, definition and pathophysiology. Clin Neurol Neurosurg. 1994;96:10-19.
86 Leitjen FS, de Weerd AW, Poortvliet DC, et al. Critical illness polyneuropathy in multiple organ dysfunction syndrome and weaning from the ventilator. Intensive Care Med. 1996;22:856-861.
87 Bolton CF, Young GB, Zochodne DW. The neurological complications of sepsis. Ann Neurol. 1993;33:94-100.
88 Murphy GS, Brull SJ. Residual neuromuscular block: lessons unlearned. Part I: definitions, incidence, and adverse physiologic effects of residual neuromuscular block. Anesth Analg. 2010;111:120-128.
89 Segredo V, Caldwell JE, Matthay MA, et al. Persistent paralysis in critically ill patients after long-term administration of vecuronium. N Engl J Med. 1992;327:524-528.
90 Hirano M, Ott MD, Raps EC, et al. Acute quadriplegic myopathy: a complication of treatment with steroids, nondepolarizing blocking agents, or both. Neurology. 1992;42:2082-2087.
91 Lacomis D, Giuliani MJ, Van Cott A, Kramer DJ. Acute myopathy of intensive care: clinical, electromyographic, and pathological aspects. Ann Neurol. 1996;40:645-654.
92 MacFarlane IA, Rosenthal FD. Severe myopathy after status asthmaticus. Lancet. 1977;2:615.
93 Barohn RJ, Jackson CE, Rogers SJ, et al. Prolonged paralysis due to nondepolarizing neuromuscular blocking agents and corticosteroids. Muscle Nerve. 1994;17:647-654.
94 Danon MJ, Carpenter S. Myopathy with thick filament (myosin) loss following prolonged paralysis with vecuronium during steroid treatment. Muscle Nerve. 1991;14:1131-1139.
95 Gooch JL. Prolonged paralysis after neuromuscular blockade. Muscle Nerve. 1995;18:937-942.
96 Kaplan PW, Rocha W, Sanders DB, et al. Acute steroid-induced tetraplegia following status asthmaticus. Pediatrics. 1986;78:121-123.
97 Lacomis D, Smith TW, Chad DA. Acute myopathy and neuropathy in status asthmaticus: case report and literature review. Muscle Nerve. 1993;16:84-90.
98 Op de Coul AAW, Lembregts PCLA, Koeman J, et al. Neuromuscular complications in patients given Pavulon (pancuronium bromide) during artificial ventilation. Clin Neurol Neurosurg. 1985;87:17-22.
99 Waclawik AJ, Sufit RL, Beinlich BR, Schutta HS. Acute myopathy with selective degeneration of myosin filaments following status asthmaticus treated with methylprednisolone and vecuronium. Neuromusc Disord. 1992;2:19.
100 Zochodne DW, Ramsay DA, Saly V, et al. Acute necrotizing myopathy of intensive care: electrophysiological studies. Muscle Nerve. 1994;17:285-292.
101 Hanson P, Dive A, Brucher JM, et al. Acute corticosteroid myopathy in intensive care patients. Muscle Nerve. 1997;20:1371-1380.
102 Norman H, Zackrisson H, Hedström Y, et al. Myofibrillar protein and gene expression in acute quadriplegic myopathy. J Neurol Sci. 2009;285:28-38.
103 Campellone JV, Lacomis D, Kramer DJ, et al. Acute myopathy after liver transplantation. Neurology. 1998;50:46-53.
104 Al-Lozi MT, Pestronk A, Yee WC, et al. Rapidly evolving myopathy with myosin-deficient muscle fibers. Ann Neurol. 1994;35:273-279.
105 Gutmann L, Blumenthal D, Schochet SS. Acute type II myofiber atrophy in critical illness. Neurology. 1996;46:819-821.
106 Sher JH, Shafiq SA, Schutta HS. Acute myopathy with selective lysis of myosin filaments. Neurology. 1979;29:100-106.
107 Van Marle W, Woods KL. Acute hydrocortisone myopathy. BMJ. 1980;281:271-272.
108 Gooch JL, Moore MH, Ryser DK. Prolonged paralysis after neuromuscular junction blockade: Case reports and electrodiagnostic findings. Arch Phys Med Rehabil. 1993;74:1007-1011.
109 Showalter CJ, Engel AG. Acute quadriplegic myopathy: Analysis of myosin isoforms and evidence for calpain-mediated proteolysis. Muscle Nerve. 1997;20:316-322.
110 Sitwell LD, Weinshenker BG, Monpetit V, Reid D. Complete ophthalmoplegia as a complication of acute corticosteroid- and pancuronium-associated myopathy. Neurology. 1991;41:921-922.
111 Du Bois DC, Almon RR. A possible role for glucocorticoid in denervation atrophy. Muscle Nerve. 1981;4:370-373.
112 Bleck TP, Brauner JS. Tetanus. In: Scheld WM, Whitley RJ, Marra CM, editors. Infections of the central nervous system. 3rd ed. New York: LWW; 2004:625-648.
113 Kuisma MJ, Saarinen KV, Teirmoa HT. Undiagnosed amyotrophic lateral sclerosis and respiratory failure. Acta Anaesthesiol Scand. 1993;37:628-630.
114 Donofrio PD, Wilbourn AJ, Albers JW, et al. Acute arsenic intoxication presenting as Guillain-Barré like syndrome. Muscle Nerve. 1987;10:114-120.
115 Greenberg C, Davies S, McGowan T, et al. Acute respiratory failure following acute arsenic poisoning. Chest. 1979;76:596-598.
116 Reddy P, Bleck TP. Clostridium botulinum. In: Mandell GM, Bennett JE, Dolin R, editors. Principles and practice of infectious diseases. 7th ed. New York: Churchill, Livingstone, Elsevier; 2010:3097-3102.
117 Peolsi G, Perili V, Sollazzi L, et al. Lambert-Eaton myasthenic syndrome: a clinical contribution. Acta Anaesthesiol Belg. 1991;42:41-44.
118 Gambling DR, Birmingham CL, Jenkins LC. Magnesium and the anaesthetist. Can J Anesth. 1988;35:644-654.
119 Swygert LA, Back EE, Auerbach SB, et al. Eosinophilia-myalgia syndrome: mortality data from the US national surveillance system. J Rheumatol. 1993;20:1711-1717.
120 Curran FJ, Colbert AP. Ventilator management in Duchenne muscular dystrophy and postpoliomyelitis syndrome: twelve years’ experience. Arch Phys Med Rehabil. 1989;70:180-185.
121 Sasaki M, Yoneyama H, Nonaka I. Respiratory involvement in nemaline myopathy. Pediatr Neurol. 1990;6:425-427.
122 Barohn RJ, McVey AL, DiMauro S. Adult acid maltase deficiency. Muscle Nerve. 1993;16:672-676.
123 Kim GW, Kim SM, Sunwoo IN, et al. Two cases of mitochondrial myopathy with predominantly respiratory dysfunction. Yonsei Med J. 1991;32:184-189.
124 Newman JH, Neff TA, Ziporin P. Acute respiratory failure associated with hypophosphatemia. N Engl J Med. 1977;296:1101-1103.
