CHAPTER 115 NEUROLOGY OF HEMATOLOGY
THE ANEMIAS
Iron deficiency from chronic blood loss is the most common form of anemia. Iron deficiency in the absence of anemia (sideropenia) may decrease the deformability of red blood cells, leading to ischemia in the distribution of small cerebral vessels. This mechanism is particularly important in the context of polycythemia, in which there are increased numbers of red blood cells, each one of which may be iron deficient. Both the polycythemia and the relative sideropenia lead to increased blood viscosity with associated neurological symptoms and signs. Iron deficiency causes a microcytic, hypochromic anemia.
The restless legs syndrome is a very common cause of insomnia. It consists of an unpleasant creeping sensation that occurs deep in the legs (and occasionally in the arms) when the person is at rest. The person feels compelled to move the legs to avoid the unpleasant feeling. Most sufferers are women, who pace the floor at night and complain of insomnia. Polysomnographic studies often reveal nocturnal myoclonus. It is likely that restless legs syndrome and nocturnal myoclonus represent various fragments of a single disorder, known as Ekbom’s syndrome.1 Many of the movement disorders associated with iron deficiency are reminiscent of those seen in basal ganglia diseases, but the precise relationship between systemic iron deficiency and these movement disorders is cryptic, although there is some reason to believe that iron is a cofactor for the enzyme tyrosine hydroxylase, which catalyzes the rate-limiting step in the biosynthesis of dopamine from tyrosine. Ekbom’s syndrome may therefore be a dopamine deficiency syndrome distinct from parkinsonism. Patients with Ekbom’s syndrome may respond to iron replacement. The rest are treated with centrally acting dopamine agonists, such as pramipexole or ropinirole. Some patients who fail to respond to a dopamine agonist derive benefit from a benzodiazepine, clonidine, or clomipramine. Patients diagnosed as having the restless legs syndrome should undergo careful evaluation for anemia, including microscopic study of the blood smear, measurements of serum iron and total iron-binding capacity, and several stool tests for occult blood. Blood and spinal fluid ferritin levels may be assayed in patients without overt evidence of iron deficiency.
Disordered DNA metabolism is clearly not confined to the blood cells, inasmuch as giant epithelial cells are found in many other organs, including the mouth, stomach, and skin. The neurological effects of the megaloblastic anemias probably result from a primary metabolic derangement in neural tissue and are clearly not directly related to the anemia per se.2 Because the blood-forming organs are particularly sensitive to the effects of cobalamin or folate deficiency, it is unusual to find the neurological effects in patients in whom no disorders of the blood are found. Anemia is, however, only one and probably a relatively late sign of cobalamin or folate deficiency, and so it is possible to find patients with the neurological effects of cobalamin or folate deficiency without anemia.3
Cobalamin (vitamin B12) deficiency may have a number of causes, including (1) defective diet (low in animal or bacterial products), (2) defective absorption (deficiency of intrinsic factor as a result of pernicious anemia, gastrectomy, or intestinal disease such as malabsorption or “blind loop” syndrome), and (3) increased metabolism (thyrotoxicoxis, pregnancy, neoplasia). Of these, the most prevalent form of cobalamin deficiency is pernicious anemia. It arises from failure of the gastric fundus to secrete adequate amounts of intrinsic factor to ensure intestinal absorption of vitamin B12. This failure of secretion of the mucoprotein intrinsic factor is caused by atrophy of the fundic glandular mucosa, a process that is usually an immune system–mediated gastritis but may be familial or senile or may result from gastric neoplasia. The presence of histamine-fast achlorhydria is a reliable method of diagnosing pernicious anemia but has often been supplanted by measurements of anti-intrinsic factor and antiparietal cell antibodies.4 Patients with autoimmune pernicious anemia often have clinical and laboratory evidence of other conditions characterized by autoimmunity, such as vitiligo and thyroiditis. Serum B12 levels have occasionally been found to be erroneously normal in documented cases, and so it is now routine to assess intracellular function by directly measuring serum homocysteine (for folate or cobalamin deficiency) and methylmalonic acid (for cobalamin deficiency).
Exposure to nitrous oxide, as in the induction of general anesthesia, may precipitate acute deterioration (anesthesia paresthetica) in patients with otherwise mild or asymptomatic cobalamin deficiency.5 The adenosylcobalamin system acts to metabolize propionic acid by converting methylmalonyl–coenzyme A to succinyl–coenzyme A. Failure of this system results in an accumulation of methylmalonic acid, which is myelinotoxic by promoting the formation of abnormal long-chain fatty acids.
Subacute combined degeneration of the spinal cord is the term used to designate the spinal cord disease caused by cobalamin deficiency.6 Patients complain of generalized weakness and paresthesias that usually begin distally in the hands. As these symptoms progress, stiffness and weakness in the limbs develop. Loss of vibration sense is the most profound sign, often joined later in the course by joint position sense loss. The Romberg sign is positive, and the gait is unsteady and awkward primarily because of proprioceptive loss (pseudotabetic gait). Weakness and spasticity are usually worse in the legs than in the arms and may progress to a spastic paraplegia if untreated. Babinski’s signs are present, but the deep tendon reflexes are variable. If a sensory level implicating the spinothalamic tracts is found on the trunk, this should always be viewed with the greatest skepticism, and the clinician should exhaustively rule out other causes of spinal cord disease. Many patients with vitamin B12 deficiency have distal symmetrical impairment of cutaneous sensation, absence of deep tendon reflexes, and even slowed nerve conduction velocities, which suggest a neuropathic component, but this is usually quite mild compared to the myelopathic illness. These manifestations may sometimes be visualized by magnetic resonance imaging and pathologically are seen as regions of spongy myelopathy, quite similar to those seen in human immunodeficiency virus–related myelopathy, raising the question of whether this virus may somehow impair the transmethylation function of cobalamin.
Optic neuropathy is the third and last major neurological complication of vitamin B12 deficiency. It is characterized by bilateral involvement of the optic nerves that results in loss of central visual acuity and depressed sensitivity, more so for color than for black and white, in the centrocecal area of the field of vision. This syndrome is clinically similar to a number of other bilateral optic neuropathy syndromes, including tobacco-alcohol amblyopia, diabetic optic neuritis, Leber’s hereditary optic atrophy, and tropical ataxic neuropathy. These syndromes may be linked to an abnormality in cyanide metabolism that results from a shortage of sulfur-donating amino acids. In the 1990s, there was an epidemic of optic neuropathy and myelopathy in Cuba, thought to be caused by multiple B-vitamin deficiency resulting from malnutrition in combination with alcohol and cyanide exposure from cigar smoking and cassava consumption. The epidemic was terminated by vitamin supplementation.7
Folic acid (folate) deficiency accounts for nearly all of the cases of megaloblastic anemia not caused by vitamin B12 deficiency. The causes of folate deficiency are (1) defective diet (low in vegetables and liver), (2) defective absorption from intestinal malabsorption as a result of sprue, steatorrhea, diverticulosis, or short circuits of the gastrointestinal tract or the “blind loop” syndrome; and (3) deranged metabolism caused by an increased requirement from hemolytic anemia, pregnancy, or neoplasia or by impairment of use as a result of liver disease or the administration of folic acid antagonists or anticonvulsants. Unlike those of vitamin B12, the bodily stores of folic acid are quite limited. A folate deficiency syndrome may commence within several months of dietary deprivation, which makes it a much more common problem among the malnourished than is vitamin B12 deficiency. Folate, once absorbed through the entire small intestine, is reduced by specific liver enzymes to tetrahydrofolic acid, a compound that plays a major role in the metabolism of one carbon fragments by its synthesis and transfer of methyl groups. Through this mechanism, folate is vital for the conversion of deoxyuridylate to thymidylate, a precursor needed for DNA synthesis. Thus, tetrahydrofolate derivations are closely linked to vitamin B12–dependent reactions, and the hematological alterations in vitamin B12 and folate deficiency are indistinguishable. Deficiencies of the two vitamins have very similar effects, and a deficiency of one may lead to faulty use of the other. Many patients with vitamin B12 deficiency have concomitant folate deficiency, but most people with the folate deficiency state, which is overwhelmingly more common, have no vitamin B12 deficiency. Folic acid deficiency is almost never pure. Because it accompanies malnutrition, it is nearly always associated with multiple vitamin deficiencies. The most common neurological manifestation of this multivitamin deficiency state is a symmetrical sensorimotor polyneuropathy. Some minor degrees of segmental demyelination may also occur, usually as a result of entrapment of metabolically weakened nerves. All the common entrapment neuropathies (e.g., carpal tunnel syndrome, meralgia paresthetica, peroneal palsy, ulnar palsy) are more frequent in patients with an underlying metabolic axonopathy such as that caused by vitamin deficiency. Folate deficiency is associated with neural tube defects, and supplements are therefore routinely prescribed during pregnancy.
THE HEMOGLOBINOPATHIES
Painful crises are among the most common clinical problems in the management of patients with sickle cell anemia. The abdominal and bone pain, so common in this disease, is probably ischemic pain related to the sickling phenomenon. The treatment for these crises consists of hydration, bed rest, and analgesia. Vascular disease is the more serious neurological aspect of this disorder and probably contributes in a major way to the decrease in life expectancy of patients with sickle cell anemia. The prevalence of overt strokes is about 20% among patients with sickle cell anemia. Most strokes are caused by small-vessel occlusions, which often result in seizures at the onset of a stroke. In some cases, progressive small-vessel occlusions with recurrent development of collateral vessels can lead to an angiographic picture similar to that seen in moyamoya disease. Hemorrhages may result from rupture of these fragile collateral vessels, leading to intracerebral, subarachnoid, spinal, and retinal hemorrhagic strokes in patients with sickle cell disease. The progressive stenosis of the supraclinoid internal carotid artery that leads to the development of the moyamoya pattern may be detected noninvasively with transcranial Doppler ultrasonography. Stroke risk is reduced dramatically through the use of prophylactic transfusions in patients in whom transcranial Doppler examinations reveal that the time-averaged mean blood velocity in the internal carotid or middle cerebral artery is 200 cm per second or higher.8 Among children younger than 15 years with strokes, sickle cell anemia is present in 7%, and thus it an important cause of stroke in childhood. Spinal cord infarction is also observed in patients with sickle cell anemia much more commonly than in the general population. Massive intracranial hemorrhage is another complication of sickle cell anemia. Large-vessel occlusions also occur in patients with the supraclinoid carotid as the site of predilection. Moyamoya disease may be treated with extracranial-intracranial arterial bypass grafting or with temporal-pial synangiosis, with the hope of reducing the likelihood of rupture of the fragile vessels of moyamoya disease.
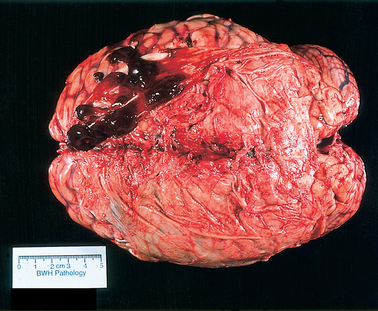
The genetic defect underlying thalassemia involves rates of synthesis of the individual polypeptide chains. Two major varieties of thalassemia exist: one involving defective α-chain synthesis, the other involving β-chain synthesis. The more common β-thalassemia may occur in the heterozygous or homozygous form to produce the syndromes of thalassemia trait or Cooley’s anemia (thalassemia major), respectively. Heterozygosity for α-thalassemia results in a very mild condition and may require an associated hemoglobin abnormality for clinical expression (thalassemia minor). Homozygous α-thalassemia is thought to be incompatible with normal fetal development. The susceptibility to infection seen in thalassemia corresponds to that seen in sickle cell anemia but is confined to patients who have undergone splenectomy for control of hemolysis. In about a third of patients with myelopathy resulting from extramedullary hematopoiesis, thalassemia is the underlying disease. The usual location for extramedullary hematopoiesis is various parts of the reticuloendothelial system, particularly the liver, spleen, and lymph nodes. However, the spinal epidural space and the intracranial subdural space (Fig. 115-1) may be involved, with consequent compression of the spinal cord or the brain or both. Most cases involving the spine occur in the thoracic segments posteriorly, usually over multiple levels. Treatment with radiotherapy is usually quite effective.
MYELOPROLIFERATIVE DISORDERS
The myeloproliferative disorders include polycythemia rubra vera, myelofibrosis with myeloid metaplasia, chronic myelogenous leukemia, and essential thrombocythemia. The proliferation in all of these diseases originates in the bone marrow, liver, or spleen, in which extramedullary blood formation may occur. Strokes are the most common neurological complication seen in polycythemia vera, occurring in 15% to 32% of the patients. As many as 15% of patients with polycythemia vera die of a stroke, five times the number in an age-matched control population. Migraine phenomena with or without headache occur when the platelet counts exceed 1,000,000/mm3, as can occur in essential thrombocythemia.
HEMORRHAGIC DIATHESIS
The major peripheral nervous system complication is intramuscular hemorrhages that may compress peripheral nerves.9 Most patients with peripheral nerve compressions may be managed with factor replacement alone, but some may require fasciotomy. Patients with hemophilia must be monitored for intracranial hemorrhage because of the more active lives enabled by vigorous replacement therapy for intra-articular hemorrhages. Bleeding may be intracerebral, subarachnoid, subdural, and epidural. In mild cases (7% to 15% of normal factor levels), intracranial bleeding occurs only after significant trauma. In patients with moderately severe disease (1% to 6% of normal factor levels), only minor trauma may produce hemorrhage, and with severe cases (<1% normal levels), it can occur after no trauma at all. The most practical and accurate method for accurate diagnosis of intracranial hemorrhage is the computed tomographic scan. Subarachnoid hemorrhage may be treated successfully with factor replacement alone, although a ventricular catheter may be necessary to treat hydrocephalus. Subdural and epidural hemorrhages often necessitate surgical therapy, which may be safely accomplished after factor replacement. Intracerebral hemorrhages are the most difficult to treat. Most patients are managed with medical therapy alone or with medical therapy plus a ventricular cannula for measurement and control of intracranial pressure.
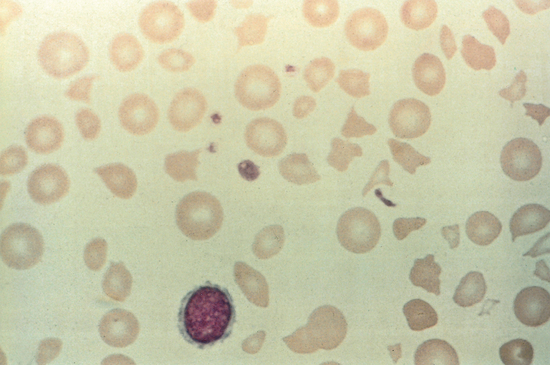
The thrombotic microangiopathies—which include thrombotic thrombocytopenic purpura (TTP); hemolytic uremic syndrome; and the syndrome of hemolytic anemia, elevated liver function test results, and low platelet levels (HELLP)—are disorders caused by aggregation of platelets, thrombocytopenia, and mechanical injury to erythrocytes.10 TTP (Moschowitz’s disease) causes neurological symptoms because of microvascular brain ischemia.11 The blood smear shows a microangiopathic hemolytic anemia (Fig. 115-2). Neurological manifestations of the disease, which reflect multifocal brain ischemia, are headache; cognitive changes, including altered states of consciousness, agitation, confusion, and delirium; hemiparesis; aphasia; syncope; visual changes; dysarthria; seizures; coma; cranial nerve palsies; paresthesias; and vertigo. TTP is caused by platelet aggregates that contain anti–von Willebrand factor antibody rather than fibrin, as is the case in disseminated intravascular coagulation (DIC). Deficiency of the enzyme that degrades large multimers of von Willebrand factor, a disintegrin and metalloprotease with thrombospondin-1–like domains (ADAMTS-13), causes TTP because the large undegraded multimers of von Willebrand factor are much more likely to produce aggregation of platelets. ADAMTS-13 may be genetically deficient, producing a familial type of TTP, or TTP may be acquired by a probable autoimmune mechanism whereby immunoglobulin G binds and disables ADAMTS-13. The diagnosis is made by identifying a typical blood smear in the appropriate clinical setting. Levels of ADAMTS-13 are less than 5% of normal in active TTP. Plasma exchange (the combination of plasmapheresis, which may remove some of the multimers of von Willebrand factor and autoantibodies against ADAMTS-13, and infusion of fresh-frozen plasma or cryosupernatant, which contains ADAMTS-13) is a treatment that leads to a survival rate of about 90%. Steroids, splenectomy, vincristine, or rituximab may be used in resistant cases.
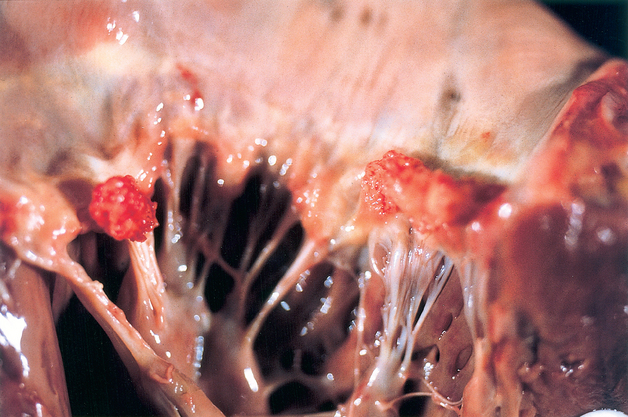
DIC is a relatively common acquired hemorrhagic thrombotic syndrome that occurs as a result of the presence of thrombin in the systemic circulation. The syndrome follows other disease states such as viral and bacterial infections, obstetrical and surgical complications, neoplasms (especially mucin-secreting adenocarcinomas), fat embolism, diabetic ketoacidosis, and head injury. DIC causes thrombosis and bleeding at multiple sites, including the nervous system. The essential neuropathological changes are multiple infarctions, petechial hemorrhages, and occasional small subdural and subarachnoid hemorrhages. Nonbacterial thrombotic (marantic) endocarditis may develop (Fig. 115-3). Fibrin thrombi are found in the cerebral vessels. The clinical syndrome depends on the particular pathology but may include seizures, mental changes, and focal findings. The management of subacute or chronic DIC with heparin may be effective, particularly in patients with thrombotic complications, but removal of the underlying cause (e.g., evacuation of the uterus in cases of abruptio placentae) is the definitive treatment.
Hypoprothrombinemia, usually caused by the use of anticoagulant drugs, is a common cause of bleeding. Heparin is a drug that inactivates thrombin, inhibits the conversion of prothrombin to thrombin, and prevents the agglutination of platelets; warfarin acts by antagonizing vitamin K. These two drugs have a similar spectrum of neurological complications. Nervous system hemorrhage in patients taking anticoagulants may involve numerous locations, including intracerebral, subarachnoid, subdural, cranial epidural, spinal epidural, spinal intramedullary, root, plexus, and peripheral nerves. Lumbar puncture is dangerous in the presence of a hemorrhagic diathesis and should be avoided in favor of computed tomography and magnetic resonance imaging as a method for making the diagnosis. In general, the incidence of serious neurological hemorrhage rises above the benefit of anticoagulation when the international normalized ratio exceeds 3.0. For neurological purposes, the target international normalized ratio is 2.0 to 3.0 for all conditions except in patients with mechanical heart valves.
PARAPROTEINEMIAS
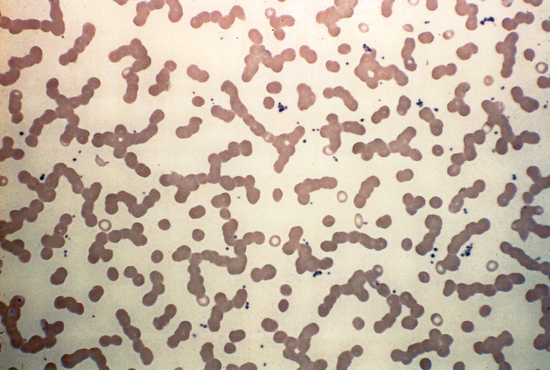
Paraparoteinemias may be seen in many conditions, including connective tissue diseases and multiple types of malignancy. The plasma cell dyscrasias are a group of disorders characterized by the uncontrolled proliferation of cells normally involved in antibody synthesis. This usually results in the elaboration of a homogeneous immunoglobulin or one of its constituent polypeptide chains. These disorders are often classified according to type of protein that is produced (myeloma, macroglobulinemia, and heavy-chain diseases). Multiple myeloma, characterized by infiltration of the bone marrow with neoplastic plasma cells, is the most common plasma cell dyscrasia. The peripheral blood smear may show abnormal clumping of erythrocytes in stacks of coinlike rows (Rouleaux). This results from the fact that the normal surface charges that cause red blood cells to repel each other are coated with the paraprotein (Fig. 115-4).
Cryoglobulinemia is the presence in the serum of proteins that precipitate in the cold and redissolve on warming. These proteins are most often associated with hepatitis C infection, myeloma, and macroglobulinemia, but they may be appear as part of a connective tissue disease or as an isolated finding in the absence of any known underlying cause. About a third of the cryoglobulins are myeloma immunoglobulin G proteins (type I), a third are immunoglobulin M macroglobulins (type II), and a third are a mixture of immunoglobulin M and G molecules (type III). The neurological syndrome associated with cryoglobulinemia is most common in type III, which is the type most often related to an underlying connection tissue disease. The most frequent neurological syndrome is a sensorimotor polyneuropathy with purpura. Many types of polyneuropathies are seen in patients with paraproteinemias, including symmetrical polyneuropathy, mononeuropathy multiplex, and various versions of chronic inflammatory demyelinating polyneuropathy, including multifocal motor neuropathy. There may be neuropathies with low levels of paraproteinemia that do not meet criteria for a frank myeloproliferative disorder (monoclonal gammopathy of unclear significance). The Bing-Neel syndrome, the central nervous system syndrome seen in macroglobulinemia, was described before Waldenström named the disease in 1944.12 It is caused in part by the hyperviscosity syndrome, but in addition, some patients exhibit a multifocal disease with a rapid downhill course that is uniformly fatal and unresponsive to plasmapheresis. The spinal fluid in these patients is abnormal, with some pleocytosis and elevated protein levels. The pathology consists of infiltration of lymphocytes and plasma cells, particularly around veins. The syndrome of polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes (POEMS) is a paraneoplastic syndrome observed in patients with osteosclerotic myeloma. Hepatosplenomegaly, sexual impotence, various skin lesions, and a painful sensorimotor polyneuropathy are usually present. The M component in the serum is produced by an osteosclerotic myeloma, which, when treated, results in improvement in all components of the syndrome.
THE HYPERCOAGULABLE STATES
Patients are considered to have hypercoagulable states if they have laboratory abnormalities or clinical conditions that are associated with an increased risk of thrombosis (prethrombotic states) or if they have recurrent thrombosis without recognizable predisposing factors (thrombosis-prone).13 The hypercoagulable states are subdivided into those in which a clearly identified, specific abnormality in hemostasis can be found (primary hypercoagulable states) and those in which various diverse clinical conditions have been associated with an increased risk of thrombosis (secondary hypercoagulable states). Cerebral thrombosis (venous and arterial) and embolism are important manifestations. The primary hypercoagulable states result from failure of one of the three physiological anticoagulant mechanisms (antithrombin III, protein C, and the fibrinolytic system) and include antithrombin III deficiency, protein C deficiency, protein C resistance caused by the factor V Leiden mutation, protein S deficiency, fibrinolytic disorders, dysfibrinogenemia, factor XII deficiency, prekallikrein deficiency, the antiphospholipid antibody syndrome, and the prothrombin gene mutation.14 The two major antiphospholipid syndromes are the lupus anticoagulant and the anticardiolipin antibody. The lupus anticoagulant is an antibody to phospholipids that interferes with the formation of the prothrombin activator, a complex of calcium ions, factors Xa and V, and a source of phospholipid, usually the platelet membrane in the coagulation cascade. This immunoglobulin G or M antiphospholipid antibody often causes prolongation of phospholipid-dependent coagulation test times, such as the activated partial thromboplastin time. This antibody is present in about 25% of patients with systemic lupus erythematosus (SLE) and may cross-react with cardiolipin, the antigen commonly used in a blood screening test for syphilis, thus producing a biological false-positive result. Despite the prolonged activated partial thromboplastin time, this antiphospholipid antibody is actually a procoagulant. The lupus anticoagulant is diagnosed through a three-step functional test. In addition to patients with SLE, other people at risk for the anticardiolipin antibody include patients taking neuroleptic drugs, those with neoplasms, and those with other autoimmune disorders. Those without underlying disease are said to have the primary antiphospholipid antibody (Hughes’) syndrome, characterized by recurrent episodes of venous and/or arterial thrombosis, recurrent midpregnancy spontaneous abortions, and thrombocytopenia. Migraine, mitral valve prolapse, and livedo reticularis are also overrepresented in these patients (Sneddon’s syndrome). Echocardiography frequently reveals the presence of vegetations on the mitral valve, presumably representing foci of nonbacterial thrombotic endocarditis (NBTE). The nervous system is commonly affected with large- and small-vessel arterial occlusions, venous occlusions, and emboli that probably arise from NBTE, which in turn results from the hypercoagulable state. Many of the neurological syndromes seen in patients with SLE are caused either by thrombosis in situ or by emboli from NBTE (known as Libman-Sacks endocarditis in patients with SLE). Treatment of patients who have the antiphospholipid antibody and who suffer recurrent thrombosis is anticoagulation with warfarin (international normalized ratio, 2.0 to 3.0).
The secondary hypercoagulable states may be divided into three major groups on the basis of the presumed predominant pathophysiological mechanism: (1) abnormalities of coagulation and fibrinolysis, such as those caused by malignancy, pregnancy, use of oral contraceptives, infusion of prothrombin complex concentrates, and nephrotic syndrome; (2) abnormalities of platelets, such as those caused by myeloproliferative disorders, paroxysmal nocturnal hemoglobinuria, hyperlipidemia, diabetes mellitus, and heparin-induced thrombocytopenia15; and (3) abnormalities of blood vessels or rheology, such as those caused by conditions promoting venous stasis (e.g., immobilization, obesity, advanced age, postoperative state), artificial surfaces, vasculitis and chronic occlusive arterial disease, homocystinemia, hyperviscosity (e.g., polycythemia, leukemia, sickle cell disease, leukoagglutination, increased serum viscosity), and TTP.
The relationship between increased tendency for thrombosis and malignancy has been known ever since Armand Trousseau described the syndrome that bears his name.16 Migratory phlebothrombosis, pulmonary emboli, and transient or permanent focal neurological deficits are known to be part of a paraneoplastic syndrome usually in patients with mucin-secreting adenocarcinomas. Some of the neurological deficits are caused by thrombosis in situ of cerebral vessels; others are caused by emboli arising from NBTE, which itself is caused by the paraneoplastic hypercoagulable state.
Pregnancy increases the risk of thrombosis, probably as a consequence of chronic low-grade DIC, which is a normal development in pregnancy, presumably in preparation for the hemostatic challenge of placental separation. Cerebral venous thrombosis is the major neurological complication of this hypercoagulable state, seen primarily in the postpartum period. It takes two major clinical forms: venous sinus occlusion and cortical vein occlusion. Although these two clinical forms often fuse as the illness progresses, venous sinus thrombosis usually manifests with increased intracranial pressure, whereas cortical vein thrombosis usually begins with partial seizures. Venous hypertension may cause hemorrhage into the brain that can often be visualized by computed tomography or magnetic resonance imaging. Magnetic resonance venography is now the “gold standard” neurodiagnostic study when venous occlusive disease is suspected. The treatment for the hypercoagulable state of pregnancy is reserved for patients with demonstrated thrombosis, and it consists of heparin, inasmuch as warfarin crosses the placenta and is possibly teratogenic. Oral contraceptives significantly increase the risk of thrombosis in a way similar to that seen in late pregnancy.
Bain BJ. Diagnosis from the blood smear. N Engl J Med. 2005;353:507-598.
Grotta JC, Manner C, Pettigrew LC, et al. Red blood cell disorders and stroke. Stroke. 1986;17:811-817.
Markus HS, Hambley H. Neurology and the blood: haematological abnormalities in ischaemic stroke. J Neurol Neurosurg Psychiatry. 1998;64:150-159.
Pollard JD, Young GAR. Neurology and the bone marrow. J Neurol Neurosurg Psychiatry. 1997;63:706-711.
Samuels MA. Neurologic aspects of hematologic disease. Curr Neurol. 1992;12:215-240.
Samuels MA. Neurologic manifestations of hematologic diseases. In: Asbury A, McKhann G, McDonald I, editors. Diseases of the Nervous System. 2nd ed. Philadelphia: WB Saunders; 1992:1510-1521.
Samuels MA, Thalinger K. Cerebrovascular manifestations of selected hematologic diseases. Semin Neurol. 1991;11:411-418.
1 Ekbom KA. Restless legs. In: Vinken PF, Gruyn GW, editors. Handbook of Clinical Neurology, vol 8: Diseases of Nerves. Amsterdam: Elsevier; 1970:311-320.
2 Healton EB, Savage DG, Brust JCM, et al. Neurologic aspects of cobalamin deficiency. Medicine. 1991;70:229-245.
3 Beck WS. Neuropsychiatric consequences of cobalamin deficiency. Adv Intern Med. 1991;36:33-56.
4 Toh B-H, VanDriel IR, Gleeson PA. Mechanisms of disease: pernicious anemia. N Engl J Med. 1997;337:1441-1448.
5 Kinsella LJ, Green R. “Anesthesia paresthetica”: nitrous oxide–induced cobalamin deficiency. Neurology. 1995;45:1608-1610.
6 Adams RD, Kubik CS. Subacute degeneration of the brain in pernicious anemia. N Engl J Med. 1944;231:1-9.
7 Roman GC. On politics and health: an epidemic of neurologic disease in Cuba. Ann Intern Med. 1995;122:530-533.
8 Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998;339:5-11.
9 Silverstein MN. Intracranial bleeding in hemophilia. Arch Neurol. 1960;3:1415-1420.
10 Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347:589-600.
11 Moschcowitz E. An acute febrile pleiochromic anemia with hyaline thrombosis of the terminal arterioles and capillaries: an undescribed disease. Arch Intern Med. 1975;35:89-95.
12 Bing J, Neel AV. Two cases of hyperglobulinemia with affection of the central nervous system on a toxic-infectious basis. Acta Med Scand. 1936;88:492-506.
13 Schafer AI. The hypercoagulable states. Ann Intern Med. 1985;102:814-828.
14 Nachman RL, Silverstein R. Hypercoagulable states. Ann Intern Med. 1993;119:819-827.
15 Warkentin TE, Chong BH, Greinacher A. Heparin-induced thrombocytopenia: towards consensus. Thromb Haemost. 1998;79:1-9.
16 Trousseau A. Phlegmasia alba dolens. Clin Med Hotel Dieu de Paris. 1865;3:94-100.






