Chapter 5 Neurocutaneous disorders
1. What are the two main forms of neurofibromatosis?
Neurofibromatosis type 1 (NF-1 or von Recklinghausen’s disease) and neurofibromatosis type 2 (NF-2). NF-1 accounts for 90% of all cases of neurofibromatosis and affects approximately 1 in 3500 individuals. NF-2 is a genetically distinct entity with a prevalence of 1 in 25,000. Both conditions have autosomal dominant inheritance with 50% of cases representing new mutations.
2. Outline the diagnostic criteria for NF-1.
The diagnosis can be made if two or more of the following criteria are present:
• Six or more café-au-lait macules >5 mm in greatest diameter in prepubertal children and >15 mm diameter in postpubertal individuals
4. What is the earliest skin sign of NF-1?
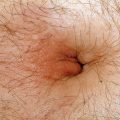
Café-au-lait macules. These sharply defined, light brown patches may be present at birth but more commonly start appearing in the first year of life (Fig. 5-1). They are noted initially by 4 years or less in all affected children and within the first year in 82% of cases.
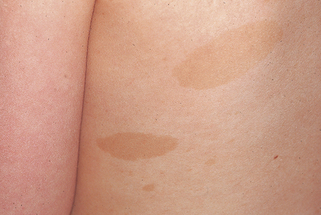
Boyd KP, Korf BR, Theos A: Neurofibromatosis type 1, J Am Acad Dermatol 61:1–14, 2009.
5. What is Crowe’s sign? When does it develop?
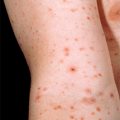
Crowe’s sign is freckling of the axillae or other body folds. It develops in 90% of NF-1 patients, usually in middle childhood. These lesions are not really freckles, but multiple small café-au-lait macules.
Key Points: Neurocutaneous Disorders
2. Multiple café-au-lait macules are the earliest skin sign of NF-1, occurring in approximately 80% of affected babies by the end of the first year of life.
3. NF-2 most commonly presents with deafness or tinnitus related to underlying vestibular schwannomas.
6. When do peripheral neurofibromas appear in NF-1?
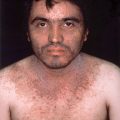
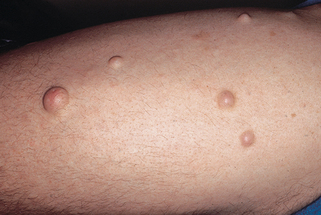
Peripheral neurofibromas usually start to develop during puberty but increase in size and number in early adult life. They are soft, pink, or flesh-colored papules, nodules, or tumors distributed mainly over the trunk and limbs (Fig. 5-2). Multiple neurofibromas can develop or existing neurofibromas may enlarge during pregnancy.
7. What is a plexiform neurofibroma?
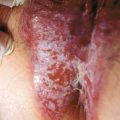
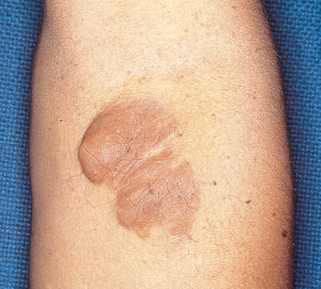
It is a diffuse, elongated neurofibroma occurring along the course of a nerve, usually the trigeminal or upper cervical nerves. Present in 30% of patients with NF-1, these lesions are mostly congenital. They are often disfiguring, associated with overlying skin hypertrophy, hyperpigmentation, and increased hair (Fig. 5-3).
9. What is the most common central nervous system (CNS) tumor occurring in NF-1?
12. How frequently should patients with NF-1 be assessed? What should this assessment include?
An annual assessment is usually sufficient. The clinical examination should include a blood pressure measurement (hypertension occurs in 6% due to renovascular stenosis or pheochromocytoma) and a full neurologic examination. Children also require a regular eye examination; surveillance for kyphoscoliosis, precocious puberty, and hypogonadism; and regular developmental assessments. Investigations should be guided by symptoms or physical signs.
15. What are the most common presenting symptoms of NF-2?
Hearing loss or tinnitus related to underlying vestibular schwannomas is the presenting symptom in the majority of patients, developing at a mean age of 20 years. Other presenting symptoms relate to underlying CNS tumors (20% to 30%), skin tumors (12.7%), and ocular abnormalities (12.7%).
Evans DGR, Sainio M, Baser ME: Neurofibromatosis type 2, J Med Genet 37:897–904, 2000.
Asthagiri AR, Parry DM, Butman JA, et al: Neurofibromatosis type 2, Lancet 373:1974–1986, 2009.
18. Tuberous sclerosis is also known as epiloia. What does this term mean?
20. Where are the genetic defects for tuberous sclerosis?
Genetic linkage studies of familial cases have demonstrated two separate genes linked to tuberous sclerosis: TSC 1, located at chromosome 9q34, encoding a protein called hamartin, and TSC 2, located at chromosome 16p13.3, encoding a protein called tuberin.
Narayanan V: Tuberous sclerosis complex: genetics to pathogenesis, Pediatr Neurol 29:404–409, 2003.
Curatolo P, Bombardieri R, Jozwiak S: Tuberous sclerosis, Lancet 372:657–668, 2008.
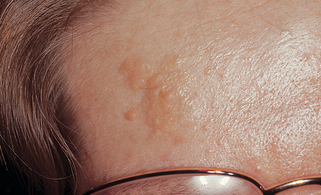
21. What is the earliest skin sign of tuberous sclerosis?
Hypomelanotic macules (Fig. 5-4). Frequently present at birth or in early infancy, these lesions are a helpful sign in infants with convulsions. Best seen with Wood’s lamp examination, they are polygonal or ash-leaf in shape, ranging in size from 1 to 3 cm and numbering 1 to 100. Occasionally, they are accompanied by 1- to 3-mm confetti-like white spots scattered over the trunk and limbs. Common skin signs of tuberous sclerosis include the following:
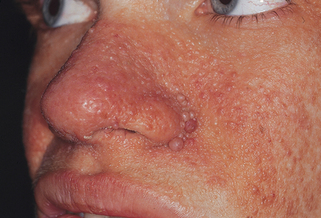
22. Adenoma sebaceum is a misnomer. What is the correct term for the facial lesions seen in tuberous sclerosis?
Angiofibromas (Fig. 5-5). These lesions consist of hyperplastic blood vessels and collagen and are not tumors of sebaceous glands. Facial angiofibromas appear at 4 to 9 years of age and increase in size and number during puberty. They are firm, discrete, reddish papules of 1 to 10 mm, developing initially in the nasolabial folds and frequently progressing over the malar region, forehead, and chin.
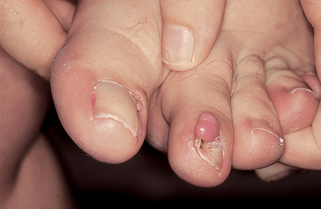
23. What are Koenen’s tumors?
Subungual and periungual fibromas (Fig. 5-6). These develop at or after puberty and present as firm, flesh-colored growths of 5 to 10 mm in length, projecting from the nail folds and beneath the nail plate.
25. What are tubers, and where do they occur?
Tubers are potato-like nodules of glial proliferation and are the characteristic CNS lesion of tuberous sclerosis. They may occur anywhere in the cerebral cortex, basal ganglia, and ventricular walls (subependymal nodules), and their number and size correlate with clinical features of seizures and mental retardation. Cortical tubers are often isodense with normal brain tissue and are best detected with magnetic resonance imaging (MRI). Subependymal nodules may calcify and are readily detectable with computed tomography (CT) scanning. Fifty percent of plain skull x-rays taken in later childhood also reveal bilateral areas of calcification in the brain.
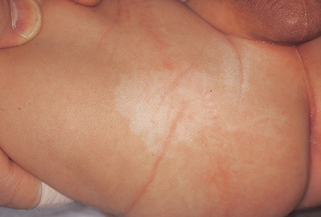
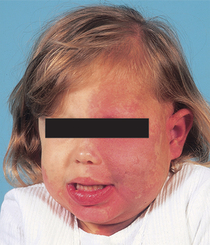
32. Where does the port wine stain most commonly occur in Sturge-Weber syndrome?
The port wine stain most commonly involves the areas innervated by the ophthalmic (V1) and maxillary (V2) divisions of the trigeminal nerve. In virtually all cases, some portion of the forehead, upper eyelid, and nasal root (V1) is involved. The port wine stain may be bilateral (Fig. 5-8) and may involve nasal or oral mucosa. Port wine stains are present on the extremities or trunk in addition to the face in 40% of cases.
33. What are the complications of leptomeningeal angiomatosis?
The vascular malformation of the cerebral meninges becomes complicated by meningeal artery calcification, calcification of the subjacent cortex, and cerebral atrophy. This results in epilepsy in 75% to 90% of cases, mental retardation (particularly in those with severe epilepsy), and, occasionally, contralateral hemiplegia.
Paller AS: The Sturge-Weber syndrome, Pediatr Dermatol 4:300–304, 1987.
35. Can the extent of neurologic involvement be predicted from the size of the facial port wine stain?
37. Do ocular complications occur in the Sturge-Weber syndrome?
Ocular complications occur in 30% to 60% of cases and include capillary malformations of the conjunctiva, iris, and choroid (ipsilateral to the facial port wine stain), glaucoma, and megalocornea. These complications may be associated only with a facial port wine stain involving the V1 distribution and do not necessarily imply CNS involvement. Glaucoma most commonly begins in the first 2 years of life; hence, regular ophthalmologic review from birth is vital in patients with V1 port wine stains.
41. What is the earliest clinical sign of ataxia-telangiectasia?
45. What type of congenital nevus is usually present?