[level-membership-for-internal-medicine-category]
84 |
The Practice of Genetics in Clinical Medicine |
APPLICATIONS OF MOLECULAR GENETICS IN CLINICAL MEDICINE
Genetic testing for inherited abnormalities associated with disease risk is increasingly used in the practice of clinical medicine. Germline alterations include chromosomal abnormalities (Chap. 83e), specific gene mutations with autosomal dominant or recessive patterns of transmission (Chap. 82), and single nucleotide polymorphisms with small relative risks associated with disease. Germline alterations are responsible for disorders beyond classic Mendelian conditions with genetic susceptibility to common adult-onset diseases such as asthma, hypertension, diabetes mellitus, macular degeneration, and many forms of cancer. For many of these diseases, there is a complex interplay of genes (often multiple) and environmental factors that affect lifetime risk, age of onset, disease severity, and treatment options.
The expansion of knowledge related to genetics is changing our understanding of pathophysiology and influencing our classification of diseases. Awareness of genetic etiology can have an impact on clinical management, including prevention and screening for or treatment of a range of diseases. Primary care physicians are relied upon to help patients navigate testing and treatment options. Consequently, they must understand the genetic basis for a large number of genetically influenced diseases, incorporate personal and family history to determine the risk for a specific mutation, and be positioned to provide counseling. Even if patients are seen by genetic specialists who assess genetic risk and coordinate testing, primary care providers should provide information to their patients regarding the indications, limitations, risks, and benefits of genetic counseling and testing. They must also be prepared to offer risk-based management following genetic risk assessment. Given the pace of genetics, this is an increasingly difficult task. The field of clinical genetics is rapidly moving from single gene testing to multigene panel testing, with techniques such as whole-exome and -genome sequencing on the horizon, increasing the complexity of test selection and interpretation, as well as patient education and medical decision making.
COMMON ADULT-ONSET GENETIC DISORDERS
INHERITANCE PATTERNS
Adult-onset hereditary diseases follow multiple patterns of inheritance. Some are autosomal dominant conditions. These include many common cancer susceptibility syndromes such as hereditary breast and ovarian cancer (due to germline BRCA1 and BRCA2 mutations) and Lynch syndrome (caused by germline mutations in the mismatch repair genes MLH1, MSH2, MSH6, and PMS2). In both of these examples, inherited mutations are associated with a high penetrance (lifetime risk) of cancer, although risk is not 100%. In other conditions, although there is autosomal dominant transmission, there is lower penetrance, thereby making the disorders more difficult to recognize. For example, germline mutations in CHEK2 increase the risk of breast cancer, but with a moderate lifetime risk in the range of 20–40%, as opposed to 50–70% for mutations in BRCA1 or BRCA2. Other adult-onset hereditary diseases are transmitted in an autosomal recessive fashion where two mutant alleles are necessary to cause disease. Examples include hemochromatosis and MYH-associated colon cancer. There are more pediatric-onset autosomal recessive disorders, such as lysosomal storage diseases and cystic fibrosis.
The genetic risk for many adult-onset disorders is multifactorial. Risk can be conferred by genetic factors at a number of loci, which individually have very small effects (usually with relative risks of <1.5). These risk loci (generally single nucleotide polymorphisms [SNPs]) combine with other genes and environmental factors in ways that are not well understood. SNP panels are available to assess risk of disease, but the optimal way of using this information in the clinical setting remains uncertain.
Many diseases have multiple patterns of inheritance, adding to the complexity of evaluating patients and families for these conditions. For example, colon cancer can be associated with a single germline mutation in a mismatch repair gene (Lynch syndrome, autosomal dominant), biallelic mutations in MYH (autosomal recessive), or multiple SNPs (polygenic). Many more individuals will have SNP risk alleles than germline mutations in high-penetrance genes, but cumulative lifetime risk of colon cancer related to the former is modest, whereas the risk related to the latter is significant. Personal and family histories provide important insights into the possible mode of inheritance.
FAMILY HISTORY
When two or more first-degree relatives are affected with asthma, cardiovascular disease, type 2 diabetes, breast cancer, colon cancer, or melanoma, the relative risk for disease among close relatives ranges from two- to fivefold, underscoring the importance of family history for these prevalent disorders. In most situations, the key to assessing the inherited risk for common adult-onset diseases is the collection and interpretation of a detailed personal and family medical history in conjunction with a directed physical examination.
Family history should be recorded in the form of a pedigree. Pedigrees should convey health-related data on first- and second-degree relatives. When such pedigrees suggest inherited disease, they should be expanded to include additional family members. The determination of risk for an asymptomatic individual will vary depending on the size of the pedigree, the number of unaffected relatives, the types of diagnoses, and the ages of disease onset. For example, a woman with two first-degree relatives with breast cancer is at greater risk for a specific Mendelian disorder if she has a total of 3 female first-degree relatives (with only 1 unaffected) than if she has a total of 10 female first-degree relatives (with 7 unaffected). Factors such as adoption and limited family structure (few women in a family) should to be taken into consideration in the interpretation of a pedigree. Additional considerations include young age of disease onset (e.g., a 30-year nonsmoking woman with a myocardial infarction), unusual diseases (e.g., male breast cancer or medullary thyroid cancer), and the finding of multiple potentially related diseases in an individual (e.g., a woman with a history of both colon and endometrial cancer). Some adult-onset diseases are more prevalent in certain ethnic groups. For instance, 2.5% of individuals of Ashkenazi Jewish ancestry carry one of three founder mutation in BRCA1 and BRCA2. Factor V Leiden mutations are much more common in Caucasians than in Africans or Asians.
Additional variables that should be documented are nonhereditary risk factors among those with disease (such as cigarette smoking and myocardial infarction; asbestos exposure and lung disease; and mantle radiation and breast cancer). Significant associated environmental exposures or lifestyle factors decrease the likelihood of a specific genetic disorder. In contrast, the absence of nonhereditary risk factors typically associated with a disease raises concern about a genetic association. A personal or family history of deep vein thrombosis in the absence of known environmental or medical risk factors suggests a hereditary thrombotic disorder. The physical examination may also provide important clues about the risk for a specific inherited disorder. A patient presenting with xanthomas at a young age should prompt consideration of familial hypercholesterolemia. The presence of trichilemmomas in a woman with breast cancer raises concern for Cowden syndrome, associated with PTEN mutations.
Recall of family history is often inaccurate. This is especially so when the history is remote and families lose contact or separate geographically. It can be helpful to ask patients to fill out family history forms before or after their visits, because this provides them with an opportunity to contact relatives. Ideally, this information should be embedded in electronic health records and updated intermittently. Attempts should be made to confirm the illnesses reported in the family history before making important and, in certain circumstances, irreversible management decisions. This process is often labor intensive and ideally involves interviews of additional family members or reviewing medical records, autopsy reports, and death certificates.
Although many inherited disorders will be suggested by the clustering of relatives with the same or related conditions, it is important to note that disease penetrance is incomplete for most genetic disorders. As a result, the pedigree obtained in such families may not exhibit a clear Mendelian inheritance pattern, because not all family members carrying the disease-associated alleles will manifest clinical evidence of the condition. Furthermore, genes associated with some of these disorders often exhibit variable disease expression. For example, the breast cancer–associated gene BRCA2 can predispose to several different malignancies in the same family, including cancers of the breast, ovary, pancreas, skin, and prostate. For common diseases such as breast cancer, some family members without the susceptibility allele (or genotype) may develop breast cancer (or phenotype) sporadically. Such phenocopies represent another confounding variable in the pedigree analysis.
Some of the aforementioned features of the family history are illustrated in Fig. 84-1. In this example, the proband, a 36-year-old woman (IV-1), has a strong history of breast and ovarian cancer on the paternal side of her family. The early age of onset and the co-occurrence of breast and ovarian cancer in this family suggest the possibility of an inherited mutation in BRCA1 or BRCA2. It is unclear however, without genetic testing, whether her father harbors such a mutation and transmitted it to her. After appropriate genetic counseling of the proband and her family, the most informative and cost-effective approach to DNA analysis in this family is to test the cancer-affected 42-year-old living cousin for the presence of a BRCA1 or BRCA2 mutation. If a mutation is found, then it is possible to test for this particular alteration in other family members, if they so desire. In the example shown, if the proband’s father has a BRCA1 mutation, there is a 50:50 probability that the mutation was transmitted to her, and genetic testing can be used to establish the absence or presence of this alteration. In this same example, if a mutation is not detected in the cancer-affected cousin, testing would not be indicated for cancer-unaffected relatives.
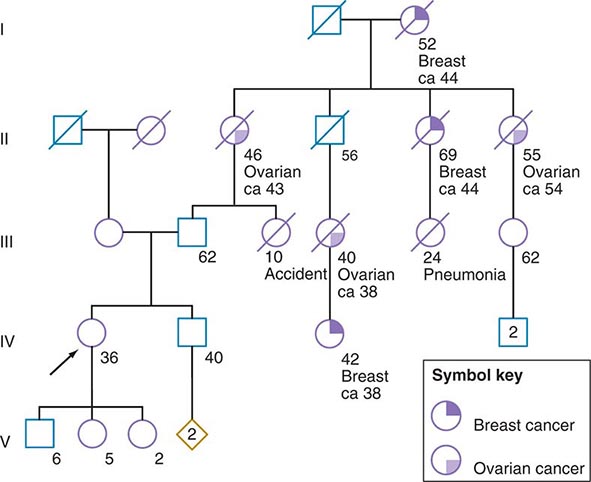
FIGURE 84-1 A 36-year-old woman (arrow) seeks consultation because of her family history of cancer. The patient expresses concern that the multiple cancers in her relatives imply an inherited predisposition to develop cancer. The family history is recorded, and records of the patient’s relatives confirm the reported diagnoses.
GENETIC TESTING FOR ADULT-ONSET DISORDERS
A critical first step before initiating genetic testing is to ensure that the correct clinical diagnosis has been made, whether it is based on family history, characteristic physical findings, pathology, or biochemical testing. Such careful clinical assessment can define the phenotype. In the traditional model of genetic testing, testing is directed initially toward the most probable genes (determined by the phenotype), which prevents unnecessary testing. Many disorders exhibit the feature of locus heterogeneity, which refers to the fact that mutations in different genes can cause phenotypically similar disorders. For example, osteogenesis imperfecta (Chap. 427), long QT syndrome (Chap. 277), muscular dystrophy (Chap. 462e), and hereditary predisposition to breast (Chap. 108) or colon (Chap. 110) cancer can each be caused by mutations in a number of distinct genes. The patterns of disease transmission, disease risk, clinical course, and treatment may differ significantly depending on the specific gene affected. Historically, the choice of which gene to test has been determined by unique clinical and family history features and the relative prevalence of candidate genetic disorders. However, rapid changes in genetic testing techniques, as discussed below, may impact this paradigm. It is now technically and financially feasible to sequence many genes (or even the whole exome) at one time. The incorporation of multiplex testing for germline mutations is rapidly evolving.
METHODOLOGIC APPROACHES TO GENETIC TESTING
Genetic testing is regulated and performed in much the same way as other specialized laboratory tests. In the United States, genetic testing laboratories are Clinical Laboratory Improvement Amendments (CLIA) approved to ensure that they meet quality and proficiency standards. A useful information source for various genetic tests is www.genetests.org. It should be noted that many tests need to be ordered through specialized laboratories.
Genetic testing is performed largely by DNA sequence analysis for mutations, although genotype can also be deduced through the study of RNA or protein (e.g., apolipoprotein E, hemoglobin S, and immunohistochemistry). For example, universal screening for Lynch syndrome via immunohistochemical analysis of colorectal cancers for absence of expression of mismatch repair proteins is under way at multiple hospitals throughout the United States. The determination of DNA sequence alterations relies heavily on the use of polymerase chain reaction (PCR), which allows rapid amplification and analysis of the gene of interest. In addition, PCR enables genetic testing on minimal amounts of DNA extracted from a wide range of tissue sources including leukocytes, mucosal epithelial cells (obtained via saliva or buccal swabs), and archival tissues. Amplified DNA can be analyzed directly by DNA sequencing, or it can be hybridized to DNA chips or blots to detect the presence of normal and altered DNA sequences. Direct DNA sequencing is frequently used for determination of hereditary disease susceptibility and prenatal diagnosis. Analyses of large alterations of the genome are possible using cytogenetics, fluorescent in situ hybridization (FISH), Southern blotting, or multiplex ligation-dependent probe amplification (MLPA) (Chap. 83e).
Massively parallel sequencing (also called next-generation sequencing) is significantly altering the approach to genetic testing for adult-onset hereditary susceptibility disorder. This technology encompasses several high-throughput approaches to DNA sequencing, all of which can reliably sequence many genes at one time. Technically, this involves the use of amplified DNA templates in a flow cell, a very different process than traditional Sanger sequencing which is time-consuming and expensive.
Multiplex panels for inherited susceptibility are commercially available and include testing of a number of genes that have been associated with the condition of interest. For example, panels are available for Brugada syndrome, hypertrophic cardiomyopathy, and Charcot-Marie-Tooth neuropathy. For many syndromes, this type of panel testing may make sense. However, in other situations, the utility of panel testing is less certain. Currently available breast cancer susceptibility panels contain six genes or more. Many of the genes included in the larger panels are associated with only a modest risk of breast cancer, and the clinical application is uncertain. An additional problem of sequencing many genes (rather than the genes for which there is most suspicion) is the identification of one or more variants of uncertain significance (VUS), discussed below.
Whole-exome sequencing (WES) is also now commercially available, although largely used in individuals with syndromes unexplained by traditional genetic testing. As cost declines, WES may be more widely used. Whole-genome sequencing is also commercially available. Although it may be quite feasible to sequence the entire genome, there are many issues in doing so, including the daunting task of analyzing the vast amount of data generated. Other issues include: (1) the optimal way in which to obtain informed consent, (2) interpretation of frequent sequence variation of uncertain significance, (3) interpretation of alterations in genes with unclear relevance to specific human pathology, and (4) management of unexpected but clinically significant genetic findings.
Testing strategies are evolving as a result of these new genetic testing platforms. As the cost of multiple gene panels and WES continue to fall, and as interpretation of such test results improve, there may be a shift from sequential single-gene (or a few genes) testing to multigene testing. For example, presently, a 30-year-old woman with breast cancer but no family history of cancer and no syndromic features would undergo BRCA1/2 testing. If negative, she would subsequently be offered TP53 testing. Notably, a reasonable number of individuals offered TP53 testing for Li-Fraumeni syndrome decline because mutations are associated with extremely high cancer risks (including childhood cancers) in multiple organs and there are no proven interventions to mitigate risk. Without features consistent with Cowden syndrome, the woman would not be routinely offered PTEN testing or testing for CHEK2, ATM, BRIP, BARD, NBN, and PALB2. However, it is now possible to synchronously analyze all of the aforementioned genes, for a nominally higher cost than BRCA1/2 testing alone. Concerns about such panels include appropriate consent strategies related to unexpected findings, VUS, and unclear clinical utility of testing moderate-penetrance genes. Thus, changes from the traditional model of single-gene genetic testing should be done with caution (Fig. 84-2).
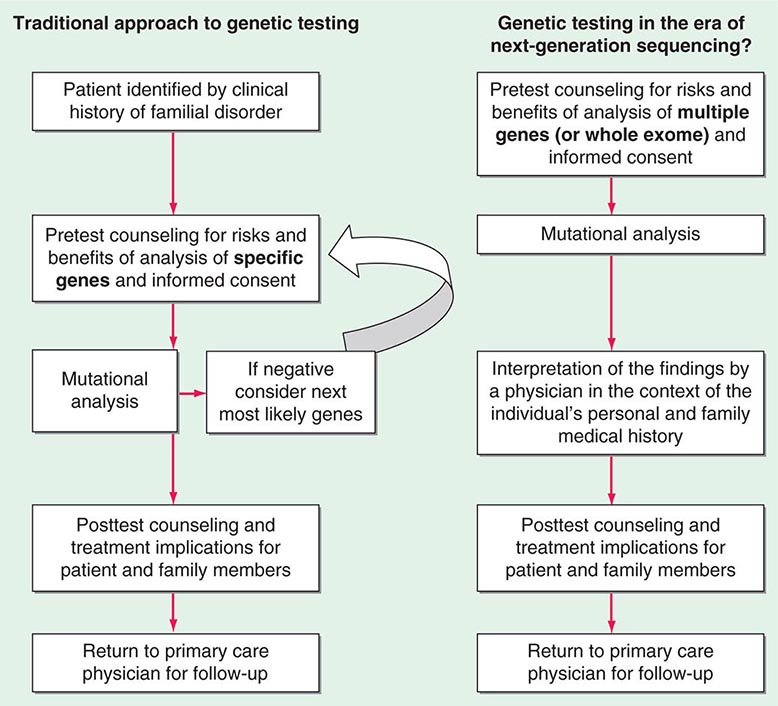
FIGURE 84-2 Approach to genetic testing.
Limitations to the accuracy and interpretation of genetic testing exist. In addition to technical errors, genetics tests are sometimes designed to detect only the most common mutations. In addition, genetic testing has evolved over time. For example, it was not possible to obtain commercially available comprehensive large genomic rearrangement testing for BRCA1 and BRCA2 until 2006. Therefore, a negative result must be qualified by the possibility that the individual may have a mutation that was not included in the test. In addition, a negative result does not mean that there is not a mutation in some other gene that causes a similar inherited disorder. A negative result, unless there is known mutation in the family, is typically classified as uninformative.
VUS are another limitation to genetic testing. A VUS (also termed unclassified variant) is a sequence variation in a gene where the effect of the alteration on the function of the protein is not known. Many of these variants are single nucleotide substitutions (also called missense mutations) that result in a single amino acid change. Although many VUSs will ultimately be reclassified as benign polymorphisms, some will prove to be functionally important. As more genes are sequenced (for example, in a multiplex panel or through WES), the percentage of individuals found to have a VUS increases significantly. The finding of a VUS is difficult for patients and providers alike and complicates decisions regarding medical management.
Clinical utility is an important consideration because genetic testing for susceptibility to chronic diseases is increasingly integrated into the practice of medicine. In some situations, there is clear clinical utility to genetic testing with significant evidence-based changes in medical management decisions based on results. However, in many cases, the discovery of disease-associated genes has outpaced studies that assess how such information should be used in the clinical management of the patient and family. This is particularly true for moderate- and low-penetrance gene mutations. Therefore, predictive genetic testing should be approached with caution and only offered to patients who have been adequately counseled and have provided informed consent.
Predictive genetic testing falls into two distinct categories. Presymptomatic testing applies to diseases where a specific genetic alteration is associated with a near 100% likelihood of developing disease. In contrast, predisposition testing predicts a risk for disease that is less than 100%. For example, presymptomatic testing is available for those at risk for Huntington’s disease; whereas, predisposition testing is considered for those at risk for hereditary colon cancer. It is important to note that for the majority of adult-onset disorders, testing is only predictive. Test results cannot reveal with confidence whether, when, or how the disease will manifest itself. For example, not everyone with the apolipoprotein E4 allele will develop Alzheimer’s disease, and individuals without this genetic marker can still develop the disorder.
The optimal testing strategy for a family is to initiate testing in an affected family member first. Identification of a mutation can direct the testing of other at-risk family members (whether symptomatic or not). In the absence of additional familial or environmental risk factors, individuals who test negative for the mutation found in the affected family member can be informed that they are at general population risk for that particular disease. Furthermore, they can be reassured that they are not at risk for passing the mutation on to their children. On the other hand, asymptomatic family members who test positive for the known mutation must be informed that they are at increased risk for disease development and for transmitting the alteration to their children.
Pretest counseling and education are important, as is an assessment of the patient’s ability to understand and cope with test results. Genetic testing has implications for entire families, and thus individuals interested in pursuing genetic testing must consider how test results might impact their relationships with relatives, partners, spouses, and children. In families with a known genetic mutation, those who test positive must consider the impact of their carrier status on their present and future lifestyles; those who test negative may manifest survivor guilt. Parents who are found to have a disease-associated mutation often express considerable anxiety and despair as they address the issue of risk to their children. In addition, some individuals consider options such as preimplantation genetic diagnosis in their reproductive decision making.
When a condition does not manifest until adulthood, clinicians and parents are faced with the question of whether at-risk children should be offered genetic testing and, if so, at what age. Although the matter is debated, several professional organizations have cautioned that genetic testing for adult-onset disorders should not be offered to children. Many of these conditions have no known interventions in childhood to prevent disease; consequently, such information can pose significant psychosocial risk to the child. In addition, there is concern that testing during childhood violates a child’s right to make an informed decision regarding testing upon reaching adulthood. On the other hand, testing should be offered in childhood for disorders that may manifest early in life, especially when management options are available. For example, children with multiple endocrine neoplasia 2 (MEN 2) may develop medullary thyroid cancer early in life and should be considered for prophylactic thyroidectomy (Chap. 408). Similarly, children with familial adenomatous polyposis (FAP) due to a mutation in APC may develop polyps in their teens with progression to invasive cancer in the twenties, and therefore, colonoscopy screening is started between the ages of 10 and 15 years (Chap. 110).
INFORMED CONSENT
Informed consent for genetic testing begins with education and counseling. The patient should understand the risks, benefits, and limitations of genetic testing, as well as the potential implications of test results. Informed consent should include a written document, drafted clearly and concisely in a language and format that is understandable to the patient. Because molecular genetic testing of an asymptomatic individual often allows prediction of future risk, the patient should understand all potential long-term medical, psychological, and social implications of testing. There have long been concerns about the potential for genetic discrimination. The Genetic Information Nondiscrimination Act (GINA) was passed in 2008 and provides some protections related to job and health insurance discrimination. It is important to explore with patients the potential impact of genetic test results on future health as well as disability and life insurance coverage. Patients should understand that alternatives remain available if they decide not to pursue genetic testing, including the option of delaying testing to a later date. The option of DNA banking should be presented so that samples are readily available for future use by family members, if needed.
FOLLOW-UP CARE AFTER TESTING
Depending on the nature of the genetic disorder, posttest interventions may include: (1) cautious surveillance and awareness; (2) specific medical interventions such as enhanced screening, chemoprevention, or risk-reducing surgery; (3) risk avoidance; and (4) referral to support services. For example, patients with known deleterious mutations in BRCA1 or BRCA2 are strongly encouraged to undergo risk-reducing salpingo-oophorectomy and are offered intensive breast cancer screening as well as the option of risk-reducing mastectomy. In addition, such women may wish to take chemoprevention with tamoxifen, raloxifene, or exemestane. Those with more limited medical management and prevention options, such as patients with Huntington’s disease, should be offered continued follow-up and supportive services, including physical and occupational therapy and social services or support groups as indicated. Specific interventions will change as research continues to enhance our understanding of the medical management of these genetic conditions and more is learned about the functions of the gene products involved.
Individuals who test negative for a mutation in a disease-associated gene identified in an affected family member must be reminded that they may still be at risk for the disease. This is of particular importance for common diseases such as diabetes mellitus, cancer, and coronary artery disease. For example, a woman who finds that she does not carry the disease-associated mutation in BRCA2 previously discovered in the family should be reminded that she still requires the same breast cancer screening recommended for the general population.
GENETIC COUNSELING AND EDUCATION
Genetic counseling should be distinguished from genetic testing and screening, although genetic counselors are often involved in issues related to testing. Genetic counseling refers to a communication process that deals with human problems associated with the occurrence of risk of a genetic disorder in a family. Genetic risk assessment is complex and often involves elements of uncertainty. Counseling, therefore, includes genetic education as well as psychosocial counseling. Genetic counseling can be useful in a wide range of situations (Table 84-1). The role of the genetic counselor includes the following:
1. Gather and document a detailed family history.
2. Educate patients about general genetic principles related to disease risk, both for themselves and for others in the family.
3. Assess and enhance the patient’s ability to cope with the genetic information offered.
4. Discuss how nongenetic factors may relate to the ultimate expression of disease.
5. Address medical management issues.
6. Assist in determining the role of genetic testing for the individual and the family.
7. Ensure the patient is aware of the indications, process, risks, benefits, and limitations of the various genetic testing options.
8. Assist the patient, family, and referring physician in the interpretation of the test results.
9. Refer the patient and other at-risk family members for additional medical and support services, if necessary.
|
INDICATIONS FOR GENETIC COUNSELING |
Genetic counseling is generally offered in a nondirective manner, wherein patients learn to understand how their values factor into a particular medical decision. Nondirective counseling is particularly appropriate when there are no data demonstrating a clear benefit associated with a particular intervention or when an intervention is considered experimental. For example, nondirective genetic counseling is used when a person is deciding whether to undergo genetic testing for Huntington’s disease. At this time, there is no clear benefit (in terms of medical outcome) to an at-risk individual undergoing genetic testing for this disease because its course cannot be altered by therapeutic interventions. However, testing can have an important impact on the individual’s perception of advanced care planning and his or her interpersonal relationships and plans for childbearing. Therefore, the decision to pursue testing rests on the individual’s belief system and values. On the other hand, a more directive approach is appropriate when a condition can be treated. In a family with FAP, colon cancer screening and prophylactic colectomy should be recommended for known APC mutation carriers. The counselor and clinician following this family must ensure that the at-risk family members have access to the resources necessary to adhere to these recommendations.
Genetic education is central to an individual’s ability to make an informed decision regarding testing options and treatment. An adequate knowledge of patterns of inheritance will allow patients to understand the probability of disease risk for themselves and other family members. It is also important to impart the concepts of disease penetrance and expression. For most complex adult-onset genetic disorders, asymptomatic patients should be advised that a positive test result does not always translate into future disease development. In addition, the role of nongenetic factors, such as environmental exposures and lifestyle, must be discussed in the context of multifactorial disease risk and disease prevention. Finally, patients should understand the natural history of the disease as well as the potential options for intervention, including screening, prevention, and in certain circumstances, pharmacologic treatment or prophylactic surgery.
THERAPEUTIC INTERVENTIONS BASED ON GENETIC RISK FOR DISEASE
Specific treatments are available for a number of genetic disorders. Strategies for the development of therapeutic interventions have a long history in childhood metabolic diseases; however, these principles have been applied in the diagnosis and management of adult-onset diseases as well (Table 84-2). Hereditary hemochromatosis is usually caused by mutations in HFE (although other genes have been less commonly associated) and manifests as a syndrome of iron overload, which can lead to liver disease, skin pigmentation, diabetes mellitus, arthropathy, impotence in males, and cardiac issues (Chap. 428). When identified early, the disorder can be managed effectively with therapeutic phlebotomy. Therefore, when the diagnosis of hemochromatosis has been made in a proband, it is important to counsel and offer testing to other family members in order to minimize the impact of the disorder.
|
EXAMPLE OF GENETIC TESTING AND POSSIBLE INTERVENTIONS |
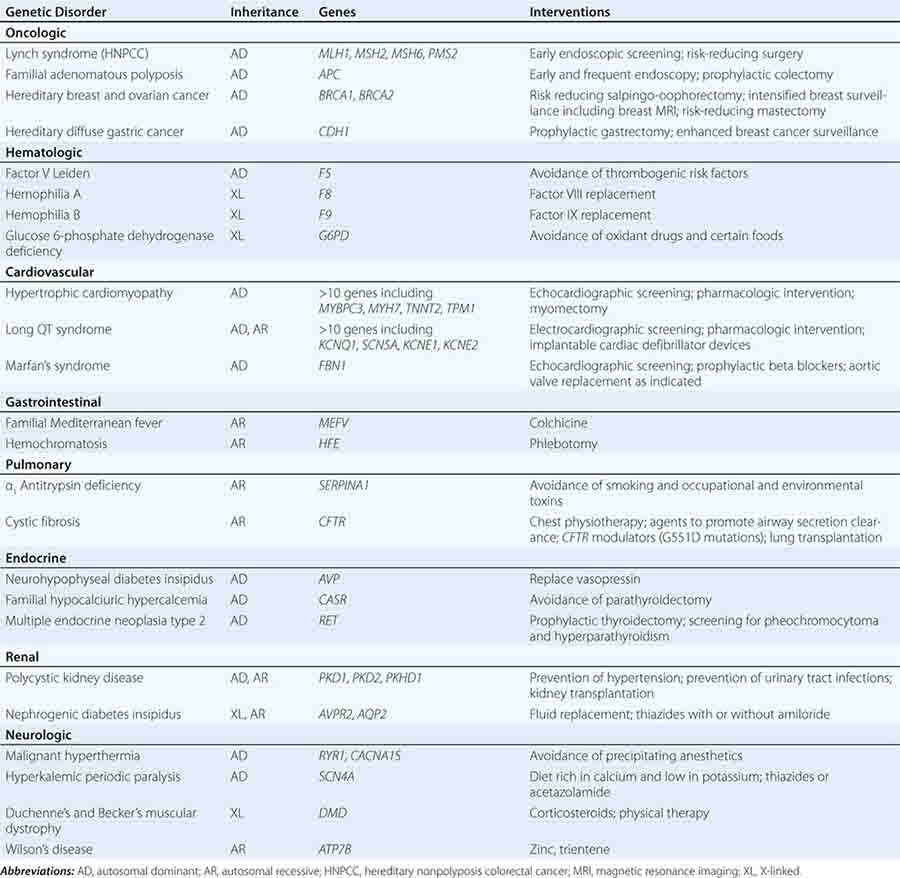
Preventative measures and therapeutic interventions are not restricted to metabolic disorders. Identification of familial forms of long QT syndrome, associated with ventricular arrhythmias, allows early electrocardiographic testing and the use of prophylactic antiarrhythmic therapy, overdrive pacemakers, or defibrillators. Individuals with familial hypertrophic cardiomyopathy can be screened by ultrasound, treated with beta blockers or other drugs, and counseled about the importance of avoiding strenuous exercise and dehydration. Those with Marfan’s syndrome can be treated with beta blockers or angiotensin II receptor blockers and monitored for the development of aortic aneurysms.
The field of pharmacogenetics identifies genes that alter drug metabolism or confer susceptibility to toxic drug reactions. Pharmacogenetics seeks to individualize drug therapy in an attempt to improve treatment outcomes and reduce toxicity. Examples include thiopurine methyltransferase (TPMT) deficiency, dihydropyrimidine dehydrogenase deficiency, malignant hyperthermia, and glucose-6-phosphate deficiency. Despite successes in this area, it is not always clear how to incorporate pharmacogenetics into clinical care. For example, although there is an association with CYP2C6 and VKORC1 genotypes and warfarin dosing, there is no evidence that incorporating genotyping into clinical practice improves patient outcomes.
The identification of germline abnormalities that increase the risk of specific types of cancer is rapidly changing clinical management. Identifying family members with mutations that predispose to FAP or Lynch syndrome leads to recommendations of early cancer screening and prophylactic surgery, as well as consideration of chemoprevention and attention to healthy lifestyle habits. Similar principles apply to familial forms of melanoma as well as cancers of the breast, ovary, and thyroid. In addition to increased screening and prophylactic surgery, the identification of germline mutations associated with cancer may also lead to the development of targeted therapeutics, for example, the ongoing development of PARP inhibitors in those with BRCA-associated cancers.
Although the role of genetic testing in the clinical setting continues to evolve, such testing holds the promise of allowing early and more targeted interventions that can reduce morbidity and mortality. Rapid technologic advances are changing the ways in which genetic testing is performed. As genetic testing becomes less expensive and technically easier to perform, it is anticipated that there will be an expansion of its use. This will present challenges, but also opportunities. It is critical that physicians and other health care professionals keep current with advances in genetic medicine in order to facilitate appropriate referral for genetic counseling and judicious use of genetic testing, as well as to provide state-of-the-art, evidence-based care for affected or at-risk patients and their relatives.
85e |
Mitochondrial DNA and Heritable Traits and Diseases |
Mitochondria are cytoplasmic organelles whose major function is to generate ATP by the process of oxidative phosphorylation under aerobic conditions. This process is mediated by the respiratory electron transport chain (ETC) multiprotein enzyme complexes I–V and the two electron carriers, coenzyme Q (CoQ) and cytochrome c. Other cellular processes to which mitochondria make a major contribution include apoptosis (programmed cell death) and additional cell type–specific functions (Table 85e-1). The efficiency of the mitochondrial ETC in ATP production is a major determinant of overall body energy balance and thermogenesis. In addition, mitochondria are the predominant source of reactive oxygen species (ROS), whose rate of production also relates to the coupling of ATP production to oxygen consumption. Given the centrality of oxidative phosphorylation to the normal activities of almost all cells, it is not surprising that mitochondrial dysfunction can affect almost any organ system (Fig. 85e-1). Thus, physicians in many disciplines might encounter patients with mitochondrial diseases and should be aware of their existence and characteristics.
|
FUNCTIONS OF MITOCHONDRIA |
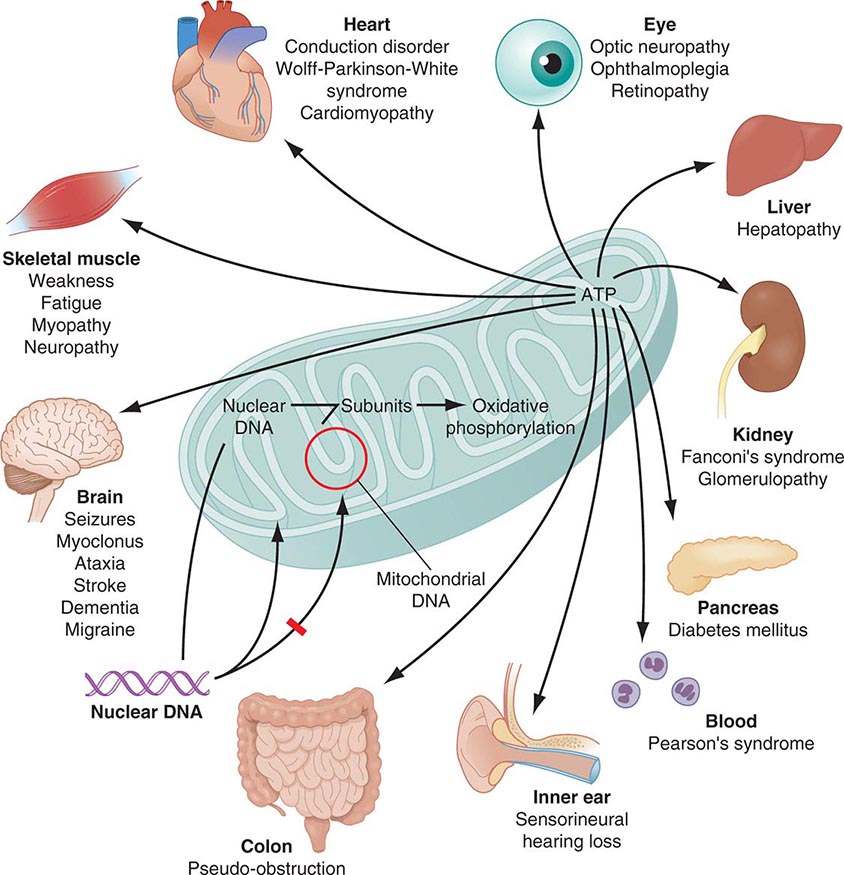
FIGURE 85e-1 Dual genetic control and multiple organ system manifestations of mitochondrial disease. (Reproduced with permission from DR Johns: Mitochondrial DNA and disease. N Engl J Med 333:638, 1995.)
The integrated activity of an estimated 1500 gene products is required for normal mitochondrial biogenesis, function, and integrity. Almost all of these are encoded by nuclear genes and thus follow the rules and patterns of nuclear genomic inheritance (Chap. 84). These nuclear-encoded proteins are synthesized in the cell cytoplasm and imported to their location of activity within the mitochondria through a complex biochemical process. In addition, the mitochondria contain their own small genome consisting of numerous copies (polyploidy) per mitochondrion of a circular, double-strand mitochondrial DNA (mtDNA) molecule comprising 16,569 nucleotides. This mtDNA sequence (also known as the “mitogenome”) might represent the remnants of endosymbiotic prokaryotes from which mitochondria are thought to have originated. The mtDNA sequence contains a total of 37 genes, of which 13 encode mitochondrial protein components of the ETC (Fig. 85e-2). The remaining 22 tRNA- and 2 rRNA-encoding genes are dedicated to the process of translating the 13 mtDNA-encoded proteins. This dual nuclear and mitochondrial genetic control of mitochondrial function results in unique and diagnostically challenging patterns of inheritance. The current chapter focuses on heritable traits and diseases related to the mtDNA component of the dual genetic control of mitochondrial function. The reader is referred to Chaps. 84 and 462e for consideration of mitochondrial disease originating from mutations in the nuclear genome. The latter include (1) disorders due to mutations in nuclear genes directly encoding structural components or assembly factors of the oxidative phosphorylation complexes, (2) disorders due to mutations in nuclear genes encoding proteins indirectly related to oxidative phosphorylation, and (3) mtDNA depletion syndromes (MDS) characterized by a reduction of mtDNA copy number in affected tissues without mutations or rearrangements in the mtDNA.
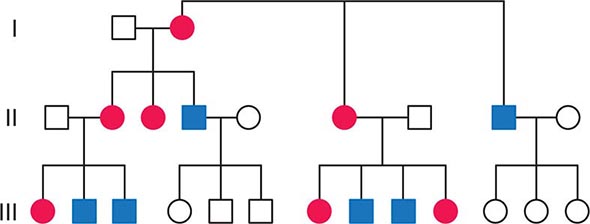
FIGURE 85e-2 Maternal inheritance of mitochondrial DNA (mtDNA) disorders and heritable traits. Affected women (filled circles) transmit the trait to their children. Affected men (filled squares) do not transmit the trait to any of their offspring.
MITOCHONDRIAL DNA STRUCTURE AND FUNCTION
As a result of its circular structure and extranuclear location, the replication and transcription mechanisms of mtDNA differ from the corresponding mechanisms in the nuclear genome, whose nucleosomal packaging and structure are more complex. Because each cell contains many copies of mtDNA, and because the number of mitochondria can vary during the lifetime of each cell, mtDNA copy number is not directly coordinated with the cell cycle. Thus, vast differences in mtDNA copy number are observed between different cell types and tissues and during the lifetime of a cell. Another important feature of the mtDNA replication process is a reduced stringency of proofreading and replication error correction, leading to a greater degree of sequence variation compared to the nuclear genome. Some of these sequence variants are silent polymorphisms that do not have the potential for a phenotypic or pathogenic effect, whereas others may be considered pathogenic mutations.
With respect to transcription, initiation can occur on both strands and proceeds through the production of an intronless polycistronic precursor RNA, which is then processed to produce the 13 individual mRNA and 24 individual tRNA and rRNA products. The 37 mtDNA genes comprise fully 93% of the 16,569 nucleotides of the mtDNA in what is known as the coding region. The control region consists of ~1.1 kilobases (kb) of noncoding DNA, which is thought to have an important role in replication and transcription initiation.
MATERNAL INHERITANCE AND LACK OF RECOMBINATION
In contrast to homologous pair recombination that takes place in the nucleus, mtDNA molecules do not undergo recombination, such that mutational events represent the only source of mtDNA genetic diversification. Moreover, with very rare exceptions, it is only the maternal DNA that is transmitted to the offspring. The fertilized oocyte degrades mtDNA carried from the sperm in a complex process involving the ubiquitin proteasome system. Thus, although mothers transmit their mtDNA to both their sons and daughters, only the daughters are able to transmit the inherited mtDNA to future generations. Accordingly, mtDNA sequence variation and associated phenotypic traits and diseases are inherited exclusively along maternal lines.
As noted below, because of the complex relationship between mtDNA mutations and disease expression, sometimes this maternal inheritance is difficult to recognize at the clinical or pedigree level. However, evidence of paternal transmission can almost certainly rule out an mtDNA genetic origin of phenotypic variation or disease; conversely, a disease affecting both sexes without evidence of paternal transmission strongly suggests a heritable mtDNA disorder (Fig. 85e-2).
MULTIPLE COPY NUMBER (POLYPLOIDY), HIGH MUTATION RATE, HETEROPLASMY, AND MITOTIC SEGREGATION
Each aerobic cell in the body has multiple mitochondria, often numbering many hundreds or more in cells with extensive energy production requirements. Furthermore, the number of copies of mtDNA within each mitochondrion varies from several to hundreds; this is true of both somatic as well as germ cells, including oocytes in females. In the case of somatic cells, this means that the impact of most newly acquired somatic mutations is likely to be very small in terms of total cellular or organ system function; however, because of the manyfold higher mutation rate during mtDNA replication, numerous different mutations may accumulate with aging of the organism. It has been proposed that the total cumulative burden of acquired somatic mtDNA mutations with age may result in an overall perturbation of mitochondrial function, contributing to age-related reduction in the efficiency of oxidative phosphorylation and increased production of damaging ROS. The accumulation of such acquired somatic mtDNA mutations with aging may contribute to age-related diseases, such as metabolic syndrome and diabetes, cancer, and neurodegenerative and cardiovascular disease in any given individual. However, somatic mutations are not carried forward to the next generation, and the hereditary impact of mtDNA mutagenesis requires separate consideration of events in the female germline.
The multiple mtDNA copy number within each cell, including the maternal germ cells, results in the phenomenon of heteroplasmy, in contrast to much greater uniformity (homoplasy) of somatic nuclear DNA sequence. Heteroplasmy for a given mtDNA sequence variant or mutation arises as a result of the coexistence within a cell, tissue, or individual of mtDNA molecules bearing more than one version of the sequence variant (Fig. 85e-3). The importance of the heteroplasmy phenomena to the understanding of mtDNA-related mitochondrial diseases is critical. The coexistence of mutant and nonmutant mtDNA and the variation of the mutant load among individuals from the same maternal sibship, and across organs and tissues within the same individual, play a pivotal role in the manifestation and severity of disease and are crucial to understanding the complexity of inheritance of mtDNA disorders. At the level of the oocyte, the percentage of mtDNA molecules bearing each version of the polymorphic sequence variant or mutation depends on stochastic events related to partitioning of mtDNA molecules during the process of oogenesis itself. Thus, oocytes differ from each other in the degree of heteroplasmy for that sequence variant or mutation. In turn, the heteroplasmic state is carried forward to the zygote and to the organism as a whole, to varying degrees, depending on mitotic segregation of mtDNA molecules during organ system development and maintenance. For this reason, in vitro fertilization, followed by preimplantation genetic diagnosis (PGD), is not as predictive of the genetic health of the offspring in the case of mtDNA mutations as in the case of the nuclear genome. Similarly, the impact of somatic mtDNA mutations acquired during development and subsequently also shows an enormous spectrum of variability.
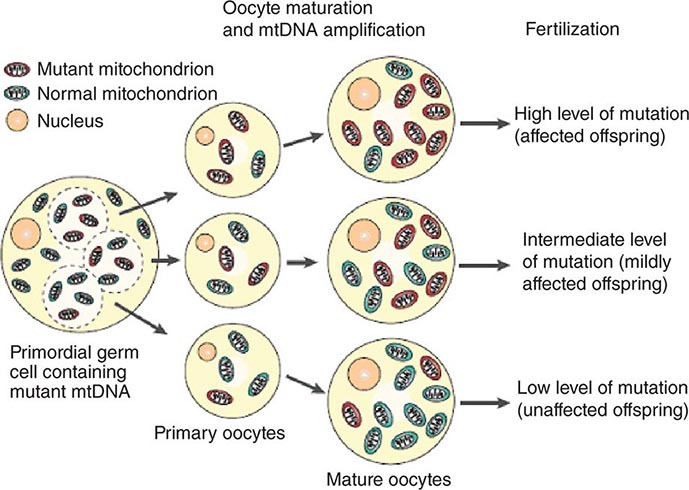
FIGURE 85e-3 Heteroplasmy and the mitochondrial genetic bottleneck. During the production of primary oocytes, a selected number of mitochondrial DNA (mtDNA) molecules are transferred into each oocyte. Oocyte maturation is associated with the rapid replication of this mtDNA population. This restriction-amplification event can lead to a random shift of mtDNA mutational load between generations and is responsible for the variable levels of mutated mtDNA observed in affected offspring from mothers with pathogenic mtDNA mutations. Mitochondria that contain mutated mtDNA are shown in red, and those with normal mtDNA are shown in green. (Reproduced with permission from R Taylor, D Turnbull: Mitochondrial DNA mutations in human disease. Nat Rev Genetics 6:389, 2005.)
Mitotic segregation refers to the unequal distribution of wild-type and mutant versions of mtDNA molecules during all cell divisions that occur during prenatal development and subsequently throughout the lifetime of an individual. The phenotypic effect or disease impact will, thus, be a function not only of the inherent disruptive effect (pathogenicity) on the mtDNA-encoded gene (coding region mutations) or integrity of the mtDNA molecule (control region mutations), but also of its distribution among the multiple copies of mtDNA in the various mitochondria, cells, and tissues of the affected individual. Thus, one consequence can be the generation of a bottleneck due to the marked decline in given sets of mtDNA variants, consequent to such mitotic segregation. Heterogeneity arises from differences in the degree of heteroplasmy among oocytes of the affected female, together with subsequent mitotic segregation of the pathogenic mutation during tissue and organ development, and throughout the lifetime of the individual offspring. The actual expression of disease might then depend on a threshold percentage of mitochondria whose function is disrupted by mtDNA mutations. This in turn confounds hereditary transmission patterns and hence genetic diagnosis of pathogenic heteroplasmic mutations. Generally, if the proportion of mutant mtDNA is less than 60%, the individual is unlikely to be affected, whereas proportions exceeding 90% cause clinical disease.
HOMOPLASMIC VARIANTS AND HUMAN MTDNA PHYLOGENY
In contrast to classic mtDNA diseases, most of which begin in childhood and are the result of heteroplasmic mutations as noted above, during the course of human evolution, certain mtDNA sequence variants have drifted to a state of homoplasmy, wherein all of the mtDNA molecules in the organism contain the new sequence variant. This arises due to a “bottleneck” effect followed by genetic drift during the very process of oogenesis itself (Fig. 85e-3). In other words, during certain stages of oogenesis, the mtDNA copy number becomes so substantially reduced that the particular mtDNA species bearing the novel or derived sequence variant may become the increasingly predominant, and eventually exclusive, version of the mtDNA for that particular nucleotide site. All of the offspring of a woman bearing an mtDNA sequence variant or mutation that has become homoplasmic will also be homoplasmic for that variant and will transmit the sequence variant forward in subsequent generations.
Considerations of reproductive fitness limit the evolutionary or population emergence of homoplasmic mutations that are lethal or cause severe disease in infancy or childhood. Thus, with a number of notable exceptions (e.g., mtDNA mutations causing Leber’s hereditary optic neuropathy; see below), most homoplasmic mutations are considered to be neutral markers of human evolution, which are useful and interesting in the population genetics analysis of shared maternal ancestry but which have little significance in human phenotypic variation or disease predisposition.
More importantly is the understanding that this accumulation of homoplastic mutations occurs at a genetic locus that is transmitted only through the female germline and that lacks recombination. In turn, this enables reconstruction of the sequential topology and radiating phylogeny of mutations accumulated through the course of human evolution since the time of the most recent common mtDNA ancestor of all contemporary mtDNA sequences, some 200,000 years ago. The term haplogroup is usually used to define major branching points in the human mtDNA phylogeny, nested one within the other, which often demonstrate striking continental geographic ancestral partitioning. At the level of the complete mtDNA sequence, the term haplotype is usually used to describe the sum of mutations observed for a given mtDNA sequence and as compared to a reference sequence, such that all haplotypes falling within a given haplogroup share the total sum of mutations that have accumulated since the most recent common ancestor and the bifurcation point they mark. The remaining observed variants are private to each haplotype. Consequentially, human mtDNA sequence is an almost perfect molecular prototype for a nonrecombining locus, and its variation has been extensively used in phylogenetic studies. Moreover, the mtDNA mutation rate is considerably higher than the rate observed for the nuclear genome, especially in the control region, which contains the displacement, or D-loop, in turn comprising two adjacent hypervariable regions (HVR-I and HVR-II). Together with the absence of recombination, this amplifies drift to high frequencies of novel haplotypes. As a result, mtDNA haplotypes are more highly partitioned across geographically defined populations than sequence variants in other parts of the genome. Despite extensive research, it has not been well established that such haplotype-based partitioning has a significant influence on human health conditions. However, mtDNA-based phylogenetic analysis can be used both as a quality assurance tool and as a filter in distinguishing neutral mtDNA variants comprising human mtDNA phylogeny from potentially deleterious mutations.
MITOCHONDRIAL DNA DISEASE
The true prevalence of mtDNA disease is difficult to estimate because of the phenotypic heterogeneity that occurs as a function of heteroplasmy, the challenge of detecting and assessing heteroplasmy in different affected tissues, and the other unique features of mtDNA function and inheritance described above. It is estimated that at least 1 in 200 healthy humans harbors a pathogenic mtDNA mutation with the potential to causes disease, but that heteroplasmic germline pathogenic mtDNA mutations actually affect up to approximately 1 in 8500 individuals.
The true disease burden relating to mtDNA sequence variation will only be known when the following capabilities become available: (1) ability to distinguish a completely neutral sequence variant from a true phenotype-modifying or pathogenic mutation, (2) accurate assessment of heteroplasmy that can be determined with fidelity, and (3) a systems biology approach (Chap. 87e) to determine the network of epistatic interactions of mtDNA sequence variations with mutations in the nuclear genome.
OVERVIEW OF CLINICAL AND PATHOLOGIC FEATURES OF HUMAN MTDNA DISEASE
Given the vital roles of mitochondria in all nucleated cells, it is not surprising that mtDNA mutations can affect numerous tissues with pleiotropic effects. More than 200 different disease-causing, mostly heteroplasmic mtDNA mutations have been described affecting ETC function. Figure 85e-4 provides a partial mtDNA map of some of the better characterized of these disorders. A number of clinical clues can increase the index of suspicion for a heteroplasmic mtDNA mutation as an etiology of a heritable trait or disease, including (1) familial clustering with absence of paternal transmission; (2) adherence to one of the classic syndromes (see below) or paradigmatic combinations of disease phenotypes involving several organ systems that normally do not fit together within a single nuclear genomic mutation category; (3) a complex of laboratory and pathologic abnormalities that reflect disruption in cellular energetics (e.g., lactic acidosis and neurodegenerative and myodegenerative symptoms with the finding of ragged red fibers, reflecting the accumulation of abnormal mitochondria under the muscle sarcolemmal membrane); or (4) a mosaic pattern reflecting a heteroplasmic state.
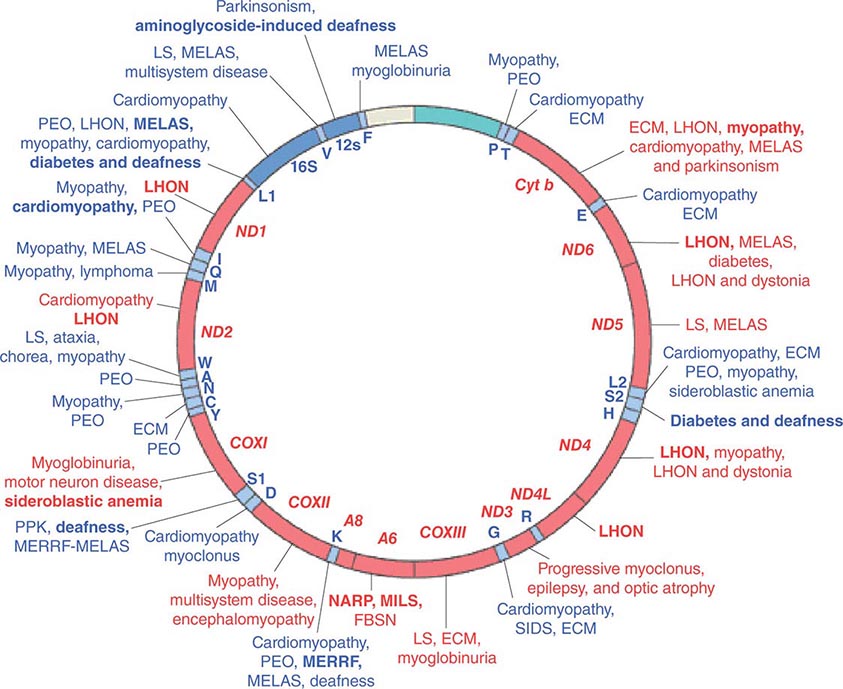
FIGURE 85e-4 Mutations in the human mitochondrial genome known to cause disease. Disorders that are frequently or prominently associated with mutations in a particular gene are shown in boldface. Diseases due to mutations that impair mitochondrial protein synthesis are shown in blue. Diseases due to mutations in protein-coding genes are shown in red. ECM, encephalomyopathy; FBSN, familial bilateral striatal necrosis; LHON, Leber’s hereditary optic neuropathy; LS, Leigh syndrome; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERRF, myoclonic epilepsy with ragged red fibers; MILS, maternally inherited Leigh syndrome; NARP, neuropathy, ataxia, and retinitis pigmentosa; PEO, progressive external ophthalmoplegia; PPK, palmoplantar keratoderma; SIDS, sudden infant death syndrome. (Reproduced with permission from S DiMauro, E Schon: Mitochondrial respiratory-chain diseases. N Engl J Med 348:2656, 2003.)
Heteroplasmy can sometimes be elegantly demonstrated at the tissue level using histochemical staining for enzymes in the oxidative phosphorylation pathway, with a mosaic pattern indicating heterogeneity of the genotype for the coding region for the mtDNA-encoded enzyme. Complex II, CoQ, and cytochrome c are exclusively encoded by nuclear DNA. In contrast, complexes I, III, IV, and V contain at least some subunits encoded by mtDNA. Just 3 of the 13 subunits of the ETC complex IV enzyme, cytochrome c oxidase, are encoded by mtDNA, and, therefore, this enzyme has the lowest threshold for dysfunction when a threshold level of mutated mtDNA is reached. Histochemical staining for cytochrome c oxidase activity in tissues of patients affected with heteroplasmic inherited mtDNA mutations (or with the somatic accumulation of mtDNA mutations, see below) can show a mosaic pattern of reduced histochemical staining in comparison with histochemical staining for the complex II enzyme, succinate dehydrogenase (Fig. 85e-5). Heteroplasmy can also be detected at the genetic level through direct Sanger-type mtDNA genotyping under special conditions, although clinically significant low levels of heteroplasmy can escape detection in genomic samples extracted from whole blood using conventional genotyping and sequencing techniques.
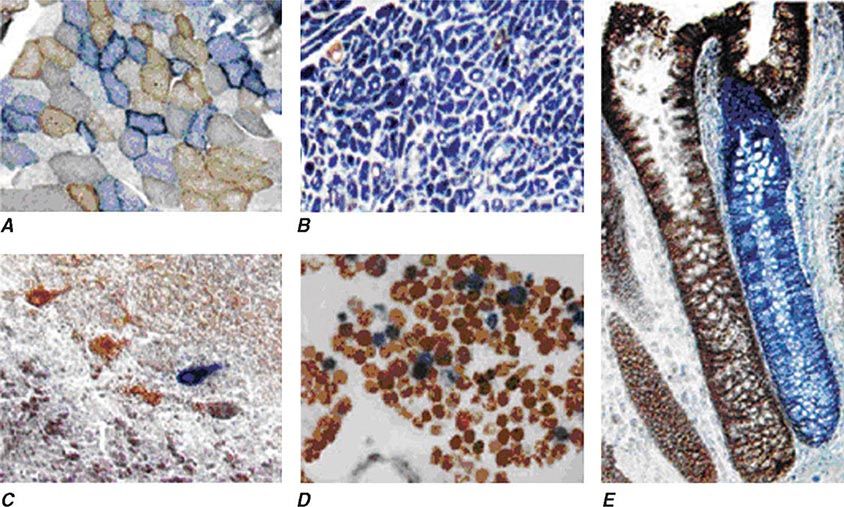
FIGURE 85e-5 Cytochrome c oxidase (COX) deficiency in mitochondrial DNA (mtDNA)–associated disease. Transverse tissue sections that have been stained for COX and succinate dehydrogenase (SDH) activities sequentially, with COX-positive cells shown in brown and COX-deficient cells shown in blue. A. Skeletal muscle from a patient with a heteroplasmic mitochondrial tRNA point mutation. The section shows a typical “mosaic” pattern of COX activity, with many muscle fibers harboring levels of mutated mtDNA that are above the crucial threshold to produce a functional enzyme complex. B. Cardiac tissue (left ventricle) from a patient with a homoplasmic tRNA mutation that causes hypertrophic cardiomyopathy, which demonstrates an absence of COX in most cells. C. A section of cerebellum from a patient with mtDNA rearrangement that highlights the presence of COX-deficient neurons. D, E. Tissues that show COX deficiency due to clonal expansion of somatic mtDNA mutations within single cells—a phenomenon that is seen in both postmitotic cells (D; extraocular muscles) and rapidly dividing cells (E; colonic crypt) in aging humans. (Reproduced with permission from R Taylor, D Turnbull: Mitochondrial DNA mutations in human disease. Nat Rev Genetics 6:389, 2005.)
The emerging next-generation sequencing (NGS) techniques and their rapid penetration and recognition as useful clinical diagnostic tools are expected to also dramatically improve the clinical genetic diagnostic evaluation of mitochondrial diseases at the level of both the nuclear genome and mtDNA. In the context of the larger nuclear genome, the ability of NGS techniques to dramatically increase the speed at which DNA can be sequenced at a fraction of the cost of conventional Sanger-type sequencing technology is particularly beneficial. Low sequencing costs and short turnaround time expedite “first-tier” screening of panels of hundreds of previously known or suspected mitochondrial disease genes or screening for the entire exome or genome in an attempt to identify novel genes and mutations affecting different patients or families. In the context of the mtDNA, NGS approaches hold the particular promise for rapid and reliable detection of heteroplasmy in different affected tissues. Although Sanger sequencing allows for complete coverage of the mtDNA, it is limited by the lack of deep coverage and low sensitivity for heteroplasmy detection when it is much less than 50%. In contrast, NGS technology is an excellent tool for rapidly and accurately obtaining a patient’s predominant mtDNA sequence and also lower frequency heteroplasmic variants. This is enabled by deep coverage of the genome through multiple independent sequence reads. Accordingly, recent studies making use of NGS techniques have demonstrated sequence accuracy equivalent to Sanger-type sequencing, but also have uncovered heretofore unappreciated heteroplasmy rates ranging between 10 and 50% and detection of single-nucleotide heteroplasmy down to levels of <10%.
Clinically, the most striking overall characteristic of mitochondrial genetic disease is the phenotypic heterogeneity associated with mtDNA mutations. This extends to intrafamilial phenotypic heterogeneity for the same mtDNA pathogenic mutation and, conversely, to the overlap of phenotypic disease manifestations with distinct mutations. Thus, although fairly consistent and well-defined “classic” syndromes have been attributed to specific mutations, frequently “nonclassic” combinations of disease phenotypes ranging from isolated myopathy to extensive multisystem disease are often encountered, rendering genotype-phenotype correlation challenging. In both classical and nonclassical mtDNA disorders, there is often a clustering of some combination of abnormalities affecting the neurologic system (including optic nerve atrophy, pigment retinopathy, and sensorineural hearing loss), cardiac and skeletal muscle (including extraocular muscles), and endocrine and metabolic systems (including diabetes mellitus). Additional organ systems that may be affected include the hematopoietic, renal, hepatic, and gastrointestinal systems, although these are more frequently involved in infants and children. Disease-causing mtDNA coding region mutations can affect either one of the 13 protein encoding genes or one of the 24 protein synthetic genes. Clinical manifestations do not readily distinguish these two categories, although lactic acidosis and muscle pathologic findings tend to be more prominent in the latter. In all cases, either defective ATP production due to disturbances in the ETC or enhanced generation of ROS has been invoked as the mediating biochemical mechanism between mtDNA mutation and disease manifestation.
MTDNA DISEASE PRESENTATIONS
The clinical presentation of adult patients with mtDNA disease can be divided into three categories: (1) clinical features suggestive of mitochondrial disease (Table 85e-2), but not a well-defined classic syndrome; (2) classic mtDNA syndromes; and (3) clinical presentation confined to one organ system (e.g., isolated sensorineural deafness, cardiomyopathy, or diabetes mellitus).
|
COMMON FEATURES OF MTDNA-ASSOCIATED DISEASES IN ADULTS |
Table 85e-3 provides a summary of eight illustrative classic mtDNA syndromes or disorders that affect adult patients and highlights some of the most interesting features of mtDNA disease in terms of molecular pathogenesis, inheritance, and clinical presentation. The first five of these syndromes result from heritable point mutations in either protein-encoding or protein synthetic mtDNA genes; the other three result from rearrangements or deletions that usually do not involve the germline.
|
MITOCHONDRIAL DISEASES DUE TO MTDNA POINT MUTATIONS AND LARGE-SCALE REARRANGEMENTS |
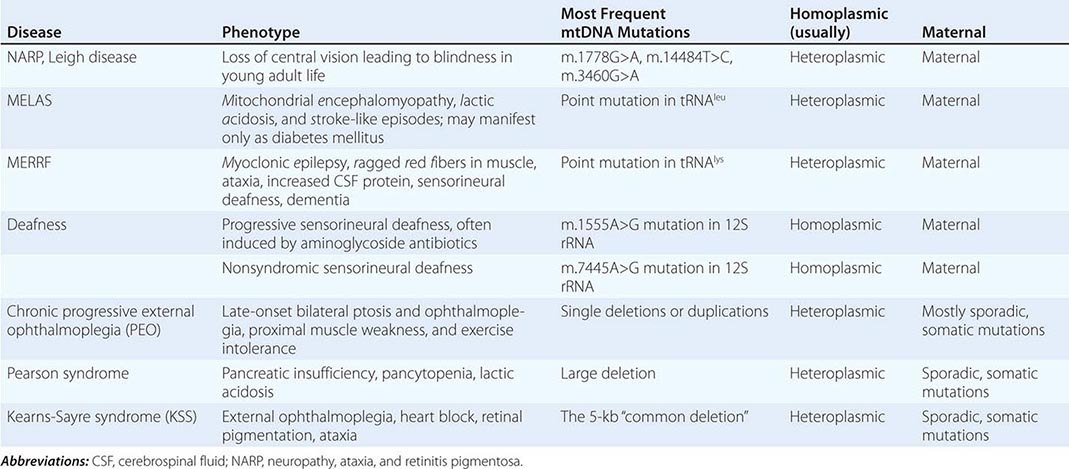
Leber’s hereditary optic neuropathy (LHON) is a common cause of maternally inherited visual failure. LHON typically presents during young adulthood with subacute painless loss of vision in one eye, with symptoms developing in the other eye 6–12 weeks after the initial onset. In some instances, cerebellar ataxia, peripheral neuropathy, and cardiac conduction defects are observed. In >95% of cases, LHON is due to one of three homoplasmic point mutations of mtDNA that affect genes encoding different subunits of complex I of the mitochondrial ETC; however, not all individuals who inherit a primary LHON mtDNA mutation develop optic neuropathy, and males are four to five times more likely than females to be affected, indicating that additional environmental (e.g., tobacco exposure) or genetic factors are important in the etiology of the disorder. Both the nuclear and mitochondrial genomic backgrounds modify disease penetrance. Indeed, a region of the × chromosome containing a high-risk haplotype for LHON was recently identified, supporting the formulation that nuclear genes act as modifiers and affording an explanation for the male prevalence of LHON. This haplotype can be used in predictive genomic testing and prenatal screening for this disease. In contrast to the other classic mtDNA disorders, it is of interest that patients with this syndrome are often homoplasmic for the disease-causing mutation. The somewhat later onset in young adulthood and modifying effect of protective background nuclear genomic haplotypes may have enabled homoplasmic pathogenic mutations to have escaped evolutionary censoring.
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) is a multisystem disorder with a typical onset between 2 to 10 years of age. Following normal early psychomotor development, the most common initial symptoms are seizures, recurrent headaches, anorexia, and recurrent vomiting. Exercise intolerance or proximal limb weakness can be the initial manifestation, followed by generalized tonic-clonic seizures. Short stature is common. Seizures are often associated with stroke-like episodes of transient hemiparesis or cortical blindness that may produce altered consciousness and may recur. The cumulative residual effects of the stroke-like episodes gradually impair motor abilities, vision, and cognition, often by adolescence or young adulthood. Sensorineural hearing loss adds to the progressive decline of these individuals. A plethora of less common symptoms have been described including myoclonus, ataxia, episodic coma, optic atrophy, cardiomyopathy, pigmentary retinopathy, ophthalmoplegia, diabetes mellitus, hirsutism, gastrointestinal dysmotility, and nephropathy. The typical age of death ranges from 10 to 35 years, but some individuals live into their sixth decade. Intercurrent infections or intestinal obstructions are often the terminal events. Laboratory investigation commonly demonstrates elevated lactate concentrations at rest with excessive increase after moderate exercise. Brain imaging during stroke-like episodes shows areas of increased T2 signal, typically involving the posterior cerebrum and not conforming to the distribution of major arteries. Electrocardiogram (ECG) may show evidence of cardiomyopathy, preexcitation, or incomplete heart block. Electromyography and nerve conduction studies are consistent with a myopathic process, but axonal and sensory neuropathy may coexist. Muscle biopsy typically shows ragged red fibers with the modified Gomori trichrome stain or “ragged blue fibers” resulting from the hyperintense reaction with the histochemical staining for succinate dehydrogenase. The diagnosis of MELAS is based on a combination of clinical findings and molecular genetic testing. Mutations in the mtDNA gene MT-TL1 encoding tRNAleu are causative. The most common mutation, present in approximately 80% of individuals with typical clinical findings, is an A-to-G transition at nucleotide 3243 (m.3243A>G). Mutations can usually be detected in mtDNA from leukocytes in individuals with typical MELAS; however, the occurrence of heteroplasmy can result in varying tissue distribution of mutated mtDNA. In the absence of specific treatment, various manifestations of MELAS are treated according to standard modalities for prevention, surveillance, and treatment.
Myoclonic epilepsy with ragged red fibers (MERRF) is a multisystem disorder characterized by myoclonus, seizures, ataxia, and myopathy with ragged red fibers. Hearing loss, exercise intolerance, neuropathy, and short stature are often present. Almost all MERRF patients have mutation in the mtDNA tRNAlys gene, and the m.8344A>G mutation in the mtDNA gene encoding the lysine amino acid tRNA is responsible for 80–90% of MERRF cases.
Neuropathy, ataxia, and retinitis pigmentosa (NARP) is characterized by moderate diffuse cerebral and cerebellar atrophy and symmetric lesions of the basal ganglia on magnetic resonance imaging (MRI). A heteroplasmic m.8993T>G mutation in the ATPase 6 subunit gene has been identified as causative. Ragged red fibers are not observed in muscle biopsy. When >95% of mtDNA molecules are mutant, a more severe clinical, neuroradiologic. and neuropathologic picture (Leigh syndrome) emerges. Point mutations in the mtDNA gene encoding the 12S rRNA result in heritable nonsyndromic hearing loss. One such mutation causes heritable ototoxic susceptibility to aminoglycoside antibiotics, which opens a pathway for a simple pharmacogenetic test in the appropriate clinical settings.
Kearns-Sayre syndrome (KSS), sporadic progressive external ophthalmoplegia (PEO), and Pearson syndrome are three disease phenotypes caused by large-scale mtDNA rearrangements including partial deletions or partial duplication. The majority of single large-scale rearrangements of mtDNA are thought to result from clonal amplification of a single sporadic mutational event, occurring in the maternal oocyte or during early embryonic development. Because germline involvement is rare, most cases are sporadic rather than inherited. KSS is characterized by the triad of onset before age 20, chronic progressive external ophthalmoplegia, and pigmentary retinopathy. Cerebellar syndrome, heart block, increased cerebrospinal fluid protein content, diabetes mellitus, and short stature are also part of the syndrome. Single deletions/duplication can also result in milder phenotypes such as PEO, characterized by late-onset progressive external ophthalmoplegia, proximal myopathy, and exercise intolerance. In both KSS and PEO, diabetes mellitus and hearing loss are frequent accompaniments. Pearson syndrome is also characterized by diabetes mellitus from pancreatic insufficiency, together with pancytopenia and lactic acidosis, caused by the large-scale sporadic deletion of several mtDNA genes.
Two important dilemmas in classic mtDNA disease have benefited from recent important research insights. The first relates to the greater involvement of neuronal, muscular, renal, hepatic, and pancreatic manifestations in mtDNA disease in these syndromes. This observation has appropriately been mostly attributed to the high energy utilization of the involved tissues and organ systems and, hence, greater dependency on mitochondrial ETC integrity and health. However, because mutations are stochastic events, mitochondrial mutations should occur in any organ during embryogenesis and development. Recently, additional explanations have been suggested based on studies of the common m.3243A>G transition. The proportion of this mutation in peripheral blood cells was shown to decrease exponentially with age. A selective process acting at the stem cell level with a strong bias against the mutated form would have its greatest effect to reduce the mutant mtDNA only in highly proliferating cells, such as those derived from the hematopoietic system. Tissues and organs with lower cell turnover, such as those involved with mtDNA mutations, would not benefit from this effect and, thus, would be the most affected.
The other dilemma arises from the observation that only a subset of mtDNA mutations accounts for the majority of the familial mtDNA diseases. The random occurrence of mutations in the mtDNA sequence should yield a more uniform distribution of disease-causing mutations. However, recent studies using the introduction of one severe and one mild point mutation into the female germline of experimental animals demonstrated selective elimination during oogenesis of the severe mutation and selective retention of the milder mutation, with the emergence of mitochondrial disease in offspring after multiple generations. Thus, oogenesis itself can act as an “evolutionary” filter for mtDNA disease.
THE INVESTIGATION OF SUSPECTED MTDNA DISEASE
The clinical presentations of classic syndromes, groupings of disease manifestations in multiple organ systems, or unexplained isolated presentations of one of the disease features of a classic mtDNA syndrome should prompt a systematic clinical investigation as outlined in Fig. 85e-6. Indeed, mitochondrial disease should be considered in the differential diagnosis of any progressive multisystem disorder. Despite the centrality of disruptive oxidative phosphorylation, an elevated blood lactate level is neither specific nor sensitive because there are many causes of blood lactic acidosis, and many patients with mtDNA defects presenting in adulthood have normal blood lactate. An elevated cerebrospinal fluid lactate is a more specific test for mitochondrial disease if there is central nervous system involvement. The serum creatine kinase may be elevated but is often normal, even in the presence of a proximal myopathy. Urinary organic and amino acids may also be abnormal, reflecting metabolic and kidney proximal tubule dysfunction. Every patient with seizures or cognitive decline should have an electroencephalogram. A brain computed tomography (CT) scan may show calcified basal ganglia or bilateral hypodense regions with cortical atrophy. MRI is indicated in patients with brainstem signs or stroke-like episodes.
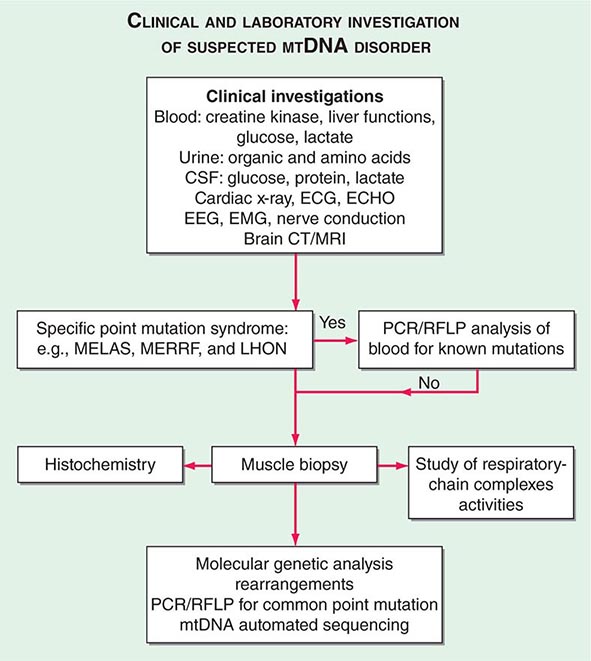
FIGURE 85e-6 Clinical and laboratory investigation of a suspected mitochondrial DNA (mtDNA) disorder. CSF, cerebrospinal fluid; CT, computed tomography; ECG, electrocardiogram; ECHO, echocardiography; EEG, electroencephalogram; EMG, electromyogram; LHON, Leber’s hereditary optic neuropathy; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stoke-like episodes; MERFF, myoclonic epilepsy with ragged red fibers; MRI, magnetic resonance imaging; PCR, polymerase chain reaction; RFLP, restriction fragment length polymorphism.
For some mitochondrial diseases, it is possible to obtain an accurate diagnosis with a simple molecular genetic screen. For examples, 95% of patients with LHON harbor one of three mtDNA point mutations (m.11778A>G, m.A3460A>G, or m.14484T>C). These patients have very high levels of mutated mtDNA in peripheral blood cells, and therefore, it is appropriate to send a blood sample for molecular genetic analysis by polymerase chain reaction (PCR) or restriction fragment length polymorphism. The same is true for most MERRF patients who harbor a point mutation in the lysine tRNA gene at position 8344. In contrast, patients with the m.3243A>G MELAS mutation often have low levels of mutated mtDNA in blood. If clinical suspicion is strong enough to warrant peripheral blood testing, then patients with a negative result should be investigated further by performing a skeletal muscle biopsy.
Muscle biopsy histochemical analysis is the cornerstone for investigation of patients with suspected mitochondrial disease. Histochemical analysis may show subsarcolemmal accumulation of mitochondria with the appearance of ragged red fibers. Electron microscopy might show abnormal mitochondria with paracrystalline inclusions. Muscle histochemistry may show cytochrome c oxidase (COX)–deficient fibers, which indicate mitochondrial dysfunction (Fig. 85e-5). Respiratory chain complex assays may also show reduced enzyme function. Either of these two abnormalities confirms the presence of a mitochondrial disease, to be followed by an in-depth molecular genetic analysis.
Recent evidence has provided important insights into the importance of nuclear-mtDNA genomic cross-talk and has provided a descriptive framework for classifying and understanding disorders that emanate from perturbations in this cross-talk. Although not strictly considered as mtDNA genetic disorders, manifestations do overlap those highlighted above (Fig. 85e-7).
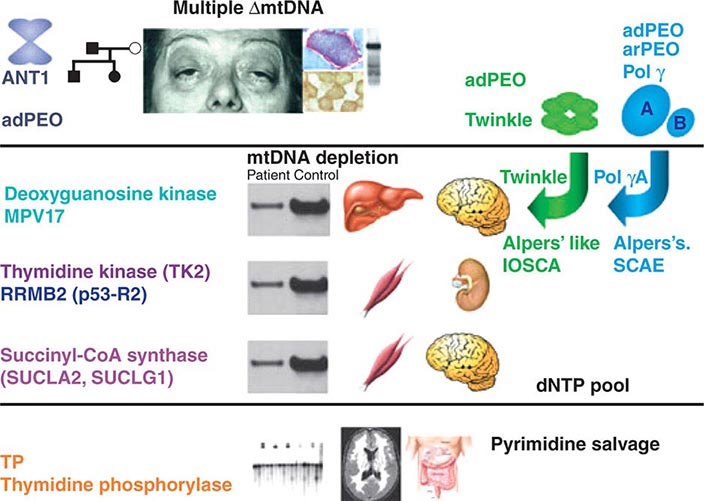
FIGURE 85e-7 Disorders associated with perturbations in nuclear-mitochondrial genomic cross-talk. Clinical features and genes associated with multiple mitochondrial DNA (mtDNA) deletions, mtDNA depletion, and mitochondrial neurogastrointestinal encephalomyopathy syndromes. ANT, adenine nucleotide translocators; adPEO, autosomal dominant progressive external ophthalmoplegia; arPEO, autosomal recessive progressive external ophthalmoplegia; IOSCA, infantile-onset spinocerebellar ataxia; SCAE, spinocerebellar ataxia and epilepsy. (Reproduced with permission from A Spinazzola, M Zeviani: Disorders from perturbations of nuclear-mitochondrial intergenomic cross-talk. J Intern Med 265:174, 2009.)
IMPACT OF HOMOPLASMIC SEQUENCE VARIATION ON HERITABLE TRAITS AND DISEASE
The relationship among the degree of heteroplasmy, tissue distribution of the mutant mtDNA, and disease phenotype simplifies inference of a clear causative relationship between heteroplasmic mutation and disease. With the exception of certain mutations (e.g., those causing most cases of LHON), drift to homoplasmy of such mutations would be precluded normally by the severity of impaired oxidative phosphorylation and the consequent reduction in reproductive fitness. Therefore, sequence variants that have reached homoplasmy should be neutral in terms of human evolution and, hence, useful only for tracing human evolution, demography, and migration, as described above. One important exception is in the case of one or more of the homoplasmic population-level variants, which designate the mtDNA haplogroup J, and the interaction with the mtDNA mutations causing LHON. Reduced disease predilection suggests that one or more of the ancient sequence variants designating mtDNA haplogroup J appears to attenuate predisposition to degenerative disease, in the face of other risk factors. Whether or not additional epistatic interactions between population-level mtDNA haplotypes and common health conditions will be found remains to be determined. If such influences do exist, then they are more likely to be relevant to health conditions in the postreproductive age groups, wherein evolutionary filters would not have had the opportunity to censor deleterious effects and interactions and wherein the effects of oxidative stress may play a role. Although much has been written about the possible associations of population-level common mtDNA variants and human health and disease phenotypes or adaptation to different environmental influences (e.g., climate), a word of caution is in order.
Many studies that purport to show such associations with phenotypes such as longevity, athletic performance, and metabolic and neurodegenerative disease are limited by small sample sizes, possible genotyping inaccuracies, and the possibility of population stratification or ethnic ancestry bias. Because mtDNA haplogroups are so prominently partitioned along phylogeographic lines, it is difficult to rule out the possibility that a haplogroup for which an association has been found is simply a marker for differences in populations with a societal or environmental difference or with different allele frequencies at other genomic loci, which are actually causally related to the heritable trait or disease of interest. The difficulty in generating cellular or animal models to test the functional influence of homoplasmic sequence variants (as a result of mtDNA polyploidy) further compounds the challenge. The most likely formulation is that the risk conferred by different mtDNA haplogroup–defining homoplasmic mutations for common diseases depends on the concomitant nuclear genomic background, together with environmental influences. Progress in minimizing potentially misleading associations in mtDNA heritable trait and disease studies should include ensuring adequate sample size taken from a large sample recruitment base, using carefully matched controls and population structure determination, and performing analysis that takes into account epistatic interactions with other genomic loci and environmental factors.
IMPACT OF ACQUIRED SOMATIC MTDNA MUTATION ON HUMAN HEALTH AND DISEASE
Studies on aging humans and animals have shown a potentially important correlation of age with the accumulation of heterogeneous mtDNA mutations, especially in those organ systems that undergo the most prominent age-related degenerative tissue phenotype. Sequencing of PCR-amplified single mtDNA molecules has demonstrated an average of two to three point mutations per molecule in elderly subjects when compared with younger ones. Point mutations observed include those responsible for known heritable heteroplasmic mtDNA disorders, such as the m.3344A>G and m.3243A>G mutations responsible for the MERRF and MELAS syndromes, respectively. However, the cumulative burden of these acquired somatic point mutations with age was observed to remain well below the threshold expected for phenotypic expression (<2%). Point mutations at other sites not normally involved in inherited mtDNA disorders have also been shown to accumulate to much higher levels in some tissues of elderly individuals, with the description of tissue-specific “hot spots” for mtDNA point mutations. Along the same lines, an age-associated and tissue-specific accumulation of mtDNA deletions has been observed, including deletions involved in known heritable mtDNA disorders, as well as others. The accumulation of functional mtDNA deletions in a given tissue is expected to be associated with mitochondrial dysfunction, as reflected in an age-associated patchy and reduced COX activity on histochemical staining, especially in skeletal and cardiac muscle and brain. A particularly well-studied and potentially important example is the accumulation of mtDNA deletions and COX deficiency observed in neurons of the substantia nigra in Parkinson’s disease patients.
The progressive accumulation of ROS has been proposed as the key factor connecting mtDNA mutations with aging and age-related disease pathogenesis (Fig. 85e-8). As noted above, ROS are a by-product of oxidative phosphorylation and are removed by detoxifying antioxidants into less harmful moieties; however, exaggerated production of ROS or impaired removal results in their accumulation. One of the main targets for ROS-mediated injury is DNA, and mtDNA is particularly vulnerable because of its lack of protective histones and less efficient injury repair systems compared with nuclear DNA. In turn, accumulation of mtDNA mutations results in inefficient oxidative phosphorylation, with the potential for excessive production of ROS, generating a “vicious cycle” of cumulative mtDNA damage. Indeed, measurement of the oxidative stress biomarker 8-hydroxy-2-deoxyguanosine has been used to measure age-dependent increases in mtDNA oxidative damage at a rate exceeding that of nuclear DNA. It should be noted that mtDNA mutation can potentially occur in postmitotic cells as well, because mtDNA replication is not synchronized with the cell cycle. Two other proposed links between mtDNA mutation and aging, besides ROS-mediated tissue injury, are the perturbations in efficiency of oxidative phosphorylation with disturbed cellular aerobic function and perturbations in apoptotic pathways, whose execution steps involve mitochondrial activity.
FIGURE 85e-8 Multiple pathways of mitochondrial DNA (mtDNA) damage and aging. Multiple factors may impinge on the integrity of mitochondria that lead to loss of cell function, apoptosis, and aging. The classic pathway is indicated with blue arrows; the generation of reactive oxygen species (ROS; superoxide anion, hydrogen peroxide, and hydroxyl radicals), as a by-product of mitochondrial oxidative phosphorylation, results in damage to mitochondrial macromolecules, including the mtDNA, with the latter leading to deleterious mutations. When these factors damage the mitochondrial energy-generating apparatus beyond a functional threshold, proteins are released from the mitochondria that activate the caspase pathway, leading to apoptosis, cell death, and aging. (Reproduced with permission from L Loeb et al: The mitochondrial theory of aging and its relationship to reactive oxygen species damage and somatic mtDNA mutations. Proc Natl Acad Sci USA 102:18769, 2005.)
Genetic intervention studies in animal models have sought to clarify the potential causative relationship between acquired somatic mtDNA mutation and the aging phenotype, and the role of ROS in particular. Replication of the mitochondrial genome is mediated by the activity of the nuclear-encoded polymerase gamma gene. A transgenic homozygous mouse knock-in mutation of this gene renders the polymerase enzyme deficient in proofreading and results in a three- to fivefold increase in mtDNA mutation rate. Such mice develop a premature aging phenotype, which includes subcutaneous lipoatrophy, alopecia, kyphonia, and weight loss with premature death. Although the finding of increased mtDNA mutation and mitochondrial dysfunction with age has been solidly established, the causative role and specific contribution of mitochondrial ROS to aging and age-related disease in humans has yet to be proved. Similarly, although many tumors display higher levels of heterogeneous mtDNA mutations, a causal relationship to tumorigenesis has not been proved.
Besides the age-dependent acquired accumulation in somatic cells of heterogeneous point mutations and deletions, a quite different effect of nonheritable and acquired mtDNA mutation has been described affecting tissue stem cells. In particular, disease phenotypes attributed to acquired mtDNA mutation have been observed in sporadic and apparently nonfamilial cases involving a single individual or even tissue, usually skeletal muscle. The presentation consists of decreased exercise tolerance and myalgias, sometimes progressing to rhabdomyolysis. As in the case of the sporadic, heteroplasmic, large-scale deletion, classic syndromes of chronic PEO, Pearson syndrome, and KSS, the absence of a maternal inheritance pattern, together with the finding of limited tissue distribution, suggests a molecular pathogenic mechanism emanating from mutations arising de novo in muscle stem cells after germline differentiation (somatic mutations that are not sporadic and occur in tissue-specific stem cells during fetal development or in the postnatal maintenance or postinjury repair stage). Such mutations would be expected to be propagated only within the progeny of that stem cell and affect a particular tissue within a given individual, without evidence of heritability.
PROSPECTS FOR CLINICAL MANAGEMENT OF MTDNA DISEASE
TREATMENT OF MTDNA DISORDERS
No specific curative treatment for mtDNA disorders is currently available; therefore, the management of mitochondrial disease is largely supportive. Management issues may include early diagnosis and treatment of diabetes mellitus, cardiac pacing, ptosis correction, and intraocular lens replacement for cataracts. Less specific interventions in the case of other disorders involve combined treatment strategies including dietary intervention and removal of toxic metabolites. Cofactors and vitamin supplements are widely used in the treatment of diseases of mitochondrial oxidative phosphorylation, although there is little evidence, apart from anecdotal reports, to support their use. This includes administration of artificial electron acceptors, including vitamin K3, vitamin C, and ubiquinone (coenzyme Q10); administration of cofactors (coenzymes) including riboflavin, carnitine, and creatine; and use of oxygen radical scavengers, such as vitamin E, copper, selenium, ubiquinone, and idebenone. Drugs that could interfere with mitochondrial function, such as the anesthetic agent propofol, barbiturates, and high doses of valproate, should be avoided. Supplementation with the nitric oxide synthase substrate, L-arginine, has been advocated as a vasodilator treatment during stroke-like episodes. The physician should also be familiar with environmental interactions, such as the strong and consistent association between visual loss in LHON and smoking. A clinical penetrance of 93% was found in men who smoked. Asymptomatic carriers of an LHON mtDNA mutation should, therefore, be strongly advised not to smoke and to moderate their alcohol intake. Although not a cure, these interventions might stave off the devastating clinical manifestations of the LHON mutation. Another example is strict avodiance of aminoglycosides in the familial syndrome of ototoxic susceptibility to aminoglycosides in the presence of the mtDNA m.1555A>G mutation of the 12SrRNA encoding gene.
GENETIC COUNSELING, PRENATAL DIAGNOSIS, AND PREIMPLANTATION GENETIC DIAGNOSIS IN MTDNA DISORDERS
The provision of accurate genetic counseling and reproductive options to families with mtDNA mutations is challenging due to the unique genetic features of mtDNA inheritance that distinguish it from Mendelian genetics. mtDNA defects are transmitted by maternal inheritance. mtDNA de novo mutations are often large deletions, affect one family member, and usually represent no significant risk to other members of the family. In contrast, mtDNA point mutations or duplications can be transmitted down the maternal line. Accordingly, the father of an affected individual has no risk of harboring the disease-causing mutation, and a male cannot transmit the mtDNA mutation to his offspring. In contrast, the mother of an affected individual usually harbors the same mutation but might be completely asymptomatic. This wide phenotypic variability is primarily related to the phenomena of heteroplasmy and the mutation load carried by different members of the same family. Consequently, a symptomatic or asymptomatic female harboring a disease-causing mutation in a heteroplasmic state will transmit to her offspring variable amounts of the mutant mtDNA molecules. The offspring will be symptomatic or asymptomatic primarily according to the mutant load transmitted via the oocyte and, to some extent, subsequent mitotic segregation during development. Interactions with the mtDNA haplotype background or nuclear human genome (as in the case of LHON) serve as an additional important determinant of disease penetrance. Because the severity of the disease phenotype associated with the heteroplasmic mutation load is a function of the stochastic differential segregation and copy number of mutant mtDNA during the oogenesis bottleneck and, subsequently, following tissue and organ development in the offspring, it is rarely predictable with any degree of accuracy. For this reason, prenatal diagnosis (PND) and PGD techniques that have evolved into integral and well-accepted standards of practice are severely hampered in the case of mtDNA-related diseases.
The value of PND and PGD is limited, partly due to the absence of data on the rules that govern the segregation of wild-type and mutant mtDNA species (heteroplasmy) among tissue in the developing embryo. Three factors are required to ensure the reliability of PND and PGD: (1) a close correlation between the mutant load and the disease severity, (2) a uniform distribution of mutant load among tissues, and (3) no major change in mutant load with time. These criteria are suggested to be fulfilled for the NARP m.8993T>G mutation but do not seem to apply to other mtDNA disorders. In fact, the level of mutant mtDNA in a chorionic villous or amniotic fluid sample may be very different from the level in the fetus, and it would be difficult to deduce whether the mutational load in the prenatal samples provides clinically useful information regarding the postnatal and adult state.
PREVENTION OF MITOCHONDRIAL DISEASE INHERITANCE BY ASSISTED REPRODUCTIVE TECHNOLOGIES
Because the treatment options for patients with mitochondrial disease are rather limited, preventive interventions that eliminate the likelihood of transmission of affected mtDNA into offspring are desirable. The lack of utility of PND and PGD techniques to reliably diagnose and predict mitochondrial disorders at preimplantation-stage products of conception has resulted in the search for alternative preventive approaches for the same problem. One possible approach to “diluting” or even entirely eliminating the mutant mtDNA is applicable only in the earliest embryonic state and in effect represents a form of germline preventive therapy (Fig. 85e-9). This possibility has been explored by using alternative assisted reproduction techniques such as ooplasmic transfer (OT), metaphase chromosome transfer (CT), pronuclear transfer (PNT), and germinal vesicle transfer (GVT) in animal models and, to an extent, in humans. OT is a technique wherein a certain volume (5–15%) of healthy donor oocyte cytoplasm with normal mitochondria is injected into the patient oocyte containing mutated mitochondria. The reasoning behind OT is to supplement the patient’s oocyte with uncompromised cytoplasmic factors such as mtDNA, mRNA, proteins, and other molecules by injecting cytoplasm from healthy oocytes. In PNT, following fertilization, pronuclei of a patient’s zygote are removed with a cytoplasm (“karyoplast”). The karyoplast is transferred to the perivitelline space of a donated zygote, which has been already enucleated. The karyoplast is then fused with enucleated zygote by electric pulses or inactivated Sendai viruses (HVJ). The reconstructed zygote contains a nucleus from the patient (patient nuclear DNA) and cytoplasm from the donor. Thus, the majority of the patient mtDNA is replaced with mtDNA from the donor oocyte. In CT, meiosis II stage of oocyte maturation provides an opportunity for the reconstruction of oocytes with different nuclear and cytoplasmic components before fertilization takes place. Reconstructed oocytes by metaphase chromosome transfer are then fertilized to produce embryos with desired mtDNA haplotypes. In GVT, replacement of compromised cytoplasm with healthy cytoplasm through germinal vesicle transfer before the start of chromosome segregation is carried out.
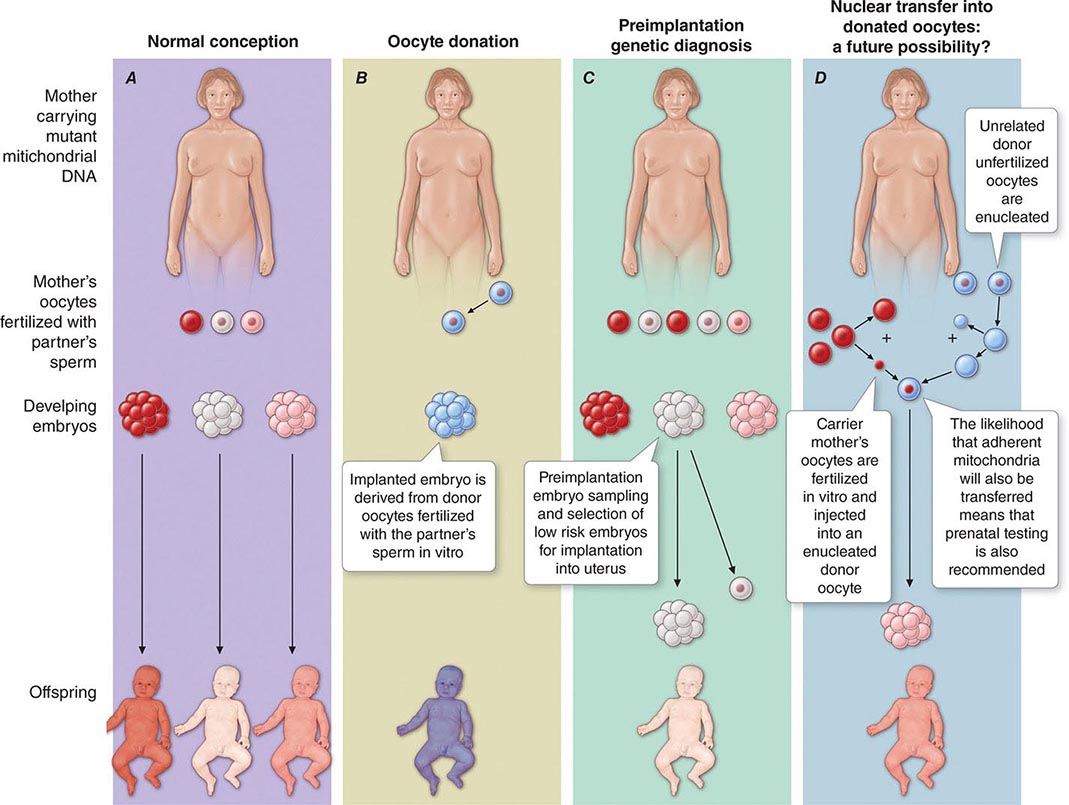
FIGURE 85e-9 Possible approaches for prevention of mitochondrial DNA (mtDNA) disease. A. No intervention: offspring’s mutant mtDNA load will vary greatly. B. Oocyte donation: currently permitted in some constituencies but limited by the availability of oocyte donors. C. Preimplantation genetic diagnosis: available for some mtDNA diseases (reliable in determining background nuclear genomic haplotype risk). D. Nuclear transfer: research stage, including initial studies in nonhuman primates. Red represents mutant mtDNA, pink and white represent successively higher proportions of normal mtDNA. Blue represents genetic material from an unrelated donor. (Adapted with permission from J Poulton et al: Preventing transmission of maternally inherited mitochondrial DNA diseases. Br Med J 338:b94, 2009.)
These approaches have not yet met with widely reported clinical success, yet there is room for optimism. As noted above, analysis of heteroplasmy and inheritance patterns indicates that even a small increase in copies of nonmutant mtDNA can exceed the threshold required to ameliorate serious clinical disease. All of the approaches described above show promise in achieving this goal and thus reducing the burden of clinical mtDNA disease in the future.
86e |
The Human Microbiome |
The technologies that allowed us to decipher the human genome have revolutionized our ability to delineate the composition and functions of the microbial communities that colonize our bodies and make up our microbiota. Each body habitat, including the skin, nose, mouth, airways, gastrointestinal tract, and vagina, harbors a distinctive community of microbes. Efforts to understand our microbiota and its collection of microbial genes (our microbiome) are changing our views of “self” and deepening our understanding of many normal physiologic, metabolic, and immunologic features and their interpersonal and intrapersonal variations. In addition, this area of research is beginning to provide new insights into diseases not previously known to have microbial “contributors” and is suggesting new strategies for treatment and prevention. Key terms used in the discussion of the human microbiome are defined in Table 86e-1.
|
GLOSSARY OF TERMS USED IN DISCUSSION OF THE HUMAN MICROBIOME |
We are holobionts—collections of human and microbial cells that function together in an elaborate symbiosis. The aggregate number of microbial cells in our microbiota exceeds the number of human cells in our adult bodies by up to 10-fold, and each healthy adult is estimated to harbor 105–106 microbial genes, in contrast to ~20,000 Homo sapiens genes. Members of our microbiota can function as mutualists (i.e., both host and microbe benefit from each other’s presence), as commensals (one partner benefits; the other is seemingly unaffected), and as potential or overt pathogens (one partner benefits; the other is harmed). Many clinicians view pathogens as individual microbial species or strains that can elicit disease in susceptible hosts. An emerging, more ecologic view is that pathogens do not function in isolation; rather, their invasion, emergence, and effects on the host reflect interactions with other members of a microbiota. An even more expansive view is that multiple organisms in a community conspire to produce pathogenic effects in certain host and environmental contexts (a pathologic community).
The ability to characterize microbial communities without culturing their component members has spawned the field of metagenomics (Table 86e-1). Metagenomics reflects a confluence of experimental and computational advances in the genome sciences as well as a more ecologic understanding of medical microbiology, according to which the functions of a given microbe and its impact on human biology depend on the context of other microbes in the same community. Traditional microbiology relies on culturing individual microbes, but metagenomics skips this step, instead sequencing DNA isolated directly from a given microbial community. The resulting datasets facilitate follow-up functional studies, such as the profiling of RNA and protein products expressed from the microbiome or the characterization of a microbial community’s metabolic activities.
Metagenomics provides insight into how microbial communities vary in several situations critical to human health. One such situation is how microbial communities are assembled following birth and how they operate over time, including responses of established communities to various perturbations. Another is how microbial communities normally vary between different anatomic sites within an individual and between different groups of people representing different ages, physiologic states, lifestyles, geographies, and gender. Yet another is how microbial communities vary in disease; whether such variations are consistent among individuals grouped according to current criteria for a disease or its subtypes; whether the microbiota or microbiome provides new ways of classifying disease states; and, importantly, whether the structural and functional configurations of microbial communities are a cause or a consequence of disease.
Analysis of our microbiomes also addresses one of the most fundamental questions in genetics: How does environment select our genes and directly influence their function? Each human encounters a unique environment during the course of his or her lifetime. Part of this personally experienced environment is incorporated into the genes and capabilities of our microbial communities. The microbiome therefore expands our conceptualization of “human” genetic potential from a single set of genes “fixed” at birth to a microbiome with additional genes and capabilities acquired via a process influenced by our family and life experiences, including modifiable lifestyle choices such as diet. This view recognizes a previously underappreciated dimension of human evolution that occurs at the level of our microbiomes and inspires us to determine how—and how fast—this microbial evolution effects changes in our human biology. For example, Westernization is associated with loss of bacterial species diversity (richness) in the microbiota, and this loss may be associated with the suite of Western diseases. The study of our microbiomes also raises important questions about personal identity, how we define the origins of health disparities, and privacy. Further, it offers the possibility of entirely new approaches to disease prevention and treatment, including regenerative medicine, which involves administration of microbial species (probiotics) to individuals harboring communities that have not developed into a mature, fully functional state or that have been perturbed in ways that can be restored by the addition of species that fill unoccupied “jobs” (niches).
This chapter provides a general overview of how human microbial communities are analyzed; reviews ecologic principles that guide our understanding of microbial communities in health and disease; summarizes recent studies that establish correlations and, in some cases, causal relationships between our microbiota/microbiomes and various diseases; and discusses challenges faced in the translation of these findings to new therapeutic interventions.
A TOOLBOX FOR METAGENOMIC ANALYSES OF HUMAN MICROBIAL COMMUNITIES
Life on Earth has been classified into three domains: Bacteria, Archaea, and Eukarya. The habitats of the surface-exposed human body harbor members of each domain plus their viruses. In large part, microbial diversity has not been characterized by culture-based approaches, partly because we do not know how to re-create the metabolic milieu fashioned by these communities in their native habitats and partly because a few organisms tend to outgrow the others. Culture-independent methods readily identify which organisms are present in a microbiota and their relative abundance. The gene widely used to identify microbes and their evolutionary relationships encodes the major RNA component of the small subunit (SSU) of ribosomes. Within each domain of life, the SSU gene is highly conserved, allowing the SSU gene sequences present in different organisms in that domain to be accurately aligned and regions of nucleotide sequence variation to be identified. Pairwise comparisons of SSU ribosomal RNA (rRNA) genes from different microbes allow construction of a phylogenetic tree that represents an evolutionary map on which previously unknown organisms can be assigned a position. This approach, known as molecular phylogenetics, permits characterization of each organism on the basis of its evolutionary distance from other organisms. Different phylogenetic types (phylotypes) can be viewed as comprising branches on an evolutionary tree.
Characterization of Bacteria Because members of the Bacteria dominate our microbiota, most studies defining our various body habitat–associated microbial communities have sequenced the bacterial SSU gene that encodes 16S rRNA. This gene has a mosaic structure, with highly conserved domains flanking more variable regions. The most straightforward way to identify bacterial taxonomic groups (taxa) in a given community is to sequence polymerase chain reaction (PCR) products (amplicons) generated from the 16S rRNA genes present in that community. PCR primers directed at the conserved regions of the gene yield PCR amplicons encompassing one or more of that gene’s nine variable regions. PCR primer design is critical: differential annealing with primer pairs designed to amplify different variable regions can lead to over- or underrepresentation of specific taxa, and different regions within the 16S rRNA gene can have different patterns of evolution. Therefore, caution must be exercised in comparisons of the relative abundance of taxa in samples characterized in different studies, as methodologic differences can lead to larger perceived differences in the inferred taxonomy than actually exist.
A key innovation is multiplex sequencing. Amplicons from each microbial-community DNA sample are tagged by incorporation of a unique oligonucleotide barcode into the PCR primer. Amplicons harboring these sample-specific barcodes can then be pooled together so that multiple samples representing multiple communities can be sequenced simultaneously (Fig. 86e-1). One important choice is the tradeoff between the number of samples that can be processed simultaneously and the number of sequences generated per sample. Interpersonal differences in the bacterial components of the microbiota are typically large, as are differences between communities occupying different body habitats in the same individual (see below); thus fewer than 1000 16S rRNA reads are characteristically required to discriminate community type. However, the identification of systematic differences in microbiota composition that correlate with physiologic status or disease state is confounded by the substantial interpersonal variation that occurs normally.
FIGURE 86e-1 Pipeline for culture-independent studies of a microbiota. (A) DNA is extracted directly from a sampled human body habitat–associated microbial community. The precise location of the community and relevant patient clinical data are collected. Polymerase chain reaction (PCR) is used to amplify portions of small-subunit (SSU) rRNA genes (e.g., the genes encoding bacterial 16S rRNA) containing one or more variable regions. Primers with sample-specific, error-correcting barcodes are designed to recognize the more conserved regions of the 16S rRNA gene that flank the targeted variable region(s). (B) Barcoded amplicons from multiple samples (communities 1–3) are pooled and sequenced in batch in a highly parallel next-generation DNA sequencer. (C) The resulting reads are then processed, with barcodes denoting which sample the sequence came from. After barcode sequences are removed in silico, reads are aligned and grouped according to a specified level of shared identity; e.g., sequences that share ≥97% nucleotide sequence identity are regarded as representing a species. Once reads are binned into operational taxonomic units (OTUs) in this fashion, they are placed on a phylogenetic tree of all known bacteria and their phylogeny is inferred. (D) Communities can be compared to one another by either taxon-based methods, in which phylogeny is not considered and the number of shared taxa are simply scored, or phylogenetic methods, in which community similarity is considered in light of the evolutionary relationships of community members. The UniFrac metric is commonly used for phylogeny-based comparisons. In stylized examples (i), (ii), and (iii), communities with varying degrees of similarity are shown. Each circle represents an OTU colored on the basis of its community of origin and placed on a master phylogenetic tree that includes all lineages from all communities. Branches (horizontal lines) are colored with each community that contains members from that branch. The three examples vary in the amount of branch length shared between the OTUs from each community. In (i), there is no shared branch length, and thus the three communities have a similarity score of 0. In (ii), the communities are identical, and a similarity score of 1 is assigned. In (iii), there is an intermediate level of similarity: communities represented in red and green share more branch length and thus have a higher similarity score than red vs. blue or green vs. blue. The amount of shared branch length in each pairwise community comparison provides a distance matrix. (E) The results of taxon- or phylogeny-based distance matrices can be displayed by principal coordinates analysis (PCoA), which plots each community spatially such that the largest component of variance is captured on the x-axis (PC1) and the second largest component of variance is displayed on the y-axis (PC2). In the example shown, the three communities in example (iii) from panel D are compared. Note that for shotgun sequencing of whole-community DNA (microbiome analysis), reads are compared with genes that are present in the genomes of sequenced cultured microbes and/or with genes that have been annotated by hierarchical functional classification schemes in various databases, such as the Kyoto Encyclopedia of Genes and Genomes (KEGG). Communities can then be compared on the basis of the distribution of functional groups in their microbiomes—an approach analogous to taxon-based methods for 16S rRNA–based comparisons—and the results plotted with PCoA.
Sequencing of bacterial 16S rRNA genes creates a challenge for medical microbiology: how to define the taxonomic groups present in a community in a systematic and informative manner, so that one community can be compared with and contrasted to another. Within each domain of life, microbes are classified in a hierarchy beginning with phylum (the broadest group) followed by class, order, family, genus, and species. To determine taxonomy, 16S rRNA sequences are aligned on the basis of their sequence similarity—a process known as picking operational taxonomic units (OTUs). Grouping of 16S rRNA sequences from a given variable region into “bins” that share ≥97% nucleotide sequence identity (97%ID OTUs) is a commonly accepted, albeit arbitrary, way to define a species.
Looking beyond the 16S rRNA gene, we find that different isolates (strains) of a given bacterial species have overlapping but not identical sets of genes in their genomes. The aggregate set of genes identified in all isolates (strains) of a given species-level phylotype represents its pan-genome. Most species are represented by multiple strains, sometimes with markedly different functions (for example, enteropathogenic versus commensal Escherichia coli). Differences in genome content among strains of a given species reflect differences in community membership as well as differences in the selective pressures these strains experience within and between habitats. Horizontal gene transfer among members of a microbiota—mediated by phage, plasmids, and other mechanisms—is a major contributor to this strain-level variation.
Strain-level diversity can be important in any consideration of how microbial communities differ between individuals and how these communities accommodate perturbations. For example, the great bacterial strain-level diversity that exists in the gut is thought to be one of the features that allows this microbiota, which occupies a constantly perfused ecosystem exposed to the complex and varying set of substances we ingest, to adapt to changing circumstances rather than depending on one strain to occupy a given niche important for proper community functioning. In ecologic studies of different environments, such as grasslands, forests, and reefs, increased diversity within a community increases its capacity to respond to disturbances and to restore itself (i.e., its resilience); the same is likely true of microbial ecosystems. When characterizing the mechanisms by which a given species produces an effect or effects on humans, it is important to consider the strain being tested; strain-level diversity has an impact on discovery and development efforts aimed at identifying next-generation probiotics that can be used therapeutically to promote health or treat disease.
Identification of Archaeal and Eukaryotic Members Surveys based on SSU rRNA gene sequencing have largely focused on Bacteria, yet the census of “who’s there” in human body habitat–associated communities must also include the other two domains of life: Archaea and Eukarya. Differences in the sequences of archaeal and bacterial 16S rRNA genes, first recognized by Carl Woese in 1977, allowed these two domains of life to be distinguished. The representation of Archaea in human microbial communities is less well defined than that of Bacteria, in part due to the difficulty in optimizing the design of PCR primers that specifically target conserved regions of archaeal (versus bacterial) 16S rRNA genes. Identifying archaeal members is important to our understanding of the functional properties of the microbiota. For example, a major challenge faced by microbial communities when breaking down polysaccharides (the most abundant biologic polymers on Earth) is the maintenance of redox balance in the setting of maximal energy production. Many microbial species have branched fermentation pathways that allow them to dispose of reducing equivalents (e.g., by the production of H2, which is energetically efficient). However, there is a caveat: the hydrogen must be removed or it will inhibit reoxidation of pyridine nucleotides. Therefore, hydrogen-consuming (hydrogenotrophic) species are key to maximizing the energy-extracting capacity of primary fermenters.
In the human gut, hydrogenotrophs include a phylogenetically diverse group of bacterial acetogens, a more limited group of sulfate-reducing bacteria that generate hydrogen sulfide, and methane-producing archaeal organisms (methanogens) that can represent up to 10% of the anaerobes present in the feces of some humans. However, the degree of archaeal diversity in the gut microbiota of healthy individuals appears to be low.
Culture-independent surveys of eukaryotic diversity are also confounded by challenges related to the design of PCR primers that target the eukaryotic SSU gene (18S rRNA) as well as the internal transcribed spacer regions of rRNA operons. Metagenomic studies of healthy human adults living in countries with distinct cultural traditions and disparate geographic features and locations have revealed that the degree of eukaryotic diversity is lower than that of bacterial diversity. In the gut, which contains far more microbes than any other body habitat, the representation of fungi is significantly lower in individuals living in Westernized societies than in those living in non-Western societies. The most abundant fungal sequences belong to the phylum-level taxa Ascomycota and Microsporidia. The phyla Ascomycota and Basidiomycota appear to be mutually exclusive, and the presence of Candida in particular correlates with recent consumption of carbohydrates.
Elucidation of Viral Dynamics Viruses are the most abundant biologic entity on Earth. Viral particles outnumber microbial cells by 10:1 in most environments. Humans are no exception in terms of viral colonization; our feces alone contain 108–109 viral particles per gram. Despite this abundance, many eukaryotic viral communities remain incompletely characterized, in part because the identification of viruses within metagenomic sequencing datasets is itself very challenging. Characterizing viral diversity requires different approaches: because no single gene is found in all viruses, no universal phylogenetic “barcode of life” equivalent to the SSU rRNA gene exists. One approach has been to selectively purify virus-like particles from community biospecimens, amplify the small amounts of DNA that are recovered, and randomly fragment the DNA and sequence the fragments (shotgun sequencing). The resulting sequences can be assembled into larger contigs whose function can be computationally predicted from homology to known genes, and the information obtained can be used to populate/expand nonredundant viral databases. These annotated nonredundant databases can then be used for more targeted mining of the rapidly expanding number of shotgun sequencing datasets generated from total-community DNA for known or putative DNA viruses.
Given the dominance of bacteria in the gut microbiota, it is not surprising that phages (viruses that infect bacteria) dominate the identifiable components of the gut’s DNA virome. Prophages are a manifestation of a so-called temperate viral–bacterial host dynamic, in which a phage is integrated into its host bacterium’s genome. This temperate dynamic provides a way to constantly refashion the genomes of bacterial species through horizontal gene transfer. Genes encoded by a prophage genome may expand the niche and fitness of their bacterial host, for example, by enabling the metabolism of previously inaccessible nutrient sources. Prophage integration can also protect the host strain from superinfection, “immunizing” the strain against infection by closely related phages. A temperate prophage life cycle allows the virus to expand in a 1:1 ratio with its bacterial host. If the integrated virus conveys increased fitness, the prevalence of the bacterial host and its phage will increase in the microbiota. Induction of a lytic cycle, where the prophage replicates and kills the host, may follow. Lytic cycles can cause high bacterial turnover. Lysis debris (e.g., components of capsules) can be used as nutrient sources by surviving bacteria; this change in the energy dynamic in a community is referred to as a phage shunt. A subpopulation of bacteria that undergoes lytic induction may sweep away other sensitive species present in the community, thus increasing the niche space available for survivors (i.e., those bacteria that already have an integrated prophage). Periodic induction of prophages leads to a “constant diversity dynamic” that helps maintain community structure and function.
Interest in viral communities has expanded in recent years, especially given a potentially therapeutic role for phages as an alternative or adjunct to antibiotics. Virome members have evolved elegant survival mechanisms that allow them to evade host defenses, diversify, and establish elaborate and mutually beneficial symbioses with their hosts. A number of recent studies have tried to adapt these mechanisms for therapeutic purposes (e.g., the use of synthetic phages to treat Pseudomonas aeruginosa infections in burn patients or in other settings). Phage therapy is not a new idea: Félix d’Herelle, co-discoverer of phages, recognized their potential medical applications nearly a century ago. However, only recently have our technologic capabilities and our knowledge of the human microbiota made phage therapy realistically attainable within our lifetimes.
ECOLOGIC PRINCIPLES AND PARAMETERS FOR COMPARING MICROBIAL COMMUNITIES
At many levels, different people are very much alike: our genomes are >99% identical, and we have similar collections of human cells. However, our microbial communities differ drastically, both between people and between habitats within a single human body. The greatest variation (beta diversity, described below) is between body sites. For example, the difference between the microbial communities residing in a person’s mouth versus the same person’s gut is comparable to the difference in communities residing in soil versus seawater. Even within a body site, the differences among people are not subtle: gut, skin, and oral communities can all differ by 80–90%, even from the broad, bacterial species–level view. The English poet John Donne said that “no man is an island”; however, from a microbial perspective, each of us consists of not just one isolated island but rather a whole archipelago of distinct habitats that exchange microbes with one another and with the outside environment at some as yet undetermined level. Before we can discuss these differences and understand their relevance to human disease, it is important to understand some basic terms and ecologic principles.
Alpha Diversity Alpha diversity is defined as the effective number of species present in a given sample. Communities that are compositionally more diverse (i.e., have more OTUs) or that are phylogenetically more diverse are defined as having greater alpha diversity. Alpha diversity can be measured by plotting the number of different types of SSU rRNA sequences identified at a given phylogenetic level (species, genera, etc.) in a sample as a function of the number of SSU rRNA gene reads collected. The most commonly used metrics of alpha diversity are Sobs (the number of species observed in a given number of sequences), Chao1 (a measure based on the number of species observed only once), the Shannon index (a measure of the number of bits of information gained by revealing the identity of a randomly chosen member of the community), and phylogenetic diversity (a measure of the total branch length of a phylogenetic tree encompassing a sample). Diversity estimators are particularly sensitive to errors introduced during PCR and sequencing.
Beta Diversity Beta diversity refers to the differences between communities and can be defined with phylogenetic or nonphylogenetic distance measurements. UniFrac is a commonly used phylogenetic metric that compares the evolutionary history of different microbial communities, noting the degree to which any two communities share branch length on a tree of microbial life: the more similar communities are to each other, the more branch length they share (Fig. 86e-1). UniFrac-based measurements of distances between communities can be visually represented with principal coordinates analysis or other geometric techniques that project a high-dimensional dataset down onto a small number of dimensions for a more approachable analysis (Fig. 86e-1). Principal coordinates analysis can also be applied to nonphylogenetic methods for comparing communities, such as Euclidean distance, Jensen-Shannon divergence, or Bray-Curtis dissimilarity, which operate independent of evolutionary tree data but can make biologic patterns more difficult to identify. The taxonomic data or distance matrices can also be used as input into a range of machine-learning algorithms (such as Random Forests) that employ supervised classification to identify differences between labeled groups of samples. Supervised classification is useful for identifying differences between cases and controls but can obscure important patterns intrinsic to the data, including confounding variables such as different sequencing runs or patient populations.
As noted above, the greatest beta diversity is that among body sites. This fact underscores the need to specify body habitat in microbiota analyses of any type, including microbial surveillance studies examining the flow of normal and pathogenic organisms into and out of different body sites in patients and their health care providers. Several other key points have emerged from beta diversity studies of human-associated microbial communities—notably, that (1) there is a high level of interpersonal variability in every body habitat studied to date, (2) intrapersonal variation in a given body habitat is less pronounced, and (3) family members have more similar communities than unrelated individuals living in separate households. Thus, a person is his/her own best control, and examination of an individual over time as a function of disease state or treatment intervention is desirable. Similarly, family members serve as logical reference controls, although age is a major covariate that affects microbiota structure.
Studies of fecal samples obtained from twins over time have shown that the overall degree of phylogenetic similarity of bacterial communities does not differ significantly between monozygotic and dizygotic twin pairs, although monozygotic twin pairs may be more similar in some populations at earlier ages. These results, together with intervention studies in mice and epidemiologic observations in humans, emphasize that early environmental exposures are a very important determinant of adult-gut microbial ecology. In humans, the initial exposures depend on delivery mode: babies sampled within 20 min of birth have relatively undifferentiated microbial communities in the mouth, the skin, and the gut. For vaginally delivered babies, these communities resemble the specific microbial communities found in the mother’s vagina. For babies delivered by cesarean section, the communities resemble skin communities. Although studies of older children and of adults stratified by delivery mode are still rare in the literature, these differences have been shown to persist until at least 4 months of age and perhaps until age 7 years. The infant gut microbiota changes to resemble the adult gut community over the first 3 years of life; comparable studies have not been done in other body habitats to date.
Exposures to environmental microbial reservoirs can continue to influence community structure. For example, unrelated cohabiting adults have more similar microbiotas in all of their body habitats than do non-cohabiting adults, and humans resemble the dogs they live with, at least in terms of skin microbiota. Gender and sexual maturation may also affect the microbiota structure, although efforts to isolate these variables are complicated by many confounding factors; any gender effect must be small compared with the effects of other variables such as diet (except in the case of the female urinary tract, which is influenced by the vaginal microbiota).
The vaginal microbiota illustrates another intriguing aspect of the contributions made by various factors to interpersonal differences in microbial community structure within a given body habitat. Bacterial 16S rRNA–based studies of the midvaginal microbiota in sexually active women have documented significant differences in community configurations between four self-reported ethnic groups: Caucasian, black, Hispanic, and Asian. Unlike most other body habitats that have been surveyed, this ecosystem is dominated by a single genus, Lactobacillus. Four species of this genus together account for more than half of the bacteria in most vaginal communities. Five community categories have been defined: four are dominated by L. iners, L. crispatus, L. gasseri, and L. jensenii, respectively, and the fifth has proportionally fewer lactobacilli and more anaerobes. The representation of these community categories is distinct within each of the four ethnic groups and correlates with vaginal pH and Nugent score (the latter being a biomarker for bacterial vaginosis). Longitudinal studies of individuals are being conducted to identify factors that determine the assembly of these distinct communities—both within and among ethnic groups—as well as their resistance to or resilience after various physiologic and pathologic disturbances. For example, the menstrual cycle and pregnancy turn out to be surprisingly significant factors (cause larger changes) compared with sexual activity.
Yet another factor affecting beta diversity is spatial location within a habitat. Several surveys show that the skin harbors bacterial communities with predictable, albeit complex, biogeographic features. To determine whether these differences are due to differences in local environmental factors, to the history of a given site’s exposure to microbes, or to a combination of the two, reciprocal microbiota transplantation has been performed. Microbial communities from one region of the skin were depleted by treatment with germicidal agents, and the region (plot) was inoculated with a “foreign” microbiota harvested from different regions of the skin or from different body habitats from the same or another individual. Community assembly at the site of transplantation was then tracked over time. Remarkably, assembly proceeded differently at different sites: forearm plots receiving a tongue microbiota remained more similar to tongue communities than to native forearm communities in terms of their composition and diversity, while forehead plots inoculated with tongue bacteria changed to become more similar to native forehead communities. Thus, in addition to the history of exposure to tongue bacteria, environmental factors operating at the forehead plot likely shape community assembly. Intriguingly, the factors that shape fungal skin communities appear to be entirely different from those that shape bacterial skin communities. The palm and forearm have high bacterial and low fungal diversity, whereas the feet have the opposite diversity pattern. Moreover, fungal communities are generally shaped by location (foot, torso, head), whereas bacterial communities are generally shaped by moisture phenotype (dry, moist, or sebaceous).
Co-Occurrence Analysis Co-occurrence analysis seeks to identify which phylotypes are co-distributed across individuals in a given body habitat and/or between habitats and to determine the factors that explain the observed patterns of co-distribution. Positive correlations tend to reflect shared preferences for certain environmental features, while negative correlations typically reflect divergent preferences or a competitive relationship. Syntrophic (cross-feeding) relationships reflect interdependent interactions based on nutrient-sharing strategies. For example, in food webs, the products of one organism’s metabolism can be used by the other for its own unique metabolic capabilities (e.g., the interactions between fermentative organisms and methanogens).
Enterotype Analysis Enterotype analysis seeks to classify individuals into discrete groups based on the configuration of their microbiotas, essentially drawing boundaries on a map defined by principal coordinates analysis or other ordination techniques. The first enterotype analysis used supervised clustering to define three major types of human-gut microbial configurations across three distinct human studies and provided a view that presupposed the existence of three clusters. Subsequent work has shown that the range of variability in the gut microbiota of children and of non-Western populations greatly exceeds the variability captured in the populations used to define the original enterotypes; in addition, even in Western populations, the variability follows more of a continuum dominated by a gradient in the abundance of the genera Bacteroides and Prevotella. Another consideration in enterotype analysis is whether location on a map defined by healthy human variation is relevant to predisposition to disease or whether instead rare species with particular functions are more important discriminants.
Functional Redundancy Functional redundancy arises when functions are performed by many bacterial taxa. Thus interpersonal differences in microbial bacterial diversity (i.e., which bacteria are present) are not necessarily accompanied by comparable degrees of difference in functional diversity (i.e., what these bacteria can do). Characterization of a microbiome by shotgun sequencing is important because, unlike SSU rRNA analyses, shotgun sequencing provides a direct readout of the genes (and, via comparative genomics, their functions) in a given community. One fundamental question is the degree to which variations in the species occupying a given body habitat correlate with variations in a community’s functional capabilities. For example, the neutral theory of community assembly developed by macroecologists suggests that species are added to the community without respect to function, automatically endowing the community with functional redundancy. If applicable to the microbial world, neutral community assembly would predict a high level of variation in the types of microbial lineages that occupy a given body habitat in different individuals, although the broad functions encoded in the microbiomes of these communities could be quite similar.
Shotgun sequencing of the fecal microbiome has revealed that different microbial communities converge on the same functional state: in other words, there is a group of microbial genes represented in the guts of unrelated as well as related individuals. The same principle holds true at other body sites (Fig. 86e-2). The “core” gut microbiome is enriched in functions related to microbial survival (e.g., translation; metabolism of nucleotides, carbohydrates, and amino acids) and in functions that benefit the host (nutrient and energy partitioning from the diet to microbes and host). The latter functions encompass the food webs mentioned above, in which products of one type of microbe become the substrates for other microbes. These webs, which can be incredibly elaborate, change as microbes adjust their patterns of gene expression and metabolism in response to alterations in nutrient availability. Thus the sum of all the activities of the members of a microbial community can be viewed as an emergent rather than a fixed property.
FIGURE 86e-2 Interpersonal variation in organismal representation in body habitat–associated communities is more extensive than interpersonal variation in gene functional features. Bacterial taxonomy and metabolic function are compared in 107 oral microbiota and microbiome samples (top) and in 139 fecal microbiota and microbiome samples (bottom). Samples represent an arbitrarily chosen subset from 242 healthy young adults living in the United States, with equal numbers of men and women. The same DNA extracts from the same samples were used for both taxonomic and functional classifications; each sample was analyzed by bacterial 16S rRNA amplicon sequencing (mean, 5400 sequences per sample) and by shotgun sequencing of community DNA (mean, 2.9 billion bases per sample). Taxonomic groups vary dramatically in their representation among different samples, with different characteristic bacterial phyla in the oral versus the fecal microbiota; e.g., members of the Actinobacteria and Fusobacteria are far more common in the mouth than in the gut, while members of Bacteroidetes are far more common in fecal samples. In contrast, metabolic pathways are far more consistently represented in different samples, even when the species that contribute to these pathways are completely different. These results suggest a high degree of functional redundancy in microbial ecosystems—similar to that observed in macroecosystems, in which many fundamentally different lineages of organisms can play the same ecologic roles (e.g., pollinator or top predator). (Adapted from Human Microbiome Project Consortium: Nature 486:207, 2012; and CA Lozupone et al: Nature 489:220, 2012.)
It is important to note that pairwise comparisons have shown that family members have functionally more similar gut microbiomes than do unrelated individuals. Thus, intrafamilial transmission of a gut microbiome within a given generation and across multiple generations could shape the biologic features of humans belonging to a kinship and modulate/mediate risks for a variety of diseases.
Stability Like other ecosystems, human body habitat–associated microbial communities vary over time, and an understanding of this variation is essential for a functional understanding of our microbiota. Few high-resolution time series of individual healthy adults have been published to date, but one available daily time series suggests that individuals tend to resemble themselves microbially day to day over a span of 6–15 months, retaining their separate identities during cohabitation. The development of low-error amplicon sequencing methods has provided a much more reliable way for defining stability at the strain level than was available in the past. Application of these methods to the guts of healthy individuals sampled over time has disclosed that a healthy adult gut harbors a persistent collection of ~100 bacterial species and several hundred strains. The stability of the bacterial components follows a power law: bacterial strains acquired early in life can persist in the gut for decades, although their proportional representation changes as a function of numerous factors, including diet. Whole-genome sequencing of culturable components of the microbiota of study participants has confirmed that strains are retained in individuals for prolonged periods and are shared among family members.
Resilience The ability of a microbiota or microbiome to rebound from a short-term perturbation, such as antibiotic administration or an infection, is defined as its resilience. This capacity can be visualized as a ball rolling over a landscape of local minima; essentially, the community moves into a new state and, to recover, must move through another, unstable state. In some cases, recovery will lead to the original stable state; in others, it will lead to a new stable state, which may be either healthy or unhealthy. Changes in, for example, diet or host physiologic status may introduce alterations into the landscape itself, making it easier to move from the initial state to any one of a number of other states, potentially with different health consequences. Microbial communities in our body habitats differ widely in resilience. For example, hand washing leads to profound changes in the microbial community, greatly increasing diversity (presumably because of the preferential removal of high-abundance, dominant phylotypes such as Propionibacterium). Within 6 h, the hand microbiota rebounds to resemble the original hand communities. The effects of repeated hand washing still need to be defined; for example, the surface microbiota on the skin (as measured by scrape biopsies) consists of ~50,000 microbial cells/cm2, whereas the subsurface microbiota (as measured by punch biopsies) consists of ~1,000,000 microbial cells/cm2.
In a study of three healthy adult volunteers given a short course of ciprofloxacin (500 mg by mouth twice a day for 5 days—a regimen commonly used against uncomplicated urinary tract infections), overall gut-community configuration came to resemble baseline within 6 months after treatment cessation, although some taxa failed to recover. However, the effects of the antibiotic perturbation were highly individualized. Administration of a second course of treatment months later led to altered-community states, relative to baseline, in all three volunteers; again, the extent of the alteration differed with the individual. Crucially, as shown in this and other studies, a given bacterial taxon can respond differently to the same antibiotic in different individuals; this observation suggests that the rest of the microbial community plays an important role in determining the effects of antibiotics on a per-individual basis.
In any body habitat, the microbial-community state after disturbance may be degraded. However, this degraded state may itself be resilient, and it may therefore be difficult to restore a more functional state. For example, Clostridium difficile infection can persist for years. The development and resilience of a degraded state may be driven by positive feedback loops, such as reactive oxygen species cascades involving host macrophages that promote the further growth of proinflammatory Proteobacteria, as well as negative-feedback loops such as depletion of the butyrate needed for promotion of a healthy gut epithelial barrier and further establishment of beneficial members of the microbiota. Consequently, microbiota-based therapies may require either (1) the elimination of a feedback loop that prevents establishment of a new community or (2) identification of a direction for change and a stimulus of sufficient magnitude (e.g., invasion and establishment of microbes from a fecal transplant or from a defined consortium of cultured, sequenced members of the human gut microbiota; see below) to overcome the resilience mechanisms inherent in the degraded state. A critical unresolved question that especially affects infants, whose microbiota is changing rapidly, is whether intervention during periods of rapid change or during periods of relative stability is generally more effective.
ESTABLISHING CAUSAL RELATIONSHIPS BETWEEN THE GUT MICROBIOTA AND NORMAL PHYSIOLOGIC, METABOLIC, AND IMMUNOLOGIC PHENOTYPES AS WELL AS DISEASE STATES
Gnotobiotic animals are raised in germ-free environments—with no exposure to microbes—and then colonized at specific stages of life with specified microbial communities. Gnotobiotic mice provide an excellent system for controlling host genotype, microbial community composition, diet, and housing conditions. Microbial communities harvested from donor mice with defined genotypes and phenotypes can be used to determine how the donors’ microbial communities affect the properties of formerly germ-free recipients. The recipients may also affect the transplanted microbiota and its microbiome. Thus gnotobiotic mice afford investigators an opportunity to marry comparative studies of donor communities to functional assays of community properties and to determine how (and for how long) these functions influence host biology.
The Cardiovascular System The gut microbiota affects the elaborate microvasculature underlying the small-intestinal epithelium: capillary network density is markedly reduced in adult germ-free animals but can be restored to normal levels within 2 weeks after gut microbiota transplantation. Mechanistic studies have shown that the microbiota promotes vascular remodeling in the gut through effects on a novel extravascular tissue factor–protease-activated receptor (PAR1) signaling pathway. Heart weight measured echocardiographically or as wet mass and normalized to tibial length or lean body weight is significantly reduced in germ-free mice; this difference is eliminated within 2 weeks after colonization with a gut microbiota. During fasting, a gut microbiota–dependent increase in hepatic ketogenesis (regulated by peroxisome proliferator–activated receptor α) occurs, and myocardial metabolism is directed to ketone body utilization. Analyses of isolated, perfused working hearts from germ-free and colonized animals, together with in vivo assessments, have shown that myocardial performance in germ-free mice is maintained by increasing glucose utilization. However, heart weight is significantly reduced in both fasted and fed mice; this heart-mass phenotype is completely reversed in germ-free mice fed a ketogenic diet. These findings illustrate how the gut microbiota benefits the host during periods of nutrient deprivation and represent one link between gut microbes and cardiovascular metabolism and health.
Conventionally raised apoE-deficient mice develop a less severe form of atherosclerosis than their germ-free counterparts when fed a high-fiber diet. This protective effect of the microbiota is obviated when animals are fed a diet low in fiber and high in simple sugars and fat. A number of the beneficial effects attributed to diets with high proportional representation of whole grains, fruits, and vegetables are thought to be mediated by end products of microbial metabolism of dietary compounds, including short-chain fatty acids and metabolites derived from flavonoids. Conversely, microbes can convert otherwise harmless dietary compounds into metabolites that increase risk for cardiovascular disease. Studies of mice and human volunteers have revealed that gut microbiota metabolism of dietary L-carnitine, which is present in large amounts in red meat, yields trimethylamine-N-oxide, which can accelerate atherosclerosis in mice by suppressing reverse cholesterol transport.
Yet another facet of microbial influence on cardiovascular physiology was revealed in a study of mice deficient in Olfr78 (a G protein–coupled receptor expressed in the juxtaglomerular apparatus, where it regulates renin secretion in response to short-chain fatty acids) or Gpr41 (another short-chain fatty acid receptor that, together with Olfr78, is expressed in the smooth muscle cells present in small resistance vessels). This study demonstrated that the microbiota can modulate host blood pressure via short-chain fatty acids produced by microbial fermentation.
Bone Adult germ-free mice have greater bone mass than their conventionally raised counterparts. This increase in bone mass is associated with reduced numbers of osteoclasts per unit bone surface area, reduced numbers of CD11b+/GR1 osteoclast precursors in bone marrow, decreased numbers of CD4+ T cells, and reduced levels of expression of the osteolytic cytokine tumor necrosis factor α. Colonization with a normal gut microbiota resolves these observed differences between germ-free and conventionally raised animals.
Brain Adult germ-free and conventionally raised mice differ significantly in levels of 38 out of 196 identified cerebral metabolites, 10 of which have known roles in brain function; included in the latter group are N-acetylaspartic acid (a marker of neuronal health and attenuation), pipecolic acid (a presynaptic modulator of γ-aminobutyric acid levels), and serine (an obligatory co-agonist at the glycine site of the N-methyl-D-aspartate receptor). Propionate, a short-chain fatty acid product of gut microbial-community metabolism of dietary fiber, affects expression of genes involved in intestinal gluconeogenesis via a gut–brain neural circuit involving free fatty-acid receptor 3; this effect provides a mechanistic explanation for the documented beneficial impact of dietary fiber in enhancing insulin sensitivity and reducing body mass and adiposity.
Studies of a mouse model (maternal immune activation) with stereotyped/repetitive and anxiety-like behaviors indicate that treatment with a member of the human gut microbiota, Bacteroides fragilis, corrects gut barrier (permeability) defects; reduces elevated levels of 4-ethylphenylsulfate, a metabolite seen in the maternal immune activation model that has been causally associated with the animals’ behavioral phenotypes; and ameliorates some behavioral effects. These observations highlight the importance of further exploration of potentially co-evolved relationships between the microbiota and host behavior.
Immune Function Many foundational studies have shown that the gut microbiota plays a key role in the maturation of the innate as well as the adaptive components of the immune system. The intestinal epithelium, which is composed of four principal cell lineages (enterocytes plus goblet, Paneth, and enteroendocrine cells), acts as a physical and functional barrier to microbial penetration. Goblet cells produce mucus that overlies the epithelium, where it forms two layers: an outer (luminal-facing) looser layer that harbors microbes and a denser lower layer that normally excludes microbes. Members of the Paneth cell lineage reside at the base of crypts of Lieberkühn and secrete antimicrobial peptides. Studies in mice have demonstrated that Paneth cells directly sense the presence of a microbiota through expression of the signaling adaptor protein MyD88, which helps transduce signals to host cells upon recognition of microbial products through Toll-like receptors (TLRs). This recognition drives expression of antibacterial products (e.g., the lectin RegIIIγ) that act to prevent microbial translocation across the gut mucosal barrier.
The intestine is enriched for B cells that produce IgA, which is secreted into the lumen; there it functions to exclude microbes from crossing the mucosal barrier and to restrict dissemination of food antigens. The microbiota plays a key role in development of an IgA response: germ-free mice display a marked reduction in IgA+ B cells. The absence of a normal IgA response can lead to a massive increase in bacterial load. B cell–derived IgA that targets specific members of the gut microbiota plays an important role in preventing activation of microbiota-specific T cells.
Gut bacterial species elicit development of protective TH17 and TH1 responses that help ward off pathogen attack. Members of the microbiota also promote the development of a specialized population of CD4+ T cells that prevent unwarranted inflammatory responses. These regulatory T cells (Tregs) are characterized by expression of the transcription factor forkhead box P3 (FOXP3) and by expression of other cell-surface markers. There is a paucity of Tregs in the colonic lamina propria of germ-free mice. Specific members of the microbiota—including a consortium of Clostridium strains isolated from the mouse and human gut as well as several human-gut Bacteroides species—expand the Treg compartment and enhance immunosuppressive functions.
The microbiota is a key trigger in the development of inflammatory bowel disease (IBD) in mice that harbor mutations in genes associated with IBD risk in humans. Moreover, components of the gut microbiota can modify the activity of the immune system to ameliorate or prevent IBD. Mice containing a mutant ATG16L1 allele linked to Crohn’s disease are particularly susceptible to IBD. Upon infection with mouse norovirus and treatment with dextran sodium sulfate, expression of a hypomorphic ATG16L1 allele leads to defects in small-intestinal Paneth cells and renders mice significantly more susceptible to ileitis than are wild-type control animals. This process is dependent on the gut microbiota and highlights how the intersection of host genetics, infectious agents, and the microbiota can lead to severe immune pathology; i.e., the pathogenic potential of a microbiota may be context-dependent, requiring a confluence of factors. An important observation is that members of the gut microbiota, including B. fragilis or members of Clostridium, prevent the severe inflammation that develops in mouse models mimicking various aspects of human IBD.
The gut microbiota has been implicated in promoting immunopathology outside of the intestine. Multiple sclerosis develops in conventionally raised mice whose CD4+ T cell compartment is reactive to myelin oligodendrocyte protein; their germ-free counterparts are completely protected from development of multiple sclerosis–like symptoms. This protection is reversed by colonization with a gut microbiota from conventionally raised animals.
Inflammasomes are cytoplasmic multiprotein complexes that sense stress and damage-associated patterns. Mice deficient in NLRP6, a component of the inflammasome, are more susceptible to colitis induced by administration of dextran sodium sulfate. This enhanced susceptibility is associated with alterations in the gut microbiota of these animals relative to that of wild-type controls. Mice are coprophagic, and co-housing of NLRP6-deficient mice with wild-type mice is sufficient to transfer the enhanced susceptibility to colitis induced by dextran sodium sulfate. Similar findings have been reported for mice deficient in the inflammasome adaptor ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain). ASC-deficient mice are more susceptible to the development of a model of nonalcoholic steatohepatitis. This susceptibility is associated with alterations in gut microbiota structure and can be transferred to wild-type animals by co-housing.
Obesity and Diabetes Germ-free mice are resistant to diet-induced obesity. Genetically obese ob/ob mice have gut microbial-community structures that are profoundly altered from those in their lean wild-type (+/+) and heterozygous +/ob littermates. Transplantation of the ob/ob mouse microbiota into wild-type germ-free animals transmits an increased-adiposity phenotype not seen in mice receiving microbiota transplants from +/+ and +/ob littermates. These differences are not attributable to differences in food consumption but rather are associated with differences in microbial community metabolism. Roux-en-Y gastric bypass produces pronounced decreases in weight and adiposity as well as improved glucose metabolism—changes that are not ascribable simply to decreased caloric intake or reduced nutrient absorption. 16S rRNA analyses have documented that changes in the gut microbiota after this surgery are conserved among mice, rats, and humans; animal studies have demonstrated these changes along the length of the gut but most prominently downstream of the site of surgical manipulation of the bowel. Notably, transplantation of the gut microbiota from mice that have undergone Roux-en-Y gastric bypass to germ-free mice that have not had this surgery produces reductions in weight and adiposity not seen in recipients of microbiotas from mice that underwent sham surgery.
The gut microbiota confers protection against the development of type 1 diabetes mellitus in the non-obese diabetic (NOD) mouse model. Disease incidence is significantly lower in conventionally raised male NOD mice than in their female counterparts, while germ-free males are as susceptible as their female counterparts. Castration of males increases disease incidence, while androgen treatment of females provides protection. Transfer of the gut microbiota from adult male NOD mice to female NOD weanlings is sufficient to reduce the severity of disease relative to that among females receiving a microbiota from an adult female or an unmanipulated female. The blocking of protection by treatment with flutamide highlights a functional role for testosterone signaling in this microbiota-mediated protection against type 1 diabetes.
NOD mice deficient in MyD88, a key component of the TLR signaling pathway, do not develop diabetes and exhibit increased relative abundance of members of the family-level taxon Lactobacillaceae. Consistent with these findings, investigators have documented lower levels of representation of members of the genus Lactobacillus in children with type 1 diabetes than in healthy controls. Components of lactobacilli have been shown to promote gut barrier integrity. Studies in various animal models indicate that translocation of bacterial components, including bacterial lipopolysaccharides, across a leaky gut barrier triggers low-grade inflammation, which contributes to insulin resistance. Mice deficient in TLR5 exhibit alterations in the gut microbiota and hyperphagia, and they develop features of metabolic syndrome, including hypertension, hyperlipidemia, insulin resistance, and increased adiposity.
The gut microbiota regulates biosynthesis as well as metabolism of host-derived products; these products can signal through host receptors to shape host physiology. An example of this symbiosis is provided by bile acids, which direct metabolic effects that are largely mediated through the farnesoid × receptor (FXR, also known as NR1H4). In leptin-deficient mice, FXR deficiency protects against obesity and improves insulin sensitivity. In mice with diet-induced obesity that are subjected to vertical sleeve gastrectomy, the surgical procedure results in elevated levels of circulating bile acids, changes in the gut microbiota, weight loss, and improved glucose homeostasis. However, weight reduction and improved insulin sensitivity are mitigated in animals with engineered FXR-deficiency.
Xenobiotic Metabolism Evidence is accumulating that pharmacogenomic studies need to consider the gene repertoire present in our H. sapiens genome as well as that in our microbiomes. For example, digoxin is inactivated by the human gut bacterium Eggerthella lenta, but only by strains with a cytochrome-containing operon. Expression of this operon is induced by digoxin and inhibited by arginine. Studies in gnotobiotic mice established that dietary protein affects (reduces) microbial metabolism of digoxin, with corresponding alterations in levels of the drug in both serum and urine. These findings reinforce the need to consider strain-level diversity in the gut microbiota when examining interpersonal variations in the metabolism of orally administered drugs.
Characterizing the Effects of the Human Microbiota on Host Biology in Mice and Humans Questions about the relationship between human microbial communities and health status can be posed in the following general format: Is there a consistent configuration of the microbiota definable in the study population that is associated with a given disease state? How is the configuration affected by remission/relapse or by treatment? If a reconfiguration does occur with treatment, is it durable? How is host biology related to the configuration or reconfiguration? What is the effect size? Are correlations robust to individuals from different families and communities representing different ages, geographic locales, and lifestyles?
As in all studies involving human microbial ecology, the issue of what constitutes a suitable reference control is extremely important. Should we choose the person himself or herself, family members, or age- or gender-matched individuals living in the same locale and representing similar cultural traditions? Critically, are the relationships observed between microbial community structure and expressed functions a response to disease state (i.e., side effects of other processes), or are they a contributing cause? In this sense, we are challenged to evolve a set of Koch’s postulates that can be applied to whole microbial communities or components of communities rather than just to a single purified organism. As in other circumstances in which experiments to determine causality of human disease are difficult or unethical, Hill’s criteria, which examine the strength, consistency, and biologic plausibility of epidemiologic data, can be useful.
Sets of mono- and dizygotic twins and their family members represent a valuable resource for initially teasing out relationships between environmental exposures, genotypes, and our own microbial ecology. Similarly, monozygotic twins discordant for various disease states enhance the ability to determine whether various diseases can be linked to a person’s microbiota and microbiome. A twin-pair sampling design rather than a conventional unrelated case–control design has advantages owing to the pronounced between-family variability in microbiota/microbiome composition and the potential for multiple states of a community associated with disease. Transplantation of a microbiota from suitable human donor controls representing different disease states and communities (e.g., twins discordant for a disease) to germ-free mice is helpful in establishing a causal role for the community in pathogenesis and for providing insights relevant to underlying mechanisms. In addition, transplantation provides a preclinical platform for identifying next-generation probiotics, prebiotics, or combinations of the two (synbiotics). Obesity and obesity-associated metabolic dysfunction illustrate these points.
The gut microbiotas (and microbiomes) of obese individuals are significantly less diverse than those of lean individuals; the implication is that there may be unfilled niches (unexpressed functions) that contribute to obesity and its associated metabolic abnormalities. Le Chatelier and colleagues observed a bimodal distribution of gene abundance in their analysis of 292 fecal microbiomes: low-gene-count (LGC) individuals averaged 380,000 microbial genes per gut microbiome, while high-gene-count (HGC) individuals averaged 640,000 genes. LGC individuals had an increased risk for type 2 diabetes and other metabolic abnormalities, whereas the HGC group was metabolically healthy. When gene content was used to identify taxa that discriminated HGC and LGC individuals, the results revealed associations between anti-inflammatory bacterial species such as Faecalibacterium prausnitzii and the HGC group and between proinflammatory species such as Ruminococcus gnavus and the LGC group. LGC microbiomes had significantly greater representation of genes assigned to tricarboxylic acid cycle modules, peroxidases, and catalases—an observation suggesting a greater capacity to handle oxygen exposure and oxidative stress; HGC microbiomes were enriched in genes involved in the production of organic acids, including lactate, propionate, and butyrate—a result suggesting increased fermentative capacity.
Transplantation of an uncultured fecal microbiota from twins stably discordant for obesity or of bacterial culture collections generated from their microbiota transmits their discordant adiposity phenotypes as well as obesity-associated metabolic abnormalities to recipient germ-free mice. Co-housing of the recipient coprophagic gnotobiotic mice results in invasion of specific bacterial species from the transplanted lean twin’s culture collection into the guts of cage mates harboring the obese twin’s culture collection (but not vice versa), thereby preventing the latter animals from developing obesity and its associated metabolic abnormalities. It is noteworthy that invasion and prevention of obesity and metabolic phenotypes are dependent on the type of human diets fed to animals: prevention is associated with a diet low in saturated fats and high in fruit and vegetable content, but not with a diet high in saturated fats and low in fruit and vegetable content.
This approach provides evidence for a causal role for the microbiota in obesity and its attendant metabolic abnormalities. It also provides a method for defining unoccupied niches in disease-associated microbial communities, the role of dietary components in determining how these niches can be filled by human gut–derived bacterial taxa, and the effects of such occupancy on microbial and host metabolism. It also provides a way to identify health-promoting diets and next-generation probiotics representing naturally occurring members of our indigenous microbial communities that are well adapted to persist in a given body habitat.
A key to this approach is the ability to harvest a microbial community from a donor representing a physiology, disease state, lifestyle, or geography of interest; to preserve the donor’s community by freezing it; and then to resurrect and replicate it in multiple recipient gnotobiotic animals that can be reared under conditions where environmental and host variables can be controlled and manipulated to a degree not achievable in clinical studies. Since these mice can be followed as a function of time prior to and after transplantation, in essence, a snapshot of a donor’s community can be converted into a movie. Transplantation of intact uncultured human (fecal) microbiota samples from multiple donors representing the phenotype of interest, with administration of the donors’ diets (or derivatives of those diets) to different groups of mice, is one way to assess whether transmissible responses are shared features of the microbiota or are highly donor specific. A second step is to determine whether the culturable component of a representative microbiota sample can transmit the phenotype(s) observed with the intact uncultured sample. Possession of a collection of cultured organisms that have co-evolved in a given donor’s body habitat sets the stage for the selection of subsets of the collection for testing in gnotobiotic mice, the determination of which members are responsible for effecting the phenotype, and the elucidation of the mechanisms underlying these effects. The models used may inform the design and interpretation of clinical studies of the very individuals and populations whose microbiota are selected for creating these models.
Human-to-human fecal microbiota transplantation (FMT) is currently the most direct way to establish proof-of-concept for a causal role for the microbiota in disease pathogenesis. A human donor’s feces are provided to a recipient via nasogastric tube or another technique. Numerous small trials have documented the effects of FMT from healthy donors to recipients with diseases ranging from C. difficile infection to Crohn’s disease, ulcerative colitis, and type 2 diabetes. Only a few of these studies have used a double-blind, placebo-controlled design.
In a double-blind, controlled trial involving men 21–65 years old with a body mass index of >30 kg/m2 and documented insulin resistance, FMT was performed using a microbiota from metabolically healthy lean donors or from the study participants themselves. A microbiota from lean donors significantly improved peripheral insulin sensitivity over that in controls. This change was associated with an increase in the relative abundance of the butyrate-producing bacteria related to Roseburia intestinalis (in the feces) and Eubacterium hallii (in the small intestine).
The efficacy of FMT for the treatment of recurrent C. difficile infection has been assessed in a number of small trials. One unblinded, placebo-controlled trial assessed the use of FMT in 42 patients with recurrent C. difficile infection (defined as at least one relapse after treatment with vancomycin or metronidazole for ≥10 d). Patients were pretreated with oral vancomycin. The experimental group then received FMT via nasoduodenal tube from healthy volunteer donors (<60 years of age) selected from the community. Controls underwent sterile lavage or received oral vancomycin alone. In 10 weeks of follow-up, infection was cured (with cure defined as three negative fecal tests for C. difficile toxin) in 81% of patients in the FMT group (13 of 16) but in only 23% (3 of 13) in the bowel-lavage control arm and 31% (4 of 13) in the vancomycin-only group. Metagenomic analysis of microbiota samples collected before and after treatment revealed an increased representation of Bacteroidetes and Clostridium clusters IV and XIVa, along with a 100-fold decrease in the relative abundance of Proteobacteria, in the FMT group.
A meta-analysis of FMT in C. difficile infection examined 20 case-series publications, 15 case reports, and the one unblinded study described above. All but one of these studies used fresh (not frozen) fecal samples. Donor selection varied, although most donors were family members or relatives and most studies excluded donors who had recently received antibiotics. It is noteworthy that the concentrations of infused donor feces varied widely (i.e., from 5 g to 200 g, resuspended in 10–500 mL); these fecal suspensions were introduced at different sites along the gastrointestinal tract, including the stomach and points throughout the small intestine and colon. Resolution of infection, which was frequently assessed on the basis of symptom resolution (with C. difficile toxin testing rarely performed), was documented in 87% (467) of 536 treated patients. The most common adverse events reported were diarrhea (94% of cases) and abdominal cramps (31%) on the day of infusion. The meta-analysis was limited to clinical outcomes and did not specifically address the role of the microbiota in disease resolution (e.g., the extent of invasion of donor taxa; their persistence; or the long-term effects of transplantation on various facets of host biology, which generally have not been evaluated).
Sober and thoughtful consideration needs to be applied to the therapeutic use of FMT, which represents an early and rudimentary approach to microbiota manipulation that very likely will be replaced by administration of defined collections of sequenced, cultured members of the human microbiota (probiotic consortia). A number of published reports on FMT have garnered significant public attention. This attention, coupled with an increasing public appreciation of the beneficial nature of our interactions with microbes, demands that the precautionary principle be honored and that risks versus benefits of such interventions be carefully evaluated.
To date, most FMT trials have failed to define (or have differed in) significant confounders, including (1) the criteria used for donor sample selection; (2) the methods used for donor sample preparation and characterization as well as the decision about whether or not to create a repository for donor and recipient samples that will permit retrospective analyses (and meta-analyses for given disease states); (3) the development of minimal standards for assessing the invasion of recipient gut communities by taxa from donor microbiota (using microbial source-tracking methods) as well as the timing, duration, nature, and breadth of sampling of the recipient as a function of transplantation; (4) the adoption of minimal standards for collection of patients’ clinical data (e.g., age, diet, antibiotic use) and the establishment of databases for entering these data (including use of a defined vocabulary for annotating the clinical data); and (5) the development of standards for informed consent in lieu of knowledge of the long-term effects of the procedure. The regulatory landscape is evolving. The U. S. Food and Drug Administration recently issued an enforcement policy specifically addressing the use of FMT for the treatment of recurrent C. difficile infection; this policy indicates that the agency intends to “exercise enforcement discretion regarding the investigational new drug (IND) requirements for the use of FMT to treat C. difficile infection not responding to standard therapies,” but it does not waive IND requirements for other FMT studies.
MOVING FORWARD
The design of human microbiome studies is rapidly evolving, in part because the data are highly multivariate, are compositional, and do not meet distributional assumptions of standard statistical tests such as analysis of variance. Consequently, the proper number of subjects to enroll and the proper populations to target remain to be established. One useful approach is to review published studies and ask whether the reported conclusion could be obtained with fewer subjects (sample rarefaction) and/or fewer sequencing reads from SSU rRNA genes, whole-community DNA (microbiomes), or expressed community mRNA (metatranscriptomes) per subject (sequence rarefaction). A common yet critical problem to avoid is under-sampling of the types of objects under study. For example, if the goal is to compare factors applying to individuals (e.g., individual diet), then dozens of individuals in each clinical category may be needed. If the goal is to compare factors applying to populations (e.g., demographic properties), then many populations may be needed.
Another key issue is whether the effect size to be studied, especially in meta-analysis, is greater than or less than technical effects. As noted above, different PCR primers will lead to different readouts of the taxonomy of a microbial community; these differences are, for example, greater than the differences between lean and obese subjects’ fecal microbiota but less than the difference between fecal communities in newborns and adults.
A central challenge in human microbiome research is establishing the extent to which diagnostic tests and therapeutic approaches are generalizable. This challenge is illustrated by studies of the capacities of gut microbiomes to metabolize orally administered drugs. The results could be very informative for the pharmaceutical industry as it seeks new and more accurate ways to predict bioavailability and toxicity. However, these studies should prompt consideration of the fact that many clinical trials are outsourced to countries where trial participants have diets and microbial community structures that differ from those of the intended initial recipients of the (marketed) drug. Capture and preservation of the wide range of microbial diversity present in different human populations—and thus of the capacity of our microbial communities to catalyze elaborate and in many respects uncharacterized biotransformations—represent potentially fertile ground for the discovery of new drugs (and new industrial processes of societal value). The chemical entities that our microbial communities have evolved to synthesize in order to support their mutually beneficial relationships and the human genes that these chemotypes influence may become new classes of drugs and new targets for drug discovery, respectively. Therefore, characterization of groups of individuals living in countries that are undergoing rapid transformations in cultural traditions and socioeconomic conditions and are witnessing the emergence of a variety of diseases associated with increasingly Western lifestyles (globalization) is a timely challenge. Birth cohort studies (including studies of twins) initiated every 10 years in these countries may be able to capture the impact of globalization, including changing diets, on human microbial ecology.
Although microbiome-associated diagnostics and therapeutics provide new and exciting dimensions for personalized medicine, attention must be paid to the potentially broad societal impact of this work. For example, studies of the human gut microbiome are likely to have a disruptive effect on current views of human nutrition, enhancing appreciation of how food and the metabolic output of interactions of dietary components with the microbiota are intimately connected to myriad features of human biology. Underlying the efforts to elucidate the relations among food, the microbiome, and human nutrition is a need to proactively develop materials for educational outreach with a narrative and vocabulary that is understandable to broad and varied consumer populations representing different cultural traditions and widely ranging degrees of scientific literacy. The results have the potential to catalyze efforts to integrate agricultural policies and practice, food production, and nutritional recommendations for consumer populations representing different ages, geographic locales, and states of health.
Defining our metagenome (the genes embedded in our H. sapiens genome plus those in our microbiome) will likely lead to an entirely new level of refinement in our description of self, our genetic evolution, our postnatal development, the microbial legacy of our connection to family, and the consequences of personal lifestyle choices. While this information can help us understand the origins of certain yet unexplained health disparities, care must be taken to avoid stigmatization of individuals or groups of individuals having different cultural norms, belief systems, or behaviors. In partnership with human microbiome researchers, anthropologists need to examine the impact of studies of the human microbiome on the participants, assessing how this field and participants’ cultural traditions interact to affect these individuals’ perceptions about the natural world, the forces that affect their lives, and their connections to one another within the context of family and community.
SUMMARY
Studies of human microbial ecology are an important manifestation of progress in the genome sciences, represent a timely step in our quest to achieve a better understanding of our place in the natural world, and reflect the evolving focus of twenty-first-century medicine on disease prevention, new definitions of health, new ways to determine the origins of individual biologic differences, and new approaches to evaluating the impact of changes in our lifestyles and biosphere on our biology. As microbiome-directed diagnostics and therapeutics emerge, we must be sensitive to the societal impact of this work.
87e |
Network Medicine: Systems Biology in Health and Disease |
The field of human biology has progressed over the last three centuries largely as a result of the reductionist approach to the scientific problems that challenge the discipline. Biologists study the experimental response of a variable of interest in a cell or organism while holding all other variables constant. In this way, it is possible to dissect the individual components of a biologic system and assume that a thorough understanding of a specific component (e.g., an enzyme or a transcription factor) will provide sufficient insight to explain the global behavior of that system (e.g., a metabolic pathway or a gene network, respectively). Biologic systems are, however, much more complex than this approach assumes and manifest behaviors that frequently (if not invariably) cannot be predicted from knowledge of their component parts characterized in isolation. Growing recognition of this shortcoming of conventional biologic research has led to the development of a new discipline, systems biology, which is defined as the holistic study of living organisms or their cellular or molecular network components to predict their response to perturbations. Concepts of systems biology can be applied readily to human disease and therapy and define the field of systems pathobiology, in which genetic or environmental perturbations produce disease and drug perturbations restore normal system behavior.
Systems biology evolved from the field of systems engineering in which a linked collection of component parts constitute a network whose output the engineer wishes to predict. The simple example of an electronic circuit can be used to illustrate some basic systems engineering concepts. All the individual elements of the circuit—resistors, capacitors, transistors—have well-defined properties that can be characterized precisely. However, they can be linked (wired or configured) in a variety of ways, each of which yields a circuit whose response to voltage applied across it is different from the response of every other configuration. To predict the circuit’s (i.e., system’s) behavior, the engineer must study its response to perturbation (e.g., voltage applied across it) holistically rather than its individual components’ responses to that perturbation. Viewed another way, the resulting behavior of the system is greater than (or different from) the simple sum of its parts, and systems engineering utilizes rigorous mathematical approaches to predict these complex, often nonlinear, responses. By analogy to biologic systems, one can reason that detailed knowledge of a single enzyme in a metabolic pathway or of a single transcription factor in a gene network will not provide sufficient detail to predict the output of that metabolic pathway or transcriptional network, respectively. Only a systems-based approach will suffice.
It has taken biologists a long time to appreciate the importance of systems approaches to biomedical problems. Reductionism has reigned supreme for many decades, largely because it is experimentally and analytically simpler than holism, and because it has provided insights into biologic mechanisms and disease pathogenesis that have led to successful therapies. However, reductionism cannot solve all biomedical problems. For example, the so-called off-target effects of new drugs that frequently limit their adoption likely reflect the failure of a drug to be studied in holistic context, i.e., the failure to explore all possible actions aside from the principal target action for which it was developed. Other approaches to understanding biology therefore are clearly needed. With the growing body of genomic, proteomic, and metabolomic data sets in which dynamic changes in the expression of many genes and many metabolites are recorded after a perturbation and with the growth of rigorous mathematical approaches to analyzing those changes, the stage has been set for applying systems engineering principles to modern biology.
Physiologists historically have had more of a (bio)engineering perspective on the conduct of their studies and have been among the first systems biologists. Yet, with few exceptions, they, too, have focused on comparatively simple physiologic systems that are tractable using conventional reductionist approaches. Efforts at integrative modeling of human physiologic systems, as first attempted by Guyton for blood pressure regulation, represent one application of systems engineering to human biology. These dynamic physiologic models often focus on the acute response of a measurable physiologic parameter to a system perturbation, and do so from a classic analytic perspective in which all the conventional physiologic determinants of the output parameter are known and can be modeled quantitatively.
Until recently, molecular systems analysis has been limited owing to inadequate knowledge of the molecular determinants of a biologic system of interest. Although biochemists have approached metabolic pathways from a systems perspective for over 50 years, their efforts have been limited by the inadequacy of key information for each enzyme (KM, kcat, and concentration) and substrate (concentration) in the pathway. With increasingly rich molecular data sets available for systems-based analyses, including genomic, transcriptomic, proteomic, and metabolomic data, biochemists are now poised to use systems biology approaches to explore biologic and pathobiologic phenomena.
PROPERTIES OF COMPLEX BIOLOGIC SYSTEMS
To understand how best to apply the principles of systems biology to human biomedicine, it is necessary to review briefly the building blocks of any biologic system and the determinants of system complexity. All systems can be analyzed by defining their static topology (architecture) and their dynamic (i.e., time-dependent) response to perturbation. In the discussion that follows, system properties are described that derive from the consequences of topology (form) or dynamic response (function). Any system of interacting elements can be represented schematically as a network in which the individual elements are depicted as nodes and their connections are depicted as links. The nature of the links among nodes reflects the degree of complexity of the system. Simple systems are those in which the nodes are linearly linked with occasional feedback or feedforward loops modulating system throughput in highly predictable ways. By contrast, complex systems are nodes that are linked in more complicated, nonlinear networks; the behavior of these systems by definition is inherently more difficult to predict owing to the nature of the interacting links, the dependence of the system’s behavior on its initial conditions, and the inability to measure the overall state of the system at any specific time with great precision. Complex systems can be depicted as a network of lower-complexity interacting components or modules, each of which can be reduced further to simpler analyzable canonical motifs (such as feedback and feedforward loops, or negative and positive autoregulation); however, a central property of complex systems is that simplifying their structures by identifying and characterizing the individual nodes and links or even simpler substructures does not necessarily yield a predictable understanding of a system’s behavior. Thus, the functioning system is greater than (or different from) the sum of its individual, tractable parts.
Defined in this way, most biologic systems are complex systems that can be represented as networks whose behaviors are not readily predictable from simple reductionist principles. The nodes, for example, can be metabolites that are linked by the enzymes that cause their transformations, transcription factors that are linked by the genes whose expression they influence, or proteins in an interaction network that are linked by cofactors that facilitate interactions or by thermodynamic forces that facilitate their physical association. Biologic systems typically are organized as scale-free, rather than stochastic, networks of nodes. Scale-free networks are those in which a few nodes have many links to other nodes (highly linked nodes, or hubs) but most nodes have only a few links (weakly linked nodes). The term scale-free refers to the fact that the connectivity of nodes in the network is invariant with respect to the size of the network. This is quite different from two other common network architectures: random (Poisson) and exponential distributions. Scale-free networks can be mathematically described by a power law that defines the probability of the number of links per node (P[k] = k–[γ], where k is the number of links per node and γ is the slope of the log P[k] versus log[k] plot); this unique property of most biologic networks is a reflection of their self-similarity or fractal nature (Fig. 87e-1).
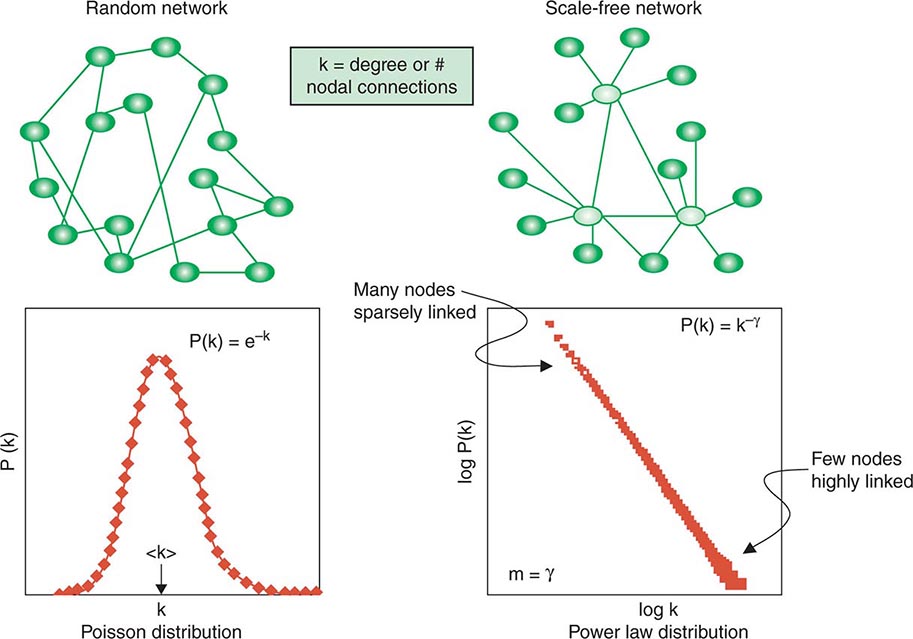
FIGURE 87e-1 Network representations and their distributions. A random network is depicted on the left, and its Poisson distribution of the number of nodal connections (k) is shown in the graph below it. A scale-free network is depicted on the right, and its power law distribution of the number of nodal connections (k) is shown in the graph below it. Highly connected nodes (hubs) are lightly shaded.
There are unique properties of scale-free biologic systems that reflect their evolution and promote their adaptability and survival. Biologic networks likely evolved one node at a time in a process in which new nodes are more likely to link to a highly connected node than to a sparsely connected node. Furthermore, scale-free networks can become sparsely linked to one another, yielding more complex, modular scale-free topologies. This evolutionary growth of biologic networks has three important properties that affect system function and survival. First, this scale-free addition of new nodes promotes system redundancy, which minimizes the consequences of errors and accommodates adverse perturbations to the system robustly with minimal effects on critical functions (unless the highly connected nodes are the focus of the perturbation). Second, this resulting network redundancy provides a survival advantage to the system. In complex gene networks, for example, mutations or polymorphisms in weakly linked genes account for biodiversity and biologic variability without disrupting the critical functions of the system; only mutations in highly linked (essential) genes (hubs) can shut down the system and cause embryonic lethality. Third, scale-free biologic systems facilitate the flow of information (e.g., metabolite flux) across the system compared with randomly organized biologic systems; this so-called “small-world” property of the system (in which the clustered nature of the highly linked hubs defines a local neighborhood within the network that communicates through weaker, less frequent links to other clusters) minimizes the energy cost for the dynamic action of the system (e.g., minimizes the transition time between states in a metabolic network).
These basic organizing principles of complex biologic systems lead to three unique properties that require emphasis. First, biologic systems are robust, which means that they are quite stable in response to most changes in external conditions or internal modification. Second, a corollary to the property of robustness is that complex biologic systems are sloppy, which means that they are insensitive to changes in external conditions or internal modification except under certain uncommon conditions (i.e., when a hub is involved in the change). Third, complex biologic systems exhibit emergent properties, which means that they manifest behaviors that cannot be predicted from the reductionist principles used to characterize their component parts. Examples of emergent behavior in biologic systems include spontaneous, self-sustained oscillations in glycolysis; spiral and scroll waves of depolarization in cardiac tissue that cause reentrant arrhythmias; and self-organizing patterns in biochemical systems governed by diffusion and chemical reaction.
APPLICATIONS OF SYSTEMS BIOLOGY TO PATHOBIOLOGY
The principles of systems biology have been applied to complex pathologic processes with some early successes. The key to these applications is the identification of emergent properties of the system under study in order to define novel, otherwise unpredictable (i.e., from the reductionist perspective) methods for regulating the system’s response. Systems biology approaches have been used to characterize epidemics and ways to control them, taking advantage of the scale-free properties of the network of infected individuals that constitute the epidemic. Through the use of a systems analysis of a neural protein-protein interaction network, unique disease-modifying proteins have been identified that are common to a wide range of cerebellar neurodegenerative disorders causing inherited ataxias. Systems analysis and disease network construction of a pulmonary arterial hypertension network led to the identification of a unique disease module involving a pathway governed by microRNA21. Systems biology models have been used to dissect the dynamics of the inflammatory response using oscillatory changes in the transcription factor nuclear factor (NF) κB as the system output. Systems biology principles also have been used to predict the development of an idiotypy–anti-idiotypy antibody network, describe the dynamics of species growth in microbial biofilms, and analyze the innate immune response. In each of these examples, a systems (patho)biology approach provided insights into the behavior of these complex systems that could not have been recognized with conventional scientific reductionism.
A unique application of systems biology to biomedicine is in the area of drug development. Conventional drug development involves identifying a potential target protein and then designing or screening compounds to identify those that inhibit the function of that target. This reductionist analysis has identified many potential drug targets and drugs, yet only when a drug is tested in animal models or humans are the systems consequences of the drug’s action revealed; not uncommonly, so-called off-target effects may become apparent and be sufficiently adverse for researchers to cease development of the agent. A good example of this problem is the unexpected outcomes of the vitamin B–based regimens for lowering homocysteine levels. In these trials, plasma homocysteine levels were reduced effectively; however, there was no effect of this reduction on clinical vascular endpoints. One explanation for this outcome is that one of the B vitamins in the regimen, folate, has a panoply of effects on cell proliferation and metabolism that probably offset its homocysteine-lowering benefits, promoting progressive atherosclerotic plaque growth and its consequences for clinical events. In addition to these types of unexpected outcomes exerted through pathways that were not considered ab initio, conventional approaches to drug development typically do not take into consideration the possibility of emergent behaviors of the organism or the metabolic pathway or the transcriptional network of interest. Thus, a systems-based analysis of potential drugs (drug-target network analysis) can benefit the development paradigm both by enhancing the likelihood that a compound of interest will not manifest unforeseen adverse effects and by promoting novel analytic methods for identifying unique control points or pathways in metabolic or genetic networks that would benefit from drug-based modulation.
SYSTEMS PATHOBIOLOGY AND HUMAN DISEASE CLASSIFICATION: NETWORK MEDICINE
Perhaps most important, systems pathobiology can be used to revise and refine the definition of human disease. The classification of human disease used in this and all medical textbooks derives from the correlation between pathologic analysis and clinical syndromes that began in the nineteenth century. Although this approach has been very successful, serving as the basis for the development of many effective therapies, it has major shortcomings. Those shortcomings include a lack of sensitivity in defining preclinical disease, a primary focus on overtly manifest disease, failure to recognize different and potentially differentiable causes of common late-stage pathophenotypes, and a limited ability to incorporate the growing body of molecular and genetic determinants of pathophenotype into the conventional classification scheme.
Two examples will illustrate the weakness of simple correlation analyses grounded in the reductionist principle of simplification (Occam’s razor) in defining human disease. Sickle cell anemia, the “classic” Mendelian disorder, is caused by a Val6Gln substitution in the β chain of hemoglobin. If conventional genetic teaching holds, this single mutation should lead to a single phenotype in patients who harbor it (genotype-phenotype correlation). This assumption is, however, false, as patients with sickle cell disease manifest a variety of pathophenotypes, including hemolytic anemia, stroke, acute chest syndrome, bony infarction, and painful crisis, as well as an overtly normal phenotype. The reasons for these different phenotypic presentations include the presence of disease-modifying genes or gene products (e.g., hemoglobin F, hemoglobin C, glucose-6-phosphate dehydrogenase), exposure to adverse environmental factors (e.g., hypoxia, dehydration), and the genetic and environmental determinants of common intermediate pathophenotypes (i.e., variations in those generic pathologic mechanisms underlying all human disease—inflammation, thrombosis/hemorrhage, fibrosis, cell proliferation, apoptosis/necrosis, immune response).
A second example of note is familial pulmonary arterial hypertension. This disorder is associated with over 100 different mutations in three members of the transforming growth factor β (TGF-β) superfamily: bone morphogenetic protein receptor-2 (BMPR-2), activin receptor-like kinase-1 (Alk-1), and endoglin. All these different genotypes are associated with a common pathophenotype, and each leads to that pathophenotype by molecular mechanisms that range from haploinsufficiency to dominant negative effects. As only approximately one-fourth of individuals in families that harbor these mutations manifest the pathophenotype, other disease-modifying genes (e.g., the serotonin receptor 5-HT2B, the serotonin transporter 5-HTT), genomic and environmental determinants of common intermediate pathophenotypes, and environmental exposures (e.g., hypoxia, infective agents [HIV], anorexigens) probably account for the incomplete penetrance of the disorder.
On the basis of these and many other related examples, one can approach human disease from a systems pathobiology perspective in which each “disease” can be depicted as a network that includes the following modules: the primary disease-determining elements of the genome (or proteome, if posttranslationally modified), the disease-modifying elements of the genome or proteome, environmental determinants, and genomic and environmental determinants of the generic intermediate pathophenotypes. Figure 87e-2 graphically depicts these genotype-phenotype relationships as modules for the six common disease types with specific examples for each type. Figure 87e-3 shows a network-based depiction of sickle cell disease using this kind of modular approach.
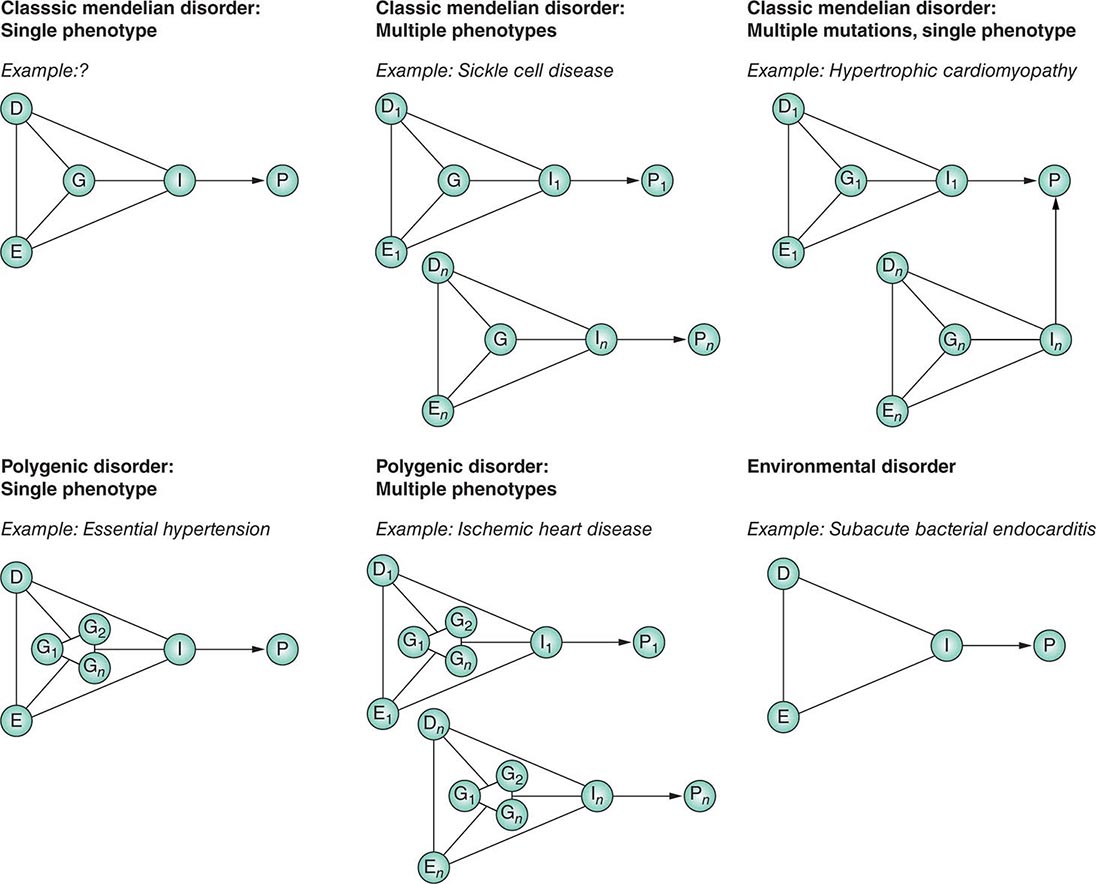
FIGURE 87e-2 Examples of modular representations of human disease. D, secondary human disease genome or proteome; E, environmental determinants; G, primary human disease genome or proteome; I, intermediate phenotype; P, pathophenotype. (Reproduced with permission from J Loscalzo et al: Molec Syst Biol 3:124, 2007.)
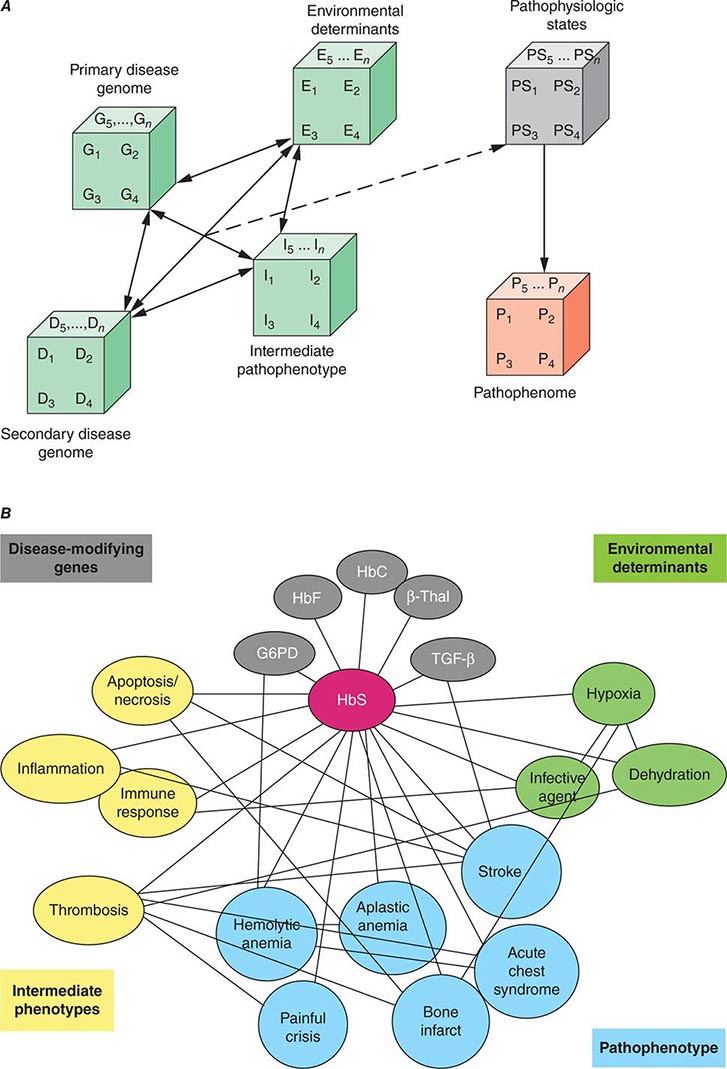
FIGURE 87e-3 A. Theoretical human disease network illustrating the relationships among genetic and environmental determinants of the pathophenotypes. Key: D, secondary disease genome or proteome; E, environmental determinants; G, primary disease genome or proteome; I, intermediate phenotype; PS, pathophysiologic states leading to P, pathophenotype. B. Example of this theoretical construct applied to sickle cell disease. Key: Red, primary molecular abnormality; gray, disease-modifying genes; yellow, intermediate phenotypes; green, environmental determinants; blue, pathophenotypes. (Reproduced with permission from J Loscalzo et al: Molec Syst Biol 3:124, 2007.)
Goh and colleagues developed the concept of a human disease network (Fig. 87e-4) in which they used a systems approach to characterize the disease-gene associations listed in the Online Mendelian Inheritance in Man database. Their analysis showed that genes linked to similar disorders are more likely to have products that physically associate and greater similarity between their transcription profiles than do genes not associated with similar disorders. In addition, proteins associated with the same pathophenotype are significantly more likely to interact with one another than with other proteins not associated with the pathophenotype. Finally, these authors showed that the great majority of disease-associated genes are not highly connected genes (i.e., not hubs) and are typically weakly linked nodes within the functional periphery of the network in which they operate.
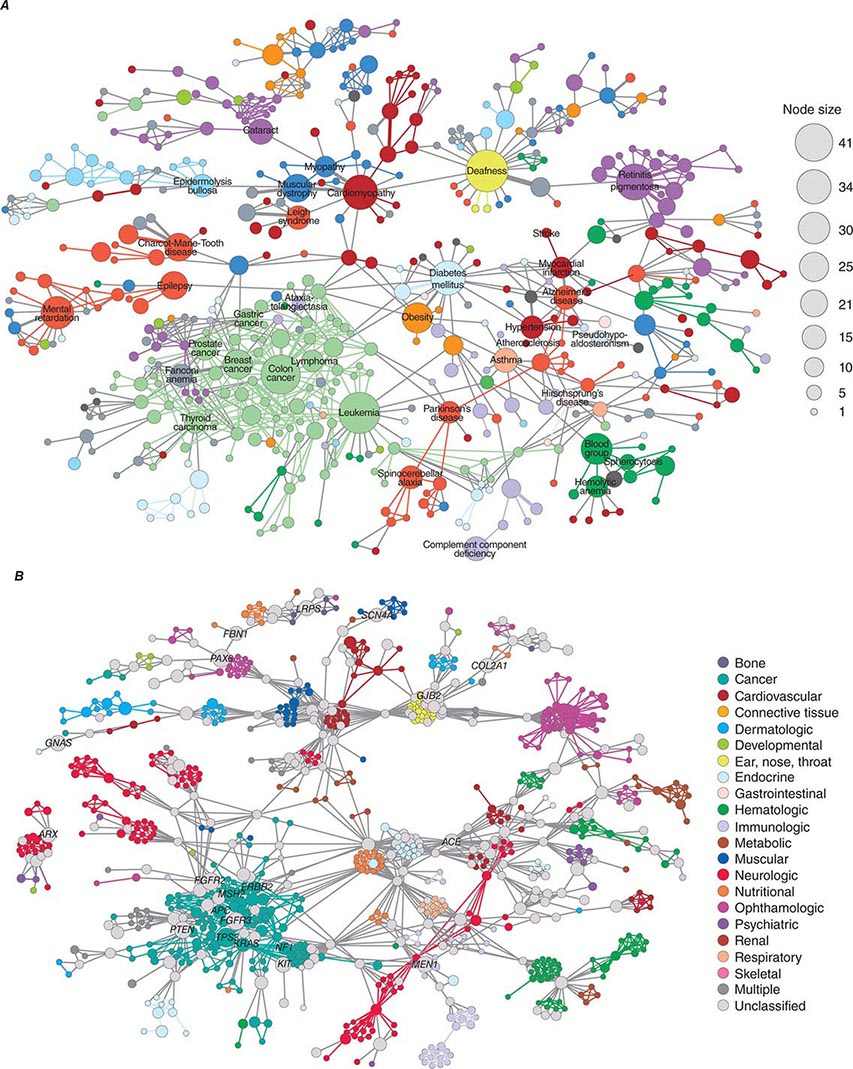
FIGURE 87e-4 A. Human disease network. Each node corresponds to a specific disorder colored by class (22 classes, shown in the key to B). The size of each node is proportional to the number of genes contributing to the disorder. Edges between disorders in the same disorder class are colored with the same (lighter) color, and edges connecting different disorder classes are colored gray, with the thickness of the edge proportional to the number of genes shared by the disorders connected by it. B. Disease gene network. Each node is a single gene, and any two genes are connected if implicated in the same disorder. In this network map, the size of each node is proportional to the number of specific disorders in which the gene is implicated. (Reproduced with permission from KI Goh et al: Proc Natl Acad Sci USA 104:8685, 2007.)
This type of analysis validates the potential importance of defining disease on the basis of its systems pathobiologic determinants. Clearly, doing this will require a more careful dissection of the molecular elements in the relevant pathways (i.e., more precise molecular pathophenotyping), less reliance on overt manifestations of disease for their classification, and an understanding of the dynamics (not just the static architecture) of the pathobiologic networks that underlie pathophenotypes defined in this way. Figure 87e-5 illustrates the elements of a molecular network within which a disease module is contained. This network is first identified by determining the interactions (physical or regulatory) among the proteins or genes that comprise it (the “interactome”). These interactions then define a topologic module within which exists functional modules (pathways) and disease modules. One approach to constructing this module is illustrated in Fig. 87e-6. Examples of the use of this approach in defining novel determinants of disease are given in Table 87e-1.
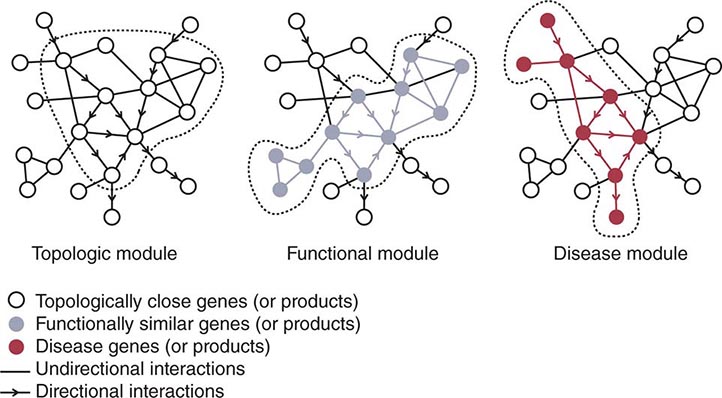
FIGURE 87e-5. The elements of the interactome. The interactome includes topologic modules (genes or gene products that are closely associated with one another through direct interactions), functional modules (genes or gene products that work together to define a pathway), and disease modules (genes or gene products that interact to yield a pathophenotype). (Reproduced with permission from AL Barabasi et al: Nat Rev Genet 12:56, 2011.)
FIGURE 87e-6. Approaches to identifying disease modules in molecular networks. A strategy for defining disease modules involves (i) reconstructing the interactome; (ii) ascertaining potential seed (disease) genes from the curated literature, the Online Mendelian Inheritance in Man (OMIM) database, or genomic analyses (genome-wide association studies [GWAS] or transcriptional profiling); (iii) identifying the disease module using different modeling or statistical approaches; (iv) identifying pathways and the role of disease genes or modules in those pathways; and (v) disease module validation and prediction. (Reproduced with permission from AL Barabasi et al: Nat Rev Genet 12:56, 2011.)
|
EXAMPLES OF SYSTEMS BIOLOGY APPLICATION TO DISEASE |
As yet another potential consideration, one can argue that disease reflects the later-stage consequences of the predilection of an organ system to manifest a particular intermediate pathophenotype in response to injury. This paradigm reflects a reverse causality view in which a disease is defined as a tendency to heightened inflammation, thrombosis, or fibrosis after an injurious perturbation. Where the process is manifest (i.e., the organ in which it occurs) is less important than that it occurs (with the exception of the organ-specific pathophysiologic consequences that may require acute attention). For example, from this perspective, acute myocardial infarction (AMI) and its consequences are a reflection of thrombosis (in the coronary artery), inflammation (in the acutely injured myocardium), and fibrosis (at the site or sites of cardiomyocyte death). In effect, the major therapies for AMI address these intermediate pathophenotypes (e.g., antithrombotics, statins) rather than any organ-specific disease-determining process. This paradigm would argue for a systems-based analysis that would first identify the intermediate pathophenotypes to which a person is predisposed, then determine how and when to intervene to attenuate that adverse predisposition, and finally limit the likelihood that a major organ-specific event will occur. Evidence for the validity of this approach is found in the work of Rzhetsky and colleagues, who reviewed 1.5 million patient records and 161 diseases and found that these disease phenotypes form a network of strong pairwise correlations. This result is consistent with the notion that underlying genetic predispositions to intermediate pathophenotypes form the predicate basis for conventionally defined end organ diseases.
Regardless of the specific nature of the systems pathobiologic approach used, these analyses will lead to a drastic revision of the way human disease is defined and treated, establishing the discipline of network medicine. This will be a lengthy and complicated process but ultimately will lead to better disease prevention and therapy and probably do so from an increasingly personalized perspective. The analysis of pathobiology from a systems-based perspective is likely to help define specific subsets of patients more likely to respond to particular interventions based on shared disease mechanisms. Although it is unlikely that the extreme of “individualized medicine” will ever be practical (or even desirable), complex diseases can be mechanistically subclassified and interventions may be tailored to those settings in which they are more likely to work.
[/level-membership-for-internal-medicine-category][not-level-membership-for-internal-medicine-category]
84 |
The Practice of Genetics in Clinical Medicine |
APPLICATIONS OF MOLECULAR GENETICS IN CLINICAL MEDICINE
Genetic testing for inherited abnormalities associated with disease risk is increasingly used in the practice of clinical medicine. Germline alterations include chromosomal abnormalities (Chap. 83e), specific gene mutations with autosomal dominant or recessive patterns of transmission (Chap. 82), and single nucleotide polymorphisms with small relative risks associated with disease. Germline alterations are responsible for disorders beyond classic Mendelian conditions with genetic susceptibility to common adult-onset diseases such as asthma, hypertension, diabetes mellitus, macular degeneration, and many forms of cancer. For many of these diseases, there is a complex interplay of genes (often multiple) and environmental factors that affect lifetime risk, age of onset, disease severity, and treatment options.
The expansion of knowledge related to genetics is changing our understanding of pathophysiology and influencing our classification of diseases. Awareness of genetic etiology can have an impact on clinical management, including prevention and screening for or treatment of a range of diseases. Primary care physicians are relied upon to help patients navigate testing and treatment options. Consequently, they must understand the genetic basis for a large number of genetically influenced diseases, incorporate personal and family history to determine the risk for a specific mutation, and be positioned to provide counseling. Even if patients are seen by genetic specialists who assess genetic risk and coordinate testing, primary care providers should provide information to their patients regarding the indications, limitations, risks, and benefits of genetic counseling and testing. They must also be prepared to offer risk-based management following genetic risk assessment. Given the pace of genetics, this is an increasingly difficult task. The field of clinical genetics is rapidly moving from single gene testing to multigene panel testing, with techniques such as whole-exome and -genome sequencing on the horizon, increasing the complexity of test selection and interpretation, as well as patient education and medical decision making.
COMMON ADULT-ONSET GENETIC DISORDERS
INHERITANCE PATTERNS
Adult-onset hereditary diseases follow multiple patterns of inheritance. Some are autosomal dominant conditions. These include many common cancer susceptibility syndromes such as hereditary breast and ovarian cancer (due to germline BRCA1 and BRCA2 mutations) and Lynch syndrome (caused by germline mutations in the mismatch repair genes MLH1, MSH2, MSH6, and PMS2). In both of these examples, inherited mutations are associated with a high penetrance (lifetime risk) of cancer, although risk is not 100%. In other conditions, although there is autosomal dominant transmission, there is lower penetrance, thereby making the disorders more difficult to recognize. For example, germline mutations in CHEK2 increase the risk of breast cancer, but with a moderate lifetime risk in the range of 20–40%, as opposed to 50–70% for mutations in BRCA1 or BRCA2. Other adult-onset hereditary diseases are transmitted in an autosomal recessive fashion where two mutant alleles are necessary to cause disease. Examples include hemochromatosis and MYH-associated colon cancer. There are more pediatric-onset autosomal recessive disorders, such as lysosomal storage diseases and cystic fibrosis.
The genetic risk for many adult-onset disorders is multifactorial. Risk can be conferred by genetic factors at a number of loci, which individually have very small effects (usually with relative risks of <1.5). These risk loci (generally single nucleotide polymorphisms [SNPs]) combine with other genes and environmental factors in ways that are not well understood. SNP panels are available to assess risk of disease, but the optimal way of using this information in the clinical setting remains uncertain.
Many diseases have multiple patterns of inheritance, adding to the complexity of evaluating patients and families for these conditions. For example, colon cancer can be associated with a single germline mutation in a mismatch repair gene (Lynch syndrome, autosomal dominant), biallelic mutations in MYH (autosomal recessive), or multiple SNPs (polygenic). Many more individuals will have SNP risk alleles than germline mutations in high-penetrance genes, but cumulative lifetime risk of colon cancer related to the former is modest, whereas the risk related to the latter is significant. Personal and family histories provide important insights into the possible mode of inheritance.
FAMILY HISTORY
When two or more first-degree relatives are affected with asthma, cardiovascular disease, type 2 diabetes, breast cancer, colon cancer, or melanoma, the relative risk for disease among close relatives ranges from two- to fivefold, underscoring the importance of family history for these prevalent disorders. In most situations, the key to assessing the inherited risk for common adult-onset diseases is the collection and interpretation of a detailed personal and family medical history in conjunction with a directed physical examination.
Family history should be recorded in the form of a pedigree. Pedigrees should convey health-related data on first- and second-degree relatives. When such pedigrees suggest inherited disease, they should be expanded to include additional family members. The determination of risk for an asymptomatic individual will vary depending on the size of the pedigree, the number of unaffected relatives, the types of diagnoses, and the ages of disease onset. For example, a woman with two first-degree relatives with breast cancer is at greater risk for a specific Mendelian disorder if she has a total of 3 female first-degree relatives (with only 1 unaffected) than if she has a total of 10 female first-degree relatives (with 7 unaffected). Factors such as adoption and limited family structure (few women in a family) should to be taken into consideration in the interpretation of a pedigree. Additional considerations include young age of disease onset (e.g., a 30-year nonsmoking woman with a myocardial infarction), unusual diseases (e.g., male breast cancer or medullary thyroid cancer), and the finding of multiple potentially related diseases in an individual (e.g., a woman with a history of both colon and endometrial cancer). Some adult-onset diseases are more prevalent in certain ethnic groups. For instance, 2.5% of individuals of Ashkenazi Jewish ancestry carry one of three founder mutation in BRCA1 and BRCA2. Factor V Leiden mutations are much more common in Caucasians than in Africans or Asians.
Additional variables that should be documented are nonhereditary risk factors among those with disease (such as cigarette smoking and myocardial infarction; asbestos exposure and lung disease; and mantle radiation and breast cancer). Significant associated environmental exposures or lifestyle factors decrease the likelihood of a specific genetic disorder. In contrast, the absence of nonhereditary risk factors typically associated with a disease raises concern about a genetic association. A personal or family history of deep vein thrombosis in the absence of known environmental or medical risk factors suggests a hereditary thrombotic disorder. The physical examination may also provide important clues about the risk for a specific inherited disorder. A patient presenting with xanthomas at a young age should prompt consideration of familial hypercholesterolemia. The presence of trichilemmomas in a woman with breast cancer raises concern for Cowden syndrome, associated with PTEN mutations.
Recall of family history is often inaccurate. This is especially so when the history is remote and families lose contact or separate geographically. It can be helpful to ask patients to fill out family history forms before or after their visits, because this provides them with an opportunity to contact relatives. Ideally, this information should be embedded in electronic health records and updated intermittently. Attempts should be made to confirm the illnesses reported in the family history before making important and, in certain circumstances, irreversible management decisions. This process is often labor intensive and ideally involves interviews of additional family members or reviewing medical records, autopsy reports, and death certificates.
Although many inherited disorders will be suggested by the clustering of relatives with the same or related conditions, it is important to note that disease penetrance is incomplete for most genetic disorders. As a result, the pedigree obtained in such families may not exhibit a clear Mendelian inheritance pattern, because not all family members carrying the disease-associated alleles will manifest clinical evidence of the condition. Furthermore, genes associated with some of these disorders often exhibit variable disease expression. For example, the breast cancer–associated gene BRCA2 can predispose to several different malignancies in the same family, including cancers of the breast, ovary, pancreas, skin, and prostate. For common diseases such as breast cancer, some family members without the susceptibility allele (or genotype) may develop breast cancer (or phenotype) sporadically. Such phenocopies represent another confounding variable in the pedigree analysis.
Some of the aforementioned features of the family history are illustrated in Fig. 84-1. In this example, the proband, a 36-year-old woman (IV-1), has a strong history of breast and ovarian cancer on the paternal side of her family. The early age of onset and the co-occurrence of breast and ovarian cancer in this family suggest the possibility of an inherited mutation in BRCA1 or BRCA2. It is unclear however, without genetic testing, whether her father harbors such a mutation and transmitted it to her. After appropriate genetic counseling of the proband and her family, the most informative and cost-effective approach to DNA analysis in this family is to test the cancer-affected 42-year-old living cousin for the presence of a BRCA1 or BRCA2 mutation. If a mutation is found, then it is possible to test for this particular alteration in other family members, if they so desire. In the example shown, if the proband’s father has a BRCA1 mutation, there is a 50:50 probability that the mutation was transmitted to her, and genetic testing can be used to establish the absence or presence of this alteration. In this same example, if a mutation is not detected in the cancer-affected cousin, testing would not be indicated for cancer-unaffected relatives.
FIGURE 84-1 A 36-year-old woman (arrow) seeks consultation because of her family history of cancer. The patient expresses concern that the multiple cancers in her relatives imply an inherited predisposition to develop cancer. The family history is recorded, and records of the patient’s relatives confirm the reported diagnoses.
GENETIC TESTING FOR ADULT-ONSET DISORDERS
A critical first step before initiating genetic testing is to ensure that the correct clinical diagnosis has been made, whether it is based on family history, characteristic physical findings, pathology, or biochemical testing. Such careful clinical assessment can define the phenotype. In the traditional model of genetic testing, testing is directed initially toward the most probable genes (determined by the phenotype), which prevents unnecessary testing. Many disorders exhibit the feature of locus heterogeneity, which refers to the fact that mutations in different genes can cause phenotypically similar disorders. For example, osteogenesis imperfecta (Chap. 427), long QT syndrome (Chap. 277), muscular dystrophy (Chap. 462e), and hereditary predisposition to breast (Chap. 108) or colon (Chap. 110) cancer can each be caused by mutations in a number of distinct genes. The patterns of disease transmission, disease risk, clinical course, and treatment may differ significantly depending on the specific gene affected. Historically, the choice of which gene to test has been determined by unique clinical and family history features and the relative prevalence of candidate genetic disorders. However, rapid changes in genetic testing techniques, as discussed below, may impact this paradigm. It is now technically and financially feasible to sequence many genes (or even the whole exome) at one time. The incorporation of multiplex testing for germline mutations is rapidly evolving.
METHODOLOGIC APPROACHES TO GENETIC TESTING
Genetic testing is regulated and performed in much the same way as other specialized laboratory tests. In the United States, genetic testing laboratories are Clinical Laboratory Improvement Amendments (CLIA) approved to ensure that they meet quality and proficiency standards. A useful information source for various genetic tests is www.genetests.org. It should be noted that many tests need to be ordered through specialized laboratories.
Genetic testing is performed largely by DNA sequence analysis for mutations, although genotype can also be deduced through the study of RNA or protein (e.g., apolipoprotein E, hemoglobin S, and immunohistochemistry). For example, universal screening for Lynch syndrome via immunohistochemical analysis of colorectal cancers for absence of expression of mismatch repair proteins is under way at multiple hospitals throughout the United States. The determination of DNA sequence alterations relies heavily on the use of polymerase chain reaction (PCR), which allows rapid amplification and analysis of the gene of interest. In addition, PCR enables genetic testing on minimal amounts of DNA extracted from a wide range of tissue sources including leukocytes, mucosal epithelial cells (obtained via saliva or buccal swabs), and archival tissues. Amplified DNA can be analyzed directly by DNA sequencing, or it can be hybridized to DNA chips or blots to detect the presence of normal and altered DNA sequences. Direct DNA sequencing is frequently used for determination of hereditary disease susceptibility and prenatal diagnosis. Analyses of large alterations of the genome are possible using cytogenetics, fluorescent in situ hybridization (FISH), Southern blotting, or multiplex ligation-dependent probe amplification (MLPA) (Chap. 83e).
Massively parallel sequencing (also called next-generation sequencing) is significantly altering the approach to genetic testing for adult-onset hereditary susceptibility disorder. This technology encompasses several high-throughput approaches to DNA sequencing, all of which can reliably sequence many genes at one time. Technically, this involves the use of amplified DNA templates in a flow cell, a very different process than traditional Sanger sequencing which is time-consuming and expensive.
Multiplex panels for inherited susceptibility are commercially available and include testing of a number of genes that have been associated with the condition of interest. For example, panels are available for Brugada syndrome, hypertrophic cardiomyopathy, and Charcot-Marie-Tooth neuropathy. For many syndromes, this type of panel testing may make sense. However, in other situations, the utility of panel testing is less certain. Currently available breast cancer susceptibility panels contain six genes or more. Many of the genes included in the larger panels are associated with only a modest risk of breast cancer, and the clinical application is uncertain. An additional problem of sequencing many genes (rather than the genes for which there is most suspicion) is the identification of one or more variants of uncertain significance (VUS), discussed below.
Whole-exome sequencing (WES) is also now commercially available, although largely used in individuals with syndromes unexplained by traditional genetic testing. As cost declines, WES may be more widely used. Whole-genome sequencing is also commercially available. Although it may be quite feasible to sequence the entire genome, there are many issues in doing so, including the daunting task of analyzing the vast amount of data generated. Other issues include: (1) the optimal way in which to obtain informed consent, (2) interpretation of frequent sequence variation of uncertain significance, (3) interpretation of alterations in genes with unclear relevance to specific human pathology, and (4) management of unexpected but clinically significant genetic findings.
Testing strategies are evolving as a result of these new genetic testing platforms. As the cost of multiple gene panels and WES continue to fall, and as interpretation of such test results improve, there may be a shift from sequential single-gene (or a few genes) testing to multigene testing. For example, presently, a 30-year-old woman with breast cancer but no family history of cancer and no syndromic features would undergo BRCA1/2 testing. If negative, she would subsequently be offered TP53 testing. Notably, a reasonable number of individuals offered TP53 testing for Li-Fraumeni syndrome decline because mutations are associated with extremely high cancer risks (including childhood cancers) in multiple organs and there are no proven interventions to mitigate risk. Without features consistent with Cowden syndrome, the woman would not be routinely offered PTEN testing or testing for CHEK2, ATM, BRIP, BARD, NBN, and PALB2. However, it is now possible to synchronously analyze all of the aforementioned genes, for a nominally higher cost than BRCA1/2 testing alone. Concerns about such panels include appropriate consent strategies related to unexpected findings, VUS, and unclear clinical utility of testing moderate-penetrance genes. Thus, changes from the traditional model of single-gene genetic testing should be done with caution (Fig. 84-2).
FIGURE 84-2 Approach to genetic testing.
Limitations to the accuracy and interpretation of genetic testing exist. In addition to technical errors, genetics tests are sometimes designed to detect only the most common mutations. In addition, genetic testing has evolved over time. For example, it was not possible to obtain commercially available comprehensive large genomic rearrangement testing for BRCA1 and BRCA2 until 2006. Therefore, a negative result must be qualified by the possibility that the individual may have a mutation that was not included in the test. In addition, a negative result does not mean that there is not a mutation in some other gene that causes a similar inherited disorder. A negative result, unless there is known mutation in the family, is typically classified as uninformative.
VUS are another limitation to genetic testing. A VUS (also termed unclassified variant) is a sequence variation in a gene where the effect of the alteration on the function of the protein is not known. Many of these variants are single nucleotide substitutions (also called missense mutations) that result in a single amino acid change. Although many VUSs will ultimately be reclassified as benign polymorphisms, some will prove to be functionally important. As more genes are sequenced (for example, in a multiplex panel or through WES), the percentage of individuals found to have a VUS increases significantly. The finding of a VUS is difficult for patients and providers alike and complicates decisions regarding medical management.
Clinical utility is an important consideration because genetic testing for susceptibility to chronic diseases is increasingly integrated into the practice of medicine. In some situations, there is clear clinical utility to genetic testing with significant evidence-based changes in medical management decisions based on results. However, in many cases, the discovery of disease-associated genes has outpaced studies that assess how such information should be used in the clinical management of the patient and family. This is particularly true for moderate- and low-penetrance gene mutations. Therefore, predictive genetic testing should be approached with caution and only offered to patients who have been adequately counseled and have provided informed consent.
Predictive genetic testing falls into two distinct categories. Presymptomatic testing applies to diseases where a specific genetic alteration is associated with a near 100% likelihood of developing disease. In contrast, predisposition testing predicts a risk for disease that is less than 100%. For example, presymptomatic testing is available for those at risk for Huntington’s disease; whereas, predisposition testing is considered for those at risk for hereditary colon cancer. It is important to note that for the majority of adult-onset disorders, testing is only predictive. Test results cannot reveal with confidence whether, when, or how the disease will manifest itself. For example, not everyone with the apolipoprotein E4 allele will develop Alzheimer’s disease, and individuals without this genetic marker can still develop the disorder.
The optimal testing strategy for a family is to initiate testing in an affected family member first. Identification of a mutation can direct the testing of other at-risk family members (whether symptomatic or not). In the absence of additional familial or environmental risk factors, individuals who test negative for the mutation found in the affected family member can be informed that they are at general population risk for that particular disease. Furthermore, they can be reassured that they are not at risk for passing the mutation on to their children. On the other hand, asymptomatic family members who test positive for the known mutation must be informed that they are at increased risk for disease development and for transmitting the alteration to their children.
Pretest counseling and education are important, as is an assessment of the patient’s ability to understand and cope with test results. Genetic testing has implications for entire families, and thus individuals interested in pursuing genetic testing must consider how test results might impact their relationships with relatives, partners, spouses, and children. In families with a known genetic mutation, those who test positive must consider the impact of their carrier status on their present and future lifestyles; those who test negative may manifest survivor guilt. Parents who are found to have a disease-associated mutation often express considerable anxiety and despair as they address the issue of risk to their children. In addition, some individuals consider options such as preimplantation genetic diagnosis in their reproductive decision making.
When a condition does not manifest until adulthood, clinicians and parents are faced with the question of whether at-risk children should be offered genetic testing and, if so, at what age. Although the matter is debated, several professional organizations have cautioned that genetic testing for adult-onset disorders should not be offered to children. Many of these conditions have no known interventions in childhood to prevent disease; consequently, such information can pose significant psychosocial risk to the child. In addition, there is concern that testing during childhood violates a child’s right to make an informed decision regarding testing upon reaching adulthood. On the other hand, testing should be offered in childhood for disorders that may manifest early in life, especially when management options are available. For example, children with multiple endocrine neoplasia 2 (MEN 2) may develop medullary thyroid cancer early in life and should be considered for prophylactic thyroidectomy (Chap. 408). Similarly, children with familial adenomatous polyposis (FAP) due to a mutation in APC may develop polyps in their teens with progression to invasive cancer in the twenties, and therefore, colonoscopy screening is started between the ages of 10 and 15 years (Chap. 110).
INFORMED CONSENT
Informed consent for genetic testing begins with education and counseling. The patient should understand the risks, benefits, and limitations of genetic testing, as well as the potential implications of test results. Informed consent should include a written document, drafted clearly and concisely in a language and format that is understandable to the patient. Because molecular genetic testing of an asymptomatic individual often allows prediction of future risk, the patient should understand all potential long-term medical, psychological, and social implications of testing. There have long been concerns about the potential for genetic discrimination. The Genetic Information Nondiscrimination Act (GINA) was passed in 2008 and provides some protections related to job and health insurance discrimination. It is important to explore with patients the potential impact of genetic test results on future health as well as disability and life insurance coverage. Patients should understand that alternatives remain available if they decide not to pursue genetic testing, including the option of delaying testing to a later date. The option of DNA banking should be presented so that samples are readily available for future use by family members, if needed.
FOLLOW-UP CARE AFTER TESTING
Depending on the nature of the genetic disorder, posttest interventions may include: (1) cautious surveillance and awareness; (2) specific medical interventions such as enhanced screening, chemoprevention, or risk-reducing surgery; (3) risk avoidance; and (4) referral to support services. For example, patients with known deleterious mutations in BRCA1 or BRCA2 are strongly encouraged to undergo risk-reducing salpingo-oophorectomy and are offered intensive breast cancer screening as well as the option of risk-reducing mastectomy. In addition, such women may wish to take chemoprevention with tamoxifen, raloxifene, or exemestane. Those with more limited medical management and prevention options, such as patients with Huntington’s disease, should be offered continued follow-up and supportive services, including physical and occupational therapy and social services or support groups as indicated. Specific interventions will change as research continues to enhance our understanding of the medical management of these genetic conditions and more is learned about the functions of the gene products involved.
Individuals who test negative for a mutation in a disease-associated gene identified in an affected family member must be reminded that they may still be at risk for the disease. This is of particular importance for common diseases such as diabetes mellitus, cancer, and coronary artery disease. For example, a woman who finds that she does not carry the disease-associated mutation in BRCA2 previously discovered in the family should be reminded that she still requires the same breast cancer screening recommended for the general population.
GENETIC COUNSELING AND EDUCATION
Genetic counseling should be distinguished from genetic testing and screening, although genetic counselors are often involved in issues related to testing. Genetic counseling refers to a communication process that deals with human problems associated with the occurrence of risk of a genetic disorder in a family. Genetic risk assessment is complex and often involves elements of uncertainty. Counseling, therefore, includes genetic education as well as psychosocial counseling. Genetic counseling can be useful in a wide range of situations (Table 84-1). The role of the genetic counselor includes the following:
1. Gather and document a detailed family history.
2. Educate patients about general genetic principles related to disease risk, both for themselves and for others in the family.
3. Assess and enhance the patient’s ability to cope with the genetic information offered.
4. Discuss how nongenetic factors may relate to the ultimate expression of disease.
5. Address medical management issues.
6. Assist in determining the role of genetic testing for the individual and the family.
7. Ensure the patient is aware of the indications, process, risks, benefits, and limitations of the various genetic testing options.
8. Assist the patient, family, and referring physician in the interpretation of the test results.
9. Refer the patient and other at-risk family members for additional medical and support services, if necessary.
|
INDICATIONS FOR GENETIC COUNSELING |
Genetic counseling is generally offered in a nondirective manner, wherein patients learn to understand how their values factor into a particular medical decision. Nondirective counseling is particularly appropriate when there are no data demonstrating a clear benefit associated with a particular intervention or when an intervention is considered experimental. For example, nondirective genetic counseling is used when a person is deciding whether to undergo genetic testing for Huntington’s disease. At this time, there is no clear benefit (in terms of medical outcome) to an at-risk individual undergoing genetic testing for this disease because its course cannot be altered by therapeutic interventions. However, testing can have an important impact on the individual’s perception of advanced care planning and his or her interpersonal relationships and plans for childbearing. Therefore, the decision to pursue testing rests on the individual’s belief system and values. On the other hand, a more directive approach is appropriate when a condition can be treated. In a family with FAP, colon cancer screening and prophylactic colectomy should be recommended for known APC mutation carriers. The counselor and clinician following this family must ensure that the at-risk family members have access to the resources necessary to adhere to these recommendations.
Genetic education is central to an individual’s ability to make an informed decision regarding testing options and treatment. An adequate knowledge of patterns of inheritance will allow patients to understand the probability of disease risk for themselves and other family members. It is also important to impart the concepts of disease penetrance and expression. For most complex adult-onset genetic disorders, asymptomatic patients should be advised that a positive test result does not always translate into future disease development. In addition, the role of nongenetic factors, such as environmental exposures and lifestyle, must be discussed in the context of multifactorial disease risk and disease prevention. Finally, patients should understand the natural history of the disease as well as the potential options for intervention, including screening, prevention, and in certain circumstances, pharmacologic treatment or prophylactic surgery.
THERAPEUTIC INTERVENTIONS BASED ON GENETIC RISK FOR DISEASE
Specific treatments are available for a number of genetic disorders. Strategies for the development of therapeutic interventions have a long history in childhood metabolic diseases; however, these principles have been applied in the diagnosis and management of adult-onset diseases as well (Table 84-2). Hereditary hemochromatosis is usually caused by mutations in HFE (although other genes have been less commonly associated) and manifests as a syndrome of iron overload, which can lead to liver disease, skin pigmentation, diabetes mellitus, arthropathy, impotence in males, and cardiac issues (Chap. 428). When identified early, the disorder can be managed effectively with therapeutic phlebotomy. Therefore, when the diagnosis of hemochromatosis has been made in a proband, it is important to counsel and offer testing to other family members in order to minimize the impact of the disorder.
|
EXAMPLE OF GENETIC TESTING AND POSSIBLE INTERVENTIONS |
Preventative measures and therapeutic interventions are not restricted to metabolic disorders. Identification of familial forms of long QT syndrome, associated with ventricular arrhythmias, allows early electrocardiographic testing and the use of prophylactic antiarrhythmic therapy, overdrive pacemakers, or defibrillators. Individuals with familial hypertrophic cardiomyopathy can be screened by ultrasound, treated with beta blockers or other drugs, and counseled about the importance of avoiding strenuous exercise and dehydration. Those with Marfan’s syndrome can be treated with beta blockers or angiotensin II receptor blockers and monitored for the development of aortic aneurysms.
The field of pharmacogenetics identifies genes that alter drug metabolism or confer susceptibility to toxic drug reactions. Pharmacogenetics seeks to individualize drug therapy in an attempt to improve treatment outcomes and reduce toxicity. Examples include thiopurine methyltransferase (TPMT) deficiency, dihydropyrimidine dehydrogenase deficiency, malignant hyperthermia, and glucose-6-phosphate deficiency. Despite successes in this area, it is not always clear how to incorporate pharmacogenetics into clinical care. For example, although there is an association with CYP2C6 and VKORC1 genotypes and warfarin dosing, there is no evidence that incorporating genotyping into clinical practice improves patient outcomes.
The identification of germline abnormalities that increase the risk of specific types of cancer is rapidly changing clinical management. Identifying family members with mutations that predispose to FAP or Lynch syndrome leads to recommendations of early cancer screening and prophylactic surgery, as well as consideration of chemoprevention and attention to healthy lifestyle habits. Similar principles apply to familial forms of melanoma as well as cancers of the breast, ovary, and thyroid. In addition to increased screening and prophylactic surgery, the identification of germline mutations associated with cancer may also lead to the development of targeted therapeutics, for example, the ongoing development of PARP inhibitors in those with BRCA-associated cancers.
Although the role of genetic testing in the clinical setting continues to evolve, such testing holds the promise of allowing early and more targeted interventions that can reduce morbidity and mortality. Rapid technologic advances are changing the ways in which genetic testing is performed. As genetic testing becomes less expensive and technically easier to perform, it is anticipated that there will be an expansion of its use. This will present challenges, but also opportunities. It is critical that physicians and other health care professionals keep current with advances in genetic medicine in order to facilitate appropriate referral for genetic counseling and judicious use of genetic testing, as well as to provide state-of-the-art, evidence-based care for affected or at-risk patients and their relatives.
85e |
Mitochondrial DNA and Heritable Traits and Diseases |
Mitochondria are cytoplasmic organelles whose major function is to generate ATP by the process of oxidative phosphorylation under aerobic conditions. This process is mediated by the respiratory electron transport chain (ETC) multiprotein enzyme complexes I–V and the two electron carriers, coenzyme Q (CoQ) and cytochrome c. Other cellular processes to which mitochondria make a major contribution include apoptosis (programmed cell death) and additional cell type–specific functions (Table 85e-1). The efficiency of the mitochondrial ETC in ATP production is a major determinant of overall body energy balance and thermogenesis. In addition, mitochondria are the predominant source of reactive oxygen species (ROS), whose rate of production also relates to the coupling of ATP production to oxygen consumption. Given the centrality of oxidative phosphorylation to the normal activities of almost all cells, it is not surprising that mitochondrial dysfunction can affect almost any organ system (Fig. 85e-1). Thus, physicians in many disciplines might encounter patients with mitochondrial diseases and should be aware of their existence and characteristics.
|
FUNCTIONS OF MITOCHONDRIA |
FIGURE 85e-1 Dual genetic control and multiple organ system manifestations of mitochondrial disease. (Reproduced with permission from DR Johns: Mitochondrial DNA and disease. N Engl J Med 333:638, 1995.)
The integrated activity of an estimated 1500 gene products is required for normal mitochondrial biogenesis, function, and integrity. Almost all of these are encoded by nuclear genes and thus follow the rules and patterns of nuclear genomic inheritance (Chap. 84). These nuclear-encoded proteins are synthesized in the cell cytoplasm and imported to their location of activity within the mitochondria through a complex biochemical process. In addition, the mitochondria contain their own small genome consisting of numerous copies (polyploidy) per mitochondrion of a circular, double-strand mitochondrial DNA (mtDNA) molecule comprising 16,569 nucleotides. This mtDNA sequence (also known as the “mitogenome”) might represent the remnants of endosymbiotic prokaryotes from which mitochondria are thought to have originated. The mtDNA sequence contains a total of 37 genes, of which 13 encode mitochondrial protein components of the ETC (Fig. 85e-2). The remaining 22 tRNA- and 2 rRNA-encoding genes are dedicated to the process of translating the 13 mtDNA-encoded proteins. This dual nuclear and mitochondrial genetic control of mitochondrial function results in unique and diagnostically challenging patterns of inheritance. The current chapter focuses on heritable traits and diseases related to the mtDNA component of the dual genetic control of mitochondrial function. The reader is referred to Chaps. 84 and 462e for consideration of mitochondrial disease originating from mutations in the nuclear genome. The latter include (1) disorders due to mutations in nuclear genes directly encoding structural components or assembly factors of the oxidative phosphorylation complexes, (2) disorders due to mutations in nuclear genes encoding proteins indirectly related to oxidative phosphorylation, and (3) mtDNA depletion syndromes (MDS) characterized by a reduction of mtDNA copy number in affected tissues without mutations or rearrangements in the mtDNA.
FIGURE 85e-2 Maternal inheritance of mitochondrial DNA (mtDNA) disorders and heritable traits. Affected women (filled circles) transmit the trait to their children. Affected men (filled squares) do not transmit the trait to any of their offspring.
MITOCHONDRIAL DNA STRUCTURE AND FUNCTION
As a result of its circular structure and extranuclear location, the replication and transcription mechanisms of mtDNA differ from the corresponding mechanisms in the nuclear genome, whose nucleosomal packaging and structure are more complex. Because each cell contains many copies of mtDNA, and because the number of mitochondria can vary during the lifetime of each cell, mtDNA copy number is not directly coordinated with the cell cycle. Thus, vast differences in mtDNA copy number are observed between different cell types and tissues and during the lifetime of a cell. Another important feature of the mtDNA replication process is a reduced stringency of proofreading and replication error correction, leading to a greater degree of sequence variation compared to the nuclear genome. Some of these sequence variants are silent polymorphisms that do not have the potential for a phenotypic or pathogenic effect, whereas others may be considered pathogenic mutations.
With respect to transcription, initiation can occur on both strands and proceeds through the production of an intronless polycistronic precursor RNA, which is then processed to produce the 13 individual mRNA and 24 individual tRNA and rRNA products. The 37 mtDNA genes comprise fully 93% of the 16,569 nucleotides of the mtDNA in what is known as the coding region. The control region consists of ~1.1 kilobases (kb) of noncoding DNA, which is thought to have an important role in replication and transcription initiation.
MATERNAL INHERITANCE AND LACK OF RECOMBINATION
In contrast to homologous pair recombination that takes place in the nucleus, mtDNA molecules do not undergo recombination, such that mutational events represent the only source of mtDNA genetic diversification. Moreover, with very rare exceptions, it is only the maternal DNA that is transmitted to the offspring. The fertilized oocyte degrades mtDNA carried from the sperm in a complex process involving the ubiquitin proteasome system. Thus, although mothers transmit their mtDNA to both their sons and daughters, only the daughters are able to transmit the inherited mtDNA to future generations. Accordingly, mtDNA sequence variation and associated phenotypic traits and diseases are inherited exclusively along maternal lines.
As noted below, because of the complex relationship between mtDNA mutations and disease expression, sometimes this maternal inheritance is difficult to recognize at the clinical or pedigree level. However, evidence of paternal transmission can almost certainly rule out an mtDNA genetic origin of phenotypic variation or disease; conversely, a disease affecting both sexes without evidence of paternal transmission strongly suggests a heritable mtDNA disorder (Fig. 85e-2).
MULTIPLE COPY NUMBER (POLYPLOIDY), HIGH MUTATION RATE, HETEROPLASMY, AND MITOTIC SEGREGATION
Each aerobic cell in the body has multiple mitochondria, often numbering many hundreds or more in cells with extensive energy production requirements. Furthermore, the number of copies of mtDNA within each mitochondrion varies from several to hundreds; this is true of both somatic as well as germ cells, including oocytes in females. In the case of somatic cells, this means that the impact of most newly acquired somatic mutations is likely to be very small in terms of total cellular or organ system function; however, because of the manyfold higher mutation rate during mtDNA replication, numerous different mutations may accumulate with aging of the organism. It has been proposed that the total cumulative burden of acquired somatic mtDNA mutations with age may result in an overall perturbation of mitochondrial function, contributing to age-related reduction in the efficiency of oxidative phosphorylation and increased production of damaging ROS. The accumulation of such acquired somatic mtDNA mutations with aging may contribute to age-related diseases, such as metabolic syndrome and diabetes, cancer, and neurodegenerative and cardiovascular disease in any given individual. However, somatic mutations are not carried forward to the next generation, and the hereditary impact of mtDNA mutagenesis requires separate consideration of events in the female germline.
The multiple mtDNA copy number within each cell, including the maternal germ cells, results in the phenomenon of heteroplasmy, in contrast to much greater uniformity (homoplasy) of somatic nuclear DNA sequence. Heteroplasmy for a given mtDNA sequence variant or mutation arises as a result of the coexistence within a cell, tissue, or individual of mtDNA molecules bearing more than one version of the sequence variant (Fig. 85e-3). The importance of the heteroplasmy phenomena to the understanding of mtDNA-related mitochondrial diseases is critical. The coexistence of mutant and nonmutant mtDNA and the variation of the mutant load among individuals from the same maternal sibship, and across organs and tissues within the same individual, play a pivotal role in the manifestation and severity of disease and are crucial to understanding the complexity of inheritance of mtDNA disorders. At the level of the oocyte, the percentage of mtDNA molecules bearing each version of the polymorphic sequence variant or mutation depends on stochastic events related to partitioning of mtDNA molecules during the process of oogenesis itself. Thus, oocytes differ from each other in the degree of heteroplasmy for that sequence variant or mutation. In turn, the heteroplasmic state is carried forward to the zygote and to the organism as a whole, to varying degrees, depending on mitotic segregation of mtDNA molecules during organ system development and maintenance. For this reason, in vitro fertilization, followed by preimplantation genetic diagnosis (PGD), is not as predictive of the genetic health of the offspring in the case of mtDNA mutations as in the case of the nuclear genome. Similarly, the impact of somatic mtDNA mutations acquired during development and subsequently also shows an enormous spectrum of variability.
FIGURE 85e-3 Heteroplasmy and the mitochondrial genetic bottleneck. During the production of primary oocytes, a selected number of mitochondrial DNA (mtDNA) molecules are transferred into each oocyte. Oocyte maturation is associated with the rapid replication of this mtDNA population. This restriction-amplification event can lead to a random shift of mtDNA mutational load between generations and is responsible for the variable levels of mutated mtDNA observed in affected offspring from mothers with pathogenic mtDNA mutations. Mitochondria that contain mutated mtDNA are shown in red, and those with normal mtDNA are shown in green. (Reproduced with permission from R Taylor, D Turnbull: Mitochondrial DNA mutations in human disease. Nat Rev Genetics 6:389, 2005.)
Mitotic segregation refers to the unequal distribution of wild-type and mutant versions of mtDNA molecules during all cell divisions that occur during prenatal development and subsequently throughout the lifetime of an individual. The phenotypic effect or disease impact will, thus, be a function not only of the inherent disruptive effect (pathogenicity) on the mtDNA-encoded gene (coding region mutations) or integrity of the mtDNA molecule (control region mutations), but also of its distribution among the multiple copies of mtDNA in the various mitochondria, cells, and tissues of the affected individual. Thus, one consequence can be the generation of a bottleneck due to the marked decline in given sets of mtDNA variants, consequent to such mitotic segregation. Heterogeneity arises from differences in the degree of heteroplasmy among oocytes of the affected female, together with subsequent mitotic segregation of the pathogenic mutation during tissue and organ development, and throughout the lifetime of the individual offspring. The actual expression of disease might then depend on a threshold percentage of mitochondria whose function is disrupted by mtDNA mutations. This in turn confounds hereditary transmission patterns and hence genetic diagnosis of pathogenic heteroplasmic mutations. Generally, if the proportion of mutant mtDNA is less than 60%, the individual is unlikely to be affected, whereas proportions exceeding 90% cause clinical disease.
HOMOPLASMIC VARIANTS AND HUMAN MTDNA PHYLOGENY
In contrast to classic mtDNA diseases, most of which begin in childhood and are the result of heteroplasmic mutations as noted above, during the course of human evolution, certain mtDNA sequence variants have drifted to a state of homoplasmy, wherein all of the mtDNA molecules in the organism contain the new sequence variant. This arises due to a “bottleneck” effect followed by genetic drift during the very process of oogenesis itself (Fig. 85e-3). In other words, during certain stages of oogenesis, the mtDNA copy number becomes so substantially reduced that the particular mtDNA species bearing the novel or derived sequence variant may become the increasingly predominant, and eventually exclusive, version of the mtDNA for that particular nucleotide site. All of the offspring of a woman bearing an mtDNA sequence variant or mutation that has become homoplasmic will also be homoplasmic for that variant and will transmit the sequence variant forward in subsequent generations.
Considerations of reproductive fitness limit the evolutionary or population emergence of homoplasmic mutations that are lethal or cause severe disease in infancy or childhood. Thus, with a number of notable exceptions (e.g., mtDNA mutations causing Leber’s hereditary optic neuropathy; see below), most homoplasmic mutations are considered to be neutral markers of human evolution, which are useful and interesting in the population genetics analysis of shared maternal ancestry but which have little significance in human phenotypic variation or disease predisposition.
More importantly is the understanding that this accumulation of homoplastic mutations occurs at a genetic locus that is transmitted only through the female germline and that lacks recombination. In turn, this enables reconstruction of the sequential topology and radiating phylogeny of mutations accumulated through the course of human evolution since the time of the most recent common mtDNA ancestor of all contemporary mtDNA sequences, some 200,000 years ago. The term haplogroup is usually used to define major branching points in the human mtDNA phylogeny, nested one within the other, which often demonstrate striking continental geographic ancestral partitioning. At the level of the complete mtDNA sequence, the term haplotype is usually used to describe the sum of mutations observed for a given mtDNA sequence and as compared to a reference sequence, such that all haplotypes falling within a given haplogroup share the total sum of mutations that have accumulated since the most recent common ancestor and the bifurcation point they mark. The remaining observed variants are private to each haplotype. Consequentially, human mtDNA sequence is an almost perfect molecular prototype for a nonrecombining locus, and its variation has been extensively used in phylogenetic studies. Moreover, the mtDNA mutation rate is considerably higher than the rate observed for the nuclear genome, especially in the control region, which contains the displacement, or D-loop, in turn comprising two adjacent hypervariable regions (HVR-I and HVR-II). Together with the absence of recombination, this amplifies drift to high frequencies of novel haplotypes. As a result, mtDNA haplotypes are more highly partitioned across geographically defined populations than sequence variants in other parts of the genome. Despite extensive research, it has not been well established that such haplotype-based partitioning has a significant influence on human health conditions. However, mtDNA-based phylogenetic analysis can be used both as a quality assurance tool and as a filter in distinguishing neutral mtDNA variants comprising human mtDNA phylogeny from potentially deleterious mutations.
MITOCHONDRIAL DNA DISEASE
The true prevalence of mtDNA disease is difficult to estimate because of the phenotypic heterogeneity that occurs as a function of heteroplasmy, the challenge of detecting and assessing heteroplasmy in different affected tissues, and the other unique features of mtDNA function and inheritance described above. It is estimated that at least 1 in 200 healthy humans harbors a pathogenic mtDNA mutation with the potential to causes disease, but that heteroplasmic germline pathogenic mtDNA mutations actually affect up to approximately 1 in 8500 individuals.
The true disease burden relating to mtDNA sequence variation will only be known when the following capabilities become available: (1) ability to distinguish a completely neutral sequence variant from a true phenotype-modifying or pathogenic mutation, (2) accurate assessment of heteroplasmy that can be determined with fidelity, and (3) a systems biology approach (Chap. 87e) to determine the network of epistatic interactions of mtDNA sequence variations with mutations in the nuclear genome.
OVERVIEW OF CLINICAL AND PATHOLOGIC FEATURES OF HUMAN MTDNA DISEASE
Given the vital roles of mitochondria in all nucleated cells, it is not surprising that mtDNA mutations can affect numerous tissues with pleiotropic effects. More than 200 different disease-causing, mostly heteroplasmic mtDNA mutations have been described affecting ETC function. Figure 85e-4 provides a partial mtDNA map of some of the better characterized of these disorders. A number of clinical clues can increase the index of suspicion for a heteroplasmic mtDNA mutation as an etiology of a heritable trait or disease, including (1) familial clustering with absence of paternal transmission; (2) adherence to one of the classic syndromes (see below) or paradigmatic combinations of disease phenotypes involving several organ systems that normally do not fit together within a single nuclear genomic mutation category; (3) a complex of laboratory and pathologic abnormalities that reflect disruption in cellular energetics (e.g., lactic acidosis and neurodegenerative and myodegenerative symptoms with the finding of ragged red fibers, reflecting the accumulation of abnormal mitochondria under the muscle sarcolemmal membrane); or (4) a mosaic pattern reflecting a heteroplasmic state.
FIGURE 85e-4 Mutations in the human mitochondrial genome known to cause disease. Disorders that are frequently or prominently associated with mutations in a particular gene are shown in boldface. Diseases due to mutations that impair mitochondrial protein synthesis are shown in blue. Diseases due to mutations in protein-coding genes are shown in red. ECM, encephalomyopathy; FBSN, familial bilateral striatal necrosis; LHON, Leber’s hereditary optic neuropathy; LS, Leigh syndrome; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes; MERRF, myoclonic epilepsy with ragged red fibers; MILS, maternally inherited Leigh syndrome; NARP, neuropathy, ataxia, and retinitis pigmentosa; PEO, progressive external ophthalmoplegia; PPK, palmoplantar keratoderma; SIDS, sudden infant death syndrome. (Reproduced with permission from S DiMauro, E Schon: Mitochondrial respiratory-chain diseases. N Engl J Med 348:2656, 2003.)
Heteroplasmy can sometimes be elegantly demonstrated at the tissue level using histochemical staining for enzymes in the oxidative phosphorylation pathway, with a mosaic pattern indicating heterogeneity of the genotype for the coding region for the mtDNA-encoded enzyme. Complex II, CoQ, and cytochrome c are exclusively encoded by nuclear DNA. In contrast, complexes I, III, IV, and V contain at least some subunits encoded by mtDNA. Just 3 of the 13 subunits of the ETC complex IV enzyme, cytochrome c oxidase, are encoded by mtDNA, and, therefore, this enzyme has the lowest threshold for dysfunction when a threshold level of mutated mtDNA is reached. Histochemical staining for cytochrome c oxidase activity in tissues of patients affected with heteroplasmic inherited mtDNA mutations (or with the somatic accumulation of mtDNA mutations, see below) can show a mosaic pattern of reduced histochemical staining in comparison with histochemical staining for the complex II enzyme, succinate dehydrogenase (Fig. 85e-5). Heteroplasmy can also be detected at the genetic level through direct Sanger-type mtDNA genotyping under special conditions, although clinically significant low levels of heteroplasmy can escape detection in genomic samples extracted from whole blood using conventional genotyping and sequencing techniques.
FIGURE 85e-5 Cytochrome c oxidase (COX) deficiency in mitochondrial DNA (mtDNA)–associated disease. Transverse tissue sections that have been stained for COX and succinate dehydrogenase (SDH) activities sequentially, with COX-positive cells shown in brown and COX-deficient cells shown in blue. A. Skeletal muscle from a patient with a heteroplasmic mitochondrial tRNA point mutation. The section shows a typical “mosaic” pattern of COX activity, with many muscle fibers harboring levels of mutated mtDNA that are above the crucial threshold to produce a functional enzyme complex. B. Cardiac tissue (left ventricle) from a patient with a homoplasmic tRNA mutation that causes hypertrophic cardiomyopathy, which demonstrates an absence of COX in most cells. C. A section of cerebellum from a patient with mtDNA rearrangement that highlights the presence of COX-deficient neurons. D, E. Tissues that show COX deficiency due to clonal expansion of somatic mtDNA mutations within single cells—a phenomenon that is seen in both postmitotic cells (D; extraocular muscles) and rapidly dividing cells (E; colonic crypt) in aging humans. (Reproduced with permission from R Taylor, D Turnbull: Mitochondrial DNA mutations in human disease. Nat Rev Genetics 6:389, 2005.)
The emerging next-generation sequencing (NGS) techniques and their rapid penetration and recognition as useful clinical diagnostic tools are expected to also dramatically improve the clinical genetic diagnostic evaluation of mitochondrial diseases at the level of both the nuclear genome and mtDNA. In the context of the larger nuclear genome, the ability of NGS techniques to dramatically increase the speed at which DNA can be sequenced at a fraction of the cost of conventional Sanger-type sequencing technology is particularly beneficial. Low sequencing costs and short turnaround time expedite “first-tier” screening of panels of hundreds of previously known or suspected mitochondrial disease genes or screening for the entire exome or genome in an attempt to identify novel genes and mutations affecting different patients or families. In the context of the mtDNA, NGS approaches hold the particular promise for rapid and reliable detection of heteroplasmy in different affected tissues. Although Sanger sequencing allows for complete coverage of the mtDNA, it is limited by the lack of deep coverage and low sensitivity for heteroplasmy detection when it is much less than 50%. In contrast, NGS technology is an excellent tool for rapidly and accurately obtaining a patient’s predominant mtDNA sequence and also lower frequency heteroplasmic variants. This is enabled by deep coverage of the genome through multiple independent sequence reads. Accordingly, recent studies making use of NGS techniques have demonstrated sequence accuracy equivalent to Sanger-type sequencing, but also have uncovered heretofore unappreciated heteroplasmy rates ranging between 10 and 50% and detection of single-nucleotide heteroplasmy down to levels of <10%.
Clinically, the most striking overall characteristic of mitochondrial genetic disease is the phenotypic heterogeneity associated with mtDNA mutations. This extends to intrafamilial phenotypic heterogeneity for the same mtDNA pathogenic mutation and, conversely, to the overlap of phenotypic disease manifestations with distinct mutations. Thus, although fairly consistent and well-defined “classic” syndromes have been attributed to specific mutations, frequently “nonclassic” combinations of disease phenotypes ranging from isolated myopathy to extensive multisystem disease are often encountered, rendering genotype-phenotype correlation challenging. In both classical and nonclassical mtDNA disorders, there is often a clustering of some combination of abnormalities affecting the neurologic system (including optic nerve atrophy, pigment retinopathy, and sensorineural hearing loss), cardiac and skeletal muscle (including extraocular muscles), and endocrine and metabolic systems (including diabetes mellitus). Additional organ systems that may be affected include the hematopoietic, renal, hepatic, and gastrointestinal systems, although these are more frequently involved in infants and children. Disease-causing mtDNA coding region mutations can affect either one of the 13 protein encoding genes or one of the 24 protein synthetic genes. Clinical manifestations do not readily distinguish these two categories, although lactic acidosis and muscle pathologic findings tend to be more prominent in the latter. In all cases, either defective ATP production due to disturbances in the ETC or enhanced generation of ROS has been invoked as the mediating biochemical mechanism between mtDNA mutation and disease manifestation.
MTDNA DISEASE PRESENTATIONS
The clinical presentation of adult patients with mtDNA disease can be divided into three categories: (1) clinical features suggestive of mitochondrial disease (Table 85e-2), but not a well-defined classic syndrome; (2) classic mtDNA syndromes; and (3) clinical presentation confined to one organ system (e.g., isolated sensorineural deafness, cardiomyopathy, or diabetes mellitus).
|
COMMON FEATURES OF MTDNA-ASSOCIATED DISEASES IN ADULTS |
Table 85e-3 provides a summary of eight illustrative classic mtDNA syndromes or disorders that affect adult patients and highlights some of the most interesting features of mtDNA disease in terms of molecular pathogenesis, inheritance, and clinical presentation. The first five of these syndromes result from heritable point mutations in either protein-encoding or protein synthetic mtDNA genes; the other three result from rearrangements or deletions that usually do not involve the germline.
|
MITOCHONDRIAL DISEASES DUE TO MTDNA POINT MUTATIONS AND LARGE-SCALE REARRANGEMENTS |
[/not-level-membership-for-internal-medicine-category]
