[level-membership-for-pediatrics-category]
Neuroblastoma
Neuroblastoma is the most common solid extracranial malignancy of childhood and the most common malignant tumor in infants.1 The overall incidence of neuroblastoma is 1 per 100,000 children in the USA, thereby comprising 7–10% of all malignancies diagnosed in patients younger than 15 years of age. Yet neuroblastoma is responsible for approximately 15% of all pediatric cancer deaths.2 Neuroblastoma is a heterogeneous disease; tumors can spontaneously regress or mature, or display a very aggressive, malignant phenotype.3 Because of these unique characteristics, neuroblastoma has been of great interest to both clinicians and basic science researchers. Progress in molecular and cellular biology in the past 30 years has contributed greatly to a better understanding of this disease. Unfortunately, this progress has not significantly altered the clinical outcome for patients with advanced-stage neuroblastoma. Although the prognosis for these patients has improved in the past three decades, the long-term outcome remains very poor.
The etiology of neuroblastoma is currently unknown, and no environmental factors have been convincingly linked to its development. The disease generally occurs sporadically, but familial neuroblastoma occurs in about 2% of the cases. The substantial biologic and clinical heterogeneity is also observed in familial cases.4 The germline mutation associated with hereditary neuroblastoma has been identified: activating mutations in the tyrosine kinase domain of the anaplastic lymphoma kinase (ALK) oncogene on the short arm of chromosome 2 (2p23).5 These mutations can also be somatically acquired, although the prevalence of ALK activation in sporadic neuroblastoma remains to be determined.
Pathology
Neuroblastoma is an embryonal tumor of the sympathetic nervous system. These tumors arise during fetal or early postnatal life from sympathetic cells (sympathogonia) derived from the neural crest. Therefore, tumors can originate anywhere along the path which neural crest cells migrate, including the adrenal medulla, paraspinal sympathetic ganglia, and sympathetic paraganglia such as the organ of Zuckerkandl. The German pathologist Rudolph Virchow is generally credited with being the first to describe the histologic appearance of what is now known as neuroblastoma in his 1864 article entitled, ‘Hyperplasia of the pineal and suprarenal glands’.6 The first to use the term ‘neuroblastoma’ was James Homer Wright in 1910 who described the classic appearance of rosettes of tumor cells around central neural fibrils.7 He also noted the association between the common sites of tumor development and the pattern of migration of primitive neural cells.
As one of the ‘small, round blue cell’ tumors of infancy and childhood, neuroblastoma, particularly when undifferentiated, must be distinguished from other neoplasms in this group (Ewing sarcoma family of tumors [ESFT], non-Hodgkin lymphoma, and rhabdomyosarcoma). Neuroblastoma can be distinguished histologically by the presence of neuritic processes (neuropil) and Homer Wright rosettes (neuroblasts surrounding eosinophilic neuropil). Scattered ganglion cells or immature chromaffin cells can also be seen. The appearance of the tumor cells may vary from undifferentiated cells to fully mature ganglion cells. In addition, neuroblastomas have variable degrees of Schwannian cell stroma, reactive non-neoplastic tissue recruited by the tumor cells. This stroma is intermixed, to a greater or lesser degree, as wavy bundles and sheets of spindle cells and produces anti-proliferative and differentiation-inducing factors that are crucial to neuronal differentiation.8,9 In addition, the Schwannian stroma appears to produce a variety of antiangiogenic factors, including pigment epithelium derived factor (PEDF)10 and secreted protein acidic and rich in cysteine (SPARC).11 Histopathologic variables among neuroblastic tumors include the degree of differentiation, maturation, lymphoid infiltration, calcification, anaplasia, necrosis, mitotic activity, neurofibrillary material (neuropil), and the presence of multinucleate cells. Finally, immunohistochemical analysis usually generates positive staining when antibodies to neuroblastoma-specific antigens such as synaptophysin, neuron-specific enolase, and chromogranin are used, and is negative when antibodies to actin, desmin, cytokeritin, leukocyte common antigen, vimentin, and CD99 are used.
Neuroblastoma is characterized by several unique clinical behaviors, including the secretion of catecholamine products and the potential to regress or mature, either spontaneously or in response to treatment. Small nodules of primitive neuroblasts are routinely found in the developing adrenal gland, even during the early postnatal period. Beckwith and Perrin described microscopic nodules that they termed ‘neuroblastoma in situ’ in the adrenals of infants undergoing autopsy following death from non-malignancy related causes.12 The incidence of this finding was more than 200-fold greater than the clinical incidence of neuroblastoma, which suggests that perhaps many neuroblastomas spontaneously regress or mature into lesions that never become clinically apparent. The process of involution is well described during embryonic life, especially in the developing central and peripheral nervous systems. Although initially thought to be mediated by the immune system, the process of involution may be the result of the withdrawal of neurotrophic maintenance factors such as nerve growth factor (NGF). Clinically apparent neuroblastoma can also regress or spontaneously mature, but the mechanism remains unknown.
Histopathologic Classification
In 1984, Shimada and colleagues first developed an age-linked classification system of neuroblastic tumors based on tumor morphology in which neuroblastomas were divided into two prognostic subgroups, favorable histology and unfavorable histology.13 In 1999, the International Neuroblastoma Pathology Classification (INPC) was devised, and then modified in 2003, and is an adaptation of the original Shimada system.14,15 The INPC is based mainly on morphologic changes associated with the maturational sequence of neuroblastic tumors. It remains an age-linked classification that depends on the differentiation grade of the neuroblasts, the cellular turnover index (mitosis-karyorrhexis index [MKI]), and the presence or absence of Schwannian stroma. The INPC classifies neuroblastic tumors into three morphologic categories: neuroblastoma, ganglioneuroblastoma, and ganglioneuroma (Fig. 66-1).
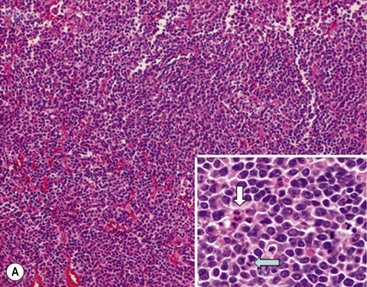
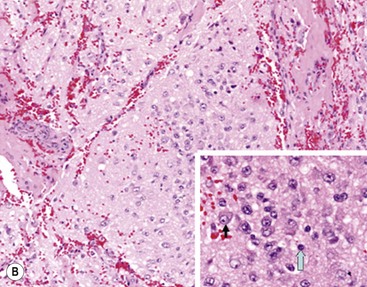
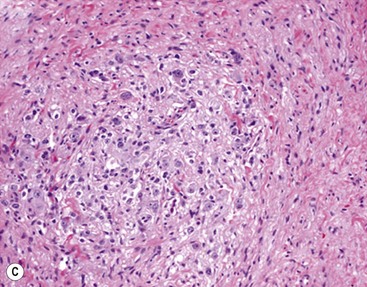
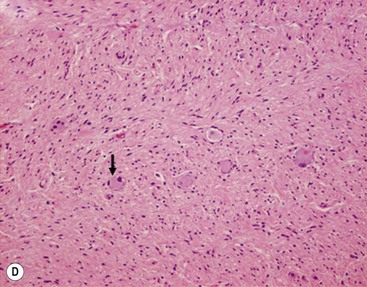
FIGURE 66-1 Histologic appearance of neuroblastic tumors. (A) An undifferentiated neuroblastoma with high MKI (10×). A clump of karyorrhectic tumor cells (white arrow) and a tumor cell undergoing mitosis (open arrow) are shown in the insert (60×). (B) A differentiating neuroblastoma, with low MKI (10×). A primitive neuroblast (gray arrow) and a differentiating tumor cell (black arrow), with features of differentiation in both the nucleus and cytoplasm, are shown in the insert (60×). Abundant neuropil is also seen. (C) A stroma-rich ganglioneuroblastoma with infrequent neuroblasts intermixed within abundant Schwannian stroma and ganglion cells (10×). (D) A stroma-rich ganglioneuroma. Ganglion cells are seen (arrow) (10×). Infiltrating lymphoid cells are also seen but no neuroblasts are present. (Courtesy of Jesse Jenkins MD and Christine Fuller MD, St. Jude Children’s Research Hospital, Memphis TN. Reprinted from Neuroblastoma, Davidoff AM. In Oldham KT, Colombani PM, Foglia RP, et al, editors. Principles and Practice of Pediatric Surgery. Philadelphia: Lippincott, Williams & Wilkins; 2005.)
Neuroblastomas are, by definition, Schwannian stroma poor (<50% of the tumor tissue) and can be subtyped as undifferentiated, poorly differentiated, or differentiating. Undifferentiated requires supplemental diagnostic methods such as immunohistochemistry, electron microscopy, or cytogenetics. Moreover, neuropil is not present. In poorly differentiated, <5% of tumor cells have features of differentiation, and neuropil is present. Differentiating tumors demonstrate >5% of tumor cells differentiating toward ganglion cells. To classify a cell as a differentiating neuroblast, there must be synchronous differentiation of the nucleus and eosinophilic cytoplasm.14
Additional factors that contribute to the prognostic distinction of stroma-poor neuroblastic tumors (neuroblastoma) as favorable or unfavorable subtypes include the MKI, which is defined as the number of tumor cells in mitosis or karyorrhexis per 5000 neuroblastic cells (i.e., low MKI, <100 cells; intermediate, 100–200 cells; high, >200 cells), and the patient’s age (<1.5 years, 1.5-5 years, >5 years) (Table 66-1). It has been hypothesized that neuroblastic cells with maturational potential require a latent period before demonstrating histologic evidence of differentiation. Therefore, there is a certain allowance for mitotic and karyorrhectic activities of neuroblastic cells in tumors in infants and younger children.16
TABLE 66-1
Prognostic Evaluation of Neuroblastic Tumors According to the International Neuroblastoma Pathology Classification

MKI, mitosis-karyorrhexis index.
Adapted from Shimada H, Ambros IM, Dehner LP, et al. The International NB Pathology Classification (the Shimada System). Cancer 1999;86:364–72.
The importance of this histopathologic classification was confirmed in a large, retrospective analysis reported by Shimada et al.17 The INPC classification of tumor histology provided independent prognostic information where tumors of favorable histology had a 90.8% probability of 5-year event-free survival [EFS] compared to 31.2% EFS for tumors of unfavorable histology. More recently, the INPC classification has been shown to add independent prognostic information beyond the prognostic contribution of age.18 Therefore, histopathology remains in the current multifactorial risk stratification for patients with neuroblastoma. This determination is particularly important in patients with MYCN non-amplified tumors who are older than 18 months and have stage 3 disease, or 12 to 18 months of age and have stage 4 disease, or have 4S disease. As the histopathologic pattern within a tumor can be heterogeneous, it is recommended that representative sections from at least 1 cm3 of viable, non-necrotic tissue be analyzed to determine histopathologic classification. The prognostic value of assessing the histopathology of a neuroblastoma after chemotherapy or radiation therapy has not been validated.
Stroma-rich neuroblastic tumors are classified as either ganglioneuroblastomas or ganglioneuromas. Ganglioneuroblastomas contain cells that are transitioning toward differentiation but are not completely differentiated/mature. Also, <50% of the total volume is made up of neuroblastic cells. Ganglioneuroblastomas can be further divided into ‘intermixed’ and ‘nodular’ subtypes, depending on the distribution of the neuroblastic cells. The distinction is important because of the significantly worse prognosis associated with the latter subtype, in which the neuroblastic clones that comprise grossly distinct nodules appear to be responsible for the aggressive phenotype for this subtype.16 Ganglioneuromas contain either maturing or mature cells, and lack any neuroblastomatous component. Most stroma-rich tumors (ganglioneuroblastoma, intermixed and ganglioneuroma, maturing subtype) are classified as ‘favorable’ by the INPC. However, the pathologic/prognostic classification of the ganglioneuroblastoma, nodular subtype is based on the morphologic evaluation of the neuroblastomatous nodule(s), and can, therefore, be unfavorable. Tumors that fit the criteria for ganglioneuroma, mature subtype, with abundant Schwannian stroma and fully mature ganglion cells, in the absence of neuroblasts, are considered benign, and are generally not considered for enrolment in protocols for neuroblastic tumors. Despite this, ganglioneuromas can be quite large and infiltrative, and attempts at removal can be associated with significant complications. In addition, survival does not seem to be influenced by extent of resection.19 Therefore, aggressive attempts at resection of ganglioneuromas are not recommended.
Molecular Biology
Advances in molecular biology research in the past three decades have resulted in an increased understanding of the genetic events in the pathogenesis and progression of many human malignancies, including those of childhood. Neuroblastoma, in particular, has served as a model for a molecular approach to treating patients with cancer, highlighting the utility of genetic analysis for diagnosis, risk stratification, and treatment planning. Chromosomal structural changes play a role in neuroblastoma, particularly those that result in the loss of tumor suppressors, or gain of oncogenes, gene amplification, and activating or inactivating mutations of relevant genes or their regulatory elements. The end result of alterations in these genetic elements, regardless of their specific mechanisms, is the disruption of the normal balance between cell proliferation and cell death.
DNA Content
Normal human cells contain two copies of each of 23 chromosomes; thus, a normal diploid cell has 46 chromosomes. The majority (55%) of primary neuroblastomas are triploid or ‘near-triploid/hyperdiploid’ and contain between 58 and 80 chromosomes; the remainder (45%) are either ‘near-diploid’ (35–57 chromosomes) or ‘near-tetraploid’ (81–103 chromosomes).20 The ‘DNA index’ of a tumor is the ratio of the number of chromosomes present to a diploid number of chromosome (i.e., 46). Therefore, diploid cells have a DNA index of 1.0, whereas near-triploid cells have a DNA index ranging from 1.26 to 1.76. Neuroblastomas that are near-diploid or near-tetraploid usually have structural genetic abnormalities, most frequently chromosome 1p deletion and MYCN amplification. Near-triploid or hyperdiploid tumors are characterized by almost three complete haploid sets of chromosomes with few structural abnormalities. Importantly, patients with near-triploid tumors typically have favorable clinical and biologic prognostic factors and excellent survival rates, as compared with those patients who have near-diploid or near-tetraploid tumors.21 This association is most important for infants with advanced disease as the prognostic significance of tumor ploidy appears to be lost in patients older than 2 years.22 Currently, ploidy only potentially impacts the risk group assessment of infants age 12–18 months with metastatic disease and infants with 4S disease in the COG risk stratification schema.
Amplification of MYCN
Investigation of the molecular biology of neuroblastoma began with the cytogenetic characterization of tumor-derived cell lines. These studies showed the frequent presence of extrachromosomal double-minute chromatin bodies (DMs) and chromosomally integrated homogeneously staining regions (HSRs) characteristic of gene amplification (Fig. 66-2).23 Since that time, it has been shown that the amplified region was derived from the distal short arm of chromosome 2 (2p24) and contained the MYCN proto-oncogene. MYCN encodes a 64 kDa nuclear phosphoprotein that forms a transcriptional complex by associating with other nuclear proteins expressed in the developing nervous system and other tissues.24 Enforced expression of MYCN increases the rates of DNA synthesis and cell proliferation, and shortens the G1 phase of the cell cycle.25 MYCN can also function as a classic dominant oncogene that cooperates with activated ras to transform normal cells.26 Targeted expression of MYCN in transgenic mice results in the development of neuroblastomas.27 This activity is potentiated when combined with mutations in ALK.28
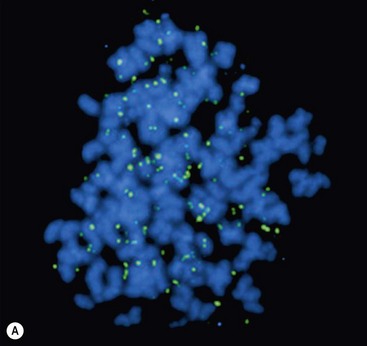
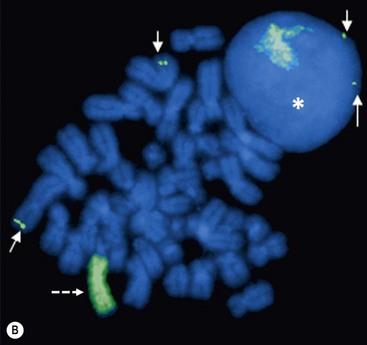
FIGURE 66-2 FISH analysis of a neuroblastoma. (A) Chromosomes in metaphase. The bright spots are double-minute chromatin bodies. (B) The metaphase chromosomes are again seen. An intact interphase nucleus is marked with an asterisk. The normal two copies of the MYCN gene are marked with solid arrows. Homogeneously staining regions (HSRs) are also seen. One is seen in the interphase nucleus, and the other is marked with a dotted arrow. (Courtesy of Marc Valentine, St. Jude Children’s Research Hospital, Memphis, TN.)
Overall, approximately 25% of primary neuroblastomas in children have MYCN amplification, with MYCN amplification being present in 40% with advanced disease but only 5–10% with low-stage disease.29 The copy number, which can range from 5- to 500-fold amplification, is usually consistent among primary and metastatic sites and at different times during tumor evolution and treatment.30 This finding suggests that MYCN amplification is an early event in the pathogenesis of neuroblastoma. Amplification of MYCN is associated with advanced stages of disease, rapid tumor progression, and poor outcome; therefore, it is a powerful prognostic indicator of tumor behavior.29,31 Amplification can be detected either by routine metaphase cytogenetics or fluorescent in situ hybridization (FISH), and current therapeutic neuroblastoma protocols have incorporated the presence or absence of MYCN amplification into their risk stratification schema.
Chromosomal Changes
Also noted on early karyotype analyses of neuroblastoma-derived cell lines were frequent deletions of the short arm of chromosome 1.32 Deletions of genetic material in tumors suggest the presence (and subsequent loss) of a tumor suppressor gene. Although no individual tumor suppressor gene has been confirmed on chromosome 1p, recent data have identified CHD5 as a strong candidate for the tumor suppressor gene that is deleted from 1p36.31 in neuroblastoma.33 Functional confirmation of the presence of a 1p tumor suppressor gene came from the demonstration that transfection of chromosome 1p into a neuroblastoma cell line resulted in morphologic changes and ultimately cell senescence.34 Approximately 20–35% of primary neuroblastomas exhibit 1p deletion, as determined by FISH, with the smallest common region of loss located within region 1p36.35 About 70% of advanced-stage neuroblastomas have 1p deletions.36 Molecular studies have shown that there is a strong correlation between 1p deletion and MYCN amplification and other high-risk features such as age older than 1 year and advanced-stage disease.35 One recent study has demonstrated that 1p deletions are independently associated with a worse outcome in patients with neuroblastoma.37
Deletion of the long arm of chromosome 11 (11q) also appears to be common in neuroblastoma, being present in about 40% of cases. Unbalanced deletion of 11q (loss with either retention or gain of 11p material) is inversely related to MYCN amplification,37,38 yet is strongly associated with other high-risk features. Recently, Attiyeh et al., on behalf of COG, showed in a large cohort of patients that unbalanced deletion of 11q and 1p36 were independently associated with a worse outcome in patients with neuroblastoma.39 Therefore, the duration of treatment for children with intermediate-risk neuroblastoma on the recent COG study was based, in part, on the 1p and 11q allelic status of the tumor.
Mutations
An example of proto-oncogene activation by point mutation involves the tyrosine kinase receptor, ALK, on the short arm of chromosome 2 (2p23). Receptor tyrosine kinases (RTK) are high-affinity cell surface receptors for many growth factors, cytokines, and hormones. When activated through ligand binding, these proteins mediate phosphorylation of tyrosine on target molecules or substrates, resulting in intracellular signaling and, ultimately, the regulation of normal cellular processes. Mutation of RTK’s can lead to constitutive activation of the signaling pathway in the absence of ligand. Recently, activating mutations of ALK have been shown to be the germline abnormality associated with hereditary neuroblastoma.40 These mutations can also be somatically acquired, as can amplification of the gene, although the prevalence of ALK activation in sporadic neuroblastoma is not known.41 Activated ALK has proven to be a targetable abnormality in neuroblastoma, with drugs such as crizotinib, an anti-ALK antibody, showing efficacy.42 Further studies have identified loss-of-function mutations in the homeobox gene PHOXB2 on 4p13 that are also associated with familial neuroblastoma, particularly when occurring together with Hirschsprung disease and/or central hypoventilation.43
Recently, inactivating mutations of ATRX, a transcriptional regulator that is part of a multiprotein complex that plays a role in regulating chromatin remodeling, nucleosome assembly, and telomere maintenance, have been found in neuroblastoma, particularly high-stage tumors in older patients.44 ATRX mutations appear to be loss-of-function mutations associated with an absence of the ATRX protein in the nucleus, and with long telomeres. How these alterations lead to lengthened telomeres is uncertain. These results may provide a molecular marker and potential therapeutic target for neuroblastoma among adolescents and young adults. It may also delineate the subset of children with neuroblastoma who have a chronic but progressive clinical course when receiving standard therapeutic approaches and who may benefit from a different treatment strategy.
Other Molecular Abnormalities
Neurotrophins and their tyrosine kinase receptors are important in the development of the sympathetic nervous system and have been implicated in the pathogenesis of neuroblastoma. Three receptor-ligand pairs have been identified: TrkA, the primary receptor for NGF; TrkB, the primary receptor of brain-derived neurotrophic factor (BDNF); and TrkC, the receptor for neurotrophin-3 (NT-3).45 TrkA appears to mediate differentiation of developing neurons or neuroblastoma in the presence of NGF ligand, and apoptosis in the absence of NGF.46 High TrkA expression is associated with favorable tumor biology and good outcome47 and is inversely correlated with MYCN amplification.48 Conversely, the TrkB/BDNF pathway appears to promote neuroblastoma survival through autocrine or paracrine signaling, especially in MYCN-amplified tumors.49 TrkB is expressed in about 40% of neuroblastomas, usually advanced-stage disease. TrkC is expressed in approximately 25% of neuroblastomas and is strongly associated with TrkA expression.50
Other molecular abnormalities frequently detected in neuroblastoma include inactivation of caspase 8, expression of CD44, and overexpression of multidrug resistance genes. Studies have demonstrated inactivation of caspase 8, a component of the Fas death-signaling complex, in MYCN-amplified neuroblastomas.51 It has been proposed that inactivation of caspase 8 renders tumor cells resistant to apoptotic signals. CD44 is a cell surface glycoprotein that appears to play a role in tumor cell adhesion.52 In neuroblastomas, CD44 expression is inversely correlated with MYCN amplification and is undetectable in most disseminated neuroblastomas.53 Multidrug resistance-associated protein (MRP) is an efflux pump whose expression in neuroblastoma appears to be correlated with MYCN amplification and poor prognosis.54,55 The presence of MRP may explain the common clinical situation in which neuroblastomas initially respond well to chemotherapy but subsequently become resistant.
Genome-Wide Association Studies
Recently, microarray technologies have generated extensive amounts of data that have aided in identifying genomic (DNA) and transcriptomic (RNA) abnormalities associated with neuroblastoma. In addition, these abnormalities have been shown to have significant predictive power when anticipating outcome for these patients.56,57 Many of these findings were generated by large scale genome-wide association studies (GWAS). This is a technique whereby all or most of the genes of patients with neuroblastoma are analyzed to find differences with the population as a whole, looking for variations that are associated with the development and aggressiveness of neuroblastoma. The causal relationship between the DNA variant associated with the cancer is uncertain but an excessive inheritance of risk variants has been postulated to increase susceptibility to the disease. Several GWAS studies have been performed in patients with neuroblastoma and have identified a number of such genetic risk variants.58 These observations suggest that developmental childhood cancers are likely influenced by common DNA variations, leading to the development of a putative genetic model.58 Recent data suggest that the higher prevalence of high-risk disease in Black and Native American patients with neuroblastoma may be associated with certain genetic variants found more commonly in these ethnic groups.59
One method for detecting CNVs is by comparative genomic hybridization (CGH). Early CGH studies showed that gain of genetic material on the long arm of chromosome 17 (17q) is perhaps the most common genetic abnormality in neuroblastomas, occurring in approximately 75% of primary tumors.60 It is unclear at this time how extra copies of 17q contribute to the malignant phenotype of neuroblastoma and which gene(s) on 17q are the critical ones. Nevertheless, gain of chromosome 17q is strongly associated with other known prognostic factors, but it may also be a powerful predictor of adverse outcome.61 More recently, GWAS studies have shown that inherited CNV at chromosome 1q21.1 is associated with neuroblastoma, implicating a neuroblastoma breakpoint family gene in early neuroblastoma genesis.62
Other studies have revealed that common genetic variation at chromosome bands 6p2263 and 2q3564 are associated with susceptibility to high-risk neuroblastoma, providing the first evidence that childhood cancers can also arise from complex interactions of polymorphic variants. More recently, a GWAS study has identified common polymorphisms including germline SNP risk alleles and somatic copy number gain, resulting in increased expression of the cysteine-rich transcriptional regulator LIM domain only 1 (LMO1) at 11p15.4. These have been shown to be strongly associated with susceptibility to developing neuroblastoma, and often are associated with advanced disease and poor survival.65
Finally, whole-genome sequencing of tumors, made possible recently by significant advances in technology, has been performed to investigate the genetic landscape of a variety of pediatric tumors.66 Initially it was felt that early alterations of genes such as MYCN may underlie the rapid acquisition of cooperating mutations in key cancer pathways through chromosome instability. However, few recurring amino acid changes have been detected in neuroblastoma specimens, suggesting that the tumor genomes were more stable than previously believed.44,67 Also, unlike the genetic landscape, the epigenetic profiles showed profound changes which suggests that epigenetic changes may have a more dominant role in pediatric tumorigenesis.
Clinical Presentation
Patients with neuroblastoma usually present with signs and symptoms that reflect the primary site and extent of disease, although localized disease is often asymptomatic. As 75% of neuroblastoma occurs in the abdominal cavity, an abdominal mass detected on physical examination is a common clinical feature, as is the complaint of abdominal pain. Other primary sites of neuroblastoma include the posterior mediastinum (20%), the cervical region (1%), and the pelvis (4%) (organ of Zuckerkandel) (Fig. 66-3). Respiratory distress or dysphagia may be a reflection of a thoracic tumor. Altered defecation or urination may be caused by mechanical compression from a pelvic tumor or by spinal cord compression by a paraspinal tumor. Spinal cord compression may also present as an altered gait. A tumor in the neck or upper thorax can produce Horner syndrome (ptosis, miosis, and anhydrosis), enophthalmos, and heterochromia of the iris. Acute cerebellar ataxia has also been observed, characterized by the dancing-eye syndrome, which includes opsoclonus, myoclonus, and chaotic nystagmus. Two-thirds of these cases occur in infants with mediastinal primary tumors.68,69 Additional signs and symptoms that reflect excessive catecholamine or vasoactive intestinal polypeptide (VIP) secretion include diarrhea, weight loss, and hypertension.
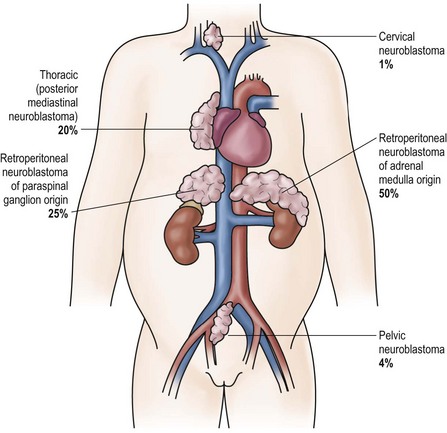
FIGURE 66-3 Primary sites for neuroblastoma are depicted in this anatomic drawing. (Reprinted from Davidoff AM. Neuroblastoma. In: Oldham KT, Colombani PM, Foglia RP, et al, editors. Principles and Practice of Pediatric Surgery. Philadelphia: Lippincott Williams & Wilkins; 2005.)
More than 40% of patients have metastatic disease at diagnosis. These patients are often quite ill and have systemic symptoms caused by widespread disease. Neuroblastoma in older patients has a pattern of metastatic disease in which metastases to the bone marrow, lymph nodes, and bone predominate. The frequency of involvement of distant sites is shown in Table 66-2. These metastases may manifest as bone pain from cortical metastases or anemia from marrow infiltration. The brain, spinal cord, heart, and lungs are rare sites of metastases, except with end-stage disease. Metastatic disease may be also associated with darkened eyes, referred to as ‘raccoon eyes,’ as a result of retroorbital venous plexus spread (Fig. 66-4A). This is an ominous physical sign, as is the presence of a limp in children without a history of head or extremity trauma. Infants with metastatic neuroblastoma can have stage 4S disease, which, by definition, is a localized primary tumor in patients younger than 1 year, with dissemination limited to skin, liver, or bone marrow (<10% of nucleated cells). These patients may present with ‘blueberry muffin’ cutaneous lesions (Fig. 66-4B), respiratory distress secondary to massive hepatomegaly, and anemia secondary to bone marrow disease. The diagnosis of neuroblastoma is generally made by histopathologic evaluation of primary or metastatic tumor tissue, or by the demonstration of tumor cells in the bone marrow together with elevated levels of urinary catecholamines.
TABLE 66-2
Sites of Metastases at Diagnosis for Patients with Evans Stage IV-S and Stage IV
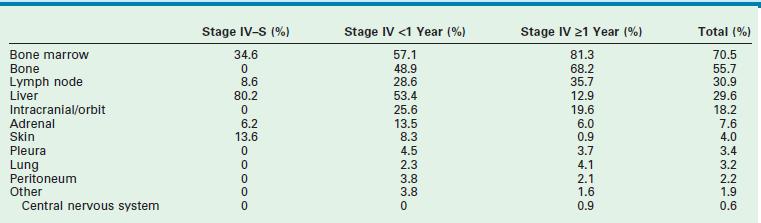
Adapted from Dubois SG, Kalika Y, Lukens JN, et al. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. Pediatr Hematol Oncol 1999;21:181–9.
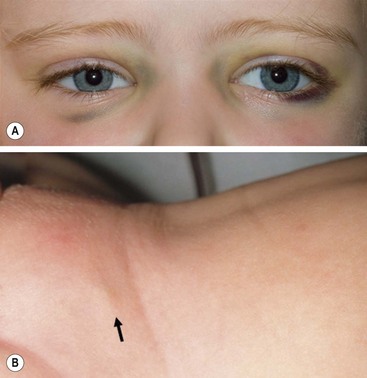
FIGURE 66-4 Clinical evidence of metastatic neuroblastoma. (A) ‘Raccoon eyes,’ characteristic of metastatic neuroblastoma in the posterior orbital venous plexus, are seen in a child with stage IV disease. (B) ‘Blueberry muffin’ spot (arrow) in the skin, characteristic of metastatic neuroblastoma, is seen in the suprapubic region of an infant with a 4S neuroblastoma. (Courtesy of Stephen Shochat, MD, St. Jude Children’s Research Hospital, Memphis, TN).
Laboratory Findings
Lactate Dehydrogenase
Despite its lack of specificity, serum lactate dehydrogenase (LDH) can have great prognostic significance. High serum levels of LDH reflect high proliferative activity or large tumor burden, and an LDH level higher than 1500 IU/L appears to be associated with a poor prognosis.70,71 Thus, LDH can be used to monitor disease activity or the response to therapy.
Ferritin
High levels of serum ferritin (>150 ng/mL) may also reflect a large tumor burden or rapid tumor progression. Elevated serum ferritin is often seen in advanced-stage neuroblastomas and indicates a poor prognosis.72 Levels often return to normal during clinical remission.
Neuron-Specific Enolase
Neuron-specific enolase (NSE) is another useful prognostic marker of advanced-stage neuroblastoma. The incidence of elevated NSE levels increases with stage.73 A serum level of NSE >100 ng/mL is associated with a poor outcome. NSE has been reported to correlate with tumor burden, suggesting its reliability as a marker of disease course.74
Catecholamine Metabolites
Neuroblastoma is characterized by the relatively unique capacity for secretion of catecholamine products, the metabolites of which can be detected in the urine of more than 90% of patients with neuroblastoma. Thus, a urine specimen is of clinical value in diagnosing neuroblastoma and determining the response to therapy. Documentation of elevated urinary catecholamines is required if the diagnosis of neuroblastoma is being made solely by the identification of neuroblasts in the bone marrow. Urinary levels of these two catabolites can also be used as markers of tumor progression or relapse, and serve as a surrogate prognostic indicator. Random urine samples are preferable to 24-hour urine estimations for younger children.75
Diagnostic Imaging
Standard Radiographs
Chest radiography can be a useful tool for demonstrating the presence of a posterior mediastinal mass, which in a child is usually a thoracic neuroblastoma. A Pediatric Oncology Group (POG) study demonstrated that a mediastinal mass was discovered on incidental chest radiographs in almost half of patients with thoracic neuroblastoma who had symptoms seemingly unrelated to their tumors.76 Abdominal radiography is less often the modality by which a neuroblastoma is discovered; however, as many as half of abdominal neuroblastomas are detectable as a mass with fine calcification.
Ultrasonography
Although ultrasonography (US) is the modality most often used during the initial assessment of a suspected abdominal mass, its sensitivity and accuracy are less than that of computed tomography (CT) or magnetic resonance imaging (MRI) for diagnosing neuoblastoma.77 These latter modalities are generally used after screening with ultrasound to assist in generating a differential diagnosis and for further anatomic definition once the presence of a mass has been confirmed.
Computed Tomography
CT can demonstrate calcification in almost 85% of neuroblastomas, and intraspinal extension of the tumor can be determined on contrast-enhanced CT.78 Overall, contrast-enhanced CT has been reported to be 82% accurate in defining neuroblastoma extent, with the accuracy increasing to nearly 97% when performed with a bone scan.79 Although some consider CT to have been supplanted by MRI, others still consider it to be the image modality of choice for patients with neuroblastoma, especially when used in conjunction with bone scintigraphy.80
Magnetic Resonance Imaging
MRI is becoming the most useful and most sensitive imaging modality for the diagnosis and staging of neuroblastoma.77,81 MRI appears to be more accurate than CT for detection of stage 4 disease. The sensitivity of MRI is 83%, and that of CT is 43%, and the specificity of MRI is 97%, and that of CT is 88%.81 Metastases to the bone and bone marrow, in particular, are better detected by MRI, as is intraspinal tumor extension (Fig. 66-5).81 When considering skeletal metastases alone, MRI and bone scan have been shown to be equivalent.81 Encasement of major vessels can be better defined by MRI than CT, especially with the use of MR angiography (see Fig. 66-5). MRI in the coronal plane is suitable for routine assessment of the whole body from the neck to the pelvis. Evaluating the utility of whole-body MRI, perhaps performed in conjunction with a functional imaging study such as positron-emission tomography (PET), is being considered for future clinical staging studies. CT and MRI are not very accurate for staging localized disease; however, the sensitivity of T1- and T2-weighted MRIs is 100% for detecting neuroblastomas in infants identified by mass screening.65
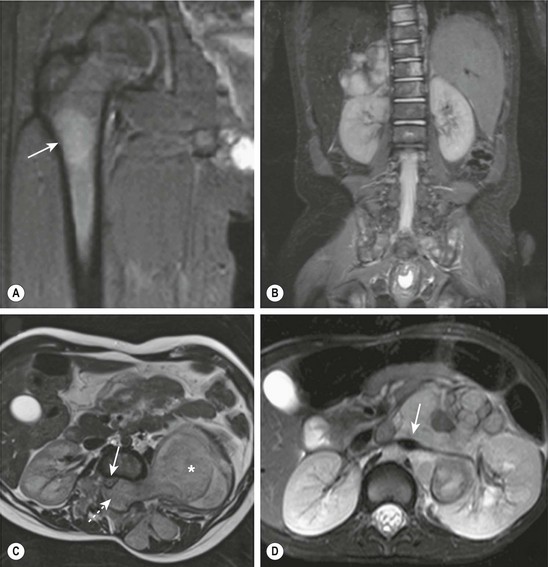
FIGURE 66-5 These MR images highlight several characteristics of high-risk neuroblastoma. (A) Bone metastasis in femur (arrow). (B) Bone marrow metastases in the vertebral bodies. (C) Intraspinal tumor extension (dotted arrow). Note displacement of spinal cord (solid arrow) from a large tumor (asterisk). (D) Encasement of major intra-abdominal vessels (arrow points to the aorta and left renal artery).
Metaiodobenzylguanidine Imaging
Metaiodobenzylguanidine (MIBG) is transported to and stored in the chromaffin cells in the same way as norepinephrine. The MIBG scintiscan is the preferred imaging study for evaluating the bone and bone marrow involvement by neuroblastoma (Fig. 66-6), having largely replaced technetium-99m methylene diphosphonate (99mTc-MDP) bone scans, which are generally inferior to MIBG in detecting skeletal or extraskeletal involvement. In addition, monitoring MDP-avid neuroblastomas by bone scintigraphy often results in false-positive imaging for months after tumor remission. Thus, 99mTc-MDP bone scanning is a second choice if MIBG imaging is not available or does not visualize known disease.82,83 Iodine-131 (131I) or iodine-123 (123I) can be used to label MIBG. 123I-MIBG supplies a reduced absorbed radiation dose and superior spatial resolution.84 The reported sensitivity of MIBG in the detection of neuroblastomas with metastases to the bone and bone marrow is 82%, and the specificity is 91%.85 Primary tumors and lymph node metastases are also detectable. MIBG can demonstrate more sites of tumor involvement in bone and bone marrow than either bone scintigraphy or standard radiography.85 However, false-negative MIBG scans have been seen in cases in which the bone scintigraphy was positive.83
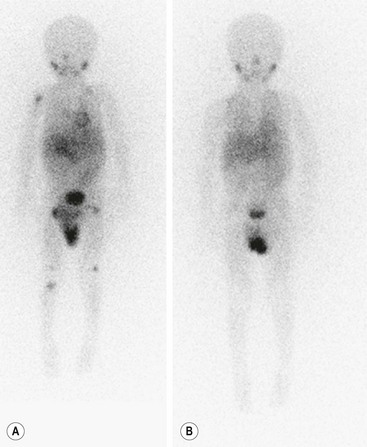
FIGURE 66-6 Imaging of neuroblastoma with MIBG scintigraphy. (A) Scan obtained at presentation of a patient with metastatic neuroblastoma. There is diffusely abnormal activity throughout much of the skeleton including the proximal right humerus, both proximal and distal femurs, and the proximal right tibia. There is also a focus of activity in the right upper retroperitoneum at the site of the primary tumor. (B) Scan obtained of the same patient after completion of therapy shows no scintigraphic evidence of MIBG-avid neuroblastoma.
Bone Marrow Examination
Marrow biopsy is a routine method for detecting bone marrow involvement. Both aspiration and trephine biopsy should be performed, although the latter has better diagnostic value. To collect more accurate information, taking specimens from multiple sites is recommended. Immunohistochemical staining with antibodies such as anti-ganglioside GD2, S-100, NSE, and ferritin is also useful for reducing the number of false-negative cases.86 Because biopsy is invasive and painful, noninvasive alternatives are being tested. Studies have suggested the superiority of MR imaging87 and MIBG scintigraphy88 over bone marrow biopsy in detecting bone marrow infiltration by neuroblastoma; however, the specificity of these modalities requires further evaluation.
Differential Diagnosis
Making a correct diagnosis of neuroblastoma can be difficult, because patients present with such diverse symptoms. For example, acute cerebellar ataxia with opsoclonus-myoclonus can be mistaken for a primary neurologic disease. Widespread bone involvement may resemble non-neoplastic bone disease such as osteomyelitis or rheumatoid arthritis, or be associated with systemic inflammatory changes. Symptoms referable to VIP secretion such as diarrhea can be misinterpreted as symptoms of an enteric infection or inflammatory bowel disease. Histologically, undifferentiated, small blue round cell neuroblastomas may be hard to distinguish from rhabdomyosarcoma, primitive neuroectodermal tumors, ESFT, or non-Hodgkin lymphoma. Use of a panel of specific antibodies, as mentioned previously, can facilitate histologic differentiation.87
Tumor Staging
International criteria for a common neuroblastoma staging system were first described in 1988, and subsequently revised in 1993.89 The International Neuroblastoma Staging System (INSS) is a surgicopathologic staging system that depends on the completeness of resection of the primary tumor, assessment of ipsilateral and contralateral lymph nodes, and the relation of the primary tumor to the midline (Table 66-3). Evaluation of the primary tumor and involvement of metastatic sites in the INSS system depends largely on imaging studies (CT or MRI) although involvement of the bone marrow continues to be an important component. MIBG scanning is also recommended as part of the initial evaluation of new patients and, subsequently, for monitoring tumor response to therapy.
TABLE 66-3
International Neuroblastoma Staging System Criteria
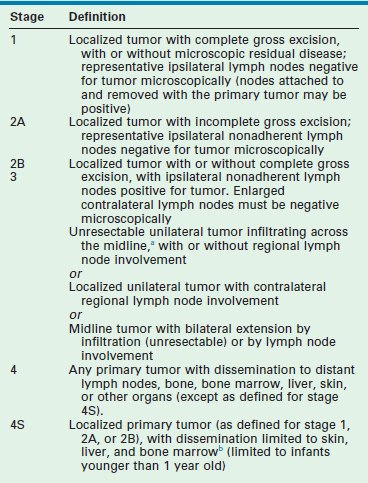
aThe midline is defined as the vertebral column. Tumors originating on one side and crossing the midline must infiltrate to or beyond the opposite side of the vertebral column.
bMarrow involvement in stage 4S should be minimal (i.e., <10% of total nucleated cells identified as malignant on bone marrow biopsy or on marrow aspirate). More extensive marrow involvement would be considered to be stage 4. The metaiodobenzylguanidine scan (if performed) should be negative in the marrow.
Although INSS has been shown to have prognostic relevance, there have been some difficulties with its widespread use. The expertise and aggressiveness of the surgeon influence tumor stage, lymph node sampling is done erratically, and patients who are simply observed without surgery cannot be appropriately staged. Therefore, a uniform, pretreatment staging system that could be used easily throughout the world, and subject to real-time central review, was sought. Montclair et al, on behalf of the International Neuroblastoma Risk Group (INRG), proposed a new staging system in 2009 based on tumor imaging rather than the extent of surgical resection.90 In this staging system, localized tumors are staged based on the absence (L1) or presence (L2) of one or more of 20 image-defined risk factors (IDRFs). Previously, Cecchetto et al. had reported that the presence of one or more of these image-defined surgical risk factors was associated with a lower complete resection rate and a greater risk of surgery-related complications when attempting an initial resection of a localized neuroblastoma.91
Metastatic tumors are defined as stage M. Stage MS, similar to INSS stage 4S, refers to disease with metastases limited to skin, liver and bone marrow (less than 10% involvement) in children less than 18 months of age at diagnosis, although the INSS 4S age cut-off is 12 months. These young patients can have L1 or L2 primary tumors. The IDRFs are listed in Box 66-1 and generally reflect encasement of vital structures, primarily vessels and nerves, as determined by diagnostic imaging studies. Absence of these factors had previously been shown to be associated with safe, complete tumor resection.91 In a review of 661 patients in the INRG database, Monclair et al. found that INRG staging had prognostic significance.90 Patients with stage L1 disease had a significantly greater 5-year EFS than those with stage L2 disease (90% ± 3% vs 78% ± 4%, p = 0.001). Although INSS is currently still the staging system used for COG patients, the INRG stage assignment is being collected prospectively on all patients for subsequent evaluation.
Biologically Based Risk Groups and Therapy
Current treatment of children with neuroblastoma is based not only on stage but also on risk stratification that takes into account clinical and biologic variables predictive of relapse. The most important clinical variables appear to be age at the time of diagnosis, with 18 months as the cut-off, and stage at diagnosis.92–94 The most powerful biologic factors at this time appear to be MYCN status and histopathologic classification.17,29,31
In addition, other biologic and molecular variables continue to be evaluated and two, the allelic status at chromosomes 1p36 and 11q23, may influence the duration of therapy for certain patients. Taken together, these variables currently define the COG risk stratification (Table 66-4). On the basis of these clinical and biological variables, infants and children with neuroblastoma are categorized into three risk groups predictive of relapse: low, intermediate, and high risk. The probability of prolonged disease-free survival for patients in each group is >95%, >90%, and <30%, respectively. Other factors are still being evaluated and may help further refine risk assessment in the future. These factors are currently being refined and augmented by analyses performed by the INRG task force. This group, which initially convened in 2004, is composed of investigators from the major pediatric cancer cooperative groups throughout the world. The main objective of this task force is to develop a consensus approach to pretreatment risk stratification for children with neuroblastoma.
TABLE 66-4
Children’s Oncology Group Risk Stratification for Children with Neuroblastoma
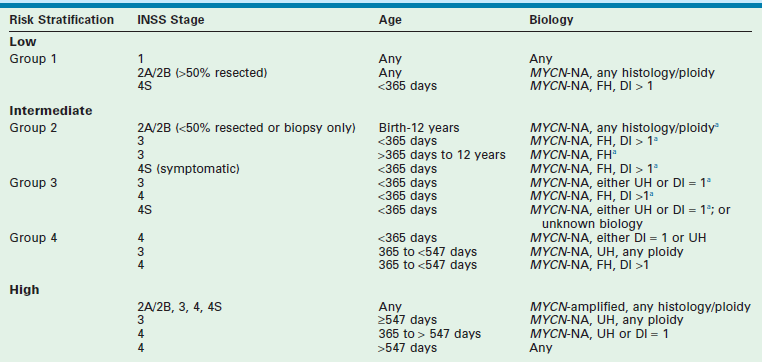
MYCN-NA, MYCN not amplified; FH, favorable histology; UH, unfavorable histology; DI, DNA index.
aIf tumor contains chromosomal 1p LOH or unbalanced 11q LOH, or if data are missing, treatment assignment is upgraded to next group.
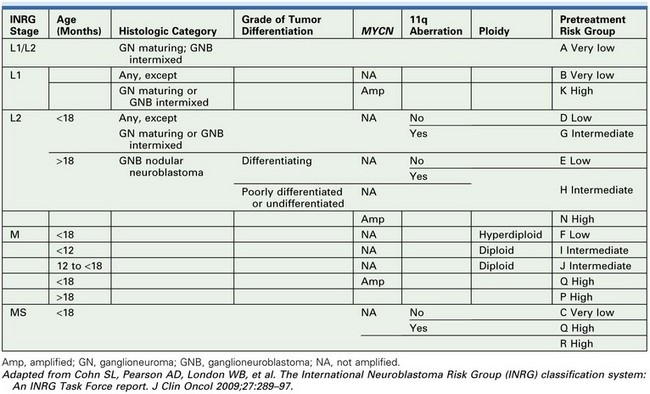
Proposed INRG Risk Stratification
In an effort to establish an international consensus on pretreatment risk stratification, the INRG task force developed the INRG Classification System based on an analysis of 8800 patients treated for neuroblastoma between 1990 and 2002. They used survival tree regression analyses with EFS as the primary endpoint to test the prognostic significance of 13 potentially prognostic factors.95 The analyses determined that seven of these prognostic variables could define 16 different pretreatment risk groups (Table 66-5). These risk groups could then be divided into four categories based on expected five-year event-free survival: very low (>85% EFS, 28.2% of patients), low (>75 to ≤85% EFS, 26.8% of patients), intermediate (≥50 to 75% EFS, 9.0% of patients) and high (<50% EFS, 36.1% of patients) risk.96 These factors are being prospectively collected on all neuroblastoma patients with the hope that these homogeneous cohorts will facilitate future comparisons of risk-based trials performed throughout the world. Of note, analysis of the prognostic importance of histopathology has been confounded by the inclusion of age, itself an independent prognostic factor, in the past. Therefore, in the INRG classification, schema tumor differentiation and MKI are separated for risk stratification.
Low-Risk Disease
Resection is currently the only therapy given to patients with low-risk disease, based on the prior experiences of each of the legacy groups (POG and Children’s Cancer Group [CCG]). The POG 8104 study found that 2-year survival was 89% for patients with POG stage A (INSS stage 1) disease, despite microscopic residual disease, when patients were treated with operation alone.96,97 In a similar cohort of patients, the CCG 3881 study found 3-year EFS and overall survival to be 94% and 99%, respectively, for patients with Evans stage I disease.98 That study also found that although patients with Evans stage II disease (similar to INSS 2A/2B) had a three-year EFS of 81% irrespective of the extent of surgical resection and subsequent treatment, the overall survival for these patients was 99%.98 This finding suggests that even if these patients experience disease relapse, most can be salvaged with additional therapy. Therefore, neither adjuvant chemotherapy nor radiation therapy appears to be necessary for the initial management of most patients with low-risk disease.
On the basis of these data, a COG study (P9641), was conducted from 1998–2006 to evaluate primary surgical therapy for biologically defined low-risk neuroblastoma. The overall strategy of this study was to treat patients with low-risk neuroblastoma with resection and supportive care only. Adjuvant therapy was given only when less than 50% of the tumor was resected or when symptoms that were life- or organ-threatening developed. A probability of 3-year survival more than 95% was predicted for these patients with low-risk disease. The results from this study were published recently and showed excellent survival rates in asymptomatic, low-risk patients with stage 2A/B neuroblastoma after operation alone and that the immediate use of chemotherapy could be restricted to a minority of patients with symptomatic low-risk disease.99 Patients with stage 2B disease who were older or had diploid or unfavorable histology tumors fared worse.
Intermediate-Risk Disease
COG study (A3961) was conducted from 1998–2006 to further refine therapy for patients with intermediate-risk disease. The overriding aim of this study was to maintain or improve survival while minimizing both acute and long-term morbidity in patients with intermediate-risk neuroblastoma. Patients received four of the most active agents against neuroblastoma: cyclophosphamide, doxorubicin, carboplatin, and etoposide, given for either four cycles (favorable biology) or eight cycles (unfavorable biology); cycles were given every 3 weeks. Radiation therapy was not used unless there was progressive disease or an unresectable primary tumor with unfavorable prognostic features at the end of chemotherapy. The outcome after reduced chemotherapy for intermediate-risk neuroblastoma was published recently.100 The 3-year overall survival for the entire group was 96%. Survival was 98% for those with favorable biologic features and 93% for those with unfavorable features.
The most recent COG protocol (ANBL0531) sought to further refine the minimal therapy needed to achieve these excellent outcomes for patients with intermediate-risk neuroblastoma. The results have not yet been published. As such, many patients, as defined by favorable clinical and biologic factors, received a further reduction in therapy. However, those patients in whom there was loss of heterozygosity (LOH, i.e., loss of one of two normally paired chromosomal regions) at chromosome 1p or 11q (unbalanced) were not eligible for this dose reduction, as these findings have been shown to be independently associated with decreased progression-free survival in patients with low- and intermediate-risk disease.37 Patients received cycles of cyclophosphamide, doxorubicin, carboplatin, and etoposide every three weeks. The duration of therapy (i.e., the number of cycles) depended upon which of three intermediate-risk groups a patient was enrolled. Group stratification again was based on clinical and biologic risk factors.
Patients >12 years of age with localized tumors have an indolent clinical course, but ultimately have an unfavorable outcome, and are being considered for more intensive therapy.101,102
High-risk Disease
The general approach to treating patients with high-risk neuroblastoma has included intensive induction chemotherapy, myeloablative consolidation therapy with stem cell rescue, and targeted therapy for minimal residual disease. Stem cell harvest is typically performed after the first two cycles of induction therapy, and resection of the primary tumor and bulky metastatic sites is attempted after the fifth cycle (Fig. 66-7). The CCG-3891 protocol enrolled patients with high-risk neuroblastoma between 1991 and 1996 and was designed to assess whether myeloablative therapy, in conjunction with autologous BMT, improved EFS when compared with chemotherapy alone, and whether subsequent treatment with 13-cis-retinoic acid would further improve EFS.103 The results from this double-randomization study demonstrated that the three-year EFS was significantly better in patients who underwent BMT during the first randomization (34%) than in those who did not (22%; p = 0.034). In the second randomization, those who received 13-cis-retinoic acid after BMT experienced a significantly better three-year EFS (46%) than those who did not receive the retinoid (29%, p = 0.027). Unfortunately, the long-term survival advantage for these patients is becoming less apparent. Nevertheless, autologous stem cell transplantation and 13-cis-retinoic acid are now part of most current high-risk neuroblastoma protocols.
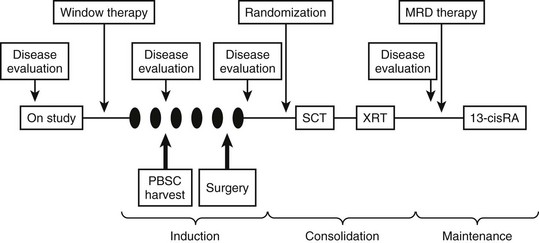
FIGURE 66-7 General schema of multimodal therapy for treating children with high-risk neuroblastoma. Variations include the testing of new drugs or combinations during ‘window therapy,’ the use of different ‘induction’ regimens, randomizing patients to receive different consolidation therapy, and testing different approaches for treating minimal residual disease. Patients generally undergo PBSC harvest after two cycles of induction therapy and undergo delayed resection of the primary tumor (and locoregional disease) after five cycles of induction therapy. SCT, stem cell transplant; XRT, radiation therapy; MRD, minimal residual disease; RA, retinoic acid; PBSC, peripheral blood stem cell.
The recently concluded COG high-risk neuroblastoma protocol (ANB0532) had as its primary goal to test whether further intensification of myeloablative therapy would improve the cure rate. Randomization to either one myeloablative consolidation with a carboplatin/etoposide/melphalan preparative regimen or two myeloablative consolidations, in which the initial regimen included thiotepa and cyclophosphamide, occurred at the completion of induction chemotherapy. Another aim of this study was to determine whether additional radiation therapy delivered to gross residual disease improved local control. Four to 6 weeks after stem cell transplantation, radiation therapy was administered to the region of the primary tumor site, including involved adjacent lymph nodes. The target volume was the area of residual disease, which was determined radiographically, after induction chemotherapy but prior to delayed surgical resection, with an additional 1.5 cm margin added, even if a complete resection was ultimately achieved. Sites of persistent active metastatic disease prior to stem cell transplantation were irradiated at the same time and with the same dose as the primary site. Those patients whose primary site achieved a complete response at the end of induction therapy received 21.6 Gy to the site of primary locoregional disease while areas with gross residual disease were treated with an additional boost of 14.4 Gy (36 Gy total).
Recurrent High-Risk Neuroblastoma
Patients with recurrent high-risk neuroblastoma have a uniformly dismal outcome. Several phase I and II trials are currently open and are testing new treatment strategies for these patients. However, although a few patients with relatively limited initial disease can be cured with salvage therapies at the time of relapse, for most stage 4 patients, disease recurrence portends a dismal outlook (<5% survival). Radiation therapy can be used for the treatment of refractory sites of metastatic disease and for palliation of painful sites of metastatic disease, with some success in providing pain relief and disease control.104
Surgery for Neuroblastoma
Complete resection of a tumor offers definitive therapy with a generally excellent outcome for most patients with localized neuroblastomas. However, the benefit of complete tumor removal may be overestimated because of the possibility that localized neuroblastomas may undergo spontaneous maturation or even regression. The role of surgery is even less clear in the curative treatment of patients with high-risk neuroblastomas.105 Unfortunately, more than half of patients with neuroblastoma present with advanced local or metastatic disease, which requires intensive multimodal therapy in addition to surgery. It is crucial that surgeons consider the heterogeneous nature of neuroblastoma, and the molecular and biologic characteristics associated with good or bad prognoses when determining the role of surgery for any specific case.
Localized Tumors
Most localized neuroblastomas have favorable biologic features and are successfully treated with excision alone.97,106–109 In addition, studies suggest that a subset of localized tumors will spontaneously regress, and that these patients can be observed without any treatment.110–113 Local recurrences, while they rarely occur, can generally be managed surgically.114 Thus, complete resection of a localized neuroblastoma may be the only therapy required for some patients.115 Regardless of the presence of microscopic or even gross residual disease with positive ipsilateral lymph nodes, no further therapy is given to patients with low-risk disease. Therefore, all patients with INSS stage 1 tumors, stage 2A/2B tumors without MYCN amplification (and >50% resection), or stage 3 tumors of the midline that undergo gross total resection with negative lymph node are treated with resection alone.
In most instances, performing a biopsy before resection is unnecessary in patients with localized tumors that are likely to be neuroblastoma, appear to be easily resectable, and have had a negative metastatic workup, especially for those whose primary site is thoracic since patients with low-risk disease will not receive adjuvant therapy. Even for patients with intermediate- or high-risk disease who will receive adjuvant chemotherapy, earlier removal of bulky disease may be feasible and potentially decrease the likelihood of developing drug-resistant tumor clones.116 However, primary resection in these patients should be done only if it can clearly be accomplished safely, without injury to adjacent organs or blood vessels and without delaying the initiation of chemotherapy. In all cases, if there is any question of resectability, pre-resection risk determination should be performed, as this may influence the operative plan. Assessment of the MYCN status can be performed on a minimal amount of tumor tissue, including tumor cells in the bone marrow. MYC-amplified tumors, unless stage 1, are all high risk. To determine the histopathologic status, COG currently requires 1 cm3 of tissue. Therefore, an open or laparoscopic biopsy is recommended, as opposed to a percutaneous core biopsy, which generally provides smaller samples.
Locoregional Disease in Patients with Metastatic Disease
More than 80% of patients who are older than 1 year of age and have metastatic neuroblastoma will have tumor in the bone marrow.117 The presence of disseminated neuroblastoma is often suggested by a patient’s ill appearance, anemia, ‘raccoon eyes,’ bone pain, or a limp, and can be confirmed by finding neuroblasts in the bone marrow. Therefore, most of these patients can have the diagnosis of neuroblastoma confirmed by bone marrow aspirate/biopsy and the demonstration of elevated catecholamines in the urine. A biopsy of the tumor is only required if there is no involvement of the bone marrow, if the histopathology of the tumor is important in determining the risk classification, or to obtain sufficient tissue to support biologic studies. Most patients with disseminated disease should receive neoadjuvant chemotherapy prior to attempted resection of the primary tumor and locoregional lymphadenopathy.
Delayed Surgical Resection of Locoregional Disease
The role of surgery in the management of children with high-risk neuroblastoma is controversial. Several reports have suggested that patients with INSS stage 3 or 4 disease who undergo gross total resection of their primary tumor and locoregional disease experience improved local tumor control and increased overall survival.118–120 However, other reports have not confirmed these observations.121–123 LaQuaglia et al. reported that the outcome for patients with stage 4 disease who were older than 1 year at the time of diagnosis was improved with gross total resection of the primary tumor, though the influence of resection was not independent of chemotherapy intensity.124 A more recent paper from the same group provided further data suggesting that local control and overall survival are correlated with gross total resection of the primary tumor in high-risk neuroblastoma.120 Similarly, Haase et al.125 supported the use of aggressive resection for patients with metastatic disease in an attempt to improve outcome, and Grosfeld et al.126 found improved survival in patients with metastatic disease who underwent complete resection of the primary tumor during delayed second-look procedures. Finally, a large meta-analysis of the outcomes for patients with stage 3 and 4 disease, and a review of the impact of the extent of primary site resection on EFS, overall survival, and local recurrence in high-risk neuroblastoma for patients treated on A3973 suggested that ≥90% resection is associated with a significant decrease in local recurrence, with a slight but sustained improvement in both EFS and overall survival, although this was not statistically significant.127,128
In contrast, several studies have found that the extent of resection does not affect the final outcome.122,129 One study found that the extent of surgical resection affected only certain subgroups of patients with high-risk disease.121 In addition, substantial complication rates have been reported after aggressive attempts at removing all gross tumor from the retroperitoneum.130 Another study noted that the outcome for patients with stage 4 neuroblastoma depended more on the biologic characteristics of the tumor than on the extent of surgical resection.131 This conclusion is further highlighted by the results of the CCG 3881 study which showed that, at least in infants with stage 4 disease and single-copy MYCN tumors, survival was excellent, regardless of whether gross total resection (3-year EFS, 91%) or incomplete resection (3-year EFS, 94%) was performed.132 However, survival was poor if the tumor was MYCN amplified regardless of the extent of resection; 3-year EFS after total resection was 14%, and 3-year EFS after incomplete resection was 10% (p = 0.18). Although the role of cytoreduction is unclear, resection of as much gross tumor as possible in patients who receive autologous or allogeneic BMT in combination with high-dose chemotherapy and total-body irradiation (TBI) may be of some benefit.133
Despite the uncertainty about the role of surgery, the COG recommends attempting gross total resection of the primary tumor and locoregional disease in patients with high-risk neuroblastoma. Most children undergo delayed operation after the completion of the fifth cycle of induction chemotherapy, even though reduction in tumor volume plateaus after the second or third cycle of chemotherapy.134 Other groups are performing surgery as soon as locoregional disease appears to be resectable on imaging studies.135 Although initial resection is often not appropriate for patients with neuroblastoma, the principle of resection at the earliest feasible time should be considered. Because no prospective, randomized studies have been performed, the influence of the extent of operative resection is unknown. However, in considering aggressive resection, the risks involved, including vascular injury and significant bleeding, kidney or bowel infarction, infection, delay in chemotherapy, and long-term complications such as renal atrophy and diarrhea, need to be weighed carefully against the uncertain benefits of extensive surgery. Certainly removal of kidneys or other organs is to be avoided as this may hinder or delay the ability to give potentially effective chemotherapy.
The age of the patient and the tumor biology are of critical importance when planning resection. For example, survival of infants with stage 4, single-copy MYCN disease is greater than 93%, regardless of the extent of surgical resection of locoregional disease.132 This favorable outcome for patients with stage 4, single-copy MYCN disease probably includes patients up to 18 months of age if the tumor’s Shimada histology is also favorable. Clearly, the excellent prognosis for these patients should not be jeopardized by overly aggressive surgery. The extent of surgery also did not appear to significantly affect the survival of infants with MYCN-amplified tumors in this nonrandomized trial as survival was very poor (<15%). Perhaps infants with stage 4 disease and MYCN-amplified tumors would benefit from more aggressive attempts at complete resection, despite the attendant risks, given that current adjuvant therapy is unlikely to cure them.
Operative Principles
More than half of neuroblastomas arise in the abdominal cavity. Tumor size, the extent of vascular encasement, and exact tumor location should be considered in selecting the approach for a retroperitoneal neuroblastoma. Options available for the abdominal incision include a transverse incision, bilateral subcostal (Chevron) incisions, or a midline incision. A transthoracic (intercostal), transdiaphragmatic extension can be added to either incision if needed. The tumor and adjacent lymphadenopathy should be carefully exposed to determine the relation between the tumor and normal organs and vessels. If encasement of major vessels such as the aorta, vena cava, or their branches is found, tumor dissection must be performed to free the vessels completely. Use of the Cavitron (Cavitron Corporation, Stamford, CT, USA) ultrasonic aspirator in selected patients may allow for better tumor dissection from the major vessels, with less blood loss and fewer complications.136 Use of the argon beam coagulator and lasers also helps to achieve complete or near-complete resection, and reduces operative complications.125,136
A detailed description of extensive surgical resection is available.122 Briefly, the major points include: (1) approach the operation as a vascular-type operation in which identification and skeletonization of the major intra-abdominal vessels is critical. If encasement of major vessels such as the aorta, vena cava, or their branches is found, tumor dissection must be performed to free the vessels completely. Generally tumor can be separated from the vessel by dissecting in a subadventitial plane. (2) The tumor should be removed piecemeal. In particular, undue torque on the renal artery in an effort to clear tumor from behind the renal hilum in one piece may result in injury to the intima of the artery with vessel spasm and/or thrombosis, leading to renal ischemia. (3) Dissection commences distal to the lower edge of the tumor, generally along the common or external iliac artery, and proceeds proximally to encounter the tumor along the aorta identifying the major arterial branches (and left renal vein). With deliberate dissection of the tumor from the mesenteric and renal vessels, injury to the liver, bowel, spleen and kidneys can be avoided (Fig. 66-8) although this frequently results in piecemeal division and excision of the tumor. (4) Right-sided tumors are managed similarly and are generally less complicated unless intimately involved with the structures of the porta hepatis.
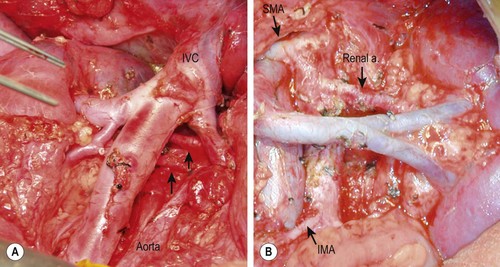
FIGURE 66-8 Intraoperative photographs after resection of retroperitoneal neuroblastomas. (A) Right-sided dissection: note the presence of two right renal arteries coming from the aorta (arrows). (B) Left-sided dissection: seen are the skeletonized aorta, mesenteric arteries, and left renal artery and vein. IVC, inferior vena cava; SMA, superior mesenteric artery; IMA, inferior mesenteric artery.
Pelvic tumors may involve the sacral plexus in addition to the iliac vessels. Thoracic tumors are usually more easily cleared from the vascular structures, but often extend into the intervertebral foramina. Extraction of tumor from this location is of uncertain benefit and can be associated with significant complications. A standard posterolateral thoracotomy can be used to expose tumors of the posterior mediastinum, and cervical incisions can be used to approach tumors of the neck. A laparoscopic or thoracoscopic approach may be used for resection of selected neuroblastomas in the abdomen and thorax.137,138
Operative Complications
Due to the extensive surgery often required, intraoperative and postoperative complications are not uncommon.139–142 As many as 80% of patients will experience significant blood loss that requires transfusion either in the operating room or in the early postoperative period. Up to 10% will suffer an injury to a major vascular structure (aorta, vena cava, or renal vessels). Injury to other viscera (stomach, bowel, liver, spleen, or kidney) occurs in approximately 5% of cases. On occasion, this necessitates removal of the injured organ, with the kidney being the most common. Postoperative complications are variable. Wound complications occur is 1–5%, as does postoperative bowel obstruction. In addition, hypertension, chyle leak into the thorax or abdomen, pleural effusion, infection and sepsis, diarrhea, and the need for prolonged total parenteral nutrition have been known to occur. Rarely, a patient will have to be emergently re-explored for postoperative hemorrhage or bowel obstruction.
Special Management Situations
Screening for Neuroblastoma
Because the two most important clinical variables for predicting outcome in patients with neuroblastoma are tumor stage and patient age at the time of diagnosis, it was hypothesized that earlier detection of neuroblastoma through mass population screening might significantly impact neuroblastoma-associated mortality. In the 1980s in Japan, mass screening of neuroblastoma was performed in infants by quantitating urinary vanillyl mandelic acid (VMA) and homovanillic acid (HVA). Initially, this mass screening showed very encouraging results.143 However, subsequent population-based studies with concurrent control groups performed in Germany and North America found that although the incidence of neuroblastoma increased, the additional cases were largely early stage, favorable biology, low-risk tumors.144,145 Because the overall mortality of patients with neuroblastoma was not affected, mass screening most likely detected tumors that would have undergone spontaneous regression and not been detected clinically. Thus, there currently appears to be no role for screening infants for neuroblastoma.
Small, localized neuroblastomas in young infants tend to regress spontaneously. Based on this observation, the COG protocol, ANBLOOP2, included an arm of expectant observation for patients with these lesions to further define their natural history. This study was designed to prove the hypothesis that close biochemical and sonographic observation could be safely applied in infants with small adrenal masses. Resection was reserved for those rare cases in which there was evidence of continued growth. To be eligible, infants with an adrenal mass had to be <6 months of age when the mass was first identified; the mass must be <16 mL in volume, if solid, or <65 mL if at least 25% cystic; and disease must be limited to the adrenal gland. The results from this study were published recently and confirmed that expectant observation of infants with small adrenal masses led to excellent three-year EFS (97.7 ± 2.3%) and 100% overall survival while avoiding operative intervention in >80% of patients.146
Stage 4S Neuroblastoma
In 1971, D’Angio, Evans, and Koop reported a number of patients with a ‘special’ variant of metastatic neuroblastoma, termed IVS (now referred to as 4S).147 These patients were infants who typically had a single, small primary tumor, but had extensive metastatic disease in the liver, skin nodules (‘blueberry muffin’ lesions), and small amounts of disease in the bone marrow (<10% of the mononuclear cells). Patients with 4S neuroblastoma were quite remarkable because the large amount of disease generally underwent spontaneous regression, even without treatment, and the infants ultimately had no evidence of disease.
Only supportive therapy has been recommended for this stage of neuroblastoma because of the high incidence of spontaneous regression and the good prognosis.148 Most of these patients have a tumor with favorable biology (single-copy MYCN, favorable Shimada histology, and DNA index >1). Therefore, they are assigned to the low-risk classification and receive no therapy. However, despite the generally benign course of their malignancy, these infants can die of complications caused by the initial bulk of their disease. Limited chemotherapy, local irradiation, or minimal resection can be used to treat infants with life-threatening symptoms of hepatomegaly. Creation of a Silastic pouch may be needed for those with significant hepatomegaly that causes either respiratory compromise secondary to diaphragmatic elevation or obstruction of the inferior vena cava. This procedure may help to avoid life-threatening events until shrinkage of the liver is achieved by either spontaneous regression or therapy. The rare infant with 4S disease and either unfavorable Shimada histology or a DNA index of 1 (or if the biology is not known) will be treated as intermediate-risk disease. Those with 4S disease that is MYCN amplified will be treated as high-risk disease.
Intraspinal Extension of Neuroblastoma
In a subset of patients with paraspinal neuroblastoma, tumor growth may extend into the spinal canal (‘dumbbell’ tumors). If neurologic symptoms result, urgent treatment is required to prevent permanent injury caused by compression of the cord. Each of the three main therapeutic modalities (surgery, radiation, and chemotherapy) has been used. A POG report showed similar rates of neurologic recovery in patients treated with surgery or chemotherapy, but significant orthopedic sequelae were seen more commonly in patients undergoing operation.149 Although chemotherapy is probably considered most appropriate for the initial management of these patients, improvements in neurosurgical techniques, including the use of laminotomy instead of laminectomy to access the intraspinal tumor, may allow reconsideration about the optimal approach, especially in patients with acutely progressive symptoms.
Opsoclonus–Myoclonus Syndrome
The opsoclonus–myoclonus syndrome (OMS) consists of myoclonic jerks and random eye movements or progressive cerebellar ataxia. OMS occurs in as many as 4% of patients, usually infants with thoracic primary tumors. Although the exact etiology of this syndrome is not known, the presence of cross-reactive autoantibodies to neural antigens in some of these patients suggests that it is it mediated by the immune system.150 Although patients generally have a good prognosis with regard to their tumor, neurologic symptoms often persist after successful removal of the tumor and can be debilitating.68,69 Some symptomatic relief can be attained by high doses of corticosteroids or adrenocorticotropic hormones. Studies have suggested that chemotherapy or intravenous IgG therapy (or both) may improve the long-term neurologic outcome for these patients.151,152 COG is currently testing this approach in a prospective clinical trial.
New Treatment Strategies
Differentiating Agents
Retinoids are vitamin A derivatives of 13-cis-retinoic acid. Retinoids decrease the proliferation and expression of MYCN of neuroblastoma cell lines in vitro and induce morphologic differentiation.153–156 Due to these attractive characteristics and the observation that resistance to chemotherapy does not result in resistance to 13-cis-retinoic acid, retinoids have been tested clinically. The CCG 3891 clinical trial randomized patients with high-risk neuroblastoma treated using chemotherapy or autologous BMT into two groups that received either 13-cis-retinoic acid for 6 months or no further therapy.103 The 3-year EFS of patients who received 13-cis-retinoic acid (46%) was significantly higher than that of patients who received no further therapy (29%; p = 0.027) and was independent of the initial randomization to either chemotherapy or autologous BMT. Thus, currently, all patients with high-risk neuroblastoma receive oral 13-cis-retinoic acid twice daily for 2 weeks and are then off therapy for 2 weeks. This treatment is continued for six cycles (6 months total). Some intermediate-risk patients also receive 13-cis-retinoic acid.
Immunotherapy
Neuroblastoma cells are sensitive to antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity.157 Immunotherapy using anti-ganglioside antibodies targeting GD2, the predominant antigen in neuroblastoma cells, appears to be a promising approach for the treatment of advanced neuroblastoma. As the induction of antibody-dependent cell-mediated cytotoxicity with anti-ganglioside GD2 antibodies is enhanced by cytokines such as GM-CSF158 and IL-2,159 a phase III trial was conducted to determine whether treatment with ch14.18 and cytokines (GM-CSF and IL-2) together with 13-cis-retinoic acid improves EFS and overall survival after autologous BMT, as compared to treatment with 13-cis-retinoic acid alone in patients with high-risk neuroblastoma. The study was stopped early because immunotherapy was superior to standard therapy (2-year EFS: 66% vs 46%, p = 0.01 and 2-year OS: 86% vs 75%, p = 0.02).160 Current trials are investigating the safety and efficacy of a ch14.18-Il-2 fusion protein, given with GM-CSF and isotretinoin.
MIBG Therapy
Refractory neuroblastoma has been treated with 131I-MIBG because it is readily taken-up by the tumor cells.161 In an investigation of patients with advanced chemoresistant neuroblastomas, response rates approached 33%.162 Studies further suggest that this treatment can be used as front-line therapy, followed by chemotherapy, without significant hematologic toxicity.163 125I-MIBG may be an even better treatment option for neuroblastomas with micrometastases or bone marrow infiltration, and is being tested for the treatment of patients with ‘ultra high-risk’ neuroblastoma (expected survival of less than 15%).164
Targeted Therapy
TRK Inhibition
As previously described, neurotrophins and their tyrosine kinase receptors are important in the development of the sympathetic nervous system and have been implicated in the pathogenesis of neuroblastoma. Studies are ongoing to test agonists of TrkA in an attempt to induce cellular differentiation. Conversely, blocking the BDNF/TrkB signaling pathway with Trk-specific tyrosine kinase inhibitors, such as CEP-751, may induce apoptosis by blocking crucial survival pathways.49,165–167
Epigenetic Targeting
Genome-wide DNA methylation analysis of neuroblastic tumors revealed that hypermethylation events are extensive and contribute to the clinicopathologic features of these tumors.168 Promoter methylation resulting in silencing of caspase 8, a protein involved in apoptosis, for example, likely contributes to the pathogenesis of MYCN-amplified neuroblastoma.169
Histones are the proteins that give structure to DNA. Both histones and DNA form the major components of chromatin. Alterations in histones can mediate changes in chromatin structure. The compacted form of DNA, termed heterochromatin, is largely inaccessible to transcription factors and, therefore, genes in the affected regions are silent. Other modifications of histones can cause DNA to take a more open or extended configuration (euchromatin), allowing for gene transcription. Histones can be modified by a number of different processes including methylation and acetylation, mediated by histone acetyl transferases and deacetylases, and histone methyltransferases. Each of these processes alters histone function, which, in turn alters the structure of chromatin and, therefore, the accessibility of DNA to transcription factors. Inhibiting the histone-mediated epigenetic processes that silence tumor suppressors in neuroblastoma is currently being tested in clinical trials.
References
1. Brodeur, GM, Castleberry, RP. Neuroblastoma. In: Pizzo PA, Poplack DG, eds. In Principles and Practice of Pediatric Oncology. Philadelphia: J. B. Lippincott, 1993.
2. Young, JL, Jr., Ries, LG, Silverberg, E, et al. Cancer incidence, survival, and mortality for children younger than age 15 years. Cancer. 1986; 58:598–602.
3. Brodeur, GM, Nakagawara, A. Molecular basis of clinical heterogeneity in neuroblastoma. Am J Pediatr Hematol Oncol. 1992; 14:111–116.
4. Maris, JM, Tonini, GP. Genetics of familial neuroblastoma. In: Brodeur GM, Sawada T, Tsudhica T, et al, eds. Neuroblastoma. 1st ed. Amsterdam: Elsevier Science; 2000:125–135.
5. Mosse, YP, Laudenslager, M, Longo, L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008; 455:930–935.
6. Virchow, R, Hyperplasie der Zirbel und der Nebenniern Vol 2 BerlinDie Krankhaften Geschwulste, ed. August Hirshwald. 1865:149–150.
7. Wright, JH. Neurocytoma or neuroblastoma, a kind of tumor not generally recognized. J Exp Med. 1910; 12:556–561.
8. Ambros, IM, Zellner, A, Roald, B, et al. Role of ploidy, chromosome 1p, and Schwann cells in the maturation of neuroblastoma. N Engl J Med. 1996; 334:1505–1511.
9. Ambros, IM, Zellner, A, Stock, C, et al. Proof of the reactive nature of the Schwann cell in neuroblastoma and its clinical implications. Prog Clin Biol Res. 1994; 385:331–337.
10. Crawford, SE, Stellmach, V, Ranalli, M, et al. Pigment epithelium-derived factor (PEDF) in neuroblastoma: A multifunctional mediator of Schwann cell antitumor activity. J Cell Sci. 2001; 114:4421–4428.
11. Chlenski, A, Liu, S, Crawford, SE, et al. SPARC is a key Schwannian-derived inhibitor controlling neuroblastoma tumor angiogenesis. Cancer Res. 2002; 62:7357–7363.
12. Beckwith, JB, Perrin, EV. In situ neuroblastoma: A contribution to the natural history of neural crest tumors. Am J Pathol. 1963; 43:1089.
13. Shimada, H, Chatten, J, Newton, WA, Jr., et al. Histopathologic prognostic factors in neuroblastic tumors: Definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. J Natl Cancer Inst. 1984; 73:405–416.
14. Shimada, H, Ambros, IM, Dehner, L, et al. Terminology and morphologic criteria of neuroblastic tumors: Recommendations by the International Neuroblastoma Pathology Committee. Cancer. 1999; 86:349–363.
15. Peuchmaur, M, d’Amore, ES, Joshi, VV, et al. Revision of the International Neuroblastoma Pathology Classification: Confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer. 2003; 98:2274–2281.
16. Shimada, H, Ambros, IM, Dehner, LP, et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer. 1999; 86:364–372.
17. Shimada, H, Umehara, S, Monobe, Y, et al. International neuroblastoma pathology classification for prognostic evaluation of patients with peripheral neuroblastic tumors: A report from the Children’s Cancer Group. Cancer. 2001; 92:2451–2461.
18. Sano, H, Bonadio, J, Gerbing, RB, et al. International neuroblastoma pathology classification adds independent prognostic information beyond the prognostic contribution of age. Eur J Cancer. 2006; 42:1113–1119.
19. De, BB, Gambini, C, Haupt, R, et al. Retrospective study of childhood ganglioneuroma. J Clin Oncol. 2008; 26:1710–1716.
20. Kaneko, Y, Kanda, N, Maseki, N, et al. Different karyotypic patterns in early and advanced stage neuroblastomas. Cancer Res. 1987; 47:311–318.
21. Look, AT, Hayes, FA, Nitschke, R, et al. Cellular DNA content as a predictor of response to chemotherapy in infants with unresectable neuroblastoma. N Engl J Med. 1984; 311:231–235.
22. Bowman, LC, Castleberry, RP, Cantor, A, et al. Genetic staging of unresectable or metastatic neuroblastoma in infants: A Pediatric Oncology Group study. J Natl Cancer Inst. 1997; 89:373–380.
23. Schwab, M, Alitalo, K, Klempnauer, KH, et al. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. 1983; 305:245–248.
24. Kohl, NE, Kanda, N, Schreck, RR, et al. Transposition and amplification of oncogene-related sequences in human neuroblastomas. Cell. 1983; 35:359–367.
25. Lutz, W, Stohr, M, Schurmann, J, et al. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene. 1996; 13:803–812.
26. Yancopoulos, GD, Nisen, PD, Tesfaye, A, et al. N-myc can cooperate with ras to transform normal cells in culture. Proc Natl Acad Sci U S A. 1985; 82:5455–5459.
27. Weiss, WA, Aldape, K, Mohapatra, G, et al. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997; 16:2985–2995.
28. Berry, T, Luther, W, Bhatnagar, N, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012; 22:117–130.
29. Brodeur, GM, Seeger, RC, Schwab, M, et al. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984; 224:1121–1124.
30. Brodeur, GM, Hayes, FA, Green, AA, et al. Consistent N-myc copy number in simultaneous or consecutive neuroblastoma samples from sixty individual patients. Cancer Res. 1987; 47:4248–4253.
31. Seeger, RC, Brodeur, GM, Sather, H, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. 1985; 313:1111–1116.
32. Brodeur, GM, Sekhon, G, Goldstein, MN. Chromosomal aberrations in human neuroblastomas. Cancer. 1977; 40:2256–2263.
33. Fujita, T, Igarashi, J, Okawa, ER, et al. CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. J Natl Cancer Inst. 2008; 100:940–949.
34. Bader, SA, Fasching, C, Brodeur, GM, et al. Dissociation of suppression of tumorigenicity and differentiation in vitro effected by transfer of single human chromosomes into human neuroblastoma cells. Cell Growth Differ. 1991; 2:245–255.
35. Fong, CT, Dracopoli, NC, White, PS, et al. Loss of heterozygosity for the short arm of chromosome 1 in human neuroblastomas: Correlation with N-myc amplification. Proc Natl Acad Sci U S A. 1989; 86:3753–3757.
36. Gilbert, F, Feder, M, Balaban, G, et al. Human neuroblastomas and abnormalities of chromosomes 1 and 17. Cancer Res. 1984; 44:5444–5449.
37. Attiyeh, EF, London, WB, Mosse, YP, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005; 353:2243–2253.
38. Takayama, H, Suzuki, T, Mugishima, H, et al. Deletion mapping of chromosomes 14q and 1p in human neuroblastoma. Oncogene. 1992; 7:1185–1189.
39. Attiyeh, EF, London, WB, Mosse, YP, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005; 353:2243–2253.
40. Mosse, YP, Laudenslager, M, Longo, L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008; 455:930–935.
41. Caren, H, Abel, F, Kogner, P, et al. High incidence of DNA mutations and gene amplifications of the ALK gene in advanced sporadic neuroblastoma tumours. Biochem J. 2008; 416:153–159.
42. Carpenter, EL, Mosse, YP. Targeting ALK in neuroblastoma-preclinical and clinical advancements. Nat Rev Clin Oncol. 2012; 9:391–399.
43. Mosse, YP, Laudenslager, M, Khazi, D, et al. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet. 2004; 75:727–730.
44. Cheung, NK, Zhang, J, Lu, C, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012; 307:1062–1071.
45. Barbacid, M. Neurotrophic factors and their receptors. Curr Opin Cell Biol. 1995; 7:148–155.
46. Levi-Montalcini, R. The nerve growth factor 35 years later. Science. 1987; 237:1154–1162.
47. Nakagawara, A, Arima-Nakagawara, M, Scavarda, NJ, et al. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med. 1993; 328:847–854.
48. Nakagawara, A, Arima, M, Azar, CG, et al. Inverse relationship between trk expression and N-myc amplification in human neuroblastomas. Cancer Res. 1992; 52:1364–1368.
49. Nakagawara, A, Azar, C, Scavarda, N. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994; 14:759–767.
50. Svensson, T, Ryden, M, Schilling, FH, et al. Coexpression of mRNA for the full-length neurotrophin receptor trk-C and trk-A in favourable neuroblastoma. Eur J Cancer. 1997; 33:2058–2063.
51. Teitz, T, Wei, T, Valentine, MB, et al. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med. 2000; 6:529–535.
52. Gunthert, U, Hofmann, M, Rudy, W, et al. A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell. 1991; 65:13–24.
53. Combaret, V, Gross, N, Lasset, C, et al. Clinical relevance of CD44 cell-surface expression and N-myc gene amplification in a multicentric analysis of 121 pediatric neuroblastomas. J Clin Oncol. 1996; 14:25–34.
54. Bradshaw, DM, Arceci, RJ. Clinical relevance of transmembrane drug efflux as a mechanism of multidrug resistance. J Clin Oncol. 1998; 16:3674–3690.
55. Norris, MD, Bordow, SB, Marshall, GM, et al. Expression of the gene for multidrug-resistance-associated protein and outcome in patients with neuroblastoma. N Engl J Med. 1996; 334:231–238.
56. Oberthuer, A, Berthold, F, Warnat, P, et al. Customized oligonucleotide microarray gene expression-based classification of neuroblastoma patients outperforms current clinical risk stratification. J Clin Oncol. 2006; 24:5070–5078.
57. Janoueix-Lerosey, I, Schleiermacher, G, Michels, E, et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol. 2009; 27:1026–1033.
58. Maris, JM. Recent advances in neuroblastoma. N Engl J Med. 2010; 362:2202–2211.
59. Henderson, TO, Bhatia, S, Pinto, N, et al. Racial and ethnic disparities in risk and survival in children with neuroblastoma: A Children’s Oncology Group study. J Clin Oncol. 2011; 29:76–82.
60. Vandesompele, J, Van Roy, N, Van Gele, M, et al. Genetic heterogeneity of neuroblastoma studied by comparative genomic hybridization. Genes Chromosomes. Cancer. 1998; 23:141–152.
61. Bown, N, Cotterill, S, Lastowska, M, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med. 1999; 340:1954–1961.
62. Diskin, SJ, Hou, C, Glessner, JT, et al. Copy number variation at 1q21.1 associated with neuroblastoma. Nature. 2009; 459:987–991.
63. Maris, JM, Mosse, YP, Bradfield, JP, et al. Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N Engl J Med. 2008; 358:2585–2593.
64. Capasso, M, Devoto, M, Hou, C, et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat Genet. 2009; 41:718–723.
65. Wang, K, Diskin, SJ, Zhang, H, et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature. 2011; 469:216–220.
66. Downing, JR, Wilson, RK, Zhang, J, et al. The pediatric cancer genome project. Nat Genet. 2012; 44:619–622.
67. Molenaar, JJ, Koster, J, Zwijnenburg, DA, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012; 483:589–593.
68. Russo, C, Cohn, SL, Petruzzi, MJ, et al. Long-term neurologic outcome in children with opsoclonus-myoclonus associated with neuroblastoma: A report from the Pediatric Oncology Group. Med Pediatr Oncol. 1997; 28:284–288.
69. Rudnick, E, Khakoo, Y, Antunes, NL, et al. Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: Clinical outcome and antineuronal antibodies-a report from the Children’s Cancer Group Study. Med Pediatr Oncol. 2001; 36:612–622.
70. Berthold, F, Kassenbohmer, R, Zieschang, J. Multivariate evaluation of prognostic factors in localized neuroblastoma. Am J Pediatr Hematol Oncol. 1994; 16:107–115.
71. Joshi, VV, Cantor, AB, Brodeur, GM, et al. Correlation between morphologic and other prognostic markers of neuroblastoma. A study of histologic grade, DNA index, N-myc gene copy number, and lactic dehydrogenase in patients in the Pediatric Oncology Group. Cancer. 1993; 71:3173–3181.
72. Silber, JH, Evans, AE, Fridman, M. Models to predict outcome from childhood neuroblastoma: The role of serum ferritin and tumor histology. Cancer Res. 1991; 51:1426–1433.
73. Berthold, F, Engelhardt-Fahrner, U, Schneider, A, et al. Age dependence and prognostic impact of neuron specific enolase (NSE) in children with neuroblastoma. In Vivo. 1991; 5:245–247.
74. Tsuchida, Y, Honna, T, Iwanaka, T, et al. Serial determination of serum neuron-specific enolase in patients with neuroblastoma and other pediatric tumors. J Pediatr Surg. 1987; 22:419–424.
75. Fitzgibbon, MC, Tormey, WP. Paediatric reference ranges for urinary catecholamines/metabolites and their relevance in neuroblastoma diagnosis. Ann Clin Biochem. 1994; 31(Pt 1):1–11.
76. Adams, GA, Shochat, SJ, Smith, EI, et al. Thoracic neuroblastoma: A Pediatric Oncology Group study. J Pediatr Surg. 1993; 28:372–377.
77. Tanabe, M, Yoshida, H, Ohnuma, N, et al. Imaging of neuroblastoma in patients identified by mass screening using urinary catecholamine metabolites. J Pediatr Surg. 1993; 28:617–621.
78. Ng, YY, Kingston, JE. The role of radiology in the staging of neuroblastoma. Clin Radiol. 1993; 47:226–235.
79. Stark, DD, Moss, AA, Brasch, RC, et al. Neuroblastoma: Diagnostic imaging and staging. Radiology. 1983; 148:101–105.
80. Cheung, NK, Kushner, BH. Should we replace bone scintigraphy plus CT with MR imaging for staging of neuroblastoma? Radiology. 2003; 226:286–287.
81. Siegel, MJ, Ishwaran, H, Fletcher, BD, et al. Staging of neuroblastoma at imaging: Report of the radiology diagnostic oncology group. Radiology. 2002; 223:168–175.
82. Brodeur, GM, Pritchard, J, Berthold, F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993; 11:1466–1477.
83. Turba, E, Fagioli, G, Mancini, AF, et al. Evaluation of stage 4 neuroblastoma patients by means of MIBG and 99mTc-MDP scintigraphy. J Nucl Biol Med. 1993; 37:107–114.
84. Paltiel, HJ, Gelfand, MJ, Elgazzar, AH, et al. Neural crest tumors: I-123 MIBG imaging in children. Radiology. 1994; 190:117–121.
85. Gelfand, MJ. Meta-iodobenzylguanidine in children. Semin Nucl Med. 1993; 23:231–242.
86. Wirnsberger, GH, Becker, H, Ziervogel, K, et al. Diagnostic immunohistochemistry of neuroblastic tumors. Am J Surg Pathol. 1992; 16:49–57.
87. Corbett, R, Olliff, J, Fairley, N, et al. A prospective comparison between magnetic resonance imaging, meta-iodobenzylguanidine scintigraphy and marrow histology/cytology in neuroblastoma. Eur J Cancer. 1991; 27:1560–1564.
88. Osmanagaoglu, K, Lippens, M, Benoit, Y, et al. A comparison of iodine-123 meta-iodobenzylguanidine scintigraphy and single bone marrow aspiration biopsy in the diagnosis and follow-up of 26 children with neuroblastoma. Eur J Nucl Med. 1993; 20:1154–11560.
89. Brodeur, GM, Pritchard, J, Berthold, F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993; 11:1466–1477.
90. Monclair, T, Brodeur, GM, Ambros, PF, et al. The International Neuroblastoma Risk Group (INRG) staging system: An INRG Task Force report. J Clin Oncol. 2009; 27:298–303.
91. Cecchetto, G, Mosseri, V, De Bernardi, B, et al. Surgical risk factors in primary surgery for localized neuroblastoma: The LNESG1 study of the European International Society of Pediatric Oncology Neuroblastoma Group. J Clin Oncol. 2005; 23:8483–8489.
92. Moroz, V, Machin, D, Faldum, A, et al. Changes over three decades in outcome and the prognostic influence of age-at-diagnosis in young patients with neuroblastoma: A report from the International Neuroblastoma Risk Group Project. Eur J Cancer. 2011; 47:561–571.
93. London, WB, Castleberry, RP, Matthay, KK, et al. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. J Clin Oncol. 2005; 23:6459–6465.
94. Evans, AE, D’Angio, GJ, Propert, K, et al. Prognostic factor in neuroblastoma. Cancer. 1987; 59:1853–1859.
95. Cohn, SL, Pearson, AD, London, WB, et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J Clin Oncol. 2009; 27:289–297.
96. Alvarado, CS, London, WB, Look, AT, et al. Natural history and biology of stage A neuroblastoma: A Pediatric Oncology Group Study. J Pediatr Hematol Oncol. 2000; 22:197–205.
97. Nitschke, R, Smith, EI, Shochat, S, et al. Localized neuroblastoma treated by surgery: A Pediatric Oncology Group Study. J Clin Oncol. 1988; 6:1271–1279.
98. Matthay, KK, Lukens, J, Haase, GM, et al, Outcome and prognostic factors for 1008 children with neuroblastoma treated from 1989-1995 on Children’s Cancer Group (CCG) protocols. Advances in Neuroblastoma Research Philadelphia, 1996.
99. Strother, DR, London, WB, Schmidt, ML, et al. Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children’s Oncology Group study P9641. J Clin Oncol. 2012; 30:1842–1848.
100. Baker, DL, Schmidt, ML, Cohn, SL, et al. Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med. 2010; 363:1313–1323.
101. Gaspar, N, Hartmann, O, Munzer, C, et al. Neuroblastoma in adolescents. Cancer. 2003; 98:349–355.
102. Franks, LM, Bollen, A, Seeger, RC, et al. Neuroblastoma in adults and adolescents: An indolent course with poor survival. Cancer. 1997; 79:2028–2035.
103. Matthay, KK, Villablanca, JG, Seeger, RC, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N Engl J Med. 1999; 341:1165–1173.
104. Paulino, AC. Palliative radiotherapy in children with neuroblastoma. Pediatr Hematol Oncol. 2003; 20:111–117.
105. Ziegler, MM. Pediatric surgical oncology. Curr Opin Pediatr. 1990; 2:580.
106. Perez, CA, Matthay, KK, Atkinson, JB, et al. Biologic variables in the outcome of stages I and II neuroblastoma treated with surgery as primary therapy: A children’s cancer group study. J Clin Oncol. 2000; 18:18–26.
107. Kushner, BH, Cheung, NK, LaQuaglia, MP, et al. International neuroblastoma staging system stage 1 neuroblastoma: A prospective study and literature review. J Clin Oncol. 1996; 14:2174–2180.
108. Evans, AE, Silber, JH, Shpilsky, A, et al. Successful management of low-stage neuroblastoma without adjuvant therapies: A comparison of two decades, 1972 through 1981 and 1982 through 1992, in a single institution. J Clin Oncol. 1996; 14:2504–2510.
109. Berthold, F, Treuner, J, Brandeis, WE, et al. [Neuroblastoma study NBL 79 of the German Society for Pediatric Oncology. Report after 2 years]. Klin Padiatr. 1982; 194:262–269.
110. Yamamoto, K, Hanada, R, Kikuchi, A, et al. Spontaneous regression of localized neuroblastoma detected by mass screening. J Clin Oncol. 1998; 16:1265–1269.
111. Suita, S, Zaizen, Y, Sera, Y, et al. Mass screening for neuroblastoma: quo vadis? A 9-year experience from the Pediatric Oncology Study Group of the Kyushu area in Japan. J Pediatr Surg. 1996; 31:555–558.
112. Nishihira, H, Toyoda, Y, Tanaka, Y, et al. Natural course of neuroblastoma detected by mass screening: A 5-year prospective study at a single institution. J Clin Oncol. 2000; 18:3012–3017.
113. Oue, T, Inoue, M, Yoneda, A, et al. Profile of neuroblastoma detected by mass screening, resected after observation without treatment: Results of the Wait and See pilot study. J Pediatr Surg. 2005; 40:359–363.
114. Maris, JM, Hogarty, MD, Bagatell, R, et al. Neuroblastoma. Lancet. 2007; 369:2106–2120.
115. Nitschke, R, Smith, EI, Shochat, S, et al. Localized neuroblastoma treated by surgery: A Pediatric Oncology Group Study. J Clin Oncol. 1988; 6:1271–1279.
116. Davidoff, AM, Corey, BL, Hoffer, FA, et al. Radiographic assessment of resectability of locoregional disease in children with high-risk neuroblastoma during neoadjuvant chemotherapy. Pediatr Blood Cancer. 2005; 44:158–162.
117. DuBois, SG, Kalika, Y, Lukens, JN, et al. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J Pediatr Hematol Oncol. 1999; 21:181–189.
118. Haase, GM, Wong, KY, deLorimier, AA, et al. Improvement in survival after excision of primary tumor in stage III neuroblastoma. J Pediatr Surg. 1989; 24:194–200.
119. Adkins, ES, Sawin, R, Gerbing, RB, et al. Efficacy of complete resection for high-risk neuroblastoma: A Children’s Cancer Group study. J Pediatr Surg. 2004; 39:931–936.
120. La Quaglia, MP, Kushner, BH, Su, W, et al. The impact of gross total resection on local control and survival in high-risk neuroblastoma. J Pediatr Surg. 2004; 39:412–417.
121. von Schweinitz, D, Hero, B, Berthold, F. The impact of surgical radicality on outcome in childhood neuroblastoma. Eur J Pediatr Surg. 2002; 12:402–409.
122. Kiely, EM. The surgical challenge of neuroblastoma. J Pediatr Surg. 1994; 29:128–133.
123. Kuroda, T, Saeki, M, Honna, T, et al. Clinical significance of intensive surgery with intraoperative radiation for advanced neuroblastoma: Does it really make sense? J Pediatr Surg. 2003; 38:1735–1738.
124. La Quaglia, MP, Kushner, BH, Heller, G, et al. Stage 4 neuroblastoma diagnosed at more than 1 year of age: Gross total resection and clinical outcome. J Pediatr Surg. 1994; 29:1162–1165.
125. Haase, G, O’Leary, M, Ramsay, N, et al. Aggressive surgery combined with intensive chemotherapy improves survival in poor-risk neuroblastoma. J Pediatr Surg. 1991; 26:1119–1123.
126. Grosfeld, JL, Baehner, RL. Neuroblastoma: An analysis of 160 cases. World J Surgery. 1980; 4:29–38.
127. LaQuaglia MP, von Allmen D, Davidoff A, et al. A comprehensive review of the role of primary tumor resection in stages 3 and 4 neuroblastoma. Presented at the Advances in Neuroblastoma Research Conference, Chiba, Japan, May 2008.
128. LaQuaglia MP, Davidoff A, London W, et al. The impact of the extent of primary site resection on event-free survival, overall survival, and local recurrence in high-risk neuroblastoma. A report from the Children’s Oncology Group A3973 Study. Presented at the Advances in Neuroblastoma Research Conference, Chiba, Japan, May 2008.
129. Castel, V, Tovar, JA, Costa, E, et al. The role of surgery in stage IV neuroblastoma. J Pediatr Surg. 2002; 37:1574–1578.
130. Azizkhan, RG, Shaw, A, Chandler, JG. Surgical complications of neuroblastoma resection. Surgery. 1985; 97:514–517.
131. Shorter, NA, Davidoff, AM, Evans, AE, et al. The role of surgery in the management of stage IV neuroblastoma: A single institution study. Med Pediatr Oncol. 1995; 24:287–291.
132. Schmidt, ML, Lukens, JN, Seeger, RC, et al. Biologic factors determine prognosis in infants with stage IV neuroblastoma: A prospective Children’s Cancer Group study. J Clin Oncol. 2000; 18:1260–1268.
133. Azizkhan, R, Haase, G. Current biologic and therapeutic implications in the surgery of neuroblastoma. Semin Surg Oncol. 1993; 9:493–501.
134. Medary, I, Aronson, D, Cheung, NK, et al. Kinetics of primary tumor regression with chemotherapy: Implications for the timing of surgery. Ann Surg Oncol. 1996; 3:521–525.
135. Davidoff, AM, Corey, BL, Hoffer, FA, et al. Radiographic assessment of resectability of locoregional disease in children with high-risk neuroblastoma during neoadjuvant chemotherapy. Medical and Pediatric Oncology. 2003.
136. Loo, R, Applebaum, H, Takasugi, J, et al. Resection of advanced stage neuroblastoma with the cavitron ultrasonic surgical aspirator. J Pediatr Surg. 1988; 23:1135–1138.
137. Iwanaka, T, Arai, M, Ito, M, et al. Surgical treatment for abdominal neuroblastoma in the laparoscopic era. Surg Endosc. 2001; 15:751–754.
138. Lobe, TE. The applications of laparoscopy and lasers in pediatric surgery. Surg Annu. 1993; 25(Pt 1):175–191.
139. Losty, P, Quinn, F, Breatnach, F, et al. Neuroblastoma–a surgical perspective. Eur J Surg Oncol. 1993; 19:33–36.
140. Azizkhan, RG, Shaw, A, Chandler, JG. Surgical complications of neuroblastoma resection. Surgery. 1985; 97:514–517.
141. Berthold, F, Utsch, S, Holschneider, AM. The impact of preoperative chemotherapy on resectability of primary tumour and complication rate in metastatic neuroblastoma. Z Kinderchir. 1989; 44:21–24.
142. Kiely, EM. Radical surgery for abdominal neuroblastoma. Semin Surg Oncol. 1993; 9:489–492.
143. Sawada, T. Past and future of neuroblastoma screening in Japan. Am J Pediatr Hematol Oncol. 1992; 14:320–326.
144. Schilling, FH, Spix, C, Berthold, F, et al. Neuroblastoma screening at one year of age. N Engl J Med. 2002; 346:1047–1053.
145. Woods, WG, Gao, RN, Shuster, JJ, et al. Screening of infants and mortality due to neuroblastoma. N Engl J Med. 2002; 346:1041–1046.
146. Nuchtern, JG, London, WB, Barnewolt, CE, et al. A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: A Children’s Oncology Group study. Ann Surg. 2012.
147. D’Angio, GJ, Evans, AE, Koop, CE. Special pattern of widespread neuroblastoma with a favourable prognosis. Lancet. 1971; 1:1046–1049.
148. Nickerson, HJ, Matthay, KK, Seeger, RC, et al. Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: A Children’s Cancer Group study. J Clin Oncols. 2000; 18:477–486.
149. Katzenstein, HM, Kent, PM, London, WB, et al. Treatment and outcome of 83 children with intraspinal neuroblastoma: The Pediatric Oncology Group experience. J Clin Oncol. 2001; 19:1047–1055.
150. Antunes, NL, Khakoo, Y, Matthay, KK, et al. Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus. J Pediatr Hematol Oncol. 2000; 22:315–320.
151. Veneselli, E, Conte, M, Biancheri, R, et al. Effect of steroid and high-dose immunoglobulin therapy on opsoclonus-myoclonus syndrome occurring in neuroblastoma. Med Pediatr Oncol. 1998; 30:15–17.
152. Borgna-Pignatti, C, Balter, R, Marradi, P, et al. Treatment with intravenously administered immunoglobulins of the neuroblastoma-associated opsoclonus-myoclonus. J Pediatr. 1996; 129:179–180.
153. Reynolds, CP, Kane, DJ, Einhorn, PA, et al. Response of neuroblastoma to retinoic acid in vitro and in vivo. Prog Clin Biol Res. 1991; 366:203–211.
154. Reynolds, CP, Schindler, P, Jones, D, et al. Comparisons of 13-cisretinoic acid to trans-retronic acid using human neuroblastoma cell lines. In: Evans AE, Biedler JL, Brodeur G, et al, eds. Advances in Neuroblastoma Research 4. New York: Wiley; 1994:237–244.
155. Sidell, N, Altman, A, Haussler, MR, et al. Effects of retinoic acid (RA) on the growth and phenotypic expression of several human neuroblastoma cell lines. Exp Cell Res. 1983; 148:21–30.
156. Thiele, CJ, Reynolds, CP, Israel, MA. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma. Nature. 1985; 313:404–406.
157. Cheung, NK. Immunotherapy. Neuroblastoma as a model. Pediatr Clin North Am. 1991; 38:425–441.
158. Ozkaynak, MF, Sondel, PM, Krailo, MD, et al. Phase I study of chimeric human/murine anti-ganglioside G(D2) monoclonal antibody (ch14.18) with granulocyte-macrophage colony-stimulating factor in children with neuroblastoma immediately after hematopoietic stem-cell transplantation: A Children’s Cancer Group Study. J Clin Oncol. 2000; 18:4077–4085.
159. Hank, JA, Surfus, J, Gan, J, et al. Treatment of neuroblastoma patients with antiganglioside GD2 antibody plus interleukin-2 induces antibody-dependent cellular cytotoxicity against neuroblastoma detected in vitro. J Immunother. 1994; 15:29–37.
160. Yu, AL, Gilman, AL, Ozkaynak, MF, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010; 363:1324–1334.
161. Gelfand, MJ. Meta-iodobenzylguanidine in children. Semin Nucl Med. 1993; 23:231–242.
162. Lashford, LS, Lewis, IJ, Fielding, SL, et al. Phase I/II study of iodine 131 metaiodobenzylguanidine in chemoresistant neuroblastoma: A United Kingdom Children’s Cancer Study Group investigation. J Clin Oncol. 1992; 10:1889–1896.
163. Haase, GM, Meagher, DP, Jr., McNeely, LK, et al. Electron beam intraoperative radiation therapy for pediatric neoplasms. Cancer. 1994; 74:740–747.
164. Hoefnagel, CA, Smets, L, Voute, PA, et al. Iodine-125-MIBG therapy for neuroblastoma. J Nucl Med. 1991; 32:361–362.
165. Evans, AE, Kisselbach, KD, Yamashiro, DJ, et al. Antitumor activity of CEP-751 (KT-6587) on human neuroblastoma and medulloblastoma xenografts. Clin Cancer Res. 1999; 5:3594–3602.
166. Acheson, A, Conover, JC, Fandl, JP, et al. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995; 374:450–453.
167. Matsumoto, K, Wada, RK, Yamashiro, JM, et al. Expression of brain-derived neurotrophic factor and p145TrkB affects survival, differentiation, and invasiveness of human neuroblastoma cells. Cancer Res. 1995; 55:1798–1806.
168. Buckley, PG, Das, S, Bryan, K, et al. Genome-wide DNA methylation analysis of neuroblastic tumors reveals clinically relevant epigenetic events and large-scale epigenomic alterations localized to telomeric regions. Int J Cancer. 2011; 128:2296–2305.
169. Teitz, T, Wei, T, Valentine, MB, et al. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med. 2000; 6:529–535.
[/level-membership-for-pediatrics-category][not-level-membership-for-pediatrics-category]
Neuroblastoma
Neuroblastoma is the most common solid extracranial malignancy of childhood and the most common malignant tumor in infants.1 The overall incidence of neuroblastoma is 1 per 100,000 children in the USA, thereby comprising 7–10% of all malignancies diagnosed in patients younger than 15 years of age. Yet neuroblastoma is responsible for approximately 15% of all pediatric cancer deaths.2 Neuroblastoma is a heterogeneous disease; tumors can spontaneously regress or mature, or display a very aggressive, malignant phenotype.3 Because of these unique characteristics, neuroblastoma has been of great interest to both clinicians and basic science researchers. Progress in molecular and cellular biology in the past 30 years has contributed greatly to a better understanding of this disease. Unfortunately, this progress has not significantly altered the clinical outcome for patients with advanced-stage neuroblastoma. Although the prognosis for these patients has improved in the past three decades, the long-term outcome remains very poor.
The etiology of neuroblastoma is currently unknown, and no environmental factors have been convincingly linked to its development. The disease generally occurs sporadically, but familial neuroblastoma occurs in about 2% of the cases. The substantial biologic and clinical heterogeneity is also observed in familial cases.4 The germline mutation associated with hereditary neuroblastoma has been identified: activating mutations in the tyrosine kinase domain of the anaplastic lymphoma kinase (ALK) oncogene on the short arm of chromosome 2 (2p23).5 These mutations can also be somatically acquired, although the prevalence of ALK activation in sporadic neuroblastoma remains to be determined.
Pathology
Neuroblastoma is an embryonal tumor of the sympathetic nervous system. These tumors arise during fetal or early postnatal life from sympathetic cells (sympathogonia) derived from the neural crest. Therefore, tumors can originate anywhere along the path which neural crest cells migrate, including the adrenal medulla, paraspinal sympathetic ganglia, and sympathetic paraganglia such as the organ of Zuckerkandl. The German pathologist Rudolph Virchow is generally credited with being the first to describe the histologic appearance of what is now known as neuroblastoma in his 1864 article entitled, ‘Hyperplasia of the pineal and suprarenal glands’.6 The first to use the term ‘neuroblastoma’ was James Homer Wright in 1910 who described the classic appearance of rosettes of tumor cells around central neural fibrils.7 He also noted the association between the common sites of tumor development and the pattern of migration of primitive neural cells.
As one of the ‘small, round blue cell’ tumors of infancy and childhood, neuroblastoma, particularly when undifferentiated, must be distinguished from other neoplasms in this group (Ewing sarcoma family of tumors [ESFT], non-Hodgkin lymphoma, and rhabdomyosarcoma). Neuroblastoma can be distinguished histologically by the presence of neuritic processes (neuropil) and Homer Wright rosettes (neuroblasts surrounding eosinophilic neuropil). Scattered ganglion cells or immature chromaffin cells can also be seen. The appearance of the tumor cells may vary from undifferentiated cells to fully mature ganglion cells. In addition, neuroblastomas have variable degrees of Schwannian cell stroma, reactive non-neoplastic tissue recruited by the tumor cells. This stroma is intermixed, to a greater or lesser degree, as wavy bundles and sheets of spindle cells and produces anti-proliferative and differentiation-inducing factors that are crucial to neuronal differentiation.8,9 In addition, the Schwannian stroma appears to produce a variety of antiangiogenic factors, including pigment epithelium derived factor (PEDF)10 and secreted protein acidic and rich in cysteine (SPARC).11 Histopathologic variables among neuroblastic tumors include the degree of differentiation, maturation, lymphoid infiltration, calcification, anaplasia, necrosis, mitotic activity, neurofibrillary material (neuropil), and the presence of multinucleate cells. Finally, immunohistochemical analysis usually generates positive staining when antibodies to neuroblastoma-specific antigens such as synaptophysin, neuron-specific enolase, and chromogranin are used, and is negative when antibodies to actin, desmin, cytokeritin, leukocyte common antigen, vimentin, and CD99 are used.
Neuroblastoma is characterized by several unique clinical behaviors, including the secretion of catecholamine products and the potential to regress or mature, either spontaneously or in response to treatment. Small nodules of primitive neuroblasts are routinely found in the developing adrenal gland, even during the early postnatal period. Beckwith and Perrin described microscopic nodules that they termed ‘neuroblastoma in situ’ in the adrenals of infants undergoing autopsy following death from non-malignancy related causes.12 The incidence of this finding was more than 200-fold greater than the clinical incidence of neuroblastoma, which suggests that perhaps many neuroblastomas spontaneously regress or mature into lesions that never become clinically apparent. The process of involution is well described during embryonic life, especially in the developing central and peripheral nervous systems. Although initially thought to be mediated by the immune system, the process of involution may be the result of the withdrawal of neurotrophic maintenance factors such as nerve growth factor (NGF). Clinically apparent neuroblastoma can also regress or spontaneously mature, but the mechanism remains unknown.
Histopathologic Classification
In 1984, Shimada and colleagues first developed an age-linked classification system of neuroblastic tumors based on tumor morphology in which neuroblastomas were divided into two prognostic subgroups, favorable histology and unfavorable histology.13 In 1999, the International Neuroblastoma Pathology Classification (INPC) was devised, and then modified in 2003, and is an adaptation of the original Shimada system.14,15 The INPC is based mainly on morphologic changes associated with the maturational sequence of neuroblastic tumors. It remains an age-linked classification that depends on the differentiation grade of the neuroblasts, the cellular turnover index (mitosis-karyorrhexis index [MKI]), and the presence or absence of Schwannian stroma. The INPC classifies neuroblastic tumors into three morphologic categories: neuroblastoma, ganglioneuroblastoma, and ganglioneuroma (Fig. 66-1).
FIGURE 66-1 Histologic appearance of neuroblastic tumors. (A) An undifferentiated neuroblastoma with high MKI (10×). A clump of karyorrhectic tumor cells (white arrow) and a tumor cell undergoing mitosis (open arrow) are shown in the insert (60×). (B) A differentiating neuroblastoma, with low MKI (10×). A primitive neuroblast (gray arrow) and a differentiating tumor cell (black arrow), with features of differentiation in both the nucleus and cytoplasm, are shown in the insert (60×). Abundant neuropil is also seen. (C) A stroma-rich ganglioneuroblastoma with infrequent neuroblasts intermixed within abundant Schwannian stroma and ganglion cells (10×). (D) A stroma-rich ganglioneuroma. Ganglion cells are seen (arrow) (10×). Infiltrating lymphoid cells are also seen but no neuroblasts are present. (Courtesy of Jesse Jenkins MD and Christine Fuller MD, St. Jude Children’s Research Hospital, Memphis TN. Reprinted from Neuroblastoma, Davidoff AM. In Oldham KT, Colombani PM, Foglia RP, et al, editors. Principles and Practice of Pediatric Surgery. Philadelphia: Lippincott, Williams & Wilkins; 2005.)
Neuroblastomas are, by definition, Schwannian stroma poor (<50% of the tumor tissue) and can be subtyped as undifferentiated, poorly differentiated, or differentiating. Undifferentiated requires supplemental diagnostic methods such as immunohistochemistry, electron microscopy, or cytogenetics. Moreover, neuropil is not present. In poorly differentiated, <5% of tumor cells have features of differentiation, and neuropil is present. Differentiating tumors demonstrate >5% of tumor cells differentiating toward ganglion cells. To classify a cell as a differentiating neuroblast, there must be synchronous differentiation of the nucleus and eosinophilic cytoplasm.14
Additional factors that contribute to the prognostic distinction of stroma-poor neuroblastic tumors (neuroblastoma) as favorable or unfavorable subtypes include the MKI, which is defined as the number of tumor cells in mitosis or karyorrhexis per 5000 neuroblastic cells (i.e., low MKI, <100 cells; intermediate, 100–200 cells; high, >200 cells), and the patient’s age (<1.5 years, 1.5-5 years, >5 years) (Table 66-1). It has been hypothesized that neuroblastic cells with maturational potential require a latent period before demonstrating histologic evidence of differentiation. Therefore, there is a certain allowance for mitotic and karyorrhectic activities of neuroblastic cells in tumors in infants and younger children.16
TABLE 66-1
Prognostic Evaluation of Neuroblastic Tumors According to the International Neuroblastoma Pathology Classification
MKI, mitosis-karyorrhexis index.
Adapted from Shimada H, Ambros IM, Dehner LP, et al. The International NB Pathology Classification (the Shimada System). Cancer 1999;86:364–72.
The importance of this histopathologic classification was confirmed in a large, retrospective analysis reported by Shimada et al.17 The INPC classification of tumor histology provided independent prognostic information where tumors of favorable histology had a 90.8% probability of 5-year event-free survival [EFS] compared to 31.2% EFS for tumors of unfavorable histology. More recently, the INPC classification has been shown to add independent prognostic information beyond the prognostic contribution of age.18 Therefore, histopathology remains in the current multifactorial risk stratification for patients with neuroblastoma. This determination is particularly important in patients with MYCN non-amplified tumors who are older than 18 months and have stage 3 disease, or 12 to 18 months of age and have stage 4 disease, or have 4S disease. As the histopathologic pattern within a tumor can be heterogeneous, it is recommended that representative sections from at least 1 cm3 of viable, non-necrotic tissue be analyzed to determine histopathologic classification. The prognostic value of assessing the histopathology of a neuroblastoma after chemotherapy or radiation therapy has not been validated.
Stroma-rich neuroblastic tumors are classified as either ganglioneuroblastomas or ganglioneuromas. Ganglioneuroblastomas contain cells that are transitioning toward differentiation but are not completely differentiated/mature. Also, <50% of the total volume is made up of neuroblastic cells. Ganglioneuroblastomas can be further divided into ‘intermixed’ and ‘nodular’ subtypes, depending on the distribution of the neuroblastic cells. The distinction is important because of the significantly worse prognosis associated with the latter subtype, in which the neuroblastic clones that comprise grossly distinct nodules appear to be responsible for the aggressive phenotype for this subtype.16 Ganglioneuromas contain either maturing or mature cells, and lack any neuroblastomatous component. Most stroma-rich tumors (ganglioneuroblastoma, intermixed and ganglioneuroma, maturing subtype) are classified as ‘favorable’ by the INPC. However, the pathologic/prognostic classification of the ganglioneuroblastoma, nodular subtype is based on the morphologic evaluation of the neuroblastomatous nodule(s), and can, therefore, be unfavorable. Tumors that fit the criteria for ganglioneuroma, mature subtype, with abundant Schwannian stroma and fully mature ganglion cells, in the absence of neuroblasts, are considered benign, and are generally not considered for enrolment in protocols for neuroblastic tumors. Despite this, ganglioneuromas can be quite large and infiltrative, and attempts at removal can be associated with significant complications. In addition, survival does not seem to be influenced by extent of resection.19 Therefore, aggressive attempts at resection of ganglioneuromas are not recommended.
Molecular Biology
Advances in molecular biology research in the past three decades have resulted in an increased understanding of the genetic events in the pathogenesis and progression of many human malignancies, including those of childhood. Neuroblastoma, in particular, has served as a model for a molecular approach to treating patients with cancer, highlighting the utility of genetic analysis for diagnosis, risk stratification, and treatment planning. Chromosomal structural changes play a role in neuroblastoma, particularly those that result in the loss of tumor suppressors, or gain of oncogenes, gene amplification, and activating or inactivating mutations of relevant genes or their regulatory elements. The end result of alterations in these genetic elements, regardless of their specific mechanisms, is the disruption of the normal balance between cell proliferation and cell death.
DNA Content
Normal human cells contain two copies of each of 23 chromosomes; thus, a normal diploid cell has 46 chromosomes. The majority (55%) of primary neuroblastomas are triploid or ‘near-triploid/hyperdiploid’ and contain between 58 and 80 chromosomes; the remainder (45%) are either ‘near-diploid’ (35–57 chromosomes) or ‘near-tetraploid’ (81–103 chromosomes).20 The ‘DNA index’ of a tumor is the ratio of the number of chromosomes present to a diploid number of chromosome (i.e., 46). Therefore, diploid cells have a DNA index of 1.0, whereas near-triploid cells have a DNA index ranging from 1.26 to 1.76. Neuroblastomas that are near-diploid or near-tetraploid usually have structural genetic abnormalities, most frequently chromosome 1p deletion and MYCN amplification. Near-triploid or hyperdiploid tumors are characterized by almost three complete haploid sets of chromosomes with few structural abnormalities. Importantly, patients with near-triploid tumors typically have favorable clinical and biologic prognostic factors and excellent survival rates, as compared with those patients who have near-diploid or near-tetraploid tumors.21 This association is most important for infants with advanced disease as the prognostic significance of tumor ploidy appears to be lost in patients older than 2 years.22 Currently, ploidy only potentially impacts the risk group assessment of infants age 12–18 months with metastatic disease and infants with 4S disease in the COG risk stratification schema.
Amplification of MYCN
Investigation of the molecular biology of neuroblastoma began with the cytogenetic characterization of tumor-derived cell lines. These studies showed the frequent presence of extrachromosomal double-minute chromatin bodies (DMs) and chromosomally integrated homogeneously staining regions (HSRs) characteristic of gene amplification (Fig. 66-2).23 Since that time, it has been shown that the amplified region was derived from the distal short arm of chromosome 2 (2p24) and contained the MYCN proto-oncogene. MYCN encodes a 64 kDa nuclear phosphoprotein that forms a transcriptional complex by associating with other nuclear proteins expressed in the developing nervous system and other tissues.24 Enforced expression of MYCN increases the rates of DNA synthesis and cell proliferation, and shortens the G1 phase of the cell cycle.25 MYCN can also function as a classic dominant oncogene that cooperates with activated ras to transform normal cells.26 Targeted expression of MYCN in transgenic mice results in the development of neuroblastomas.27 This activity is potentiated when combined with mutations in ALK.28
FIGURE 66-2 FISH analysis of a neuroblastoma. (A) Chromosomes in metaphase. The bright spots are double-minute chromatin bodies. (B) The metaphase chromosomes are again seen. An intact interphase nucleus is marked with an asterisk. The normal two copies of the MYCN gene are marked with solid arrows. Homogeneously staining regions (HSRs) are also seen. One is seen in the interphase nucleus, and the other is marked with a dotted arrow. (Courtesy of Marc Valentine, St. Jude Children’s Research Hospital, Memphis, TN.)
Overall, approximately 25% of primary neuroblastomas in children have MYCN amplification, with MYCN amplification being present in 40% with advanced disease but only 5–10% with low-stage disease.29 The copy number, which can range from 5- to 500-fold amplification, is usually consistent among primary and metastatic sites and at different times during tumor evolution and treatment.30 This finding suggests that MYCN amplification is an early event in the pathogenesis of neuroblastoma. Amplification of MYCN is associated with advanced stages of disease, rapid tumor progression, and poor outcome; therefore, it is a powerful prognostic indicator of tumor behavior.29,31 Amplification can be detected either by routine metaphase cytogenetics or fluorescent in situ hybridization (FISH), and current therapeutic neuroblastoma protocols have incorporated the presence or absence of MYCN amplification into their risk stratification schema.
Chromosomal Changes
Also noted on early karyotype analyses of neuroblastoma-derived cell lines were frequent deletions of the short arm of chromosome 1.32 Deletions of genetic material in tumors suggest the presence (and subsequent loss) of a tumor suppressor gene. Although no individual tumor suppressor gene has been confirmed on chromosome 1p, recent data have identified CHD5 as a strong candidate for the tumor suppressor gene that is deleted from 1p36.31 in neuroblastoma.33 Functional confirmation of the presence of a 1p tumor suppressor gene came from the demonstration that transfection of chromosome 1p into a neuroblastoma cell line resulted in morphologic changes and ultimately cell senescence.34 Approximately 20–35% of primary neuroblastomas exhibit 1p deletion, as determined by FISH, with the smallest common region of loss located within region 1p36.35 About 70% of advanced-stage neuroblastomas have 1p deletions.36 Molecular studies have shown that there is a strong correlation between 1p deletion and MYCN amplification and other high-risk features such as age older than 1 year and advanced-stage disease.35 One recent study has demonstrated that 1p deletions are independently associated with a worse outcome in patients with neuroblastoma.37
Deletion of the long arm of chromosome 11 (11q) also appears to be common in neuroblastoma, being present in about 40% of cases. Unbalanced deletion of 11q (loss with either retention or gain of 11p material) is inversely related to MYCN amplification,37,38 yet is strongly associated with other high-risk features. Recently, Attiyeh et al., on behalf of COG, showed in a large cohort of patients that unbalanced deletion of 11q and 1p36 were independently associated with a worse outcome in patients with neuroblastoma.39 Therefore, the duration of treatment for children with intermediate-risk neuroblastoma on the recent COG study was based, in part, on the 1p and 11q allelic status of the tumor.
Mutations
An example of proto-oncogene activation by point mutation involves the tyrosine kinase receptor, ALK, on the short arm of chromosome 2 (2p23). Receptor tyrosine kinases (RTK) are high-affinity cell surface receptors for many growth factors, cytokines, and hormones. When activated through ligand binding, these proteins mediate phosphorylation of tyrosine on target molecules or substrates, resulting in intracellular signaling and, ultimately, the regulation of normal cellular processes. Mutation of RTK’s can lead to constitutive activation of the signaling pathway in the absence of ligand. Recently, activating mutations of ALK have been shown to be the germline abnormality associated with hereditary neuroblastoma.40 These mutations can also be somatically acquired, as can amplification of the gene, although the prevalence of ALK activation in sporadic neuroblastoma is not known.41 Activated ALK has proven to be a targetable abnormality in neuroblastoma, with drugs such as crizotinib, an anti-ALK antibody, showing efficacy.42 Further studies have identified loss-of-function mutations in the homeobox gene PHOXB2 on 4p13 that are also associated with familial neuroblastoma, particularly when occurring together with Hirschsprung disease and/or central hypoventilation.43
Recently, inactivating mutations of ATRX, a transcriptional regulator that is part of a multiprotein complex that plays a role in regulating chromatin remodeling, nucleosome assembly, and telomere maintenance, have been found in neuroblastoma, particularly high-stage tumors in older patients.44 ATRX mutations appear to be loss-of-function mutations associated with an absence of the ATRX protein in the nucleus, and with long telomeres. How these alterations lead to lengthened telomeres is uncertain. These results may provide a molecular marker and potential therapeutic target for neuroblastoma among adolescents and young adults. It may also delineate the subset of children with neuroblastoma who have a chronic but progressive clinical course when receiving standard therapeutic approaches and who may benefit from a different treatment strategy.
Other Molecular Abnormalities
Neurotrophins and their tyrosine kinase receptors are important in the development of the sympathetic nervous system and have been implicated in the pathogenesis of neuroblastoma. Three receptor-ligand pairs have been identified: TrkA, the primary receptor for NGF; TrkB, the primary receptor of brain-derived neurotrophic factor (BDNF); and TrkC, the receptor for neurotrophin-3 (NT-3).45 TrkA appears to mediate differentiation of developing neurons or neuroblastoma in the presence of NGF ligand, and apoptosis in the absence of NGF.46 High TrkA expression is associated with favorable tumor biology and good outcome47 and is inversely correlated with MYCN amplification.48 Conversely, the TrkB/BDNF pathway appears to promote neuroblastoma survival through autocrine or paracrine signaling, especially in MYCN-amplified tumors.49 TrkB is expressed in about 40% of neuroblastomas, usually advanced-stage disease. TrkC is expressed in approximately 25% of neuroblastomas and is strongly associated with TrkA expression.50
Other molecular abnormalities frequently detected in neuroblastoma include inactivation of caspase 8, expression of CD44, and overexpression of multidrug resistance genes. Studies have demonstrated inactivation of caspase 8, a component of the Fas death-signaling complex, in MYCN-amplified neuroblastomas.51 It has been proposed that inactivation of caspase 8 renders tumor cells resistant to apoptotic signals. CD44 is a cell surface glycoprotein that appears to play a role in tumor cell adhesion.52 In neuroblastomas, CD44 expression is inversely correlated with MYCN amplification and is undetectable in most disseminated neuroblastomas.53 Multidrug resistance-associated protein (MRP) is an efflux pump whose expression in neuroblastoma appears to be correlated with MYCN amplification and poor prognosis.54,55 The presence of MRP may explain the common clinical situation in which neuroblastomas initially respond well to chemotherapy but subsequently become resistant.
Genome-Wide Association Studies
Recently, microarray technologies have generated extensive amounts of data that have aided in identifying genomic (DNA) and transcriptomic (RNA) abnormalities associated with neuroblastoma. In addition, these abnormalities have been shown to have significant predictive power when anticipating outcome for these patients.56,57 Many of these findings were generated by large scale genome-wide association studies (GWAS). This is a technique whereby all or most of the genes of patients with neuroblastoma are analyzed to find differences with the population as a whole, looking for variations that are associated with the development and aggressiveness of neuroblastoma. The causal relationship between the DNA variant associated with the cancer is uncertain but an excessive inheritance of risk variants has been postulated to increase susceptibility to the disease. Several GWAS studies have been performed in patients with neuroblastoma and have identified a number of such genetic risk variants.58 These observations suggest that developmental childhood cancers are likely influenced by common DNA variations, leading to the development of a putative genetic model.58 Recent data suggest that the higher prevalence of high-risk disease in Black and Native American patients with neuroblastoma may be associated with certain genetic variants found more commonly in these ethnic groups.59
One method for detecting CNVs is by comparative genomic hybridization (CGH). Early CGH studies showed that gain of genetic material on the long arm of chromosome 17 (17q) is perhaps the most common genetic abnormality in neuroblastomas, occurring in approximately 75% of primary tumors.60 It is unclear at this time how extra copies of 17q contribute to the malignant phenotype of neuroblastoma and which gene(s) on 17q are the critical ones. Nevertheless, gain of chromosome 17q is strongly associated with other known prognostic factors, but it may also be a powerful predictor of adverse outcome.61 More recently, GWAS studies have shown that inherited CNV at chromosome 1q21.1 is associated with neuroblastoma, implicating a neuroblastoma breakpoint family gene in early neuroblastoma genesis.62
Other studies have revealed that common genetic variation at chromosome bands 6p2263 and 2q3564 are associated with susceptibility to high-risk neuroblastoma, providing the first evidence that childhood cancers can also arise from complex interactions of polymorphic variants. More recently, a GWAS study has identified common polymorphisms including germline SNP risk alleles and somatic copy number gain, resulting in increased expression of the cysteine-rich transcriptional regulator LIM domain only 1 (LMO1) at 11p15.4. These have been shown to be strongly associated with susceptibility to developing neuroblastoma, and often are associated with advanced disease and poor survival.65
Finally, whole-genome sequencing of tumors, made possible recently by significant advances in technology, has been performed to investigate the genetic landscape of a variety of pediatric tumors.66 Initially it was felt that early alterations of genes such as MYCN may underlie the rapid acquisition of cooperating mutations in key cancer pathways through chromosome instability. However, few recurring amino acid changes have been detected in neuroblastoma specimens, suggesting that the tumor genomes were more stable than previously believed.44,67 Also, unlike the genetic landscape, the epigenetic profiles showed profound changes which suggests that epigenetic changes may have a more dominant role in pediatric tumorigenesis.
Clinical Presentation
Patients with neuroblastoma usually present with signs and symptoms that reflect the primary site and extent of disease, although localized disease is often asymptomatic. As 75% of neuroblastoma occurs in the abdominal cavity, an abdominal mass detected on physical examination is a common clinical feature, as is the complaint of abdominal pain. Other primary sites of neuroblastoma include the posterior mediastinum (20%), the cervical region (1%), and the pelvis (4%) (organ of Zuckerkandel) (Fig. 66-3). Respiratory distress or dysphagia may be a reflection of a thoracic tumor. Altered defecation or urination may be caused by mechanical compression from a pelvic tumor or by spinal cord compression by a paraspinal tumor. Spinal cord compression may also present as an altered gait. A tumor in the neck or upper thorax can produce Horner syndrome (ptosis, miosis, and anhydrosis), enophthalmos, and heterochromia of the iris. Acute cerebellar ataxia has also been observed, characterized by the dancing-eye syndrome, which includes opsoclonus, myoclonus, and chaotic nystagmus. Two-thirds of these cases occur in infants with mediastinal primary tumors.68,69 Additional signs and symptoms that reflect excessive catecholamine or vasoactive intestinal polypeptide (VIP) secretion include diarrhea, weight loss, and hypertension.
FIGURE 66-3 Primary sites for neuroblastoma are depicted in this anatomic drawing. (Reprinted from Davidoff AM. Neuroblastoma. In: Oldham KT, Colombani PM, Foglia RP, et al, editors. Principles and Practice of Pediatric Surgery. Philadelphia: Lippincott Williams & Wilkins; 2005.)
More than 40% of patients have metastatic disease at diagnosis. These patients are often quite ill and have systemic symptoms caused by widespread disease. Neuroblastoma in older patients has a pattern of metastatic disease in which metastases to the bone marrow, lymph nodes, and bone predominate. The frequency of involvement of distant sites is shown in Table 66-2. These metastases may manifest as bone pain from cortical metastases or anemia from marrow infiltration. The brain, spinal cord, heart, and lungs are rare sites of metastases, except with end-stage disease. Metastatic disease may be also associated with darkened eyes, referred to as ‘raccoon eyes,’ as a result of retroorbital venous plexus spread (Fig. 66-4A). This is an ominous physical sign, as is the presence of a limp in children without a history of head or extremity trauma. Infants with metastatic neuroblastoma can have stage 4S disease, which, by definition, is a localized primary tumor in patients younger than 1 year, with dissemination limited to skin, liver, or bone marrow (<10% of nucleated cells). These patients may present with ‘blueberry muffin’ cutaneous lesions (Fig. 66-4B), respiratory distress secondary to massive hepatomegaly, and anemia secondary to bone marrow disease. The diagnosis of neuroblastoma is generally made by histopathologic evaluation of primary or metastatic tumor tissue, or by the demonstration of tumor cells in the bone marrow together with elevated levels of urinary catecholamines.
TABLE 66-2
Sites of Metastases at Diagnosis for Patients with Evans Stage IV-S and Stage IV
Adapted from Dubois SG, Kalika Y, Lukens JN, et al. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. Pediatr Hematol Oncol 1999;21:181–9.
FIGURE 66-4 Clinical evidence of metastatic neuroblastoma. (A) ‘Raccoon eyes,’ characteristic of metastatic neuroblastoma in the posterior orbital venous plexus, are seen in a child with stage IV disease. (B) ‘Blueberry muffin’ spot (arrow) in the skin, characteristic of metastatic neuroblastoma, is seen in the suprapubic region of an infant with a 4S neuroblastoma. (Courtesy of Stephen Shochat, MD, St. Jude Children’s Research Hospital, Memphis, TN).
Laboratory Findings
Lactate Dehydrogenase
Despite its lack of specificity, serum lactate dehydrogenase (LDH) can have great prognostic significance. High serum levels of LDH reflect high proliferative activity or large tumor burden, and an LDH level higher than 1500 IU/L appears to be associated with a poor prognosis.70,71 Thus, LDH can be used to monitor disease activity or the response to therapy.
Ferritin
High levels of serum ferritin (>150 ng/mL) may also reflect a large tumor burden or rapid tumor progression. Elevated serum ferritin is often seen in advanced-stage neuroblastomas and indicates a poor prognosis.72 Levels often return to normal during clinical remission.
Neuron-Specific Enolase
Neuron-specific enolase (NSE) is another useful prognostic marker of advanced-stage neuroblastoma. The incidence of elevated NSE levels increases with stage.73 A serum level of NSE >100 ng/mL is associated with a poor outcome. NSE has been reported to correlate with tumor burden, suggesting its reliability as a marker of disease course.74
Catecholamine Metabolites
Neuroblastoma is characterized by the relatively unique capacity for secretion of catecholamine products, the metabolites of which can be detected in the urine of more than 90% of patients with neuroblastoma. Thus, a urine specimen is of clinical value in diagnosing neuroblastoma and determining the response to therapy. Documentation of elevated urinary catecholamines is required if the diagnosis of neuroblastoma is being made solely by the identification of neuroblasts in the bone marrow. Urinary levels of these two catabolites can also be used as markers of tumor progression or relapse, and serve as a surrogate prognostic indicator. Random urine samples are preferable to 24-hour urine estimations for younger children.75
Diagnostic Imaging
Standard Radiographs
Chest radiography can be a useful tool for demonstrating the presence of a posterior mediastinal mass, which in a child is usually a thoracic neuroblastoma. A Pediatric Oncology Group (POG) study demonstrated that a mediastinal mass was discovered on incidental chest radiographs in almost half of patients with thoracic neuroblastoma who had symptoms seemingly unrelated to their tumors.76 Abdominal radiography is less often the modality by which a neuroblastoma is discovered; however, as many as half of abdominal neuroblastomas are detectable as a mass with fine calcification.
Ultrasonography
Although ultrasonography (US) is the modality most often used during the initial assessment of a suspected abdominal mass, its sensitivity and accuracy are less than that of computed tomography (CT) or magnetic resonance imaging (MRI) for diagnosing neuoblastoma.77 These latter modalities are generally used after screening with ultrasound to assist in generating a differential diagnosis and for further anatomic definition once the presence of a mass has been confirmed.
Computed Tomography
CT can demonstrate calcification in almost 85% of neuroblastomas, and intraspinal extension of the tumor can be determined on contrast-enhanced CT.78 Overall, contrast-enhanced CT has been reported to be 82% accurate in defining neuroblastoma extent, with the accuracy increasing to nearly 97% when performed with a bone scan.79 Although some consider CT to have been supplanted by MRI, others still consider it to be the image modality of choice for patients with neuroblastoma, especially when used in conjunction with bone scintigraphy.80
Magnetic Resonance Imaging
MRI is becoming the most useful and most sensitive imaging modality for the diagnosis and staging of neuroblastoma.77,81 MRI appears to be more accurate than CT for detection of stage 4 disease. The sensitivity of MRI is 83%, and that of CT is 43%, and the specificity of MRI is 97%, and that of CT is 88%.81 Metastases to the bone and bone marrow, in particular, are better detected by MRI, as is intraspinal tumor extension (Fig. 66-5).81 When considering skeletal metastases alone, MRI and bone scan have been shown to be equivalent.81 Encasement of major vessels can be better defined by MRI than CT, especially with the use of MR angiography (see Fig. 66-5). MRI in the coronal plane is suitable for routine assessment of the whole body from the neck to the pelvis. Evaluating the utility of whole-body MRI, perhaps performed in conjunction with a functional imaging study such as positron-emission tomography (PET), is being considered for future clinical staging studies. CT and MRI are not very accurate for staging localized disease; however, the sensitivity of T1- and T2-weighted MRIs is 100% for detecting neuroblastomas in infants identified by mass screening.65
FIGURE 66-5 These MR images highlight several characteristics of high-risk neuroblastoma. (A) Bone metastasis in femur (arrow). (B) Bone marrow metastases in the vertebral bodies. (C) Intraspinal tumor extension (dotted arrow). Note displacement of spinal cord (solid arrow) from a large tumor (asterisk). (D) Encasement of major intra-abdominal vessels (arrow points to the aorta and left renal artery).
Metaiodobenzylguanidine Imaging
Metaiodobenzylguanidine (MIBG) is transported to and stored in the chromaffin cells in the same way as norepinephrine. The MIBG scintiscan is the preferred imaging study for evaluating the bone and bone marrow involvement by neuroblastoma (Fig. 66-6), having largely replaced technetium-99m methylene diphosphonate (99mTc-MDP) bone scans, which are generally inferior to MIBG in detecting skeletal or extraskeletal involvement. In addition, monitoring MDP-avid neuroblastomas by bone scintigraphy often results in false-positive imaging for months after tumor remission. Thus, 99mTc-MDP bone scanning is a second choice if MIBG imaging is not available or does not visualize known disease.82,83 Iodine-131 (131I) or iodine-123 (123I) can be used to label MIBG. 123I-MIBG supplies a reduced absorbed radiation dose and superior spatial resolution.84 The reported sensitivity of MIBG in the detection of neuroblastomas with metastases to the bone and bone marrow is 82%, and the specificity is 91%.85 Primary tumors and lymph node metastases are also detectable. MIBG can demonstrate more sites of tumor involvement in bone and bone marrow than either bone scintigraphy or standard radiography.85 However, false-negative MIBG scans have been seen in cases in which the bone scintigraphy was positive.83
FIGURE 66-6 Imaging of neuroblastoma with MIBG scintigraphy. (A) Scan obtained at presentation of a patient with metastatic neuroblastoma. There is diffusely abnormal activity throughout much of the skeleton including the proximal right humerus, both proximal and distal femurs, and the proximal right tibia. There is also a focus of activity in the right upper retroperitoneum at the site of the primary tumor. (B) Scan obtained of the same patient after completion of therapy shows no scintigraphic evidence of MIBG-avid neuroblastoma.
