Neuromuscular diseases
ANN HALLUM, PT, PhD and DIANE D. ALLEN, PT, PhD
After reviewing this chapter the student or therapist will be able to:
1. Describe the basic pathology and medical treatment of amyotrophic lateral sclerosis, Guillain-Barré syndrome, and Duchenne muscular dystrophy.
2. Describe the current goals and interventions for each condition.
3. Describe the “safe” exercise windows related to disuse atrophy and exercise (overwork) damage.
4. Be able to apply intervention concepts discussed in this chapter to other neuromuscular diseases.
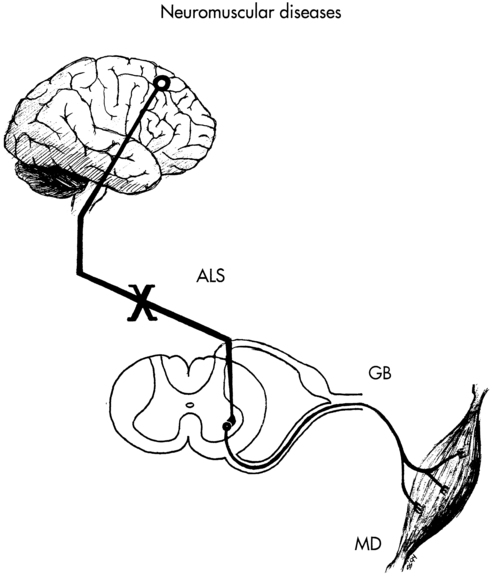
Neuromuscular diseases encompass disorders of upper or lower motor nerves or the muscles they innervate. This chapter traces the connections among the central nervous system (CNS), peripheral nervous system (PNS), and musculoskeletal system through the disordered functioning associated with three neuromuscular diseases: amyotrophic lateral sclerosis (ALS), which damages upper and lower motor neurons; Guillain-Barré syndrome (GBS), which compromises lower motor neurons and the PNS; and Duchenne muscular dystrophy (DMD), which affects the muscles themselves. To review the normal connections, upper motor neurons originate in the motor cortex of the brain (Betz cells). Axons from these upper motor neurons descend by means of the corticobulbar and corticospinal tracts to synapse with lower motor neurons in the brain stem (neurons of the cranial nerves with motor functions) and spinal cord (anterior horn cells or alpha motor neurons). Simultaneously, corticobulbar tract fibers innervate neurons originating within the brain stem and descending through the spinal cord to provide additional input to lower (alpha) motor neurons. Axons from the lower motor neurons within both the brain stem and spinal cord run within the peripheral nerves, which include motor and sensory fibers, to synapse with muscle fibers. The muscle fibers respond to excitation by contracting. Depending on the site of the pathology, neuromuscular diseases can be classified as neurogenic or myopathic. ALS and GBS are neurogenic disorders; DMD is a primary myopathy (Figure 17-1).

In considering the movement dysfunction associated with these diseases, strength and endurance are most affected, with flexibility deficits resulting from these.1 All three disorders decrease a person’s ability to generate force in the affected muscles, with weakness as a primary symptom. Loss of muscle strength can lead to speech, swallowing, and respiratory difficulties along with functional limitations. Fatigue is another primary deficit, although the neurogenic disorders tend to result in central fatigue (deficit in ability to recruit motor units) as opposed to the peripheral fatigue of the myopathies (deficit in ability of muscle fibers to contract forcefully).2 Secondary movement problems include loss of range of motion (ROM) in immobile muscles and joints, and pain or muscle spasms. Adaptability, the ability to sense obstacles or changes in the environment and change the course of a movement in response,1 may be affected with the sensory loss in GBS but is not typically a problem in ALS or DMD.
Amyotrophic lateral sclerosis
Pathology and medical diagnosis
The cause of ALS is unknown; however, numerous theories have been proposed. Ninety percent of the cases of ALS are sporadic without a known genetic component; however, most neurodegenerative diseases are now thought to be related to complex protein misfolding disorders. The latest research suggests that ALS and other neurodegenerative disorders are related to TDP-43 proteinopathy.3 Approximately 5% to 10% of the cases seem to have a complex genetic basis coded on ALS1 through ALS8 and other mutations that are associated with frontal lobe dementias. Twenty percent of genetic causes of ALS are thought to be related to mendelian mutations in the superoxide dismutase–1 (SOD1) gene (ALS1). Other factors considered in the genesis of ALS are vascular endothelial growth factors, toxicity leading to motor neuron death, oxidative stress and mitochondrial dysfunction related to microglial inflammation,4,5 and environmental factors.6
The differential diagnosis for ALS is extensive. The possibility of cervical or lumbar spondylosis, syringomyelia, multiple sclerosis, primary lateral sclerosis, and diseases associated with lower motor neuron pathology, among other diagnoses, needs to be excluded before the diagnosis of ALS is made.7 Currently, no single laboratory test is available to confirm a diagnosis of ALS, although creatine phosphokinase levels are elevated in approximately 70% of patients and tend to be higher in patients with limb onset ALS rather than bulbar onset.8 Genetic testing to identify the mutations in the Cu,Zn SOD1 gene is available when a family history of ALS is present. Other laboratory tests, such as identification of biochemical markers in the blood and cerebrospinal fluid, are used to exclude other neurological diseases. Electromyography (EMG) and nerve conduction studies can be helpful to confirm the presence of widespread lower motor neuron disease without peripheral neuropathy or polyradiculopathy. Neuroimaging studies are used to rule out conditions that may have clinical signs similar to those of ALS.9
Because of the absence of clear laboratory markers of ALS, the clinical diagnosis must be made on the basis of recognition of a pattern of observed and reported symptoms of both upper and lower motor neuron disease and persistent declines in physical functions supported by inclusionary and exclusionary diagnostic testing. Because of the overlap of symptoms with other neuromuscular disorders, misdiagnosis is not uncommon.10
ALS is the most common form of motor neuron disease, with an incidence of approximately three to five cases per 100,000 persons. Mean age at onset is 57 years, with two thirds of patients aged 50 to 70 years old at time of onset.11 Men are affected approximately 1.3 to two times more frequently than are women, although the differences are less with late onset of disease (ages 70+).12
Clinical presentation
The World Federation of Neurology (WFN) has developed suggested diagnostic criteria (suspected, possible, probable, and definite) for patients with ALS entering clinical research trials. Essentially, a patient with “definite” ALS must show concomitant upper motor neuron and lower motor neuron signs in three spinal regions or in two spinal regions with bulbar signs. Either upper or lower motor signs must also be evident in other regions of the body.13 Exclusionary criteria are oculomotor nerve pathway abnormalities (the oculomotor nerve is spared in ALS), significant movement disorder patterns, sphincter control problems, the presence of sensory and autonomic nervous system (ANS) dysfunction, and cognitive deterioration.14 (Refer to the WFN ALS website [www.wfnals.org] section on ALS education for up-to-date criteria used for clinical studies.)
Although a consistent diagnostic criterion for ALS has been the absence of sensory involvement, some evidence exists that there is a progressive functional deficit in sensation, perhaps related to ongoing immobility.15
Similarly, cognitive deficits are considered exclusionary criteria for an ALS diagnosis. However, a small subgroup of patients with both familial and sporadic forms of ALS has been identified as having concomitant evidence of frontotemporal dementia (FTD), showing lower scores on executive cognitive functions, word finding, and phrase length.16 A combination of ALS and FTD suggests a common cause may be possible.17 Because of these findings, therapists should be aware of the possibility of cognitive deficits in their patients with ALS, manifested as a decrement in executive skills such as planning and organization and language problems. Such patients may have more difficulty following through on medication and therapeutic recommendations, and their families may need more support. Unassociated with overall cognitive impairment, some deficits in action knowledge as opposed to object knowledge have been noted in patients with ALS, correlating with atrophy in the motor and premotor cortex.18 Specific cognitive deficits, therefore, may be more common than previously noted.
The earliest clinical markers heralding ALS are fasciculations (especially unequivocal fasciculation in the tongue), muscle cramps, fatigue, weakness, and atrophy.13,19 During initial diagnostic visits, patients frequently report to their physicians a profound sense of fatigue or the loss of exercise tolerance.19 Ninety percent of patients report weakness occurring in a striated muscle or group of muscles. Because the onset of ALS is insidious, most patients are not aware of the strength changes, or they have adjusted to the changes until they have difficulty with a functional activity such as tying shoes or climbing stairs. Physical examination usually demonstrates more widespread weakness and atrophy than reported by the patient. By the time most patients report weakness, they have lost approximately 80% of their motor neurons in the areas of weakness. This demonstrates the plasticity of the nervous system and its drive to adapt to meet functional goals. The weakness spreads over time to include musculature throughout the body. Succeeding symptoms of weakness in other muscles depends on the continued loss of motor neurons to the 20% threshold needed for perception of weakness.20,21 A typical, but not absolute, pattern of motor progression is early distal involvement followed by proximal limb involvement. In some cases bulbar symptoms herald the onset of ALS, but bulbar symptoms more commonly occur later in the disease. Flexor muscles tend to be weaker than extensor muscles.22
Although the atrophy and weakness component of ALS is most obvious, 80% or more of patients show early clinical evidence of pyramidal tract dysfunction (e.g., hyperreflexia in the presence of weakness and atrophy, spasticity, and Babinski and Hoffmann reflexes).13 Although in some cases the upper motor neuron signs may be absent clinically, Chou23 has shown on autopsy that significant involvement may be present despite the lack of clinical evidence.
The pattern of ALS onset is highly varied, with several patterns identified by primary area of onset. Lower-extremity onset is slightly more common than upper-extremity onset, which is more common than bulbar onset. Some patients show initial symptoms in distal musculature of upper and lower extremities. A significant diagnostic feature of the pattern of disease is the asymmetry of the weakness and the sparing of some muscle fibers even in highly atrophied muscles. For example, a patient may have weakness of the right intrinsics and shoulder musculature or weakness of the left anterior tibial muscles. Bulbar symptoms are presaged by tongue fasciculations and weakness, facial and palatal weakness, and swallowing difficulties, which result in dysphagia and dysarthria. Pseudobulbar palsy is sometimes present in ALS, manifested by spontaneous laughing or crying unrelated to the situation.24 Despite the pattern of onset, however, the eventual course of the illness is similar in most patients, with an unremitting spread of weakness to other muscle groups leading to total paralysis of spinal musculature and muscles innervated by the cranial nerves. Death is usually related to respiratory failure.25
In a longitudinal study using monthly questionnaires, direct patient interviews, record reviews, physician interviews, and family member interviews, Brooks and colleagues20 followed 702 patients with ALS. Their findings suggest that spread of neuronal degeneration occurred more quickly to adjacent areas than to noncontiguous areas. The spread to adjacent areas was more rapid at the brain stem, cervical, and lumbar regions. Limb involvement after bulbar onset was more aggressive in men than in women.20
One study focused on developing methods to assess the natural history of the progression of ALS so that medical and supportive treatment planning and interventions could be instituted.26 Hillel and colleagues27 have developed the ALS severity scale for rapid functional assessment of disease stage. Their 10-point ordinal scale allows clinicians and therapists to score patients in four categories: speech, swallowing, and lower-extremity and upper-extremity function (Box 17-1).
BOX 17-1  AMYOTROPHIC LATERAL SCLEROSIS SEVERITY SCALE: LOWER EXTREMITY, UPPER EXTREMITY, SPEECH, SWALLOWING
AMYOTROPHIC LATERAL SCLEROSIS SEVERITY SCALE: LOWER EXTREMITY, UPPER EXTREMITY, SPEECH, SWALLOWING
| LOWER EXTREMITIES (WALKING) | ||
| Normal | ||
| 10 | Normal ambulation | Patient denies any weakness or fatigue; examination reveals no abnormality. |
| 9 | Fatigue suspected | Patient experiences sense of weakness or fatigue in lower extremities during exertion. |
| Early Ambulation Difficulties | ||
| 8 | Difficulty with uneven terrain | Difficulty and fatigue when walking long distances, climbing stairs, and walking over uneven ground (even thick carpet). |
| 7 | Observed changes in gait | Noticeable change in gait; pulls on railings when climbing stairs; may use leg brace. |
| Walks with Assistance | ||
| 6 | Walks with mechanical device | Needs or uses cane, walker, or assistant to walk; probably uses wheelchair away from home. |
| 5 | Walks with mechanical device and assistant | Does not attempt to walk without attendant; ambulation limited to less than 50 ft; avoids stairs. |
| Functional Movement Only | ||
| 4 | Able to support | At best, can shuffle a few steps with the help of an attendant for transfers. |
| 3 | Purposeful leg movements | Unable to take steps but can position legs to assist attendant in transfers; moves legs purposefully to maintain mobility in bed. |
| No Purposeful Leg Movement | ||
| 2 | Minimal movement | Minimal movement of one or both legs; cannot reposition legs independently. |
| 1 | Paralysis | Flaccid paralysis; cannot move lower extremities (except, perhaps, to close inspection). |
| UPPER EXTREMITIES (DRESSING AND HYGIENE) | ||
| Normal Function | ||
| 10 | Normal function | Patient denies any weakness or unusual fatigue of upper extremities; examination demonstrates no abnormality. |
| 9 | Suspected fatigue | Patient experiences sense of fatigue in upper extremities during exertion; cannot sustain work for as long as normal; atrophy not evident on examination. |
| Independent and Complete Self-Care | ||
| 8 | Slow self-care | Dressing and hygiene performed more slowly than usual. |
| 7 | Effortful self-care performance | Requires significantly more time (usually double or more) and effort to accomplish self-care; weakness is apparent on examination. |
| Intermittent Assistance | ||
| 6 | Mostly independent | Handles most aspects of dressing and hygiene alone; adapts by resting, modifying (e.g., use of electric razor), or avoiding some tasks; requires assistance for fine motor tasks (e.g., buttons, ties). |
| 5 | Partial independence | Handles some aspects of dressing and hygiene alone; however, routinely requires assistance for many tasks such as applying makeup, combing, and shaving. |
| Needs Attendant for Self-Care | ||
| 4 | Attendant assists patient | Attendant must be present for dressing and hygiene; patient performs the majority of each task with the assistance of the attendant. |
| 3 | Patient assists attendant | The attendant directs the patient for almost all tasks; the patient moves in a purposeful manner to assist the attendant; does not initiate self-care. |
| Total Dependence | ||
| 2 | Minimal movement | Minimal movement of one or both arms; cannot reposition arms. |
| 1 | Paralysis | Flaccid paralysis; unable to move upper extremities (except, perhaps, to close inspection). |
| SPEECH | ||
| Normal Speech Processes | ||
| 10 | Normal speech | Patient denies any difficulty speaking; examination demonstrates no abnormality. |
| 9 | Nominal speech abnormalities | Only the patient or spouse notices speech has changed; maintains normal rate and volume. |
| Detectable Speech Disturbance | ||
| 8 | Perceived speech changes | Speech changes are noted by others, especially during fatigue or stress; rate of speech remains essentially normal. |
| 7 | Obvious speech abnormalities | Speech is consistently impaired; rate, articulation, and resonance are affected; remains easily understood. |
| Intelligible with Repeating | ||
| 6 | Repeats message on occasion | Rate is much slower, repeats specific words in adverse listening situation; does not limit complexity or length of messages. |
| 5 | Frequent repeating required | Speech is slow and labored; extensive repetition or a “translator” is commonly used; patient probably limits the complexity or length of messages. |
| Speech Combined with Nonvocal Communication | ||
| 4 | Speech plus nonverbal communication | Speech is used in response to questions; intelligibility problems need to be resolved by writing or a spokesperson. |
| 3 | Limits speech to one-word responses | Vocalizes one-word responses beyond yes and no; otherwise writes or uses a spokesperson; initiates communication nonvocally. |
| Loss of Useful Speech | ||
| 2 | Vocalizes for emotional expression | Uses vocal inflection to express emotion, affirmation, and negation. |
| 1 | Nonvocal | Vocalization is effortful, limited in duration, and rarely attempted; may vocalize for crying or pain. |
| X | Tracheostomy | |
| SWALLOWING | ||
| Normal Eating Habits | ||
| 10 | Normal swallowing | Patient denies any difficulty chewing or swallowing; examination demonstrates no abnormality. |
| 9 | Nominal abnormality | Only patient notices slight indicators such as food lodging in the recesses of the mouth or sticking in the throat. |
| Early Eating Problems | ||
| 8 | Minor swallowing problems | Reports some swallowing difficulties; maintains essentially a regular diet; isolated choking episodes. |
| 7 | Prolonged times, smaller bite size | Meal time has significantly increased and smaller bite sizes are necessary; must concentrate on swallowing thin liquids. |
| Dietary Consistency Changes | ||
| 6 | Soft diet | Diet is limited primarily to soft foods; requires some special meal preparation. |
| 5 | Liquefied diet | Oral intake adequate; nutrition limited primarily to liquefied diet; adequate thin liquid intake usually a problem; may force self to eat. |
| Needs Tube Feeding | ||
| 4 | Supplemental tube feedings | Oral intake alone no longer adequate; patient uses or needs a tube to supplement intake; patient continues to take significant (greater than 50%) nutrition orally. |
| 3 | Tube feeding with occasional oral nutrition | Primary nutrition and hydration accomplished by tube; receives less than 50% of nutrition orally. |
| No Oral Feeding | ||
| 2 | Secretions managed with aspirator and/or medications | Cannot safely manage any oral intake; secretions managed with aspirator and/or medications; swallows reflexively. |
| 1 | Aspiration of secretions | Secretions cannot be managed noninvasively; rarely swallows. |
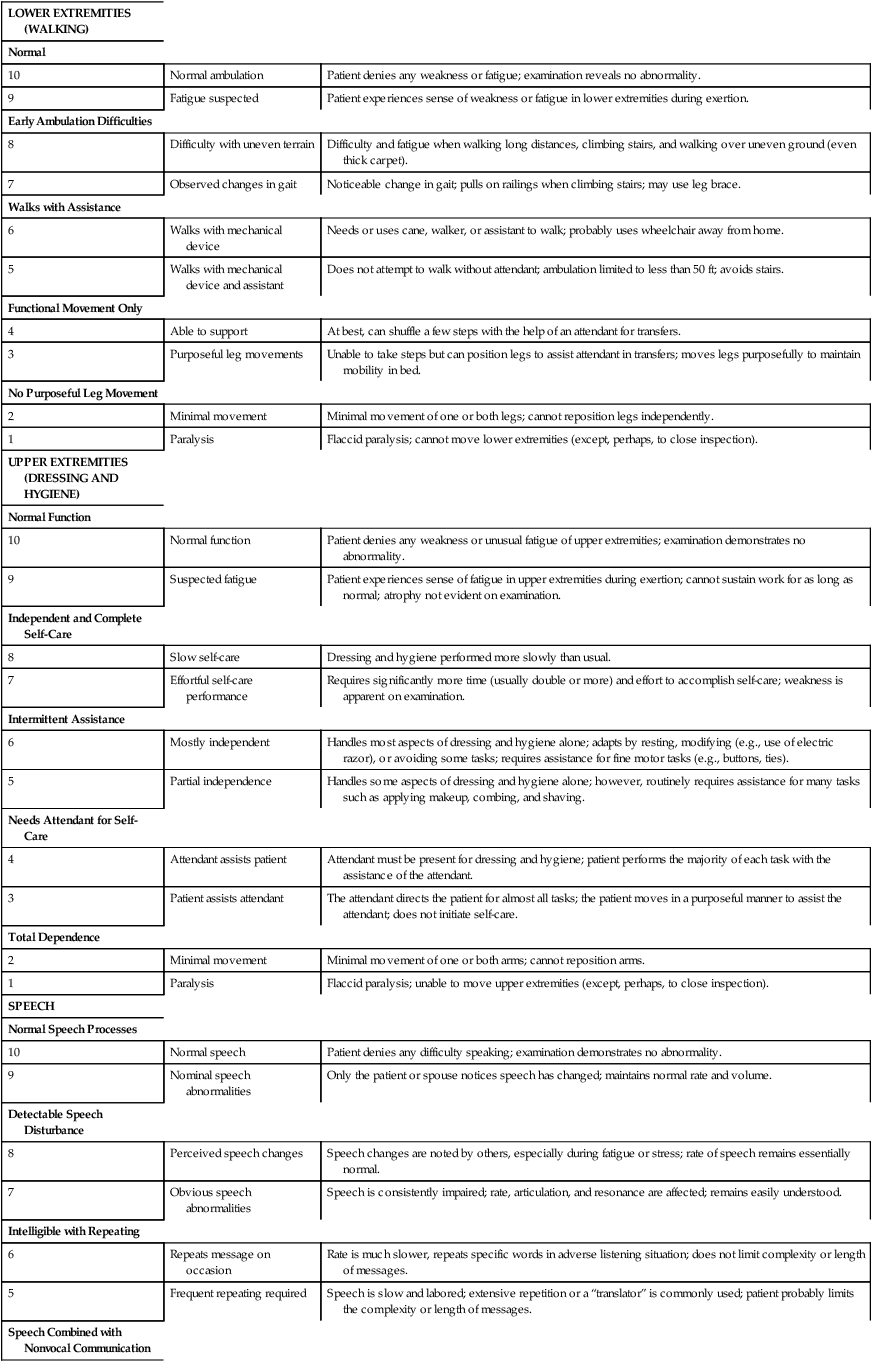
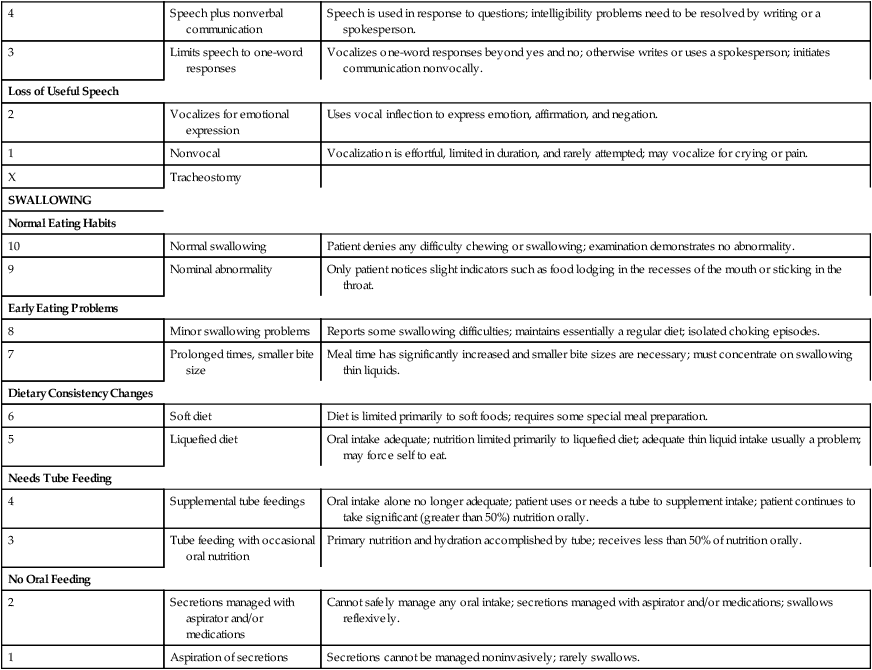
Adapted with permission from Hillel AD, Miller RM, Yorkston K, et al: Amyotrophic lateral sclerosis severity scale. Neuroepidemiology 8:142, 1989.
A five-point scale of severity is currently being used in ALS clinical drug trials. Patients in stage 1 (mild disease) have a recent diagnosis and are functionally independent in ambulation, activities of daily living (ADLs), and speech. Stage 2 (moderate) identifies patients with mild deficits in function in three regions or a moderate to severe deficit in one region and mild or normal function in two other regions. Stage 3 (severe) defines patients who need assistance because of deficits in two or three regions; for example, the patient needs assistance to walk or transfer, needs help with upper-extremity activities, and/or is dysarthric or dysphasic. Stage 4 identifies patients with nonfunctional movement of at least two regions and moderate or nonfunctional movement of a third area. Stage 5 is death.22 (See Brooks and colleagues14 and Pradas and colleagues28 for information on the natural history of ALS and its importance in the design of clinical treatment trials.)
Along with the primary impairments of weakness and fatigue affecting body structure and function in ALS, patients also have progressive limitations in activity and participation.29 Activity limitations result in gradual loss of independence in community and then household tasks. Mechanical and electronic adaptive devices can help extend independence in some ADLs past the initial strength losses. Participation limitations result in progressive isolation from the community and family unless extraordinary efforts persist to retain a communication system at home and through electronic media.
Medical prognosis
In almost all cases ALS progresses relentlessly and leads to death from respiratory failure. The rate of progression seems to be consistent for each patient but varies considerably among patients. Patients with an initial onset of bulbar weakness (dysarthria, dysphagia) and respiratory weakness (dyspnea) tend to have a more rapid progression to death than patients whose weakness begins in the distal extremities.30 Death usually follows within 2 to 4 years after diagnosis, with a small number of patients living for 15 to 20 years.10
Years of survival after diagnosis may change as drug therapies are developed.31 In addition, increasing numbers of patients are electing to prolong life with home-based mechanical ventilation as opposed to palliative or comfort care only.
Medical management
ALS has no known cure and minimal effective disease-slowing treatments. Mitchell and Borasio24 have created a table (see Table 2 in their study) that summarizes the results of trials of the many putative ALS-modifying pharmaceuticals. Only riluzole has been approved for treatment of ALS. Riluzole provides very modest improvement over a placebo in both bulbar and limb function, but not in actual strength of muscles.32 The drug extended lifespan an average of 2 to 3 months. The side effects were minimal in some studies, but fatigue and weakness have been noted in 26% and 18% of patients taking riluzole compared with a placebo.33
The popular press has reported on nutritional cures for ALS, including regular use of vitamin E. However, Orrell and colleagues34 found insufficient evidence to support clinical use of vitamin E supplements in ALS as an additive to riluzole treatment or as adjunctive therapy, although no apparent contraindication was found to taking the supplement. Other nutritional and nonpharmaceutical supplements have had some success in animal models of ALS, but this has not yet been confirmed in humans.35
Cannabis has been studied for its effect on spasticity in patients with multiple sclerosis and spinal cord injury. In a study of 131 people with ALS, 13 used cannabis, with reports of reduction in spasticity, pain, and depression.36 Because of the apparent hopelessness of the diagnosis, many physicians, especially those not associated with major medical centers having neuromuscular disease units, do not refer patients with ALS for services, yet few primary care physicians or neurologists have extensive experience in the care of patients and families coping with ALS because of the low incidence of the disease. Yet, referral of patients with ALS to a multidisciplinary clinic typically extends the patient’s lifespan, especially patients with bulbar onset of ALS.25,37
Muscle spasms and pain
Some patients experience muscle cramps and spasms related to upper motor neuron pathology, and up to 73% of patients complain of pain, typically in the later stages.24 Although most spasms can be relieved with stretching or increased movement, some patients require medications such as quinine or baclofen to relieve symptoms (see Chapter 36 for information on drug therapies). In a review of studies on the treatment of spasticity in ALS, Ashworth and colleagues38 found only one randomized study addressing spasticity: a moderate-endurance exercise regimen decreased spasticity at 3 months after initiation of the program. Stretching and massage may prove helpful for nocturnal muscle cramps.25 Kesiktas and colleagues39 report that in a controlled study of spasticity in patients after spinal cord injury, adding hydrotherapy to a program of medication and exercise decreased severity of spasms and decreased the amount of medication required. A similar response could be hypothesized in patients with ALS. In addition to muscle spasms, patients report nonspecific aching and muscle soreness, probably related to immobility and trauma to paralyzed muscles during caregiving procedures. However, many patients do not receive adequate pain medication, or the pain is not controlled by the medication taken.40 A Cochrane review in 2008 found no randomized or quasi-randomized controlled trials of drug therapy for pain in ALS, although several case series reported the use of acetaminophen, nonsteroidal antiinflammatory drugs (NSAIDs), or opioids.41 Careful administration of medications such as baclofen, tizanidine, dantrolene sodium, and diazepam is useful for some patients with spasticity. Because each has a different action and side effects, the medications may have to be adjusted to find the right dosage and combination. In some patients with severe cramping, botulinum toxin injections might be helpful, but they must be carefully administered to prevent further weakness. Because many patients have compromised respiratory function, the physician must take great care when prescribing pain medication, especially opiates, which are often used when antispasmodics or antiinflammatory pain medications no longer work.25 Patients should be instructed to keep a daily reporting log of the effectiveness of the medication so that the dosage can be adjusted if necessary.
Dysphagia
Dysphagia, a difficulty swallowing liquids, foods, or saliva, accounts for considerable misery in the patient with advanced ALS, and it must be dealt with aggressively. Patients with dysphagia have both nutritional and swallowing problems associated with weakness of the lips, tongue, palate, and mastication muscles.42 As the progressive loss of swallowing develops, patients are also at extreme risk for aspiration. Most patients with dysphagia also have severe problems with management of their saliva (sialorrhea). If a patient has difficulty transporting saliva back to the oropharynx for swallowing, choking and drooling are common.43 This condition is disconcerting to the affected person, who must constantly wipe the mouth or have someone do it for him or her.
In addition, secretions are often thickened because of dehydration. With pooling of the thickened saliva, the possibility of aspiration is increased. Viscosity of saliva can best be treated by hydration and, in some cases, pharmaceuticals. Drugs, such as decongestants, antidepressant drugs with anticholinergic side effects, and atropine-type drugs, can help control the amount of saliva, provided the patient is well hydrated.44 In extreme cases, various surgical procedures such as ligation of the salivary gland ducts, severing the parasympathetic supply to the salivary glands, and excision of the salivary glands have been used effectively.45 Newer treatments to decrease excessive secretions are radiotherapy and botulinum A toxin injections into salivary glands.46
Although dietary treatment is not known to be effective in changing the course of the disease, a nutritious diet to meet caloric, fluid, vitamin, and mineral needs must be maintained. Seventy-three percent of patients with ALS have difficulty bringing food to the mouth, making them dependent on others for their dietary needs. Because of the time it takes to be fed, many patients decrease their intake. All patients with dysphagia should be referred for a dietary consultation to determine the choice and progression of solid and liquid foods and supplements.47 Appel and colleagues47 describe nutritional plans to maintain nutrition and hydration in patients with motor neuron diseases. Patients with bulbar symptoms and severe dysphagia who are no longer able to consume nutrients orally because of motor control problems and recurrent aspiration may need a percutaneous endoscopic gastrostomy (PEG) for feeding, depending on the patient’s wishes for long-term care. Some evidence exists that the PEG should be performed early in the disease process to prevent severe weight loss and aspiration.48 Although a PEG does not appreciably lengthen survival time,49 patients may have less fear of choking or aspiration. Receiving nourishment from a PEG does not prevent the person from taking food orally if desired.
Dysarthria
Dysarthria, impairment in speech production, is the result of abnormal function of the muscles and nerves associated with coordinated functions of the tongue and lips, larynx, soft palate, and respiratory system. Speech impairments are the initial symptom in most patients with bulbar involvement. Speech intelligibility is compromised by hypernasality, abnormalities of speed and cadence of speech, and reduced vocal volume. Speech is further compromised by inadequate breath volumes for normal phrasing. A possible option to help patients with severe hypernasality is a palatal lift prosthesis to augment velopharyngeal function.50,51 Because little can be done medically to delay the loss of speech control, early referral to a speech therapist is essential. Numerous augmentative and alternative communication systems are now available, the simplest being voice amplification systems or homemade point boards and computer-based head or eye tracking text-to-speech systems that can be modified as the patient status changes. The type of communication system should be chosen with awareness of the patient-caregiver environment.52
Respiratory management
Progressive respiratory failure is the primary cause of death in ALS patients. Respiratory failure is related to primary diaphragmatic, intercostal, and accessory respiratory muscle weakness.53 Respiratory failure should be anticipated and discussed early following the diagnosis of ALS so that patients and their caregivers can express their wishes and develop an advanced directive for care in the terminal phase of the disease.54
Physiological tests used to indicate respiratory dysfunction include vital capacity, sniff nasal pressure, and nocturnal oximetry.10 Clinical signs of increased respiratory dysfunction are dyspnea with exertion or lying supine; hypoventilation; weak or ineffective cough; increased use of auxiliary respiratory muscles; tachycardia (also a sign of pulmonary infection with fever and tachypnea); changes in sleep pattern; daytime sleepiness and concentration problems; mood changes; and morning headaches.55
In early stages of patient care, physical therapists (PTs) may help manage respiratory dysfunction by providing postural drainage with cough facilitation (suctioning if necessary), especially during acute respiratory illnesses. The patient and care providers should also be taught breathing exercises, chest stretching, and incentive spirometry techniques, as well as postural drainage techniques if the caregivers are prepared to provide such support. Although breathing exercises consisting of resisted inspiratory muscle training can facilitate functional respiration, even practicing unresisted breathing for 10 minutes three times a day has been shown to result in improved function.56 An assessment of the home environment is imperative to identify sleeping positions and energy conservation techniques that can be incorporated into the patient’s daily life.
As respiratory symptoms increase, oxygen at 2 L/min or less can be used intermittently at home. When hypoventilation with a decline in oxygen saturation becomes common during sleep, resulting in morning confusion and irritability, patients have the option to initiate noninvasive, positive-pressure ventilation (NIV) such as bilevel positive airway pressure (BiPAP). BiPAP, which provides greater inspiratory pressure than expiratory pressure to decrease the effort of breathing, can be administered by either mask or contoured nasal delivery systems. Some evidence indicates that early use of NIV can increase survival time by several months and increase quality of life.57 When a patient can no longer benefit from NIV, a decision must be made about initiating ventilation by tracheostomy or palliative care.58 (See also Miller and colleagues59 for an excellent discussion of practice parameters in the decision-making process related to ventilatory support.) Although in the initial stages of ALS most patients indicate they would not want prolonged respirator dependence at home, patients may change their minds as they adapt to the disease restrictions.60 A small study of patients who started tracheostomy intermittent positive-pressure ventilation (TIPPV) demonstrated increased long-term survival (2 to 64 months).54 In another series of 70 patients on long-term TIPPV, 50% of the patients were living after 5 years; however, 11.4% of these patients had entered a “locked-in state in which they were unable to communicate in any manner.”61 Decisions about long-term respirator use should be made by the patient and involved family members or partners, with input from the interdisciplinary team caring for the patient. Discussions of preferred long-term care options should be revisited as the patient’s condition changes.
If a patient decides that home ventilation is a reasonable option, those involved in the decision should visit another patient who is using in-home mechanical ventilation, if possible. Because the decision for home mechanical ventilation (HMV, NIV, or TIPPV) also affects the life of the patient’s spouse, children, and extended family who may be responsible for some aspects of home care, or whose lives may be affected by the presence of in-home nurses or attendants, the decision for HMV should not be taken lightly. Extensive preparation, ongoing support, and respite options for caregivers are necessary if HMV is to be successful. Success of HMV also depends on such variables as third-party payment for home care equipment and nurse or attendant staffing, working status of the partner or spouse, age and physical fitness of the spouse and children, pre-ALS family psychosocial interactions, and financial factors. HMV should be viewed as long term, often extending for more than 1 year. Initiation of HMV results in a reasonable perceived quality of life for the patient, yet caregivers report that their quality of life may be lower than the patient’s because of the burden of care that must be provided.62
With chronic respiratory insufficiency, the patient and family must be involved in the long-term care decisions related to instituting mechanical assistance under either emergency situations or in response to gradual deterioration. This discussion should occur before the patient develops respiratory failure. Acute respiratory failure can be frightening, and few patients or family members are prepared to forego intubation and artificial ventilation during the emergency. Patients and caregivers should understand that not making a decision about mechanical ventilation, noninvasive or invasive, is a decision to support mechanical ventilation.63
Physicians and health care workers who work with the patient and family must be aware of their own feelings and beliefs about prolonging life. For example, a healthy physician or therapist who values control and an active lifestyle may envision a life on a ventilator as intolerable and pass that value on to the patient, who may or may not have the same needs. The patient’s decision, or change in decision, must be respected by the medical team involved in care.64 In medical centers that use a team approach, patients and families may find support by meeting with counselors or peers with ALS who are making or have made decisions about long-term ventilator care.
Therapeutic management of movement dysfunction associated with ALS
Perhaps because of the multitude of issues to consider when managing the impairments and limitations associated with ALS, evidence suggests that patients treated by a specialized ALS multidisciplinary team fare better than do those treated by single-source providers,65 or in general neurology clinics.33 A Cochrane review of the evidence for multidisciplinary care advantages in this population concluded that the evidence is of low quality, so far, with no controlled trials identified.66 Whether administered through an ALS-specific team or not, therapeutic management will necessitate examination of the patient’s current status, evaluation of the deficits in relation to patient preferences and needs, and establishment of a plan based on mutually determined and realistic goals. The rate of the patient’s disease progression, the areas and extent of involvement, and the stage of illness must be considered. A patient at the initial stages will have different needs than a patient at later stages who has chosen NIV or tracheostomy ventilation that may extend life span at a markedly reduced mobility level. The goal at all stages is to optimize health and increase the quality of life. With guidance and environmental adaptations, patients with slowly progressing weakness may be able to continue many of their ADLs for an extended number of years. In the final stages of the disease, when the patient is bedridden, programs to increase strength or endurance are not appropriate, and interventions such as stretching may not effectively control contracture development. However, patients may still benefit from positioning and range-of-motion (ROM) exercises to decrease muscle and joint pain related to immobility. The prescription of assistive devices and training of caregivers will also be needed. The efficacy of therapeutic interventions will be related to the timing of interventions, the motivation and persistence of the patient in carrying out the program, and support from family members or caregivers.67 Objective documentation of outcome measures will help justify the usefulness of therapeutic interventions at all stages of this disease.
Examination
If possible, before the patient’s initial visit, the therapist should contact the patient and request that he or she keep an activity log for several days. If an early contact is not possible, the therapist can assign that task during the initial session. The log should include 15-minute time increments in which the patient or caregiver can record what she or he was doing during a specific period. The log should also indicate whether the patient was experiencing fatigue or pain during the activity and how the patient perceived her or his respiratory status. An example of an activity log and how it is used is shown in Figure 17-2. The sense of fatigue with repetitive muscle activity or functional activity should be specifically tracked by the patient.

 Example of a log for monitoring activity level of patients with amyotrophic lateral sclerosis.
Example of a log for monitoring activity level of patients with amyotrophic lateral sclerosis.Weakness will be the primary deficit, with other problems following depending on the location of strength loss. Muscle weakness and the experience of fatigue may be independent measures of ALS pathology, however.68 Although weakness may affect balance during gait, patients with ALS have not shown deficits in postural control during quiet stance despite significant paresis or tone changes, possibly because sensation is relatively preserved.69
The therapist’s examination will vary depending on the patient’s situation29; however, a typical initial assessment may include the following:
 Review of the patient’s medical and activity records, especially time since diagnosis, time course of disease progression to date, current medications, concurrent medical issues, current activities and participation and tolerance for them.
Review of the patient’s medical and activity records, especially time since diagnosis, time course of disease progression to date, current medications, concurrent medical issues, current activities and participation and tolerance for them.



 Assessment of functional activity level (using a standardized test or assessment tool whenever possible) to include, as appropriate: transfers, gait, upper-extremity function, postural control, and assistive devices; suggested tools include the ALS functional rating scale (ALSFRS),70 the ALS severity scale (ALSSS),27 timed walk test, or Purdue Pegboard.70
Assessment of functional activity level (using a standardized test or assessment tool whenever possible) to include, as appropriate: transfers, gait, upper-extremity function, postural control, and assistive devices; suggested tools include the ALS functional rating scale (ALSFRS),70 the ALS severity scale (ALSSS),27 timed walk test, or Purdue Pegboard.70

 Assessment of bulbar and respiratory function. (For an in-depth evaluation of bulbar function, the patient should be referred to an ear, nose, and throat clinic or communications disorders clinic unless full evaluation is available in a comprehensive ALS clinic. See Table 17-1 for bulbar and respiratory evaluation suggestions.)
Assessment of bulbar and respiratory function. (For an in-depth evaluation of bulbar function, the patient should be referred to an ear, nose, and throat clinic or communications disorders clinic unless full evaluation is available in a comprehensive ALS clinic. See Table 17-1 for bulbar and respiratory evaluation suggestions.)
TABLE 17-1 
COMMON PHYSICAL FINDINGS IN BULBAR AMYOTROPHIC LATERAL SCLEROSIS
| ANATOMICAL SITE | INNERVATION | METHOD OF EVALUATION | PROGRESSION OF FINDINGS | PROGRESSION OF SYMPTOMS |
| GROUP I | ||||
| Tongue | XII | Inspect for fasciculations at rest | Fasciculations evident | Dysarthria (disturbance of lingual-alveolar consonants t, d, l, and so on) |
| Range of motion | Slow, incomplete lateral movementsLoss of lateral forceUnable to reach palate with mouth open | Inability to clear buccal sulcus of foodMarked dysarthria (slow rate and slurring of consonants) | ||
| Protrusion | Unable to protrude beyond lips | Oral transport difficultiesDietary changes | ||
| Perform rapid lateral motion | Unable to protrude beyond incisorsAtrophy evidentParalysis | Speech intelligibility problems | ||
| Lips | VII | Suck on gloved fingerSmile or curl lips over teethHold seal and blow out cheeks | Lack of suctionInability to complete a sealInability to purse lips | Inability to whistleInability to use a strawDysarthria (loss of bilabial consonants p and b)Drooling |
| GROUP 2 | ||||
| Palate | V, X, XI | Visual examination during phonation and stimulation of gagPuff out cheeks to check for nasal air leak (hold lips closed if necessary) | Unsustained or slow palatal elevationSoft palate fails to reach Passavant ridgeAbsence of palatal movement | Dysarthria (hypernasal speech)Inability to use a strawNasal air emission during speechNasopharyngeal reflex on swallowing |
| Muscles of mastication | V | Palpate during bite | Noticeable wasting | Chewing fatigue |
| Masseter, temporalis | Visual inspection for wasting | Unable to palpate contraction | Elimination of specific, tough foods from dietDietary changes (soft foods and liquids)Mouth breathing and drying of secretions | |
| Pterygoids | Move jaw from side to side | No observable lateral jaw movement | Unable to use dentures | |
| GROUP 3 | ||||
| Neck and shoulder | XI | |||
| Trapezius | Hold arm in coronal plane, hand externally rotated, as patient elevates arm against resistance while the trapezius is palpated | Progressive inability to raise the arm (often asymmetrical weakness) | Inability to comb hairInability to perform facial grooming | |
| Sternocleidomastoid or mounted head support | Turn the head against resistance applied to opposite side of patient’s chin | Progressive weakness in turning the head against resistance (often asymmetrical) | Inability to lift head when supineInability to support head while sitting; wears neck collar, has weakness | |
| Vocal cords | X | Mirror or fiberoptic laryngoscopy | Progressive loss of abduction of vocal cords: mild abductor weakness, near-midline paralysisParadoxical vocal cord movement | Strained or strangled voiceShort of breath (stridor usually not present because of impaired respiratory function) |
| GROUP 4 | ||||
| Extraocular muscles | III, IV, VI | Assessment of extraocular movements | Limitation of extraocular movement | Limitation of gaze |
| Respiratory group | ||||
| Diaphragm | C3-5 | Pulmonary function test or handheld respirometer for vital capacity | Diminishing vital capacity: 1.5-2.0 L | Shortness of breath during exertion if patient has remained active |
| Intercostal | C7-L3 | |||
| Accessory muscles of respiration | VII, XI, XII, C5-8 | CoughSustain a vowelBlow against a tissue | 1.0-1.5 L | Weak coughChange in speech phrasing (5-10 syllables per breath) |
| 0.5-1.0 L | Speech produced in syllable-by-syllable fashion (if vocal)Shortness of breath on swallowing |
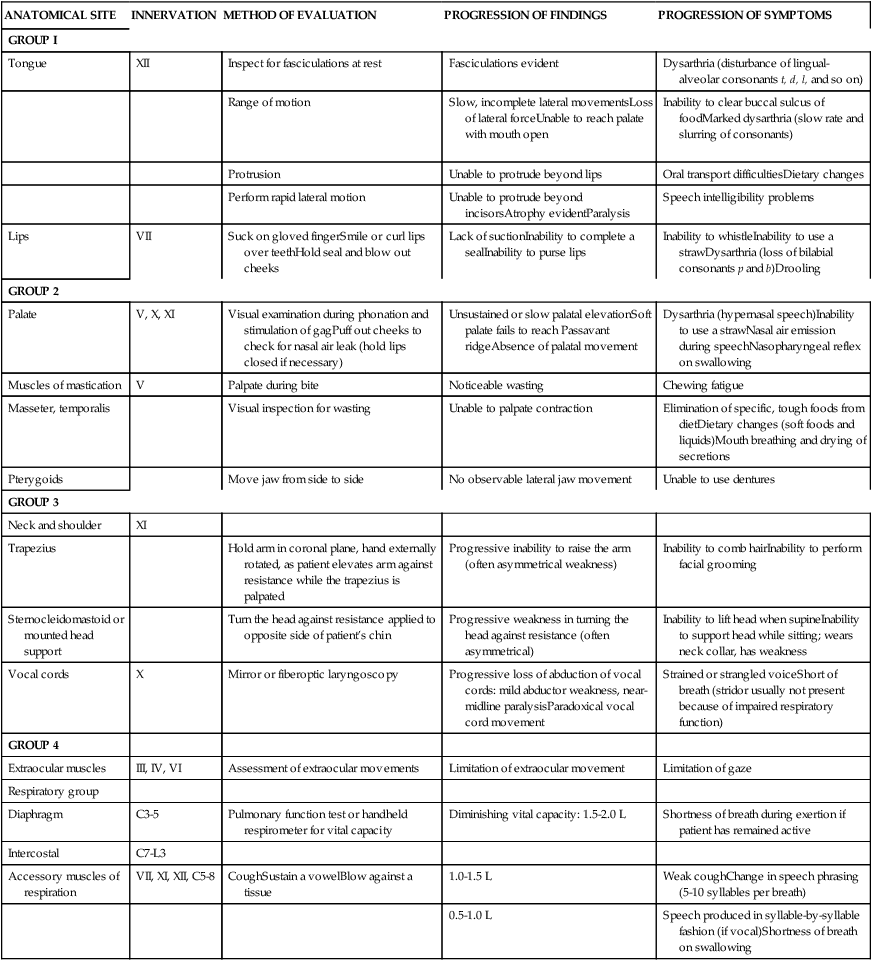
Modified with permission from Hillel AD, Miller RM: Bulbar amyotrophic lateral sclerosis: patterns of progression and clinical management. Head Neck 11:51–59, 1989. Copyright 1989. Reprinted by permission of John Wiley & Sons, Inc.

Brinkmann and colleagues70 identify standards for assessment of patients with ALS in clinical trials. The review and description of standardized methods for performing recommended tests and measurements is extremely valuable for any therapist assessing and treating patients with ALS.




Goals of therapeutic intervention
Intervention goals and the recommended exercise and activity program designed by PTs or OTs must be based on the patient’s personal goals. Goals are often a difficult area for therapists to discuss with the patient because the disease is progressive despite intervention. Patients, therapists, and physicians commonly assume that because nothing can be done to “cure” the disease, not making additional demands on a patient who is already coping with daily loss is somehow kinder. Some believe that exercise programs may create false hopes that exercise will delay progression. Others believe that exercise will hasten progression.71 The literature on rehabilitation in neuromuscular disorders, however, suggests that patients with ALS can benefit from carefully designed exercise and activity programs. Active participation in determining goals for therapy can provide the patient and the family with some sense of control over a difficult situation.7
The general, broad goals for both patient and therapist are related to maintaining maximal independence in daily living and a positive quality of life for as long as possible. More specific therapeutic goals are (1) maintenance of mobility and independent functioning, to include safe mobility for patient and caregiver; (2) maintenance of maximal muscle strength and endurance within limits imposed by ALS; (3) prevention and minimization of secondary consequences of the disease, such as contractures, thrombophlebitis, decubitus ulcers, and respiratory infections7,67; (4) management of energy conservation techniques and respiratory comfort; (5) determination of adaptive equipment needs to include mobility, self-help and feeding devices, augmentative communication units, and hygiene equipment that supports both patient and caregiver7; and (6) eliminating or preventing pain.72
Therapeutic considerations
To prevent more rapid functional loss than expected from the natural history of the disease, both the patient and therapist must delicately balance the level of activity between the extremes of inadequate exercise and excessive exercise. Exercise has been recommended for the general public for its many benefits.73 Inadequate exercise may result in loss of strength and endurance from disuse, as well as secondary problems such as loss of ROM, muscle cramping, and pain. Excessive exercise may result in excessive fatigue and consequent inability to perform ADLs during recovery periods. Overuse injury with excessive strengthening exercise may also lead to unnecessary pain and loss of strength. The next two sections review the evidence for the optimum amount of activity or exercise.
Disuse atrophy.
Because ALS is a disease of older adults, patients may not have maintained their aerobic fitness or muscle strength before the onset of their neuromuscular problem. Newly diagnosed patients also commonly report that they had markedly decreased their activity level in the months before diagnosis because of a sense of fatigue or increasing clumsiness from increasing weakness. If the patient had led a sedentary lifestyle before diagnosis, the additional decrease in activity level after the onset of ALS can lead quickly to marked cardiovascular deconditioning and disuse weakness. The disuse weakness lowers muscle force production and reduces muscle endurance.74
Exercise or overwork damage.
Anecdotal evidence that muscle activity or overwork exercise can lead to a loss of muscle strength has been reported since the poliomyelitis epidemic of the 1940s and 1950s.75 During that epidemic, physicians and therapists noted that patients with poor- and fair-grade muscles who exercised repeatedly or with heavy resistance after reinnervation often lost the ability to contract the muscle at all76 (see Chapter 35). Controlled testing of this observation suggests that overwork damage occurs in mostly denervated muscles, not in all muscles. Reitsma77 noted that vigorous exercise damaged muscles in rats if less than one third of motor units were functional. If more than one third of the motor units remained, exercise led to hypertrophy. An additional mechanism of potential overwork damage is inhibition of the collateral sprouting of intact axons to innervate “orphaned” muscle fibers when other axons degenerate. Yuen and Olney78 provided evidence that collateral sprouting of intact axons can partially reinnervate orphaned muscle fibers in ALS. In a rat model, highly intensive activity reduced the ability of adjacent axons to sprout after fewer than 20% of intact motor units remained.79 In contrast, vigorous exercise in a mouse model had no adverse effect on the course of ALS.80 Lui and Byl81 systematically reviewed the literature reporting exercise effects in animal models of ALS and calculated an effective size of 1.39 (where numbers over 0.8 are considered large) in favor of exercise. The few negative effects they noted were associated with either very-high–intensity exercise or a slow rate of exercise (slower than usual activity for animals when unrestricted in activity). In addition to generic overwork, evidence exists that repeated maximal eccentric contractions may specifically damage even normal muscle fibers, resulting in muscle weakness of several weeks’ duration.82 Although normal muscle eventually adapts to repeated eccentric exercise, whether the reparative effect is possible in patients with neuromuscular diseases is uncertain. Aboussouan55 reviews some of the specific mechanisms of exercise intolerance in neuromuscular diseases, including mitochondrial dysfunction, abnormal muscle metabolism, impaired muscle activation, and central activation failure.
Many researchers have expressed concern about the possible relation between high-resistance exercise and muscle fiber degeneration in humans with motor neuron disease.83,84 Because of the concerns about damage from stressing substantially denervated muscles, Sinaki and Mulder85 published recommendations in 1978 that patients with ALS not engage in any vigorous exercise and focus instead on exercise associated with walking and daily activities. On the other hand, McCrate and Kaspar86 review the possible mechanisms by which exercise protects nerves from more rapid degeneration. Evidence regarding the positive benefits of exercise in ALS has been accumulating, with fewer adverse effects than some expected.
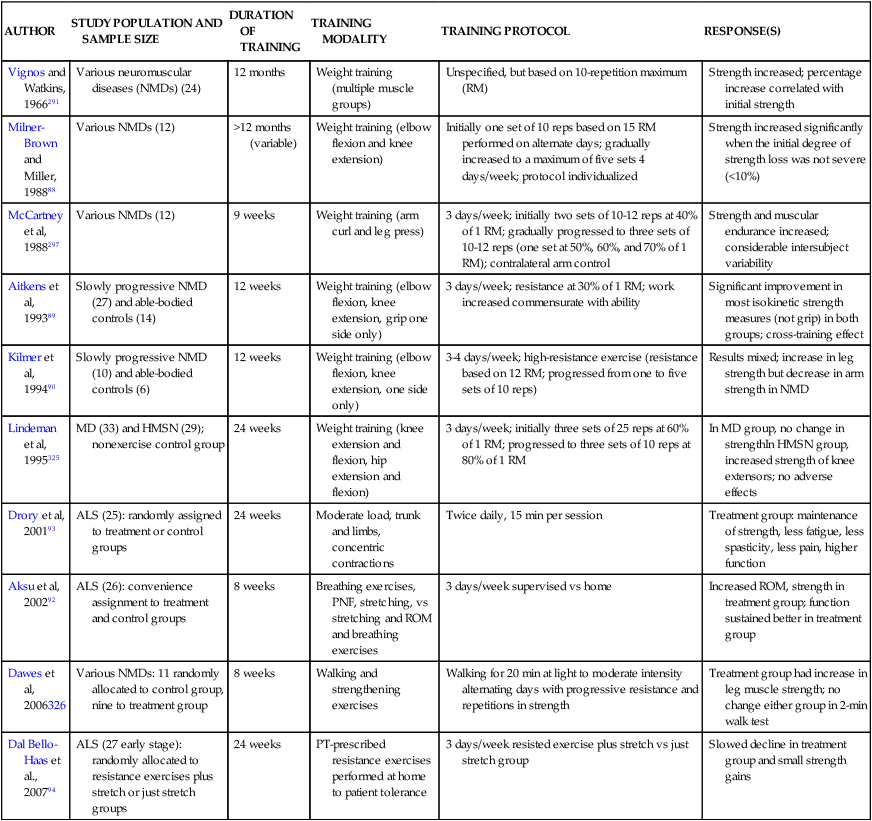
Sanjak and colleagues87 reported that muscle damage does not necessarily result from resistance exercise testing or training, although fatigue occurs more easily during both anaerobic and aerobic exercise. Milner-Brown and Miller88 found that mild progressive resistance exercise was helpful in neuromuscular disorders if the patient had muscle strength in the good (4/5) to normal (5/5) range. They determined that patients should begin their exercise program early because strength training of muscles with less than 10% of normal function was generally not effective. Aitkens and colleagues89 noted strength gains of 4% to 20% without deleterious effects after a 12-week program of moderate-resistance (30% of maximum isometric force) exercises in patients with slowly progressive neuromuscular diseases. Kilmer and colleagues,90 in the same population, found no additional advantage to high-resistance training (12 weeks of exercise using the maximum isometric force the individual was able to lift 12 times) and noted evidence of overwork in some subjects. In a case report of a patient with ALS, strengthening 6 days a week for 10 weeks with proprioceptive neuromuscular facilitation (PNF) patterns using maximal resistance applied manually or with tubing resulted in strengthening of 14 muscle groups out of 18 with no adverse effects.91 Aksu and colleagues92 compared a supervised versus home exercise protocol in 26 ambulatory ALS patients. They noted that supervised breathing exercises, stretching, manually applied resistance exercise with PNF, and functional mobility training 3 days a week for 8 weeks resulted in small gains in function in the first 4 weeks and a slower decline over the subsequent 10 months compared with home-based breathing, stretching, and active ROM exercises. The groups were not randomly allocated but were not significantly different in the measured variables at baseline.92 In a randomized controlled trial, Drory and colleagues93 assigned 25 patients with ALS to a group continuing their normal daily activities or a group participating in a moderate daily program of exercise individualized for each patient. The primary exercise focus was to have muscles of the trunk and limbs work against “modest” loads while undergoing significant shortening (not lengthening or eccentric contractions). The exercises were completed twice daily for 15 minutes at home with phone contact by the treating therapist every 14 days. Data were evaluated for 3 and 6 months after initial assessment. All patients showed continued disease progression; however, in all cases, at the 6-month assessment patients who exercised showed positive effects in maintenance of muscle strength, less fatigue, less spasticity, less pain, and higher functional ratings.93 In another randomized controlled trial, moderate load and moderate-intensity resistance exercises prescribed individually to patients with ALS in the early stages resulted in significantly less decline in function, small improvements in strength, and no reported adverse effects, compared with patients who performed stretching exercises alone.94 A Cochrane review designated the quality of the Drory and colleagues (2001) study as “fair” and the Dal Bello-Haas and colleagues94 study as “adequate.”95 Table 17-2 summarizes some of the studies of strength training in neuromuscular diseases.
TABLE 17-2 
SUMMARY OF STRENGTH TRAINING STUDIES IN NEUROMUSCULAR DISEASES
| AUTHOR | STUDY POPULATION AND SAMPLE SIZE | DURATION OF TRAINING | TRAINING MODALITY | TRAINING PROTOCOL | RESPONSE(S) |
| Vignos and Watkins, 1966291 | Various neuromuscular diseases (NMDs) (24) | 12 months | Weight training (multiple muscle groups) | Unspecified, but based on 10-repetition maximum (RM) | Strength increased; percentage increase correlated with initial strength |
| Milner-Brown and Miller, 198888 | Various NMDs (12) | >12 months (variable) | Weight training (elbow flexion and knee extension) | Initially one set of 10 reps based on 15 RM performed on alternate days; gradually increased to a maximum of five sets 4 days/week; protocol individualized | Strength increased significantly when the initial degree of strength loss was not severe (<10%) |
| McCartney et al, 1988297 | Various NMDs (12) | 9 weeks | Weight training (arm curl and leg press) | 3 days/week; initially two sets of 10-12 reps at 40% of 1 RM; gradually progressed to three sets of 10-12 reps (one set at 50%, 60%, and 70% of 1 RM); contralateral arm control | Strength and muscular endurance increased; considerable intersubject variability |
| Aitkens et al, 199389 | Slowly progressive NMD (27) and able-bodied controls (14) | 12 weeks | Weight training (elbow flexion, knee extension, grip one side only) | 3 days/week; resistance at 30% of 1 RM; work increased commensurate with ability | Significant improvement in most isokinetic strength measures (not grip) in both groups; cross-training effect |
| Kilmer et al, 199490 | Slowly progressive NMD (10) and able-bodied controls (6) | 12 weeks | Weight training (elbow flexion, knee extension, one side only) | 3-4 days/week; high-resistance exercise (resistance based on 12 RM; progressed from one to five sets of 10 reps) | Results mixed; increase in leg strength but decrease in arm strength in NMD |
| Lindeman et al, 1995325 | MD (33) and HMSN (29); nonexercise control group | 24 weeks | Weight training (knee extension and flexion, hip extension and flexion) | 3 days/week; initially three sets of 25 reps at 60% of 1 RM; progressed to three sets of 10 reps at 80% of 1 RM | In MD group, no change in strengthIn HMSN group, increased strength of knee extensors; no adverse effects |
| Drory et al, 200193 | ALS (25): randomly assigned to treatment or control groups | 24 weeks | Moderate load, trunk and limbs, concentric contractions | Twice daily, 15 min per session | Treatment group: maintenance of strength, less fatigue, less spasticity, less pain, higher function |
| Aksu et al, 200292 | ALS (26): convenience assignment to treatment and control groups | 8 weeks | Breathing exercises, PNF, stretching, vs stretching and ROM and breathing exercises | 3 days/week supervised vs home | Increased ROM, strength in treatment group; function sustained better in treatment group |
| Dawes et al, 2006326 | Various NMDs: 11 randomly allocated to control group, nine to treatment group | 8 weeks | Walking and strengthening exercises | Walking for 20 min at light to moderate intensity alternating days with progressive resistance and repetitions in strength | Treatment group had increase in leg muscle strength; no change either group in 2-min walk test |
| Dal Bello-Haas et al., 200794 | ALS (27 early stage): randomly allocated to resistance exercises plus stretch or just stretch groups | 24 weeks | PT-prescribed resistance exercises performed at home to patient tolerance | 3 days/week resisted exercise plus stretch vs just stretch group | Slowed decline in treatment group and small strength gains |
Fewer researchers have considered endurance in neuromuscular disorders.73 Sanjak and colleagues87 noted that exercise energy requirements during bicycle ergometry testing were greater than expected, possibly because of motor inefficiency caused by weakness. Work capacity and maximal oxygen consumption were decreased, but heart rate, respiratory responses, and blood pressure were within normal limits. Wright and colleagues96 found small positive physiological effects from an aerobic walking program in patients with slowly progressive neuromuscular disorders. Pinto and colleagues97 provided eight ALS patients with NIV during exercise to compensate for respiratory insufficiency. Patients walked on a treadmill for 10 to 15 minutes to the point of subjective fatigue, leg pain, heart rate above 75% of resting value, or desaturation of oxygen not correctable with NIV. In comparison to a nonexercising control group, the exercising group had a significant reduction in the rate of decline of respiratory function test results, strength, and function over the 1-year training period.97
Endurance training for longer than 10 to 15 minutes in patients with ALS may be restricted by central fatigue, the decreased ability to recruit all motor units or develop high discharge rates,98 and not merely respiratory function. Sharma and colleagues99 explored the mechanism of fatigue in ALS. Both maximum voluntary contraction and tetanic force decreased in patients with ALS compared with controls following a 25-minute low-intensity intermittent exercise, but with similar recovery. Fatigue may thus be a consequence of chronic denervation resulting in secondary muscle changes such as altered muscle metabolism and impaired calcium kinetics along with the loss of motor unit activation.99
In addition to strength and endurance gains from exercise, ongoing, gentle exercise programs may also help decrease persistent pain and muscle stiffness that often accompany weakened, overtaxed muscle groups.100 A case study of a patient with ALS undergoing a focused exercise program revealed a positive psychological effect on the patient’s coping strategies.101 Besides exercise programs, some preliminary evidence exists to suggest that creatine supplementation may increase isometric power in patients with ALS over the short term.102 Modafinil has been noted to have potential in helping with severe fatigue in ALS.103
Many studies focus on the impact of exercise on muscle strength; however, knowledge of impairments does not necessarily correlate directly with functional status. Although some research has shown improvements in muscle force production with strengthening and endurance training, associated functional improvements were evident in some studies92 but not others.104 Jette and colleagues82 calculated the percentage of predicted normal maximal isometric force (%PMF) relative to four walking levels in patients with ALS: unable to walk, walking within the home only, walking in the community with assistance, and independent walking in the community. Although they found great variation in muscle force production between and within the different levels of walking for each patient, they demonstrated that relatively small changes in force production were associated with losses of functional levels. For example, on average, when an independent ambulator began to need assistance in the community, the lower-extremity strength dropped to less than 54%PMF. When the patient became an in-home ambulator only, the average strength dropped to approximately 37%PMF, and it was approximately 19%PMF when the patient was no longer able to walk. Jette and colleagues82 acknowledge that many factors need to be considered when interpreting their work; however, their study relates functional skills to isometric muscle force production in a concrete way. Factors such as spasticity, age at onset of ALS, prior levels of fitness and activity, and psychological factors, including past responses to extremely challenging situations and satisfaction with social support, must also be considered.
Based on the evidence and current practice, exercise prescription in the early stages of ALS should address the following72:
1. To improve compliance, include both a formal exercise program and enjoyable physical activities.
2. Include activities with opportunities for social development and personal accomplishment.
3. Strengthening programs should emphasize concentric rather than eccentric muscle contractions; use moderate resistance rather than high resistance; and focus on muscles that have at least antigravity strength.
4. Endurance programs should be monitored for signs of fatigue, more so when continuous activity lasts longer than about 15 minutes. Activity programs should include rest periods.
5. Patients should ensure that they have adequate oxygenation, aeration, and carbohydrate loads73 as well as adequate fluids before exercising.
6. Muscle strength must be monitored to assess for possible overwork weakness; in unsupervised programs, patients must be instructed about signs and symptoms that indicate overwork, including feeling weaker within 30 minutes after exercise, having excessive soreness 24 to 48 hours after exercise, and experiencing severe muscle cramping, heaviness in the extremities, or prolonged shortness of breath105; and therapists should check with an independently exercising patient regularly to assess whether any deterioration in strength may be from progression of the disease or overwork weakness.
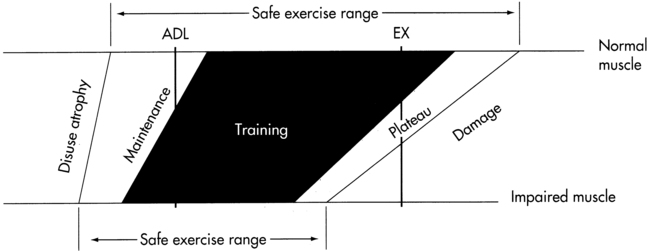
If a patient shows evidence of significant, persistent weakness after institution of an exercise program or persistent morning fatigue after exercise on the previous day, the therapist must carefully redesign the patient’s exercise program and activity level and increase the frequency of monitoring the patient’s program. The program must be adjusted as the disease progresses. Figure 17-3 is a diagram showing the appropriate exercise “window” for use in working with a patient with a neuromuscular disorder.

Therapeutic interventions
Maintenance of strength and endurance requires daily activity and repetitive muscle contractions. In normal persons, absence of muscle contraction can result in decreases of 3% to 5% in muscle strength per day. If the patient’s exercise level requires less than 20% of the maximal voluntary contraction of the muscles, a decrease in strength will occur; yet overwork must be avoided.106
Sinaki107 has described three phases and six substages of ALS with recommended exercise levels (Box 17-2). Although therapists should not assume that all patients will fit precisely within the stages as described, the stages do provide suggestions for interventions on the basis of degree of impairment, functional limitations, and level of disability. In the following section, staging patterns are used as the framework for therapy interventions. Staging information is particularly helpful to therapists who do not have the opportunity to work with large numbers of patients with ALS.




Continue stage 2 program as tolerated; use caution not to fatigue to point of decreasing patient’s ADL independence
Keep patient physically independent as long as possible through pleasurable activities such as walking
Encourage deep breathing exercises, chest stretching, postural drainage if needed
Prescribe wheelchair, standard or motorized, with modifications to allow eventual reclining back with head rest, elevating legs
Phase II (partially independent)
Heat, massage as indicated to control spasm
Active assisted passive ROM exercises to the weakly supported joints; caution to support, rotate shoulder during abduction and joint accessory motions
Encourage isometric contractions of all musculature to tolerance
Try arm slings, overhead slings, or wheelchair arm supports
Motorized chair if patient wants to be independently mobile; adapt controls as needed
Encourage family to learn proper transfer, positioning principles, and turning techniques
Encourage modifications at home to aid patient’s mobility and independence
Electric hospital bed with antipressure mattress
If patient elects home mechanical ventilation (HMV), adapt chair to hold respirator unit
For dysphagia: soft diet, long spoons, tube feeding, percutaneous gastrostomy
To decrease flow of accumulated saliva: medication, suction, surgery
For dysarthria: palatal lifts, electronic speech amplification, eye-pointing electronics
For breathing difficulty: clear airway, tracheostomy, respirator if patient elects HMV
Modified with permission from Sinaki M: Exercise and rehabilitation measures in amyotrophic lateral sclerosis. In Yase Y, Tsubaki T, editors: Amyotrophic lateral sclerosis: recent advances in research and treatment, Amsterdam, 1988, Elsevier Science.
Most patients need specific guidance about what type of activities and exercises they should do.61 Although many physicians may suggest to patients that they increase their activity level, their suggestions are seldom specific. Examples of exercise advice that patients have recalled are “Try to move around as much as possible,” “Walk some more,” and “Be active, but don’t overdo it.” Because changing their typical exercise pattern is difficult for most patients, even when they know doing so is important, referral for a physical therapy consultation can be helpful.108
Phase I (independent): stages 1 to 3.
A program to increase activity must be specifically designed, with input from the patient about willingness to participate and knowledge of the patient’s environmental situations and social support systems. In the early stages of the disease, patients should be encouraged to continue as many prediagnosis activities as tolerated. For example, a golfer should continue to golf for as long as possible. Walking the course should be encouraged if it is not too fatiguing. When walking or balance becomes difficult on uneven terrain, the golfer can use a golf cart, decrease the number of holes played, move to a par 3 course, or hit balls at a driving range. If upper-extremity weakness is a major problem that interferes with swinging the club for distance shots, the player can continue playing the greens or on putting courses. Some golfers may need adaptations to club handles with nonskid material such as Dycem (Dycem Non-Slip products, www.dycem.com) or Scoot-Gard (Vantage Industries Product) to prevent the club from rotating on impact.
Patients with newly diagnosed ALS who had a sedentary lifestyle before diagnosis should be encouraged to increase their activity level. This may include activities that require muscular effort within or around the home, such as sharing household and gardening tasks or beginning a walking program around the neighborhood. After diagnosis, some patients begin searching for in-home exercise devices such as bicycles and rowing machines. As with healthy persons who start an exercise program after the purchase of exercise equipment, patients with ALS are not likely to use the equipment consistently if they did not before a diagnosis. The search for a “perfect” exercise machine may reflect the patient’s desperation to do something tangible. Without taking away the patient’s motivation to exercise, therapists can encourage participation in exercise programs that do not require expensive equipment, such as walking or working out to specific exercise routines. A clever therapist can make a video for each patient that includes stretching and gentle exercise programs that elicit muscle contractions from all functional muscle groups (by using inexpensive elastic bands or small weights) with follow-up breathing, “warm down,” and relaxation exercises. Patients could follow a program of six maximal isometric contractions held for 6 seconds and isotonic elastic band exercises at submaximal levels to maintain and improve muscle strength.109 Patients should exercise for short periods several times a day rather than attempting to exercise all muscle groups in one session.
For most patients in the early stages of ALS, pleasurable, natural activities such as swimming, bowling (can gradually decrease weight of ball if shoulder strength is a problem), walking, bicycling (three-wheeler may be needed or in-home stationary bicycle, either of which must be evaluated for easy mounting and dismounting), or tai chi should be recommended. Some patients prefer to exercise alone, whereas others will gain confidence and companionship by joining a group activity. Listening to the patient’s desires related to group activities is important. The dropout rate is high among those who have been pressured to participate. Some spouses or family members are supportive of the patient’s activity needs and will join the patient in his or her regimen. If possible, the spouse and family members should be engaged in the treatment planning process.110
The therapist must observe the patient completing her or his entire recommended activity program. The patient’s response to the program must be monitored because fatigue from exercise sessions can interfere with the ability to carry out other normal daily activities. If the patient becomes too exhausted at the end of a session, he or she may learn to fear exercise and may become depressed about the decreased activity status. This depression may lead to decreased activity and further deconditioning (see Chapter 6).
Phase II (partially independent): stages 4 and 5.
During late phase I and through phase II, many patients show significant weakness of both upper- and lower-extremity musculature, but each patient has his or her own pattern and rate of progression of weakness and onset of spasticity, bulbar, and respiratory symptoms. A typical patient at this time may have marked weakness of the intrinsic muscles, shoulder muscle weakness (in some cases “hanging arm” syndrome) with shoulder pain, and generalized lower-extremity weakness (in some cases more severe distally). Patients may be able to walk within the home environment, but many patients have precarious balance and fall easily because of muscle weakness. At this stage, most patients report fatigue with minimal work and have to rest frequently when carrying out ADLs. ROM can deteriorate quickly in this phase of the disease, requiring daily stretching to end range for the calf, quadriceps, hip adductors, trunk lateral flexors, and long finger flexors.29 Moderate exercise can have a modest effect in reducing spasticity.93
Patients at this point, even if ambulatory, should consider using a wheelchair outside the home to conserve energy.72 Factors to consider in choosing a wheelchair include extent of insurance coverage or financial assistance programs for purchase of wheelchair (some policies or programs may provide only one type of wheelchair or only one wheelchair, either motorized or manual); transportability of motorized chair from home to community and work (few motorized wheelchair brands fold for stowing in car trunk, and few families can afford to purchase a van that will allow the patient to drive or be driven while in a motor chair); reclining potential of chair back and headrest (preferably electric) to allow the patient to shift weight and rest while in the chair during later stages of the disease; removable arm rests for ease of transfer; potential for headrest attachment or extension; potential mounting area for portable respirator equipment if needed; and ease with which caregiver can help patient with chair mobility transfers.72 Chairs should have lumbar support and appropriate cushioning to prevent pressure ulcers.105
At this stage, patients with more advanced bulbar symptoms begin to experience dysarthria and may need guidance in dealing with communication issues. Murphy111 indicated four major reasons for communication: to identify needs or request help, share information, respond politely in social situations, and maintain social closeness. The primary focus of communication for the study participants was to maintain social closeness. Although few patients had any instruction in ways to deal with communication problems, most patients and caregivers created ways to make themselves understood, such as giving cues about the topic and context, creating a “shorthand” language, and checking with the dysarthric speaker to ensure that the listener understands the patient correctly. A number of patients in the study who had significant dysarthria commented that attempting to communicate socially was extremely tiring. Therapists who are guiding patients with energy conservation techniques should be aware of the exhaustion that can be associated with communication. A number of strategies recommended by the American Speech-Language-Hearing Association112 can be used by the person with ALS to deal with the effects of dysarthria, including the following:
 Reduce background noise in the room.
Reduce background noise in the room.
 Face the person while talking.
Face the person while talking.
 Use short, simple phrases rather than long, complicated ones.
Use short, simple phrases rather than long, complicated ones.
 Take the time to say what needs to be said; do not allow people to rush conversation.
Take the time to say what needs to be said; do not allow people to rush conversation.


Also in this stage, some patients and families may need support to identify adapted feeding systems (special utensils, adapted plates, adjustable tables) and hygiene equipment if transfers within the family bathroom are problematic.113
Because Mr. Turner in Case Study 17-1 was cared for in a neuromuscular disease clinic, he benefited from input from multiple specialists working as a team to help him maintain his independence. Unfortunately, many patients do not have the benefit of such a coordinated treatment environment. Therefore, when necessary, the therapist must be in a position to provide input on adaptive and safety devices and bulbar issues if other specialist input is not available. Therapists working in smaller communities and rural areas most likely need to be chameleon-like to play many therapeutic roles when working with the patient with ALS.
CASE STUDY 17-1  MR. TURNER
MR. TURNER
With input from the therapist, Mr. Turner and his wife identified the following general goals:
1. Increase mobility while conserving energy
2. Control fatigue and pain of upper extremities and neck during computer work
3. Maintain maximal muscle strength and ROM (patient reported that he felt stiff)
4. Identify safety issues within the home and work environment and adjust household and work environment to prepare for the time when Mr. Turner could not ascend and descend stairs safely
A treatment plan was discussed to achieve the following:
1. Increase mobility. Because of his increased walking difficulties, Mr. Turner decided to use a front-wheeled walker with a seat attachment at home. Because of his hand grip weakness, he felt most stable using attached forearm troughs. For his worksite, he selected a motorized wheelchair so that he could maintain his independence at work. Although he found that he could push an ultralight manual chair, his upper-extremity strength was clearly decreasing. Mr. Turner decided that he preferred the motorized chair to an electric scooter because of the financial cost of switching devices when the scooter no longer provided adequate postural support.
Because Mr. Turner’s insurance and Medicare would not fund an additional manual chair and because the family had no way to transport the electric wheelchair, the ALS Society loaned the family a manual wheelchair for home use. Although not ideal, it was functional. Mr. Turner’s son made some inexpensive adjustments to adapt the chair for a headrest, and his daughter and grandchildren repainted the chair to his specifications.
Because Mr. Turner wanted to keep as active as possible and use his walker within the home, he was fitted with bilateral ankle-foot orthoses (AFOs) with a flexible ankle joint and pretibial shell to facilitate knee extension. Straps were simple overlap style because Mr. Turner had poor thumb and grasp control.
2. Decrease fatigue and pain of upper extremities. Mr. Turner was taught some simple ROM exercises of the neck and arms to perform every half hour while working at the computer. In a simulated work environment the therapist noted that Mr. Turner had a forward head position when working at a computer similar to his workstation. The height of the computer was adjusted to decrease his neck strain, and the desk height was adjusted to allow his wheelchair to fit under the desk so that his arms could rest fully on the surface. He felt immediate relief with the adaptations. He was also fitted for a soft neck collar to wear when he felt he needed more neck support. (As his condition worsened, he learned to rest his head on the headrest of his chair and recline slightly for a few minutes every 15 minutes.)
3. Maintain maximal muscle strength and ROM. Mr. Turner was taught as many self-ranging maneuvers as possible, which he was encouraged to do in small segments frequently throughout the day. For example, his series of motions included neck rotations, side bends, and flexion and extension within strength limits; upper-extremity motions with the exception of shoulder flexion and abduction past 90 degrees; hip flexion, abduction, and rotations; full knee extension; and all ankle motions. When using the walker, Mr. Turner was encouraged to extend each hip fully and to stretch his heel cords. Mrs. Turner and their adult children were taught to administer full ROM exercises, including trunk rotations, with special attention to ranging of the shoulder to prevent impingement. Simple massage techniques were also taught to all family members who felt comfortable with the task.
Mr. Turner had been active before the onset of ALS and he liked to exercise. He rented a portable pedaling unit to attach to a chair at home. He pedaled two to four times a day, with no additional resistance, to the point at which he felt fatigue (usually 3 to 5 minutes at this stage). He carefully monitored his soreness and fatigue level after exercise and increased and decreased his pedaling depending on how he felt immediately and several days after exercise. Mr. Turner felt invigorated by this exercise, which he usually did while watching television. He was also taught a series of simple elastic band exercises, with tensile strength adjusted according to his ability to contract his muscles without fatigue. Mr. Turner was also shown a series of isometric exercises for all muscle groups to do throughout the work day. Because he had some foot and ankle edema, he was encouraged to wear lightweight pressure stockings while sitting. Mr. Turner also had access to a swimming pool, and he was encouraged to carry out walking and upper-extremity exercises as long as another adult was with him in the water at all times.
4. Assess environment of home and work. Occupational therapy input was requested to help with ADL aids such as reachers, utensil adaptors to facilitate grip, rubber pen grippers, key adaptors to permit turning, and thumb abduction splints to assist in pincer grasp. Mr. Turner’s OT made several visits to his worksite and home to identify adaptations of the environment for safety and independence. His wheelchair was eventually adapted with universal joint arm troughs to decrease his effort during self-feeding and basic upper-body hygiene. Ramps were recommended for home entry, and nonpermanent safety rails were placed in the bathroom. Mr. Turner was able to assist with transfer to a shower chair, and the shower head was replaced with a handheld unit.
A speech pathology consultation was also requested. Using information from the PT’s manual muscle testing, the speech pathologist carried out a thorough bulbar evaluation and provided information about swallowing techniques. The speech therapist focused on ways to decrease drooling and ways to cope with food pocketing (tongue mobility was impaired) by using techniques such as hand pressure on the cheek to push food back to the center of the mouth. The therapist also instructed Mr. Turner and his wife how to prepare foods with textures that were easily swallowed and manipulated. Mr. Turner had lost 5 pounds during the last 6 months, so he was also referred to the dietician for information about how to maintain nutritious calorie intake.
Phase III (dependent): stage 6.
PTs and OTs are usually less involved in the care of the patient in phase III, and nursing personnel become more active. During this phase, therapists make home visits to support caregivers and respond to questions about pain control, bed mobility, positioning to prevent pressure ulcers, ROM, and equipment adaptations.29,72,105 Therapists should be sure to teach all caregivers some basic body mechanics to use during lifting and patient care activities. If possible, caregivers should be taught how to safely move the person with ALS from the bed to a reclining wheelchair or other reclining chair during specific times of the day so that the person can continue to be part of the family activities. However, the ease of caregivers in transferring and caring for the person in the wheelchair must also be considered. Although some patients want to be in the midst of family activities even when dependent on HMV, other patients feel uncomfortable with their dependency and appearance and are reasonably content to stay in their room with television and visits from family members. This highly personal decision by patients must be respected. The therapist should review ROM procedures with family and professional caregivers and provide splinting or positioning devices if spasticity or paralysis leads to caregiving difficulties (e.g., excessive adductor tone and contractures interfering with hygiene and bowel care) or tissue damage and pain. If nursing care providers do not give advice on pressure relief beds or mattresses of air or foam,105 therapists should be prepared to do so. Unfortunately, many insurance providers and Medicare may not fund special mattresses, and they can be costly. Therapists may also need to review postural drainage techniques with caregivers.
Of greatest importance in phase III, and sometimes in earlier stages, is the patient’s ability to communicate. In the earliest manifestation of dysarthria, therapists train patients to slow the speech rate and cadence, exaggerate lip and tongue movements, and manage phrasing through breath control.83 Although spouses and caregivers can often interpret their partner’s or patient’s severely dysarthric speech (see earlier discussion of phase II), most patients who use NIV or invasive ventilation for a prolonged period need to find nonverbal methods to communicate. If severe bulbar impairments precede extremity paralysis, paper and pencil, alphabet and word boards, and adapted computer keyboards can be used with minimal upper-extremity or finger strength for pointing. The American Speech-Language-Hearing Association provides suggestions for developing communication boards with the specific language most appropriate for the patient’s situation.112 For example, the board may be designed with commonly needed sentences, words used in the person’s daily life, and the alphabet. As the person’s ability to finger point decreases, the language board can be redesigned. When no extremity movement is possible, subtle neck movements or pressures, eye gaze, eye blink, upper facial movements, and electroencephalographic activity can be harnessed to operate communication devices.114,115 Learning to use electroencephalographic interfaces, however, takes months of intense training and may not provide a reasonable system for communication for most patients with ALS.116
When selecting a communication device, therapists must work closely with the patient and family members to ensure that the system is compatible with patient skills and communication needs and preferences. Expensive systems commonly lie unused because of simple factors such as lack of proximity to the patient, interference of the unit with personal care, increased caregiver workload to manage the unit, and slowness of communication processing. The best systems are tailored to the precise needs of the patient; however, many patients do not have the financial or insurance support to purchase the device, and many patients in the end stages of ALS do not have the time to wait for systems designed for their specific needs. Therefore commercially manufactured systems may be most appropriate. (See Cook and Hussey114 for a comprehensive list of communication devices and control interfaces.)
Some patients and caregivers learn to communicate effectively with simple eye gaze, eye blinking, and clicking techniques with Morse code or self-developed codes. At minimum, patients with no ability to communicate or move and their caregivers must have some system to communicate emergency needs; for example, looking to the right means “help” and looking to the left means “pain.” Therapists should help patients develop alternative modes of communication before intelligible speech becomes impossible. (See also Cobble117 for information on language impairments.)
In addition to communication systems, environmental control systems can be programmed to turn on and off television, lights, and other electronic units with the same type of switching units used for communication (e.g., eye blink, infrared beam, head movement pressure). Unfortunately, these devices are often expensive and may not be available to all patients. (See Cook and Hussey114 for a comprehensive review of environmental control systems.) Financial support is often not extended for high-tech equipment by third-party payers because of the patient’s limited life expectancy. The ability to communicate and call for help, however, is of paramount importance with completely dependent patients.
By phase III most patients have significant problems eating and maintaining nutrition, although these problems may manifest in earlier stages. Patients often report choking or coughing after swallowing liquids or problems moving food around in the mouth or to the back of the throat for swallowing. These problems are best handled medically and can be assessed with videofluoroscopy or videoendoscopy. The aggressiveness of treatment intervention depends on the patient’s preference and whether she or he still wants to attempt any oral feeding (e.g., syringe feeding, oral gastric tubes) or wishes to have a PEG or another alternative to oral feedings implemented. Therapists, however, can help patients and caregivers develop strategies that improve eating and nutrition, such as adjusting eating position, changing head and neck alignments, adding thickeners to liquids, and adjusting portion sizes and texture of foods.7
Psychosocial issues
Giving the bad news of a terminal diagnosis is difficult for even the most experienced clinician. In dealing with the diagnosis of ALS, most physicians now believe that the diagnosis, prognosis, and possible patterns of progression should be shared with the patient and family or partners and caregiving friends. Only by knowing the truth can patients and families deal openly with one another and make plans for the future.118 McCluskey and colleagues119 suggest that those giving the medical or therapeutic diagnosis should attend to good practice parameters when giving bad news, such as creating the appropriate setting, identifying patient and caregiver needs, asking what patients and caregivers want to know, providing knowledge, exploring feelings of the patients and caregivers, and formulating a strategy for dealing with the situation. Patients and family members seldom remember what they are told when first given a terminal diagnosis. They do, however, remember how the information was given. Therefore information should be given honestly but with a sense of hope. All information need not be given at the time of diagnosis. Rather, the patient and family can be exposed to more in-depth information over a number of sessions when they have the opportunity to ask questions that occur during the assimilation process. Therapists, especially those working in isolation from a comprehensive clinic, should also follow these guidelines by providing information, helping the patient and family identify goals, and establishing a plan for intervention. Patients should know that the goals will have to be adjusted and plans reset as the disease process continues. If patients and families know that they can contact the therapist for support and advice, many of the negative aspects of the illness can be confronted in a positive manner. Preferably, an appointment for a follow-up visit will be set so patients and family members feel that contact with the care provider is expected.
Information about transitions related to nutrition, communication, and respiratory functions should be delivered to patients and families in time to make thoughtful decisions rather than just before a time of crisis, such as after a choking episode or during a respiratory arrest. Care should also be taken to respect the cultural and spiritual views of the patient and family.58 Preferably, patients and family members will prepare an advance medical directive that should be reviewed with the physician at least every 6 months.120
Purtilo and Haddad121 identified four major fears of the patient who has a terminal condition: fear of isolation, fear of pain, fear of dependence, and fear of death itself. Patients with progressive diseases often see their social contacts decrease. Mr. Turner in Case Study 17-1 was concerned when he was no longer able to join his colleagues in the company cafeteria. After he received his motorized wheelchair he was able to continue his social contacts until his bulbar symptoms progressed to a point that he chose not to eat in public. When Mr. Turner lost the ability to speak and had to use his computerized speech system, he noticed that fewer colleagues stopped by his office to talk because of the slowness of the communication process. Although he understood the problem, Mr. Turner mourned the loss of friendship and his loss of standing as a competent computer expert. Because of his need for social contact, Mr. Turner continued to work until he could no longer tolerate the sitting position. His fear of isolation increased when he became homebound. Although colleagues came for visits regularly at first, as Mr. Turner progressed to a near locked-in state only a few close friends came by for brief visits. Mr. Turner’s greatest fear was being separated from his family and abandoned to hospital care with inconsistent staffing patterns. Fortunately, in his community, Mrs. Turner was able to set up visitations from several church members, clerics, and hospice volunteers.
Fear of uncontrolled pain is common among people with terminal diseases. Patients need assurance that their pain will be controlled. Fortunately, today pain medications can be administered in many forms, dosages, and frequencies that can be tailored to the patient’s specific needs. In a study of the final month of life with ALS, caregivers reported that a major emphasis of care was to eliminate as much pain and discomfort as possible, even if it shortened the patient’s life.122 Keeping a pain log of intensity, type, location, and time of pain may provide the physician with information necessary to best prescribe dosages. Many patients with ALS do experience significant pain from musculoskeletal sources, persistent spasms, or spasticity and pressure sores. Most of these problems can be handled with appropriate pain medications, muscle relaxants, careful positioning, frequent ROM exercises, and tissue massage. Undertreated and uncontrolled pain is associated with a patient’s seeking information on assisted suicide.123 Some patients who expressed interest in assisted suicide options did not follow up because of religious beliefs and concerns about possible loss of life insurance coverage for surviving family members.124
A major concern of patients with ALS is the dependence necessary for ADLs associated with late phase II and phase III of the disease. Because the process is gradual, most patients have the opportunity to make adjustments. The dependency issues and resulting privacy issues are more uncomfortable for some patients than for others, especially for the person who has always valued self-control and independence. Some patients are concerned about their increasing dependence because of the consequences of increasing burden of care on spouses or other caregivers.125 That concern for others sometimes causes patients to choose hospital, nursing home, or in-patient hospice care over home care during the terminal stage of the disease. Not all patients with terminal illness react the same way during the dying process. Throughout the process, patients and family members may cycle back and forth through a range of different emotional and coping reactions: depression, anger, hostility, bargaining, and acceptance and adaptation (order is not implied).121 How the patient coped with life’s difficulties before the illness and her or his prior relationship patterns often direct how the patient will deal with the terminal illness. In one study, patients adjusted most successfully to the changes in their functional status if they did not look back to the past and compare their losses to their future.126
Health care providers and family members often have great difficulty coping with a patient who is depressed; they may make repeated efforts to “talk the person out of” the depression. Medical professions must be able to distinguish between depression that can be destructive and the mourning or grieving that is a necessary and vital response to dealing with loss. In both states the person may feel a level of withdrawal, sadness, apathy, loss of interest in activities, and cognitive distortions. In a depressive state, however, the patient experiences an accompanying loss of self-esteem. A person in mourning rarely experiences that loss of self-esteem essential to a diagnosis of depression. The grieving person’s feelings are congruent with the degree of loss experienced.127 A person who grieves for what is lost but who has adapted to the prognosis may make plans for the impending death. Such behaviors are positive coping strategies. However, depressive symptoms related to hopelessness, uncontrolled suffering, and perceived burden on caregivers are more related to a choice for treatment discontinuance of feeding or ventilatory support.124
The issue of depression is complicated by the pseudobulbar effect of emotional lability (inappropriate laughing and crying), which is manifested by approximately 50% of patients with ALS. This emotional lability is not under complete control of the patient and is often misunderstood by family members and caregivers. Although current treatment is antidepressant medications, underlying clinical depression may or may not be present that would respond to higher doses of antidepressant medication and counseling.120
Caregiver issues
The entire family is affected by the sick person’s increasing dependency and impending death. In a small study of 11 family caregivers, many caregivers felt frustrated and resentful because their lives were consumed with the caregiving responsibilities. Most caregivers had adjusted to some degree after 2 to 4 years. Caregivers who adjusted most successfully learned to take time for themselves without guilt and to tap their social support systems for help.126,128 Similarly, 40 caregivers of young adults with severe disabilities reported being overwhelmed by the physical requirements of daily care and felt a severe loss of spontaneity in their lives.129 They also reported a sense of isolation from everyday social interactions. Although they highly valued their social support systems, they expressed frustration that few people offered instrumental or direct service support, such as respite care or help with medical appointments, housekeeping, or shopping. Despite the stresses of caregiving, the caregivers felt positive about their roles in helping the dependent adult by finding meaning in their acts of caregiving.129 Fortunately, most families manage to cope with the process—the major contributing factor being the coping ability of families before the illness. To be really effective, the therapist working with the patient with ALS must be prepared to help families and caregivers find appropriate ways of coping with the emotional, social, and physical stress of caregiving. For example, therapists should present, without pressing, adaptive equipment options to patients when they first start to show impairment in functional ability. If shown how the equipment will help them maintain independence, most patients are receptive to its use. Even when presented in a positive way, however, a wheelchair or adaptive devices may be resisted long after the adaptations would facilitate mobility and ADLs. Therapists must be attentive to patients’ feelings and fears at this time because use of a wheelchair heralds to many patients the beginning of the end.
Other factors that affect the family of a patient with ALS include medical insurance and differing levels of long-term care coverage. Some families are fortunate to have excellent coverage that provides extensive home nursing support, whereas other families are unable to cope with the financial stresses and must accept public assistance during the final stages of the disease. As opposed to Germany and Japan, which provide long-term nursing care insurance, in the United States financial stress on patients with ALS can reach more than $150,000 per year for ventilation support at home.63 Financial burden significantly impacts patient and caregiver decisions. (See Case Study 17-1 and end-of-life issues resources at www.nlm.nih.gov/medlineplus/endoflifeissues.html#cat1.)
Guillain-barré syndrome
Pathology and medical diagnosis
In the past 15 years a broad spectrum of inflammatory demyelinating polyradiculoneuropathies has been identified. GBS, or acute inflammatory demyelinating immune-mediated polyneuropathy, is the most common form of the disease. GBS affects nerve roots and peripheral nerves, leading to motor neuropathy and flaccid paralysis with possible sensory and ANS effects.130 Purely motor forms and mixed motor and sensory forms of GBS have been identified.131 Unlike ALS, GBS usually has a good prognosis, with most patients returning to their prior functional status by 1 year after onset.
The incidence of GBS is approximately one to four cases per 100,000 persons. A variant form is acute motor axonal neuropathy, which, like GBS, has a good prognosis. Less common forms are acute motor and sensory axonal neuropathy, which has a less positive prognosis (and which some consider to be a distinct type of peripheral neuropathy); Miller-Fisher syndrome, with primarily cranial nerve symptoms, ataxia, and areflexia132; and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), which causes progressive or relapsing and remitting numbness and weakness.133 Epidemiological studies show that males are affected by GBS twice as often as are females.134
Approximately 27% of patients with GBS have no identified preceding illness; however, more than two thirds had symptoms of an infectious disease 2 weeks before the onset of GBS symptoms. Although no consistent predisposing factors are known, evidence exists to support connections with Campylobacter jejuni, Mycoplasma pneumoniae, cytomegalovirus, and Epstein-Barr virus. In GBS the spinal roots and peripheral nerves are infiltrated with macrophages and T lymphocytes. Macrophages then attack and strip the myelin sheaths. In milder cases of GBS the axons are left intact and the nerves are remyelinated, typically in a matter of weeks.135 However, in some cases, the axons also degenerate, with recovery dependent on axonal regeneration from intact elements, which takes months and may be incomplete.135 In acute axonal motor neuropathy, macrophages invade the axon directly, leaving the myelin intact.136 Some evidence exists in a substantial number of patients with GBS that axonal loss is related to long-lasting or permanent muscle weakness.137,138
Because of damage to the myelin sheath, saltatory propagation of the action potential is disturbed, resulting in slowed conduction velocity, dyssynchrony of conduction, disturbed conduction of higher frequency impulses, or complete conduction block.139 Partial conduction block is most often seen in the early stages of GBS, and the conduction block increases as the patient reaches a plateau. The most common conduction block findings are in the peroneal nerve, followed by the tibial nerve. Proximal conduction block is evident more often than distal conduction block. In axonal neuropathy, conduction block is more severe, and the number of functional motor units is decreased (Figure 17-4).140 The diagnostic criteria for GBS are detailed in Box 17-3.
BOX 17-3  COMMON DIAGNOSTIC FEATURES OF GUILLAIN-BARRÉ SYNDROME
COMMON DIAGNOSTIC FEATURES OF GUILLAIN-BARRÉ SYNDROME
B Mild sensory symptoms or signs, particularly paresthesias and hypesthesias
C Autonomic dysfunction such as tachycardia and arrhythmias, vasomotor symptoms
D Absence of fever at onset of symptoms; history of flulike illness common
E Laboratory test results nonspecific but may show elevation of cerebrospinal fluid protein; cerebrospinal fluid cells at 10 or fewer mononuclear leukocytes per cubic millimeter of cerebrospinal fluid
F Electrodiagnostic testing, nerve conduction velocities usually abnormal
G Recovery usually begins 2 to 4 weeks after plateau of disease process
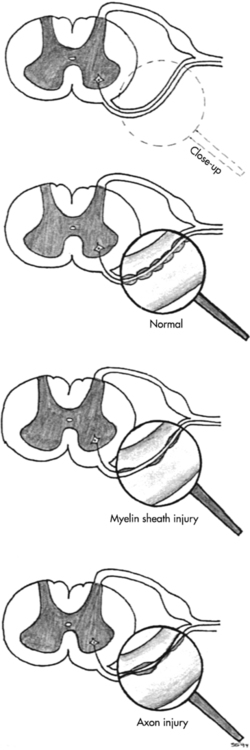

Clinical presentation
GBS in both children and adults is characterized by a rapidly evolving, relatively symmetrical ascending weakness or flaccid paralysis. Motor impairment may vary from mild weakness of distal lower-extremity musculature to total paralysis of the peripheral, axial, facial, and extraocular musculature. Severe fatigue is present in 38% to 86% of patients with GBS, depending on the cutoff point used to define severity and the age of the sample, with a positive correlation between severe fatigue and age.141 Tendon reflexes are usually diminished or absent. Twenty percent to 38% of patients may require assisted ventilation because of paralysis or weakness of the intercostal and diaphragm musculature.142,143 Impaired respiratory muscle strength may lead to an inability to cough or handle secretions and to decreased vital capacity, tidal volume, and oxygen saturation. Secondary complications such as infections or organ system failure lead to death in approximately 5% of patients with GBS.144 Approximately 35% to 50% of patients develop some cranial nerve involvement, primarily facial muscle weakness, although patients may also develop oropharyngeal and oculomotor involvement.143,145
ANS symptoms are noted in approximately 50% of patients. Low cardiac output, cardiac dysrhythmias, and marked fluctuations in blood pressure may compromise management of respiratory function and can lead to sudden death. Other typical ANS symptoms may result in peripheral pooling of blood, poor venous return, ileus, and urinary retention.146
Sensory symptoms such as distal hyperesthesias, paresthesias (tingling, burning), numbness, and decreased vibratory or position sense are common. The sensory disturbances often have a stocking-and-glove pattern rather than the dermatomal distribution of loss. Although the sensory problems are seldom disabling, they can be disconcerting and upsetting to patients, especially during the acute stage.147,148
Pain was identified as a significant presenting symptom reported in the original articles describing GBS. When pain was prominent, patients spontaneously revealed its presence during a medical history. Therefore therapists who may be working with patients with an onset of low back pain not associated with known injury or stress and reports of paresthesias (pins and needles) and vibratory or decreased tendon reflexes should evaluate or monitor for possible GBS.149,150
The most common description of presenting pain was of muscle aching typically associated with vigorous or excessive exercise. Pain was usually symmetrical and reported most frequently in the large-bulk muscles such as the gluteals, quadriceps, and hamstrings and less often in the lower leg and upper-extremity muscles. Some pain reported during late stages of the illness was described as “stiffness.” Pain was consistently more disturbing at night.150 As the disease progresses, some patients experience severe burning or hypersensitivity to touch or even air movement, which can interfere with nursing care and limit therapy interventions. The types of pain reported include paresthesias, dysesthesias, axial and radicular pain, joint pain, and myalgias.151 Dysautonomia (orthostatic hypotension, blood pressure instability, cardiac arrhythmias and sometimes bowel and bladder dysfunction) is relatively common in patients with GBS requiring ventilatory support; in one prospective study of 297 patients, cardiac arrest associated with dysautonomia was the leading cause of death.137 In patients with paraplegia or quadriplegia, approximately one fourth had problems with urinary retention caused by detrusor areflexia or overactivity, overactive urethral sphincter, and disturbed bladder sensation.134 The possibility of deep vein thrombosis (DVT) and pulmonary embolus must also be monitored and prophylactic treatment used.152
Medical prognosis
Although some patients have a fulminating course of progress with maximal paralysis within 1 to 2 days of onset, 50% of patients reach the nadir (the point of greatest severity) of the disease within 1 week, 70% by 2 weeks, and 80% by 3 weeks.145 In some cases the process of increasing weakness continues for 1 to 2 months. Onset of recovery is varied, with most patients showing gradual recovery of muscle strength 2 to 4 weeks after progression has stopped or the condition has plateaued. Although 50% of the patients may show minor neurological deficits (e.g., diminished or absent tendon reflexes) and 15% may show persistent residual deficits in function, approximately 80% become ambulatory within 6 months of onset of symptoms. The most common long-term deficits are weakness of the anterior tibial musculature and, less often, weakness of the foot and hand intrinsics, quadriceps, and gluteal musculature. Three percent to 5% of patients die of secondary cardiac, respiratory, or other systemic organ failure.134,151 Fatigue or poor endurance was also noted as a long-term consequence of GBS, possibly attributable to deconditioning and peripheral fatigue related to muscle fatigue during the healing process.141,153 Vasjar and colleagues154 also report that fatigue and poor exercise tolerance were common persisting symptoms in children who appeared to have fully recovered from acute GBS.
Although often not the focus of most studies on the long-term impact of GBS, sensory deficits (impaired response to pinprick, light touch, and vibration and proprioception in combination with other sensory losses) are an ongoing problem for patients 3 to 6 years after recovery from acute GBS. In a study of 122 subjects, 38% showed sensory deficits in the upper extremities155 and 66% had ongoing sensory deficits of the lower extremities.151 The muscle aches and cramps experienced by some of these patients appeared to be related to sensory rather than persistent motor dysfunctions as usually thought.
Overall, factors associated with a poor prognosis are severity of muscle weakness (especially quadriplegia), the need for respiratory support, cranial nerve involvement associated with loss of eye movement and swallowing, rapid rate of progression from onset, length of time to nadir, older age at onset, history of gastrointestinal illness, and recent cytomegalovirus infections.134,142 In a prospective study of 297 patients with GBS in Italy, disease severity was not associated with time to clinical recovery, but it did predict ultimate outcome, along with shorter length of time to nadir, older age at onset, evidence of axon damage, and recent gastroenteritis.137
Medical management
Medical treatment depends on the rate and degree of ascending paralysis. Because most patients return to their prior functional status, excellent supportive care during the acute stage is imperative. Respiratory compromise should be expected, and all patients, including those with limited paralysis and sensory dysfunction, must be closely monitored for the rapid onset of pulmonary and cardiac decompensation or cardiac arrhythmias, paroxysmal or orthostatic hypotension, urinary retention, and paralytic ileus caused by dysautonomia.152 Because of the possibility of sudden respiratory failure, patients with evidence of GBS must be hospitalized so that immediate cardiorespiratory support can be given if functional vital capacity (FVC) falls below 20 mL/kg or oxygen saturation falls below 75%.144 Patients who progress to respiratory paralysis must be treated in an intensive care environment where adequate respiratory function can be maintained, secondary infections can be prevented or limited, and metabolic functions can be carefully monitored. The patient should be intubated if the FVC falls below 12 mL/kg or if the patient is increasingly dyspneic even if FVC is above the cutoff level.145,156 Twenty-five percent of patients who experience respiratory failure will develop pneumonia.151 Even if daytime respiration seems adequate, night-time respiratory insufficiency (sleep-disordered breathing) should be ruled out if patients have persistent sleepiness or fatigue.141
Patients with GBS in the intensive care unit (ICU) on ventilation and with varying levels of paralysis and sensory dysfunction feel trapped and out of control because they cannot express their needs. These patients can usually hear well and most can see what is happening around them. They benefit from being oriented to time, having the personnel explain all procedures, and having some means of obtaining help. Therapists can work with the ICU staff to provide the patient with alternative forms of communication, such as eye blink, clicking, and communication boards designed for their needs. Having some form of communication and knowing that they will not be left alone will help prevent traumatic stress reactions.152
In addition to the intensive monitoring of progression and supportive care required for patients with GBS, two specific immunotherapy-based treatments—plasma exchange (removal of plasma from withdrawn blood with retransfusion of the formed elements back into the blood) and intravenous immunoglobulin (IVIg) (taking blood from a vein, separating plasma, and returning the blood cells with a plasma substitute)—have been under investigation for their ability to decrease the duration of respirator dependence and the time to onset of improvement. Systematic reviews of these interventions as of 2010 have found that plasma exchange decreases recovery time and is most beneficial if begun within the first week of diagnosis and can be beneficial up to 30 days after diagnosis.157 Plasma exchange is also cost-effective as used in patients with mild, moderate, or severe courses of GBS.158 IVIg is somewhat safer and easier to administer than plasma exchange; IVIg speeds recovery by the same amount of time as plasma exchange and is more effective than supportive care only. Adding IVIg to plasma exchange did not improve time to recovery any more than either treatment alone.159 High-quality evidence is available to support IVIg use in adults with GBS; the quality of evidence is slightly less high to support its use in children with GBS.160
Although corticosteroids have been used to decrease the inflammatory process in GBS since the 1960s, a review of clinical studies of corticosteroid effectiveness showed that corticosteroid treatment alone does not hasten recovery from GBS.161 Hughes and colleagues have developed practice parameters associated with these findings.162
Therapeutic management of movement dysfunction associated with guillain-barré syndrome
Therapeutic management of the movement deficits associated with GBS includes supportive management during the acute phase, prevention of long-term medical comorbidities during the acute through early recovery stages, and rehabilitation throughout recovery.163 With the assumption that the patient will have significant return of function within months, therapists must help maintain the integrity of functioning systems, address pain, teach compensatory strategies, and appropriately promote increasing activity after the plateau. The immediate needs of the patient will change as the patient moves through the acute stage, the plateau at the nadir, and the recovery stage of GBS before and after muscles attain antigravity strength. Transitioning between the changes in immediate therapeutic goals necessitates careful examination of the current status, progression of disease, and needs of the patient.
Examination
A comprehensive examination of the patient’s movement and function includes factors shown in Box 17-4. The extent of the examination in any one session depends on the patient’s condition and ability to participate. History taking should include the course of the disease, along with any recent illness, preexisting neuromotor or other medical conditions, current concerns, and the patient’s immediate goals. Screening tests can help determine whether sensory and autonomic systems are involved along with motor systems. Checking vital signs at rest and immediately after activity, assessing skin integrity especially in immobile patients, screening cranial nerve performance, and noting communication ability are all important components. Additional testing of sensation (and documentation on a body chart, for example) or autonomic systems may be required if the screening tests indicate.
BOX 17-4  FACTORS TO CONSIDER IN THE EXAMINATION OF PATIENTS WITH GUILLAIN-BARRÉ SYNDROME
FACTORS TO CONSIDER IN THE EXAMINATION OF PATIENTS WITH GUILLAIN-BARRÉ SYNDROME
Patterns and sequence of symptom onset
Recent illness or injury, prior episodes of sensorimotor problems
Visual inspection to identify symmetry of muscle bulk and function
Myotatic reflexes, rule out tonic reflexes
Manual muscle testing, carefully identifying pattern of weakness (testing should be as muscle specific as possible rather than assessing muscle groups only; use form for serial recording)
Presence of muscle fasciculations
Range of motion (use form for serial recording)
Equilibrium reactions sitting and standing (if testable)
Current functional status (activities of daily living, including bowel and bladder function, ambulation)
Identify pattern of sensory loss or changes (use body chart)
Identify specific type of sensory change (e.g., paresthesias, anesthesia, hypesthesias) (use body chart)
Identify pain type and location (use body chart): what makes it better, what makes it worse?
Identify pressure points or areas that might lead to pressure sores
Identify patient and family concerns in acute circumstances and concerns about long-term issues that may affect patient and family. Assessment need not be extensive if referral can be made for social service evaluation of patient and family financial concerns, day-to-day living problems (e.g., transportation, child care), support systems, and coping strategies.
Fatigue and respiratory difficulties may also preclude complete strength assessment in a single session. Fatigue may result from deconditioning, increased effort required to perform similarly with weakened muscles, and inability to recruit sufficient motor units to maintain contractions.164 Fatigue can be documented in relation to amount of activity tolerated (with specific symptoms noted before rest is required) or with a questionnaire such as the Fatigue Severity Scale (FSS), Fatigue Impact Scale (FIS), or the Visual Analogue Scale for Fatigue (VAS-F).141 Functional tests may include standardized scales of independence in ADLs or balance, tests of manual dexterity, and temporal measures of gait. Chehebar and colleagues163 review some of the pros and cons of standardized tests such as the Barthel Index, modified Hughes scale of GBS disability, and the Functional Independence Measure. Health-related quality-of-life measures used in related populations include the Nottingham Health Profile and the SF-36.164 Forsberg and colleagues165 provide a comprehensive list of tests they administered in a prospective study of 42 patients followed for 2 years after the onset of GBS. At 2 weeks postonset, 40 of 42 patients had submaximal scores on total muscle strength, grip strength, balance, and gait speed testing. At 2 months, total muscle strength was still most affected, whereas 25% of the patients had regained maximal grip strength, balance, and gait speed (designated as 1.4 to 1.5 m/sec). By 2 years, over half of the subjects still lacked the maximum total muscle score, and 40% claimed fatigue. Sensory deficits were claimed by up to 36% of patients at 2 years.165
Changes in the patient’s condition should be monitored with serial MMT, ROM assessments, sensory testing, and functional status examinations. See Karni and colleagues166 for suggestions on serial functional assessments. Before the patient is discharged from the hospital or rehabilitation unit, therapists should complete an assessment of the patient’s home environment so that appropriate safety and adaptive equipment can be in place in time for the patient’s return home.
Respiratory and dysphagia examination
Therapists are usually involved early in the care of patients with GBS. For patients with respiratory or bulbar paralysis, the therapist’s initial contact may be in the ICU. Although most hospitals have fully equipped ICUs, a therapist working in a rural or smaller community hospital may be the first person to note a patient’s changing respiratory status during an evaluation and treatment session for muscle weakness or back pain. Therefore the therapist must be prepared to advise nursing and medical staff about the need to test oxygen saturation levels and FVC. Therapist attention to respiratory complications is particularly important in the managed care environment, which discourages hospitalization if presenting symptoms are not life endangering.145 A simple estimate of FVC can be done at bedside. If after taking a large breath the patient can count out loud only to 10, the forced vital capacity is approximately 1 L and intubation should be considered. Complete information on the PT’s evaluation of patients in acute respiratory failure is provided by Irwin and Tecklin.167
Patients who have been intubated or who have cranial nerve involvement with oral motor weakness commonly have a high incidence of aspiration. Patients with severe oral-motor problems and dysphagia should be evaluated thoroughly and treated by a therapist skilled in oral-motor dysfunction and feeding. This may be a speech therapist, OT, or PT depending on the facility. Patients with a feeding tube (PEG) should receive their feedings in a relatively upright position and should remain in that position for 30 to 60 minutes after feeding to decrease the chance of aspiration. According to Logemann,168 approximately 40% of patients receiving bedside swallowing assessments have undetected aspiration. Therefore the bedside evaluation should be considered only a preliminary step in the diagnostic process. In addition to careful assessment of oral-motor control, some clinicians recommend cervical auscultation to listen to swallowing sounds, particularly during the acute phase of the illness.
With evidence of swallowing difficulties and possible aspiration, the patient should be referred for comprehensive testing with videofluoroscopy. Swallowing also can be assessed by techniques such as fiberoptic endoscopy, ultrasound, electroglottography to determine laryngeal movement, and scintigraphy, which involves scanning a radioactive bolus during swallowing.169 (Refer to section on medical management of ALS for suggestions for dealing with dysphagia.)
Intervention goals
 Facilitate resolution of respiratory problems and dysphagia
Facilitate resolution of respiratory problems and dysphagia

 Prevent contractures, decubitus ulcers, and injury to weakened or denervated muscles
Prevent contractures, decubitus ulcers, and injury to weakened or denervated muscles
 Introduce a graduated program of active exercise while monitoring overuse and fatigue
Introduce a graduated program of active exercise while monitoring overuse and fatigue

Therapeutic interventions
In a Cochrane review of exercise in people with peripheral neuropathies, no randomized or quasi-randomized controlled trials were identified for patients with GBS as of September 2009.135 However, some treatment programs used for patients with other neuromotor dysfunctions can be adapted for use with patients with GBS.
Respiratory and cranial nerve dysfunction
Depending on the facility, PTs may be involved in the respiratory care of patients with GBS. PTs may conduct chest percussion, breathing exercises, resistive inspiratory training, or strict protocols to prevent overfatigue of respiratory muscles while weaning patients from mechanical ventilation.170 Goals of treatment are related to increasing ventilation or oxygenation, decreasing oxygen consumption, controlling secretions, and improving exercise tolerance. See Irwin and Tecklin167 for coverage of treatment programs and techniques appropriate for the GBS patient with acute or residual respiratory dysfunction.
When patients are placed on mechanical ventilators, communication can be difficult and frustrating.171 The rehabilitation team can help develop and execute alternative means of communication.
In the more severe cases of GBS, cranial nerve involvement can lead to multiple complications such as dysphagia and vocal cord paralysis. In many facilities, speech pathologists or OTs are responsible for establishing a dysphagia treatment program. Therapists responsible for treatment of patients with dysphagia and swallowing problems should refer to Logemann’s classic text on the evaluation and treatment of swallowing disorders.168 Therapeutic goals are the prevention of choking and aspiration and the stimulation of effective swallowing and eating. The act of chewing and swallowing is complex and requires coordinated reflexive and conscious action. Intervention is focused on positioning (upright with head tilted slightly forward),171 head control, and oral-motor coordination (e.g., sucking an ice cube, stimulating the gag response, facilitating swallowing with quick pressure on the neck and thyroid notch timed with intent to swallow). A conscious swallowing technique is introduced with thick liquids and progressed to thinner liquids after the patient’s oral-motor coordination response is enough to control movement of fluids. Once the patient has good lip closure, fluids should be introduced one sip at a time from a straw cut to a short length to minimize effort. Semisoft, moist foods are gradually introduced (pasta, mashed potatoes, squash, gelatin). Any crumbly or stringy foods (coffee cakes, cookies, snack chips, celery, cheeses) should be avoided, and the patient should not attempt to talk or be interrupted during eating until choking does not occur and swallowing is comfortable and consistent.172 Feeding training should occur during frequent, short sessions to prevent fatigue. Therapists should be prepared to use the Heimlich maneuver if choking occurs or have a suction machine available at bedside.
Pain
If pain seems to be a major factor limiting the patient’s passive or active motion, the treatment team should determine the best approach to alleviate pain. According to one study, patients with GBS did not seem to show a consistent response to any specific pain medication, although six of the 13 patients seemed to have a positive response to codeine, oxycodone plus acetaminophen (Percocet), and oxycodone plus aspirin (Percodan).147 Some patients may find relief with medications used to treat neurogenic pain, such as the tricyclic antidepressants, carbamazepine, or gabapentin (anticonvulsants).151 For patients who do not respond to conventional analgesics or tricyclic antidepressants, a short course of high-dose corticosteroids can lead to pain relief.144
Some patients with neuropathy have noted decreased pain after using transcutaneous electrical nerve stimulation (TENS).173,174 Although no study has examined the effect of TENS specifically on pain associated with GBS, it might be a treatment option to help with desensitization in patients whose pain is not controlled with passive movement or pain medications.
Another option is capsaicin, the active ingredient in chili peppers, which when applied topically interacts with the sensory neurons to relieve pain from peripheral neuropathies.151 Therapists, wearing gloves, apply a topical anesthetic until the area is numb. The capsaicin is then applied topically. The capsaicin remains on the skin until the patient starts to feel the heat, at which point it is promptly removed. Because the nerves are overstimulated by the burning sensation, the sensory gateway is unable to report pain for an extended period.175
Some patients who experience extreme sensitivity to light touch, such as from movement of sheets, air flow, and intermittent touch contact, benefit from a “cradle” that holds sheets away from the body. Some find relief if the limbs are wrapped snuggly with elastic bandages, which provide continuous low pressure while warding off light and intermittent stimuli. Alternatively, the patient’s pain response can be desensitized through methodical stimulation with frequent, consistent stimuli to the affected area for short durations to allow acclimatization.170
Contractures, decubitus ulcers, and injury to weakened or denervated muscles
Positioning.
In the acute stage of GBS, rehabilitation will focus on positioning and passive ROM to prevent contractures and decubitus ulcers.176 Preventing pressure sores starts within the first few days of hospitalization, especially for the patient who has complete or nearly complete paralysis. A positioning program for the dependent patient is the first line of defense, with turning at least every 2 hours for both pressure relief and lung drainage.171 In addition, the patient should have a special mattress or unit that constantly changes the pressure within the mattress to shift the patient’s position or is designed to spread pressure over wide surfaces. Patients who are slender or who have lost significant muscle mass from GBS-induced atrophy will have prominent bony surfaces; the therapist may need to fashion foam “doughnuts” or pads or use sheepskin-type protection for pressure relief. Patients who have muscle pain may prefer to have their hips and knees flexed. If so, the patient must be taken out of the flexed position for part of each hour to avoid muscle shortening.
As part of a complete positioning program, therapists should consider how best to maintain the physiological position of the hands and feet. Research has shown that mild continuous stretch maintained for at least 20 minutes is more beneficial than stronger, brief stretching exercises.177 Thus the use of splints for prolonged positioning is superior to the use of short bursts of intermittent, manually applied passive stretching for maintaining functional range. Although some facilities still use a footboard to control passive ankle plantarflexion, most therapists now use moldable plastic splints that can be worn when the patient is in any position. Because ankle-foot splints often prevent visual inspection of the heel position, care must be taken to ensure that the heel is firmly down in the orthosis and that the strapping pattern is adequate to secure the foot. The strap system must be simple enough to be positioned properly by all staff and family members caring for the patient. The ankle-foot splint should extend slightly beyond the end of the toes to prevent toe flexion and skin breakdown from the toes rubbing on sheeting. Care should be taken not to compress the peroneal nerve with the splint as it crosses the fibula,148 a particularly vulnerable area after the loss of muscle mass in the lower legs from the GBS.139 Wrist and hand splints may be prefabricated, resting-style splints, or molded to meet the patient’s specific needs. Because spasticity is not a problem in the patient with GBS, a simple cone or rolled cloth may be adequate to maintain good wrist, thumb, and finger alignment for short-term immobility.
Range of motion.
ROM can usually be maintained with standard positioning and ROM programs. Nevertheless, some patients, especially those who have reported severe extremity and axial pain early during the disease process and those who have been quadriplegic and respirator dependent for prolonged periods, may develop significant joint contractures despite preventive interventions. As with patients with spinal cord or severe head injuries, heterotopic ossification has been reported in patients with GBS.178 Meythaler and colleagues179 note that early mobilization was related to therapeutic decreases in serum calcium levels and suggest that aggressive ROM (but not hard or abrupt movements that may injure the muscle) may impede the effects of heterotopic bone overgrowth, which can have a severe impact on ROM. Once heterotopic ossification has been identified, treatment includes modification of ROM exercise to use only active and passive motion within the pain-free arc.170
Soryal and colleagues180 reported on three patients with GBS who had marked residual contractures that limited function after strength improved. None of the patients had radiological signs of erosive arthropathy or inflammatory joint disease. Soryal hypothesized a number of possible mechanisms for the limitations in ROM: (1) therapists and nurses may have been reluctant to take patients who reported marked pain during passive movement through the full ROM; (2) the contractures may have been a result of pain or damage caused by inappropriate excessive passive movement of hypotonic and sensory-impaired joints and muscles (often caused by poor movement of the patient in bed or by poorly trained staff or family members moving limbs); (3) the paralysis may have resulted in lymphatic stasis with accumulation of fluid in tissue spaces and nutritional disturbances; and (4) vasomotor disturbances resulting from autonomic neuropathy may have led to adhesions and fibrosis. Although the authors found few reports describing contractures as a significant residual problem, they suggested that ROM programs must be defined precisely as to frequency and duration, particularly for patients reporting early joint pain.180
Some patients will prefer to position their limbs so muscle and tendons are in the shortened range in an attempt to decrease muscle pain. This may lead to capsular contractures. The therapist should be aware of changes in “end feel” over time when testing ROM of each joint to determine if capsular and ligamentous structures are also becoming more restricted as the muscle and tendon tissue shortens. Patients who have intact sensation of pain and temperature may respond positively to the use of heat (up to approximately 45° C or 113° F) before stretching to decrease muscle pain and facilitate tissue elongation before stretching. Several basic studies of rat tail tendon and the relation between load and heat have shown that attaining permanent length increases in collagenous tissue is possible with a combination of heat and stretch.181–184 (Caution: Heat should not be used on a patient with a sensory deficit that inhibits ability to distinguish differences in temperature.)
On the basis of evidence that continuous passive motion (CPM) is effective in maintaining joint range in both rabbits and human beings,185 Mays186 described a case study of a patient with GBS (quadriplegia with 7 days of mechanical ventilation) who had persistent pain and stiffness of the upper extremities and fingers approximately 3 months after the onset of GBS. CPM of the hands and fingers was added to a program of occupational therapy that included ROM, splinting, and ADLs. The author reported an increase in the rate of recovery of finger range and a decrease in pain after use of CPM. Numerous other studies have reported the value of CPM in maintaining or increasing ROM after hip and knee surgery. It may be a useful adjunct to traditional therapy for patients with GBS, especially those who continue to develop contractures with standard, intermittent ROM programs. Patients with severe paresthesias or dysesthesias may not be able to tolerate CPM equipment.
Progressive program of active exercise while monitoring for overuse and fatigue
In the acute stage of GBS, active exercise is limited to whatever the patient can move without pain or excessive fatigue. Slings or adaptive devices may help support the weight of a limb to continue active movement in a gravity-eliminated plane for those muscles that have lost antigravity strength. As the disease reaches its nadir, activity remains limited. Once weakness stops progressing, passive maintenance of ROM may be the only activity possible for immobile patients. As strength begins to return after the plateau, therapists must prescribe limited amounts of low-resistance activities, with strict avoidance of antigravity strain on the muscles until strength reaches the 3/5 (Fair) range of MMT. Active exercise can be added very slowly, with frequent rest periods and monitoring to avoid fatigue.177,187 Activity should be halted at the first point of fatigue or muscle ache; abnormal sensations (tingling, paresthesias) that persist for prolonged periods after exercise may also indicate that the exercise or activity level was excessive. Any progression of resistance or repetitions of strengthening exercises should be monitored for 3 to 7 days for increase in weakness, muscle spasms, or soreness before exercises are progressed further.188 If additional weakness or soreness ensues, the additional activity must be eliminated for several days, with reinitiation at a lower level of resistance or number of repetitions and more gradual increase. Work simplification and energy conservation strategies may be useful to improve function in the recovery stage of GBS.170 As strength increases, additional resistance may be applied to those muscles showing good recovery while avoiding strain on muscles that have not yet reached the same level, frequently the most distal musculature. Even when strength has returned throughout, rehabilitation and exercise may need to continue to address fatigue that may persist at each of the International Classification of Functioning, Disability and Health (ICF) levels: body function and structure, activity, and participation.141 For an example of treatment progression during the acute stage from week 1 through week 12, see Table 17-3.
TABLE 17-3 
MEDICAL STATUS OF PATIENTS WITH GUILLAIN-BARRÉ SYNDROME AND POSSIBLE TREATMENT OUTLINE
| MEDICAL STATUS | TREATMENT* |
| Tracheostomy | Week 1: Postural drainage every 3 hours around the clock Passive ROM exercises to all joints Splinting (molded plastic) of hands and feet to maintain functional position Positioning, splinting, and ROM program schedule posted at bedside |
| Respirator dependent | |
| Complete cranial nerve paralysis | |
| Quadriplegia | |
| Weeks 2-5: Postural drainage decreased to two times each shift (every 8 hours) Passive ROM exercises, physiological and accessory motions, gentle stretching of intercostal musculature, trunk rotations Continue splinting and positioning program Family education: family members taught gentle physiological ROM techniques, with attention to correct shoulder patterns and simple massage techniques |
|
| Respirator set on intermittent mandatory ventilation | Weeks 6-7: Postural drainage two times each shift (every 8 hours) Continue ROM program, splinting, and positioning Begin to build tolerance of upright sitting with good trunk alignment Begin facilitation of active facial and tongue muscle activity in patterns necessary for swallowing, eating, and speaking; speech pathology, occupational therapy consultation for dysphagia training Family members active in care, helping with ROM, splinting, and positioning schedule as they choose |
| Weaning to respirator at night by end of week 7 | |
| No active muscle contractions except eye opening and lip movements | |
| Dysphagia | |
| Palpable muscle activity in neck, trunk, proximal musculature of upper and lower extremities | Weeks 8-12: Postural drainage one time each shift Chest stretching, breathing exercises Dysphagia program in collaboration with speech consultant Muscle reeducation program with electromyographic biofeedback progressing to gravity-eliminated exercises using suspension slings attached to bed Tilt-table standing program to increase tolerance to upright (wearing positioning splints if necessary) Collaborate with occupational therapist for treatment in wheelchair with suspension slings to facilitate active arm motion in gravity-limited position Exercise, rest, positioning schedule posted Family, patient educated about stimulating activity level to prevent fatigue, overuse of reinnervating muscles |
In the initial stages of upright activity after any period of bed rest, therapists must progress patients with GBS very carefully because 19% to 50% of this population show orthostatic hypotension along with dysautonomia.139,146 A program to improve tolerance to upright position can be started in the ICU if the patient is on a circle electric or Nelson standing bed. If a standing bed is not available, a sitting program can be initiated as soon as it is tolerated. A progressive standing program can be instituted when the patient’s respiratory system and ANS are no longer unstable and the patient can be moved to a tilt table. Caution should be taken to stabilize the patient fully to maintain alignment and to limit activity in muscles having strength below the fair range. When beginning training, some patients benefit from using an abdominal binder or foot-to-thigh compression stockings if tolerated. Because of the relation between poor hydration and hypotension, therapists must ensure the patient is well hydrated before beginning upright or standing tolerance programs.139
As was discussed in the section on therapeutic considerations for patients with ALS, a muscle that has significant denervation is more likely to respond to exercise with overwork fatigue (see Figure 17-3 for the therapeutic window for exercise). Studying the effect of exercise on rat muscle after nerve injury, Herbison and colleagues187 identified a loss of contractile proteins during initial reinnervation. After reinnervation the same amount of exercise resulted in muscle hypertrophy. Bensman188 reported on eight patients who had stabilized after acute polyradiculoneuritis (among them patients with GBS). All eight patients had a temporary loss of function after strenuous physical exercise. Three patients apparently had significant decreases in strength. All patients were then placed on a program of passive ROM exercises, and an increase in muscle strength was noted. Recurring episodes of a temporary loss of function appeared to be related to strenuous exercise and fatigue. The current position for patients with GBS, then, is that excessive exercise during early reinnervation when only a few functioning motor units are present can lead to further damage rather than to the expected exercise-induced hypertrophy of muscle.
During the initial stages of exercise, the repetitions per exercise period should be low and the frequency of short periods of exercise should be high.177 As reinnervation occurs and motor units become responsive, the early process of muscle reeducation exercise used by the therapist may be similar to that used after polio. To encourage active contraction of the muscle the therapist should carefully demonstrate to the patient the expected movement. The therapist then passively moves the patient’s limb while the patient observes. After gaining a clear picture of what movement is expected, the patient is encouraged to contract muscles. Facilitatory techniques such as skin stroking, brushing, vibration, icing, and tapping may be used in conjunction with the muscle reeducation process if the sensory and pain status of the patient permits. The patient is taught to reassess his or her movements and make corrective responses. As the patient gains strength, the movements are translated into functional activities.187
As reinnervation progresses and strength and exercise tolerance increases, the therapist may choose to use facilitative exercise techniques such as neurodevelopmental sequencing189 or PNF190,191 to recruit maximal desired contraction of specific muscle groups. Although PNF techniques are excellent for eliciting maximal contraction, care must be taken not to overwork the weaker components of the movement pattern. A positive aspect of PNF techniques is that they can be tied in with functional patterns such as rolling, which is necessary for bed mobility, transitions to quadruped, kneeling, sitting, standing, and gait.
Because patients with GBS are transferred from acute care facilities to rehabilitation, skilled nursing, or home environments more quickly than in the past, therapists must be careful to document any serial negative changes or plateaus in motor, sensory, or respiratory impairments or functional status that may herald a relapse.139 Although 65% to 75% or more of patients with GBS show a return to clinically normal motor function, 2% to 5% of patients have a recurrence of symptoms similar in onset and pattern to the original illness.192 Recurrence of symptoms should trigger immediate cessation of activity and possibly medical reassessment in case of respiratory insufficiency.
Anecdotal and empirical evidence shows that patients with GBS can continue to show deficits during strenuous exercises that require maximal endurance. Four soldiers who were considered clinically recovered from GBS (normal motor power with or without reappearance of reflexes and the absence of sensory impairment) were unable to pass the Army Physical Fitness Test (APFT), which is designed to measure a minimal acceptable age-related level of physical fitness for military duty (maximal effort to challenge respiratory and muscular endurance, strength, and flexibility). Before onset of GBS, the four patients had all exceeded the APFT standards. None was able to pass the APFT as long as 4 years after the illness, indicating that the persistent deficit interfered with their ability to continue their military careers.193 The possibility of long-term endurance deficits should be considered when patients appear to have reached full recovery but report difficulty when returning to work or activities that require sustained maximal effort.194,195
So far, no pharmaceutical agents have been helpful in alleviating fatigue in this population. In a study of the use of amantadine to relieve severe fatigue in 74 patients with GBS randomly allocated to treatment or placebo groups, the groups showed no difference in any of the primary or secondary measures recorded.196 Determining the effectiveness of interventions to affect fatigue may be complicated by differences in measures of experienced fatigue (subjectively reported) versus physiological fatigue (central or peripheral reduction in voluntary muscle force production) and the weak relationship between these in many neuromuscular disorders.197
Cardiovascular fitness may also be compromised after recovery from GBS. This may be caused by altered muscle function, but it is also related to deconditioning from an imposed sedentary lifestyle.154 Several studies have reported the effect of endurance exercise training after GBS. In one case study a 23-year-old woman with a chronic-relapsing form of GBS with onset at age 15 years was placed on a walking and cycling program at 45% or less of her predicted maximal heart rate reserve. The low-intensity exercise program was selected to prevent possible fatigue-related relapse. After the program, the subject had improved her walking time 37%, walking distance approximately 88%, and cycle ride time more than 100%. Although no standardized or formalized recording of functional level was recorded before and after the exercise program, the patient reported that her energy level for ADLs was a “little higher” and that stair walking was easier.194 In another single-subject study of a 54-year-old man 3 years after onset of GBS with residual weakness, the authors demonstrated similar improvements in cardiopulmonary and work capacities as well as leg strength after a 16-week course of a thrice-weekly aerobic exercise program. The subject also reported expanded ADL capabilities. The authors suggested that their training regimen may disrupt the cycle of inactivity after recovery from GBS that leads to disuse atrophy and further deconditioning in patients with mild residual weakness.198 Fehlings and colleagues199 tested muscle strength and endurance in a group of children at least 2 years after acute onset of GBS. Although the children appeared essentially recovered, endurance of the arm muscles was lower than that of the lower extremities. They hypothesize that the typical walking, running, and cycling activities that the children participated in were sufficient to improve strength and endurance of lower-extremity muscles, and they recommended that children be encouraged to participate in activities such as swimming to improve upper-extremity endurance. Controlled tetherball and volleyball activities are also appropriate. Tuckey and Greenwood200 reported positive results of treatment with partial body-weight support (PBWS) treadmill exercise for a patient with severe GBS. Garssen and colleagues201 reported a 20% reduction in fatigue levels, along with improved physical condition and strength, after a 12-week intensive bicycling exercise program for patients several years after the onset of GBS.
Improvements in strength and endurance after GBS may continue for months to years. A prospective study following 6 patients for 18 months after onset of GBS recorded continuing improvement of muscle strength on average throughout the assessment period, and yet the average strength of major muscle groups had not yet reached that of healthy controls.202 Although the traditional thought has been that little clinical improvement occurs after 2 to 3 years, Bernsen and colleagues203 found that 21% of the patients in a study of 150 patients after recovery from acute GBS reported improvement after 2.5 to 6.5 years, although the authors thought the perception of improvement was related to improved sensory function. Of future research and clinical interest are the long-term consequences of GBS and how the normal aging process will affect patients who have some mild residual effects—for example, whether some patients will develop increasing weakness over time similar to persons with postpolio syndrome.139
For those patients who experience significant losses in proprioception after GBS, sensory reintegration activities and high repetitions of task practice may help to redevelop motor engrams that are based on the altered sensory perception.139
Patients with GBS have a significantly reduced health-related quality of life compared with control subjects at approximately 1 year after onset, associated with decreased functional scores and changes in work status.204 Although physical training may be expected to improve functional scores and work capabilities, Bussmann and colleagues205 found little correlation between physical fitness and other domains. They hypothesized that training has psychological components, such as positive effect on mood and self-confidence, that influence quality of life in addition to physical changes.
Adaptive equipment and orthoses
Most therapy units today have access to varied sizes of plastic, fixed-ankle AFOs that can be used until a decision is made to have the patient fitted with custom AFOs. A newer system of prefabricated AFOs with adjustable ankle motion cams has been developed that allows the therapist to limit plantar flexion and dorsiflexion to the specific needs of the patient. For patients with reasonable control of plantar flexion and dorsiflexion but with lateral instability because of peroneal weakness, a simple ankle stirrup device such as the AirCast Air-Stirrup Ankle Brace (AirCast, Summit, NJ) can be used temporarily to provide lateral ankle stability. Although few patients with GBS need knee-ankle-foot orthoses (KAFOs) on a long-term basis, inexpensive air splints or adjustable long-leg metal splints to control knee position are sometimes helpful when working on standing weight bearing and during initial gait training. See Chapter 34 for additional information on orthotics.
Psychosocial issues
Although most patients with GBS have a good recovery over a period of 2 or more years, the acute stage of the disease can be frightening, especially to patients who progress to complete paralysis and respiratory failure. Nancy, in Case Study 17-2, reported that she was terrified during the time she was totally paralyzed (including eyelid movement) and on a respirator. She said that nurses, doctors, and hospital staff seemed to assume she could not hear because she was unable to respond in any manner. In her words,

Skirrow and colleagues206 remind clinicians that the “intensive care patient is plunged into a world of machines that flash and beep; of tubes and wires that seem to spring from almost every orifice; and of mind-numbing sedative and analgesic medications.” Needless to say, evidence is increasing that patients treated in acute trauma rooms or ICUs can have posttraumatic stress disorder (PTSD). Particularly vulnerable are patients who have had previous traumatic experiences. PTSD places patients at marked risk for increased startle responses, extreme vigilance or anticipation of painful events, sleep disorders, terrifying dreams, and dissociative flashbacks after leaving the ICU; sometimes these symptoms are left untreated for years after the experience.152,207 Patients discharged from prolonged ICU experiences, especially those who had respiratory failure, have an increased incidence of anxiety, depression, and panic disorders years after discharge.
In a nursing study of patient experiences in the ICU, researchers found that patients often felt anxious, apprehensive, and fearful. The patients expected ICU nurses to be experienced and technically adept, but those who felt most secure despite the traumatic ICU experiences felt that the nurses were vigilant to their needs and offered personalized care,152,208 a point clearly made by Nancy in the case study. Although one might expect ICU staff to be carefully tuned in to patient needs, the highly technical nature of modern ICUs may attract personnel less focused on individual patient care, or it may prevent caring staff from attending to the little kindnesses that are so comforting to critically ill patients. Baxter207 suggests that caregivers in the ICU try to orient patients to what is being done, to approach the patients within their field of vision, and to minimize unexpected noises and sudden touching.
Although most patients recover well from GBS, 3 to 6 years after onset of GBS 38% of patients in a Dutch study had to make a job change to accommodate their physical status, 44% had to alter their leisure activities, and nearly 50% described ongoing psychosocial changes.203 Similar findings were reported in a study of Japanese patients recovering from GBS.209,210
In summary, the rehabilitation program for a person with GBS must be graded carefully according to the stage of illness. In the acute care environment when respiratory deficits are present, the initial emphasis is directed toward support of maximal respiratory status through postural drainage, chest stretching, and breathing exercises. Because of prolonged bed rest and immobility related to weakness, accessory and physiological ROM must be maintained with around-the-clock efforts. Splinting or positioning devices are recommended to maintain functional positions during prolonged periods of immobility. A gradual program to increase upright tolerance is begun when respiratory and autonomic functions have stabilized. Therapists must keep in mind the potential to damage denervated muscles with aggressive strengthening programs when developing a rehabilitation plan and a home-based conditioning program. Perhaps as a result of cautious exercise programs, cardiovascular conditioning appears to lag significantly behind strengthening, so endurance training should specifically follow the return of strength. Adaptive equipment and orthoses should be used as needed to protect weakened muscles, facilitate normal movement, and prevent fatigue during the reinnervation process. Although a rehabilitation program has been found to make a measurable difference in patient long-term recovery, many patients are being discharged without follow-up care.211 Therefore therapists should be assertive in ensuring that their patients with GBS have ongoing contact with rehabilitation specialists who can guide the recovery process (see Case Study 17-2).