14 Neurology and the senses
Examination
Movement and coordination
Cranial nerve examination
I – Olfactory nerve
• Ask whether the child can smell their mother’s cooking or perfume. Formal examination requires use of a panel of different scents but is rarely necessary. Note that very pungent smells like ammonia can still be detected in olfactory nerve dysfunction as the trigeminal nerve also conveys olfactory fibres that detect noxious substances.
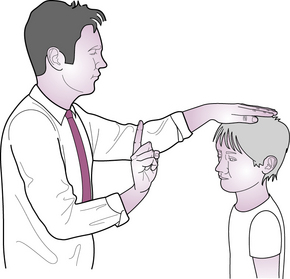
III, IV and VI – Occulomotor, trochlear and abducens nerves
V – Trigeminal nerve
• Ask children to close their eyes and say ‘yes’ when you touch their forehead, cheek and chin on both sides of the face (ophthalmic, maxillary and mandibular divisions of the trigeminal nerve)
• Can they clench the teeth (palpate over the angles of the mandible) and open their mouth against resistance (deviation to the weaker side)? This tests the muscles of mastication.
VIII – Auditory or vestibulocochlear nerve
• How well do the parents think their child can hear?
• Can the child hear your wrist watch, or fingers gently rubbing? Formal hearing tests require special equipment and expertise
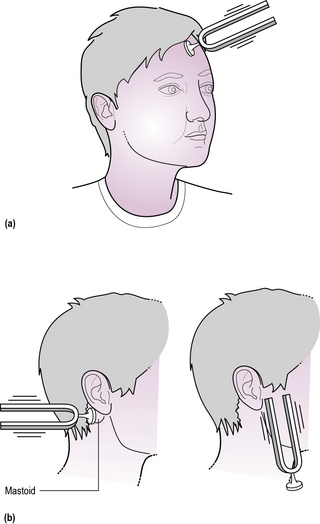
• A tuning fork may help diagnose conductive hearing loss (Rinne and Weber’s test, Figure 14.2), but an age-appropriate hearing assessment and tympanometry (to look for normal movement of the tympanic membrane) is essential if there is any significant concen regarding hearing.
Headaches
Causes of headache in children are summarized in Box 14.1.
Tension headache
Case 14.1 gives an example of tension headache which is the commonest cause of headache in children. The key features are:
• Headache is present ‘all the time’
• Frontal or occipital headaches are described as ‘like a tight band round the head’
• Long (though commonly intermittent) history
• Pain worse as the day goes by
Migraine
• An episodic disorder with defined attacks
• Attacks may occur in clusters
• Visual disturbance (‘aura’), prior to onset in about 30%. Sparkling lights, coloured lines, hemianopia and micropsia are typical. They precede the headache, and typically last for 5–20 minutes. Sensory symptoms such as loss of sensation, and focal motor deficits such as hemiplegia occur uncommonly.
• Incapacitating throbbing headache which may be localized to one side. In young children, headache may be absent or minor, with abdominal pain and vomiting being the most prominent symptoms (‘abdominal migraine’)
• Nausea, vomiting and abdominal pain
Medication, particularly the oral contraceptive pill, may also be a migraine trigger.
Raised intracranial pressure
• Often a short history (less than 3 months)
• Morning or nocturnal headache
• Headache on moving and bending. A degree of neck stiffness may be present
• Behavioural change – irritability, lack of energy, lethargy
• Large head circumference, especially in those under 2 years of age
• Impaired visual acuity or other visual impairment
• Papilloedema – note that this is a comparatively late sign
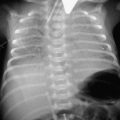
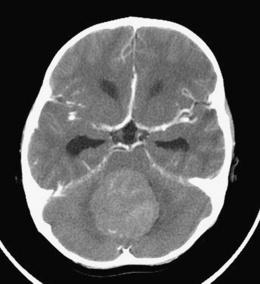
In children over 1 year of age, the commonest cause is a tumour (as in Case 14.2). Brain tumours are collectively the second commonest malignancy in childhood, although still rare (see Chapter 7, pp. 72–74). Other intracranial space-occupying lesions may cause raised pressure, including head injury resulting in subdural bleeding or effusion, particularly in infants and young children, when non-accidental injury must be considered (see also Chapter 5). Infectious causes include postmeningitic hydrocephalus and brain abscess. Cerebral oedema may also cause acutely raised pressure, typically after severe head injury or encephalitis. These conditions frequently present with altered consciousness rather than headache. An example of raised ICP secondary to a medulloblastoma is shown in Figure 14.3.
Management of raised intracranial pressure
In cases of raised ICP where CSF flow is obstructed within the brain (e.g. by congenital aqueduct stenosis in an infant or a posterior fossa tumour in an older child) a ventriculoperitoneal shunt is usually required to relieve the obstruction and prevent brain or retinal damage. In other cases, caused, for example, by meningitis or haemorrhage, the obstruction is within the basal cisterns (as in Case 14.3), the subarachnoid space or the arachnoid villi. These blockages are commonly transient. Repeated CSF drainage by lumbar puncture will reduce CSF protein and may control CSF pressure and head growth for a few days while the blockage resolves spontaneously.
Epileptic and non-epileptic seizures
For management of the child with status epilepticus, see Appendix I, p. 291.
In Case 14.4 the diagnosis of day-dreaming was made because the turns were indistinct with a gradual onset and lasted a long time if the boy was not stimulated. The first-hand account is vital and can avoid the need for investigation.
History
Onset
How did the fit start? Was there an aura? What was the first thing the child or witnesses noticed?
This detailed description of the episode can then be compared with the seizures and turns of childhood described in Tables 14.1 and 14.2.
| Seizure type | Clinical features |
|---|---|
| Tonic-clonic seizure | Rapidly unconscious. Initial tonic (stiff) phase followed by irregular jerking movements (clonic) |
| Focal motor seizure | Clonic movements starting in one limb or one side of the body. May spread and become generalized |
| Absence | Abrupt onset of altered consciousness with postural control maintained. Usually brief (less than 10 seconds) and with no motor features |
| Focal seizures with temporal lobe automatisms | Onset with disturbing or indescribable sensations. Children often announce the onset (‘I’ve got my funny feelings’) and seek the support of a carer. Symptoms include lip smacking, mouthing, repetitive hand movements or stereotyped behaviours |
| Atonic or drop attacks | Abrupt loss of postural tone leading to falls, often with facial injury |
| Myoclonic jerks | Sudden, brief involuntary single or multiple contractions of muscles or muscle groups |
Table 14.2 Non-epileptic seizures
| Seizure type | Clinical features |
|---|---|
| Infants | |
| Apnoea | Includes benign sleep apnoea, apnoea of prematurity and apnoea associated with illness, e.g. RSV (respiratory syncytial virus) infection |
| Laryngeal spasm | Reflex closure of vocal cords as a response to trivial aspiration or reflux |
| Benign sleep | Rhythmic symmetrical jerking of limbs, myoclonus during sleep |
| Preschool children | |
| Reflex anoxic seizure | Vagally mediated bradycardia in response to painful or shocking stimuli. Typically a fall is followed within a few seconds by pallor and collapse, usually with stiffening followed by hypotonia. There may be a few rhythmic limb jerks before recovery, which is rapid and complete |
| Self-gratification (masturbation) | Rhythmic movements of trunk often with limb flexion. Often flushed. Occurs when bored. Can be distracted |
| Tantrums | Need no description |
| Blue breath holding | Starts to cry and gets respiratory arrest at full expiration. Goes blue and stiff or floppy. May suffer an anoxic seizure |
| Night terrors | Children disturbed at night and seem disorientated and confused. Children have no recall the next day |
| Nightmares | Bad dreams. May still be frightened the next day |
| School children | |
| Reflex anoxic seizures | May be precipitated by disgust or revulsion as well as painful stimuli |
| Vasovagal syncope | Typically, tall thin girls in hot school assemblies have these attacks, but they can occur at any age. Key features are pallor and an aura of light-headedness. There may be some jerking while unconscious |
| Day-dreaming | Gradual onset of vague distant behaviour with a vacant stare. May last several minutes. Associated with boredom |
| Non-epileptic attacks | These children are able to mimic an epileptic seizure, producing with variable accuracy the clinical features but the EEG is normal |
Diagnosis
1. Is it really an epileptic seizure? Common patterns of seizures and non-epileptic events at different ages are illustrated in Tables 14.2 and 14.3. Most non-epileptic events can be managed with simple explanation and reassurance.
2. Is it an acute symptomatic seizure? These are seizures caused by an acute insult to a previously normal brain, such as encephalitis, hypoglycaemia or fever. Febrile seizures are a special category of symptomatic seizures (see below). Look for evidence of acute illness. If the child is unconscious see Appendix I, pp. 291–292. Check blood glucose in all children presenting with a seizure. (See Table 14.3 for the causes of acute symptomatic seizures.)
3. Is it a febrile seizure? In young children (6 months to 6 years) seizures may be precipitated by fever alone. Five per cent of children are affected. Typically the child has a brief, generalized, tonic-clonic seizure early in the course of an illness as the temperature is rising rapidly. It is important to exclude meningitis clinically – a lumbar puncture is rarely needed except in those under 1 year. Half of children will have a further febrile seizure but only 10% have three or more. The risk of future epilepsy is 3%. Anticonvulsants are of limited value. Traditionally families have been advised to use paracetamol early in the course of subsequent febrile illnesses. NICE guidance in 2009 made it clear that there is no evidence to support this advice. Tepid sponging is largely impractical and may do more harm than good.
4. Is it an isolated epileptic seizure? Single unprovoked seizures are not uncommon in mid-childhood. They carry a 50% risk of developing epilepsy characterized by further seizures. Normally isolated seizures do not demand investigation, treatment or follow-up.
| Classification | Cause |
|---|---|
| Infections | Meningitis, encephalitis, cerebral abscess |
| Trauma | Head injury, non-accidental injury, post-traumatic intracerebral bleeding |
| Metabolic | Hypoglycaemia, hyponatraemia, hypocalcaemia usually complicating an acute illness |
| Vascular | Spontaneous intracerebral bleeding and stroke Hypertension |
Epilepsy
Such a complex condition demands classification to define clinically important syndromes. The broadest classification is between generalized and partial epilepsies. Generalized epilepsies typically produce seizures such as absences, drop attacks and myoclonic jerks which are associated with a generalized disturbance of brainwave activity. Focal epilepsies on the other hand are characterized by an irritable focus in one part of the brain from which abnormal brainwaves may spread to produce secondary generalization. Either pattern of EEG abnormality can produce a tonic-clonic seizure. If the cause of the epilepsy is unknown, it is described as idiopathic, but if the cause can be defined, e.g. scarring from an old head injury, it is said to be a symptomatic epilepsy. Certain patterns of epilepsy have been defined within this basic classification. These epilepsy syndromes define the aetiology, natural history, EEG appearance and therapy for many childhood epilepsies (see Table 14.4).
| Syndrome | Clinical features | |
|---|---|---|
| Generalized | Infantile spasms | See Case 14.5 |
| Childhood absence | See Case 14.6 | |
| Juvenile myoclonic | Adolescents with early morning myoclonus and tonic-clonic seizures after late nights or alcohol excess | |
| Partial | Temporal lobe epilepsy | See Case 14.7 |
| Symptomatic focal | Epilepsy following a brain insult, e.g. brain injury or meningitis | |
| Benign childhood epilepsy with centrotemporal spikes | Infrequent nocturnal partial seizures in mid-childhood Temporal spikes on EEG |
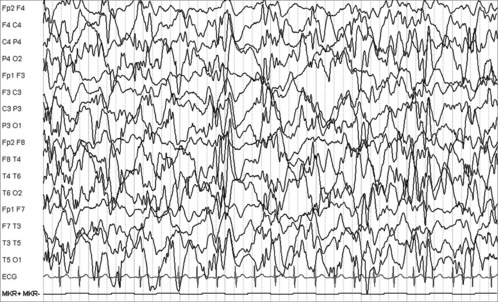
Infantile spasms (Case 14.5) are the clinical manifestation of a severe epileptic encephalopathy characterized by a grossly disordered EEG – hypsarrhythmia (Figure 14.4). The syndrome is treated with steroids, unless there is coexisting tuberous sclerosis (see p. 204), in which case vigabatrin is usually more effective. Lack of response indicates a poor neurodevelopmental outcome, as in this case. Infantile spasms may occur secondary to an underlying congenital or metabolic disorder.
The boy in Case 14.6 has childhood absence epilepsy – an idiopathic generalized epilepsy. Simple absence seizures develop between 5 and 10 years. The treatment of choice would be sodium valproate, lamotrigine or ethosuximide and the condition often resolves in later childhood.
The girl in Case 14.7 is having focal seizures with temporal lobe automatisms (semi-purposeful, repetitive movements like fumbling with buttons). Her awareness at the onset and subsequent tonic-clonic seizure illustrate the progression of the EEG abnormality across the brain. There is an epileptic EEG focus in the right temporal area and an MRI scan shows right-sided temporal lobe changes (mesial temporal sclerosis affecting the right hippocampus). If these seizures fail to respond promptly to anti-epilepsy medications then a referral for epilepsy surgery should be considered.
Case 14.6
A boy with blank spells
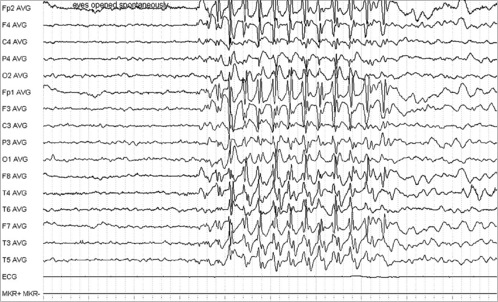
Joshua, a 7-year-old boy, presented after his teacher expressed concern about his concentration. Parents described absence attacks. The onset was abrupt. He stopped what he was doing and stared blankly. The turns lasted about 5 seconds and there was instant recovery. There were no abnormal movements. It was possible to trigger an attack in clinic on getting Joshua to hyperventilate. The EEG demonstrated bursts of 3 per second spike wave activity (see Figure 14.5).
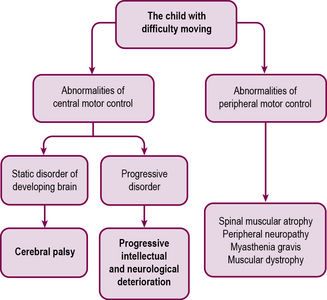
Chronic neurological disability
Difficulties with movement
Difficulties in control of movement may arise in the brain (upper motor neurone) or peripheral nervous system (lower motor neurone) (Figure 14.6). These can usually be distinguished clinically (Table 14.5). In either case, deteriorating conditions must be detected by careful history taking and investigation if necessary.
| Upper motor neurone | Lower motor neurone | |
|---|---|---|
| Power | Normal or reduced | Reduced |
| Tone | Typically increased but can be normal or may reduce with repetition | Reduced |
| Reflexes | Increased | Reduced or absent |
| Plantar responses | Usually up-going | Down-going or absent |
| Chorea or athetosis | Sometimes present | Never present |
| Ataxia | Occasional | Never – but may be confused with weakness |
Abnormalities of central motor control – cerebral palsy
Cerebral palsy is classified by the type of motor abnormality observed (spasticity, chorea, athetosis, dystonia, ataxia) and, if relevant, the parts of the body involved, e.g. spastic diplegia (see Table 14.6).
| By type of movement disorder | |
| Spasticity | Stiff muscles with high tone |
| Athetosis | Unwanted writhing movements |
| Chorea | Unwanted jerky movements |
| Dystonia | Variable tone associated with athetosis |
| Ataxia | Shaky uncoordinated movements |
| By parts of the body involved | |
| Hemiplegia | Involves the arm and leg on one side |
| Quadriplegia | Also called whole-body cerebral palsy |
| Diplegia | Involves four limbs but legs are more severely affected |
| Monoplegia | Rare – look carefully for abnormalities in the other limbs |
There are four major types of cerebral palsy as illustrated in Cases 14.8 to 14.11.
Hemiplegia (65% of CP) typically presents between 6 and 10 months. Birth history is usually normal – it is thought the unilateral brain abnormality develops in early pregnancy, perhaps as a result of a vascular occlusion. In Case 14.8, George will learn to walk – even if this is a little delayed. Associated problems, including learning difficulties, seizures and behaviour problems affect about a third of children and may result in complex disability.
Spastic quadriplegia (10% of CP), or whole-body cerebral palsy (Case 14.9), may result from birth events, although severe congenital brain anomalies cause a similar clinical picture. The associated problems of learning difficulties and epilepsy affect most children when the disability is profound. Long-term complications include a ‘windswept posture’, hip dislocation, scoliosis, gastro-oesophageal reflux and recurrent chest infections.
Spastic diplegia (20% of CP) is the classic abnormality after preterm delivery (Case 14.10), although most children with diplegia have an unknown cause. Inherited forms (X-linked and dominant) are recognized.
Choreoathetoid, or dystonic cerebral palsy (as in Case 14.11) (5% of CP), is rare. Neonatal bilirubin encephalopathy is the classic cause of this type of cerebral palsy but many cases are idiopathic and a few are familial. Deafness is a common association. Intelligence is often normal.
Progressive intellectual and neurological deterioration
Progressive central nervous system disease may present in infancy (as in Case 14.12), childhood or adolescence. Earlier presentations are more severe. Intellect, motor skills, vision and behavioural control may all be lost. Seizures may occur. The cause is often a metabolic or storage disease but some are idiopathic. The prion disease, new variant Creutzfeldt–Jakob disease, remains very rare.
Abnormalities of peripheral motor control – the weak and floppy child
When the pathology of a movement disorder lies in the muscles, the peripheral nerves, the motor end plate or the anterior horn cell within the spinal cord, the resultant clinical picture is of weakness and low tone. The key clinical features in these cases are weakness, which is often progressive, and absence of limb reflexes. Many cases are inherited (see Figure 14.7).
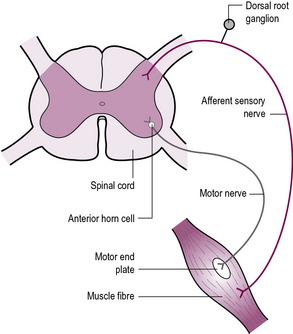
In Case 14.13 the diagnosis of infantile-onset spinal muscular atrophy, also known as Werdnig–Hoffman disease (a recessive condition), was made on DNA analysis. It is incurable and invariably fatal in the first 3 years of life, with the majority dying before their first birthday. Caitlyn quickly deteriorated and died less than a month later of respiratory failure. There may be a 1 in 4 recurrence risk, and genetic counselling is essential. Note that cervical fracture secondary to non-accidental injury may present similar clinical features.
Duchenne muscular dystrophy (Case 14.14) is a slowly and inexorably progressive condition. It is an X-linked recessive condition due to large mutations in the dystrophin gene and so only affects boys. Presentation is typically between 3 and 7 years of age, with lordosis, waddling gait and Gower’s sign. Calf muscle hypertrophy follows within 1–2 years. The majority of boys need a wheelchair by 12 years. About 1 in 3500 boys are affected, worldwide, with 20% of cases arising as de novo mutations. Diagnosis can be quickly obtained by measuring creatine kinase (CK) which is hugely elevated. Genetic confirmation and counselling is necessary for carrier females. Twenty per cent of boys have associated learning difficulties. Careful physical therapy and support is needed to prevent postural deformity and maintain function. Oral steroids have been shown to significantly slow the progression of muscle weakness, and maintain ambulation, on average by an additional 2–3 years.
Trials of novel gene therapies are in the very early stages.
Myasthenia gravis (Case 14.15) is rare. Think of it when weakness is variable, with deterioration during the day, especially with intermittent diplopia and difficulty chewing or swallowing. It is often associated with other autoimmune diseases.
Neurocutaneous syndromes
Two types of neurofibromatosis are recognized. Both are inherited in an autosomal dominant fashion.
Hearing
The child at school
Poor school performance or behaviour may indicate a problem. Speech delay is another common presentation. Screening at school entry picks up some cases. See Table 14.7 for assessment and diagnosis of hearing loss.
| Age | Diagnosis | Assessment |
|---|---|---|
| Neonate | Aminoglycoside toxicity Congenital infection – CMV, rubella Hypoxia associated with birth or prematurity Genetic |
Otoacoustic emission Brain stem evoked potential |
| Child | Otitis media/chronic secretory otitis media (glue ear) Trauma Meningitis |
Parental questionnaire Visual reinforcement audiometry Pure tone audiometry Speech discrimination test |
Chronic secretory otitis media (glue ear)
Deafness (as in Case 14.16) is classified according to the level of impairment (Table 14.8). The impact of this loss on a child is not always easily determined.
| Diagnosis | dB loss |
|---|---|
| Mild | 30–50 |
| Moderate | 50–70 |
| Severe | 70–90 |
| Profound | >90 |
Vision
Squint
Squints are common. They can be divided into paralytic and non-paralytic types.
Diagnosing squints
Managing non-paralytic squints
Visual impairment affects many aspects of development (as shown in case 14.17). The motor skills may be delayed as children are unaware of their surroundings and have no desire to explore. Language development suffers because there is limited association between what the child hears and the object the child perceives. Parents find it difficult to know how best to interact and play with their child and this may further slow developmental progress. Subsequently the assessment and management of children with a severe visual impairment requires a specialist service. Developmental assessments are difficult. Accurate diagnosis is vital. It is unlikely that the impairment can be treated, but other health or developmental problems may be identified as a result.
The white eye
Retinoblastoma
Retinoblastoma is a tumour of photoreceptor cells with an incidence of 5 per million, almost all cases developing before the age of 4 years. The majority occur as sporadic cases but an autosomal-dominant inherited form exists due to deletion of the Rb gene on chromosome 13. Bilateral disease is uncommon in the sporadic form but occurs in up to 70% of genetic cases. Diagnosis is suggested by the finding of a white pupil (‘leukocoria’) and confirmed by detailed ophthalmoscopy under general anaesthetic (as in Case 14.18). Treatment involves localized cryotherapy with or without lens-sparing radiotherapy for small lesions and enucleation of the orbit for more extensive disease. Adjunctive chemotherapy is required for advanced disease.
The red eye
Iritis
Inflammation of the anterior eye may accompany a range of connective tissue diseases, most commonly juvenile idiopathic arthritis (see Chapter 6, p. 55). Presentation may be acute with a painful red eye, photophobia and lacrimation, or chronic with insidious loss of vision. The pupil may appear irregular, and the anterior chamber may appear cloudy or contain debris on slit-lamp examination. Complications include cataract and glaucoma. Treatment is with steroid eye drops with or without antiviral or antibiotic treatment.