17 Neurological Alterations and Management
After reading this chapter, you should be able to:
• differentiate cerebral hypoxia from cerebral ischaemia and focal from global ischaemia
• differentiate between primary and secondary brain injuries due to brain injury
• relate the procedures of selected neurodiagnostic tests to nursing implications for patient care
• discuss the rationale for medical and nursing management in the care of the brain-injured patient.
Concepts of Neurological Dysfunction
Alterations in Consciousness
Altered Cognition and Coma
Coma is a state of unresponsiveness from which the patient, who appears to be asleep, cannot be aroused by verbal and physical stimuli to produce any meaningful response; therefore, the diagnosis of coma implies the absence of both arousal and content of consciousness.1 Coma must be considered a symptom with numerous causes, different natural modes, and several management modes.
Somnolence is a state of unconsciousness from which the patient can be fully awakened. Although there are many specific causes of unconsciousness, the sites of cerebral affection are either the bilateral cerebral cortex or the brainstem reticular activating system. The commonest causes of bilateral cortical disease are deficiencies of oxygen, metabolic disorders, physical injury, toxins, postconvulsive coma and infections.2 The reticular activating system maintains the state of wakefulness through continuous stimulation of the cortex. Any interruption may lead to unconsciousness. The reticular activating system can be affected in three principal ways: by supratentorial pressure, by infratentorial pressure, and by intrinsic brainstem lesions. Supratentorial lesions produce impaired consciousness by enlarging and displacing tissue. Lesions that affect the brainstem itself damage the reticular activating system directly.
Aetiology of altered cognition
Recently gained confusion, severe apathy, stupor or coma implies dysfunction of the cerebral hemispheres, the diencephalon and/or the upper brainstem.3 Focal lesions in supratentorial structures may damage both hemispheres, or may produce swelling that compresses the diencephalic activating system and midbrain, causing transtentorial herniation and brainstem damage. Primary subtentorial (brainstem or cerebellar) lesions may compress or directly damage the reticular formation anywhere between the level of the midpons and, (by upward pressure), the diencephalon. Metabolic or infectious diseases may depress brain functions by a change in blood composition or the presence of a direct toxin. Impaired consciousness may also be due to reduced blood flow (as in syncope or severe heart failure) or a change in the brain’s electrical activity (as in epilepsy). Concussion, anxiolytic drugs and anaesthetics impair consciousness without producing detectable structural changes in the brain.
Many of the enzymatic reactions of neurons, glial cells, and specialised cerebral capillary endothelium in the brain must be catalysed by the energy-yielding hydrolysis of adenosine triphosphate (ATP) to adenosine diphosphate (ADP) and inorganic phosphate. Without a constant and generous supply of ATP, cellular synthesis slows or stops, neuronal functions decline or cease, and cell structures quickly fall apart.4 The brain depends entirely on the process of glycolysis and respiration within its own cells to provide its energy needs. Even a short interruption of blood flow or oxygen supply threatens tissue vitality.
Seizures
A seizure is an uninhibited, abrupt discharge of ions from a group of neurons resulting in epileptic activity.5 The majority of patients experiencing seizures in the ICU do not have preexisting epilepsy, and their chances of developing epilepsy in the future are usually more dependent on the cause than on the number or intensity of seizures that they experience. However, because of other deleterious neuronal and systemic effects of seizures, their rapid diagnosis and suppression during a period of critical illness is necessary.
Aetiology of seizures
Seizures may either prompt the patient’s admission to ICU (because of status epilepticus) or develop as a complication of another illness.6 Seizures can be due to vascular, infectious, neoplastic, traumatic, degenerative, metabolic, toxic or idiopathic causes. Factors influencing the development of posttraumatic epilepsy include an early posttraumatic seizure, depressed skull fracture, intracranial haematoma, dural penetration, focal neurological deficit and posttraumatic amnesia (PTA) over 24 hours with the presence of a skull fracture or haematoma. Seizures in critically ill patients are most commonly due to drug effects; metabolic, infectious or toxic disorders; and intracranial mass lesions although they may be due to trauma or neoplasm.7 Conditions producing seizures tend either to increase neuronal excitation or to impair neuronal inhibition. A few generalised disorders (e.g. non-ketotic hyperglycaemia) may produce partial or focal seizures.
Alterations in Motor and Sensory Function
Muscle tone, which is a necessary component of muscle movement, is a function of the muscle spindle (myotatic) system and the extrapyramidal system, which monitors and buffers input to the lower motor neurons by way of the multisynaptic pathways.8 Upper motor neuron lesions produce spastic paralysis, and lower motor neuron lesions produce flaccid paralysis. Damage to the upper motor and sensory neurons of the corticospinal, corticobulbar and spinothalamic tracts is a common component of stroke.9 Polyneuropathies involve multiple peripheral nerves and produce symmetrical sensory, motor, and mixed sensorimotor deficits:
• Lesions of the corticospinal and corticobulbar tracts: result in weakness or total paralysis of predominantly distal voluntary movement, Babinski’s sign (i.e. dorsiflexion of the big toe and fanning of the other toes in response to stroking the outer border of the foot from heel to toe), and often spasticity (increased muscle tone and exaggerated deep tendon reflexes).
• Disorders of the basal ganglia: (extrapyramidal disorders) do not cause weakness or reflex changes. Their hallmark is involuntary movement (dyskinesia), causing increased movement (hyperkinesias) or decreased movement (hypokinesia) and changes in muscle tone and posture.
• Cerebellar disorders: cause abnormalities in the range, rate and force of movement. Strength is minimally affected.
Autonomic Nerve Dysfunction
There is strong evidence for numerous interactions among the central nervous system (CNS), peripheral nervous system (both sympathetic and parasympathetic branches), the endocrine system, and the immune system, hence ANS dysfunction is related to that complex triad.10 Autonomic nerve (AN) dysfunction ranges from alterations in the sympathetic–parasympathetic balance to almost complete cessation as occurs in spinal cord injury. As the ANS controls organ function AN dysfunction is related to all-organ alteration and failure. The immune system is connected to the nervous system through the ANS with many of the patients with infections, systemic inflammatory response and multi-organ failure exhibiting AN dysfunction. AN dysfunction is closely related to systemic inflammation hence those with conditions with increased levels of inflammatory markers such as chronic disease and obesity have predisposing AN dysfunction. AN dysfunction is assessed by time and spectral domain heart rate variability and is currently being researched as a neurological assessment technique.11
Alterations in Cerebral Metabolism and Perfusion
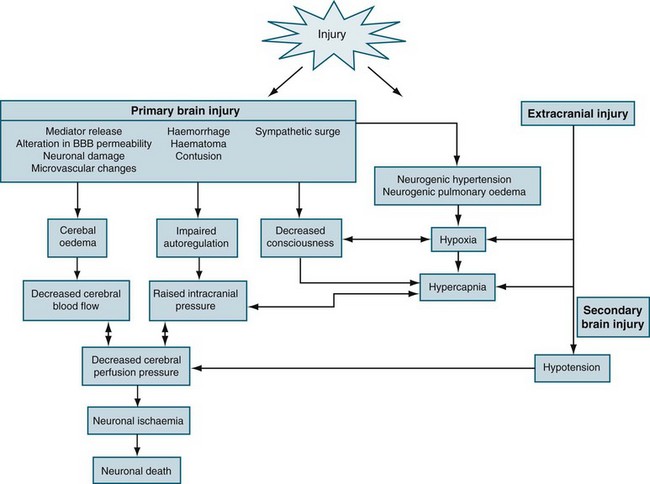
For decades, impairment of cerebral metabolism has been attributed to impaired oxygen delivery, mediated by reduced cerebral perfusion in the swollen cerebral parenchyma. Accordingly, reduction of ICP is usually argued for restoration of previously compromised cerebral perfusion for improvement of cerebral metabolism. Although uncontrolled ICP elevation has been shown to be responsible for reduced oxygen delivery, non-ischaemic impairment of oxidative metabolism and mitochondrial damage has only recently been recognised as a prominent source of energy crisis triggered by brain injury in the presence of adequate cerebral blood flow.12 Accumulating evidence has shown that the mitochondrion has a pivotal role in post traumatic neuronal death by integrating numerous noxious signals responsible for both structural and functional damage on one hand and by amplifying these signals through activation of several cellular signalling events leading to cell death. In addition, more complex processes with the alteration of cerebral perfusion, such as cerebral hypoperfusion, ischaemia, reperfusion injury, inflammation and oedema result in increased intracranial pressure (ICP).
Cerebral Ischaemia
Ischaemia is the inadequate delivery of oxygen, the inadequate removal of carbon dioxide from the cell, and an increase in the production of intracellular lactic acid. Ischaemia can be caused by an increase in nutrient utilisation by the brain in a hyperactive state, a decrease in delivery related to either cerebral or systemic complications, and/or a mismatch between delivery and demand.13 The ischaemic cascade is described in Figure 17.1. Inflammation, together with oxidative stress, excitotoxicity, disrupted calcium homeostasis and energy failure, is one of the key pathological changes in ischaemic brain damage.14 There is a significant inflammatory response in ischaemic brains, including leucocyte and monocyte infiltration into the brain, activation of microglia and astrocytes, elevated production of inflammatory cytokines and chemokines and increased expression and activity of adhesion molecules, complement and metalloproteinases. Of importance, brain ischaemia can lead to significant inflammatory responses in the central nervous system and can also cause significant changes in the peripheral immune system. There are two phases. In the relatively early phase, activated spleen cells and lymph nodes and blood mononuclear cells secrete significantly enhanced levels of TNF-α, IL-6 and IL-2. This then results in global immunosuppression affecting the spleen, lymph nodes, thymus and a significant decrease in the number of immune cells in the circulation.15 When cerebral blood flow (CBF) falls to about 40% of normal, EEG slowing occurs. When CBF falls below 10 mL/100 g/min (20%), the function of ionic pumps fails, which leads to membrane depolarisation. Cerebral ischaemia and reperfusion injury contribute to the cascade of physiological events, termed secondary brain injury. Recent studies have shown that low-dose paracetamol reduces inflammatory protein release from brain endothelial cells exposed to oxidant stress16 and that propofol protects against neuronal apotosis.17
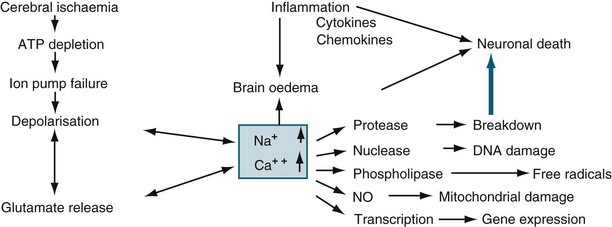
Cerebral Oedema
Cerebral oedema is defined as increased brain water content. The brain is particularly susceptible to injury from oedema, because it is located within a confined space and cannot expand, and because there are no lymphatic pathways within the CNS to carry away the fluid that accumulates. The white matter is usually much more involved, as myelinated fibres have a loose extracellular space, while the grey matter has a much higher cell density with many connections and much less loose extracellular space.18 The two main subdivisions of cerebral oedema are extracellular and intracellular.
Intracellular (cytotoxic) oedema
Cellular swelling, usually of astrocytes in the grey matter, is generally seen after cerebral ischaemia caused by cardiac arrest or minor head injury.19 The blood–brain barrier (BBB) is intact and capillary permeability is not impaired. The cause of intracellular oedema is anoxia and ischaemia; it is usually not clinically significant, and is reversible in its early phases.
Extracellular (vasogenic) oedema
Extracellular oedema involves increased capillary permeability, and had been termed ‘BBB breakdown’.20 Rises in brain water content with extracellular oedema are often quite dramatic, because the fluid that results from increased capillary permeability is usually rich in proteins, resulting in the spread of oedema and brain ischaemia. This can lead to cytotoxic oedema, and to the progressive breakdown of both astrocytes and neurons.19 While the classification of oedema is useful to define specific treatments, it is somewhat arbitrary, as cytotoxic and vasogenic oedema often occur concurrently. In fact, each of these processes may cause the other. Ultimately, these changes can lead to raised intracranial pressure and herniation.
Hydrocephalus
Hydrocephalus is the result of an imbalance between the formation and drainage of cerebrospinal fluid (CSF). Reduced absorption most often occurs when one or more passages connecting the ventricles become blocked, preventing movement of CSF to its drainage sites in the subarachnoid space just inside the skull.21 This type of hydrocephalus is called ‘non-communicating’. Reduction in absorption rate, called ‘communicating hydrocephalus’ can be caused by damage to the absorptive tissue. Both types lead to an elevation of the CSF pressure within the brain. A third type of hydrocephalus, ‘normal pressure hydrocephalus’, is marked by ventricle enlargement without an apparent rise in CSF pressure, which mainly affects the elderly.
Hydrocephalus may be caused by: congenital brain defects; haemorrhage, in either the ventricles or the subarachnoid space; CNS infection (syphilis, herpes, meningitis, encephalitis or mumps); and tumours. Irritability is the commonest sign of hydrocephalus in infants and, if untreated, may lead to lethargy. Bulging of the fontanelle, the soft spot between the skull bones, may also be an early sign. Hydrocephalus in infants prevents fusion of the skull bones, and causes expansion of the skull. Symptoms of normal pressure hydrocephalus include dementia, gait abnormalities and incontinence.22 Treatment includes ventriculostomy drainage of CSF in the short term, or a surgical shunt for those with chronic conditions. Either is predisposed to blockage and infection.
Intracranial Hypertension
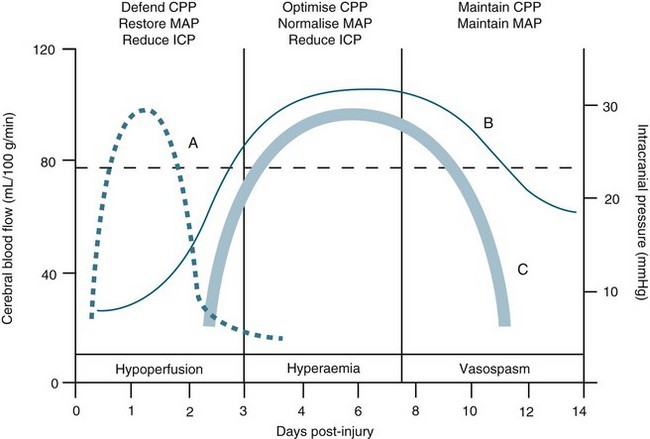
Intracranial pressure is the pressure exerted by the contents of the brain within the confines of the skull and the BBB. The Munro–Kelly hypothesis states that the contents of the cranium (60% water, 40% solid) are not compressible and thus an increase in volume causes a rapid rise in pressure and changes to the compensatory reserve and pulse amplitude, as illustrated in Figure 17.2.23 Normal ICP is 0–10 mmHg, and a sustained pressure of >15 mmHg is termed intracranial hypertension, with implications for CBF.24 Areas of focal ischaemia appear when ICP is >20 mmHg and global ischaemia occurs at >50 mmHg. ICP waveform contains valuable information about the nature of cerebrospinal pathophysiology. ICP increased to the level of systemic arterial pressure extinguishes cerebral circulation, which will restart only if arterial pressure rises sufficiently beyond the ICP to restore cerebral blood flow. Autoregulation of cerebral blood flow and compliance of the cerebrospinal system are both expressed in ICP. Methods of waveform analysis are useful, both to derive this information and to guide the management of patients.25
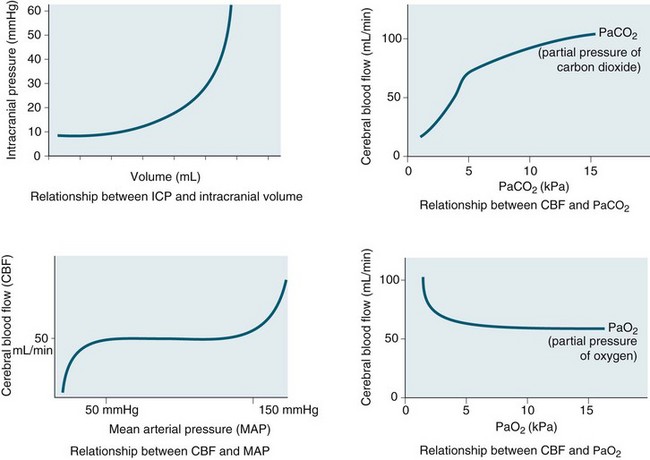
Initially, intracranial compliance allows compensation for rises in intracranial volume due to autoregulation. During a slow rise in volume in a continuous mode, the ICP rises to a plateau level at which the increased level of CSF absorption keeps pace with the rise in volume with ample compensatory reserve. This is expressed as an index, as shown in Figure 17.3.26 Intermittent expansion causes only a transient rise in ICP at first. When sufficient CSF has been absorbed to accommodate the volume, the ICP returns to normal. The ICP finally rises to the level of arterial pressure which itself begins to rise, accompanied by bradycardia or other disturbances of heart rhythm (termed the Cushing’s response). This is accompanied by dilation of the small pial arteries and some slowing of venous flow, which is followed by pulsatile venous flow.
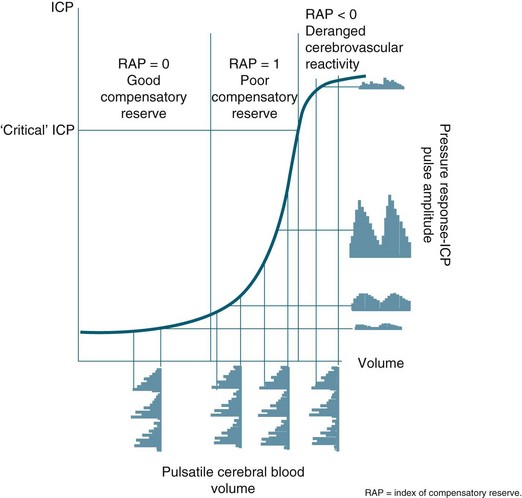
FIGURE 17.3 In a simple model, pulse amplitude of intracranial pressure (ICP) (expressed along the y-axis on the right side of the panel) results from pulsatile changes in cerebral blood volume (expressed along the x-axis) transformed by the pressure–volume curve. This curve has three zones: a flat zone, expressing good compensatory reserve, an exponential zone, depicting poor compensatory reserve, and a flat zone again, seen at very high ICP (above the ‘critical’ ICP) depicting derangement of normal cerebrovascular responses. The pulse amplitude of ICP is low and does not depend on mean ICP in the first zone. The pulse amplitude increases linearly with mean ICP in the zone of poor compensatory reserve. In the third zone, the pulse amplitude starts to decrease with rising ICP. RAP, index of compensatory reserve.26
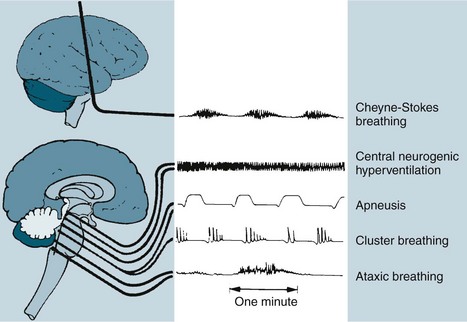
The respiratory changes depend on the level of brainstem involved. A midbrain involvement results in Cheyne-Stokes respiration. When the midbrain and pons are involved, there is sustained hyperventilation. There are rapid and shallow respirations with upper medulla involvement, with ataxic breathing in the final stages (see Figure 17.4).27
Often, neurogenic pulmonary oedema may occur due to increased sympathetic activity as a result of the effects of elevated ICP on the hypothalamus, medulla or cervical spinal cord. The causes of intracranial hypertension are classified as acute or chronic. Acute causes include brain trauma, ischaemic injury and intracerebral haemorrhage. Infections such as encephalitis or meningitis may also lead to intracranial hypertension. Chronic causes include many intracranial tumours, such as ependymomas, or subdural bleeding that may gradually impinge on CSF pathways and interfere with CSF outflow and circulation. As the ICP continues to increase, the brain tissue becomes distorted, leading to herniation and additional vascular injury.28
Neurological Therapeutic Management
Optimising Cerebral Perfusion and Oxygenation
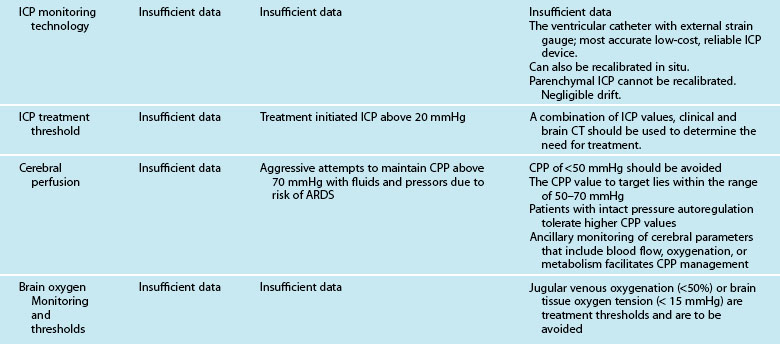
Intracranial hypertension and cerebral ischaemia are the two most important secondary injury processes that can be anticipated, monitored and treated in the ICU. This applies to all aetiologies of brain injury including trauma. This section discusses the modalities of neuroprotection, including the management of intracranial hypertension, vasospasm and cerebral ischaemia. Nursing interventions for the prevention of secondary insults and promotion of cerebral perfusion are described in Table 17.1. Importantly, the aims of nursing management are based on published guidelines and are directed at optimising cerebral perfusion and metabolism by various initiatives.
TABLE 17.1 Nursing interventions for the promotion of cerebral perfusion in acute brain injury
• Effectively reduce ICP while preserving or improving CPP
• Position body with neck straight and no knee elevation in order to maintain venous outflow.
• Make sure cervical collar and endotracheal tube ties are not too tight, especially behind the neck.
• If patient has a ventriculostomy, drain per doctor’s orders.
• Elevate head of bed above the level of the heart to obtain optimal level of ICP and CPP. Monitor ICP, CPP and PbtO2 to ensure optimal level for your patient (15–30°).
• Sedate using propofol, morphine, fentanyl and/or lorazepam/midazolam
• Mannitol prescription at 0.25–1.0 g/kg IV for ICP sustained at less than 20 mmHg (watch serum osmolality and consider holding for values >320 mOsmol/kg) OR Hypertonic Saline 7.5% prescription
• Consider paralytics if positioning, cooling, sedation and mannitol does not resolve increased ICP.
• Maintain the brain temperatures at 36–37°C, using cooling measures; prevent shivering (increases cerebral metabolic demands)
• Optimise CPP to prescribed levels (60–70 mm Hg).
• Optimise PaCO2 as indicated to increase CBF.
• Optimise sedation and consider paralytics.
• Consider barbiturate prescription if above measures are not successful.
• Maintain CVP of 5–10 mmHg, and a PCWP of 10–15 mmHg.
• Administer normal saline and/or colloids as prescribed to maintain parameters.
• Transfuse to haematocrit of 33% or haemoglobin content 80–100 g/L. (Prescription to correct coagulopathies).
• Monitor closely for signs and symptoms of neurogenic pulmonary oedema, especially in patients with cardiac history.
• Maintenance of brain temperature at 36–37°C, with active cooling if necessary.
• Transcranial Doppler image to check for vasospasm.
• Non-traumatic SAH, administer IV nimodipine or magnesium infusion to prevent vasospasm as prescribed; consider components of HHH therapy.
• Ischaemic stroke, administer tPA within 3 hours of event.
• ICH, prevent rebleeding; administer prescribed recombinant factor VII, reduce hypertension.
Management of Cerebral Oxygenation and Perfusion
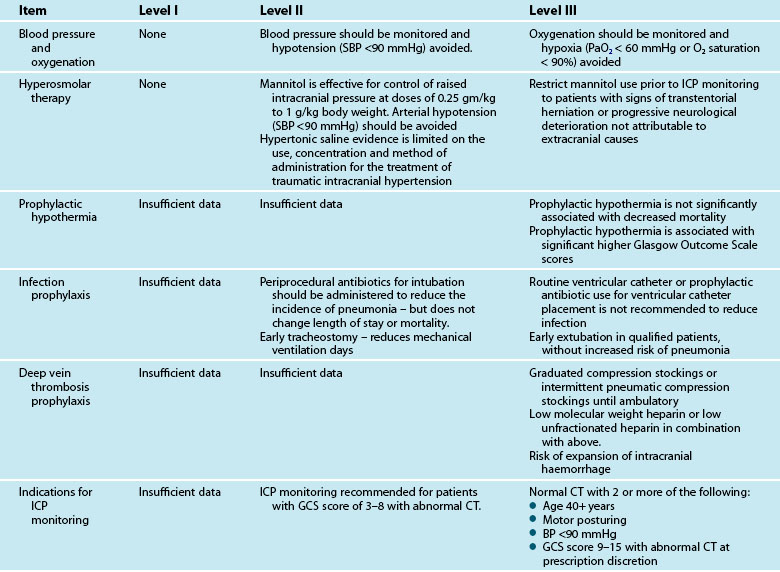
Cerebral monitoring in brain-injured patients has focused on the prevention of secondary injury to the brain owing to impaired perfusion. However, ICP monitoring and ICP manipulation does not equal cerebral oxygenation.29 There are currently four techniques that can be used to assess cerebral oxygenation: jugular venous oxygen saturation, positron emission tomography, near-infrared spectroscopy, and brain tissue oxygenation monitoring (PbtO2). Their strengths and weaknesses are the subject of several recent reviews.30,31 The selection among these forms of oxygenation monitoring is focused on the appropriateness of focal or global monitoring, the location of the monitor in relation to the injury, and the intermittent or continuous nature of the monitoring. The use of PbtO2, as assessed by the intraparenchymal polarographic oxygen probe, has the advantage of directly monitoring the zone of injury and thus earlier detection of perfusion abnormalities that may impact global cerebral oxygenation later. This may also allow the rescue of watershed areas of perfusion. However, there is controversy regarding the appropriate placement of such monitors. Insertion of the probe into non injured areas yields data equivalent to global assessments of cerebral oxygenation. Consequently, close attention should be paid to the location of the catheter in relation to the injury in interpretation and use of PbtO2 results. Jugular venous oxygen saturation (SjO2) is representative of global cerebral oxygen metabolism, but technically it is difficult to obtain reproducible results. Cerebral tissue oxygenation values of <20 mmHg are targeted for intervention based on Brain Trauma Foundation (BTF) guidelines but only at level III evidence.32 PbtO2 can be increased by increasing the FiO2/PaO2 ratio and by reducing cerebral metabolic requirements for oxygen (CMRO2) using brain temperature control with active cooling and metabolic rate control with sedation and adequate feeding. Additional interventions such as volume infusion, transfusion, and inotropic support directed at improving cardiac output can also be used to increase oxygen delivery.33
Brain inflammation after injury contributes to impaired oxygenation and perfusion, but currently its management has not translated to successful clinical management. However, the use of cerebral microdialysis (MD) and the measurement of biochemical markers (lactate, glutamate, pyruvate, glycerol and glucose) of cerebral inflammation and metabolism do contribute towards early warnings of impending hypoxia/ischaemia and neurological deterioration, and this may allow timely implementation of neuroprotective strategies. Elevation of the lactate/pyruvate ratio is typically seen in cerebral ischaemia and mitochondrial dysfunction, and has been used to tailor therapy.34 However, MD reflects only local tissue biochemistry and the accurate placement of the catheter is crucial. Furthermore, because there are wide variations in measured variables, trend data are more important than absolute values. Although MD is used routinely in a few centres it has not yet been introduced into widespread clinical practice and, at present, should be considered a research tool for use in specialist centres. MD has the potential to become established as a key component of multi-modality monitoring during management of acute brain injury during neurointensive care.
Management of Intracranial Hypertension
1. CSF by ventricular drainage (as discussed previously)
2. cerebral blood volume by hyperventilation, osmotic diuretic therapy or hypothermia
3. brain tissue water content by osmotic diuretic therapy
Hyperventilation
Hyperventilation reduces PaCO2 and will reduce ICP by vasoconstriction induced by alkalosis but it also decreases cerebral blood flow.35 The fall in ICP parallels the fall in CBV. Hyperventilation decreases regional blood flow to hypoperfused areas of the brain. Thus, generally PaCO2 should be maintained in the low normal range of about 35 mmHg. Hyperventilation should be utilised only when ICP elevations are refractory to other methods and when brain tissue oxygenation is in the normal range.36 The BTF Guidelines recommend hyperventilation therapy only for brief periods when there is no neurological deterioration or for longer periods when ICP is refractory to other therapies.32
Osmotherapy
Acute administration of an osmotic such as mannitol or hypertonic saline produces a potent antioedema action, primarily on undamaged brain regions with an intact BBB. This treatment causes the movement of water from the interstitial and extracellular space into the intravascular compartment, thereby improving intracranial compliance or elastance. In addition to causing ‘dehydration’ of the brain, osmotic agents have been shown to exert beneficial non-osmotic cerebral effects, such as augmentation of cerebral blood flow (by reducing blood viscosity, resulting in enhanced oxygen delivery), free radical scavenging, and diminishing CSF formation and enhancing CSF reabsorption.37
The BTF recommends mannitol in intracranial hypertension in bolus administration, keeping the serum osmolarities greater than 320 mOsm/L, plasma Na+ <160 mmol/L and avoiding hypovolaemia. Urine output after mannitol administration needs to be replaced, generally with normal saline. Brain free water is increased with 5% dextrose and hyperglycaemia; hence these need to be avoided. The use of frusemide in conjunction with mannitol promotes a synergistic action, particularly in patients refractory to mannitol alone. Recent studies now suggest that mannitol and frusemide have antiepileptic properties38 and that mannitol has a role in ischaemic stroke.
Intravenous hypertonic saline (HTS) increases cerebral perfusion and decreases brain swelling and inflammation more effectively than conventional resuscitation fluids. HTS behaves like 20% mannitol in acute cerebral oedema but maintains haemodynamic status. However, unlike HTS, mannitol induces a diuresis, which is relatively contraindicated in patients with both TBI and hypovolaemia as it may worsen intravascular volume depletion and decrease cerebral perfusion. Therefore, despite theoretical advantages of HTS resuscitation in patients with TBI, an Australian randomised controlled trial39 found no difference in outcome between HTS and other resuscitation fluids in prehospital resuscitation. However, in many Australian and New Zealand intensive care units, HTS is used as a preferred alternative to mannitol in patients with raised ICP.
Normothermia
Hyperthermia occurs in up to 40% of patients with ischaemic stroke and intracerebral haemorrhage and in 40–70% of patients with severe TBI or aneurysmal subarachnoid haemorrhage. Hyperthermia is independently associated with increased morbidity and mortality after ischaemic and haemorrhagic stroke, and in subarachnoid haemorrhage and TBI patients temperature elevation has been linked to raised intracranial pressure. Temperature elevations as small as 1–2°C above normal can aggravate ischaemic neuronal injury and exacerbate brain oedema.40 Mild hypothermia protects numerous tissues from damage during ischaemic insult.41 The use of paracetamol, cooling blankets, ice packs, evaporative cooling and new cooling technologies may be useful in maintaining normothermia. Hyperaemia (increased blood flow) may occur during rewarming, resulting in acute brain swelling and rebound intracranial hypertension.42 In an original study, Marion and colleagues.43 demonstrated a higher mortality rate than in more recent trials,44 possibly due to rapid rewarming and rebound hyperaemia and cerebral oedema.
Maintenance of body temperature at 35°C may be optimal.45 Intracranial pressure falls significantly at brain temperatures below 37°C but no difference was observed at temperatures below 35°C. Cerebral perfusion pressure peaks at 35–36°C and decreases with further falls in temperature.45 At a temperature below 35°C, both oxygen delivery and oxygen consumption decrease. Cardiac output decreases progressively with hypothermia. Therefore, cooling to 35°C may reduce intracranial hypertension and maintain sufficient CPP without associated cardiac dysfunction or oxygen debt.46 As the temperature is lowered from 34°C to 31°C, the volume of IV fluid infusion and inotrope requirements increase substantially and, despite such interventions, mean arterial pressure decreases. At 31°C serum potassium, white blood cell count and platelet counts are diminished.47 Thus, it seems that hypothermia to 35°C may be optimal.
Corticosteroids
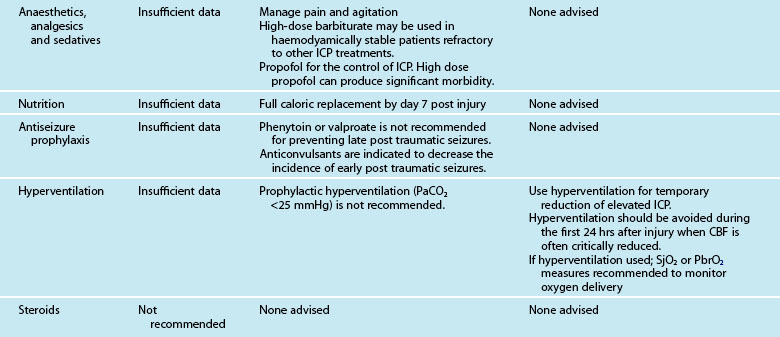
Excessive inflammation has been implicated in the progressive neurodegeneration that occurs in multiple neurological diseases, including cerebral ischaemia. The efficacy of glucocorticoids is well established in ameliorating oedema associated with brain tumours and in improving the outcome in subsets of patients with bacterial meningitis. Despite encouraging experimental results, clinical trials of glucocorticoids in ischaemic stroke, intracerebral haemorrhage, aneurysmal subarachnoid haemorrhage and traumatic brain injury have not shown a definite therapeutic effect. Furthermore, the CRASH (corticosteroid randomisation after significant head injury) trial demonstrated an increased risk of death from use of steroids from all causes within two weeks of injury, and was stopped early.48 Consequently, the BTF Guidelines state that the use of steroids is not recommended for TBI.32
Barbiturates and sedatives
The BTF Guidelines state that high-dose barbiturate therapy may be considered in haemodynamically-salvageable severe TBI patients with intracranial hypertension refractory to maximal medical and surgical interventions.49 The utilisation of barbiturates for the prophylactic treatment of ICP has not been indicated. Barbiturates exert cerebral protective and ICP-lowering effects through alteration in vascular tone, suppression of metabolism and inhibition of free radical-mediated lipid peroxidation. Barbiturates may effectively lower cerebral blood flow and regional metabolic demands. The lower metabolic requirements decrease cerebral blood flow and cerebral volume. This results in beneficial effects on ICP and global cerebral perfusion. Barbiturates within the BTF guidelines are now included under the heading of Anaesthetics, Analgesics and Sedatives and these also recommend (Level II) that it is beneficial to minimise painful or noxious stimuli as well as agitation as they may potentially contribute to elevations in ICP. Therefore propofol is recommended for the control of ICP, but does not improve mortality or six-month outcome. High dose propofol should be avoided as it can produce significant morbidity.49
Surgical interventions
The European TBI Guidelines suggest that operative management be considered for large intracerebral lesions within the first four hours of injury. The use of unilateral craniectomy after the evacuation of a mass lesion, such as an acute subdural haematoma or traumatic intracerebral haematoma, is accepted practice. Surgery is also recommended for open compound depressed skull fractures that cause a mass effect.50
Decompressive craniectomy, for refractory intracranial hypertension, has been performed since 1977, with a significant reduction in ICP for both TBI50 and ischaemic stroke.51 In 2011 a multi-centre prospective randomised trial of early decompressive craniectomy in patients with severe traumatic brain injury reported that in adults with severe diffuse traumatic brain injury and refractory intracranial hypertension, early bifrontotemporoparietal decompressive craniectomy decreased intracranial pressure and the length of stay in the ICU but surprisingly was associated with more unfavorable outcomes at both 6 and 12 months using the Extended Glasgow Outcome Scale.52
Prevention of Cerebral Vasospasm
Cerebral vasospasm is a self-limited vasculopathy that develops 4–14 days after subarachnoid haemorrhage (SAH) and/or TBI (see Figure 17.5). Oxyhaemoglobin, a product of haemoglobin breakdown, probably initiates vasoconstriction, leading to smooth-muscle proliferation, collagen remodelling and cellular infiltration of the vessel wall. The resulting vessel narrowing can lead to ischaemia. SAH patients develop cerebral vasospasm, and about one-third develop symptomatic vasospasm, which is associated with neurological signs and symptoms of ischaemia. Posttraumatic brain injury cerebral vasospasm occurs in approximately 10–15% of patients.
Calcium antagonists, such as nimodipine, have not been effective in TBI subarachnoid haemorrhage with vasospasm, and recent studies have suggested that calcium antagonists even prevent neurogenesis after TBI. Nimodipine has demonstrated effectiveness in the treatment of vasospasm in aneurysmal SAH and is now an option for recommended practice. An initial study of nimodipine in patients with TBI demonstrated no difference in outcome, and a Cochrane Systematic Review supports this conclusion.53
Magnesium may prevent cerebral vasospasm through several mechanisms. Increased ATP entry into cells could decrease ischaemic depolarisation and limit infarction size. Magnesium also both inhibits the presynaptic release of excitatory amino acids and is a non-competitive antagonist to postsynaptic NMDA receptors. The drug can also cause vasodilation by inhibiting calcium channel-mediated smooth muscle contraction. Finally, magnesium increases cardiac contractility, which may improve cerebral perfusion in dysautoregulated brain tissue. TBI animal studies have demonstrated promising neuroprotection, but this is still to be confirmed in clinical trials.54 Magnesium, however, does not cross the intact BBB easily, limiting its effect to injury and disease with leaky BBB. A randomised clinical trial of aneurysmal SAH patients receiving magnesium found that IV magnesium infusion reduced the frequency of delayed cerebral ischaemia in patients with aneurysmal SAH and subsequent poor outcome.55
In SAH, more aggressive intravascular volume expansion and induced hypertension are used in conjunction with haemodynamic monitoring. By maintaining haematocrit at 30–33%, a shift in the oxygen dissociation curve is avoided.56 Haemodilutional therapy increases collateral circulation at the site of haemorrhage, while reducing aggregation of erythrocytes where small vessel spasm has occurred. However, there is some emerging physiological data suggesting that normovolaemic hypertension may be the component most likely to increase cerebral blood flow after subarachnoid haemorrhage. In contrast, hypervolaemic haemodilution is associated with increased complications and might also lower the haemoglobin to excessively low levels.56 Also in aneurysmal SAH, endovascular therapies, such as intra-arterial papaverine infusion, are employed. Papaverine acts immediately and increases arterial calibre and cerebral blood flow, but its effects are short-lived. Balloon angioplasty is particularly effective as a durable means of alleviating arterial narrowing and preventing stroke in patients with symptomatic vasospasm after aneurysmal SAH. The timing of endovascular intubation and use of inotropes in patients with cardiac dysfunction are unresolved issues.57
Central Nervous System Disorders
Traumatic Brain Injury
Head injury is a broad classification that includes injury to the scalp, skull or brain. Traumatic brain injury (TBI) is the most serious form of head injury. The range of severity of TBI is broad, from concussion through to post coma unresponsiveness. The Australian age-standardised incidence rate of TBI in 2004/5 was about 150 per 100,000 population. Approximately 90,211 Australians58,59 and 16,000–22,500 New Zealanders60 are hospitalised for TBI every year. Males aged 15–19 years have the highest incidence rates and suffer TBI at a rate almost three times that of women. The very young (0–4 years) and the very old (over 85 years) are also at increased risk.61 Indigenous Australians suffer TBI at almost three times the rate (410 per 100,000) of non-Indigenous Australians.62 It is estimated that 40,000 Australians are living with a disability as a result of TBI.63 Despite definition issues relating to TBI epidemiology, there was an average annual decline of 5% in the TBI rate to 1997/98 but the incidence has increased since then. An Australian and new Zealand epidemiological study64 of TBI (see Research vignette) found that the mean age was 41.6 years; 74.2% were men; 61.4% were due to vehicular trauma, 24.9% were falls in elderly patients, and 57.2% had severe TBI (Glasgow Coma Scale score ≤8). Twelve-month mortality was 26.9% in all patients and 35.1% in patients with severe TBI.
Aetiology
In Australia, motor vehicle-related trauma accounts for about two-thirds of moderate and severe TBI, with falls and assaults being the next most common causes. New Zealand has a higher proportion of recreational injuries compared to vehicle-related trauma. Sporting accidents and falls account for a far greater percentage of mild injuries. Alcohol is associated with up to half of all cases of TBI. In Australia and New Zealand, blunt trauma (falls and vehicle-related), rather than penetrating (stabbing and firearms) or blast, is the predominant mechanism of injury.63 The transfer of energy to the brain tissue actually causes the damage and is a significant determinant in the severity of injury (and routinely noted in ED on admission). In the past 10 years, the introduction of safer car designs, airbags and other road traffic initiatives (e.g. redesigning hazardous intersections, driver education campaigns, random breath testing and reducing speed limits) have decreased the overall number of road fatalities; improvements in retrieval, neurosurgery and intensive care in the past few decades have enabled many people to survive injuries that would previously have been fatal. Research into and prevention of falls and shaken-baby syndromes has had a small impact on incidence reduction.65,66
Pathophysiology of TBI
TBI is a heterogeneous pathophysiological process (see Figure 17.5). The mechanisms of injury forces inflicted on the head in TBI produce a complex mixture of diffuse and focal lesions within the brain.67 Damage resulting from an injury can be immediate (primary) or secondary in nature. Secondary injury results from disordered autoregulation and other pathophysiological changes within the brain in the days immediately after injury. Urgent neurosurgical intervention for intracerebral, subdural or extradural haemorrhages can mitigate the extent of secondary injury. Scalp lesions can bleed profusely and quickly lead to hypovolaemic shock and brain ischaemia. Cerebral oedema, haemorrhage and biochemical response to injury, infection and increased ICP are among the commonest physiological responses that can cause secondary injury. Tissue hypoxia is also of major concern and airway obstruction immediately after injury contributes significantly to secondary injury. Poor cerebral blood flow, as a result of direct (primary) vascular changes or damage, can lead to ischaemic brain tissue, and eventually neuronal cell death.68 Systemic changes in temperature, haemodynamics and pulmonary status can also lead to secondary brain injury (Figure 17.6). In moderate to severe and, occasionally in mild, injury, cerebral blood flow is altered in the initial 2–3 days, followed by a rebound hyperaemic stage (days 4–7) leading to a precarious state (days 8–14) of cerebral vessel unpredictability and vasospasm.64 More than 30% of TBI patient have AN dysfunction characterised by episodes of increased heart rate, respiratory rate, temperature, blood pressure, muscle tone, decorticate or decerebrate posturing, and profuse sweating.70 Lack of insight into these processes and implementing early weaning of supportive therapies can lead to significant secondary insults.
Focal injury
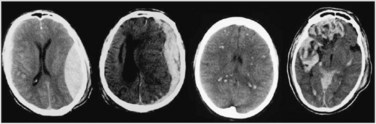
Because of the shape of the inner surface of the skull, focal injuries are most commonly seen in the frontal and temporal lobes, but they can occur anywhere. Contact phenomena are commonly superficial and can generate superficial or contusional haemorrhages through coup and contrecoup mechanisms.71 Cerebral contusions are readily identifiable on CT scans, but may not be evident on day 1 scans, becoming visible only on days 2 or 3. Deep intracerebral haemorrhages can result from either focal or diffuse damage to the arteries.
Diffuse injury
Diffuse (axonal) injury (DAI) refers to the shearing of axons and supporting neuroglia; it may also traumatise blood vessels and can cause petechial haemorrhages, deep intracerebral haematomas and brain swelling.71 DAI results from the shaking, shearing and inertial effects of a traumatic impact. Mechanical damage to small venules as part of the BBB can also trigger the formation of haemorrhagic contusions. This vascular damage may increase neuronal vulnerability, causing post-traumatising perfusion deficits and the extravasation of potentially neurotoxic blood-borne substances. The most consistent effect of diffuse brain damage, even when mild, is the presence of altered consciousness. The depth and duration of coma provide the best guide to the severity of the diffuse damage. The majority of patients with DAI will not have any CT evidence to support the diagnosis. Other clinical markers of DAI include the high speed or force strength of injury, absence of a lucid interval, and prolonged retrograde and anterograde amnesia. Figure 17.7 contrasts CT scans with haematoma formation and DAI.
Mild TBI
Mild TBI often presents as a component of multitrauma or sports injury and can be overlooked at the expense of other peripheral injuries. Risk factors such as vomiting, dizziness, facial and skull fractures; including the loss of CSF from the nose or the ear, will categorise those needing further surveillance. Routine head CT and assessment of PTA are recommended to exclude mass lesions and DAI. Diagnosis and management in the acute phase of mild TBI is as crucial to functional outcome and rehabilitation as in moderate-to-severe TBI.72
Skull fractures
Skull fractures are present on CT scans in about two-thirds of patients after TBI. Skull fractures can be linear, depressed or diastatic, and may involve the cranial vault or skull base. In depressed skull fractures the bone fragment may cause a laceration of the dura mater, resulting in a cerebrospinal fluid leak.73 Basal skull fractures include fractures of the cribriform plate, frontal bones, sphenoid bones, temporal bone and occipital bones. The clinical signs of a basal skull fracture may include: CSF otorrhoea or rhinorrhoea, haemotympanum, postauricular ecchymoses, periorbital ecchymoses, and injury to the cranial nerves: VII (weakness of the face), VIII (loss of hearing), olfactory (loss of smell), optic (vision loss) and VI (double vision).
Nursing Practice
The surveillance and prevention of secondary injury is the key to improving morbidity and mortality outcomes69 (see Table 17.1). It should be noted that in a post hoc in analysis of saline critically ill patients with TBI, fluid resuscitation with albumin was associated with higher mortality rates than was resuscitation with saline.74 Interventions are targeted at maintaining adequate cerebral blood flow and minimising oxygen consumption by the brain in order to prevent ischaemia. The anticipation and prevention of systemic complications are also of vital importance. Assessment is vital to establish priorities in care and is discussed in Chapter 16.
Nursing management of the neurologically impaired, immobilised, mechanically ventilated patient is described in Table 17.2 and is an adaptation of the current guidelines32 (see Table 17.3) to clinical practice (see Online resources for TBI-related protocols). In all TBI multitrauma patients, disability and exposure/environmental control assessment includes the routine trauma series of X-rays, namely chest, pelvis and cervical spine (lateral, anteroposterior and odontoid peg views). These should be reviewed by a radiologist and areas of concern, particularly in the upper and lower regions of the cervical spine, should be clarified with further investigations such as CT scans. Isolated TBI requires CT scanning of the head and upper spine. The management of TBI should include spinal precautions until spinal injury is definitively excluded.
TABLE 17.2 Nursing management of the neurologically impaired, immobilised, mechanically-ventilated patient
| Nursing domain | Nursing outcome | Nursing interventions |
|---|---|---|
| Ventilation and oxygenation |
• Assess ventilation parameters: ensure ET patency and position.
• Assess bilateral chest movement: listen for airway obstruction or ET cuff leak; auscultate for air entry.
• Adequate sedation and ventilation to maintain PbtO2, ICP, CPP.
• Suction only as necessary: preoxygenate, avoid prolonged coughing; effective technique.
• Avoid ICP complications of PEEP.
• Cerebral blood flow uncompromised.
• Minimal and transient changes in PbtO2–ICP–CPP and return to desired parameters within 5 min of nursing intervention.
• Patient integument maintained and infection free: skin, mucous membranes, cornea, wounds, invasive lines
• Complications of immobility prevented: DVT, pneumonia, muscle strength.
• Patient safety enabled, preventing nosocomial infection, secondary brain injury, self-harm.
• Haemodynamic stability maintained. Brain ischaemia and intracranial hypertension controlled.
• Nursing interventions planned for minimal disturbance; efficient intervention.
• Pressure-relieving mattress: allows minimal position changes for integument protection, with minimal CMRO2 requirement, sequential compression device for venous return.
• Hygiene maintained: assess integument, assess cornea, assess mucous membranes.
• Maintain infection control interventions with invasive devices and wounds.
• Administer preventive plan of treatment with vigilance and prediction.
• Enable communication with other health professionals.
• Chemical and physical restraint applied per assessment and prescription, within institutional policy.
• Refer and coordinate information and service provision from other health professionals.
• The provision of quality, informed and inclusive care to the patient provides family and significant others with the confidence that the nurse advocates for the patient in their place.
• Ensure psychological assessment and administer prescribed therapy for delirium and post traumatic stress.
• Nursing interventions planned to allow for rest and recovery.
Spinal Cord Trauma
In Australia, nearly 11,000 people live with a disability from spinal cord injury (SCI), with an age-adjusted incidence rate of 13.6 per million of the population.75 In 2007–08 there were 362 new spinal cord injuries, the majority of which (79%) were due to traumatic causes. SCI were most frequent in the 15–24 year age group (30%), although trends show a significant increase in the average age at injury from 38 years in 1995–96 to 42 years in 2007–08. Males accounted for 84% of traumatic SCI. Transport-related injuries (46%) and falls (28%) were the main contributors to traumatic SCI.
In 2001–02 New Zealand had an unadjusted rate of 27 per million and has one of the highest SCI incidences in the Western world, related mostly to snowboarding and rugby.60 SCI occurs three times more often in men, and the incidence among those aged 15–34 years is roughly double the rate in those 35 years and over. More than half of the SCIs are due to vehicular trauma and a quarter due to motorcycle crashes. Falls account for nearly a third of the injuries, with nearly half occurring in older people. Recreational and sporting injuries account for 15% of SCI, and 19% are work-related. Of all SCI cases, 51% resulted in complete tetraplegia (loss of function in the arms, legs, trunk and pelvic organs). The predominant risk factors for SCI include age, gender, and alcohol and drug use. The vertebrae most often involved in SCI are the 5th, 6th and 7th cervical (neck), the 12th thoracic, and the 1st lumbar. These vertebrae are the most susceptible because there is a greater range of mobility in the vertebral column in these areas.76 Damage to the spinal cord ranges from transient concussion or stunning (from which the patient fully recovers) to contusion, laceration and compression of the cord substance (either alone or in combination), to complete transection of the cord (which renders the patient paralysed below the level of the injury).
Mechanisms of Injury
Cervical injury can occur from both blunt and penetrating trauma but in reality is a combination of different mechanisms of acceleration and deceleration with and without rotational forces and axial loading.77 An illustrative example is a diving injury, caused by a direct load through the head and cervical spine. In reality, cervical trauma is produced by a combination of these mechanisms as listed below.
• Hyperflexion: These injuries usually result from forceful decelerations and are often seen in patients who have sustained trauma from a head-on motor vehicle collision (MVC) or diving accident. The cervical region is most often involved, especially at the C5–C6 level.
• Vertical compression or axial loading: This typically occurs when a person lands on the feet or buttocks after falling or jumping from a height. The vertebral column is compressed, causing a fracture that result in damage to the spinal cord.
• Hyperextension: This is the most common type of injury. Hyperextension injuries can be caused by a fall, a rear-end MVC, or hit on the head (e.g. during a boxing match). Hyperextension of the head and neck may cause contusion and ischaemia of the spinal cord without vertebral column damage. Whiplash injuries are the result of hyperextension. Violent hyperextension with fracture of the pedicles of C2 and forward movement of C2 on C3 produces the ‘Hangman’s fracture’.
• Extension–rotation: Rotational injuries result from forces that cause extreme twisting or lateral flexion of the head and neck. Fracture or dislocation of vertebrae may also occur. The spinal canal is narrower in the thoracic segment relative to the width of the cord, so when vertebral displacement occurs it is more likely to damage the cord. Until the age of 10, the spine has increased physiological mobility due to lax ligaments, which affords some protection against acute SCI. Elderly patients are at a higher risk due to osteophytes and narrowing of the spinal canal.
Classification of Spinal Cord Injuries
SCIs can be broadly classified as complete or incomplete.78 The diagnosis of complete SCI cannot be made until spinal cord shock resolves. If the bulbocavernosus reflex (BCR) is present (involuntary contraction of the rectal sphincter after squeezing the glans penis or clitoris or tugging on an indwelling urinary catheter) it indicates a complete injury. If, after the return of the BCR, the patient has some sensation below the level of injury, he/she is considered to be sensory-incomplete. If the BCR has returned and the patient has some motor function and sensation below the level of injury, he/she is considered to be sensory- and motor-incomplete. There are four incomplete SCI syndromes as follows:
• Anterior cord syndrome: Injury to the motor and sensory pathways in the anterior parts of the spine; thus patients are able to feel crude sensation, but movement and detailed sensation are lost in the posterior part of the spinal cord. Clinically, the patient usually has complete motor paralysis below the level of injury (corticospinal tracts) and loss of pain, temperature, and touch sensation (spinothalamic tracts), with preservation of light touch, proprioception and position sense. The prognosis for anterior cord syndrome is the worst of all the incomplete syndrome prognoses.
• Posterior cord syndrome: This is usually the result of a hyperextension injury at the cervical level and is not commonly seen. Position sense, light touch and vibratory sense are lost below the level of the injury.
• Central cord syndrome: Injury to the centre of the cervical spinal cord, producing weakness, paralysis and sensory deficits in the arms but not the legs. Hyperextension of the cervical spine is often the mechanism of injury, and the damage is greatest to the cervical tracts supplying the arms. Clinically, the patient may present with paralysed arms but with no deficit in the legs or bladder.
• Brown-Séquard syndrome: This involves injury to the left or right side of the spinal cord. Movements are lost below the level of injury on the injured side, but pain and temperature sensation are lost on the opposite side of injury. The clinical presentation is one in which the patient has either increased or decreased cutaneous sensation of pain, temperature and touch on the same side of the spinal cord at the level of the lesion. Below the level of the lesion on the same side, there is complete motor paralysis. On the patient’s opposite side, below the level of the lesion, there is loss of pain, temperature and touch, because the spinothalamic tracts cross soon after entering the cord.
Pathophysiology
SCIs can be separated into two categories: primary injuries and secondary injuries. Primary injuries are the result of the initial insult or trauma, and are usually permanent. The force of the primary insult produces its initial damage in the central grey matter of the cord. Secondary injuries are usually the result of a contusion or tear injury, in which the nerve fibres begin to swell and disintegrate. Secondary neural injury mechanisms include ischaemia, hypoxia and oedema. Ischaemia, the most prominent post-SCI event, may occur up to 2 hours post-injury and is intensified by the loss of autoregulation of the spinal cord microcirculation.78 This will decrease blood flow, which is then dependent on the systemic arterial pressure in the presence of hypotension or vasogenic spinal shock. Oedema develops at the injured site and spreads into adjacent areas. Hypoxia may occur as a result of inadequate airway maintenance and ventilation. Immune cells, which normally do not enter the spinal cord, engulf the area after a spinal cord injury and release regulatory chemicals, some of which are harmful to the spinal cord. Highly reactive oxidising agents (free radicals) are produced, which damage the cell membrane and disrupt the sodium–potassium pump.
Free-radical production and lipid peroxidation lead to vasoconstriction, increased endothelial permeability and increased platelet activation. A secondary chain of events produces ischaemia, hypoxia, oedema and haemorrhagic lesions, which in turn result in the destruction of myelin and axons. Autoregulation of spinal cord blood flow may be impaired in patients with severe lesions or substantial oedema formation. These secondary reactions, believed to be the principal causes of spinal cord degeneration at the level of injury, are now thought to be reversible 4–6 hours after injury. Therefore, if the cord has not suffered irreparable damage, early intervention is needed to prevent partial damage from developing into total and permanent damage.80
Spinal shock occurs with physiological or anatomical transection or near-transection of the spinal cord; it occurs immediately or within several hours of a spinal cord injury and is caused by the sudden cessation of impulses from the higher brain centres.79 It is characterised by the loss of motor, sensory, reflex and autonomic function below the level of the injury, with resultant flaccid paralysis. Loss of bowel and bladder function also occurs. In addition, the body’s ability to control temperature (poikilothermia) is lost and the patient’s temperature tends to equilibrate with that of the external environment.
Neurogenic spinal shock occurs as a result of mid- to upper-level cervical injuries and is the result of sympathetic vascular denervation and peripheral vasodilation. The loss of spinal cord vasculature autoregulation occurs, causing the blood flow to the spinal cord to be dependent on the systemic blood pressure. Signs and symptoms include hypotension, severe bradycardia, and loss of the ability to sweat below the level of injury. The same clinical findings pertaining to disruption of the sympathetic transmissions in spinal shock occur in neurogenic shock.78
Nursing Practice
Spinal cord injury should be suspected in patients with neck pain, sensory and motor deficits, unconsciousness, intoxication, spondylitis or rheumatoid arthritis, head injury and facial fractures. If spinal cord injury is suspected or cannot be excluded, the patient must be placed on a spine board with the head and neck immobilised in a neutral position using a rigid collar to reduce the risk of neurological deterioration from repeated mechanical insults. Spinal injury patients are susceptible to pressure insults, so time must be considered when hard surfaces are used for immobilisation. Total neck immobilisation should not interfere with maintenance of the airway, and inadequate respiratory function must be avoided.82
Resuscitation
Initial treatment aims for decompression of the spinal cord and reversal of neurogenic shock and respiratory failure. Spinal shock is associated with decreases in systemic vascular resistance, arterial hypotension, venous pooling, severe bradycardia and decreased myocardial contractility. Consequently, treatment of neurogenic shock includes fluid replacement using crystalloid or colloid solutions to maintain arterial blood pressure, circulatory volume, renal function and tissue oxygenation. Infusion of free water must be avoided, as this decreases plasma osmolarity and promotes spinal cord oedema. Atropine may be administered to reverse bradycardia and increase cardiac output. Administration of vasopressors (e.g. noradrenaline) prior to correction of the intravascular volume status may increase systemic vascular resistance (left ventricular afterload) and further impair myocardial contractility. Therefore, volume replacement is the first step, and administration of vasopressors the second step in the treatment of arterial hypotension and low cardiac output after acute cervical spinal cord injury.79
The major early cause of death in patients with acute cervical SCI is respiratory failure. Tracheal intubation may be indicated in unconscious patients, during shock, in patients with other major associated injuries, and during cardiovascular and respiratory distress. It is also indicated in conscious patients presenting with the following criteria: maximum expiratory force below +20 cmH2O, maximum inspiratory force below −20 cmH2O, vital capacity below 1000 mL, and presence of atelectasis, contusion and infiltrate.81
Investigations and alignment
Following the initial assessment of the patient, detailed diagnostic radiography defines the bone damage and compression of the spinal cord. First, lateral, anteroposterior, odontoid and possibly oblique cervical spine radiographs are obtained. If there is no evidence of injury, flexion and/or extension views may be considered. If any of these radiographs suggest cervical spine abnormalities, specific radiological procedures such as cervical myelography, high-resolution CT scan or magnetic resonance imaging will identify fractures, dislocation of bony fragments, and spinal cord contusion.82 In patients with a dislocated cervical fracture, decompression and anatomical bony realignment may be achieved with traction forces applied manually, or with halo or Gardner–Wells systems under radiological control. If the anatomical bony alignment procedures and traction forces fail to decompress the cord, surgical intervention to remove the lesion is required. The timing of surgical intervention remains controversial. While urgent surgical decompression or internal stabilisation should be performed in all patients with deteriorating neurological status, some centres tend to defer surgical treatment in patients with spinal cord injury but stable neurological deficit.
Concepts of Neuroprotection and Regeneration
There have been many negative SCI clinical trials in regard to neuroprotection with the exception of methylprednisolone within 8 hours after SCI, which has shown some beneficial effect.77 The failure of these neuroprotective agents has been attributed to the attempt of blocking only one molecular pathway of a complex range of SCI molecular mechanisms. However, there has been renewed interest in regeneration which involves stem cell transplantation or similar restorative approaches designed to optimise spontaneous axonal growth and myelination but is still in its infancy in Australia and NZ due to limiting legislation in regard to stem cell research.
Collaborative Management
• Tracheostomy is indicated in high cervical spine injury and ischaemia, sometimes only while the early oedema is resolving.
• Spinal alignment and immobilisation requires careful positioning with dedicated neck support by experienced clinicians.
• Shoulder and lumbar support pillows are often prescribed. Pressure-relief mattresses must be suitably designed for spine immobilisation and when prescribed can be tilted to facilitate ventilation.
• Meticulous integument and bowel care are indicated with daily protocols for regular stool softeners and peristaltic stimulants essential for the prevention of autonomic dysreflexia and autonomic nerve dysfunction.
• Early nutritious feeding is essential, whether oral or enteric; however, aspiration must be prevented. The supplementation of feeding with high-energy protein fluids to match the catabolic state assists with recovery (see Chapter 19).
• Hyperglycaemia is associated with increased inflammation and must be controlled to less than 10 mmol/Hg, avoiding hypoglycaemia.84
• The concept of pain relief and sedation in patients with spinal cord injury is based on the maintenance of coupling between metabolism and spinal cord blood flow while achieving hypnosis, analgesia and a ‘relaxed cord’. This concept includes maintenance of normal to high systemic perfusion pressures, normoxia and normocapnia.
• Psychological and empathetic support is essential and appropriate referral for grieving and stress is paramount. Rehabilitation counselling and planning starts at the acute stage in order to give the family unit some future focus and hope.
See the Online resources for specific protocols related to spinal injury.
Cerebrovascular Disorders
Stroke
Stroke is the primary cerebrovascular disorder in Australia and New Zealand and is still the third-leading cause of death. Every year approximately 40,000 people in Australia are admitted to hospital with a diagnosis of stroke; approximately 6000 New Zealanders suffer from a stroke every year and approximately 2000 deaths each year are attributable to stroke.85,86 The prevalence of stroke is higher among men than women (1.4% versus 1.0%). Almost 60% of people who have had a stroke are aged 65 years and over, while 18% are under the age of 55 years. Indigenous Australians have higher rates of death and illness from heart, stroke and vascular diseases than other Australians. In 2007–08, death rates were 2.6 times as high and hospitalisation rates 1.4 times as high as for other Australians.85 Stroke is currently the biggest single cause of adult disability in Australasia. Strokes can be divided into two major categories: ischaemic (85%), in which vascular occlusion and significant hypoperfusion occur; and haemorrhagic (15%), in which there is extravasation of blood into the brain. Although there are some similarities between the two broad types of stroke, the aetiology, pathophysiology, medical management, surgical management and nursing care differ.
Aetiology
Hypertension is the leading risk factor for stroke. Other risk factors include diabetes, cardiac disease, previous cerebrovascular disease (transient ischaemic attack or stroke or myocardial infarction), age, sex, lipid disorders, excessive ethanol ingestion, elevated hematocrit, elevated fibrinogen and cigarette smoking. Cerebral arteriosclerosis predisposes indiuiduals to both ischaemic and haemorrhagic stroke. Smoking is the strongest risk factor for aneurysmal SAH. Atrial fibrillation, endocarditis and medications containing supplemental oestrogen are risk factors for embolic stroke. Seizures develop in approximately 10% of cases, usually appearing in the first 24 hours and more likely to be focal than generalised. Most patients with aphasia will have a cerebral infarction in the distribution of the left middle cerebral artery.87
Ischaemic Stroke
Ischemic stroke compromises blood flow and energy supply to the brain, which triggers mechanisms that lead to cell death. Infarction occurs rapidly in the region of most severe ischaemia (termed ischaemic penumbra) and expands at the expense of the surrounding hypoxic tissue, from the centre to the periphery. Therapeutic strategies in acute ischaemic stroke are based on the concept of arresting the transition of the penumbral region into infarction, thereby limiting ultimate infarct size and improving neurological and functional outcome. Ischaemic stroke can be further categorised as middle cerebral artery occlusion, acute basilar occlusion, and cerebellar infarcts.88
The management of an ischaemic stroke comprises four primary goals: restoration of cerebral blood flow (reperfusion), prevention of recurrent thrombosis, neuroprotection, and supportive care. The timing of each element of clinical management needs to be implemented in a decisive manner. Refer to Table 17.4 for classification and treatment strategies and to Online resources for specific ischaemic stroke protocols.
TABLE 17.4 Classification and type of ischaemic stroke and treatment options
| Classification | Treatment options |
|---|---|
| Middle cerebral artery occlusion |
Haemorrhagic Stroke
Haemorrhagic strokes are caused by bleeding into the brain tissue, the ventricles or the subarachnoid space.89 Primary intracerebral haemorrhage from a spontaneous rupture of small vessels accounts for approximately 80% of haemorrhagic strokes and is primarily caused by uncontrolled hypertension. Secondary intracerebral haemorrhage is associated with arteriovenous malformations (AVMs), intracranial aneurysms, or certain medications (e.g. anticoagulants and amphetamines). Symptoms are produced when an aneurysm or arteriovenous malformation (AVM) enlarges and presses on nearby cranial nerves or brain tissue or, more dramatically, when a blood vessel, aneurysm or AVM ruptures, causing intracerebral or subarachnoid haemorrhage. When an aneurysm ruptures, arterial pressure forces blood into the subarachnoid space between the arachnoid mater and the surface of the brain. Free blood then travels through the fissures into the basal cisterns and across the surface of the brain. When clotted, this blood can interfere with the circulation and reabsorption of cerebrospinal fluid (CSF), potentially causing obstructive hydrocephalus and raised intracranial pressure. The commonest cause is a leaking aneurysm in the area of the circle of Willis or a congenital AVM of the brain. Blood in the subarachnoid space is a powerful meningeal irritant, and it is this irritation that causes most of the initial signs and symptoms of SAH.
In intracerebral haemorrhage the bleeding is usually arterial and occurs most commonly in the cerebral lobes, basal ganglia, thalamus, brainstem (mostly the pons) and cerebellum. Occasionally, the bleeding ruptures the wall of the lateral ventricle and causes intraventricular haemorrhage, which is often fatal.89
The Factor Seven for Acute Hemorrhagic Stroke (FAST) multicentre international clinical trial recently reported that haemostatic therapy with recombinant activated factor VII (rFVIIa) reduced growth of the haematoma but did not improve survival or functional outcome after intracerebral haemorrhage.90
Subarachnoid Haemorrhage
Admission to ICU is indicated for subarachnoid haemorrhage Hunt-Hess SAH severity Scale III (see Table 17.5) and greater to manage systemic complications, recognise and treat clinical deterioration, investigate the cause of the haemorrhage and to treat any underlying aneurysm or arteriovenous malformation. Resuscitation is directed towards maintaining cerebral perfusion pressure by ensuring adequate arterial blood pressure (often with the use of inotropes to produce relative hypertension although reactive hypertension is often present), ensuring a relatively high circulating blood volume (hypervolaemia), and producing relative haemodilution (’triple H therapy’).91
| Score | Description |
|---|---|
| 0 | Unruptured; asymptomatic discovery |
| I | Asymptomatic or minimal headache with slight nuchal rigidity |
| II | Moderate to severe headache, nuchal rigidity; no neurological deficit other than cranial nerve deficit |
| III | Drowsiness, confusion, or mild focal deficit (e.g. hemiparesis), or a combination of these findings |
| IV | Stupor, moderate to severe deficit, possibly early decerebrate rigidity and vegetative disturbances |
| V | Deep coma, decerebrate rigidity, moribund appearance |
Hypovolaemia occurs in 30–50% of patients, as does excessive hyponatraemia in 30% of patients. In the first six days, plasma volume decreases of greater than 10% can occur following SAH, thus increasing the risk of vasospasm and ischaemia. Women have been found to have more significant drops in blood volume than men following SAH.92 ‘Third space’ loss, insensible losses and blood loss account for this drop in fluid volume, as well as electrolyte disturbances.
Other aspects of management in the acute stages include suitable analgesia, seizure control, and treatment with nimodipine to prevent secondary ischaemia caused by vasospasm. Vasospasm often occurs 4–14 days after initial haemorrhage when the clot undergoes lysis (dissolution), increasing the chances of rebleeding. It is believed that early surgery to clip the aneurysm prevents rebleeding and that removal of blood from the basal cisterns around the major cerebral arteries may prevent vasospasm.93,94 (See previous section on Management of vasospasm.)
ICP monitoring and drainage of CSF via ventriculostomy is indicated in SAH but not in cerebral haemorrhage.89 SAH causes increased sympathetic activation from the presence of haemoglobin in the subarachnoid space. This results in elevated catecholamine levels, which may result in focal myocardial necrosis, explaining the presence of inverted T waves, ST depression, prominent U waves, and Q-T intervals in more than 50% of patients. As cardiac function is one of the determinants for adequate cerebral blood flow, it is essential to identify such occurrences early and treat them accordingly.95 Hyponatraemia occurs from alterations in atrial natriuretic factor (ANF) in response to sympathetic nervous system activation. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) is primarily responsible for hyponatraemia in those with SAH, as is cerebral salt-wasting syndrome; however, both mechanisms are still relatively misunderstood.90
Cerebral Venous Thrombosis
Cerebral venous thrombosis is particularly important to recognise because there is general consensus that early anticoagulation can result in good clinical outcomes.96 MR and CT vascular imaging has made it easier to establish the diagnosis, but close monitoring of the patient is essential, as late deterioration can occur.
Collaborative Management of Stroke
Expected outcomes for patients with acute ischaemic and haemorrhagic stroke include prevention of secondary injury, of airway and respiratory complications, and the maintenance of haemodynamic stability. Timely assessment and intervention is paramount in the management of ischaemic stroke, especially regarding interventional pharmacology and prevention of cerebral haemorrhage. See Online resources for specific protocols related to stroke.
Infection and Inflammation
Meningitis
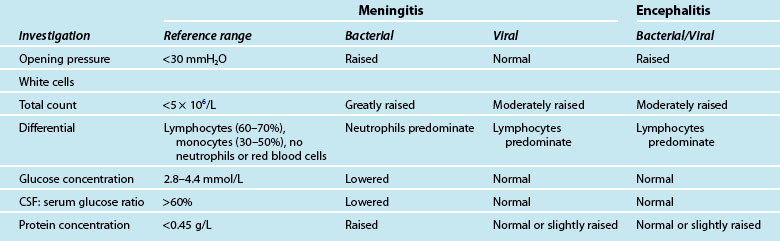
The incidence of disease caused by Neisseria meningitidis remains an issue of public health concern in Australia and New Zealand. The introduction of a publicly funded program of selective vaccination with conjugate serogroup C meningococcal vaccine in 2004 has resulted in a significant reduction in the number of cases of meningococcal disease.97 Nationally in 2008 only 15 serogroup C infections were identified and serogroup B accounted for 88% of all infections. New Zealand has one of the highest rates of meningococcal B disease in the developed world but the incidence has declined. There were 132 cases of meningococcal disease notified in 2009, which equates to a rate of 3.8 per 100,000 population. The number of confirmed cases was 117, giving a confirmation rate of 88.6% which is the third-equal-highest confirmation rate since 1991. Five deaths occurred in 2009, giving a case-fatality rate of 3.8%. Since 1991 a total of 265 deaths have been recorded, an overall case-fatality rate of 4.2%. The policy of giving antibiotics prior to hospital admission, implemented in 1995, reduced the case-fatality rate for those receiving antibiotics. In addition this rate has reduced from 470 cases in 2001, prior to the immunisation for meningococcal B commencing in 2004.98 The incidence of meningococcal disease varies seasonally, rising in June and peaking in October each year. The highest incidence of meningococcal disease was for children aged 4 years and under. A secondary peak in the incidence of meningococcal disease is seen in adolescents and young adults.99 However, during the H1N1 influenza epidemic there were several cases of H1N1 influenza-related meningitis. See Table 17.6 for CSF profiles for acute meningitis and encephalitis and Table 17.7 for the classification, treatment and clinical presentation of meningitis.
| Acute meningitis | Bacterial – notifiable disease | Viral |
|---|---|---|
| Aetiology |
Rapid recognition and diagnosis of meningitis is imperative.
Quick and insidious progress of disease
Colonisation of mucosal surfaces (nasopharynx)
Haematogenous or contiguous spread
Specific antibodies important defence
Bacterial invasion of meninges: inflammatory response, breakdown of the BBB, cerebral oedema, intracranial hypertension
The physical signs are not so marked and the illness is not as severe and prolonged as bacterial meningitis.
Viral infection of mucosal surfaces of respiratory or gastrointestinal tract
Virus replication in tonsillar or gut lymphatics
Viraemia with haematogenous dissemination to the CNS
Meningeal inflammation, BBB breakdown, cerebral oedema, vasculitis and spasm
Presents with sepsis: headache, fever, photophobia, vomiting, neck stiffness, alteration of mental status.
Meningococcaemia is characterised by an abrupt onset of fever (with petechial or purpuric rash).
Progresses to purpura fulminans, associated with the rapid onset of hypotension, acute adrenal haemorrhage syndrome, and multiorgan failure.
Cranial nerve palsy (III, IV, VI, VII) uncommon and develop after several days
Focal neurological signs in 10–20% cases
Signs of intracranial hypertension: coma, altered respiratory status
Leads to hypertension and bradycardia before herniation, or brain death, leads to irreversible septic shock
If meningococcal infection is suspected, the best way to reduce mortality is to administer
Empirical IV therapy immediately
Cefatoxime 2g IV 6hrly or immediately
Consequent dose, times and type of antibiotic need to be modified after full investigation and a detailed examination have taken place.
Dexamethasone may be prescribed: Needs to be at same time of antibiotic as outcome neurologically is reduced if given after antibiotic. Reduces BBB permeability.
Complications
Complications of meningitis vary according to the aetiological organism, the duration of symptoms prior to initiation of appropriate therapy, and the age and immune status of the patient.100 Temporary problems include development of haemodynamic instability and disseminated intravascular coagulopathy, particularly in meningococcal infection, SIADH or other dysregulation of the hypothalamic–pituitary axis (e.g. diabetes insipidus) and an acute rise in ICP.
Collaborative care
Neurological derangement often coexists with circulatory insufficiency, impaired respiration, metabolic derangement and seizures. Protecting the patient from injury secondary to raised ICP and seizure activity is essential. Prevention in relation to complications associated with immobility, such as decubitus and pneumonia, is required. It is important to institute droplet infection control precautions in those attending the patient until 24 hours after the initiation of antibiotic therapy (oral and nasal discharge is considered infectious). See Online resources for infection control protocols relating specifically to meningitis.
Encephalitis
Aetiology
Herpes simplex virus (HSV) is the commonest cause of non-seasonal encephalitis in Australia. Without treatment, HSV encephalitis is fatal in up to 80% of cases, and leaves up to 50% of survivors with long-term sequelae.101
• In the absence of particular risk factors, other common causes are enteroviruses, influenza virus and Mycoplasma pneumoniae. However, the likely pathogens in encephalitis are dramatically influenced by geographic location, history of travel and animal exposure, and vaccination.
• Murray Valley encephalitis (MVE) virus causes seasonal epidemics of encephalitis at times of high regional rainfall. This arthropod-borne virus is the commonest flavivirus to cause encephalitis in Australia.
• Since 2005, the distribution of Japanese B encephalitis virus has expanded into Australia via the Torres Strait Islands.102 It causes disease clinically similar to MVE. In addition, two novel encephalitis viruses were recently identified in Australia, the Hendra virus and Australian bat lyssavirus. These should be considered if there is a history of animal exposure, or if no other pathogen can be implicated.
• Mycobacterium tuberculosis, the yeast Cryptococcus neoformans and Treponema pallidum (syphilis) may also affect the brain parenchyma but usually produce chronic or subacute meningitis in such circumstances.
Pathophysiology
In the majority of encephalitis cases, the offending organism finds access to the brain via the nasopharyngeal epithelium and the olfactory nerve system. Arboviruses are transmitted from infected animals to human through bite of infected animals.103 The cytokine storm results in neural cell damage, as well as the apoptosis of astrocytes. The disruption of the blood–brain barrier progresses to the systemic cytokine storm, resulting in septic shock, disseminated intravascular coagulopathy (DIC) and multiorgan failure (MOF).
Clinical features and diagnosis
Encephalitis may present with progressive headache, fever and alterations in cognitive state (confusion, behavioural change, dysphasia) or consciousness. Focal neurological signs (paresis) or seizures (focal or generalised) may also occur. Upper motor signs (hyperreflexia and extensor–plantar responses) are often present, but flaccid paralysis and bladder symptoms may occur if the spinal cord is involved. Associated movement disorders or the SIADH secretion may be seen. In northern Australia, it may be desirable to distinguish MVE from Japanese encephalitis clinically. Both conditions often affect the brainstem and basal ganglia, but MVE often involves the spinal cord, while Japanese encephalitis may produce striking meningeal signs, with or without thalamic involvement. Both have high mortality (25–33%) and high rates of chronic sequelae in survivors (~50%).101
The most sensitive type of imaging for diagnosis of encephalitis is MRI; in HSV encephalitis, CT scans may initially appear normal, but MRI usually shows involvement of the temporal lobes and thalamus.103,104 Examination of CSF can assist in differential diagnosis. Electroencephalography is less sensitive but may be helpful if it shows characteristic features (e.g. lateralising periodic sharp and slow-wave patterns). Refer to Table 17.6 for CSF profiles.
Collaborative management
Support in an ICU is often required in encephalitis to maintain ventilation, protect the airway and manage complications, such as cerebral oedema. The management of acute viral encephalitis includes aggressive airway, ventilation, sedation, seizure, haemodynamic, and fluid and nutritional support. Clinical deterioration in encephalitis is usually the result of severe cerebral oedema with diencephalic herniation or systemic complications, including generalised sepsis and multiple organ failure. The use of ICP monitoring in acute encephalitis remains controversial but should be considered if there is a rapid deterioration in the level of consciousness, and if imaging suggests raised ICP. Prolonged sedation may be necessary. Decompressive craniotomy may be successful in cases where there is rapid swelling of a non-dominant temporal lobe, as poor outcome is otherwise likely.105
Neuromuscular Alterations
Guillain–Barré Syndrome
Guillain–Barré syndrome (GBS) is an immune-mediated disorder resulting from generation of autoimmune antibodies and/or inflammatory cells which cross-react with epitopes on peripheral nerves and roots, leading to demyelination or axonal damage or both, and autoimmune insult to the peripheral nerve myelin. In Australia, Guillain–Barré has an average incidence of about 1.5 per 100,000, in men slightly higher than in women.106 Of all patients, 85% recover with minimal residual symptoms; severe residual deficits occur in up to 10%. Residual deficits are most likely in patients with rapid disease progression, those who require mechanical ventilation, or those 60 years of age or over. Death occurs in 3–8% of cases, resulting from respiratory failure, autonomic dysfunction, sepsis or pulmonary emboli.107
Aetiology
The diagnosis of GBS is confirmed by the findings of cytoalbuminological dissociation (elevation of the CSF protein without concomitant CSF pleocytosis), and by neurophysiological findings suggestive of an acute (usually demyelinating) neuropathy. These abnormalities may not be present in the early stages of the illness.106 There are two forms of GBS. The demyelinating form, the more common one, is characterised by demyelination and inflammatory infiltrates of the peripheral nerves and roots. In the axonal form the nerves show Wallerian degeneration with an absence of inflammation. Discrimination between the axonal and demyelinating forms relies mainly on electrophysiological methods. There is a close association between GBS and a preceding infection, suggesting an immune basis for the syndrome. The commonest infections are due to Cambylobacter jejuni, cytomegalovirus and Epstein–Barr virus.
Clinical manifestations
Onset is rapid, and approximately 20% of cases lead to total paralysis, requiring prolonged intensive therapy with mechanical ventilation. The therapeutic window for GBS is short, and the current optimal treatment with whole plasma exchange or IV immunoglobulin (IVIg) therapy lacks immunological specificity and only halves the severity of the disease.108 GBS has three phases – acute, plateau, and recovery – each stage lasting from days to weeks and in recovery to months and years. The patient presents with:
• symmetrical weakness, diminished reflexes, and upward progression of motor weakness. A history of a viral illness in the previous few weeks suggests the diagnosis
• changes in vital capacity and negative inspiratory force, which are assessed to identify impending neuromuscular respiratory failure.
Indications for ICU admission include the following: ventilatory insufficiency, severe bulbar weakness threatening pulmonary aspiration, autonomic instability, or coexisting general medical factors,109 and often a combination of factors, are present. The incidence of respiratory failure requiring mechanical ventilation in GBS is approximately 30%.
Recently sensory involvement in relation to pain has been studied asserting the clinical observation of pain ranging from mild to severe in the acute and rehabilitant phases. Chronic pain is often present in survivors of GBS.110
There may be total paralysis of all voluntary muscles of the body, including the cranial musculature, the ocular muscles and the pupils. Prolonged paralysis and incomplete recovery are more likely, and prolonged ventilatory support may be necessary. Walgaard and colleagues found that GBS patients who experience rapid disease progression, bulbar dysfunction, bilateral facial weakness or autonomic nerve dysfunction were more likely to require mechanical ventilation.111 Tracheostomy is usually performed within 2 weeks, and mechanical ventilation is delivered in a supportive mode with minimal yet adequate sedation and pain management.
Nursing practice
• Comprehensive respiratory assessment (level of overall patient comfort, frequency and depth of breathing, forced vital capacity, use of accessory muscles, presence of paradoxical respiration and integrity of upper airway reflexes), ABG data and chest radiography determine levels of fatigue in both the acute stage (for intubation and ventilation) and rehabilitation (weaning) stage. Long-term ventilation increases the risk of ventilator-acquired pneumonia (VAP), and routine surveillance for VAP is vital.
• Cardiovascular assessment is important, as serious tachyarrhythmias and bradyarrhythmias and destabilising fluctuations in blood pressure caused by autonomic impairment are prevalent. This feature is common during fatigue, sleep and states of dehydration. Often, autonomic dysfunction is worst in the early stages of a nosocomial infection.112
• Cranial nerve assessment and dermatome (for sensory) and muscle strength assessment assist in mapping the progression, severity and rehabilitation of the disease and determining the risk of aspiration. Pain (especially neuropathic) is particularly common in GBS during changes in myelination, and can be difficult to treat.113 Assessment will include all aspects as indicated for the long-term immobile, intubated, ventilated and neuromuscular-impaired patient.
Independent practice
• Endotracheal and pharyngeal suction can be demanding (weakened upper airway reflexes), and sputum plugging and retention requires frequent repositioning and physiotherapy.
• Routine daily gentle exercise as part of a flexible program improves wellbeing and strength.
• Fatigue must be avoided, as autonomic nerve dysfunction, deafferent pain syndromes, muscle pain and depression can be exacerbated.
• Suctioning, coughing, bladder distension, constipation and the Valsalva manoeuvre can also aggravate autonomic nerve dysfunction instability.
• Therapeutic massage, warm and cold packs and careful positioning contribute to comfort and pain management.
• The patient’s surroundings should be pleasant and presentable, especially during long recovery.
• Communication techniques need to be refined to prevent fatigue and frustration.
• Patience, tolerance, empathy, humour and family involvement assist the patient in psychological resilience and recovery.
Myasthenia gravis
Myasthenia gravis is an autoimmune disorder caused by autoantibodies against the nicotinic acetylcholine receptor on the postsynaptic membrane at the neuromuscular junction. It is characterised by weakness and fatiguability of the voluntary muscles. It peaks in the third and sixth decades of life. Its prevalence in Western countries is 14.2/100,000.114 Prevalence rates have been rising steadily over the past decades, probably due to decreased mortality, longer survival, and higher rates of diagnosis. The development of respiratory failure, progressive bulbar weakness leading to failure of airway protection and severe limb and truncal weakness causing extensive paralysis, as in a myasthenic crisis, all may result in admission to ICU.
Pathophysiology
In myasthenia gravis both structural changes in the architecture of the neuromuscular junction and dynamic alterations in the turnover of acetylcholine receptors erode the safety margin and efficiency of neuromuscular transmission. Of all patients with myasthenia gravis, 80–85% have an identifiable and quantifiable antibody found in the IgG fraction of plasma, which is responsible for blocking receptors to the action of acetylcholine at the neuromuscular junction.113 Therefore, successful neuromuscular transmission is markedly affected by small and subtle changes in acetylcholine release and other triggers (as above), and this gives rise to the decrement in transmission with repetitive stimulation and the characteristic fatiguable muscle weakness. Pharmacological management for myasthenia gravis includes the use of anticholinesterases (pyridostigmine), steroids, azathioprine and cyclophosphamide. Thymectomy reduces the antibodies responsible for acetylcholine blockade and is often performed early in the disease.115 Plasmapheresis and IVIg are used in the short term for myasthenic crisis and are especially useful for preventing respiratory collapse or assisting with weaning.
Clinical manifestations
In a myasthenic crisis, vital capacity falls, cough and sigh mechanisms deteriorate, atelectasis develops and hypoxaemia results.115 Ultimately, fatigue, hypercarbia and ventilatory collapse occur. Commonly superimposed pulmonary infections lead to increased morbidity and mortality. Assessment for triggers begins with a careful review of systems, with attention to recent fevers, chills, cough, chest pain, dysphagia, nasal regurgitation of liquids and dysuria. Detailed history-taking should note any trauma, surgical procedures and medication use. General assessment includes vital signs; ear, nose and throat inspection; chest auscultation; and abdominal check. In addition to supportive care and the removal of triggers, management of myasthenic crisis includes treatment of the underlying myasthenia gravis. An experienced neurologist, who will ultimately provide the patient’s care outside the ICU, should be part of the care team. Options for treatment during crisis include: use of AChE inhibitors, plasma exchange, IV immunoglobulins, and immunosuppressive drugs, including corticosteroids. Median duration of hospitalisation for crisis is 1 month. The patient usually spends half of this time intubated in the ICU. About 25% of patients are extubated on hospital day 7, 50% by hospital day 13, and 75% by hospital day 31. The mortality rate during hospitalisation for crisis has fallen from nearly 50% in the early 1960s to between 3% and 10% today. With the incidence of crisis remaining stable over the past 30 years, this fall in mortality rates probably reflects improvements in the intensive care assessment and management of these patients.114
Nursing practice
Careful and accurate assessment by the nurse in the presenting myasthenic crisis patient determines the triggers of the event and incorporates a history, including infections and prescribed medications. These medications can exacerbate the acetylcholine receptor blockade, and respiratory demand proves too much for the myasthenic patient. Awareness by the nurse of trigger medications ensures advocacy for the patient when the prescription is uncertain.114
• Respiratory and cardiovascular assessment incorporates upper and lower airway muscle weakness. ABGs are a poor marker for intubation and ventilation because these values change late in the decompensation cycle. Being able to recognise fatigue (inability to speak, poor lung expansion, VC below 1 L, shoulder and arm weakness) in patients with neuromuscular weakness and air hunger is important.117
• Non-invasive ventilation can be difficult to administer safely, with the potential for aspiration due to insidious upper airway weakness, however the option should be considered with careful assessment of gag, swallow and cough reflexes in order to prevent intubation.116
• Neuromuscular blockade should be avoided (residual long-term paralysis), with the use of glottal local anaesthetic spray for emergency intubation and ventilation.
• Placement of small-bore duodenal tubes decreases the risk of aspiration and may be more comfortable than regular nasogastric tubes for the patient.
• Tracheostomy is generally not needed, as the duration of intubation is often less than 2 weeks.
• Cardiac assessment needs to include assessment for arrhythmias of both atrial and ventricular origin due to autonomic nerve dysfunction.112 These can be insidious and can be provoked by subtle changes in electrolytes.
• Nursing care will relate to the needs of long-term immobilised, intubated, ventilated patients with neuromuscular alterations.
Myasthenia gravis patients have similar care needs to those of patients with GBS (refer to Independent practice for GBS). Fatigue and the structure and timing of care are very important. Flexibility of care is important, as energy fluctuates on an hourly basis.117 Despite having a shorter recovery time than GBS, weaning and recovery in myasthenia gravis is a still a slow process and impulsive extubation is discouraged.118 Therapy should be tailored on an individual basis using best clinical judgment.
Selected Neurological Cases
Status Epilepticus
Status epilepticus (SE) has been generally defined as enduring seizure activity that is not likely to stop spontaneously. The traditional SE definition is 30 minutes of continuous seizure activity (which has recently been updated due to neurological alteration to 5 minutes only)or 2 or more seizures without full recovery of consciousness between the seizures.119 There are as many types of SE as there are types of seizures. The distinction between convulsive and nonconvulsive SE depends on clinical observation and on a clear understanding of several SE types. Estimates of the overall incidence of SE have varied from 10 to 60 per 100,000 person-years, depending on the population studied and the definitions used.120 Over half the cases of SE are acute symptomatic, emphasising the importance in management of identifying an acute precipitant. Infections with fever are the major cause of SE, accounting for 52% of cases, while in adults low antiepileptic drug levels, cerebrovascular accident, hypoxia, metabolic causes and alcohol represent the main acute causes. The mortality in status epilepticus is about 20%; most patients die of the underlying condition rather than the status epilepticus itself. SE can result in permanent neurological and mental deterioration, particularly in young children; the risks of morbidity greatly increase with longer duration of the status epilepticus episode. SE in the intensive care setting falls into two main groups: those transferred to the ICU because of uncontrolled SE (refractory SE), and those who are admitted to the ICU for another reason and have SE as an additional finding.121
Pathophysiology
At a cellular level, status epilepticus results from a failure of normal inhibitory pathways, primarily mediated by gamma-aminobutyric acid (GABA) acting via GABA (A) receptors. This loss of inhibitory drive allows the activation of excitatory feedback loops, leading to repetitive, synchronous firing of large groups of neurons. As seizure activity continues, there is further decline in GABAergic function. Continued excitatory input mediated primarily by glutamate leads to neuronal cell death.119
Clinical Manifestations
Convulsive SE is a medical emergency. The initial consequence of a prolonged convulsion is a massive release of plasma catecholamines, which results in a rise in heart rate, blood pressure and plasma glucose. During this stage cardiac arrhythmias are often seen, and may be fatal. Cerebral blood flow is greatly increased, and thus glucose delivery to active cerebral tissue is maintained. As the seizure continues, hyperthermia above 40°C with lactic and respiratory acidosis continues to intensify especially without adequate resuscitation and control of the seizure.122
The SE may then enter a second, late phase in which cerebral and systemic protective measures progressively fail. The main characteristics of this phase are: a fall in blood pressure; a loss of cerebral autoregulation, resulting in the dependence of cerebral blood flow on systemic blood pressure; and hypoglycaemia due to the exhaustion of glycogen stores and increased neurogenic insulin secretion. Intracranial pressure can rise precipitously in SE. The combined effects of systemic hypotension and intracranial hypertension may result in a compromised cerebral circulation and cerebral oedema. Intracranial pressure monitoring is advisable in prolonged severe SE when raised intracranial pressure is suspected. Further complications that can occur include rhabdomyolysis leading to acute tubular necrosis, hyperkalaemia and hyponatraemia.122
Nursing Practice
The following nursing practice should be undertaken.
Resuscitation
SE requires control of the seizure and then investigation regarding the cause. Airway protection is often difficult in the seizing patient, so the first line of treatment includes basic life-support measures followed by the administration of IV propofol, midazolam or, in refractory cases, phenytoin. Neuromuscular blockade will be required to facilitate intubation in patients who continue to have tonic–clonic seizure activity despite these pharmacological interventions. Rocuronium (1 mg/kg), a short-acting, non-depolarising muscle relaxant that is devoid of significant haemodynamic effects and does not raise intracranial pressure, is the preferred agent. Succinylcholine should be avoided if possible, as the patient may be hyperkalaemic as a consequence of possible rhabdomyolysis. Prolonged neuromuscular blockade should be avoided as it only stops the motor response hence masking the altered neuronal activity.123 Once the seizures are controlled, intubation and ventilation can protect the airway and potentially reverse the acidosis. In the patient who already has an airway secured, urgent IV administration of propofol, midazolam or phenytoin is indicated.124
Specific post-SE patient assessment
EEG monitoring in the ICU for refractory SE is essential, as a patient may enter a drug-induced coma with little outward sign of convulsions yet have ongoing electrographic epileptic activity. Furthermore, continuous recording will give an indication of worsening of generalised convulsive status epilepticus regardless of the presence or absence of sedating drugs or paralysing agents. This can be monitored only by EEG and manifests as bilateral EEG ictal discharges. Deeper sedation and anaesthesia is then indicated and can be titrated to EEG results.124
Collaborative practice
Because only a small fraction of seizures go on to become SE, the probability that a given seizure will proceed to SE is small at the start of the seizure and increases with seizure duration. The goal of pharmacological therapy is to achieve the rapid and safe termination of the seizure, and to prevent its recurrence without adverse effects on the cardiovascular and respiratory systems or without altering the level of consciousness. Diazepam, lorazepam, midazolam, phenytoin and phenobarbitone have all been used as first-line therapy for the termination of SE.125 The antiseizure activity of phenytoin is complex; however, its major action appears to block the voltage-sensitive, use-dependent sodium channels. Once SE is controlled, attention turns to preventing its recurrence. The best regimen for an individual patient will depend on the cause of the seizure and any history of antiepileptic drug therapy. A patient who develops SE in the course of alcohol withdrawal may not need antiepileptic drug therapy once the withdrawal has run its course. In contrast, patients with new, ongoing epileptogenic stimuli (e.g. encephalitis or trauma) may require high doses of antiepileptic medication to control their seizures.
Intracerebral Haemorrhage
Intracerebral haemorrhage (ICH) is an acute and spontaneous extravasation of blood into the brain parenchyma and is one of the most serious subtypes of stroke, affecting over a million people worldwide each year, most of whom live in Asia. About one-third of people with ICH die early after onset. The majority of survivors are left with major long-term disability. ICH accounts for 10–30% of all stroke admissions to hospital, and leads to catastrophic disability, morbidity, and a 6-month mortality of 30–50%.126 Long-term outcomes are poor: only 20% of patients regain functional independence at 6 months. ICH is most common in men, in elderly people, and in Asians and African–Americans. The annual crude incidence of stroke in Australia has been estimated at 17.8 per 100,000 126 and in 2006 there were 8484 deaths attributable to stroke.127
There are several modifiable risk factors for spontaneous ICH. Hypertension is by far the most important and prevalent risk factor, directly accounting for about 60–70% of cases.128 Chronic hypertension causes degeneration, fragmentation, and fibrinoid necrosis of small penetrating arteries in the brain, which can eventually result in spontaneous rupture. Hypertensive ICH typically occurs in the basal ganglia (putamen, thalamus or caudate nucleus), pons, cerebellum, or deep hemispheric white matter.
Pathophysiology
Understanding of the pathophysiology of ICH has changed in recent years. What was thought to be a simple and rapid bleeding event is now understood to be a dynamic and complex process that involves several distinct phases. The two most important new concepts are that many haemorrhages continue to grow and expand over several hours after onset of symptoms – a process known as early haematoma growth – and that most of the brain injury and swelling that happens in the days after ICH is the result of inflammation caused by thrombin and other coagulation end-products.129
On rupture of a pathologically altered artery, blood extravasates into the surrounding parenchyma. The blood appears to dissect tissue planes, compressing adjacent structures. Serial imaging has shown that 20–38% of ICH haematomas enlarge within 36 hours of onset. Haematomas larger than 25 cm3 are more likely to grow in the first six hours after symptom onset. In addition, elevated systolic blood pressure and serum glucose levels are independently associated with enlargement of the haematoma. About half of spontaneous ICH cases originate in the basal ganglia, a third in the cerebral hemispheres, and a sixth in the brainstem or cerebellum.130
Clinical manifestations
In 40% of cases, ICH is accompanied by intraventricular haemorrhage, which can cause acute hydrocephalus and high ICP, and lessens the chance of a good outcome. Rapid onset of a focal neurological deficit with clinical signs of high ICP – such as an abrupt change in level of consciousness, headache, and vomiting – suggest a diagnosis of ICH. However, these symptoms can also take place after acute ischaemic stroke. For this reason, CT or MRI is essential for confirming dziagnosis. Rapid progression to coma with motor posturing suggests massive supratentorial haemorrhage, bleeding into the brainstem or diencephalon, or acute obstructive hydrocephalus due to intraventricular haemorrhage. Over 90% of patients have acute hypertension exceeding 160/100 mmHg, whether or not there is a history of pre-existing hypertension. Dysautonomia in the form of central fever, hyperventilation, hyperglycaemia, and tachycardia or bradycardia is also common.131
Nursing Practice
The following nursing practice should be undertaken.
Specific blood pressure management
There is a high risk of deterioration, death or dependency with raised blood pressure after ICH; thus it and should be corrected immediately to minimise the potential for haematoma expansion and to maintain adequate cerebral perfusion pressure.132 Extreme hypertension within the first six hours is common and should be aggressively but carefully treated to avoid excessive reduction of the cerebral perfusion pressure, which might precipitate ischaemia in the region surrounding the haematoma.
The Australian Stroke Foundation’s current guidelines recommend a target systolic BP below 180 mmHg or a mean arterial blood pressure of 130 mmHg.133 Management of BP is particularly important in ICH and is currently the subject of a large Australian RCT (Interact-2).134
Prevention of cerebral ischaemia and secondary brain injury
Intravenous therapy should be aimed at maintaining euvolaemia with an isotonic fluid, such as normal saline. Potassium supplementation is often necessary, although glucose should be avoided, except in rare cases of hypoglycaemia. Emergency measures for ICP control are appropriate for stuporous or comatose patients, or those who present acutely with clinical signs of brainstem herniation (see the section on Management of intracranial hypertension and ischaemia). The head should be elevated to 30 degrees for optimal balance between perfusion and intracranial pressure and to prevent aspiration. Warfarin increases the risk of ICH 5–10 times, and presenting patients should have this reversed with fresh frozen plasma, prothrombin-complex concentrates and vitamin K. Early in the course of patients with ICH, even with exclusion of coagulopathy, injection of activated factor VII results in significant reduction in the rate of haematoma expansion.90
Summary
Case study
Research vignette
Critique
Interestingly, this study did not suggest a substantial improvement in outcomes following dissemination of evidence-based guidelines for the management of TBI in comparison to historical controls in America, Europe and Australia, despite during the ICU admission, there was concordance with evidence-based guidelines concerning systemic monitoring and supportive measures such as nutrition, thromboprophylaxis and gastric ulcer prophylaxis. Similarly, there were consistent practices in the participating ICUs concordant with management guidelines for TBI. This was typified by the low incidence of the use of ‘brain-specific therapies’ such as osmotherapy, barbiturates, hypothermia, hyperventilation and corticosteroids. However ICP monitoring was employed in less than half of patients admitted with severe TBI, for which intraparenchymal pressure tipped catheters were most commonly used. It should be noted that since then improvement has been noted in an Australasian study with the SAFE study in patients with TBI72 demonstrating higher ICP monitoring rates more in line with the TBI guidelines, using ventricular catheters (~75%) and conversely lower mortality (24.56%) overall and (29.24%) in severe TBI.
Learning activities
1. What clinical signs are indicative of a fractured base of skull? Are the injuries noted on CT focal or diffuse?
2. Reading the Case Study, interpret Sam’s vital signs in relation to cerebral perfusion. Are management changes required?
3. Ischaemia prevention requires a PbtO2>20. How can this be achieved?
4. What is the pathophysiological basis for the rise in ICP? How would this manifest on the ICP waveform?
5. A 20-year-old man suffered spinal cord injury at the C2–C3 level as the result of a motorcycle accident. Explain the effects of this man’s injury on ventilation and communication; sensorimotor function; autonomic nervous system function; bowel, bladder and sexual function; and temperature regulation.
6. A 25-year-old-man is an unbelted driver involved in a motor vehicle accident and presents in a coma.
7. A child is taken to the emergency room with lethargy, fever and a stiff neck on examination.
8. Your patient had symptoms of an ischaemic stroke approximately 2 hours ago and is undergoing a confirmatory CT scan in 30 minutes. You know tPA must be administered within 3 hours of the symptoms. What actions would you take? What is your rationale for these actions?
American Association of Spinal Cord Injury Nurses (AASCIN). http://www.aascin.org.
The Brain Trauma Foundation. http://www.braintrauma.org.
Centers for Disease Control: Traumatic Brain Injury. www.cdc.gov/traumaticbraininjuy/index.html.
Cerebral Spinal Fluid Drainage. http://intensivecare.hsnet.nsw.gov.au/five/doc/evd_csfspecimen_S_n_liverpool.pdf.
Cervical Collars. http://intensivecare.hsnet.nsw.gov.au/five/doc/cervical_collars_care_fitting_S_n_stgeorge.pdf.
Australian Institute of Health and Welfare publication. [Stroke] www.aihw.gov.au/cvd/stroke.cfm.
Australian & NZ Traumatic Brain Injury Study Results (ATBIS). www.anzics.com.au/ctg/completed-studies/50-atbis- or Brain Injury Association, Inc
Cervical Traction. http://intensivecare.hsnet.nsw.gov.au/five/doc/cervical_traction_S_n_nepean.pdf.
Ethical guidelines for the care of people in post-coma unresponsiveness http://www.nhmrc.gov.au/_files_nhmrc/file/publications/synopses/e81.pdf
External Ventricular Drains. http://intensivecare.hsnet.nsw.gov.au/five/doc/evd_guideline_S_n_liverpool.pdf.
External Ventricular Drain Removal. http://intensivecare.hsnet.nsw.gov.au/five/doc/evd_removalof_S_n_liverpool.pdf.
Hypertonic Saline Protocol. http://www.ambulance.qld.gov.au/medical/pdf/Hypertonic_Saline_7.5_DTP_1.048_Ver_1.1.1.pdf.
Meningitis. http://netsvic.org.au/clinicalguide/cpg.cfm?doc_id=5179.
Model of Stroke Care Western Australia. http://www.healthnetworks.health.wa.gov.au/modelsofcare/docs/Stroke_Model_of_Care.pdf.
National Resource Centre for Traumatic Brain Injury. www.brainlink.org.au.
www.anzics.monash.org/atbis.html.
National Stroke Foundation of Australia publication. Did you know that? http://www.strokefoundation.com.au.
Neurological Foundation of New Zealand. http://www.neurological.org.nz/.
Post Coma Unresponsiveness Guidelines. http://www.nhmrc.gov.au/_files_nhmrc/file/publications/synopses/e81.pdf.
Spinal Injury Log Roll Protocol. http://intensivecare.hsnet.nsw.gov.au/five/doc/logroll_guideline_R_cp_rnsh.pdf.
Spinal Injury Methylprednisolone Protocol. http://intensivecare.hsnet.nsw.gov.au/five/doc/methylprednisolone_spinalcord_D_svh.pdf.
Stroke Foundation of New Zealand. http://www.stroke.org.nz/.
Stroke Management Guidelines. http://www.strokesociety.com.au/index.php?option=com_docman&Itemid=196.
Stroke Thrombolytic Protocol. http://www.mja.com.au/public/issues/187_10_191107/bat11279_fm.pdf.
Sports injuries: head and spine www.injuryupdate.com.au/injuries/head_&_neck/spinal_injuries.php
Traumatic Brain Injury National Data Centre. http://www.tbindc.org/.
1 Olson D, Graffagnino C. Consciousness, coma, and caring for the brain-injured patient. AACN Clin Iss. 2005;16(4):441–455.
2 Duffau H. Does post-lesional subcortical plasticity exist in the human brain? Neurosci Res. 2009;65(2):131–135.
3 Cavanna AE, Cavanna SL, Servo S, Monaco F. The neural correlates of impaired consciousness in coma and unresponsive states. Discov Med. 2010;9(48):431–438.
4 Strosznajder RP, Czubowicz K, Jesko H, Strosznajder JB. Poly(ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol Neurobiol. 2010;41(2–3):187–196.
5 Abou Khaled KJ, Hirsch LJ. Updates in the management of seizures and status epilepticus in critically ill patients. Neurol Clin. 2008;26(2):385–408.
6 Varelas P, Mirski M. Management of seizures in critically ill patients. Curr Neurol Neurosci Rep. 2004;4(6):489–496.
7 Veening JG, Barendregt HP. The regulation of brain states by neuroactive substances distributed via the cerebrospinal fluid; a review. Cerebrospinal Fluid Res. 2010;6(7):1.
8 Winhammar J, Rowe D, Henderson R, Kiernan M. Assessment of disease progression in motor neuron disease. Lancet Neurol. 2005;4(4):229–238.
9 Hallett M. Plasticity of the human motor cortex and recovery from stroke. Brain Res Rev. 2001;36(2–3):169–174.
10 Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol. 2009;5(7):374–381.
11 Tanaka M, Mizuno K, Tajima S, Sasabe T, Watanabe Y. Central nervous system fatigue alters autonomic nerve activity. Life Sci. 2009;84(7–8):235–239.
12 Soustiel JF, Larisch S. Mitochondrial damage: a target for new therapeutic horizons. Neurotherapeutics. 2010;7(1):13–21.
13 Back T, Schuler O. The natural course of lesion development in brain ischemia. Acta Neurochir Suppl. 2004;89:55–61.
14 Offner H, Vandenbark AA, Hurn PD. Effect of experimental stroke on peripheral immunity: CNS ischemia induces profound immunosuppression. Neuroscience. 2009;158:1098–1111.
15 Trendelenburg G, Dirnagl U. Neuroprotective role of astrocytes in cerebral ischemia: focus on ischemic preconditioning. Glia. 2005;50(4):307–320.
16 Tripathy D, Grammas P. Acetaminophen inhibits neuronal inflammation and protects neurons from oxidative stress. J Neuroinflammation. 2009;6:10.
17 Li Jun, Han Baoqing, Ma Xuesong, Qi Sihua. The effects of propofol on hippocampal caspase-3 and Bcl-2 expression following forebrain ischemia–reperfusion in rats. Brain Research. 2010;1356:11–23.
18 Benarroch E. Neuron–astrocyte interactions: partnership for normal function and disease in the central nervous system. Mayo Clin Proc. 2005;80(10):1326–1338.
19 Kahle KT, Simard JM, Staley KJ, Nahed BV, Jones PS, Sun D. Molecular mechanisms of ischemic cerebral edema: role of electroneutral ion transport. Physiology. 2009;24(4):257–265.
20 Strbian D, Durukan A, Pitkonen M, Marinkovic I, Tatlisumak E, et al. The blood–brain barrier is continuously open for several weeks following transient focal cerebral ischemia. Neuroscience. 2008;153(1):175–181.
21 Owler BK, Pitham T, Wang D. Aquaporins: relevance to cerebrospinal fluid physiology and therapeutic potential in hydrocephalus. Cerebrospinal Fluid Res. 2010;7(15):7–15.
22 Edwards R, Dombrowski S, Luciano M, Pople I. Chronic hydrocephalus in adults. Brain Pathol. 2004;14(3):325–336.
23 McLeod A. Traumatic injuries to the head and spine, 2: nursing considerations. Br J Nurs. 2004;13(17):1041–1049.
24 Wolfe TJ, Torbey MT. Management of intracranial pressure. Curr Neurol Neurosci Rep. 2009;9(6):477–485.
25 Hickey JV, Olson DM, Turner DA. Intracranial pressure waveform analysis during rest and suctioning. Biol Res Nurs. 2009;11(2):174–186.
26 Czosnyka M, Pickard J. Monitoring and interpretation of intracranial pressure. J Neurol Neurosurg Psychiat. 2004;75(6):813–821.
27 Porth C, Martin G. Essentials of pathophysiology: concepts of altered health states, 3rd edn. Philadelphia: Lippincott: Williams & Wilkins; 2011.
28 Lavinio A, Rasulo FA, De Peri E, Czosnyka M, Latronico N. The relationship between the intracranial pressure-volume index and cerebral autoregulation. Intensive Care Med. 2009;35(3):546–549.
29 Stiefel MF, Udoetuk JD, Spiotta AM, Gracias VH, Goldberg A, et al. Conventional neurocritical care and cerebral oxygenation after traumatic brain injury. J Neurosurg. 2006;105:568–575.
30 Rahlwink UK, Figaji AA. Methods of monitoring brain oxygenation. Childs Nerv Syst. 2010;26(4):453–464.
31 Jaeger M, Dengl M, Meixensberger J, Schuhmann MU. Effects of cerebrovascular pressure reactivity-guided optimization of cerebral perfusion pressure on brain tissue oxygenation after traumatic brain injury. Crit Care Med. 2010;38(5):1343–1347.
32 Brain Trauma Foundation, American Association of Neurological Surgeons, Joint Section on Neurotrauma and Critical Care. Guidelines for management of severe head injury. New York: Brain Trauma Foundation; 2007.
33 Haitsma IK, Maas AIR. Monitoring cerebral oxygenation in traumatic brain injury. Prog Brain Res. 2007;161:207–216.
34 Bhatia A, Gupta AK. Neuromonitoring in the intensive care unit. II. Cerebral oxygenation monitoring and microdialysis. Intensive Care Med. 2007;33(8):1322–1328.
35 Curley G, Kavanagh BP, Laffey JG. Hypocapnia and the injured brain: more harm than benefit. Crit Care Med. 2010;38(5):1348–1359.
36 Fletcher JJ, Bergman K, Blostein PA, Kramer AH. Fluid balance, complications, and brain tissue oxygen tension monitoring following severe traumatic brain injury. Neurocrit Care. 2010;13(1):47–56.
37 Toung TJK, Yi Chang, Lin J, Bhardwaj A. Increases in lung and brain water following experimental stroke: effect of mannitol and hypertonic saline. Crit Care Med. 2005;33(1):203–208.
38 Haglund MM, Hochman DW. Furosemide and mannitol suppression of epileptic activity in the human brain. J Neurophysiol. 2005;94(2):907–918.
39 Cooper DJ, Myles PS, McDermott FT, Murray L, Laidlaw J, et al. Prehospital hypertonic saline resuscitation of patients with hypotension and severe traumatic brain injury: a randomised controlled trial. JAMA. 2004;291:1350–1357.
40 Dietrich WD, Bramlett HM. The evidence for hypothermia as a neuroprotectant in traumatic brain injury. Neurotherapeutics. 2010;7(1):43–50.
41 Liu L, Yenari MA. Clinical application of therapeutic hypothermia in stroke. Neurol Res. 2009;31(4):331–335.
42 Povlishock JT, Wei EP. Posthypothermic rewarming considerations following traumatic brain injury. J Neurotrauma. 2009;26(3):333–340.
43 Marion DW, Penrod LE, Kelsey SF, et al. Treatment of traumatic brain injury with moderate hypothermia. New Engl J Med. 1997;336:540–546.
44 Zhao QJ, Zhang XG, Wang LX. Mild hypothermia therapy reduces blood glucose and lactate and improves neurologic outcomes in patients with severe traumatic brain injury. J Crit Care. 2011. In press
45 Masaoka H. Cerebral blood flow and metabolism during mild hypothermia in patients with severe traumatic brain injury. J Med Dent Sci. 2010;57(2):133–138.
46 Sydenham E, Roberts I, Alderson P. Hypothermia for traumatic head injury. Cochrane Database Syst Rev. 2009 (2): CD001048.
47 Clifton GL, Drever P, Valadka A, Zygun D, Okonkwo D. Multicenter trial of early hypothermia in severe brain injury. J Neurotrauma. 2009;26(3):393–397.
48 CRASH trial collaborators. Final results of MRC CRASH, a randomised placebo controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet. 2005;365(9475):1957–1959.
49 Brain Trauma Foundation. Anesthetics, analgesics, and sedatives. J Neurotrauma. 2007;24(Supp 1):S71–S76.
50 Li LM, Timofeev I, Czosnyka M, Hutchinson PJ. Review article: the surgical approach to the management of increased intracranial pressure after traumatic brain injury. Anesth Analg. 2010;111(3):736–748.
51 Vibbert M, Mayer SA. Early decompressive hemicraniectomy following malignant ischemic stroke: the crucial role of timing. Curr Neurol Neurosci Rep. 2010;10(1):1–3.
52 Cooper James D, Rosenfeld JV, Murray L, Arabi YM, Davies AR, D’Urso P, Kossmann T, Ponsford J, Seppelt I, Reilly P, Wolfe R. Decompressive craniectomy in diffuse traumatic brain injury. N Engl J Med. 2011;364(16):1493–1502.
53 Rinkel GJ, Feigin VL, Algra A, van den Bergh WM, Vermeulen M, van Gijn J. Calcium antagonists for aneurysmal subarachnoid haemorrhage. Cochrane Database Syst Rev 2005; (1): CD000277.
54 Temkin NR, Anderson GD, Winn HR, Ellenbogen RG, Britz GW, et al. Magnesium sulfate for neuroprotection after traumatic brain injury: a randomised controlled trial. Lancet Neurol. 2007;6(1):29–38.
55 Van den Bergh W, the MASH Study Group. Magnesium sulfate in aneurysmal subarachnoid hemorrhage: a randomized controlled trial. Stroke. 2005;36(5):1011–1015.
56 Muench E, Horn P, Bauhuf C, Roth H, Philipps M, et al. Effects of hypervolemia and hypertension on regional cerebral blood flow, intracranial pressure, and brain tissue oxygenation after subarachnoid hemorrhage. Crit Care Med. 2007;35(8):1844–1851.
57 Eddleman CS, Hurley MC, Naidech AM, Batjer HH, Bendok BR. Endovascular options in the treatment of delayed ischemic neurological deficits due to cerebral vasospasm. Neurosurg Focus. 2009;26(3):E6.
58 Australian Institute of Health and Welfare (AIHW). Australian hospital statistics 2008–09 [Health Services Series no. 34. AIHW cat. no. HSE 37]. AIHW: Canberra, 2010.
59 Helps Y, Henley G, Harrison JE. Hospital separations due to traumatic brain injury, Australia 2004–05 [Injury research and statistics series number 45. (Cat no. INJCAT 116)]. AIHW: Adelaide, 2008.
60 New Zealand Health Information Service. Selected morbidity data for public funded hospitals 2001/2002. Wellington: New Zealand Ministry of Health; 2004.
61 Fortune N, Wen X. The definition, incidence and prevalence of acquired brain injury in Australia [AIHW cat. no. DIS 15]. Australian Institute of Health and Welfare: Canberra, 1999.
62 Australian Institute of Health and Welfare (AIHW). Health and welfare of Australia’s Aboriginal and Torres Strait Islander peoples [AIHW cat. no. IHW-14]. AIHW: Canberra, 2005.
63 Bradley C, Harrison JE. Injury risk factors, attitudes and awareness [Submission to the CATI-TRG. Injury Technical Paper Series no. 3. AIHW cat. no. INJCAT 61]. AIHW: Adelaide, 2003.
64 Myburgh JA, Cooper DJ, Finfer SR, Venkatesh B, Jones D, et al. Epidemiology and 12-month outcomes from traumatic brain injury in Australia and New Zealand. J Trauma-Injury Infect Crit Care. 2008;64(4):854–862.
65 Luukinen H, Viramo P, Herala M, Kervinen K, Kesaniemi YA, et al. Fall-related brain injuries and the risk of dementia in elderly people: a population-based study. Eur J Neurol. 2005;12(2):86–92.
66 Keenan HT, Runyan DK, Marshall SW, Nocera MA, Merten DF. A population-based comparison of clinical and outcome characteristics of young children with serious inflicted and noninflicted traumatic brain injury. Pediatrics. 2004;114(3):633–639.
67 Gaetz M. The neurophysiology of brain injury. Clin Neurophysiol. 2004;115(1):4–18.
68 Soustiel JF, Sviri GE, Mahamid E, Shik V, Abeshaus S, Zaaroor M. Cerebral blood flow and metabolism following decompressive craniectomy for control of increased intracranial pressure. Neurosurgery. 2010;67(1):65–72.
69 Badruddin A, Taqi MA, Abraham MG, Dani D, Zaidat OO. Neurocritical care of a reperfused brain. Curr Neurol Neurosci. 2011;11(1):104–110.
70 Hendricks HT, Heeren AH, Vos PE. Dysautonomia after severe traumatic brain injury. Eur J Neurol. 2010;17(9):1172–1177.
71 Andriessen TM, Jacobs B, Vos PE. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J Cell Mol Med. 2010;14(10):2381–2392.
72 SAFE Study Investigators; Australian and New Zealand Intensive Care Society Clinical Trials Group; Australian Red Cross Blood Service; George Institute for International HealthMyburgh J, et al. Saline or albumin for fluid resuscitation in patients with traumatic brain injury. N Engl J Med. 2007;357(9):874–884.
73 Jagoda AS. Mild traumatic brain injury: key decisions in acute management. Psychiatr Clin North Am. 2010;33(4):797–806.
74 Donovan D. Simple depressed skull fracture causing sagittal sinus stenosis and increased intracranial pressure: case report and review of the literature. Surg Neurol. 2005;63(4):380–383.
75 Norton L. Spinal cord injury, Australia 2007–08 [Injury research and statistics series no. 52. Cat. no. INJCAT 128]. AIHW: Canberra, 2010.
76 Russ SA, Hancock SW, Quinton M, Moore R, Harrison P. Patterns and risks in spinal trauma: the emergency transport perspective. Arch Dis Child. 2005;90(9):985.
77 Robertson A, Branfoot T, Barlow I, Giannoudis P. Spinal injury patterns resulting from car and motorcycle accidents. Spine. 2002;27(24):2825–2830.
78 Gala V, Voyadzis JM, Kim D, Asir A, Fessler RG. Trauma of the nervous system, spinal cord trauma. Bradley W, Daroff D, Fenichel D, Jankovic J. Neurology in clinical practice, 5th edn, Philadelphia: Butterworth-Heinemann Elsevier, 2008.
79 Sheerin F. Spinal cord injury: causation and pathophysiology. Emerg Nurse. 2005;12(9):29–38.
80 Pimentel L, Diegelmann L. Evaluation and management of acute cervical spine trauma. Emerg Med Clin North Am. 2010;28(4):719–738.
81 Ahn H, Singh J, Nathens A, Macdonald RD, Travers A, Tallon J, Fehlings M, Yee A. Pre-hospital care management of a potential spinal cord injured patient: a systematic review of the literature and evidence-based guidelines. J Neurotrauma. 2011. In press
82 Gupta R, Bathen ME, Smith JS, Levi AD, Bhatia NN, Steward OJ. Advances in the management of spinal cord injury. Am Acad Orthop Surg. 2010;18(4):210–222.
83 Hawryluk GW, Rowland J, Kwon BK, Fehlings MG. Protection and repair of the injured spinal cord: a review of completed, ongoing, and planned clinical trials for acute spinal cord injury. Neurosurg Focus. 2008;25(5):E14.
84 Godoy DA, Di Napoli M, Rabinstein AA. Treating hyperglycemia in neurocritical patients: benefits and perils. Neurocrit Care. 2010;13(3):425–438.
85 Australian Institute of Health and Welfare. Cardiovascular disease mortality: trends at different ages [Cardiovascular series no. 31. Cat. no.47]. AIHW: Canberra, 2010.
86 Stroke Foundation of New Zealand and New Zealand Guidelines Group. Clinical Guidelines for Stroke Management 2010. Wellington: Stroke Foundation of New Zealand; 2010.
87 Benejam B, Sahuquillo J, Poca MA, Frascheri L, Solana E, et al. Quality of life and neurobehavioral changes in survivors of malignant middle cerebral artery infarction. Neurol. 2009;256(7):1126–1133.
88 Runchey S, McGee S. Does this patient have a hemorrhagic stroke? Clinical findings distinguishing hemorrhagic stroke from ischemic stroke. JAMA. 2010;303(22):2280–2286.
89 Oliveria-Filho J, Koroshetz WJ. Hemorrhagic stroke. In: O’Donnell J, Nácul FE. Surgical intensive care medicine. New York: Springer, 2010.
90 Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2008;358(20):2127–2137.
91 Dankbaar JW, Slooter AJ, Rinkel GJ, Schaaf IC. Effect of different components of triple-H therapy on cerebral perfusion in patients with aneurysmal subarachnoid haemorrhage: a systematic review. Crit Care. 2010;14(1):R23.
92 Jost SC, Diringer MN, Zazulia AR, Videen TO, et al. Effect of normal saline bolus on cerebral blood flow in regions with low baseline flow in patients with vasospasm following subarachnoid haemorrhage. J Neurosurg. 2005;103(1):25–30.
93 Hinson HE, Hanley DF, Ziai WC. Management of intraventricular hemorrhage. Curr Neurol Neurosci Rep. 2010;10(2):73–82.
94 Bederson JB, Connolly ES, Jr., Batjer HH, Dacey RG, Dion JE, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke. 2009;40(3):994–1025.
95 Coghlan LA, Hindman BJ, Bayman EO, Banki NM, Gelb AW, et al. Independent associations between electrocardiographic abnormalities and outcomes in patients with aneurysmal subarachnoid hemorrhage: findings from the intraoperative hypothermia aneurysm surgery trial. Stroke. 2009;40(2):412–418.
96 Agostoni E, Aliprandi A, Longoni M. Cerebral venous thrombosis. Expert Rev Neurother. 2009;9(4):553–564.
97 Huppatz C, Kelly PM, Levi C, Dalton C, Williams D, Durrheim DN. Encephalitis in Australia, 1979–2006: trends and aetiologies. Commun Dis Intell. 2009;33(2):192–197.
98 Martin D, Lopez L. The epidemiology of meningococcal disease in New Zealand in 2009. [Cited December 2010]. Available from http://www.surv.esr.cri.nz/PDF_surveillance/MeningococcalDisease/2009/2009AnnualRpt.pdff
99 Communicable Diseases Network Australia, Meningococcal Disease Sub-Committee. Guidelines for the early clinical and public health management of meningococcal disease in Australia 2007. [Cited December 2010]. Available from http://www.health.gov.au/internet/main/publishing.nsf/content/BC329B583B663546CA25736D007674AA/$File/meningococcal-guidelines.pdf
100 Markey PG, Davis JS, Harnett GB, Williams SH, Speers DJ. Meningitis and a febrile vomiting illness caused by echovirus type 4, northern territory, Australia. Emerg Infect Dis. 2010;16(1):63–68.
101 Huppatz C, Durrheim DN, Levi C, Dalton C, Williams D, et al. Etiology of encephalitis in Australia, 1990–2007. Emerg Infect Dis. 2009;15(9):135–165.
102 Wilder-Smith A, Halstead S. Japanese encephalitis: Update on vaccines and vaccine recommendations. Curr Opin Infect Dis. 2010;23(5):426–431.
103 Playford EG, McCall B, Smith G, Slinko V, Allen G, et al. Human hendra virus encephalitis associated with equine outbreak, Australia, 2008. Emerg Infect Dis. 2010;16(2):219–223.
104 Einsiedel L, Kat E, Ravindran J, Slavotinek J, Gordon DL. MR findings in Murray Valley encephalitis. Am J Neuroradiol. 2003;24(7):1379–1382.
105 Douglas MW, Stephens DP, Burrow JNC, Anstey NM, Talbot K, Currie BJ. Murray valley encephalitis in an adult traveller complicated by long-term flaccid paralysis: Case report and review of the literature. Trans R Soc Trop Med Hyg. 2007;101(3):284–288.
106 Roberts JA, Grant KA, Yoon YK, Polychronopoulos S, Ibrahim A, Thorley BR. Annual report of the Australian national poliovirus reference laboratory, 2008. Commun Dis Intell. 2009;33(3):291–297.
107 Sederholm BH. Treatment of acute immune-mediated neuropathies: Guillain–Barré syndrome and clinical variants. Semin Neurol. 2010;30(4):365–372.
108 Fergusson D, Hutton B, Sharma M, Tinmouth A, Kumanan Wilson D, et al. Use of intravenous immunoglobulin for treatment of neurologic conditions: a systematic review. Transfusion. 2005;45(10):1640–1657.
109 Orlikowski D, Prigent H, Sharshar T, Lofaso F, Raphael JC. Respiratory dysfunction in Guillain–Barré syndrome. Neurocrit Care. 2004;1(4):415–422.
110 Ruts L, Drenthen J, Jongen JLM, Hop WCJ, Visser GH, et al. Pain in Guillain–Barré syndrome: A long-term follow-up study. Neurology. 2010;75(16):1439–1447.
111 Walgaard C, Lingsma HF, Ruts L, Drenthen J, Van Koningsveld R, et al. Prediction of respiratory insufficiency in Guillain–Barré syndrome. Ann Neurol. 2010;67(6):781–787.
112 Flachenecker P. Autonomic dysfunction in guillain-barré syndrome and multiple sclerosis. J Neurol. 2007;254(2 Supp):96–101.
113 Gregory MA, Gregory RJ, Podd JV. Understanding Guillain–Barré syndrome and central nervous system involvement. Rehabil Nurs. 2005;30(5):207–212.
114 Romi F, Gilhus NE, Aarli JA. Myasthenia gravis: clinical, immunological, and therapeutic advances. Acta Neurol Scand. 2005;111(2):134–141.
115 Bershad EM, Feen ES, Suarez JI. Myasthenia gravis crisis. South Med J. 2008;101(1):63–69.
116 Seneviratne J, Mandrekar J, Wijdicks EFM, Rabinstein AA. Noninvasive ventilation in myasthenic crisis. Arch Neurol. 2008;65(1):54–58.
117 Kittiwatanapaisan W, Gauthier DK, Williams AM, Oh SJ. Fatigue in myasthenia gravis patients. J Neurosci Nurs. 2003;35(2):87–93. 106
118 Seneviratne J, Mandrekar J, Wijdicks EFM, Rabinstein AA. Predictors of extubation failure in myasthenic crisis. Arch Neurol. 2008;65(7):929–933.
119 Huff JS, Fountain NB. Pathophysiology and definitions of seizures and status epilepticus. Emerg Med Clin North Am. 2011;29(1):1–13.
120 Chin RF, Neville BG, Scott RC. A systematic review of the epidemiology of status epilepticus. Eur J Neurol. 2004;11(12):800–810.
121 Marik PE. Seizures and Status Epilepticus. In: Marik E, ed. Handbook of evidence-based critical care. New York: Springer; 2010:503–516. Part 6
122 Bleck TP. Refractory status epilepticus. Curr Opin Crit Care. 2005;11(2):117–120.
123 Shearer P, Riviello J. Generalized convulsive status epilepticus in adults and children: Treatment guidelines and protocols. Emerg Med Clin North Am. 2011;29(1):51–64.
124 Rossetti AO, Oddo M. The neuro-ICU patient and electroencephalography paroxysms: If and when to treat. Curr Opin Crit Care. 2010;16(2):105–109.
125 Claassen J, Hirsch LJ, Emerson RG, Mayer SA. Treatment of refractory status epilepticus with pentobarbital, propofol, or midazolam: a systematic review. Epilepsia. 2002;43(2):146–153.
126 Jamrozik K, Dobson A, Hobbs M, et al. Monitoring the incidence of cardiovascular disease in Australia [AIHW cat. no. CVD 16. Cardiovascular Disease Series no. 17]. AIHW: Canberra, 2001.
127 Australian Institute of Health and Welfare (AIHW). How we manage stroke in Australia [AIHW cat. no. CVD 31.Series S]. AIHW: Canberra, 2006.
128 National Heart Foundation of Australia (National Blood Pressure and Vascular Disease Advisory Committee). Guide to management of hypertension 2008. Updated August 2009. Web version 2009. [Cited April 2011]. Available from www.heartfoundation.org.au
129 Fischbein NJ, Wijman CAC. Nontraumatic intracranial hemorrhage. Neuroimaging Clin N Am. 2010;20(4):469–492.
130 Qureshi AI, Mendelow AD, Hanley DF. Intracerebral haemorrhage. Lancet. 2009;373(9675):1632–1644.
131 Elliott J, Smith M. The acute management of intracerebral hemorrhage: a clinical review. Anesth Analg. 2010;110(5):1419–1427.
132 Tetri S, Juvela S, Saloheimo P, Pyhtinen J, Hillbom M. Hypertension and diabetes as predictors of early death after spontaneous intracerebral hemorrhage. J Neurosurg. 2009;110(3):411–417.
133 National Stroke Foundation. Clinical Guidelines for Stroke Management 2010. Melbourne: National Stroke Foundation; 2010.
134 Interact 2 Clinical trial. [Cited Dec 2010]. Available from http://clinicaltrials.gov/ct2/show/NCT00716079?term=NCT00716079&rank=1