[level-membership-for-opthalmology-category]
19 Neuro-ophthalmology
OCULAR MOVEMENT
SUPRANUCLEAR GAZE CONTROL
Saccadic movements are rapid and relocate fixation of gaze, either reflexly or voluntarily. They are initiated in the contralateral premotor frontal cortex and, once initiated, the movement is irrevocable and ocular position cannot again be modified until the saccade has been completed. A saccade occurs after a latent period of about 200 ms following initiation and has a high velocity of up to 700°/sec. Saccades are tested clinically by instructing the patient to look first at one stationary target and then at another, or to look right and left or up and down with no target present (see Fig. 19.4).
Pursuit movements are slower and are concerned with keeping the target at the fovea. They appear to be generated in the ipsilateral occipital cortex but little is known about the supranuclear pathway. Pursuit movements have a latency of about 125 ms from initiation and a maximum velocity of less than 50°/sec. The movement is smooth and modified continuously according to the speed of the target if the pursuit movement lags behind the target position a corrective saccade is inserted to keep up. Pursuit movements are tested by asking the patient to follow a slowly moving target. It is of great importance that the target is clearly visible and that it is not moved too fast (see Fig. 19.5).
Vestibular ocular movements may be tested by a ‘doll’s head’ manoeuvre, in which the patient is asked to fixate on a target while the examiner rotates the patient’s head (see Fig. 19.6). The doll’s head manoeuvre tests both the left and right labyrinths and may be normal even if one labyrinth is totally nonfunctioning. To test each labyrinth separately, caloric stimulation can be employed which induces nystagmus by syringing the external auditory meatus with cold or warm water. Recently a bedside test has been described, based on the observation that the initial component of a rapid head rotation depends upon the integrity of the labyrinth towards which the head is turned (see Fig. 19.8).
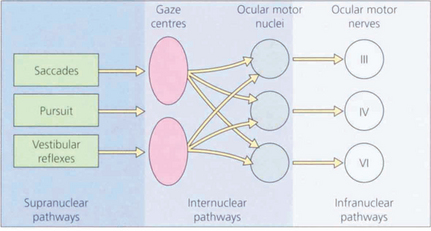
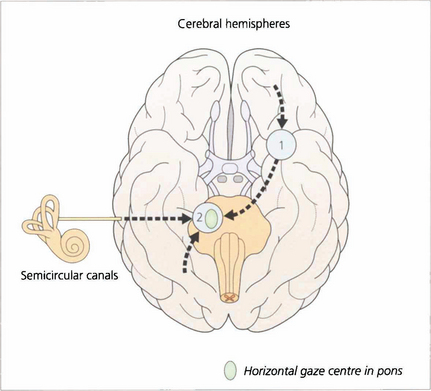
Fig. 19.1 The major inputs to the vertical and horizontal gaze centres are saccadic and pursuit commands from the cerebral hemispheres and the vestibular ocular reflexes from the vestibular nuclei in the medulla. These impulses are then transmitted to the individual ocular motor nuclei in the midbrain and pons. Internuclear pathways coordinate the movements generated in the gaze centres with each other and with the third, fourth and sixth nerve nuclei. Excitatory impulses to an agonist muscle are accompanied by inhibitory impulses to its antagonist.
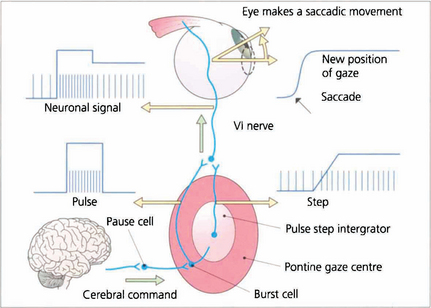
Fig. 19.2 Saccadic movements have been shown biomechanically to be generated by impulses in the form of a pulse and step. To change eye position a rapid burst of neuronal signals (the pulse) is fired by the gaze centre and this rapidly moves the eye to the new position. This rapid burst is integrated in the ‘pulse-step generator’ (a neuronal complex within the gaze centre) to produce an increased tonic discharge (the step) which holds the eye in its new position. Similar responses take place in the agonist of the fellow eye and reciprocal impulses relax the antagonist muscles. These changes are reflected by the EMG recording in an individual muscle. Neurophysiologically saccades are initiated by burst cells in the paramedian pontine reticular formation (PPRF) which are inhibited from firing by pause cells at all times except during a saccade. Thus the saccadic input suppresses the pause cell which permits the burst cell to discharge immediately.
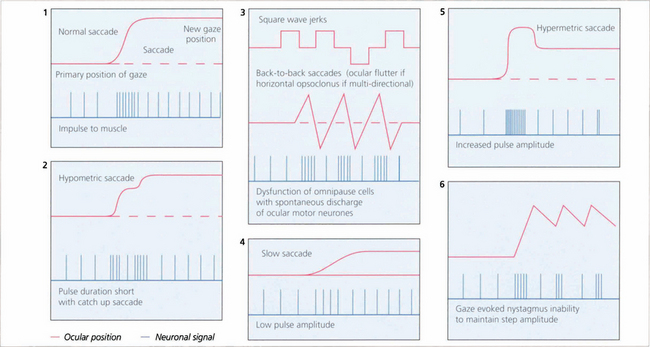
Fig. 19.3 The pulse–step generator exists physiologically in the horizontal gaze centre—otherwise known as the paramedian pontine reticular formation (PPRF)— in the pons and various mismatches of the pulse to step signals have been shown to explain some types of nystagmus and eye movement abnormalities seen in clinical practice.
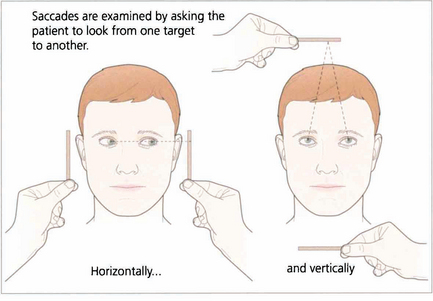
Fig. 19.4 Saccadic eye movements can be elicited simply by asking patients to look right and left or up and down. This is a useful strategy where the observation of large amplitude saccades may reveal subtle abnormalities of saccadic velocity. An alternative strategy is to ask patients to shift fixation from one target to another, in this way saccades of large and small amplitude and involving a variety of gaze shifts can be observed. The accuracy of saccades (under- or over-shooting) can be tested. Slowing of the velocity of saccades may be conjugate (as in a gaze palsy), dysconjugate (as in a lateral rectus paresis or internuclear ophthalmoplegia) or global (as in an ocular myopathy or some forms of cerebellar disease).
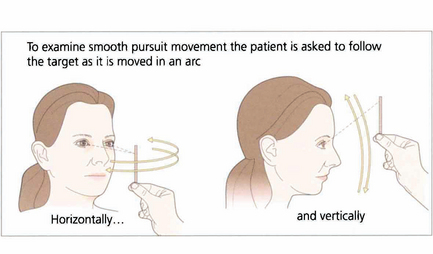
Fig. 19.5 When asking a subject to follow a target it is essential that the target is clearly visible and is moved neither too slowly nor too rapidly. The reader should experiment by trying to follow his or her own finger as it is moved in a 180° arc from one side of the head to the other at arm’s length. A comfortable speed is about 30° per seconds (6 seconds from ear to ear). Pursuit movements are used to examine ductions and versions but, instead of looking at the range of movement, we are now interested in the accuracy of the following movement itself. The observer must watch for saccadic intrusions which will indicate that the eye is not keeping up with the target (in other words, there is reduced gain of the smooth pursuit reflex). This is a universal finding in cerebellar disease.
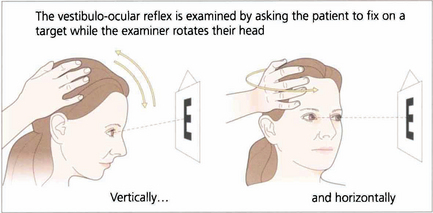
Fig. 19.6 Vestibular ocular reflexes (VORs) are tested by asking the patient to fixate a distant target (such as an acuity chart) and then gently moving the patient’s head from side to side or up and down with increasing frequency until the patient reports movement of the target or the subjective acuity falls, indicating retinal slip of the image. Alternatively a retinal landmark can be fixed using the direct ophthalmoscope while the examiner rocks the head to and fro. Loss of the VOR causes symptoms such as instability of the horizon as the patient walks and is seen not uncommonly with multiple sclerosis.
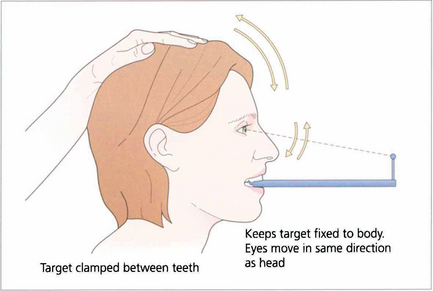
Fig. 19.7 Suppression of the VOR can be tested by asking the patient to grip a target mounted on a spatula between the teeth and to move the head from side to side or up and down with increasing rapidity until the patient reports an inability to keep the image ‘still’ or saccadic eye movements are observed as fixation is lost and taken up again. The mechanism used to suppress the VOR is the pursuit mechanism; if smooth pursuit is abnormal then so will be VOR suppression, and vice versa. Patients who are able to suppress their VOR complain of unusual symptoms such as difficulty with reading when travelling.
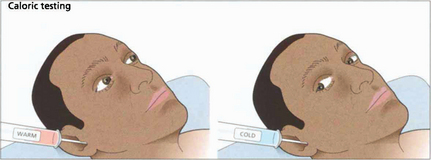
Fig. 19.8 Only one functioning labyrinth is required for the tests described in Fig. 19.6 to be normal. Caloric testing can be used to examine each labyrinth separately. Caloric reflexes are produced by stimulating the semicircular canals and vestibular nuclei with warm or cold water and can provide useful information on the integrity of these pathways in the brainstem; this is especially useful in the neurological assessment of brainstem damage in an unconscious patient. In a conscious patient cold water in the external auditory meatus generates a nystagmus of both eyes with a fast phase to the opposite side but in unconscious patients the saccadic phase is lost and a tonic deviation to the same side is seen. This indicates an intact pons but the results must be interpreted with caution after acute drug overdoses when false-negative responses may be seen. Calorics can be adapted to test vertical gaze (and therefore the integrity of the midbrain) by syringing both ears.
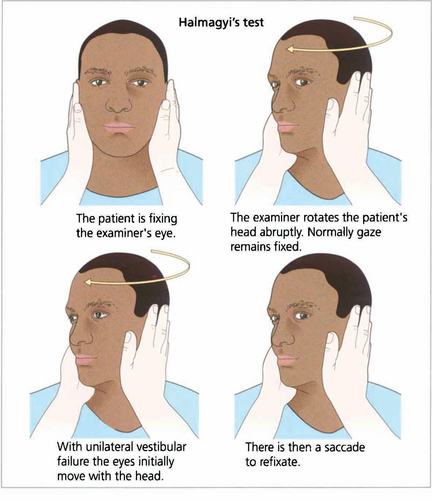
Fig. 19.9 Unilateral labyrinthine function can be tested at the bedside. If the head is rotated abruptly, the initial phase of the VOR is mediated by the labyrinth towards which the head is turned. If that labyrinth is not functioning initially the eyes move with the head and there is a corrective saccade as fixation is taken up again.
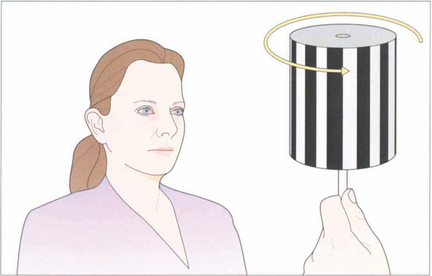
Fig. 19.10 The vestibular system responds only to acceleration and slow head movements (less than 1° per second) stimulate the optokinetic reflex (OKN). The hand-held OKN drum is useful as a bedside test of the pursuit and saccadic systems. If there is a saccadic palsy the eyes will deviate tonically in the direction of the stripe movement: the pursuit mechanism has been elicited but there is no saccade to return gaze towards primary position. Turning the drum to the right elicits an ipsilateral pursuit movement to the right and a contralateral saccade to the left. In essence turning the drum to the right tests the right hemisphere and vice versa to the left.
ANATOMY OF THE OCULAR MOTOR PATHWAYS
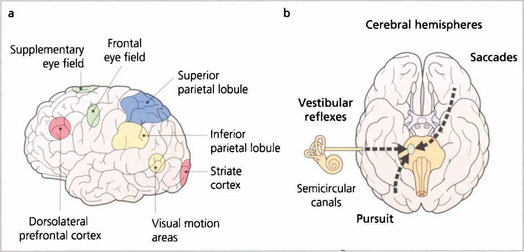
Fig. 19.11 Saccades are generated in a number of areas in the frontal premotor area of the contralateral hemisphere including the frontal eye fields; thus a saccade to the right is generated in the left frontal lobe, passes through the anterior limb of the internal capsule and decussates in the midbrain to the horizontal gaze centre in the pons. The incoming visual information that controls smooth pursuit is analysed in an occipitoparietal motion area. Pursuit movements are then generated in the ipsilateral superior parietal lobule so that a pursuit movement to the right is controlled by the right hemisphere. Little is known of the neuronal pathway, but the final common pathways for saccades and pursuit are the conjugate horizontal and vertical gaze mechanisms in the pons and midbrain. The inferior parietal lobule may be concerned with shifts of attention, which may also generate saccades.
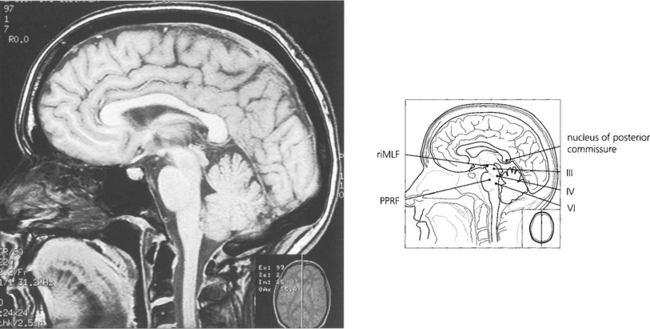
Fig. 19.12 The final common pathway for all horizontal movements are the sixth nerve nuclei to which the PPRF, pursuit mechanisms and vestibular nuclei project. Interneurones in the sixth nerve nucleus project through the medial longitudinal fasciculus (MLF) to the contralateral medial rectus subunit of the oculomotor nucleus. For vertical saccades, the rostral interstitial nucleus of the MLF (riMLF) projects to the third and fourth nerve nuclei. Vestibular inputs controlling vertical eye movements ascend from the lower brainstem to the fourth and third nerve nuclei via the MLF. The nucleus of the posterior commissure projects on to both riMLFs and is particularly important for upgaze.
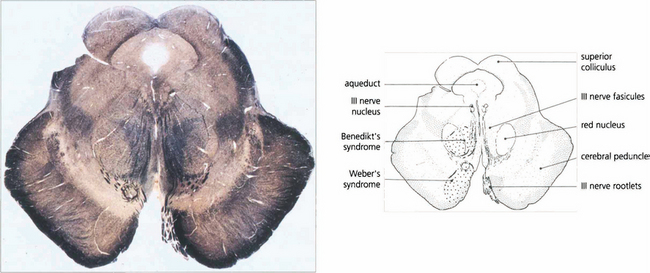
Fig. 19.13 A transverse histological section of the midbrain at the level of the superior colliculus demonstrates the third nerve nucleus inferior to the aqueduct with its efferent fibres. The medial longitudinal fasciculus (MLF) in connecting the third and fourth nerve nuclei and contralateral horizontal gaze centres is responsible for coordinating horizontal and vertical movements. Infarcts in this region cause Weber’s syndrome (unilateral third nerve palsy and contralateral hemiplegia) and Benedikt’s syndrome (unilateral third nerve palsy, contralateral tremor and hyperaesthesia). (Stained to show myelin.)
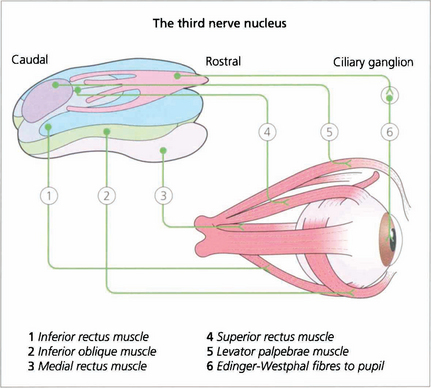
Fig. 19.14 The third nerve nucleus is a complex assemblage of motor neurones. The parasympathetic nucleus (Edinger–Westphal nucleus) of the pupil lies rostrally and superiorly. The inferior rectus, medial rectus and inferior oblique muscles are all supplied from ipsilateral subnuclei, but the superior rectus subnucleus is contralateral. Both levator palpebrae muscles have a single caudal superior nucleus. A complete ‘nuclear’ unilateral third nerve palsy is therefore characterized by ipsilateral inferior oblique, medial and inferior rectus weakness with bilateral ptosis and contralateral superior rectus weakness.
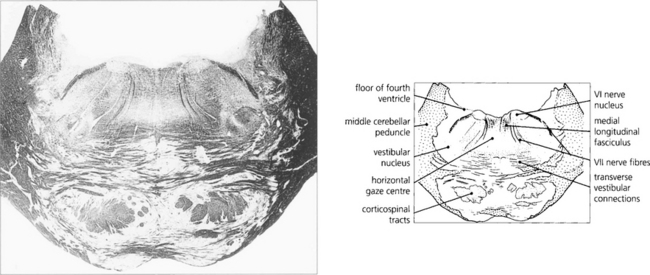
Fig. 19.15 A transverse histological section through the pons at the level of the sixth nerve nuclei demonstrates the medial longitudinal fasciculus (MLF) and the centre for horizontal gaze (PPRF). Interneurones from the PPRF pass initially to the sixth nerve nucleus where they synapse with the MLF (see Fig. 19.34). (Stained to show myelin.)
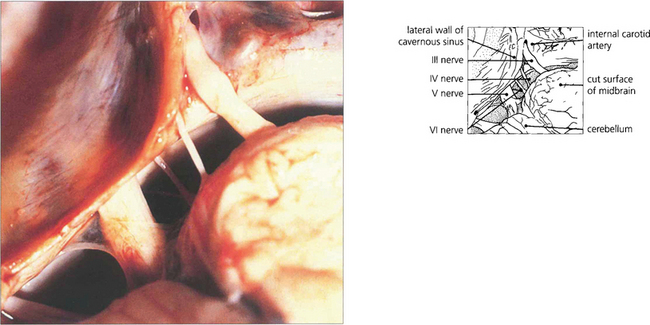
Fig. 19.16 The third and sixth nerves leave the ventral surface of the brain and pass through the interpeduncular cistern before entering the cavernous sinus. The sixth nerve passes almost vertically upwards and bends forwards under the petroclinoid ligament at the apex of the petrous temporal bone. Here it can be compressed against the bone by raised CSF pressure or damaged by fractures or disease in the bone (hence producing the sixth nerve palsy associated with raised intracranial pressure). The fourth nerve leaves the midbrain dorsally, decussates in the superior medullary velum and passes through the subarachnoid space around the midbrain between the superior cerebellar and posterior cerebral arteries before entering the cavernous sinus.
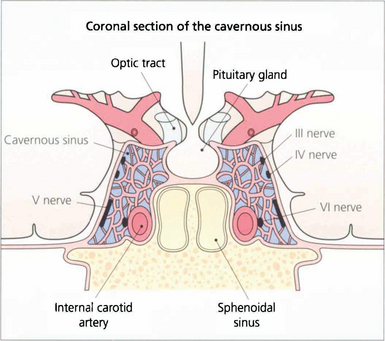
Fig. 19.17 In the cavernous sinus the third and fourth nerves lie superiorly in the lateral wall; at the anterior end the third nerve divides into superior (superior rectus and lid levator) and inferior (inferior and medial rectus, inferior oblique, pupillary and ciliary muscles) branches. Sensory fibres from the eye and forehead enter the cavernous sinus anteriorly in the ophthalmic division of the fifth nerve, lying inferiorly in the lateral wall to join the trigeminal ganglion at the posterior limit. The internal carotid artery and sixth nerve lie within the sinus.
CONJUGATE GAZE PALSIES
HORIZONTAL SUPRANUCLEAR PALSY
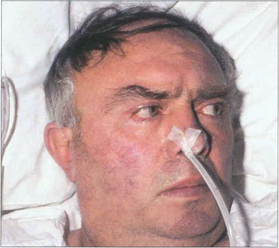
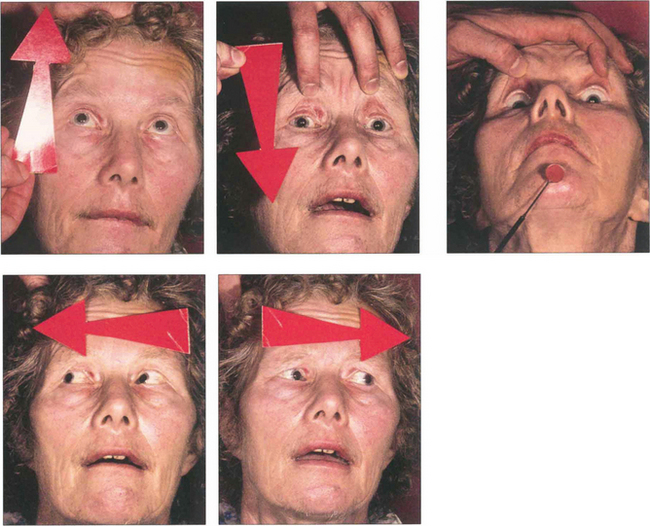
Fig. 19.18 Following an acute cerebrovascular accident involving supranuclear pathways, saccadic gaze is lost in the direction opposite to the side of the lesion and input from the remaining hemisphere deviates the eyes towards the side of the lesion. (Occasionally, irritative lesions, such as cerebral tumours or abscesses, produce deviation of the eyes to the contralateral side.)
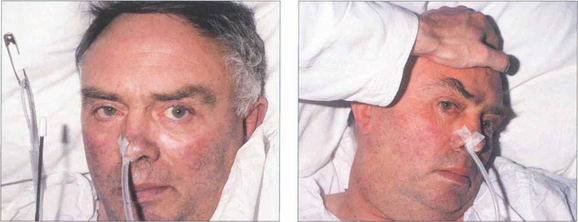
Fig. 19.19 (Left) On command the patient is unable to make a gaze movement to the target on his right. (Right) On doll’s head rotation to the left, however, the eyes deviate fully to the right demonstrating intact pontine reflexes and the supranuclear nature of the lesion.
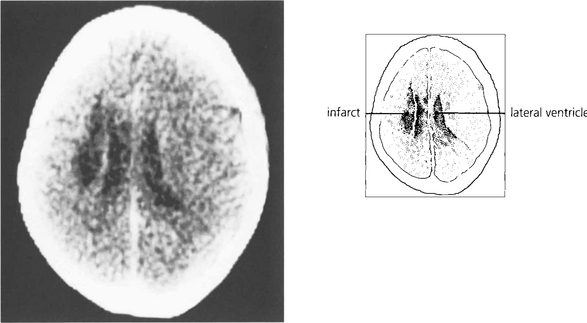
Fig. 19.20 A CT scan of the patient seen in Fig. 19.18, showing a large infarct in the internal capsule due to the occlusion of middle cerebral artery branches, corresponding to lesion 1 in Fig. 19.21. If the patient survives the acute ocular deviation recovers rapidly. Horizontal gaze becomes full and saccades return to normal. This is probably due to a restoration of control mechanisms by the superior colliculus.
VERTICAL GAZE PALSY
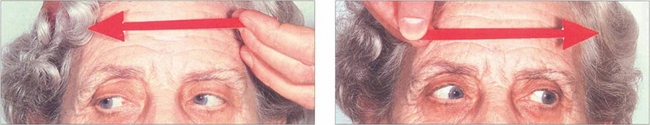
Fig. 19.22 This elderly woman suffered a cerebrovascular accident in the upper midbrain. She retained the ability to make rapid and full horizontal saccades.
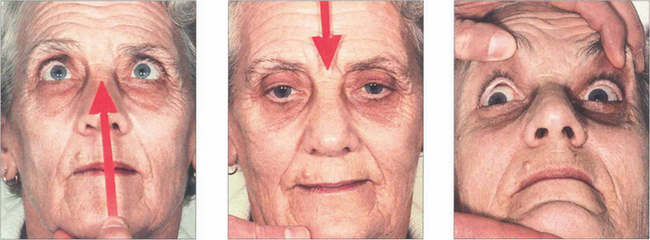
Fig. 19.23 In the same patient, voluntary up and down gaze to saccadic command is lost and compensated for by head movement. ‘Doll’s head’ rotation shows that vertical movements can be produced and that pontine reflexes are intact.

Fig. 19.24 Convergence retraction nystagmus is an interesting ocular movement abnormality sometimes seen with vertical gaze palsies. Bursts of convergence with retraction of both eyes into the orbits are seen either spontaneously or on attempting an upward saccade. These movements may also be accompanied by accommodative spasm which accounts for the frequent complaint of blurred vision. Convergence retraction nystagmus is shown particularly well if the patient views a downward rotating optokinetic drum which stimulates upward refixational saccades.
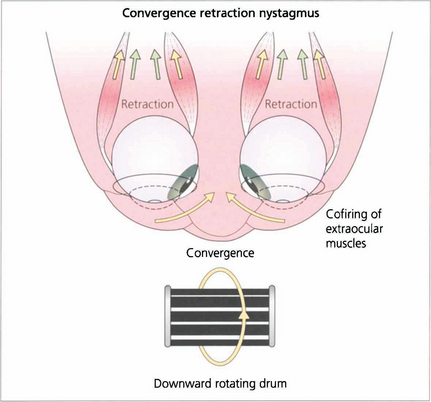
Fig. 19.25 In convergence retraction nystagmus the lesion is thought to cause disinhibition of the ocular motor nuclei allowing bursts of co-firing of the external ocular muscles. The medial recti muscles are the most powerful of the external ocular muscles and consequently the globes converge and retract into the orbits.
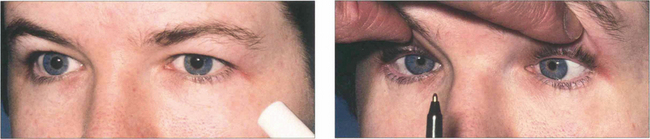
Fig. 19.26 Light–near dissociation of the pupils is the third sign of the triad of Parinaud’s syndrome. The pupils are normally moderately dilated and show a poor or absent light reaction but preserve the near reaction. It has been suggested that light reflex fibres are more dorsal in the midbrain and are therefore selectively damaged by lesions in this area with comparative sparing of the more ventrally sited near reflex pathways.
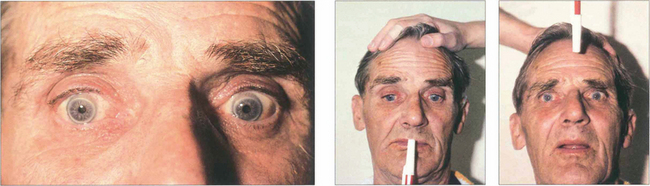
Fig. 19.27 Collier’s sign is a bilateral retraction of the upper eyelids sometimes seen with acute upper midbrain lesions. This elderly man was initially referred with a diagnosis of ‘acute dysthyroid eye disease’. He had, in fact, suffered a cerebrovascular accident of the midbrain region. Horizontal gaze was entirely normal but there was a partial vertical gaze palsy and retraction of both upper eyelids. The patient had dubious long tract signs. The vertical gaze palsy and lid retraction rapidly recovered over 2–3 weeks.
INTERNUCLEAR OPHTHALMOPLEGIA
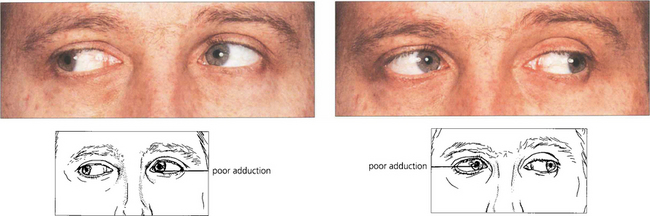
Fig. 19.29 Bilateral internuclear ophthalmoplegia in young people is almost always associated with demyelination from multiple sclerosis. Vertical nystagmus is usually present on upgaze and there may be a skew deviation. In this patient, the skew deviation alternates, there being a left-over-right on right gaze and a right-over-left on left gaze. Subtle lesions can be demonstrated by asking the patient to make rapid horizontal movements to show the slowness of adduction in each eye.
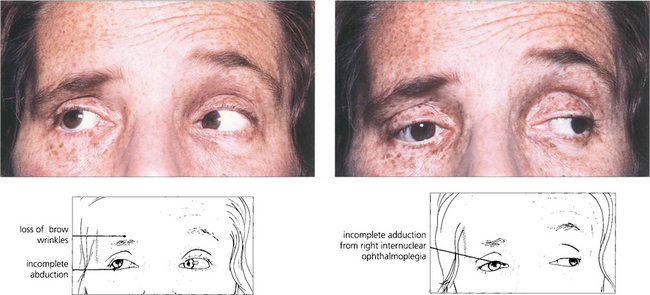
Fig. 19.30 Unilateral internuclear ophthalmoplegia is more frequently due to a vascular or ischaemic lesion as the basilar artery supplies the pons by lateralizing perforating branches. This hypertensive patient shows a right-sided lower motor neurone seventh nerve palsy, internuclear ophthalmoplegia and partial sixth palsy following a pontine infarct.
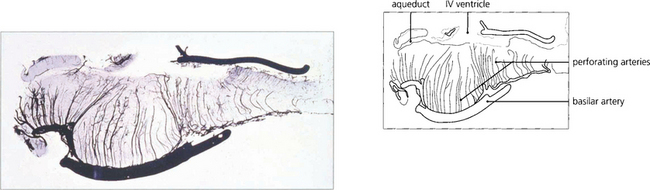
Fig. 19.31 The basilar artery supplies the pons by lateralizing perforating branches. Occlusion of one of these produces an unilateral INO.
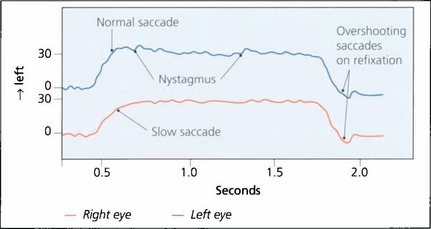
Fig. 19.32 In this recording of a right unilateral internuclear ophthalmoplegia a 30° saccadic movement to the left demonstrates slow adduction of the right eye and abducting nystagmus of the left. Return to the primary position shows normal saccadic velocities in each eye but hypermetric overshooting saccades are seen, which are then corrected to hold the primary position of gaze. Such overshooting saccades are a common feature of internuclear ophthalmoplegia and can be recognized clinically.
By courtesy of Dr M Gresty.
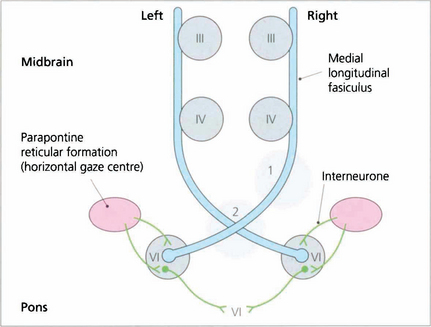
Fig. 19.33 A ‘wiring diagram’ shows that lesion 1 produces a right unilateral internuclear ophthalmoplegia whereas lesion 2 causes bilateral internuclear ophthalmoplegia. Note that interneurones from the PPRF pass into the sixth nerve nucleus and synapse with interneurones that travel in the MLF to innervate the contralateral medial rectus subunit of the oculomotor nucleus
THE ‘ONE AND A HALF’ SYNDROME (PARALYTIC PONTINE EXOTROPIA)
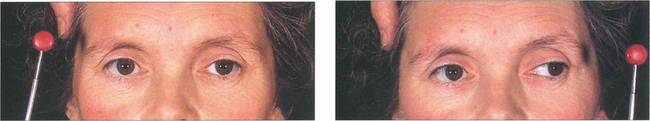
Fig. 19.34 This patient with multiple sclerosis shows a right gaze palsy and right internuclear ophthalmoplegia, with only abduction of the left eye on horizontal gaze remaining. Vertical movements were intact.
By courtesy of Mr M D Sanders.
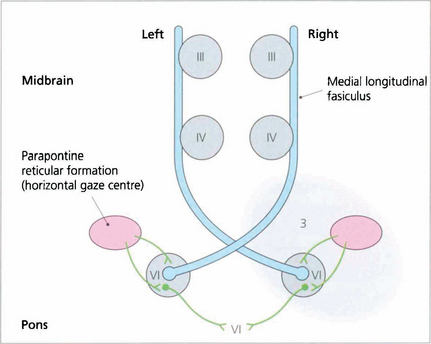
Fig. 19.35 A wiring diagram shows that lesion 3 prevents stimulation of the right PPRF producing a right pontine gaze palsy. Impulses from the cortex to the left PPRF result in stimulation of the left sixth nucleus and abduction of that eye but stimulation of the right medial rectus through the MLF is blocked. Complete ‘1½ syndromes’ are uncommon but the combination of a partial gaze palsy with partial ipsilateral internuclear ophthalmoplegia is not uncommon and is readily observed by asking the patient to attempt rapid horizontal saccadic movements.
THE PUPIL
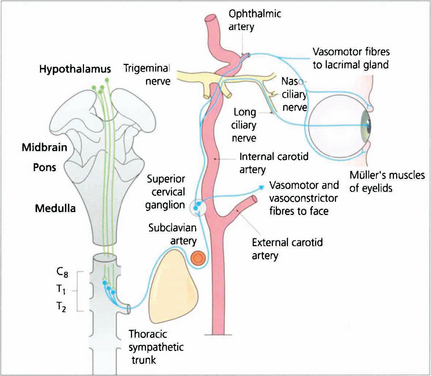
Fig. 19.36 Sympathetic impulses are generated in the region of the hypothalamus and transmitted down the spinal cord to synapse at the ciliospinal centre in the lateral grey column. Pupillary impulses leave the cord in myelinated preganglionic fibres at T1 (some also at C8 and T2) and pass upwards in the sympathetic chain to synapse in the superior cervical ganglion lying at the level of the C1 and C2 vertebrae. Nonmyelinated postganglionic fibres form a plexus on the common carotid artery; vasomotor fibres to the face leave on the external carotid artery at the bifurcation. The internal carotid artery carries sympathetic innervation into the cavernous sinus where the pupillary fibres join the nasociliary branch of the ophthalmic division of the fifth nerve and enter the eye by two or three long ciliary nerves at the optic disc to supply the dilator pupillae muscle. Other sympathetic branches are taken by branches of the ophthalmic artery to the lacrimal glands, Müller’s muscles and the orbital vessels.
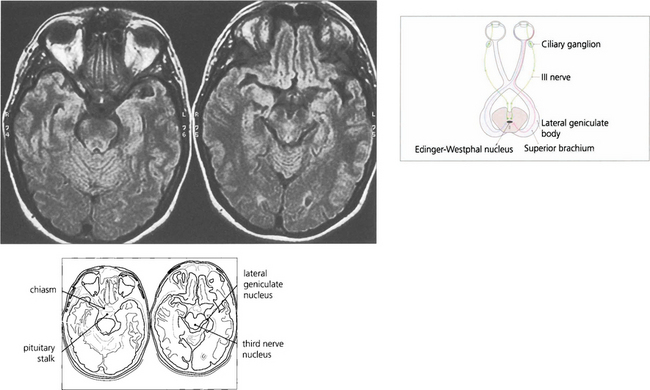
Fig. 19.37 The afferent pupillary light reflexes are mediated through axons from ganglion cells in the retina that pass back in the optic nerve and decussate in the chiasm with the other visual fibres. The pupillary fibres pass through the optic tract and superior brachium to the Edinger–Westphal nucleus of the third nerve; here, they synapse to produce a simultaneous and bilateral response in each third nerve through interneuronal connections. Efferent parasympathetic preganglionic axons run forward in each third nerve and pass into the inferior division at the anterior aspect of the cavernous sinus to the ciliary ganglion, where they synapse to supply the constrictor pupillae by the short ciliary nerves. The ratio of light fibres to accommodative fibres in the third nerve is said to be 1:30.
RELATIVE AFFERENT PUPILLARY DEFECT
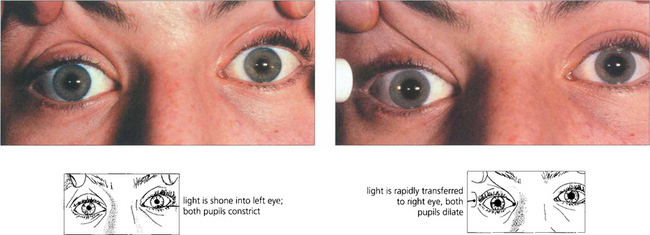
Fig. 19.38 A right afferent pupillary defect is seen in this patient with retrobulbar neuritis of the right optic nerve. Stimulation of the left eye produces bilateral pupillary constriction. Transfer of the light to the right eye produces a relative dilatation of the pupil in both eyes. If one pupil is damaged or paralysed, an afferent pupillary defect can still be diagnosed by observing whether the size of the response varies in the functioning pupil with alternate stimulation of the two eyes. The density of an afferent defect can be quantified by placing neutral density filters of increasing darkness in front of the good eye until the pupillary responses are balanced.
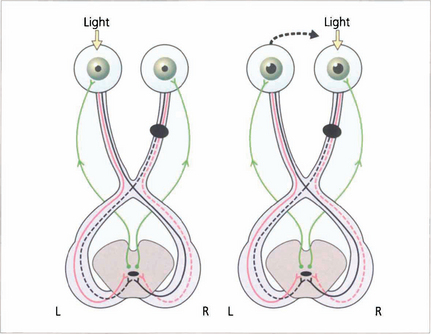
Fig. 19.39 The ‘wiring diagram’ demonstrates the neurological basis of a RAPD. Stimulation of the normal left eye produces brisk bilateral and equal pupillary constriction by the direct and consensual light reflexes. Rapid transfer to the right eye, where there is an optic nerve lesion, removes some relative input on the Edinger–Westphal nucleus and both pupils dilate. Swinging the light from left to right to left to right is the best way to demonstrate RAPD.
ANISOCORIA
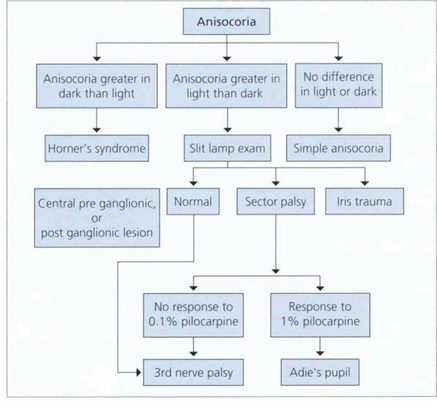
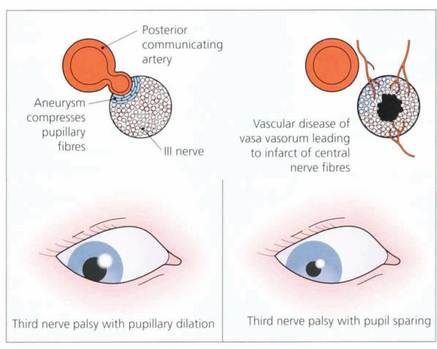
Fig. 19.41 In the interpeduncular cistern the third nerve passes inferior to the posterior cerebral artery and lateral to the posterior communicating artery. At this position the pupillary fibres lie on the dorsomedial aspect of the nerve; they are readily compressed by a posterior communicating artery aneurysm to produce a third nerve palsy with pupillary dilatation. In contrast, vascular disease such as diabetes or hypertension affects the vasa vasorum of the nerve, infarcting central nerve fibres and paralysing the external ocular muscles but sparing the pupil (see Ch. 18).
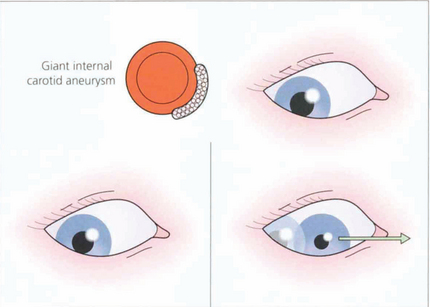
Fig. 19.42 Two less common abnormalities of the pupil associated with oculomotor nerve damage are illustrated here. (Left) If the oculomotor nerve is compressed in the cavernous sinus, for example by a meningioma or giant aneurysm of the intracavernous carotid, there is often the combination of a parasympathetic and sympathetic lesion of the pupil. The pupil is midrange and neither dilates in the dark nor reacts to light. (Right) Finally, with chronic compression there may be aberrant reinnervation of the pupil, which will constrict when an attempt is made to move the eye in the direction of action of one of the muscles innervated by the oculomotor nerve (the drawing illustrates the medial rectus) (see also Ch. 18).
HORNER’S SYNDROME
Pharmacological testing can be used to diagnose and localize the defect in Horner’s syndrome but this has become of less importance with the advent of magnetic resonance imaging (MRI) which gives definitive information. The tests are, however, important because they illustrate basic pharmacological principles (Table 19.1). The diagnosis can be confirmed by instilling G 4.0% cocaine in both eyes. This blocks the reuptake of norepinephrine into the presynaptic vesicles. The pupils are observed 15 min later the normal pupil dilates and eyelids retract as reuptake of norepinephrine at the synapse is blocked by the cocaine. The affected eye has no norepinephrine release to be blocked and so the pupil size does not alter. Failure to dilate to G hydroxyamphetamine which stimulates release of norepinephrine from the presynaptic vesicles demonstrates a postganglionic lesion as the nerve is already depleted of norepinephrine; the importance of this is that preganglionic lesions are often sinister and may accompany lesions such as a Pancoast tumour of the lung.

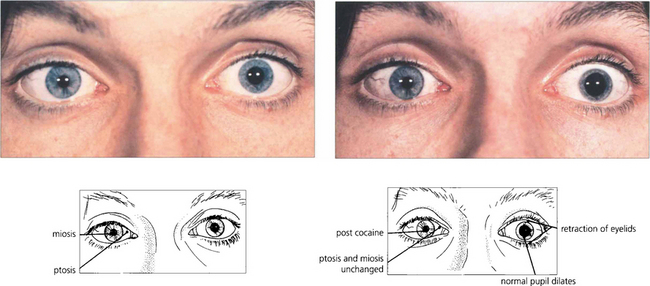
Fig. 19.43 Miosis and ptosis are seen in the affected right eye of a patient with acquired Horner’s syndrome. Some 15 min after a drop of G 4.0% cocaine to each eye the left pupil dilates and the palpebral aperture widens but the right is unchanged confirming the diagnosis. Subsequent failure of the right pupil to dilate with G hydroxyamphetamine 1–2 days later (after the effect of the cocaine has worn off) would show that the localization of the lesion was postganglionic. Horner’s syndrome does not produce more than about 2 mm of ptosis which is due to involvement of Müller’s muscles in the upper lid, involvement in the lower lid gives rise to ‘upside down’ ptosis.
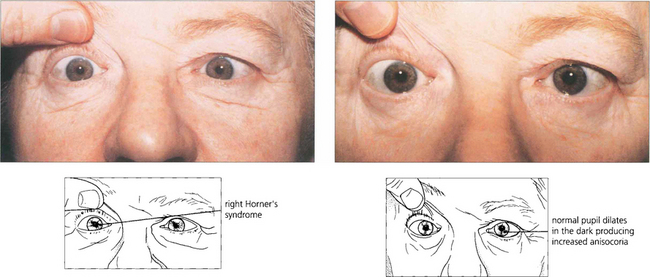
Fig. 19.44 Horner’s syndrome shows greater aniscoria in the dark than in the light as the affected eye fails to dilate. This can be useful as a simple bedside diagnostic test.
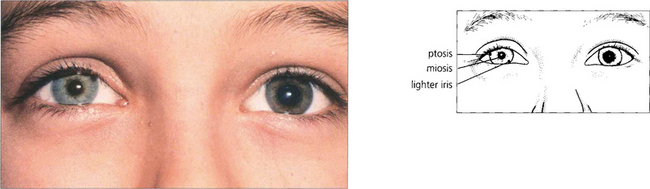
Fig. 19.45 Heterochromia is a feature of congenital or very longstanding Horner’s syndrome, the affected iris being lighter in colour. Maintenance of melanin in the iris stroma (not the pigment epithelium) is dependent on an intact sympathetic supply.
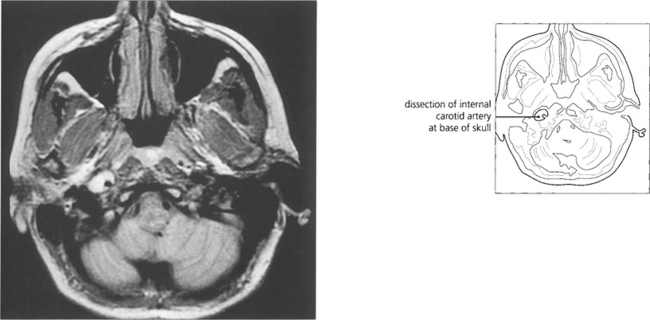
Fig. 19.46 An acute painful Horner’s syndrome may be caused by a carotid dissection which can occur after minimal trauma in susceptible individuals. This is an extremely important diagnosis to make as there is a substantial risk of thromboembolism in the hours and days following the dissection leading to a catastrophic stroke. The diagnosis is made by requesting T2-weighted axial MR images of the skull base. The scan shown here is of a patient who developed an painful Horner’s syndrome following a bout of vomiting.
PUPILLARY LIGHT–NEAR DISSOCIATION
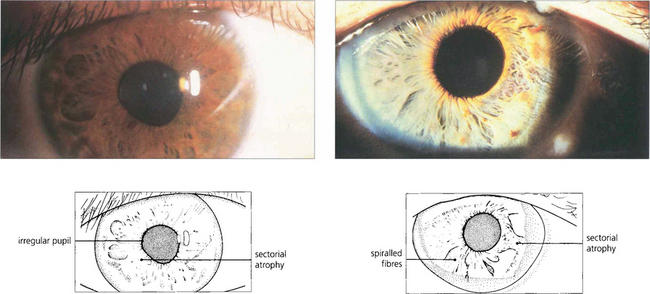
Fig. 19.47 Typical sectoral iris palsies seen in two cases of Adie’s pupil. Constriction of the pupil to G 0.1% pilocarpine due to postganglionic denervation hypersensitivity confirms the diagnosis.
By courtesy of Professor H S Thompson.
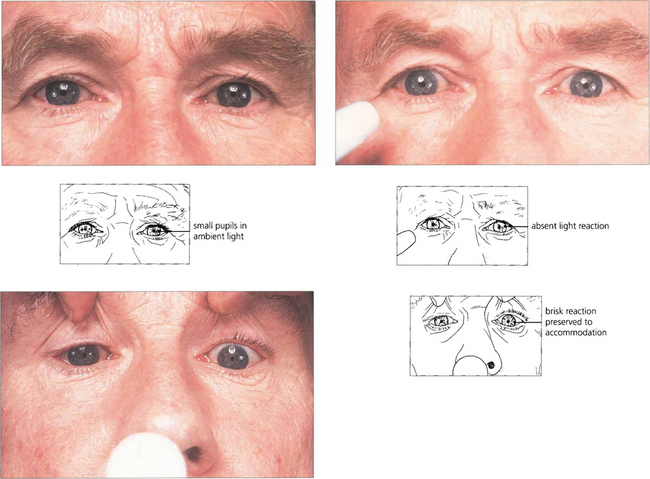
Fig. 19.48 Argyll Robertson pupils are irregular, miosed and asymmetrical in size with a poor light reflex and brisk near reflex. They are the hallmark of neurosyphilis. However, similar pupils are seen on very rare occasions in diabetics. The lesion is located in the region of the Edinger–Westphal nucleus.
VISUAL FIELD LOSS
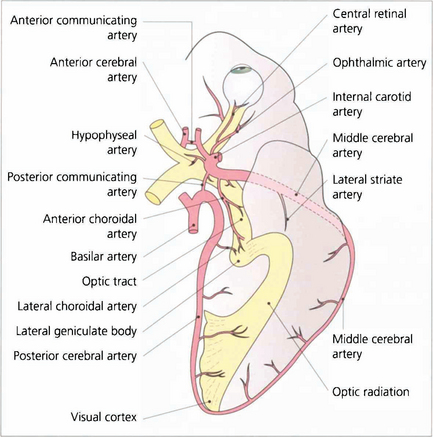
Fig. 19.49 A knowledge of the anatomy of the blood supply of the visual pathways is essential in understanding the causes of many types of field defect. This is derived from the internal carotid (anterior and middle cerebral arteries) and the vertebral (posterior cerebral arteries) with multiple areas of anastomosis between both systems. These are particularly rich in the region of the chiasm, optic tract and lateral geniculate nucleus so that infarcts in these areas are extremely rare. The most common infarct of the visual pathways is an isolated lesion of the occipital cortex involving the posterior cerebral artery. The anastomosis between this and the middle cerebral artery at the posterior pole of the occipital lobe produces the typical macular sparing pattern of homonymous hemianopia seen with lesions of this area.
PRECHIASMAL FIELD DEFECTS
Axons from the retinal ganglion cells converge on the optic disc; they are divided horizontally by the horizontal raphe on the temporal side but enter radially on the nasal side. Damage to a bundle of axons at the vertical margins of the disc will produce characteristic uniocular ‘arcuate’ field defects (see Chs 7 and 17). If this affects the superior or inferior optic disc margin the field loss will occur below or above the horizontal meridian respectively, producing an ‘altitudinal’ field defect. Compressive and inflammatory optic nerve lesions tend to produce central scotomas (see Ch. 17).
The retinotopic arrangement of fibres in the optic nerve is not as precise as has been suggested in anatomy texts. Nonetheless, it is the case that fibres in three major groupings, those from the upper hemi-retina, the lower hemi-retina and those forming the centrocaecal projection, do not intermingle until decussation begins as the chiasm is approached (see Fig. 19.50).
Optic nerve disease produces a group of typical physical signs, no matter what the cause of the lesion (Table 19.2). The severity of an afferent pupillary defect will depend on the density of the lesion and degree of asymmetry of involvement in the two optic nerves.
| Signs of optic nerve disease | Causes of optic nerve lesions |
|---|---|
| Reduced acuity | Demyelinating disease |
| Reduced colour vision | Compression |
| Afferent pupillary defect | Ischaemia |
| Central scotoma | Inflammation or vasculitis |
| Optic disc changes (may be normal, pallor or swollen) | Genetically inherited Nutritional or toxic |
| Trauma | |
| Neoplastic infiltration |
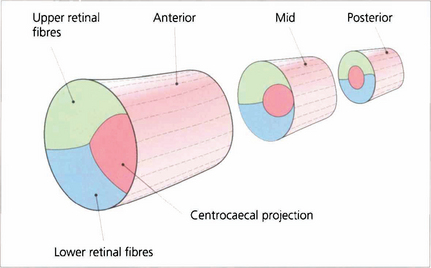
Fig. 19.50 The retinotopic projection of nerve fibres in the optic nerve. There is considerable intermingling of the axons in the optic nerve and there is no precise topographic organization. However the centrocaecal fibres and the fibres from the upper and lower hemi-retinae do remain distinct as shown above until decussation. The centrocaecal projection enters the nerve on its temporal side.
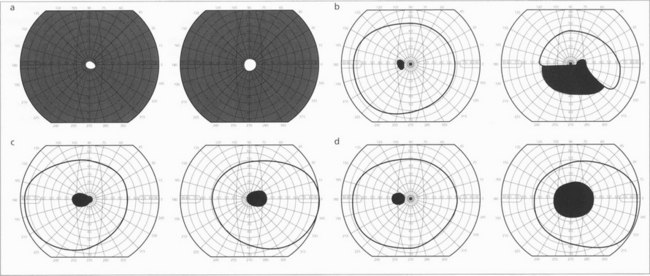
Fig. 19.51 Patterns of visual field loss with retinal, optic disc and optic nerve disease. Constriction of both visual fields (a) is seen with retinal dystrophies (e.g. retinitis pigmentosa) and occasionally from optic disc disease (e.g. longstanding papilloedema). Altitudinal defects (b) indicate optic disc disease, an inferior altitudinal defect is particularly a feature of anterior ischaemic optic neuropathy. (c) Centrocaecal scotomas are a feature of toxic, nutritional and genetic inherited optic neuropathies. (d) Central scotomas are typically seen with optic nerve lesions that are compressive or inflammatory rather than ischaemic.
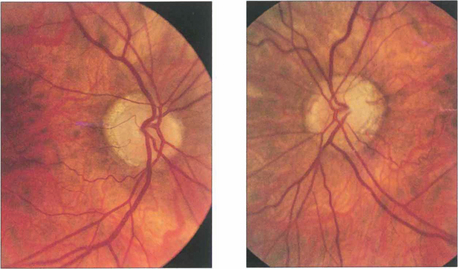
Fig. 19.52 Optic atrophy is an important physical sign that always requires investigation and explanation. This patient with a longstanding asymmetrical bilateral neuropathy from sarcoidosis shows bitemporal pallor, more marked in the left eye than the right (as well as myopic crescents).
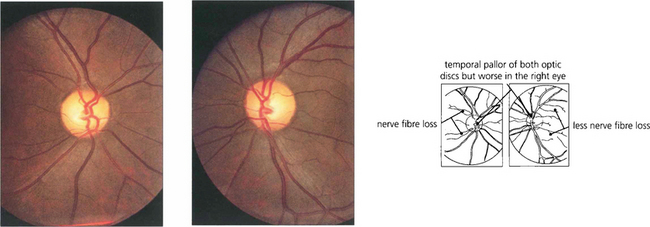
Fig. 19.53 Loss of nerve fibres can be visualized in the retina. Destruction of nerve fibres in the anterior visual pathway (from the retina to the lateral geniculate body) results in atrophy and loss of retinal nerve fibres about 6 weeks later. Nerve fibre defects are most easily observed by using red-free light (see Ch. 7) and can be seen more easily in relatively pigmented fundi as in this patient with a pituitary adenoma. Small defects appear as ‘grooves’ in the nerve fibre area, larger defects as areas of nerve fibre loss. Confirmatory signs of optic nerve damage are found in a reduced visual acuity, reduced colour and brightness sensation, a relative afferent pupillary defect and corresponding visual field defect.
THE CHIASM
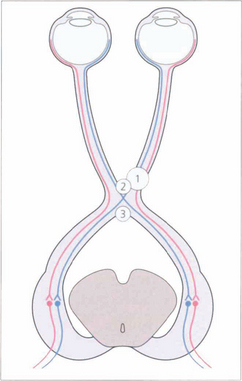
Fig. 19.54 Nasal retinal fibres (temporal field) decussate to the opposite optic tract, whereas temporal retinal fibres (nasal field) remain uncrossed. Figs 19.57–19.59 relate lesions at sites 1, 2 and 3 to field defects.
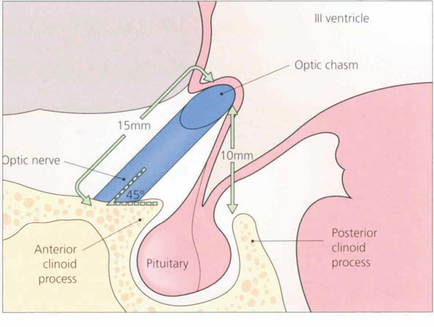
Fig. 19.55 The optic nerves exit from the optic canals at an angle of about 45° to the horizontal and have an intracranial course of about 15 mm. The chiasm lies about 10 mm above the roof of the pituitary fossa. It follows, therefore, that a pituitary tumour must grow to a considerable extent above the fossa before it compresses the chiasm.

Fig. 19.56 For this reason, patients with endocrine secreting tumours, such as this patient with acromegaly, usually present before they develop field defects whereas prolactinomas in men and postmenopausal women or nonhormone-secreting chromophobe adenomas commonly present with visual failure.
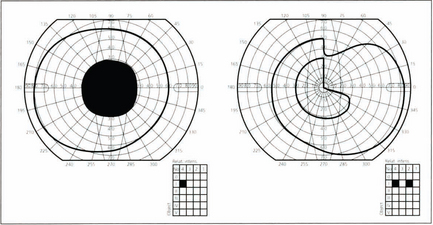
Fig. 19.57 A lesion on the outer lateral aspect of the tuberculum sella, commonly a meningioma (lesion 1 in Fig. 19.54), may compress not only the optic nerve on the same side but crossing fibres from the other eye at the chiasm. This produces field loss known as a ‘junctional defect’: a unilateral central scotoma and a superotemporal defect in the other eye caused by to a lesion at the junction of the optic nerve and chiasm. It used to be thought that this was due to crossing fibres looping forwards into the contralateral optic nerve (Willbrand’s knee) but this is now considered to be artefactual.
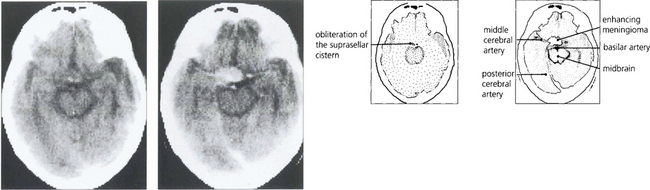
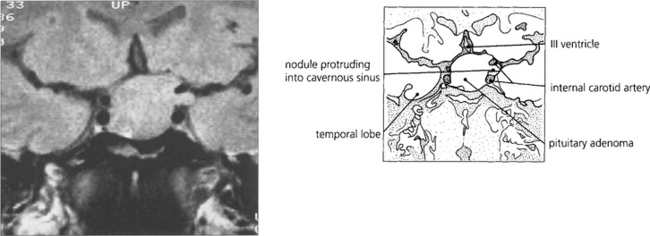
Fig. 19.58 Enhanced computed tomography demonstrates a suprasellar meningioma that produced this unusual ‘junctional’ pattern of field defect. The patient also had a partial sixth nerve palsy and a third nerve palsy with aberrant regeneration from invasion of the cavernous sinus.
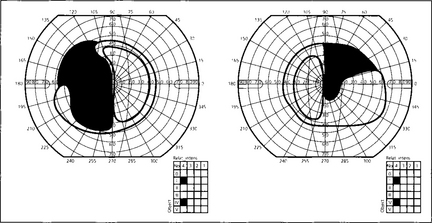
Fig. 19.59 This diagram shows a typical superior bitemporal field defect that would be produced by a lesion at site 2 in Fig. 19.54 (a lesion at site 3 would produce an inferior bitemporal defect). The patient had acuities of 20/60 right and 20/80 left, a left relative afferent pupillary defect, poor colour vision in both eyes but worse in the left and bilateral optic disc pallor which was greater in the left eye.
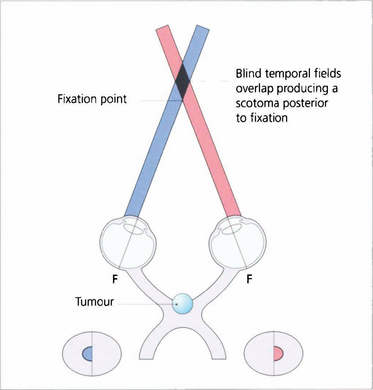
Fig. 19.60 Dense central bitemporal field loss produces the phenomenon of postfixational blindness. If the patient focuses on a near object the bitemporal field defects overlap producing a scotoma posterior to fixation. Patients find that small objects disappear into this area and this produces interesting symptoms; for example, an architect may have difficulty in putting a pencil on a line or someone may notice difficulty in cutting their fingernails.
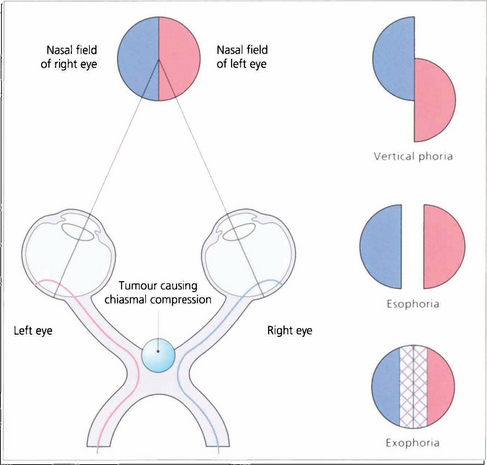
Fig. 19.61 With dense bitemporal hemianopias there may be little overlap between the remaining nasal fields to produce enough binocular vision to control a phoria. This may break down, often only transiently, causing diplopia in the absence of an apparent ocular motility defect. This phenomenon is known as hemifield slide.
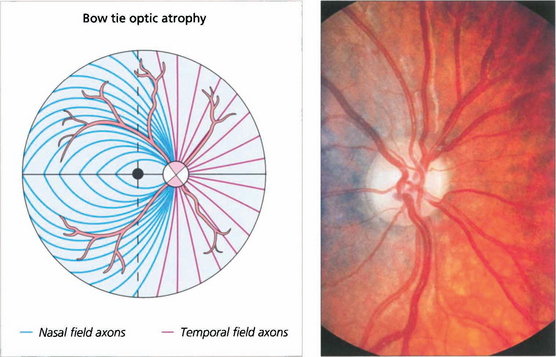
Fig. 19.62 Where crossing fibres are predominantly affected and where there is also extensive axonal loss a characteristic pattern of optic atrophy can be seen. Temporal retinal nerve fibres (nasal visual field) arch around the macula and enter the disc vertically, superiorly and inferiorly. Correspondingly, although nasal retinal fibres (temporal field) enter the disc around its entire circumference but horizontally on both sides they enter the disc without intermingling with temporal retinal fibres. The ‘bow tie’ pattern of atrophy results from exclusive loss of nasal retinal fibres; the arcuate bundles superiorly and inferiorly are thinned but still present while there is total atrophy in a horizontal band across the disc. This pattern is seen most dramatically in cases of chiasmal transection; the effects of compressive lesions are rarely as selective as this.
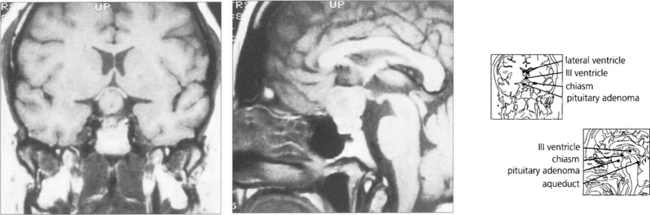
Fig. 19.63 T1-weighted MRI is the investigation of choice to confirm the presence of a pituitary lesion. Scans should be performed in the sagittal and coronal planes to define the anatomical boundaries of the lesion. The optic nerves and chiasm can be identified as anatomical structures and the precise area of compression can be seen.
OPTIC TRACT LESIONS
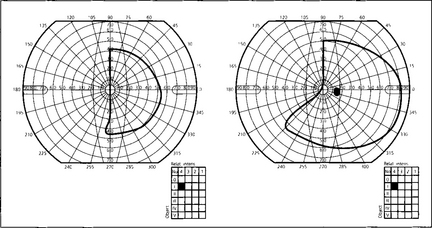
Fig. 19.65 Optic tract defects are uncommon and usually result from lesions causing chiasmal compression that extend posteriorly, especially craniopharyngiomas or intrinsic gliomas of the chiasm. There is often a combination of optic nerve, chiasm and tract features. Pure tract hemianopias may be seen with trauma, following surgery, congenitally and in multiple sclerosis. When incomplete, the field defects are often incongruous (difference in size and density between the two affected hemifields); this arises because of the complex anatomy of the lateral geniculate nucleus (see Fig. 19.67). There are two other useful signs which help to decide whether a hemianopia is due to a tract or postgeniculate lesion. These are due to the fact that the nerve fibres are still retinal ganglion cell axons: therefore an afferent pupillary defect is found in the eye with the temporal field defect. Secondly, although there will be bilateral disc pallor a longstanding lesion will produce ‘bow-tie’ atrophy in the eye with the temporal hemianopia because the crossing fibres are affected.
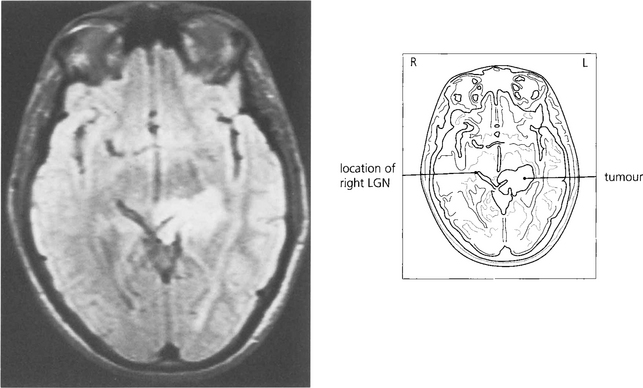
Fig. 19.66 The patient presented with an asymptomatic complete right homonymous hemianopia. Fundus-copy revealed a bow-tie atrophy in the right eye (with the temporal hemianopia). Imaging showed a long-standing lesion (possibly a hamartoma) in the region of the left optic tract/LGN/early optic radiation.
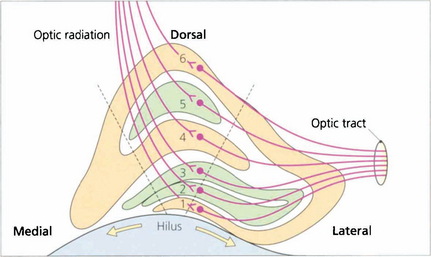
Fig. 19.67 Some 90 per cent of retinal ganglion cells project to the lateral geniculate nucleus (LGN); the remainder project to the superior colliculus and the pupil centres in the midbrain. The LGN is a complex structure that is easiest to visualize in coronal section. Centrally it is made up of six layers: layers 2, 3 and 5 receive the axons of uncrossed temporal retinal fibres and layers 1, 4 and 6 receive the axons of the crossed fibres. The central part is concerned with macular vision, the medial part with upper retinal quadrants (inferior field) and the lateral part with lower retinal quadrants, so that the representation of the visual field at this unique junction has been rotated through 90°. Any point in the visual field is represented by a vertical column of cells passing perpendicularly through each lamina. Laminae 1 and 2 contain cell bodies with a larger diameter and are known as the ‘magnocellular’ layers, in contrast to the ‘parvocellular’ layers superiorly. The magnocellular projection (10 per cent of cells) is concerned with movement detection, and the parvocellular (80 per cent of cells) with colour vision and acuity. The remaining 10 per cent of cells pass through the interlaminar zones, known as the koniocellular projection; these cells are of uncertain function. The LGN receives its blood supply from the anterior choroidal artery (a branch of the carotid artery) and the lateral choroidal artery (a branch of the posterior cerebral artery) which anastomose on its surface. The dashed lines indicate the extent of a lesion which would give rise to the field defect shown in Fig. 19.68 from infarction of the lateral choroidal artery.
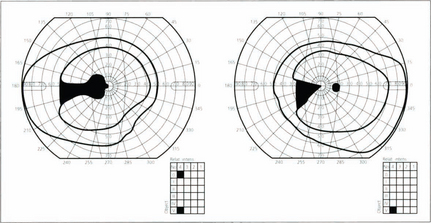
Fig. 19.68 Lesions of the lateral geniculate nucleus are excessively rare but can be diagnosed from the clinical features. The LGN itself is difficult to visualize on imaging but its location can be easily inferred as they sit atop the ambient cisterns. The typical field defect is a wedge-shaped hemianopic defect that is incongruous or alternatively has preservation of a central wedge in each of hemianopic field. There is no relative afferent pupillary defect as the pupillary fibres have already left to reach the third nerve nucleus by the superior brachium but optic disc pallor is seen because of damage to the retinal ganglion cell axons.
OPTIC RADIATIONS
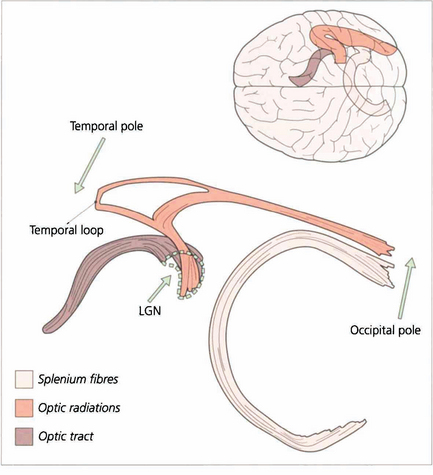
Fig. 19.69 These diagrams are taken from a study employing a novel technique known as tractography where white matter bundles can be ‘virtually dissected’ from MRI. The optic radiation tract is shaped like an elongated ‘S’. The optic tract fibres enter the LGN anteroventrally and the optic radiation fibres leave laterodorsally. The optic radiation fibres divide into a small ventral temporal loop, which is now referred to as the Fleschig–Meyer loop, and the dorsal optic radiation. Feschig–Meyer’s loop is damaged in temporal lobe lesions because it loops forward towards the temporal pole and performs a hairpin bend around the temporal horn of the lateral ventricle.
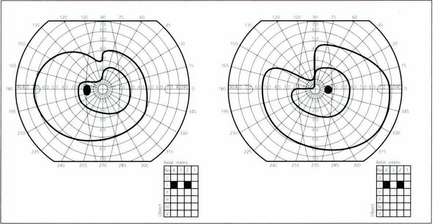
Fig. 19.70 It is unusual for temporal lobe lesions to produce a field defect but if Fleschig–Meyer’s loop is damaged an upper quadrantic hemianopia will be produced, denser in the eye with the nasal defect. Pure temporal lobe field defects must, by definition, have normal pupillary responses as the pupillary axons have already separated from the visual pathway. However, the temporal lobe and optic tracts lie in close proximity and share a similar blood supply so it is not uncommon for temporal lobe lesions to involve the optic tracts. This would be shown by the presence of an afferent pupillary defect in the eye on the side of the lesion with pallor of both optic discs.
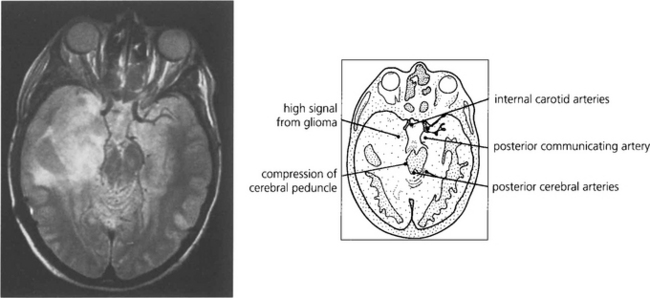
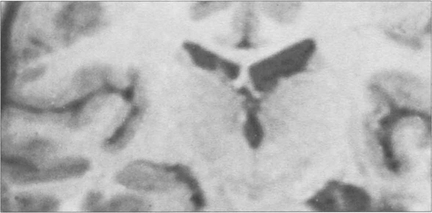
Fig. 19.72 ‘Purer’ examples of damage to Fleschig–Meyer’s loop are seen following hippocampal resections for intractable epilepsy due to hippocampal sclerosis, such as is shown here.
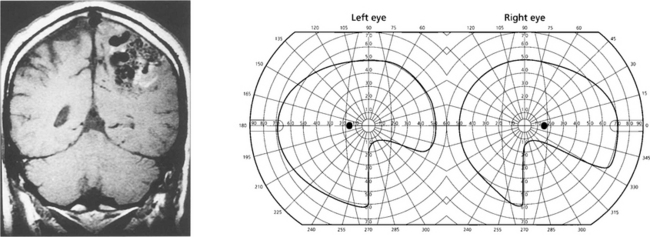
Fig. 19.73 Inferior homonymous quadrantanopias can occur following damage to the dorsal occipital lobe but may also occur with parietal lobe lesions affecting the dorsal optic radiations. The pathway for the smooth pursuit component of the optokinetic reflex (OKN) response projects from the parietal lobe ipsilaterally to the pons (see Fig. 19.11). In patients with hemianopia or inferior quadrantanopia abnormalities of OKN may be used to demonstrate whether or not the parietal cortex is damaged. This is important as parietal lobe lesions are frequently due to tumours whereas occipital lesions tend to be vascular. If the damage is purely occipital OKN will be normal; if it is parietal then OKN will not be generated when the stripes are moving towards the side of the lesion as there will be no following response. This can also be shown by carefully comparing pursuit of a target to left and right, when the OKN drum is rotated towards the side of the lesion pursuit will be broken up by ‘catch-up’ saccades. The scan here shows a parietal lobe vascular malformation.
THE VISUAL CORTEX
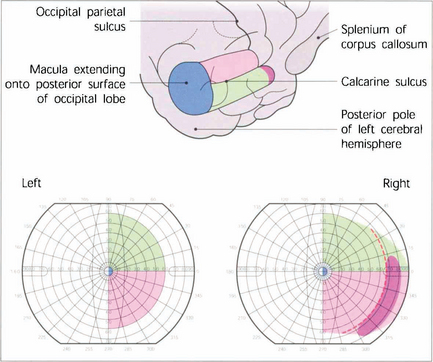
Fig. 19.74 V1 lies mostly within the calcarine sulcus on the medial surface of the occipital lobe extending as far as the occipitoparietal sulcus. The total length is about 5 cm and approximately half of this is concerned with central vision with the macula itself being represented most posteriorly and extending on to the hemispheric surface at the pole of the occipital lobe. The outer temporal 30° of visual field of each eye is represented uniocularly at the extreme anterior pole of the calcarine sulcus and the representation of the horizontal meridian of the visual field follows the apex of the calcarine sulcus with the lower contralateral quadrant represented above this line and the upper quadrant below it.
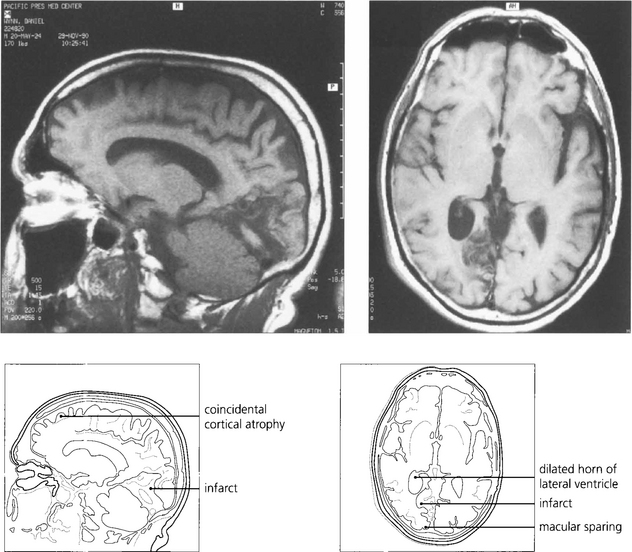
Fig. 19.75 The most common cause of an isolated homonymous hemianopia in the absence of other neurological signs is an occipital lobe infarct from an occlusion in the posterior cerebral artery territory. Such patients often present to the eye department because they have no symptoms other than loss of vision. These are frequently embolic and a cardiac examination is mandatory. The figure shows sagittal (left) and axial (right) views of a right posterior cerebral artery occlusion. The patient had a complete hemianopia.
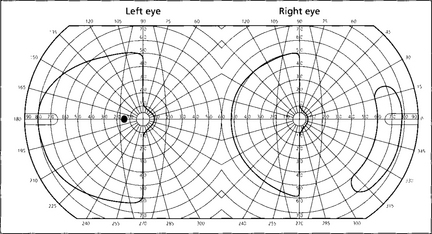
Fig. 19.76 If macular sparing is present (preservation of the central 5–10° of visual field from collateral circulation to the occipital pole by the middle cerebral artery), this is diagnostic of an occipital lobe lesion. Sparing of the outer temporal field in the eye on the side of the hemianopia is also a feature of some relatively localized occipital lobe infarcts and is due to unilateral representation of the outer temporal field in the depths of the calcarine fissure.
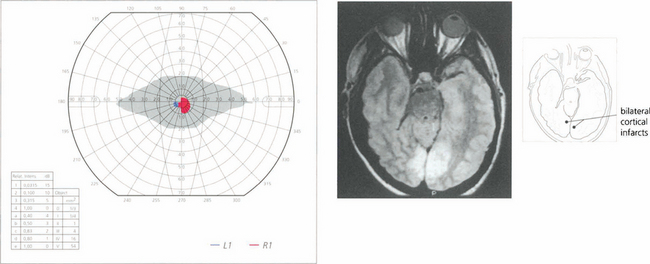
Fig. 19.77 A patient who suffers from bilateral posterior cerebral artery occlusion will be cortically blind. Recovery is often partial. This patient had bilateral macular sparing, presumably because there was extensive supply of both occipital poles from middle cerebral territory. Such patients are often considered to have a nonorganic visual loss because visual acuity, pupil reactions and fundus examination are normal. Note that there is a vertical midline split preserved between the residual hemianoptic fields.
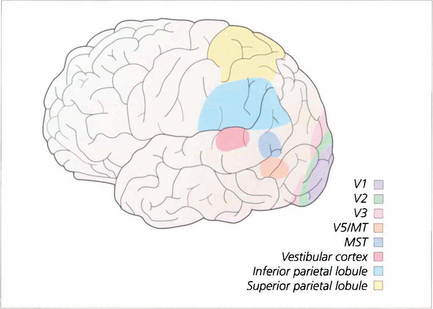
Fig. 19.78 Extra-striate visual cortex, previously grouped together as ‘visual association cortex’, is now known to be a rich matrix of multiple representations of the visual field. Areas specialized for movement perception, colour vision, spatial localization, face recognition and other visual submodalities have been identified. This, much simplified, sketch gives some idea of the complexity of these areas.
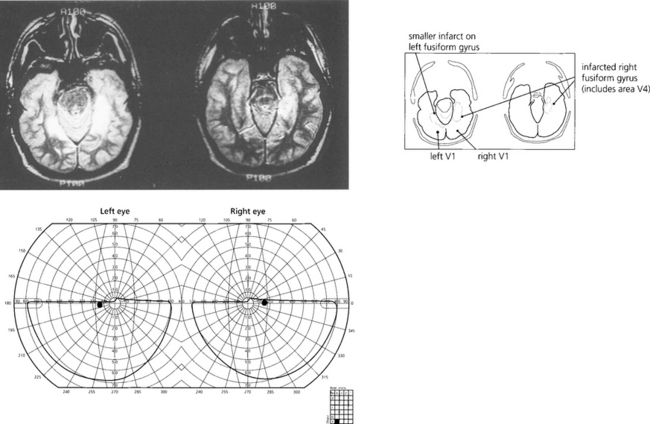
Fig. 19.79 This patient cannot recognize faces—even his own in photographs—(prosopagnosia) and has loss of colour vision (cerebral achromatopsia) due to bilateral ventral occipitotemporal infarction. Field defects were bilateral superior homonymous quadrantanopias. Note the field defect is dense and very congruous with a vertical midline split.
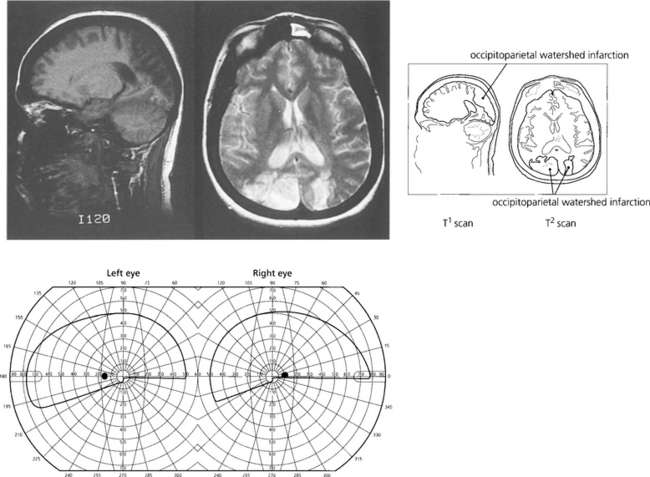
Fig. 19.80 Another common clinical syndrome, because it follows bilateral watershed occipitoparietal infarction, is characterized by loss of visuospatial functions (Balint–Holmes syndrome of visual disorientation). Such patients cannot localize objects in space, for example, objects in a photograph of a steet scene. Bilateral lower homonymous quadrantanopias would be commonly seen. Watershed infarcts commonly occur after severe hypotension such as following a major haemorrhage or anaesthetic hypoxia.
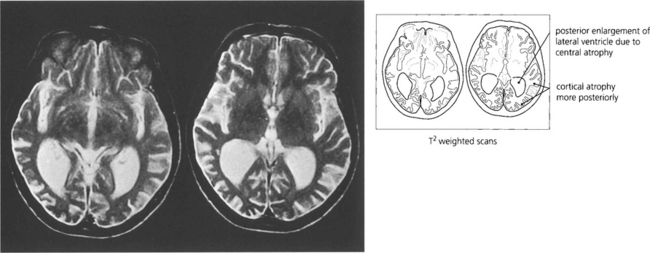
Fig. 19.81 Balint–Holmes syndrome is also seen in a variant of Alzheimer’s disease where the process begins posteriorly in the occipitoparietal cortex. Patients present with difficulty in reading and subtle visual complaints but often have normal acuity and visual fields early in the process; the diagnosis is often delayed considerably.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
19 Neuro-ophthalmology
OCULAR MOVEMENT
SUPRANUCLEAR GAZE CONTROL
Saccadic movements are rapid and relocate fixation of gaze, either reflexly or voluntarily. They are initiated in the contralateral premotor frontal cortex and, once initiated, the movement is irrevocable and ocular position cannot again be modified until the saccade has been completed. A saccade occurs after a latent period of about 200 ms following initiation and has a high velocity of up to 700°/sec. Saccades are tested clinically by instructing the patient to look first at one stationary target and then at another, or to look right and left or up and down with no target present (see Fig. 19.4).
Pursuit movements are slower and are concerned with keeping the target at the fovea. They appear to be generated in the ipsilateral occipital cortex but little is known about the supranuclear pathway. Pursuit movements have a latency of about 125 ms from initiation and a maximum velocity of less than 50°/sec. The movement is smooth and modified continuously according to the speed of the target if the pursuit movement lags behind the target position a corrective saccade is inserted to keep up. Pursuit movements are tested by asking the patient to follow a slowly moving target. It is of great importance that the target is clearly visible and that it is not moved too fast (see Fig. 19.5).
Vestibular ocular movements may be tested by a ‘doll’s head’ manoeuvre, in which the patient is asked to fixate on a target while the examiner rotates the patient’s head (see Fig. 19.6). The doll’s head manoeuvre tests both the left and right labyrinths and may be normal even if one labyrinth is totally nonfunctioning. To test each labyrinth separately, caloric stimulation can be employed which induces nystagmus by syringing the external auditory meatus with cold or warm water. Recently a bedside test has been described, based on the observation that the initial component of a rapid head rotation depends upon the integrity of the labyrinth towards which the head is turned (see Fig. 19.8).
Fig. 19.1 The major inputs to the vertical and horizontal gaze centres are saccadic and pursuit commands from the cerebral hemispheres and the vestibular ocular reflexes from the vestibular nuclei in the medulla. These impulses are then transmitted to the individual ocular motor nuclei in the midbrain and pons. Internuclear pathways coordinate the movements generated in the gaze centres with each other and with the third, fourth and sixth nerve nuclei. Excitatory impulses to an agonist muscle are accompanied by inhibitory impulses to its antagonist.
Fig. 19.2 Saccadic movements have been shown biomechanically to be generated by impulses in the form of a pulse and step. To change eye position a rapid burst of neuronal signals (the pulse) is fired by the gaze centre and this rapidly moves the eye to the new position. This rapid burst is integrated in the ‘pulse-step generator’ (a neuronal complex within the gaze centre) to produce an increased tonic discharge (the step) which holds the eye in its new position. Similar responses take place in the agonist of the fellow eye and reciprocal impulses relax the antagonist muscles. These changes are reflected by the EMG recording in an individual muscle. Neurophysiologically saccades are initiated by burst cells in the paramedian pontine reticular formation (PPRF) which are inhibited from firing by pause cells at all times except during a saccade. Thus the saccadic input suppresses the pause cell which permits the burst cell to discharge immediately.
Fig. 19.3 The pulse–step generator exists physiologically in the horizontal gaze centre—otherwise known as the paramedian pontine reticular formation (PPRF)— in the pons and various mismatches of the pulse to step signals have been shown to explain some types of nystagmus and eye movement abnormalities seen in clinical practice.
Fig. 19.4 Saccadic eye movements can be elicited simply by asking patients to look right and left or up and down. This is a useful strategy where the observation of large amplitude saccades may reveal subtle abnormalities of saccadic velocity. An alternative strategy is to ask patients to shift fixation from one target to another, in this way saccades of large and small amplitude and involving a variety of gaze shifts can be observed. The accuracy of saccades (under- or over-shooting) can be tested. Slowing of the velocity of saccades may be conjugate (as in a gaze palsy), dysconjugate (as in a lateral rectus paresis or internuclear ophthalmoplegia) or global (as in an ocular myopathy or some forms of cerebellar disease).
Fig. 19.5 When asking a subject to follow a target it is essential that the target is clearly visible and is moved neither too slowly nor too rapidly. The reader should experiment by trying to follow his or her own finger as it is moved in a 180° arc from one side of the head to the other at arm’s length. A comfortable speed is about 30° per seconds (6 seconds from ear to ear). Pursuit movements are used to examine ductions and versions but, instead of looking at the range of movement, we are now interested in the accuracy of the following movement itself. The observer must watch for saccadic intrusions which will indicate that the eye is not keeping up with the target (in other words, there is reduced gain of the smooth pursuit reflex). This is a universal finding in cerebellar disease.
Fig. 19.6 Vestibular ocular reflexes (VORs) are tested by asking the patient to fixate a distant target (such as an acuity chart) and then gently moving the patient’s head from side to side or up and down with increasing frequency until the patient reports movement of the target or the subjective acuity falls, indicating retinal slip of the image. Alternatively a retinal landmark can be fixed using the direct ophthalmoscope while the examiner rocks the head to and fro. Loss of the VOR causes symptoms such as instability of the horizon as the patient walks and is seen not uncommonly with multiple sclerosis.
Fig. 19.7 Suppression of the VOR can be tested by asking the patient to grip a target mounted on a spatula between the teeth and to move the head from side to side or up and down with increasing rapidity until the patient reports an inability to keep the image ‘still’ or saccadic eye movements are observed as fixation is lost and taken up again. The mechanism used to suppress the VOR is the pursuit mechanism; if smooth pursuit is abnormal then so will be VOR suppression, and vice versa. Patients who are able to suppress their VOR complain of unusual symptoms such as difficulty with reading when travelling.
Fig. 19.8 Only one functioning labyrinth is required for the tests described in Fig. 19.6 to be normal. Caloric testing can be used to examine each labyrinth separately. Caloric reflexes are produced by stimulating the semicircular canals and vestibular nuclei with warm or cold water and can provide useful information on the integrity of these pathways in the brainstem; this is especially useful in the neurological assessment of brainstem damage in an unconscious patient. In a conscious patient cold water in the external auditory meatus generates a nystagmus of both eyes with a fast phase to the opposite side but in unconscious patients the saccadic phase is lost and a tonic deviation to the same side is seen. This indicates an intact pons but the results must be interpreted with caution after acute drug overdoses when false-negative responses may be seen. Calorics can be adapted to test vertical gaze (and therefore the integrity of the midbrain) by syringing both ears.
Fig. 19.9 Unilateral labyrinthine function can be tested at the bedside. If the head is rotated abruptly, the initial phase of the VOR is mediated by the labyrinth towards which the head is turned. If that labyrinth is not functioning initially the eyes move with the head and there is a corrective saccade as fixation is taken up again.
Fig. 19.10 The vestibular system responds only to acceleration and slow head movements (less than 1° per second) stimulate the optokinetic reflex (OKN). The hand-held OKN drum is useful as a bedside test of the pursuit and saccadic systems. If there is a saccadic palsy the eyes will deviate tonically in the direction of the stripe movement: the pursuit mechanism has been elicited but there is no saccade to return gaze towards primary position. Turning the drum to the right elicits an ipsilateral pursuit movement to the right and a contralateral saccade to the left. In essence turning the drum to the right tests the right hemisphere and vice versa to the left.
ANATOMY OF THE OCULAR MOTOR PATHWAYS
Fig. 19.11 Saccades are generated in a number of areas in the frontal premotor area of the contralateral hemisphere including the frontal eye fields; thus a saccade to the right is generated in the left frontal lobe, passes through the anterior limb of the internal capsule and decussates in the midbrain to the horizontal gaze centre in the pons. The incoming visual information that controls smooth pursuit is analysed in an occipitoparietal motion area. Pursuit movements are then generated in the ipsilateral superior parietal lobule so that a pursuit movement to the right is controlled by the right hemisphere. Little is known of the neuronal pathway, but the final common pathways for saccades and pursuit are the conjugate horizontal and vertical gaze mechanisms in the pons and midbrain. The inferior parietal lobule may be concerned with shifts of attention, which may also generate saccades.
Fig. 19.12 The final common pathway for all horizontal movements are the sixth nerve nuclei to which the PPRF, pursuit mechanisms and vestibular nuclei project. Interneurones in the sixth nerve nucleus project through the medial longitudinal fasciculus (MLF) to the contralateral medial rectus subunit of the oculomotor nucleus. For vertical saccades, the rostral interstitial nucleus of the MLF (riMLF) projects to the third and fourth nerve nuclei. Vestibular inputs controlling vertical eye movements ascend from the lower brainstem to the fourth and third nerve nuclei via the MLF. The nucleus of the posterior commissure projects on to both riMLFs and is particularly important for upgaze.
Fig. 19.13 A transverse histological section of the midbrain at the level of the superior colliculus demonstrates the third nerve nucleus inferior to the aqueduct with its efferent fibres. The medial longitudinal fasciculus (MLF) in connecting the third and fourth nerve nuclei and contralateral horizontal gaze centres is responsible for coordinating horizontal and vertical movements. Infarcts in this region cause Weber’s syndrome (unilateral third nerve palsy and contralateral hemiplegia) and Benedikt’s syndrome (unilateral third nerve palsy, contralateral tremor and hyperaesthesia). (Stained to show myelin.)
Fig. 19.14 The third nerve nucleus is a complex assemblage of motor neurones. The parasympathetic nucleus (Edinger–Westphal nucleus) of the pupil lies rostrally and superiorly. The inferior rectus, medial rectus and inferior oblique muscles are all supplied from ipsilateral subnuclei, but the superior rectus subnucleus is contralateral. Both levator palpebrae muscles have a single caudal superior nucleus. A complete ‘nuclear’ unilateral third nerve palsy is therefore characterized by ipsilateral inferior oblique, medial and inferior rectus weakness with bilateral ptosis and contralateral superior rectus weakness.
Fig. 19.15 A transverse histological section through the pons at the level of the sixth nerve nuclei demonstrates the medial longitudinal fasciculus (MLF) and the centre for horizontal gaze (PPRF). Interneurones from the PPRF pass initially to the sixth nerve nucleus where they synapse with the MLF (see Fig. 19.34). (Stained to show myelin.)
Fig. 19.16 The third and sixth nerves leave the ventral surface of the brain and pass through the interpeduncular cistern before entering the cavernous sinus. The sixth nerve passes almost vertically upwards and bends forwards under the petroclinoid ligament at the apex of the petrous temporal bone. Here it can be compressed against the bone by raised CSF pressure or damaged by fractures or disease in the bone (hence producing the sixth nerve palsy associated with raised intracranial pressure). The fourth nerve leaves the midbrain dorsally, decussates in the superior medullary velum and passes through the subarachnoid space around the midbrain between the superior cerebellar and posterior cerebral arteries before entering the cavernous sinus.
Fig. 19.17 In the cavernous sinus the third and fourth nerves lie superiorly in the lateral wall; at the anterior end the third nerve divides into superior (superior rectus and lid levator) and inferior (inferior and medial rectus, inferior oblique, pupillary and ciliary muscles) branches. Sensory fibres from the eye and forehead enter the cavernous sinus anteriorly in the ophthalmic division of the fifth nerve, lying inferiorly in the lateral wall to join the trigeminal ganglion at the posterior limit. The internal carotid artery and sixth nerve lie within the sinus.
CONJUGATE GAZE PALSIES
HORIZONTAL SUPRANUCLEAR PALSY
Fig. 19.18 Following an acute cerebrovascular accident involving supranuclear pathways, saccadic gaze is lost in the direction opposite to the side of the lesion and input from the remaining hemisphere deviates the eyes towards the side of the lesion. (Occasionally, irritative lesions, such as cerebral tumours or abscesses, produce deviation of the eyes to the contralateral side.)
Fig. 19.19 (Left) On command the patient is unable to make a gaze movement to the target on his right. (Right) On doll’s head rotation to the left, however, the eyes deviate fully to the right demonstrating intact pontine reflexes and the supranuclear nature of the lesion.
Fig. 19.20 A CT scan of the patient seen in Fig. 19.18, showing a large infarct in the internal capsule due to the occlusion of middle cerebral artery branches, corresponding to lesion 1 in Fig. 19.21. If the patient survives the acute ocular deviation recovers rapidly. Horizontal gaze becomes full and saccades return to normal. This is probably due to a restoration of control mechanisms by the superior colliculus.
VERTICAL GAZE PALSY
Fig. 19.22 This elderly woman suffered a cerebrovascular accident in the upper midbrain. She retained the ability to make rapid and full horizontal saccades.
Fig. 19.23 In the same patient, voluntary up and down gaze to saccadic command is lost and compensated for by head movement. ‘Doll’s head’ rotation shows that vertical movements can be produced and that pontine reflexes are intact.
Fig. 19.24 Convergence retraction nystagmus is an interesting ocular movement abnormality sometimes seen with vertical gaze palsies. Bursts of convergence with retraction of both eyes into the orbits are seen either spontaneously or on attempting an upward saccade. These movements may also be accompanied by accommodative spasm which accounts for the frequent complaint of blurred vision. Convergence retraction nystagmus is shown particularly well if the patient views a downward rotating optokinetic drum which stimulates upward refixational saccades.
Fig. 19.25 In convergence retraction nystagmus the lesion is thought to cause disinhibition of the ocular motor nuclei allowing bursts of co-firing of the external ocular muscles. The medial recti muscles are the most powerful of the external ocular muscles and consequently the globes converge and retract into the orbits.
Fig. 19.26 Light–near dissociation of the pupils is the third sign of the triad of Parinaud’s syndrome. The pupils are normally moderately dilated and show a poor or absent light reaction but preserve the near reaction. It has been suggested that light reflex fibres are more dorsal in the midbrain and are therefore selectively damaged by lesions in this area with comparative sparing of the more ventrally sited near reflex pathways.
