[level-membership-for-pediatrics-category]
Neurological disorders
Headache
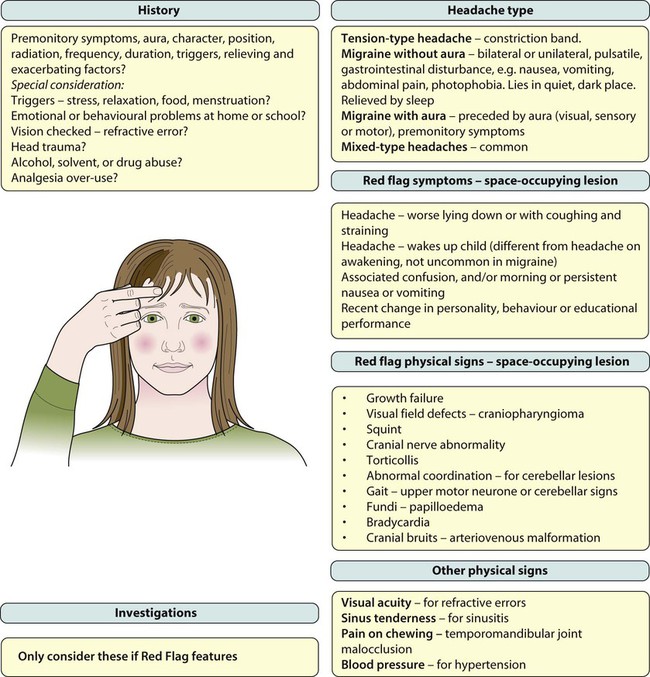
Headache is a frequent reason for older children and adolescents to consult a doctor. The International Headache Society (IHS) has devised a classification, as shown in Box 27.1, which defines:
• Primary headaches: four main groups, comprising migraine, tension-type headache, cluster headache (and other trigeminal autonomic cephalalgias); and other primary headaches (such as cough or exertional headache). They are thought to be due to a primary malfunction of neurones.
• Secondary headaches: symptomatic of some underlying pathology, e.g. from raised intracranial pressure and space-occupying lesions
• Trigeminal and other cranial neuralgias, and other headaches including root pain from herpes zoster.
Primary headaches
Tension-type headache
Migraine with aura
The most common aura comprises visual disturbance, which may include:
• Negative phenomena, such as hemianopia (loss of half the visual field) or scotoma (small areas of visual loss)
• Positive phenomena such as fortification spectra (seeing zigzag lines).
Uncommon forms of migraine
• Familial – linked to a calcium channel defect, dominantly inherited
• Sporadic hemiplegic migraine
• Basilar-type migraine – vomiting with nystagmus and/or cerebellar signs
• Periodic syndromes – often precursors of migraine and include:
– Cyclical vomiting – recurrent stereotyped episodes of vomiting and intense nausea associated with pallor and lethargy. The child is well in between.
– Abdominal migraine – an idiopathic recurrent disorder characterised by episodic midline abdominal pain in bouts lasting 1–72 h. Pain is moderate to severe in intensity and associated with vasomotor symptoms, nausea and vomiting. The child is well in between episodes.
– Benign paroxysmal vertigo of childhood – a heterogeneous disorder characterised by recurrent brief episodes of vertigo occurring without warning and resolving spontaneously in otherwise healthy children. Between episodes, neurological examination, audiometric and vestibular function tests are normal.
Secondary headaches
Raised intracranial pressure and space-occupying lesions
• Visual field defects – from lesions pressing on the optic pathways, e.g. craniopharyngioma (a pituitary tumour)
• Cranial nerve abnormalities causing diplopia, new-onset squint or facial nerve palsy. The VIth (abducens) cranial nerve has a long intracranial course and is often affected when there is raised pressure, resulting in a squint with diplopia and inability to abduct the eye beyond the midline. It is a false localising sign. Other nerves are affected depending on the site of lesion, e.g. pontine lesions may affect the VIIth (facial) cranial nerve and cause a facial nerve palsy
• Torticollis (tilting of the head)
• Growth failure, e.g. craniopharyngioma or hypothalamic lesion
• Papilloedema – a late feature
• Cranial bruits – may be heard in arteriovenous malformations but these lesions are rare.
Management
Rescue treatments
• Analgesia – paracetamol and non-steroidal anti-inflammatory drugs (NSAIDs), taken as early as possible in an individual troublesome episode
• Anti-emetics – prochlorperazine and metoclopramide
• Serotonin (5-HT1) agonists, e.g. sumatriptan. A nasal preparation of this is licensed for use in children over 12 years of age.
Seizures
The causes of seizures are listed in Box 27.2.
Febrile seizures
The acute management of seizures is described in Chapter 6. Examination should focus on the cause of the fever, which is usually a viral illness, but a bacterial infection including meningitis should always be considered. The classical features of meningitis such as neck stiffness and photophobia may not be as apparent in children <18 months of age, so an infection screen (including blood cultures, urine culture and lumbar puncture for CSF) may be necessary. If the child is unconscious or has cardiovascular instability, lumbar puncture is contraindicated and antibiotics should be started immediately.
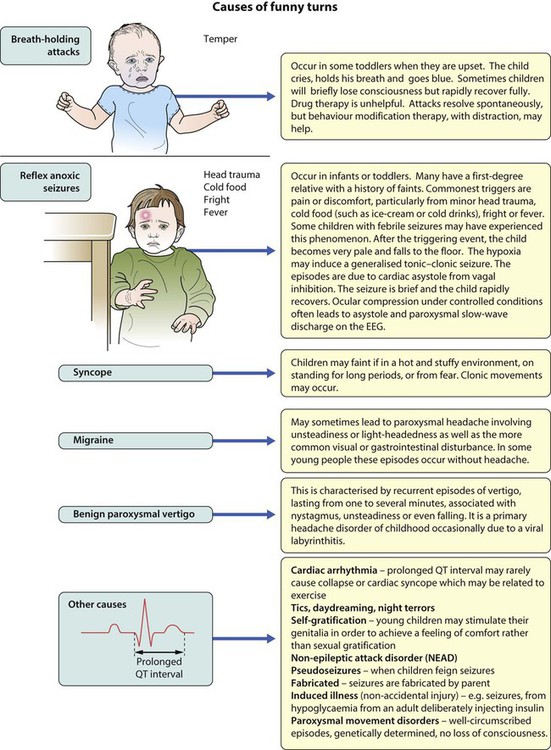
Paroxysmal disorders
There is a broad differential diagnosis for children with paroxysmal disorders (‘funny turns’). Epilepsy is a clinical diagnosis based on the history from eyewitnesses and the child’s own account. If available, videos of the seizures or suspected seizures can be of great help. The diagnostic question is whether the paroxysmal events are that of an epilepsy of childhood or one of the many conditions which mimic it (Fig. 27.1). The most common pitfall is that of syncope leading to an anoxic (non-epileptic) tonic-clonic seizure.
Epilepsies of childhood
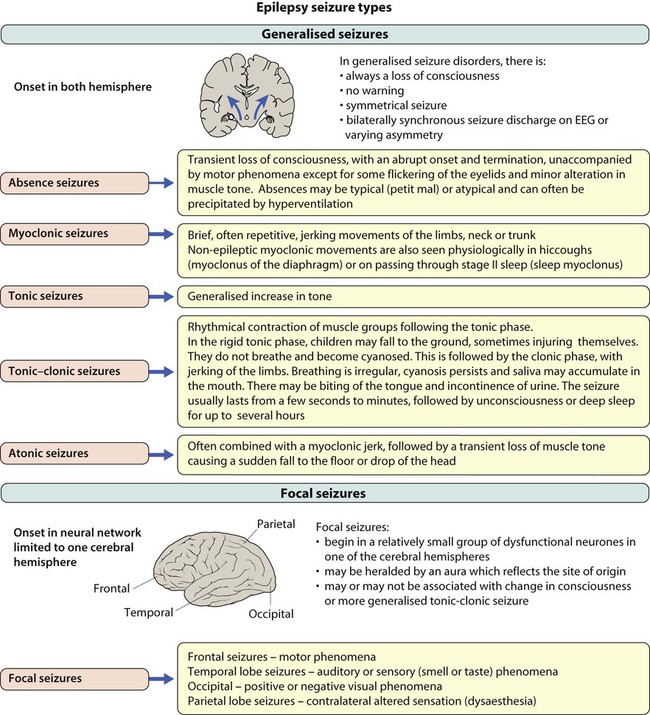
Epilepsy has an incidence of about 0.05% (after the first year of life when it is even more common) and a prevalence of 0.5%. This means that most large secondary schools will have about six children with an epilepsy. Epilepsy is a chronic neurological disorder characterised by recurrent unprovoked seizures, consisting of transient signs and/or symptoms associated with abnormal, excessive or synchronous neuronal activity in the brain. Most epilepsy is idiopathic but other causes of seizures are listed in Box 27.2.
• Generalised – discharge arises from both hemispheres. They may be – absence, myoclonic, tonic, tonic-clonic and atonic
• Focal – where seizures arise from one or part of one hemisphere.
Focal seizure manifestations will depend on the part of the brain where the discharge originates:
• Frontal seizures – involve the motor or premotor cortex. May lead to clonic movements, which may travel proximally (Jacksonian march). Asymmetrical tonic seizures can be seen, which may be bizarre and hyperkinetic and can be mistakenly dismissed as non-epileptic events. Atonic seizures may arise from mesial frontal discharge.
• Temporal lobe seizures, the most common of all the epilepsies – may result in strange warning feelings or aura with smell and taste abnormalities and distortions of sound and shape. Lip-smacking, plucking at one’s clothing and walking in a non-purposeful manner (automatisms) may be seen, following spread to the pre-motor cortex. Déjà-vu and jamais-vu are described (intense feelings of having been, or never having been, in the same situation before). Consciousness can be impaired and the length of event is longer than a typical absence.
• Occipital seizures – cause distortion of vision.
• Parietal lobe seizures – cause contralateral dysaesthesias (altered sensation), or distorted body image.
The main seizure types are summarised in Figure 27.2 and the epilepsy syndromes in Table 27.1.
Table 27.1
Some epilepsy syndromes – arranged by age of onset
| Name | Age | Seizure pattern | Comments |
| West syndrome | 4–6 months | Violent flexor spasms of the head, trunk and limbs followed by extension of the arms (so-called ‘salaam spasms’). Flexor spasms last 1–2 s, often multiple bursts of 20–30 spasms, often on waking, but may occur many times a day. May be misinterpreted as colic. Social interaction often deteriorates – a useful marker in the history |
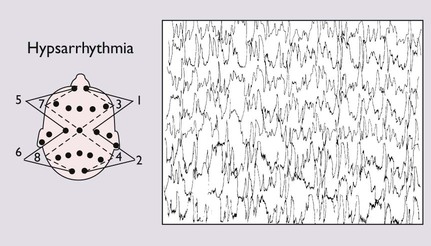
Many causes; two-thirds have underlying neurological cause. The EEG shows hypsarrhythmia, a chaotic pattern of high-voltage slow waves, and multi-focal sharp wave discharges (Fig. 27.3). Treatment is with vigabatrin or corticosteroids; good response in 30–40%, but unwanted effects are common. Most will subsequently lose skills and develop learning disability or epilepsy |
| Lennox–Gastaut syndrome | 1–3 years | Multiple seizure types, but mostly drop attacks (astatic seizures), tonic seizures and atypical absences. Also neurodevelopmental arrest or regression and behaviour disorder | Often other complex neurological problems or history of infantile spasms. Prognosis is poor |
| Childhood absence epilepsy | 4–12 years | Stare momentarily and stop moving, may twitch their eyelids or a hand minimally. Lasts only a few seconds and certainly not longer than 30 s. Child has no recall except realises they have missed something and may look puzzled or say ‘pardon’ on regaining consciousness. Developmentally normal but can interfere with schooling. Accounts for only 2% of childhood epilepsy |
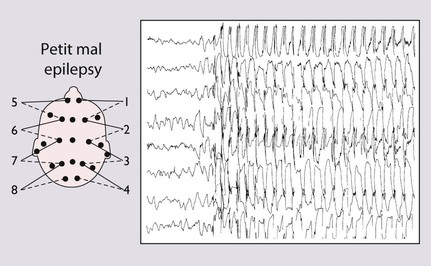
Two-thirds are female. The episodes can be induced by hyperventilation, the child being asked to blow on a piece of paper or windmill for 2–3 min, a useful test in the outpatient clinic. The EEG shows generalised 3/second spike and wave discharge, which is bilaterally synchronous during and sometimes between episodes (Fig. 27.4). Prognosis is good, with 95% remission in adolescence; 5–10% may develop tonic-clonic seizures in adult life |
| Benign* epilepsy, with centrotemporal spikes (BECTS) | 4–10 years | Tonic-clonic seizures in sleep, or simple focal seizures with awareness of abnormal feelings in the tongue and distortion of the face (supplied by the Rolandic area of the brain) | Comprises 15% of all childhood epilepsies. EEG shows focal sharp waves from the Rolandic or centrotemporal area. Important to recognise as it is benign and does not always require treatment. Almost all remit in adolescence |
| Early-onset benign* childhood occipital epilepsy (Panayiotopoulos type) | 1–14 years | Younger children – periods of unresponsiveness, eye deviation, vomiting and autonomic features. Older children – headache and visual disturbance including distortion of images and hallucinations | Uncommon. EEG shows occipital discharges. Remit in childhood |
| Juvenile myoclonic epilepsy | Adolescence-adulthood | Myoclonic seizures, but generalised tonic-clonic seizures and absences may occur, mostly shortly after waking. A typical history is throwing drinks or cornflakes about in the morning as myoclonus occurs at this time. Learning is unimpaired | Characteristic EEG. Response to treatment is usually good but lifelong. A genetic linkage has been identified. Remission unlikely |
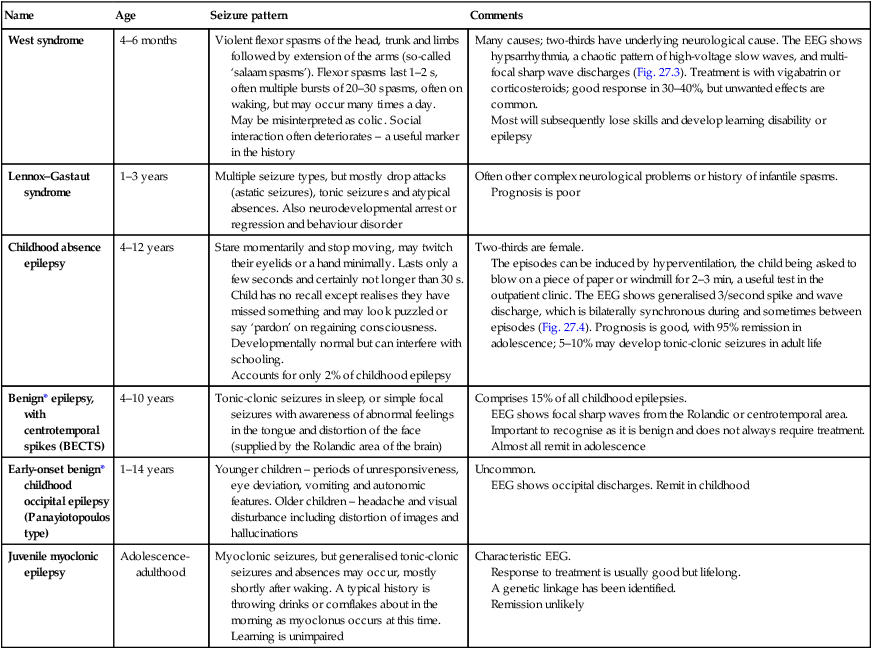
*Although called benign, may be specific learning difficulties in some children.
Investigation of seizures
Imaging
• Structural. MRI and CT brain scans are not required routinely for childhood generalised epilepsies. They are indicated if there are neurological signs between seizures, or if seizures are focal, in order to identify a tumour, vascular lesion, or area of sclerosis which could be treatable. MRI FLAIR (fluid-attenuated inversion recovery) sequences better detect mesial temporal sclerosis in temporal lobe epilepsy.
• Functional scans. While it is not always possible to see structural lesions, techniques have advanced to allow functional imaging to detect areas of abnormal metabolism suggestive of seizure foci. These include PET (positron emission tomography) and SPECT (single positron emission computed tomography), which use isotopes and ligands, injected and taken up by metabolically active cells. Both can be used between seizures to detect areas of hypometabolism in epileptogenic lesions. SPECT can also be used to capture seizures and areas of hypermetabolism. Functional MRI can be used alongside psychological testing –including memory assessment – to minimise the risk of postoperative impairment.
Management
Anti-epileptic drug (AED) therapy
• Not all seizures require AED therapy. This decision should be based on the seizure type, frequency and the social and educational consequences of the seizures set against the possibility of unwanted effects of the drugs.
• Choose the appropriate drug for the seizure. Inappropriate AEDs may be detrimental, e.g. carbamazepine can make absence and myoclonic seizures worse.
• Monotherapy at the minimum dosage is the desired goal, although in practice more then one drug may be required.
• All AEDs have potential unwanted effects and these should be discussed with the child and parent.
• Drug levels are not measured routinely, but may be useful to check for adherence to advice or with some drugs with erratic pharmacokinetics, e.g. phenytoin.
• Children with prolonged seizures are given rescue therapy to keep with them. This is usually a benzodiazepine, e.g. rectal diazepam or buccal midazolam.
• AED therapy can usually be discontinued after 2 years free of seizures.
Guidance regarding treatment options for different seizure types are shown in Table 27.2. Common unwanted effects of AEDs are shown in Table 27.3.
Table 27.2
Choice of anti-epileptic drugs (NICE 2004)
| Seizure type | First-line | Second-line |
| Generalised seizures | ||
| Tonic-clonic | Valproate, carbamazepine | Lamotrigine, topiramate |
| Absence | Valproate, ethosuximide | Lamotrigine |
| Myoclonic | Valproate | Lamotrigine |
| Focal seizures | Carbamazepine, valproate Lamotrigine shown since to be most effective – but slow titration |
Topiramate, levetiracetam, oxcarbazepine, gabapentin, tiagabine, vigabatrin |
Table 27.3
Common or important unwanted effects of anti-epileptic drugs
| Drug | Side-effects |
| Valproate | Weight gain, hair loss |
| Rare idiosyncratic liver failure | |
| Carbamazepine/oxcarbazepine | Rash, neutropenia, hyponatraemia, ataxia |
| Liver enzyme induction, can interfere with other medication | |
| Vigabatrin | Restriction of visual fields, which has limited its use |
| Sedation | |
| Lamotrigine | Rash |
| Ethosuximide | Nausea and vomiting |
| Topiramate | Drowsiness, withdrawal and weight loss |
| Gabapentin | Insomnia |
| Levetiracetam | Sedation – rare |
| Benzodiazepines – clobazam, clonazepam, diazepam, nitrazepam | Sedation, tolerance to effect, increased secretions |
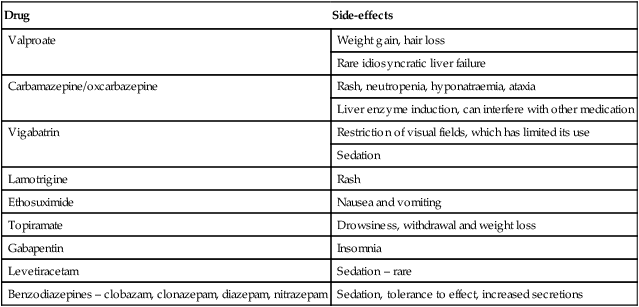
All the above may cause drowsiness and occasional skin rashes.
Other treatment options
In children with intractable seizures, there are a number of radical treatment options.
• Ketogenic (fat-based) diets may be helpful in some children. Its mechanism of action is poorly understood.
• Vagal nerve stimulation, delivered using externally programmable stimulation of a wire implanted around the vagal nerve, may possibly be useful; trials are being conducted.
• Surgery. Cessation of seizures and drug therapy may be achieved in some children whose clinical seizures are well-localised as demonstrated by good concordance between EEG, MRI and functional imaging findings. The main procedure is temporal lobectomy for mesial temporal sclerosis, but other procedures include hemispherectomy or hemispherotomy (isolation of hemisphere which is not removed so as avoid post-operative shifts in space) and other focal resections. Detailed assessment is required to ensure that the benefits outweigh the risks.
Central motor disorders
The three central movement control centres are:
• Motor cortex, lying along the pre-central gyrus (the homunculus reflects the body upside down, legs superiorly and face inferiorly, just above the Sylvian fissure; with large areas to govern fine movements of the tongue, fingers and thumb). Information from here passes down the corticospinal (pyramidal) tracts to link with the basal ganglia.
• Basal ganglia, deep grey matter structures, store patterns of movement so that we need not put conscious effort into every movement we make.
• Cerebellum, acting as the ‘air-traffic controller’; receiving feedback on joint position nanosecond by nanosecond to keep us on course as we execute movements.
Disorders of these central movement control centres are:
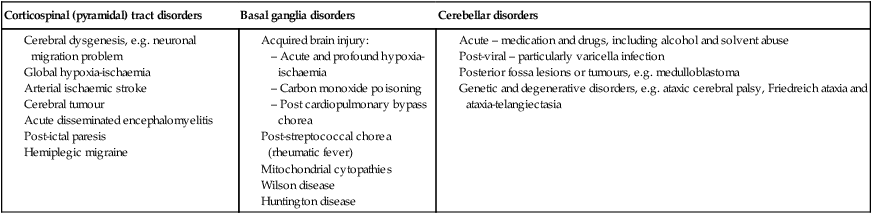
• Corticospinal (pyramidal) tract disorders – there is weakness with a pattern of adduction at the shoulder, flexion at the elbow and pronation of the forearm; adduction and internal rotation at the hip, flexion at hip, knee and plantar flexion at the ankle with brisk hyper-reflexia and extensor plantars. Fine finger movement will be lost.
• Basal ganglia disorders – will lead either to difficulty initiating movement, with fluctuating (largely increased) tone – a ‘dystonia’, or a ‘dyskinesia’, where packets of movement information are released to give jerky movement (chorea) or writhing movement (athetosis).
• Cerebellar disorders – will lead to difficulty holding a posture (particularly with eyes closed); past-pointing (dysmetria); poor alternating movements (dysdiadochokinesis); and a characteristic scanning dysarthria. Posterior sensory pathway problems may give a similar clinical picture but are much rarer in childhood. The gait is wide-based. Associated nystagmus may be seen. Causes of these disorders are listed in Table 27.4.
Table 27.4
| Corticospinal (pyramidal) tract disorders | Basal ganglia disorders | Cerebellar disorders |
Peripheral motor disorders: the neuromuscular disorders
Any part of the lower motor pathway can be affected in a neuromuscular disorder, so that anterior horn cell disorders, peripheral neuropathies, disorders of neuromuscular transmission and primary muscle diseases can all occur. The causes of neuromuscular disorders are shown in Figure 27.5. The key clinical feature of a neuromuscular disorder is weakness, which may be progressive or static. Affected children may present with:
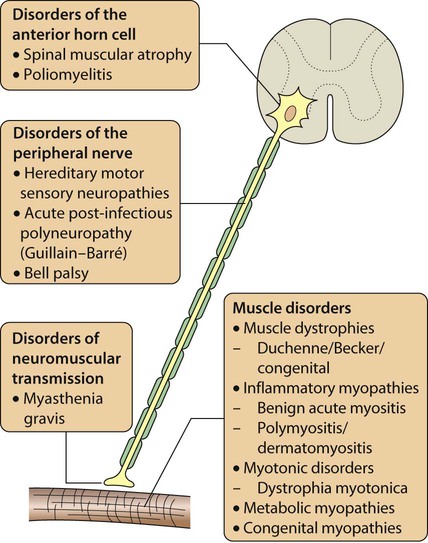
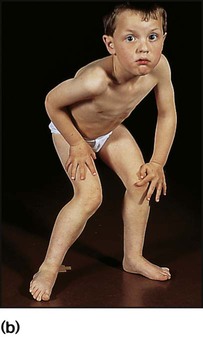
History and examination may provide useful clues. Children with myopathy often show a waddling gait or positive Gowers sign suggestive of proximal muscle weakness. Gowers sign is the need to turn prone to rise to a standing from a supine position. This is normal until the age of 3 years. It is only when children have become very weak that they ‘climb up the legs with the hands’ to gain the standing position (Fig. 27.6). A pattern of more distal wasting and weakness, particularly in the presence of pes cavus, suggests an hereditary motor sensory neuropathy. Increasing fatiguability through the day, often with ophthalmoplegia and ptosis, would be more consistent with depletion at the motor end-plate and a diagnosis of myasthenia gravis.

• Anterior horn cell – there are signs of denervation: weakness, loss of reflexes, fasciculation and wasting as the nerve supply to the muscle fails.
• Neuropathy – often distal nerves affected. Motor neuropathy will give weakness, sensory neuropathy will give impaired perception of pain and temperature, or touch, with a loss of reflexes in either.
• Myopathy – there is weakness (often proximal), wasting, gait disturbance.
• Neuromuscular junction – as end-plate acetylcholine stores become depleted, there is diurnal worsening through the day, leading to fatiguability.
Investigations
• Serum creatine phosphokinase – markedly elevated in Duchenne and Becker muscular dystrophy and inflammatory myopathies
• Muscle biopsy, needle or open – modern histochemical techniques often enable a definitive diagnosis
• DNA testing – to identify abnormal genes
• Ultrasound and MRI of muscles – used in specialist centres to diagnose and monitor progress.
• Nerve conduction studies – to identify delayed motor and sensory nerve conduction velocities seen in neuropathy
• DNA testing – for abnormal genes
• Nerve biopsy – rarely performed
• EMG (electromyography) helps in differentiating myopathic from neuropathic disorders, e.g. fatiguability on repetitive nerve stimulation in myasthenia. However, it should be used selectively in children, as the nerve conduction studies cause a tingling sensation and electromyography requires insertion of fine needle electrodes.
Disorders of the anterior horn cell
Presentation is with weakness, wasting and absent reflexes. The features of poliomyelitis are described in Chapter 14.
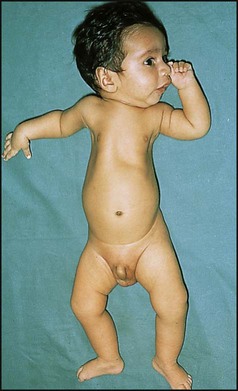
Spinal muscular atrophy type 1 (Werdnig–Hoffmann disease)
A very severe progressive disorder presenting in early infancy (Fig. 27.7). Diminished fetal movements are often noticed during pregnancy and there may be arthrogryposis (positional deformities of the limbs with contractures of at least two joints) at birth. Typical signs include:
Peripheral neuropathies
The hereditary motor sensory neuropathies (HMSN)
Bell palsy and facial nerve palsies
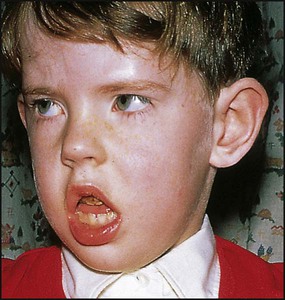
Bell palsy is an isolated lower motor neurone paresis of the VIIth cranial nerve leading to facial weakness (Fig. 27.8). Although the aetiology is unclear, it is probably post-infectious with an association with herpes simplex virus in adults. Corticosteroids may be of value in reducing oedema in the facial canal during the first week; no benefit from aciclovir has been demonstrated. Recovery is complete in the majority of cases but may take several months. The main complication is conjunctival infection due to incomplete eye closure on blinking. This may require the eye to be protected with a patch or even tarsorrhaphy.
Disorders of neuromuscular transmission
Myasthenia gravis
This presents as abnormal muscle fatiguability which improves with rest or anticholinesterase drugs.
Juvenile myasthenia
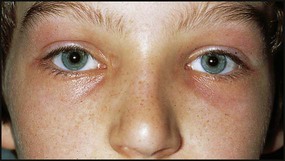
This is similar to adult autoimmune myasthenia and is due to binding of antibody to acetylcholine receptors on the post-junctional synaptic membrane. This gives a reduction of the number of functional receptors. Presentation is usually after 10 years of age with ophthalmoplegia and ptosis, loss of facial expression and difficulty chewing (Fig. 27.9). Generalised, especially proximal, weakness may be seen.
Muscle disorders
The muscular dystrophies
This is a group of inherited disorders with muscle degeneration, often progressive.
Metabolic myopathies
• Glycogen storage disorders (see Ch. 25).
• Disorders of lipid metabolism. Fatty acids are important muscle fuel. Fatty acid oxidation occurs in the mitochondria and defects in this pathway can result in weakness. Carnitine is essential to supply long-chain fatty acids to the mitochondria for breakdown, and carnitine deficiency causes weakness.
• Mitochondrial cytopathies. Rare disorders which are coded as maternally inherited mitochondrial DNA. Myopathy may be the major manifestation or the disorder may be multisystem, with lactic acidosis and encephalopathy. Mitochondrial DNA testing is available.
The inflammatory myopathies
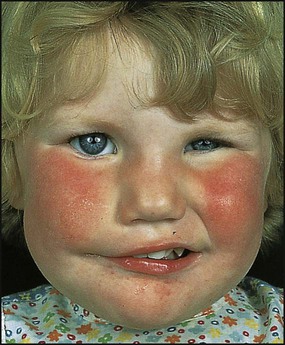
Dermatomyositis
This is a systemic illness, probably due to an angiopathy. Usual onset is between 5 and 10 years. This can be acute, but more typically is insidious with fever, misery, and eventually symmetrical muscle weakness, which is mainly proximal. Sometimes pharyngeal muscle involvement affects swallowing. There is also a characteristic violaceous (heliotrope) rash to the eyelids, and periorbital oedema (Fig. 27.10). The rash may also affect the extensor surfaces of joints, e.g. elbow, and with time subcutaneous calcification can appear. Inflammatory markers (CRP, ESR) can be raised but not invariably. Muscle biopsy shows an inflammatory cell infiltrate and atrophy. Physiotherapy is needed to prevent contractures. Corticosteroids are the standard treatment, and continue at a tailored dose for 2 years. Other immunosuppressants, e.g. methotrexate, ciclosporin, may be needed. Mortality is 5–10%.
Myotonic disorders
Dystrophia myotonica
This relatively common illness is dominantly inherited and caused by a nucleotide triplet repeat expansion, so this means there can be anticipation through generations, especially when maternally transmitted (see Ch. 8). Newborns can present with hypotonia and feeding and respiratory difficulties due to muscle weakness. It is then useful to examine the mother for myotonia. This manifests as slow release of handshake or difficulty releasing the tightly clasped fist. This may be mild and not have been appreciated. Sensitivity is required as diagnosis in the neonate may have repercussions for the family. Older children can present with myopathic facies (Fig. 27.11), learning difficulties and myotonia. Adults develop cataracts and males develop baldness and testicular atrophy. Death is usually due to cardiomyopathy.
The ‘floppy infant’
Persisting hypotonia in infants can be readily felt on picking up the infant, who tends to slip through the fingers or hang like a rag doll when suspended prone. There will be marked head lag when the head is lifted by the arms from supine. The causes are listed in Box 27.3. The clinical examination may help determine the site of the lesion, whether cortical or neuromuscular. Central hypotonia is associated with poor truncal tone but preserved limb tone. Dysmorphic features suggest a genetic cause. Lower motor neurone lesions are suggested by a frog-like posture (Fig. 27.7), poor antigravity movements and absent reflexes.
Ataxia
Friedreich ataxia
Ataxia telangiectasia
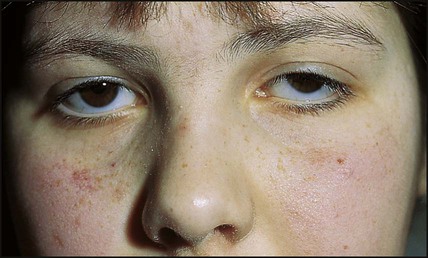
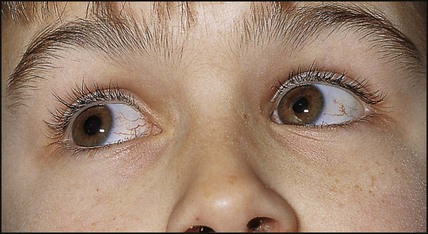
This disorder of DNA repair is an autosomal recessive condition. The gene (ATM) has been identified. There may be mild delay in motor development in infancy and oculomotor problems with incoordination and delay in ocular pursuit of objects (oculomotor dyspraxia), with difficulty with balance and coordination becoming evident at school age. There is subsequent deterioration, with a mixture of dystonia and cerebellar signs. Many children require a wheelchair for mobility in early adolescence. Telangiectasia develops in the conjunctiva (Fig. 27.12), neck and shoulders from about 4 years of age. These children:
• Have an increased susceptibility to infection, principally from an IgA surface antibody defect
• Develop malignant disorders, principally acute lymphoblastic leukaemia (about 10%)
• Have a raised serum alpha-fetoprotein
• Have an increased white cell sensitivity to irradiation, which can be used diagnostically, but the ATM gene test is now mostly used.
Cerebrovascular disease
Stroke
Infantile stroke is dealt with in Chapter 10. Childhood stroke may be due to vascular, thromboembolic or haemorrhagic disease. The clinical presentation is determined by the vascular territory involved. There is compromise of the anterior circulation (internal carotid, anterior and middle cerebral arteries), which leads to hemiparesis with or without speech disturbance. Less common is compromise of the posterior circulation (vertebrobasilar arteries) with associated visual or cerebellar signs.
• Cardiac: congenital cyanotic heart disease, e.g. Fallot tetralogy, endocarditis
• Haematological: sickle cell disease; deficiencies of anti-thrombotic factors, e.g. protein S
• Post-infective: following varicella or other viral infection
• Inflammatory: damage to vessels in autoimmune disease, e.g. SLE (systemic lupus erythematosus)
• Metabolic/genetic: homocystinuria, mitochondrial disorders, e.g. MELAS (myoclonic epilepsy, lactic acidosis and stroke); CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy), the most common form of hereditary stroke disorder
• Vascular malformations: moyamoya disease. These children have abnormal vasculature. Moyamoya comes from the Japanese for ‘puff of smoke’, similar to the blurred appearance seen on angiography.
Neural tube defects and hydrocephalus
Neural tube defects
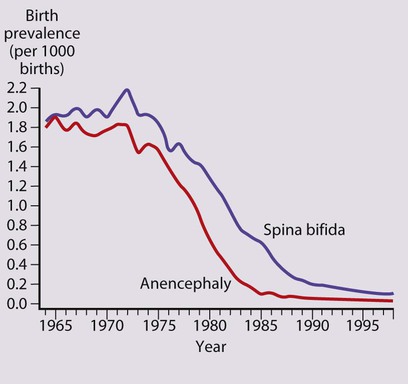
Neural tube defects result from failure of normal fusion of the neural plate to form the neural tube during the first 28 days following conception. Their birth prevalence in the UK has fallen dramatically from 4 per 1000 live births in the 1970s to 0.15 per 1000 live births in 1998 and to 0.11 per 1000 live births in 2005 (Fig. 27.13). This is mainly because of a natural decline, as well as antenatal screening.
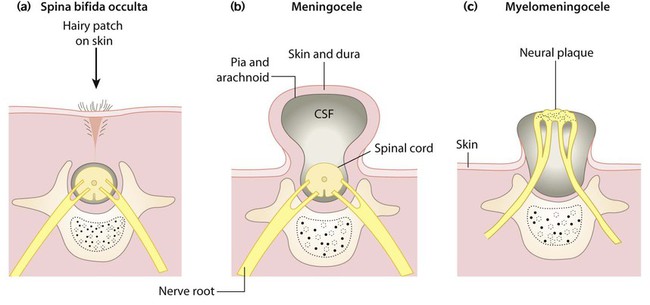
Spina bifida occulta
This failure of fusion of the vertebral arch (Fig. 27.14a) is often an incidental finding on X-ray, but there may be an associated overlying skin lesion such as a tuft of hair, lipoma, birth mark or small dermal sinus, usually in the lumbar region. There may be underlying tethering of the cord (diastematomyelia), which, with growth, may cause neurological deficits of bladder function and lower limbs. The extent of the underlying lesion can be delineated using ultrasound and/or MRI scans. Neurosurgical relief of tethering is usually indicated.
Meningocele and myelomeningocele
Meningoceles (Fig. 27.14b) usually have a good prognosis following surgical repair.
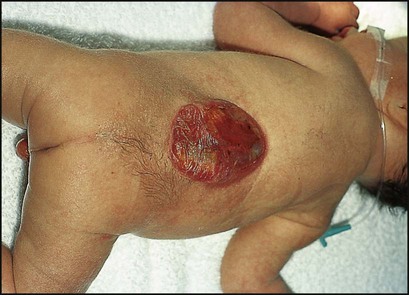
Myelomeningoceles (Figs 27.14c, 27.15) may be associated with:
• Variable paralysis of the legs
• Muscle imbalance, which may cause dislocation of the hip and talipes
• Bladder denervation (neuropathic bladder)
• Bowel denervation (neuropathic bowel)
• Hydrocephalus from the Chiari malformation (herniation of the cerebellar tonsils and brainstem tissue through the foramen magnum), leading to disruption of CSF flow.
Management – The back lesion is usually closed soon after birth.
For sensory loss – skin care is required to avoid the development of skin damage and ulcers.
Hydrocephalus
In hydrocephalus, there is obstruction to the flow of cerebrospinal fluid, leading to dilatation of the ventricular system proximal to the site of obstruction. The obstruction may be within the ventricular system or aqueduct (non-communicating or obstructive hydrocephalus), or at the arachnoid villi, the site of absorption of CSF (communicating hydrocephalus) (Box 27.4).
Clinical features
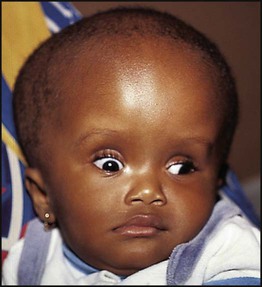
In infants with hydrocephalus, as their skull sutures have not fused, the head circumference may be disproportionately large or show an excessive rate of growth. The skull sutures separate, the anterior fontanelle bulges and the scalp veins become distended. An advanced sign is fixed downward gaze or sun setting of the eyes (Fig. 27.16). Older children will develop signs and symptoms of raised intracranial pressure.
Hydrocephalus
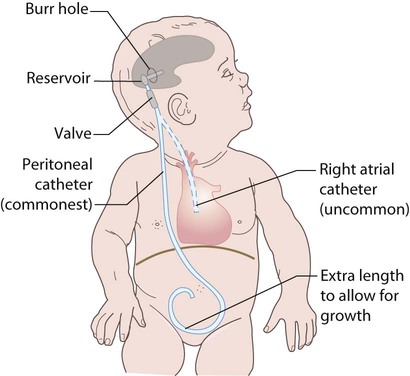
Treatment is required for symptomatic relief of raised intracranial pressure and to minimise the risk of neurological damage. The mainstay is the insertion of a ventriculoperitoneal shunt (Fig. 27.17), but endoscopic treatment to create a ventriculostomy can now be performed. Shunts can malfunction due to blockage or infection (usually with coagulase-negative staphylococci). They then need replacing or revising. Overdrainage of fluid can cause low-pressure headaches but the insertion of regulatory valves can help avoid this.
The neurocutaneous syndromes
Neurofibromatosis type 1 (NF1)
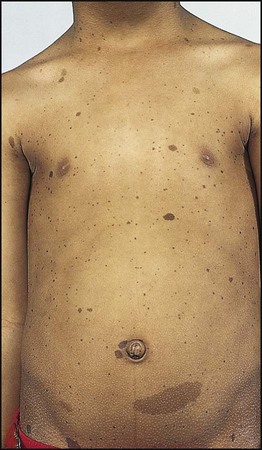
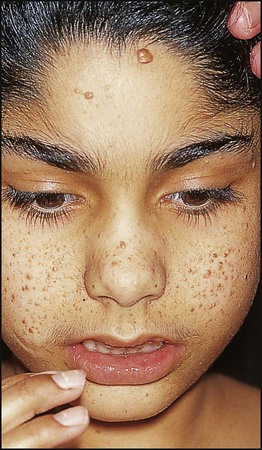
In order to make the diagnosis, two or more of these criteria need to be present:
• Six or more café-au-lait spots >5 mm in size before puberty, >15 mm after puberty (Fig. 27.18)
• More than one neurofibroma, an unsightly firm nodular overgrowth of any nerve
• Axillary freckles (Fig. 27.18)
• Optic glioma which may cause visual impairment
• One Lisch nodule, a hamartoma of the iris seen on slit-lamp examination
• Bony lesions from sphenoid dysplasia, which can cause eye protrusion
Tuberous sclerosis
The cutaneous features consist of:
• Depigmented ‘ash leaf’-shaped patches which fluoresce under ultraviolet light (Wood’s light)
• Roughened patches of skin (shagreen patches) usually over the lumbar spine
• Adenoma sebaceum (angiofibromata) in a butterfly distribution over the bridge of the nose and cheeks, which are unusual before the age of 3 years (Fig. 27.19).
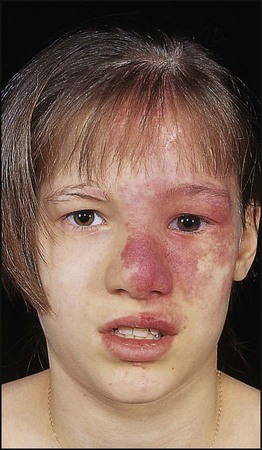
Sturge–Weber syndrome
This is a sporadic disorder with a haemangiomatous facial lesion (a port-wine stain) in the distribution of the trigeminal nerve associated with a similar lesion intracranially. The ophthalmic division of the trigeminal nerve is always involved (Fig. 27.20). Calcification of the gyri used to show characteristic ‘rail-road track’ calcification on skull X-ray, but MRI is the imaging modality of choice nowadays. In the most severe form, it may present with epilepsy, learning disability and hemiplegia. Children presenting with intractable epilepsy in early infancy may benefit from hemispherectomy. For children who are less severely affected, deterioration is unusual after the age of 5 years, although there may still be seizures and learning difficulties. There is a high risk of glaucoma, which should be assessed in the neonatal period.
Neurodegenerative disorders
• Lysosomal storage disorders, e.g. lipid storage disorders and mucopolysaccharidoses, in which absence of an enzyme leads to accumulation of a harmful metabolite
• Peroxisomal enzyme defects, e.g. X-linked adrenoleucodystrophy. Peroxisomes are catalase- and oxidase-containing organelles involved in long-chain fatty acid oxidation. Enzyme deficiencies can lead to accumulation of very long-chain fatty acids (VLCFAs)
• Heredodegenerative disorders, e.g. Huntington disease, which presents with progressive dystonia, dementia, seizures and corticospinal tract signs
• Wilson disease, from the accumulation of copper, may cause changes in behaviour and additional involuntary movements or a mixture of neurological and hepatic symptoms
• Subacute sclerosing panencephalitis (SSPE), a delayed response in adolescence to previous measles infection causing neurological regression with a characteristic EEG, but has become rare since measles immunisation.
Lysosomal storage disorders
In lipid storage disorders (Table 27.5), which are sphingolipidoses, there is an accumulation of sphingolipids, essential components of CNS membranes. They are diagnosed on testing white cell enzymes.
Table 27.5
| Disorder | Enzyme defect | Clinical features |
| Tay–Sachs disease | Hexosaminidase A | Autosomal recessive disorder |
| Most common among Ashkenazi Jews | ||
| Developmental regression in late infancy, exaggerated startle response to noise, visual inattention and social unresponsiveness | ||
| Severe hypotonia, enlarging head | ||
| Cherry red spot at the macula | ||
| Death by 2–5 years | ||
| Diagnosis – measurement of the specific enzyme activity | ||
| Carrier detection of high-risk couples is practised | ||
| Prenatal detection is possible | ||
| Gaucher disease | Beta-glucosidase | Occurs in 1 in 500 Ashkenazi Jews |
| Chronic childhood form – splenomegaly, bone marrow suppression, bone involvement, normal IQ | ||
| Splenectomy may alleviate hypersplenism | ||
| Enzyme replacement therapy is available, but is expensive | ||
| Acute infantile form – splenomegaly, neurological degeneration with seizures | ||
| Carrier detection and prenatal diagnosis are possible | ||
| Niemann–Pick disease | Sphingomyelinase | At 3–4 months, feeding difficulties and failure to thrive, hepatosplenomegaly, developmental delay, hypotonia and deterioration of hearing and vision |
| Cherry red spot in macula affects 50% | ||
| Death by 4 years |
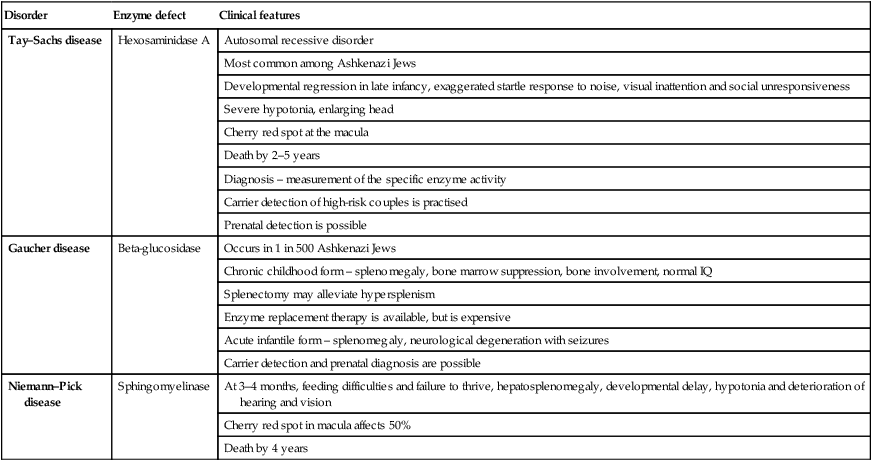
The mucopolysaccharidoses are progressive multisystem disorders which may affect the neurological, ocular, cardiac and skeletal systems (Table 27.6). Hepatosplenomegaly is usually present. Most children present with developmental delay following a period of essentially normal growth and development up to 6–12 months of age. Developmental attainment then slows and children may show some loss of skills. It is only in the second 6 months of life that the characteristic facies begin to emerge, with coarsening of the facial features and prominent forehead due to frontal bossing (Fig. 27.21).
Mucopolysaccharidoses
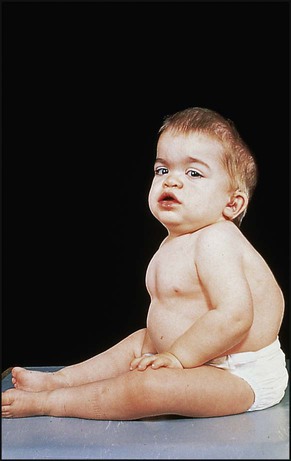
Table 27.6
Clinical features of mucopolysaccharidoses
| Eyes | Corneal clouding |
| Retinal degeneration | |
| Glaucoma | |
| Skin | Thickened skin |
| Coarse facies | |
| Heart | Valvular lesions |
| Cardiac failure | |
| Neurology | Developmental regression |
| Skeletal | Thickened skull |
| Broad ribs | |
| Claw hand | |
| Thoracic kyphosis | |
| Lumbar lordosis | |
| Other | Hepatosplenomegaly |
| Carpal tunnel syndrome | |
| Conductive deafness | |
| Umbilical and inguinal hernias |
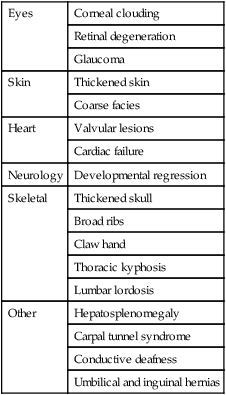
Table 27.7
Types of mucopolysaccharidoses
| Type | Inheritance | Cornea | Heart | Brain | Skeletal |
| MPS I (Hurler) | AR | +++ | ++ | +++ | ++ |
| MPS II (Hunter) | X-linked | − | + | ++ | + |
| MPS III (Sanfilippo) | AR | ± | − | + | + |
| MPS IV (Morquio) | AR | + | + | − | +++ |
| MPS VI (Maroteaux–Lamy) | AR | +++ | ++ | − | ++ |
AR, autosomal recessive.
The characteristics of five of the varieties are shown in Table 27.7. The diagnosis is made by identifying the enzyme defect and the excretion in the urine of the major storage substances, the glycosaminoglycans (GAGs). Treatment is supportive according to the child’s needs. Successful enzyme replacement by bone marrow transplantation has been performed but cannot reverse any established neurological abnormality.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Neurological disorders
Headache
Headache is a frequent reason for older children and adolescents to consult a doctor. The International Headache Society (IHS) has devised a classification, as shown in Box 27.1, which defines:
• Primary headaches: four main groups, comprising migraine, tension-type headache, cluster headache (and other trigeminal autonomic cephalalgias); and other primary headaches (such as cough or exertional headache). They are thought to be due to a primary malfunction of neurones.
• Secondary headaches: symptomatic of some underlying pathology, e.g. from raised intracranial pressure and space-occupying lesions
• Trigeminal and other cranial neuralgias, and other headaches including root pain from herpes zoster.
Primary headaches
Tension-type headache
Migraine with aura
The most common aura comprises visual disturbance, which may include:
• Negative phenomena, such as hemianopia (loss of half the visual field) or scotoma (small areas of visual loss)
• Positive phenomena such as fortification spectra (seeing zigzag lines).
Uncommon forms of migraine
• Familial – linked to a calcium channel defect, dominantly inherited
• Sporadic hemiplegic migraine
• Basilar-type migraine – vomiting with nystagmus and/or cerebellar signs
• Periodic syndromes – often precursors of migraine and include:
– Cyclical vomiting – recurrent stereotyped episodes of vomiting and intense nausea associated with pallor and lethargy. The child is well in between.
– Abdominal migraine – an idiopathic recurrent disorder characterised by episodic midline abdominal pain in bouts lasting 1–72 h. Pain is moderate to severe in intensity and associated with vasomotor symptoms, nausea and vomiting. The child is well in between episodes.
– Benign paroxysmal vertigo of childhood – a heterogeneous disorder characterised by recurrent brief episodes of vertigo occurring without warning and resolving spontaneously in otherwise healthy children. Between episodes, neurological examination, audiometric and vestibular function tests are normal.
Secondary headaches
Raised intracranial pressure and space-occupying lesions
• Visual field defects – from lesions pressing on the optic pathways, e.g. craniopharyngioma (a pituitary tumour)
• Cranial nerve abnormalities causing diplopia, new-onset squint or facial nerve palsy. The VIth (abducens) cranial nerve has a long intracranial course and is often affected when there is raised pressure, resulting in a squint with diplopia and inability to abduct the eye beyond the midline. It is a false localising sign. Other nerves are affected depending on the site of lesion, e.g. pontine lesions may affect the VIIth (facial) cranial nerve and cause a facial nerve palsy
• Torticollis (tilting of the head)
• Growth failure, e.g. craniopharyngioma or hypothalamic lesion
• Papilloedema – a late feature
• Cranial bruits – may be heard in arteriovenous malformations but these lesions are rare.
Management
Rescue treatments
• Analgesia – paracetamol and non-steroidal anti-inflammatory drugs (NSAIDs), taken as early as possible in an individual troublesome episode
• Anti-emetics – prochlorperazine and metoclopramide
• Serotonin (5-HT1) agonists, e.g. sumatriptan. A nasal preparation of this is licensed for use in children over 12 years of age.
Seizures
The causes of seizures are listed in Box 27.2.
Febrile seizures
The acute management of seizures is described in Chapter 6. Examination should focus on the cause of the fever, which is usually a viral illness, but a bacterial infection including meningitis should always be considered. The classical features of meningitis such as neck stiffness and photophobia may not be as apparent in children <18 months of age, so an infection screen (including blood cultures, urine culture and lumbar puncture for CSF) may be necessary. If the child is unconscious or has cardiovascular instability, lumbar puncture is contraindicated and antibiotics should be started immediately.
Paroxysmal disorders
There is a broad differential diagnosis for children with paroxysmal disorders (‘funny turns’). Epilepsy is a clinical diagnosis based on the history from eyewitnesses and the child’s own account. If available, videos of the seizures or suspected seizures can be of great help. The diagnostic question is whether the paroxysmal events are that of an epilepsy of childhood or one of the many conditions which mimic it (Fig. 27.1). The most common pitfall is that of syncope leading to an anoxic (non-epileptic) tonic-clonic seizure.
Epilepsies of childhood
Epilepsy has an incidence of about 0.05% (after the first year of life when it is even more common) and a prevalence of 0.5%. This means that most large secondary schools will have about six children with an epilepsy. Epilepsy is a chronic neurological disorder characterised by recurrent unprovoked seizures, consisting of transient signs and/or symptoms associated with abnormal, excessive or synchronous neuronal activity in the brain. Most epilepsy is idiopathic but other causes of seizures are listed in Box 27.2.
• Generalised – discharge arises from both hemispheres. They may be – absence, myoclonic, tonic, tonic-clonic and atonic
• Focal – where seizures arise from one or part of one hemisphere.
Focal seizure manifestations will depend on the part of the brain where the discharge originates:
• Frontal seizures – involve the motor or premotor cortex. May lead to clonic movements, which may travel proximally (Jacksonian march). Asymmetrical tonic seizures can be seen, which may be bizarre and hyperkinetic and can be mistakenly dismissed as non-epileptic events. Atonic seizures may arise from mesial frontal discharge.
• Temporal lobe seizures, the most common of all the epilepsies – may result in strange warning feelings or aura with smell and taste abnormalities and distortions of sound and shape. Lip-smacking, plucking at one’s clothing and walking in a non-purposeful manner (automatisms) may be seen, following spread to the pre-motor cortex. Déjà-vu and jamais-vu are described (intense feelings of having been, or never having been, in the same situation before). Consciousness can be impaired and the length of event is longer than a typical absence.
• Occipital seizures – cause distortion of vision.
• Parietal lobe seizures – cause contralateral dysaesthesias (altered sensation), or distorted body image.
The main seizure types are summarised in Figure 27.2 and the epilepsy syndromes in Table 27.1.
Table 27.1
Some epilepsy syndromes – arranged by age of onset
| Name | Age | Seizure pattern | Comments |
| West syndrome | 4–6 months | Violent flexor spasms of the head, trunk and limbs followed by extension of the arms (so-called ‘salaam spasms’). Flexor spasms last 1–2 s, often multiple bursts of 20–30 spasms, often on waking, but may occur many times a day. May be misinterpreted as colic. Social interaction often deteriorates – a useful marker in the history |
Many causes; two-thirds have underlying neurological cause. The EEG shows hypsarrhythmia, a chaotic pattern of high-voltage slow waves, and multi-focal sharp wave discharges (Fig. 27.3). Treatment is with vigabatrin or corticosteroids; good response in 30–40%, but unwanted effects are common. Most will subsequently lose skills and develop learning disability or epilepsy |
| Lennox–Gastaut syndrome | 1–3 years | Multiple seizure types, but mostly drop attacks (astatic seizures), tonic seizures and atypical absences. Also neurodevelopmental arrest or regression and behaviour disorder | Often other complex neurological problems or history of infantile spasms. Prognosis is poor |
| Childhood absence epilepsy | 4–12 years | Stare momentarily and stop moving, may twitch their eyelids or a hand minimally. Lasts only a few seconds and certainly not longer than 30 s. Child has no recall except realises they have missed something and may look puzzled or say ‘pardon’ on regaining consciousness. Developmentally normal but can interfere with schooling. Accounts for only 2% of childhood epilepsy |
Two-thirds are female. The episodes can be induced by hyperventilation, the child being asked to blow on a piece of paper or windmill for 2–3 min, a useful test in the outpatient clinic. The EEG shows generalised 3/second spike and wave discharge, which is bilaterally synchronous during and sometimes between episodes (Fig. 27.4). Prognosis is good, with 95% remission in adolescence; 5–10% may develop tonic-clonic seizures in adult life |
| Benign* epilepsy, with centrotemporal spikes (BECTS) | 4–10 years | Tonic-clonic seizures in sleep, or simple focal seizures with awareness of abnormal feelings in the tongue and distortion of the face (supplied by the Rolandic area of the brain) | Comprises 15% of all childhood epilepsies. EEG shows focal sharp waves from the Rolandic or centrotemporal area. Important to recognise as it is benign and does not always require treatment. Almost all remit in adolescence |
| Early-onset benign* childhood occipital epilepsy (Panayiotopoulos type) | 1–14 years | Younger children – periods of unresponsiveness, eye deviation, vomiting and autonomic features. Older children – headache and visual disturbance including distortion of images and hallucinations | Uncommon. EEG shows occipital discharges. Remit in childhood |
| Juvenile myoclonic epilepsy | Adolescence-adulthood | Myoclonic seizures, but generalised tonic-clonic seizures and absences may occur, mostly shortly after waking. A typical history is throwing drinks or cornflakes about in the morning as myoclonus occurs at this time. Learning is unimpaired | Characteristic EEG. Response to treatment is usually good but lifelong. A genetic linkage has been identified. Remission unlikely |
*Although called benign, may be specific learning difficulties in some children.
Investigation of seizures
Imaging
• Structural. MRI and CT brain scans are not required routinely for childhood generalised epilepsies. They are indicated if there are neurological signs between seizures, or if seizures are focal, in order to identify a tumour, vascular lesion, or area of sclerosis which could be treatable. MRI FLAIR (fluid-attenuated inversion recovery) sequences better detect mesial temporal sclerosis in temporal lobe epilepsy.
• Functional scans. While it is not always possible to see structural lesions, techniques have advanced to allow functional imaging to detect areas of abnormal metabolism suggestive of seizure foci. These include PET (positron emission tomography) and SPECT (single positron emission computed tomography), which use isotopes and ligands, injected and taken up by metabolically active cells. Both can be used between seizures to detect areas of hypometabolism in epileptogenic lesions. SPECT can also be used to capture seizures and areas of hypermetabolism. Functional MRI can be used alongside psychological testing –including memory assessment – to minimise the risk of postoperative impairment.
Management
Anti-epileptic drug (AED) therapy
• Not all seizures require AED therapy. This decision should be based on the seizure type, frequency and the social and educational consequences of the seizures set against the possibility of unwanted effects of the drugs.
• Choose the appropriate drug for the seizure. Inappropriate AEDs may be detrimental, e.g. carbamazepine can make absence and myoclonic seizures worse.
• Monotherapy at the minimum dosage is the desired goal, although in practice more then one drug may be required.
• All AEDs have potential unwanted effects and these should be discussed with the child and parent.
• Drug levels are not measured routinely, but may be useful to check for adherence to advice or with some drugs with erratic pharmacokinetics, e.g. phenytoin.
• Children with prolonged seizures are given rescue therapy to keep with them. This is usually a benzodiazepine, e.g. rectal diazepam or buccal midazolam.
• AED therapy can usually be discontinued after 2 years free of seizures.
Guidance regarding treatment options for different seizure types are shown in Table 27.2. Common unwanted effects of AEDs are shown in Table 27.3.
Table 27.2
Choice of anti-epileptic drugs (NICE 2004)
| Seizure type | First-line | Second-line |
| Generalised seizures | ||
| Tonic-clonic | Valproate, carbamazepine | Lamotrigine, topiramate |
| Absence | Valproate, ethosuximide | Lamotrigine |
| Myoclonic | Valproate | Lamotrigine |
| Focal seizures | Carbamazepine, valproate Lamotrigine shown since to be most effective – but slow titration |
Topiramate, levetiracetam, oxcarbazepine, gabapentin, tiagabine, vigabatrin |
Table 27.3
Common or important unwanted effects of anti-epileptic drugs
| Drug | Side-effects |
| Valproate | Weight gain, hair loss |
| Rare idiosyncratic liver failure | |
| Carbamazepine/oxcarbazepine | Rash, neutropenia, hyponatraemia, ataxia |
| Liver enzyme induction, can interfere with other medication | |
| Vigabatrin | Restriction of visual fields, which has limited its use |
| Sedation | |
| Lamotrigine | Rash |
| Ethosuximide | Nausea and vomiting |
| Topiramate | Drowsiness, withdrawal and weight loss |
| Gabapentin | Insomnia |
| Levetiracetam | Sedation – rare |
| Benzodiazepines – clobazam, clonazepam, diazepam, nitrazepam | Sedation, tolerance to effect, increased secretions |
[/not-level-membership-for-pediatrics-category]
