CHAPTER 118 NEUROLOGY OF ENDOCRINOLOGY
CLASSIFICATION OF HORMONES
Multicellular organisms have developed complex mechanisms to ensure regulation of metabolism. Although the nervous system and endocrine system were classically considered to play crucial and largely independent roles in this process, studies over several decades demonstrated that these systems converge to maintain homeostasis and various physiological functions. In its simplest form, the endocrine system may be viewed as consisting of an endocrine gland: that is, a collection of specialized cells that synthesize and secrete a hormone and a target tissue that responds to this hormone. Hormones are chemical substances, produced by endocrine organs or cells dispersed in major organ systems, that act at distant tissues through the blood to exert their biological actions.
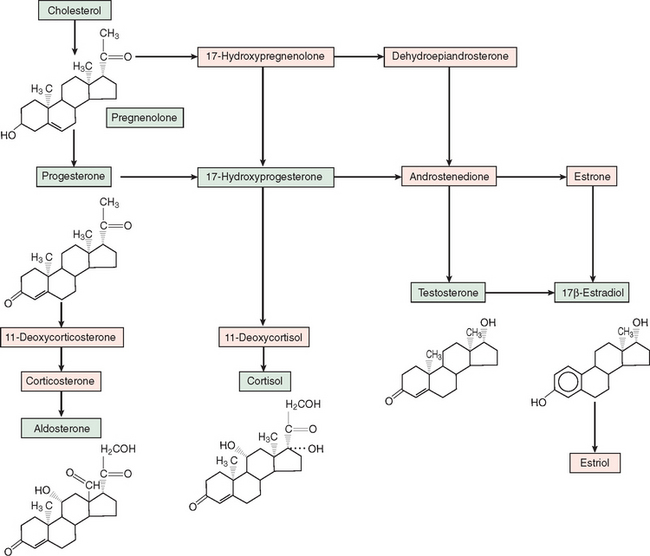
Steroid hormones are derived from enzymatic processing of a cholesterol precursor (Fig. 118-1). Conversion of cholesterol to pregnenolone through side-chain cleavage is the first step in the steroidogenic pathway and occurs in all steroidogenic tissues (i.e., adrenal cortex, testes, ovaries, and placenta). Further metabolism is determined by specific enzymes that mediate hydroxylation, methylation, and demethylation. Corticosteroids and progestins contain 21 carbons; androgens, 19 carbons; and estrogens, 18 carbons and aromatic A-ring. Because 11- and 21-hydroxylases are expressed only in the adrenal cortex, the production of glucocorticoids and mineralocortiocoids occurs only in this gland. Cortisol, the principal glucocorticoid, is synthesized mostly by zona fasciculata cells and hydroxylated at the 17-carbon position and exerts major effects on glucose and various metabolic processes. Aldosterone, the principal mineralocorticoid, is a 17-deoxycorticosteroid produced in the zona glomerulosa of the adrenal cortex. Progesterone is the main steroid secreted by the placenta and ovaries and also serves as a precursor for corticosteroids and other sex steroid hormones.
MECHANISMS OF HORMONE ACTION
Hormones by definition are transported in the blood, although the methods for transporting peptides, steroids, and catecholamines vary, depending on solubility. Peptide and protein hormones are hydrophilic and dissolve directly or associate with albumin in the plasma. Insulin circulates as monomeric and polymeric forms, whereas some anterior pituitary hormones circulate as dissociated subunits. Steroid and thyroid hormones, in contrast, are insoluble and bind to transport proteins (corticosteroid-binding globulin in the case of cortisol and thyroid-binding globulin and transthyretin in the case of thyroxine). The hormone bound to the carrier protein cannot interact with the receptor, but it serves as a reserve to replenish the free (bioactive) form. Nonetheless, the bound hormone exists in equilibrium with free hormone and receptor-bound fractions. The free hormone level increases as the rate of hormone degradation and clearance rises. On the other hand, conditions that increase the amount of carrier proteins, such as pregnancy and liver disease, elevate total hormone levels, although the balance between free and bound hormone is preserved.
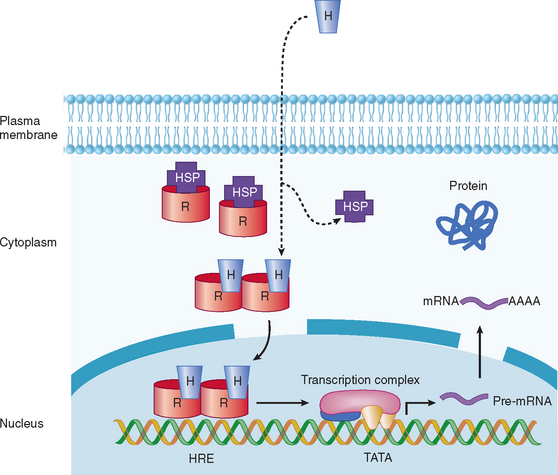
In view of the fact that hormone concentrations are very low, in the range of 10−15 to 10−9 M, and more than 100-fold lower than levels of similar sterols, amino acids, and polypeptides, how do target cells identify hormones to initiate specific biological effects? In general, receptors for peptide, amine, and steroid hormones have a domain that recognizes and binds the hormone, with subsequent activation of a signaling mechanism that transduces the information into an intracellular action. Steroid and thyroid hormones and retinoic acid are lipophilic; associate with transport proteins in the blood, which prolongs their plasma half-life; and readily cross the plasma membrane in target tissues and bind to receptors in the nucleus or cytoplasm (Table 118-1; Fig. 118-2).1,2 The hormone-receptor complex undergoes activation, which leads to changes in chromatin, binding to specific regions of DNA, and transcription or inactivation of target genes. Studies since the 1980s have led to greater understanding of steroid-regulated genes, hormone response elements (i.e., short DNA segments that bind to specific steroid receptor-hormone complexes), and how these cis-acting DNA elements interact with transacting factors.3–5 Transacting factors include coactivator and corepressor molecules that modulate transcription.5 For example, in the absence of hormone, thyroid and retinoic acid receptors associate with NcoR, SMRT and other proteins, which results in repression of target genes. Binding by thyroxine dissociates the complex culminating in gene activation. Members of the p160 family of coactivators (e.g., SRC-1, NCoA-1, GRIP 1, TIF2, and p/CIP) have been implicated in the function of corticosteroids and other steroids.
| Steroid Family | Seven-Transmembrane Domain | Single Transmembrane |
|---|---|---|
ACTH, adrenocorticotrophic hormone; FSH, follicle-stimulating hormone; LH, luteinizing hormone; PPAR, peroxisome proliferator-activated receptor; TRH, thyrotropin-releasing hormone; TSH, thyroid-stimulating hormone.
Polypeptides, glycoprotein hormones, and catecholamines are water soluble, have no transport proteins, and possess a relatively short half-life. These hormones bind to surface receptors on the plasma membrane and generate second-messenger molecules (Fig. 118-3). Epinephrine, as well as several neuropeptides and gut and pituitary hormones, including neuropeptide Y, cholecystokinin, vasopressin, glucagon, ACTH, thyroid-stimulating hormone (TSH), and luteinizing hormone, bind to their respective membrane receptors, stimulate adenylate cyclase located on the inner plasma membrane, and catalyze the formation of cyclic adenosine monophosphate (cAMP) from adenosine triphosphate.6 Activation or inactivation of adenylate cyclase occurs through the guanosine triphosphate–dependent regulator proteins (e.g. Gs [stimulatory] and Gi [inhibitory]).7 Changes in cAMP exert diverse effects on metabolism through various substrates, such as the transcription factor cAMP response element binding protein (CREB). CREB phosphorylation by cAMP leads to interaction with the coactivator CREB-binding protein, which results in stimulation of gene transcription.8 These effects can be terminated by hydrolysis of cAMP by phosphodiesterases or dephosphorylation by phosphoprotein phosphatases.9
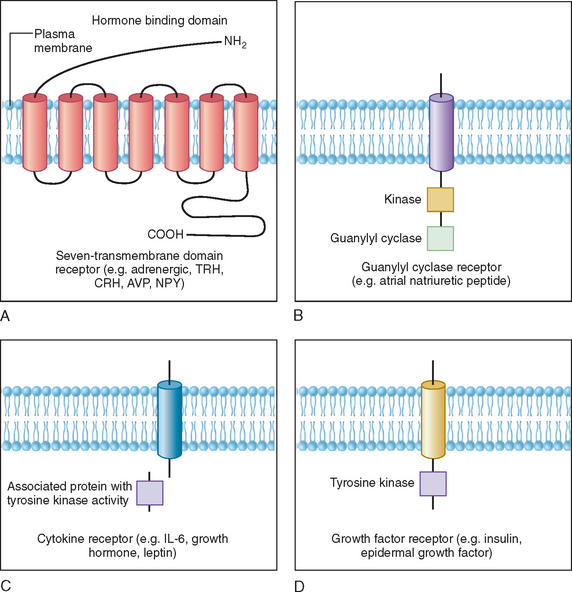
Some polypeptide hormones produced by vascular tissues, such as atrial natriuretic factor, stimulate guanosine triphosphate by guanylate cyclase and increase cyclic guanosine monophosphate (cGMP), which leads to activation of cGMP-dependent protein kinase, phosphorylation, and alteration in the of smooth muscle proteins.10 This effect is terminated by specific cGMP phosphodiesterase. Other hormones signal through phosphatidylinositides and calcium. The actions of ACTH in the adrenal cortex, luteinizing hormone in the ovaries and Leydig cells of the testes, and angiotensin II in vascular tissues have been associated with activation of phospholipase C, which mediates the hydrolysis of phosphatidylinositol-4,5-biphosphate (PIP2) to 1,4,5-triphosphate (PIP3) and diacylglycerol. Binding of PIP3 to organelles increases intracellular Ca2+.9 Diacylglycerol activates protein kinase C, which phosphorylates various substrates involved in metabolism.
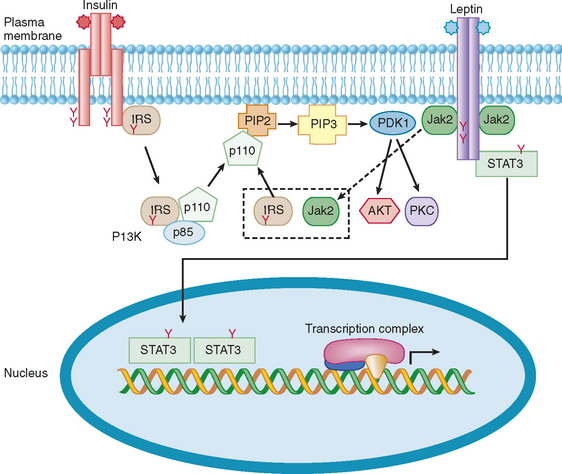
Cytokine receptors lack intrinsic tyrosine kinase activity but associate with proteins that are tyrosine kinases.11,12 For example, growth hormone, prolactin, inflammatory cytokines, and leptin bind to their receptors, which activate cytoplasmic protein tyrosine kinases, such as Jak1 and Jak2.11–13 These then phosphorylate other proteins, such as signal transducers and activators of transcription (STAT) proteins and docking proteins containing Src homology 2 (SH2) domains, culminating in transcriptional activation of neuropeptides and various target genes. In contrast, the insulin receptor possesses intrinsic tyrosine kinase activity in the cytoplasmic domain that is activated upon binding of insulin to the extracellular domain of the receptor. This autophosphorylation of insulin receptor leads to phosphorylation of insulin receptor substrates, activation of PI-3 kinase, and a cascade of biochemical events underlying insulin’s effects on glucose uptake, lipid and protein metabolism, and growth.14
REGULATION OF HORMONE LEVELS AND RHYTHMS
Hormones generally regulate existing reactions instead of initiating de novo reactions. In contrast to the rapid time course of nervous activity, which lasts from milliseconds to seconds, the action of hormones is often prolonged and may persist for some time after the hormone is withdrawn. However, as discussed in detail later, the neurosecretory system allows the neural pathways in the hypothalamus, brainstem, and other regions of the central nervous system to interact with peripheral endocrine tissues, such as the thyroid, adrenal gland, and gonads.
NEUROENDOCRINOLOGY
Development and Structure of Hypothalamic-Pituitary Unit
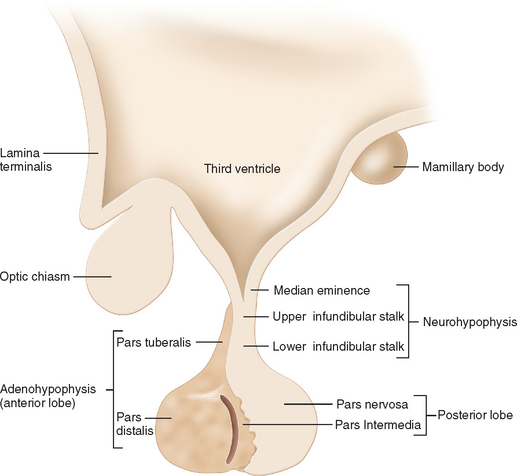
The hypothalamic-pituitary unit is derived from the ventral diencephalons.15 Between the third and fourth weeks of embryonic development, the sulcus limitans divides the alar (dorsal) and basal (ventral) plates of the neural tube. At about the fifth week, the hypothalamic sulcus divides the alar plate into a dorsal portion, which gives rise to the thalamus, and a ventral portion, which forms the hypothalamus, infundibulum, and posterior pituitary gland.16 The anterior pituitary gland begins as a diverticulum of the ectodermal lining in the roof of the buccal cavity (Rathke’s pouch) and expands dorsally to invest the infundibular stalk by the eighth week. In the adult, the hypothalamus lies in the ventral aspect of the brain directly above the pituitary gland, which lies within the sella turcica in the sphenoid bone.
The median eminence is located at the base of the hypothalamus.17 The arcuate (infundibular) nucleus overlies the median eminence. This structure, along with other circumventricular organs (i.e., organum vasculosum of the lamina terminalis, subfornical, choroid plexus, pineal gland, subfornical and area postrema) located in the walls of the lateral third, and fourth ventricles, have a rich capillary plexus with fenestrated capillaries, rendering adjacent regions outside the blood-brain barrier.18 These structures provide a window through which polypeptide and protein hormones in the blood can communicate with neurons in the hypothalamus and other brain areas.
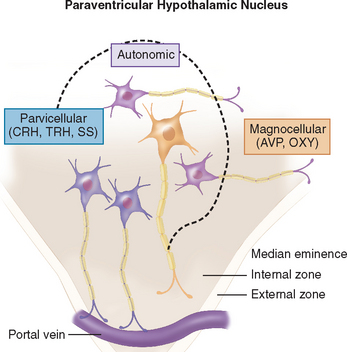
The hypothalamus is divided into nine zones containing various neuronal groups.16,19 In the longitudinal plane, the hypothalamus is divided into a midline zone adjacent to the third ventricle; a medial zone containing preoptic, anterior, ventromedial, dorsomedial, paraventricular, posterior, and mammillary neuronal groups; and a lateral zone separated from the medial zone by the fornix. Three hypothalamic zones are defined in the horizontal plane: supraoptic, tuberal, and mammillary, in a rostral-to-caudal direction. Histologically, the hypothalamus contains large (magnocellular) neurons that produce vasopressin and oxytocin in the lateral subdivision of the paraventricular nucleus (Fig. 118-4) and supraoptic nucleus. The axons of these neurons collect to form the posterior pituitary gland. Magnocellular neurons co-localize angiotensin II, corticotropin-releasing hormone (CRH), enkephalins, and cholecystokinin. Parvicellular neurons are located in the periventricular and paraventricular hypothalamic areas, and synthesize peptides (e.g., thyrotropin-releasing hormone [TRH], CRH, and somatostatin) that are released into the portal system surrounding the infundibular stalk and regulate the secretion of hormones by anterior pituitary cells. These components are discussed in the next sections in regard to the synthesis and secretion of hormones, their physiological functions, and their roles in disease states.
The anterior pituitary gland receives arterial blood from the internal carotid arteries via the hypophysial arteries.20 The superior hypophysial artery forms a capillary plexus in the median eminence that drains into long portal veins along the infundibular stalk. These portal veins divide into another capillary plexus and then re-form into venous channels that drain into the cavernous sinus. The infundibular stalk and posterior pituitary are supplied by branches of the middle and inferior hypophysial arteries, and the latter form a capillary plexus in the posterior pituitary gland, where axons from magnocellular neurons in the paraventricular nucleus and supraoptic nucleus terminate.20 Veins from the posterior lobe drain to the cavernous sinus and into the systemic circulation. The hypophysial portal system allows the hypothalamus to control anterior pituitary function and enable blood flow between the posterior and anterior lobes, as well as retrograde flow from the pituitary gland to hypothalamus.
Neurohypophysial Hormones (Table 118-2)
Arginine Vasopressin
Arginine vasopressin (AVP) also called antidiuretic hormone, and oxytocin are cyclic nonapeptides produced by distinct magnocellular neurons in the paraventricular nucleus and supraoptic nucleus and released in the capillary plexus surrounding the posterior pituitary gland.21 The preprohormones are converted to prohormones by cleavage of the signal peptide. Subsequent cleavage of the prohormones yields one molecule each of AVP (molecular weight, 1084) and oxytocin (molecular weight, 1007), and a large protein called neurophysin (molecular weight, 10,000). AVP causes contraction of vascular smooth muscle and stimulates water absorption by the kidneys. Although the pressor effect of AVP does not appear to be crucial under normal physiological conditions, the antidiuretic effect of AVP is crucial for maintaining water balance. AVP binds to V1 and V2 receptors, stimulates adenylate cyclase, and increases cAMP levels in target tissues, which culminate in its pressor and antidiuretic effects.21,22 AVP secretion is increased when plasma osmolality increases. The latter is monitored by osmoreceptors located in the anterior hypothalamus, which activate magnocellular neurons, increase plasma AVP, and reduce free water clearance by the kidneys.
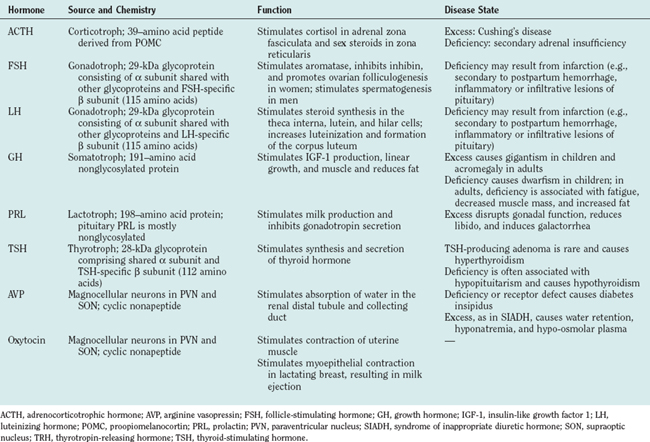
AVP deficiency causes the production of large volumes of very dilute urine (polyuria), resulting in polydipsia. This condition, known as diabetes insipidus, may result from central (hypothalamic) or peripheral (renal) causes.21,22 Central diabetes insipidus may be secondary to traumatic injury, granulomatous disease, or other infiltrative diseases that affect axons traversing the infundibular stalk or terminals in the neurohypophysis. In contrast, nephrogenic diabetes insipidus results from genetic or acquired defects of AVP receptors. Compulsive water drinking and use of ethanol and drugs that interfere with V2 receptors in the collecting duct epithelium all produce polyuria. The distinction between central and nephrogenic diabetes insipidus is facilitated by administering AVP or a longer acting analog, desmopressin, which reverses central but not nephrogenic diabetes insipidus.21
Hypersecretion of AVP may occur in the syndrome of inappropriate secretion of antidiuretic hormone (SIADH), which is characterized by hypervolemia or euvolemia, hyponatremia, and reduced plasma osmolality. SIADH may be caused by central nervous system pathology such as brain tumors, abscesses and inflammation, drugs (including nicotine, phenothiazines, tricyclic antidepressants, and vincristine), pulmonary diseases (such as tuberculosis and bacterial or viral pneumonia), and acquired immunodeficiency syndrome. The primary treatment of SIADH is water restriction.21,22
Adenohypophysial Hormones (Table 118-2)
Anterior pituitary cells were first described as acidophils, basophils, and chromophobes, on the basis of hematoxylin-eosin staining (Fig. 118-5). The development of electron microscopy and immunohistochemistry enabled anterior pituitary cells to be classified according to secretory products: somatotrophs (growth hormone), lactotrophs (prolactin), thyrotrophs (TSH), corticotrophs (ACTH), and gonadotrophs (luteinizing hormone and follicle-stimulating hormone).23 These cells derive from a common primordium, and their development is regulated by transcription factors. Thyrotrophs, lactotrophs, and somatotrophs derive from a common lineage, determined by Prop-1 and Pit-1, whereas corticotrophs and gonadotrophs develop from independent lineages.24
Growth Hormone
Growth hormone–producing cells constitute about 50% of anterior pituitary cells and are typically located in the lateral portion of the gland.25 Growth hormone–releasing hormone is produced by neurons in the arcuate nucleus and stimulates the secretion of growth hormone. In contrast, somatostatin produced by neurons in the periventricular region inhibits growth hormone secretion. The primary function of growth hormone is to promote linear growth, mostly through stimulating the level of insulin-like growth factor 1, which increases amino acid uptake, transcription, and protein synthesis. Growth hormone prevents protein catabolism while enhancing lipolysis. Moreover, growth hormone decreases carbohydrate use and glucose uptake, which may result in glucose intolerance and diabetes. Growth hormone is secreted episodically and has a short half-life (20 to 50 minutes); thus, insulin-like growth factor 1 measurement is a better assessment of growth hormone function. Early morning growth hormone level is typically less than 2 ng/mL, and the hormone circulates mostly bound to protein.
Congenital growth hormone deficiency may be familial or sporadic and leads to dwarfism.26 Functional growth hormone deficiency may occur in malnutrition and emotional deprivation, leading to growth retardation. In adults, the loss of growth hormone from mass lesions and infiltrative diseases of the sella or pituitary infarction (e.g., Sheehan’s syndrome) has been associated with fatigue, hypoglycemia, reduction in muscle, and increased adiposity.26 Hormone replacement with recombinant human growth hormone ameliorates these symptoms. Growth hormone–secreting adenomas are the second most common functional pituitary tumors (after prolactinomas).27 Excess growth hormone leads to gigantism in children and acromegaly in adults. Typical manifestations of acromegaly include enlargement of the hands and feet, gnathopathy in association with malocclusion of the teeth, soft tissue overgrowth involving the skin (papillomas and tags) and colonic mucosa, cardiomegaly, hypertension, hyperphydrosis, glucose intolerance, irregular menses, decreased libido, goiter with or without hypothyroidism, headache, and visual defect. The diagnosis is based on the history and physical examination findings, elevation of insulin-like growth factor 1 level, failure of growth hormone to be suppressed by an oral glucose load, and imaging of the sella with magnetic resonance imaging or computed tomographic scan. The standard treatment for acromegaly is transsphenoidal resection.27 In rare cases, a craniotomy and transtemporal approach may be needed for major suprasellar extension of the tumor. Residual tumor is treated medically with octreotide and occasionally with bromocriptine, which is less effective. Gamma knife radiotherapy has been used to control tumor localized to the sella, and conventional irradiation has been used for recurrent extensive disease, although there is higher risk for hypopituitarism and cognitive deficits.
Prolactin
Prolactin is synthesized and secreted by lactotrophs of the anterior pituitary and stimulates lactation in the postpartum period.28 Prolactin secretion is pulsatile and peaks between 4:00 and 7:00 A.M. Although TRH and serotonin increase prolactin level, it is unlikely that this serves a physiological role. Rather, prolactin is subject mostly to inhibition by dopamine produced by arcuate hypothalamic neurons. The most common hypersecreting pituitary tumor is a prolactin adenoma. The typical features include galactorrhea, gonadal dysfunction (oligorrhea or amenorrhea, infertility), decreased libido and impotence in men, and manifestations of estrogen deficiency, such as vaginal dryness and osteopenia, in women.
The diagnosis of prolactinoma is based on history and physical examination findings, and a basal prolactin level greater than 200 ng/mL virtually establishes the diagnosis of a prolactin tumor. Magnetic resonance imaging frequently reveals a microadenoma (<1 cm in diameter). Bromocriptine, a dopamine agonist, is effective therapy for prolactin tumors but is frequently associated with dizziness, postural hypotension, nausea, and vomiting. Cabergoline is a nonergot dopamine agonist that can be administered once or twice weekly and has fewer side effects than does bromocriptine.27 Transsphenoidal surgery is rarely indicated for prolactinomas, because drug therapy is highly effective (80% to 90%). However, surgery by a skilled practitioner is a treatment option in the presence of microadenoma and drug intolerance.
Adrenocorticotropic Hormone
ACTH is processed from a precursor molecule, proopiomelanocortin in the corticotroph, and stimulates the secretion of glucocorticoids and, to a lesser degree, mineralocorticoids and androgenic steroids from the adrenal cortex.25 The physiological secretion of ACTH is regulated by CRH in a pulsatile manner. ACTH levels peak before sleep ends and decreases as the day progresses. This diurnal rhythm precedes and is coupled to cortisol levels. In addition, both ACTH and cortisol levels are subject to stimulation by stress, depression, interleukin-1, and AVP. Negative feedback regulation of ACTH occurs through a long loop from cortisol, a short loop from ACTH itself at the pituitary gland, and an ultrashort loop from ACTH at CRH-producing neurons in the hypothalamus.
ACTH deficiency manifests as adrenal insufficiency, often in the setting of hypopituitarism, although isolated ACTH deficiency may result from lymphocytic hypophysitis. Pituitary ACTH hypersecretion (Cushing’s disease) is the commonest cause of hypercortisolism (Cushing’s syndrome).29 Excess ACTH may be produced by hyperplastic or adenomatous corticotrophs. The onset of Cushing’s disease is often insidious, and the disease may progress over months to years. As with most endocrinopathies, this condition is more prevalent in women. Typically, patients develop central obesity, moon facies, malar plethora, proximal myopathy, hypertension, glucose intolerance, amenorrhea or impotence, violaceous striae, easy bruising, and neuropsychiatric symptoms. Poor wound healing and osteoporosis may also develop. Virilization in women points more to adrenal carcinoma.
The diagnosis of Cushing’s syndrome involves a documentation of endogenous hypercortisolemia through 24-hour urine collection or through failure of suppression of cortisol by overnight dexamethasone treatment. Cushing’s disease must be distinguished from exogenous glucocorticoid treatment; pseudo-Cushing’s disease as seen in obesity, depression, and alcoholism; ectopic ACTH production (e.g., from lung tumors), and rare CRH-producing tumors in the hypothalamus, lungs, and gastrointestinal tract. In all cases, biochemical evaluation is crucial. Imaging of the pituitary gland and other organs and petrosal venous sampling are used to confirm and localize the lesion.29 Transsphenoidal resection is the treatment of choice for ACTH-secreting pituitary adenomas.27 In rare cases, adrenalectomy is performed to correct excess cortisol when the ACTH lesion cannot be located. Medical treatment with ketoconazole, metyrapone, and aminoglutethimide has been used as adjunctive therapy, and mitotane is used to reduce tumor bulk in adrenal carcinoma.
Adipose-Brain Axis
The notion that adipose tissue is merely a specialized depot for storing energy in the form of fat is no longer valid.30 Indeed, it has been known since the 1970s that adipose tissue is a major site for metabolism of sex steroids. The discovery of leptin in 1994 confirmed the active role of adipose tissue as a source of hormones.31 In addition to leptin, adipose tissue secretes bioactive peptides, including adiponectin, angiotensin, proinflammatory cytokines, and proteins involved in coagulation, which act locally through autocrine/paracrine mechanisms and systemically to regulate energy balance, in addition to regulating neuroendocrine, cardiovascular, and immune functions.30
Leptin is secreted in proportion to fat mass. Thus, leptin concentration is higher in obese than lean individuals. Moreover, leptin responds to nutritional status: The level falls during fasting and increases over several hours after feeding. These changes are mediated, at least in part, by insulin and glucose. Congenital deficiency of either leptin or leptin receptors causes hyperphagia, morbid obesity, insulin resistance, immunodeficiency, and a variety of endocrine deficits, most notably hypothalamic hypogonadism, mild tertiary hypothyroidism, and growth hormone deficiency.32 These abnormalities are reversed by leptin treatment, which confirms a causal role. Interestingly, the loss of adipose tissue in lipodystrophic humans and rodents produces metabolic changes identical to those of leptin deficiency. Furthermore, the fall in leptin level during fasting triggers changes in the neuroendocrine axis and immune function similar to those of congenital leptin deficiency and lipodystrophy. In all cases, leptin replacement ameliorates the metabolic and hormonal abnormalities.30
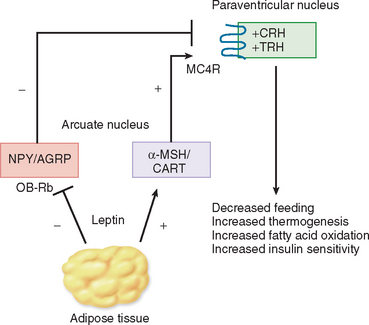
Studies so far suggest that the primary site of leptin action is the hypothalamus.33 Leptin enters the brain via a saturable process and binds to receptors in the hypothalamus, brainstem, and other regions of the brain. Leptin target neurons in the arcuate nucleus express orexigenic peptides, such as neuropeptide Y and agouti-related peptide, and anorexigenic peptides, such as proopiomelanocortin (the precursor of α-melanocyte–stimulating hormone) and cocaine- and amphetamine-regulated transcript (CART) (Fig. 118-6). Leptin inhibits feeding by suppressing the levels of neuropeptide Y and agouti-related peptide and by increasing proopiomelanocortin and CART. This net reduction in orexigenic peptides results in weight loss, caused by appetite suppression and increase in metabolic rate. Leptin also controls the synthesis and release of hypophysiotropic hormones, such as TRH and CRH, and may regulate gonadal function at the levels of the hypothalamus and gonadotrophs within the pituitary gland.33
The leptin receptor belongs to the family of class 1 cytokine receptors that includes interleukin-6, prolactin, and growth hormone (see Fig. 118-3).13 Binding of leptin to its receptor stimulates phosphorylation of tyrosine residues in the intracellular domain, leading to activation of Jak2, and phosphorylation and activation of STAT3 (Fig. 118-7). The latter is translocated to the nucleus, where it acts in concert with various transcription factors to regulate the expression of neuropeptides and other genes. The leptin signal is terminated through induction of suppressor of cytokine signaling 3 (SOCS3).34,35 As predicted, the loss of the leptin receptor in humans and rodents, or the targeted ablation of STAT3, recapitulates the obese phenotype of leptin deficiency. In contrast, haploinsufficiency of SOCS3 and, more specifically, ablation of SOCS3 in neurons, enhances leptin sensitivity, leading to inhibition of feeding, weight loss, and improvement in glucose and lipid levels.34,35 Studies have also demonstrated an interaction between the signal transduction of leptin and insulin.36 Both these hormones act through PI-3 kinase to regulate metabolism (see Fig. 118-7).
Despite these advances, the role of leptin in polygenic and dietinduced obesity remains unclear, as the rise in endogenous leptin is unable to prevent weight gain in most cases of obesity. Potential mechanisms include an impairment of hypothalamic leptin transport or subtle disruptions in signaling mechanisms distal to the leptin receptor, leading to leptin resistance.37 The disruption in signaling mechanisms may involve changes in the levels and function of SOCS3 and various neurotransmitters.
Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116:337-350.
Niswender KD, Baskin DG, Schwartz MW. Insulin and its evolving partnership with leptin in the hypothalamic control of energy homeostasis. Trends Endocrinol Metab. 2004;15:362-369.
Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639-650.
Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45-71.
Shimon I, Melmed S. Management of pituitary tumors. Ann Intern Med. 1998;129:472-483.
1 Bhargava A, Pearce D. Mechanisms of mineralocorticoid action: determinants of receptor specificity and actions of regulated gene products. Trends Endocrinol Metab. 2004;15:147-153.
2 Zhang J, Lazar MA. The mechanism of action of thyroid hormones. Annu Rev Physiol. 2000;62:439-466.
3 Gruber CJ, Gruber DM, Gruber IM, et al. Anatomy of the estrogen response element. Trends Endocrinol Metab. 2004;15:73-78.
4 Smith CL, O’Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45-71.
5 Torchia J, Glass C, Rosenfeld MG. Coactivators and corepressors in the integration of transcriptional responses. Curr Opin Cell Biol. 1998;10:373-383.
6 Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639-650.
7 Gainetdinov RR, Premont RT, Bohn LM, et al. Desensitization of G protein–coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107-144.
8 Farfel Z, Bourne HR, Iiri T. The expanding spectrum of G protein diseases. N Engl J Med. 1999;340:1012-1020.
9 Meyer TE, Habener JF. Cyclic adenosine 3′,5′-monophosphate response element binding protein (CREB) and related transcription-activating deoxyribonucleic acid–binding proteins. Endocr Rev. 1993;14:269-290.
10 Drewett JG, Garbers DL. The family of guanylyl cyclase receptors and their ligands. Endocr Rev. 1994;15:135-162.
11 Carter-Su C, Smit LS. Signaling via JAK tyrosine kinases: growth hormone receptor as a model system. Recent Prog Horm Res. 1998;53:61-82.
12 Pazin MJ, Williams LT. Triggering signaling cascades by receptor tyrosine kinases. Trends Biochem Sci. 1992;17:374-378.
13 Tartaglia LA. The leptin receptor. J Biol Chem. 1997;272:6093-6096.
14 Cheatham B, Kahn CR. Insulin action and the insulin signaling network. Endocr Rev. 1995;16:117-142.
15 Sheng HZ, Westphal H. Early steps in pituitary organogenesis. Trends Genet. 1999;15:236-240.
16 Markakis EA. Development of the neuroendocrine hypothalamus. Front Neuroendocrinol. 2002;23:257-291.
17 Knigge KM, Scott DE. Structure and function of the median eminence. Am J Anat. 1970;129:223-244.
18 Johnson AK, Gross PM. Sensory circumventricular organs and brain homeostatic pathways. FASEB J. 1993;7:678-686.
19 Swanson LW, Mogenson GJ. Neural mechanisms for functional coupling of autonomic, endocrine and somatomotor responses in adaptative behavior. Brain Res. 1981;228:1-34.
20 Leclercq T, Grisoli F. Arterial blood supply of the normal human pituitary gland. J Neurosurg. 1983;58:678-681.
21 Roberston GL. Diabetes insipidus. Endocrinol Metab Clin North Am. 1995;24:549-572.
22 Bourque CW, Oliet SHR, Richard D. Osmoreceptors, osmoreception and osmoregulation. Front Neuroendocrinol. 1994;15:231-274.
23 Nakane PK. Classifications of anterior pituitary cell types with immunoenzyme histochemistry. J Histochem Cytochem. 1970;18:9-20.
24 Cohen LE, Radovick S. Molecular basis of combined pituitary hormone deficiencies. Endocr Rev. 2002;23:431-442.
25 Fitzgerald KT. The structure and function of the pars tuberalis of the vertebrate adenohypophysis. Gen Comp Endocrinol. 1979;37:383-399.
26 Vance ML, Mauras N. Growth hormone therapy in adults and children. N Engl J Med. 1999;341:1206-1216.
27 Shimon I, Melmed S. Management of pituitary tumors. Ann Intern Med. 1998;129:472-483.
28 Ben-Jonathan N, Hnasko R. Dopamine as a prolactin (PRL) inhibitor [Review]. Endocr Rev. 2001;22:724-763.
29 Findling JW, Raff H. Screening and diagnosis of Cushing’s syndrome. Endocrinol Metab Clin North Am. 2005;34:385-402.
30 Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116:337-350.
31 Zhang Y, Proenca R, Maffei M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425-432.
32 Ahima RS, Flier JS. Leptin. Annu Rev Physiol. 2000;62:413-437.
33 Ahima RS, Saper CB, Flier JS, et al. Lepin regulation of neuroendocrine systems. Front Neuroendocrinol. 2000;21:263-307.
34 Howard JK, Cave BJ, Oksanen LJ, et al. Enhanced leptin sensitivity and attenuation of dietinduced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004;10:734-738.
35 Mori H, Hanada R, Hanada T, et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to dietinduced obesity. Nat Med. 2004;10:739-743.
36 Niswender KD, Baskin DG, Schwartz MW. Insulin and its evolving partnership with leptin in the hypothalamic control of energy homeostasis. Trends Endocrinol Metab. 2004;15:362-369.
37 Munzberg H, Myers MGJr. Molecular and anatomical determinants of central leptin resistance. Nat Neurosci. 2005;8:566-570.






