[level-membership-for-surgery-category]
Chapter 57 Neurofibromatosis 2
NEUROFIBROMATOSIS 2 DIFFERENTIATED FROM NEUROFIBROMATOSIS 1
CLINICAL CHARACTERISTICS OF NEUROFIBROMATOSIS 2
Definition
The NIH Consensus Development Conference also developed guidelines for the diagnosis of NF2. NF2 is distinguished by bilateral vestibular schwannomas with multiple meningiomas, cranial tumors, optic gliomas, and spinal tumors. A definite diagnosis is made on the basis of the presence of bilateral vestibular schwannomas or developing a unilateral vestibular schwannoma by age 30 and a first-degree blood relative with NF2, or developing at least two of the following conditions known to be associated with NF2: meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacity/juvenile cortical cataract (Table 57-1).1
| INDIVIDUALS WITH THE FOLLOWING CLINICAL FEATURES HAVE CONFIRMED (DEFINITE) NF2 |
| Bilateral VS or family history of NF2 (first-degree family relative) plus |
| Unilateral VS <30 years or |
| Any two of the following: meningioma, glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract |
| INDIVIDUALS WITH THE FOLLOWING CLINICAL FEATURES SHOULD BE EVALUATED FOR NF2 (PRESUMPTIVE OR PROBABLE NF2) |
| Unilateral VS <30 years plus at least one of the following: meningioma, glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract |
| Multiple meningiomas (≥2) plus unilateral VS <30 years or one of the following: glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract |
VS, vestibular schwannomas.
There may be significant heterogeneity in the presentation of the disease from one individual to the next. Some individuals may have a very mild form of the disease with small vestibular schwannomas manifesting in an older individual. Meanwhile, some children present with multiple intracranial tumors at a very young age. Despite the heterogeneity of the disease within a family, the expression of NF2 tends to be very similar.2 There is a significant genetic component to the disease with much variability within the parameters of the observed phenotype. Studies have shown that a truncating mutation (nonsense and frame shift) may be linked with a more severe form of NF2.3–5 The more severe form of NF2 is termed Wishart form. Individuals with this severe form present with early onset of the disease with multiple intracranial schwannomas and meningiomas that result in blindness, deafness, paralysis, and death by age 40. Despite the strong genotype-phenotype correlation, individual differences in tumor growth occur within subjects, making it difficult to predict how an individual will change over time even when the genotype is known.
The milder form, or Gardner form, of NF2 is less debilitating. The schwannomas may remain stable for many years, few meningiomas develop, and patients may not develop symptoms until later in life and often have fewer disabilities. The genetic basis of the mild form has not been well characterized. Many of these may be mosaic forms of the disease, however.2-4,6-12
Prevalence and Incidence
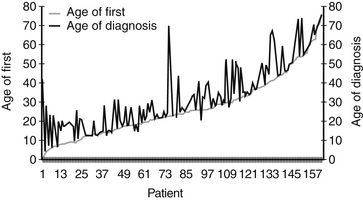
The average age of diagnosis of NF2 is 25 years; however, many patients present with symptoms before the diagnosis. There is an average delay of diagnosis of approximately 7 years (Fig. 57-1). There is no difference in the proportion of men versus women who develop NF2, and no prevalence has been described based on ethnicity. Epidemiologic studies place the incidence of NF2 between 1 in 40,000 live births13 and 1 in 87,410 live births.14
Molecular Genetics
The NF2 gene was mapped to chromosome 22 q 12-2in 1993.15–17 The NF2 gene located at chromosome 22 codes for a tumor suppressive protein termed Merlin or Schwannomin. This protein negatively regulates Schwann cell production. The loss of this protein allows overproduction of Schwann cells. The mutation in the NF2 chain predisposes individuals to developing a schwannoma when the second hit occurs to the gene; control of Schwann cells is lost or mutated within the cell. Various types of mutations have been identified, including single base substitutions, insertions, and deletions.4,18–20 The mild, or Gardner, type of NF2 may be associated with missense mutations, whereas associations between the other mutations and phenotypes are not as clear.21 The occurrence of NF2 is not restricted to families known to carry the mutation. Frequently, genetic mosaicism occurs, which may not be detected by common mutation analysis techniques.22 Unilateral vestibular schwannomas may exhibit the same type of genetic markers as NF2.23 The mutations in unilateral vestibular schwannomas are confined to the affected tumor tissue. In patients with NF2, the mutation is present in all cell types.22
Tumor Types
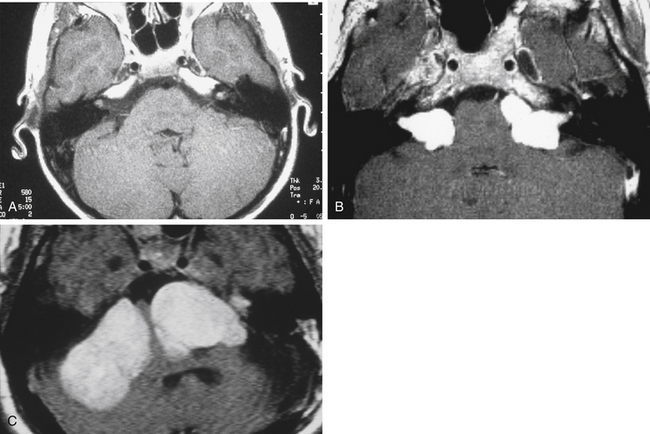
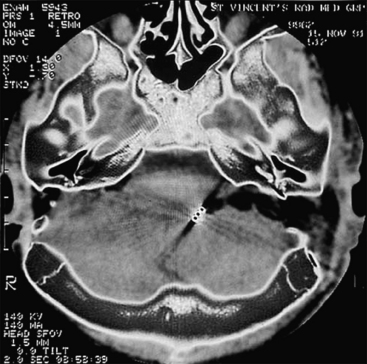
Bilateral vestibular schwannomas (acoustic neuromas) are benign neoplasms of the acoustic or eighth cranial nerve (Fig. 57-2).24 The tumors are thought to arise at the glioma–Schwann’s cell junction within the internal auditory meatus. The tumors most commonly arise from the superior vestibular nerve, although with NF2, tumors may be found on the cochlear and facial nerves within the internal auditory meatus. The consequences of a vestibular schwannoma are numerous, including hearing loss progressing to deafness, dizziness and balance problems, tinnitus, facial nerve paralysis, brainstem compression, and, if left untreated, death.
Despite the strong genetic effect in NF2, there is enormous variability in the number of tumor types, the rate of progression, and the disabilities experienced. This enormous variability is also found in patient presentation. Some patients may be asymptomatic. Patients who have no symptoms when diagnosed have generally been identified on the basis of genetic analysis conducted because a blood relative has NF2 or presymptomatic screening. Although the NIH criteria for NF2 require the presence of bilateral vestibular schwannomas for diagnosis, patients may first develop unilateral schwannomas as a young child with no other tumors, or adult patients may present with multiple meningiomas (cranial and spinal) and no vestibular schwannomas.9,25 Although the NIH criteria for NF2 imply that all NF2 patients develop bilateral vestibular schwannomas, some researchers are not convinced of this.36 Evans and colleagues26 based their conclusion on the observation of a possible variant form of NF2 manifesting with skin and spinal tumors in the absence of vestibular schwannomas. Nonetheless, the phenotype generally is reflective of the underlying disorder.
Intracranial Schwannomas
Vestibular schwannomas are the most common intracranial schwannoma associated with NF2. The most frequently identified nonvestibular schwannomas are schwannomas of CN III and V. Bilateral CN III or V schwannomas are the most common additional schwannomas seen. It is important to identify these lesions on MRI. Lower cranial nerve schwannomas may also be identified, but are much less frequently seen. A vestibular schwannoma rarely turns malignant, and sometimes the unilateral vestibular schwannoma may regress in size altogether. Growth of the tumors does not seem to be related either to loss of heterozygosity (genetic level of analysis) or to auditory functioning (phenotype level of analysis). For this reason, it is recommended that a patient have at least yearly MRI scans to track changes in size.33–40 All newly diagnosed patients should have a full head and spine study to stage their disease. After the disease is staged, a 6 month study is performed to determine if the tumor is fast-growing or slow-growing.41
Hearing Changes in Patients with Vestibular Schwannomas
Hearing loss is well documented as the most common presenting symptom in patients who have vestibular schwannomas.42–51 Auditory changes over time in vestibular schwannoma patients are less well known. Rosenberg52 studied the natural history of 80 patients with non-NF2 unilateral vestibular schwannomas for an average of 4.4 years. Rosenberg52 found a positive correlation between tumor growth and worsening pure tone average. There was no statistically significant correlation, however, between positive tumor growth and speech discrimination, change in ABR, and bithermal caloric electronystagmography test result over time.
Lalwani and colleagues53 reported that pure tone pattern speech receptive thresholds and word recognition scores were significantly worse in NF2 patients who had a mild form of NF2 and large tumors compared with patients with mild NF2 with small tumors. Loss of acoustic reflexes and prolonged wave III and V were also associated with larger tumors. In contrast, patients with severe NF2 showed no relationship among tumor size and pure tone levels, speech discrimination thresholds, or word recognition scores. The lack of association may have been due to complete loss of hearing in severe NF2 patients at the time of the assessment. The larger tumors were also associated with prolonged ABR waves III and V latencies. No data across time were reported. Generally, hearing is progressively impaired with increasing growth of vestibular schwannomas necessitating the need for surgical intervention or medical treatments in NF2 patients.
The natural history study evaluated hearing changes prospectively in 63 newly diagnosed NF patients, and found that hearing was stable after diagnosis. During the first 2 years after diagnosis, 27% of patients had a significant change in their hearing, and 73% had very stable hearing during the 2 year period. Patients with a family history of NF2 had more stable hearing after an initial diagnosis compared with patients without a family history. Patients with a family history are usually diagnosed at a younger age; the stability of hearing in this group may represent the younger age group. The better the hearing in the newly diagnosed patients, the more stable the hearing. Hearing changes did not vary between ears with each ear acting independently.54
Other Tumor Types in Neurofibromatosis 2
Meningiomas
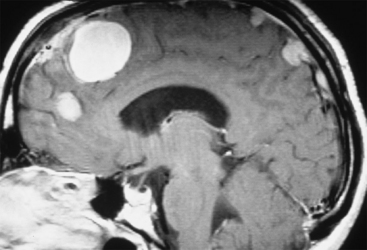
NF2 has been associated with multiple central nervous system tumors, the most common of which are intracranial meningiomas (Fig. 57-3).55 Nearly all NF2 patients develop these tumors in time. Fifty percent of NF2 patients present with schwannomas and meningiomas; 90% present with spine tumors in addition to schwannomas. The presence of more than one type of tumor in a patient usually indicates a more aggressive disease course. The co-occurrence of vestibular schwannomas and meningiomas has been linked to a synergistic effect of increase in growth rate of the schwannoma and the meningioma beyond that expected of a sporadic schwannoma or meningioma.56,57 Despite the high number of patients with multiple tumors initially, most meningiomas and spinal tumors are asymptomatic and are first seen on MRI. In addition, multiple skin tumors may be found in patients with NF2 (Table 57-3).
Spinal Tumors
Various spinal tumors may occur in NF2 patients, and can be found in the cervical, thoracic, and lumbar region. These tumors are categorized further as either extramedullary or intramedullary tumors depending on their presentation relative to the spinal cord. Extramedullary tumors are commonly schwannomas or meningiomas, whereas intramedullary tumors are often ependymomas, but these can be astrocytomas or schwannomas.58 Spinal tumors may often be numerous small tumors in the cauda equina. Tumors located in the cervical or thoracic region are usually solitary tumors, and these may grow to cause spinal cord compression. These tumors may have a solid or cystic component similar to schwannomas and meningiomas seen intracranially. These tumors may extend out the spinal foramen into the soft tissue causing spinal cord compression.59
Eye Findings
NF2 subjects tend also to develop cortical and posterior subcapsular cataracts, which can lead to blindness (see Table 57-2).27 Retinal hamartomas have been observed in a few cases,2,28–30 but are not as frequent. Some subjects (2% to 3% of subjects)31,32 present with numbness or tingling in their arms or legs. Almost 30% of NF2 subjects may have surgery to remove spinal tumors, but the progression of spinal tumors associated with NF2 is not well described. At this time, the presence of vestibular schwannoma in NF2 and the consequences of not treating them are well known, and these tumors may be the most debilitating.
| Symptoms | Patients (%) |
|---|---|
| Neurologic | 17.5 |
| Skin tumor | 11.7 |
| Vision loss | 10.7 |
| Asymptomatic | 10.7 |
| Tinnitus | 7.8 |
| Weakness | 2.9 |
| Vertigo | 1 |
| Other/unspecified | 4.9 |
| Tumor Type | Patients (%) |
|---|---|
| Bilateral vestibular schwannoma | 99 |
| Skin | 50 |
| Meningioma | 46 |
| Spinal | 60 |
TREATMENT OPTIONS FOR VESTIBULAR SCHWANNOMAS IN NEUROFIBROMATOSIS 2
Hearing Preservation
Hearing preservation rates for small unilateral tumors have approached 70%.51 The results in NF2 patients seem to be worse, however, than the results reported in patients with unilateral vestibular schwannomas.63 Doyle and Shelton64 found that 67% of NF2 patients underwent hearing presentation surgery using the middle fossa approach, and 38% of those had serviceable hearing postoperatively. Improvements in the middle fossa surgical approach were introduced in 1992 that led to a significant improvement in the outcomes of NF2 tumor removal. A more recent review of 18 NF2 patients who had middle fossa removal of their tumors reported that approximately 50% of patients had hearing preserved at the preoperative level; this compares with 25% in the previous study. The overall ability to preserve any hearing was still close to 68%.65 The results of this series have led to a more aggressive attempt to preserve hearing in NF2 patients. Patients who present with small tumors and good hearing are now routinely offered an attempt at hearing preservation. Long-term follow-up is still needed in NF2 patients because additional tumors may arise in the facial or cochlear nerves.
We have published our results treating children with NF2 with small tumors and good hearing. This retrospective chart review reported on 35 children with NF2 who had undergone middle fossa resection (47 surgeries) between 1992-2004. In 55% of surgeries, hearing of less than or equal to 70 dB pure tone average was maintained postoperatively. It is now our practice to perform middle fossa resection in children with NF2, and the sooner in the disease process, the better. Our results indicate that hearing and facial nerve function can be successfully preserved using this approach. Factors to consider include patient age, severity of additional NF2-related symptoms, and obtaining high-quality, thin-slice MR images before surgery. Bilateral middle fossa resection after hearing preservation on the first side is also successful and potentially preserves hearing in both ears.66
Retrosigmoid Craniotomy with Partial Removal
Retrosigmoid craniotomy with partial removal in NF2 patients carries a significant risk because the cochlear fibers are dispersed throughout the tumor, in contrast to unilateral vestibular schwannomas (Fig. 57-4). The risk of hearing loss with partial removal is much higher, and this procedure is typically not recommended.
Auditory Brainstem Implant
The auditory brainstem implant was developed at the House Ear Institute to allow electric stimulation of the cochlear nucleus after bilateral vestibular schwannoma removal. The device is placed on the brainstem (Fig. 57-5) during translabyrinthine vestibular schwannoma removal. This device is indicated in individuals who have no serviceable hearing and are undergoing vestibular schwannoma removal. Most patients obtain enhanced communication skills with the device.
Stereotactic Irradiation
Stereotactic irradiation has been recommended for some NF2 patients, but its use must be carefully considered because radiation exposure may induce or accelerate tumors in a patient with an inactivated tumor suppressor gene. It was reported more recently that two of four patients who had previously received radiation therapy developed a malignancy in the irradiated ear.67
1. Gutmann D.H., Aylsworth A., Carey J.C., et al. The diagnostic evaluation and multidisciplinary management of NF1 and NF2. JAMA. 1997;278:51-57.
2. Parry D.M., Eldridge R., Kaiser-Kupfer M.I., et al. Neurofibromatosis 2 (NF2): Clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet. 1994;52:450-461.
3. Evans D.G., Trueman L., Wallace A., et al. Genotype/phenotype correlations in type 2 neurofibromatosis (NF2): Evidence for more severe disease associated with truncating mutations. J Med Genet. 1998;35:450-455. [published erratum appears in J Med Genet 361(1):87, 1999]
4. Kluwe L., Mautner V.F. A missense mutation in the NF2 gene results in moderate and mild clinical phenotypes of neurofibromatosis type 2. Hum Genet. 1996;97:224-227.
5. Ruttledge M.H., Andermann A.A., Phelan C.M., et al. Type of mutation in the NF2 gene frequently determines severity of disease. Am J Hum Genet. 1996;59:331-342.
6. Baser M.E., Mautner V.F., Ragge N.K., et al. Presymptomatic diagnosis of neurofibromatosis 2 using linked genetic markers, neuroimaging, and ocular examinations. Neurology. 1996;47:1269-1277.
7. Bijlsma E.K., Merel P., Fleury P., et al. Family with neurofibromatosis type 2 and autosomal dominant hearing loss: Identification of carriers of the mutated NF2 gene. Hum Genet. 1995;96:1-5.
8. Gardner W.J., Frazier C.H. Bilateral acoustic neurofibromas: A clinical study and field survey of a family of five generations with bilateral deafness in thirty-eight members. Arch Neurol Psychiatry. 1930;23:266-302.
9. Mautner V.F., Baser M.E., Kluwe L. Phenotype variability in two families with novel splice-site and frameshift NF2 mutations. Hum Genet. 1996;98:203-206.
10. Sainio M., Strachan T., Blomstedt G., et al. Presymptomatic DNA and MRI diagnosis of neurofibromatosis 2 with mild clinical course in an extended pedigree. Neurology. 1995;45:1314-1322.
11. Welling D.B. Clinical manifestations of mutations in the neurofibromatosis type 2 gene in vestibular schwannomas (acoustic neuromas). Laryngoscope. 1998;108:178-189.
12. Wishart J.H. Case of tumors of the skull, dura mater and brain. Edinburgh Med Surg J. 1822;18:393-397.
13. Evans D.G., Huson S.M., Donnai D., et al. A genetic study of type 2 neurofibromatosis in the United Kingdom, I: Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29:841-846.
14. Antinheimo J., Sankila R., Carpen O., et al. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology. 2000;54:71-76.
15. Rouleau G.A., Wertelecki W., Haines J.L., et al. Genetic linkage of bilateral acoustic neurofibromatosis to a DNA marker on chromosome 22. Nature. 1987;329:246-248.
16. Rouleau G.A., Merel P., Lutchman M., et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neurofibromatosis type 2. Nature. 1993;363:515-521.
17. Trofatter J.A., MacCollin M.M., Rutter J.L., et al. A novel moesin-ezrin-, radixin-like gene is a candidate for the neurofibromatosis type 2 tumor suppressor. Cell. 1993;72:791-800.
18. Merel P., Haong-Xuan K., Sanson M., et al. Predominant occurrence of somatic mutations of the NF2 gene in meningiomas and schwannomas. Genes Chromosomes Cancer. 1995;13:211-216.
19. Merel P., Hoang-Xuan K., Sanson M., et al. Screening for germ-line mutations in the NF2 gene. Genes Chromosomes Cancer. 1995;12:117-127.
20. Welling D.B., Guida M., Goll F., et al. Mutational spectrum in the neurofibromatosis type 2 gene in sporadic and familial schwannomas. Hum Genet. 1996;98:189-193.
21. Welling D.B. Clinical manifestations of mutations in the neurofibromatosis type 2 gene in vestibular schwannomas (acoustic neuromas). Laryngoscope. 1998;108:178-189.
22. Wu C.L., Thakker N., Neary W., et al. Differential diagnosis of type 2 neurofibromatosis: Molecular discrimination of NF2 and sporadic vestibular schwannomas. J Med Genet. 1998;35:973-977.
23. Irving R.M., Harada T., Moffat D.A., et al. Somatic neurofibromatosis type 2 gene mutations and growth characteristics in vestibular schwannoma. Am J Otol. 1997;18:754-760.
24. Cushing H. Bilateral Acoustic Tumors, Generalized Neurofibromatosis and the Meningeal Endotheliomata. Tumors of the Nervous Acoustics and the Syndrome of the Cerebellopontine Angle. Philadelphia: Saunders; 1963. (1917 original edition)
25. Mautner V.F., Lindenau M., Koppen J., et al. Type 2 neurofibromatosis without acoustic neuroma. Zentralbl Neurochir. 1995;56:83-87.
26. Evans D.G., Lye R., Neary W., et al. Probability of bilateral disease in people presenting with a unilateral vestibular schwannoma. J Neurol Neurosurg Psychiatry. 1999;66:764-767.
27. Bouzas E.A., Freidlin V., Parry D.M., et al. Lens opacities in neurofibromatosis 2: Further significant correlations. Br J Ophthalmol. 1993;77:354-357.
28. Good W.V., Erodsky M.C., Edwards M.S., Hoyt W.F. Bilateral retinal hamartomas in neurofibromatosis type 2. Br J Ophthalmol. 1991;75:190.
29. Landau K., Yasargil G.M. Ocular fundus in neurofibromatosis type 2. Br J Ophthalmol. 1993;77:646-649.
30. Ragge N.K., Baser M.E., Klein J., et al. Ocular abnormalities in neurofibromatosis 2. Am J Ophthalmol. 1995;120:634-641.
31. Evans D.G., Huson S.M., Donnai D., et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84:603-618.
32. Lim D.J., Rubenstein A.E., Evans D.G., et al. Advances in neurofibromatosis 2 (NF2): A workshop report. J Neurogenet. 2000;14:63-106.
33. Mathies C., Samii M., Krebs S. Management of vestibular schwannomas (acoustic neuromas): Radiological features in 202 cases—their value for diagnosis and their predictive importance. Neurosurgery. 1997;40:469-481.
34. Burkey J.M., Rizer F.M., Schuring A.G., et al. Acoustic reflexes, auditory brainstem response, and MRI in the evaluation of acoustic neuromas. Laryngoscope. 1996;106:839-841.
35. Curati W.L., Graif M., Kingsley D.P., et al. MRI in acoustic neuroma: A review of 35 patients. Neuroradiology. 1986;28:208-214.
36. Duffner P.K., Cohen M.E., Seidel F.G., Shucard D.W. The significance of MRI abnormalities in children with neurofibromatosis. Neurology. 1989;39:373-378.
37. Levine S.C., Antonelli P.J., Le C.T., Haines S.J. Relative value of diagnostic tests for small acoustic neuromas. Am J Otol. 1991;12:341-346.
38. Lhullier F.M., Doyon D.L., Halimi P.M., et al. Magnetic resonance imaging of acoustic neuromas: Pitfalls and differential diagnosis. Neuroradiology. 1992;34:144-149.
39. Long S.A., Arriaga M., Nelson R.A. Acoustic neuroma volume: MRI-based calculations and clinical implications. Laryngoscope. 1993;103:1093-1096.
40. Modugno G.C., Pirodda A., Ferri G.G., et al. Small acoustic neuromas: Monitoring the growth rate by MRI. Acta Neurochir. 1999;141:1063-1067.
41. Irving R.M., Moffat D.A., Hardy D.G., et al. A molecular, clinical, and immunohistochemical study of vestibular schwannoma. Otolaryngol Head Neck Surg. 1997;116:426-430.
42. Strasnick B., Glasscock M.E.III, Haynes D., et al. The natural history of untreated acoustic neuromas. Laryngoscope. 1994;104:1115-1119.
43. Fucci M.J., Buchman C.A., Brackmann D.E., Berliner K.I. Acoustic tumor growth: Implications for treatment choices. Am J Otol. 1999;20:495-499.
44. Brackmann D.E., Owens R.M., Friedman R.A., et al. Prognostic factors for hearing preservation in vestibular schwannoma surgery. Am J Otol. 1999;20:495-499.
45. Briggs R.J., Brackmann D.E., Baser M.E., Hitselberger W.E. Comprehensive management of bilateral acoustic neuromas: Current perspectives. Arch Otolaryngol Head Neck Surg. 1994;120:1307-1314.
46. Doyle K.J., Nelson R.A. Bilateral acoustic neuromas (NF2). In: House W.F., Luetje C.M., Doyle K.J., editors. Acoustic Tumors: Diagnosis and Management. San Diego: Singular Publishing Group, 1997.
47. Evans D.G., Huson S.M., Donnai D., et al. A genetic study of type 2 neurofibromatosis in the United Kingdom, I: Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29:841-846.
48. Gadre A.K., Kwartler J.A., Brackmann D.E., et al. Middle fossa decompression of the internal auditory canal in acoustic neuroma surgery: A therapeutic alternative. Laryngoscope. 1990;100:948-952.
49. Kesterson L., Shelton C., Dressler L., Berliner K.I. Clinical behavior of acoustic tumors: A flow cytometric analysis. Arch Otolaryngol Head Neck Surg. 1993;119:269-271.
50. Saunders J.E., Luxford W.M., Devgan K.K., Fetterman B.L. Sudden hearing loss in acoustic neuroma patients. Otolaryngol Head Heck Surg. 1995;113:23-31.
51. Slattery W.H.III, Brackmann D.E., Hitselberger W. Middle fossa approach for hearing preservation with acoustic neuromas. Am J Otol. 1997;18:596-601.
52. Rosenberg S.I. Natural history of acoustic neuromas. Laryngoscope. 2000;110:497-508.
53. Lalwani A.K., Abaza M.M., Makariou E.V., Armstrong M. Audiologic presentation of vestibular schwannoma in neurofibromatosis type 2. Am J Otol. 1998;19:352-357.
54. Masuda A, Fischer LM, Oppenheimer ML, et al. 2004
55. Bouzas E.A., Parry D.M., Eldridge R., Kaiser-Kupfer M.I. Visual impairment in patients with neurofibromatosis 2. Neurology. 1993;43:622-623.
56. Pallini R., Tancredi A., Cassalbore P., et al. Neurofibromatosis type 2: Growth stimulation of mixed acoustic schwannoma by concurrent adjacent meningioma: Possible role of growth factors. Case report. J Neurosurug. 1998;89:149-154.
57. Antinheimo J., Haappasalo H., Haltia M., et al. Proliferation potential and histological features in neurofibromatosis 2-associated and sporadic meningiomas. J Neurosurg. 1997;87:610-614.
58. Patronas N.J., Courcoutsakis N., Bromley C.M., et al. Intramedullary and spinal canal tumors in patients with neurofibromatosis 2: MR imaging findings and correlation with genotype. Radiology. 2001;218:434-442.
59. Gillepsie J.E. Imaging in neurofibromatosis 2: Screening using magnetic resonance imaging. Ear Nose Throat J. 1999;78:102-103.
60. Delleman J.W., De Jong J.G., Bleeker G.M. Meningiomas in five members of a family over two generations, in one member simultaneously with acoustic neurinomas. Neurology. 1978;28:567-570.
61. King A., Gutmann D.H. The question of familial meningiomas and schwannomas: NF2B or not to be? Neurology. 2000;54:4-5.
62. Wiebe S., Munoz D.G., Smith S., Lee D.H. Meningioangiomatosis: A comprehensive analysis of clinical and laboratory features. Brain. 1999;122:709-726.
63. Slattery W.H.III, Brackmann D.E. Hearing preservation and restoration in CPA tumor surgery. Neurosurg Q. 1997;7:169-182.
64. Doyle K.J., Shelton C. Hearing preservation in bilateral acoustic neuroma surgery. Am J Otol. 1993;14:562-565.
65. Slattery W.H., Brackmann D.E., Hitzelburger W. Hearing Preservation in NF-2. Am Journal Otology. 1998;19:638-643.
66. Slattery W.H., Fischer L.M., Hitzelburger W., et al. Hearing Preservation Surgery for NF-2 related Vestibular Schwannomas in Pediatric Patients. J Neurosurg. 2007;106:255-260.
67. Baser M.E., Ragge N.K., Riccardi V.M., et al. Phenotype variability in monozygotic twins with neurofibromatosis 2. Am J Med Genet. 1996;64:563-567.
[/level-membership-for-surgery-category][not-level-membership-for-surgery-category]
Chapter 57 Neurofibromatosis 2
NEUROFIBROMATOSIS 2 DIFFERENTIATED FROM NEUROFIBROMATOSIS 1
CLINICAL CHARACTERISTICS OF NEUROFIBROMATOSIS 2
Definition
The NIH Consensus Development Conference also developed guidelines for the diagnosis of NF2. NF2 is distinguished by bilateral vestibular schwannomas with multiple meningiomas, cranial tumors, optic gliomas, and spinal tumors. A definite diagnosis is made on the basis of the presence of bilateral vestibular schwannomas or developing a unilateral vestibular schwannoma by age 30 and a first-degree blood relative with NF2, or developing at least two of the following conditions known to be associated with NF2: meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacity/juvenile cortical cataract (Table 57-1).1
| INDIVIDUALS WITH THE FOLLOWING CLINICAL FEATURES HAVE CONFIRMED (DEFINITE) NF2 |
| Bilateral VS or family history of NF2 (first-degree family relative) plus |
| Unilateral VS <30 years or |
| Any two of the following: meningioma, glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract |
| INDIVIDUALS WITH THE FOLLOWING CLINICAL FEATURES SHOULD BE EVALUATED FOR NF2 (PRESUMPTIVE OR PROBABLE NF2) |
| Unilateral VS <30 years plus at least one of the following: meningioma, glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract |
| Multiple meningiomas (≥2) plus unilateral VS <30 years or one of the following: glioma, schwannoma, juvenile posterior subcapsular lenticular opacities/juvenile cortical cataract |
VS, vestibular schwannomas.
There may be significant heterogeneity in the presentation of the disease from one individual to the next. Some individuals may have a very mild form of the disease with small vestibular schwannomas manifesting in an older individual. Meanwhile, some children present with multiple intracranial tumors at a very young age. Despite the heterogeneity of the disease within a family, the expression of NF2 tends to be very similar.2 There is a significant genetic component to the disease with much variability within the parameters of the observed phenotype. Studies have shown that a truncating mutation (nonsense and frame shift) may be linked with a more severe form of NF2.3–5 The more severe form of NF2 is termed Wishart form. Individuals with this severe form present with early onset of the disease with multiple intracranial schwannomas and meningiomas that result in blindness, deafness, paralysis, and death by age 40. Despite the strong genotype-phenotype correlation, individual differences in tumor growth occur within subjects, making it difficult to predict how an individual will change over time even when the genotype is known.
The milder form, or Gardner form, of NF2 is less debilitating. The schwannomas may remain stable for many years, few meningiomas develop, and patients may not develop symptoms until later in life and often have fewer disabilities. The genetic basis of the mild form has not been well characterized. Many of these may be mosaic forms of the disease, however.2-4,6-12
Prevalence and Incidence
The average age of diagnosis of NF2 is 25 years; however, many patients present with symptoms before the diagnosis. There is an average delay of diagnosis of approximately 7 years (Fig. 57-1). There is no difference in the proportion of men versus women who develop NF2, and no prevalence has been described based on ethnicity. Epidemiologic studies place the incidence of NF2 between 1 in 40,000 live births13 and 1 in 87,410 live births.14
Molecular Genetics
The NF2 gene was mapped to chromosome 22 q 12-2in 1993.15–17 The NF2 gene located at chromosome 22 codes for a tumor suppressive protein termed Merlin or Schwannomin. This protein negatively regulates Schwann cell production. The loss of this protein allows overproduction of Schwann cells. The mutation in the NF2 chain predisposes individuals to developing a schwannoma when the second hit occurs to the gene; control of Schwann cells is lost or mutated within the cell. Various types of mutations have been identified, including single base substitutions, insertions, and deletions.4,18–20 The mild, or Gardner, type of NF2 may be associated with missense mutations, whereas associations between the other mutations and phenotypes are not as clear.21 The occurrence of NF2 is not restricted to families known to carry the mutation. Frequently, genetic mosaicism occurs, which may not be detected by common mutation analysis techniques.22 Unilateral vestibular schwannomas may exhibit the same type of genetic markers as NF2.23 The mutations in unilateral vestibular schwannomas are confined to the affected tumor tissue. In patients with NF2, the mutation is present in all cell types.22
Tumor Types
Bilateral vestibular schwannomas (acoustic neuromas) are benign neoplasms of the acoustic or eighth cranial nerve (Fig. 57-2).24 The tumors are thought to arise at the glioma–Schwann’s cell junction within the internal auditory meatus. The tumors most commonly arise from the superior vestibular nerve, although with NF2, tumors may be found on the cochlear and facial nerves within the internal auditory meatus. The consequences of a vestibular schwannoma are numerous, including hearing loss progressing to deafness, dizziness and balance problems, tinnitus, facial nerve paralysis, brainstem compression, and, if left untreated, death.
Despite the strong genetic effect in NF2, there is enormous variability in the number of tumor types, the rate of progression, and the disabilities experienced. This enormous variability is also found in patient presentation. Some patients may be asymptomatic. Patients who have no symptoms when diagnosed have generally been identified on the basis of genetic analysis conducted because a blood relative has NF2 or presymptomatic screening. Although the NIH criteria for NF2 require the presence of bilateral vestibular schwannomas for diagnosis, patients may first develop unilateral schwannomas as a young child with no other tumors, or adult patients may present with multiple meningiomas (cranial and spinal) and no vestibular schwannomas.9,25 Although the NIH criteria for NF2 imply that all NF2 patients develop bilateral vestibular schwannomas, some researchers are not convinced of this.36 Evans and colleagues26 based their conclusion on the observation of a possible variant form of NF2 manifesting with skin and spinal tumors in the absence of vestibular schwannomas. Nonetheless, the phenotype generally is reflective of the underlying disorder.
Intracranial Schwannomas
Vestibular schwannomas are the most common intracranial schwannoma associated with NF2. The most frequently identified nonvestibular schwannomas are schwannomas of CN III and V. Bilateral CN III or V schwannomas are the most common additional schwannomas seen. It is important to identify these lesions on MRI. Lower cranial nerve schwannomas may also be identified, but are much less frequently seen. A vestibular schwannoma rarely turns malignant, and sometimes the unilateral vestibular schwannoma may regress in size altogether. Growth of the tumors does not seem to be related either to loss of heterozygosity (genetic level of analysis) or to auditory functioning (phenotype level of analysis). For this reason, it is recommended that a patient have at least yearly MRI scans to track changes in size.33–40 All newly diagnosed patients should have a full head and spine study to stage their disease. After the disease is staged, a 6 month study is performed to determine if the tumor is fast-growing or slow-growing.41
[/not-level-membership-for-surgery-category]
