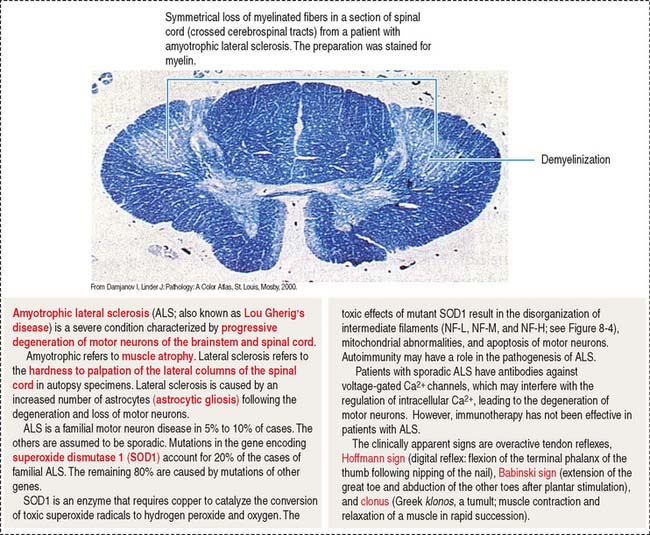
8 NERVOUS TISSUE
DEVELOPMENT OF THE NERVOUS SYSTEM
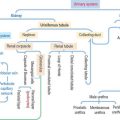
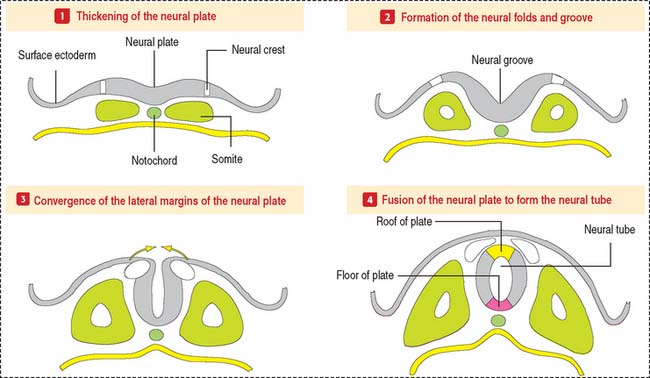
The CNS develops from the primitive ectoderm (Figure 8-1 and Box 8-A). A simple epithelial disk—the neural plate—rapidly rolls into a hollow cylinder—the neural tube. This process is known as neurulation. During this process, a specialized portion of the neural plate—the neural crest—separates from both the neural tube and the overlying ectoderm. In later development, the neural crest forms the neurons of the peripheral ganglia and other components of the PNS. A defect in the closing of the neural tube causes different congenital malformations (see Box 8-B).
Box 8-A Three sources of the CNS
Box 8-B Neural tube defects
Some of these cells invade developing visceral organs and form the parasympathetic and enteric ganglia and the chromaffin cells of the adrenal medulla.
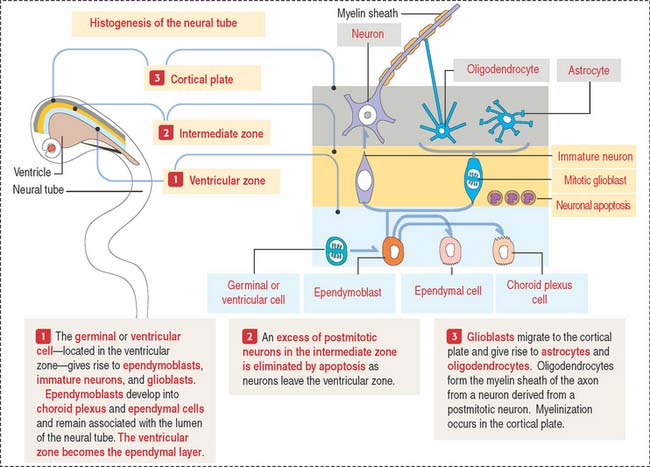
The early neural tube consists of a pseudostratified columnar epithelium formed by three zones (Figure 8-2): (1) the ventricular zone—the zone where progenitor cells give rise to most cells of the nervous tissue (except microglial cells); (2) the intermediate zone—where neurons migrate toward the cortical plate and where excess neurons are destroyed by apoptosis; and (3) the cortical plate—the future gray matter of the cerebral cortex.
Immature neurons leave the ventricular zone, migrate to the intermediate zone, lose their capacity to undergo cell division, and differentiate into functional neurons. The neuronal migration mechanism and the consequences of abnormal migration are highlighted in Box 8-C.
Box 8-C Neuronal migration
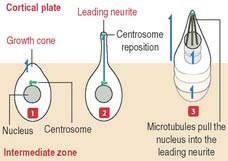
During this differentiation process, a selection process—similar to that in the thymus for T cells (see Chapter 10, Immune-Lymphatic System)—results in either neuronal heterogeneity or death. Neurons that become postmitotic in the intermediate zone reach the outer layers of the cortical plate and continue their differentiation.
Once the production of immature neurons is complete, the germinal or ventricular cells produce glioblasts, which differentiate into astrocytes, oligodendrocytes, and ependymoblasts. Ependymoblasts give rise to ependymal cells, lining the ventricular cavities of the CNS, and choroid epithelial cells, which are components of the choroid plexus.
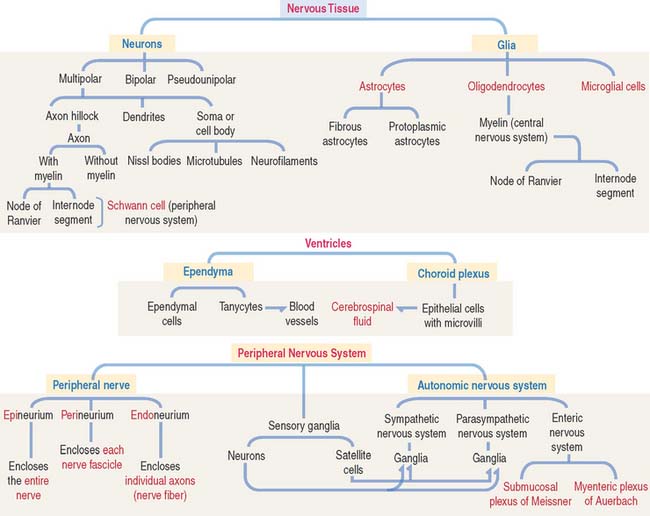
CELL TYPES: NEURONS AND GLIA
Neuron
The functional unit of the nervous system is a highly specialized, excitable cell, the nerve cell or neuron. Neurons usually consist of three principal components (Figures 8-3 and 8-4): (1) soma or cell body, (2) dendrites, and (3) axon.
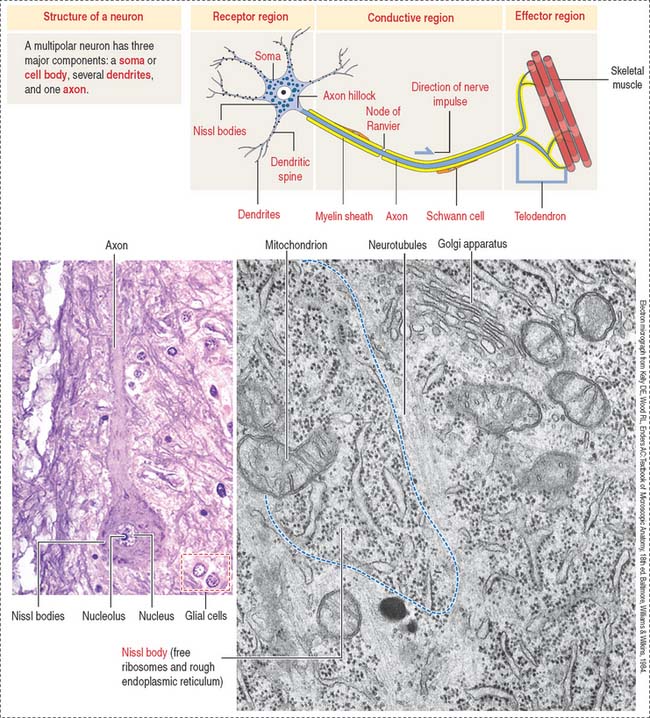
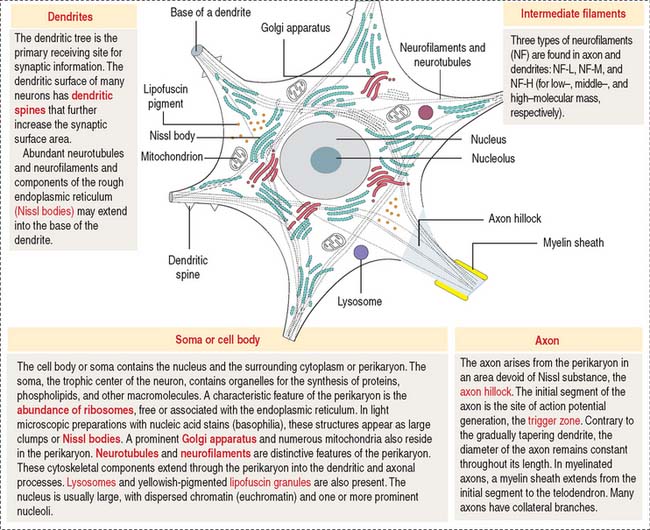
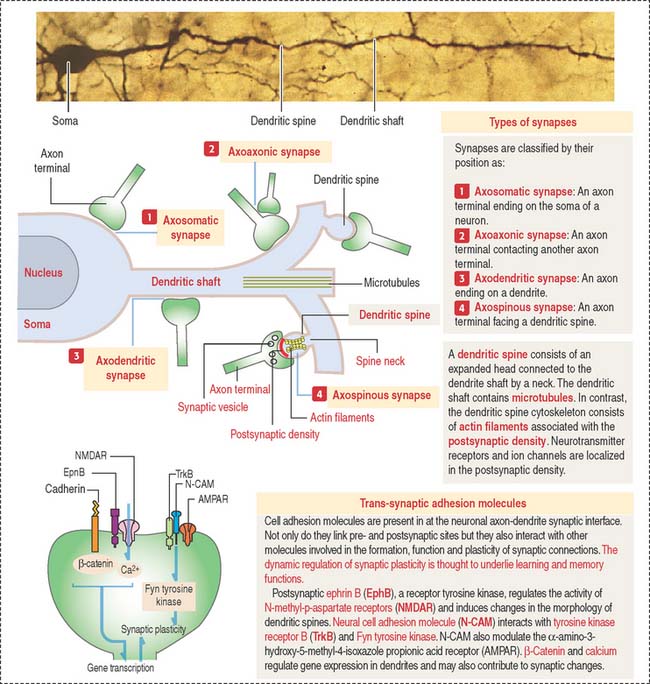
The dendrites are processes that arise as multiple treelike branches of the soma, forming a dendritic tree collectively. The entire surface of the dendritic branches is covered by small protrusions called dendritic spines. Dendritic spines establish numerous axonal synaptic connections, as we will see later (see Figure 8-7).
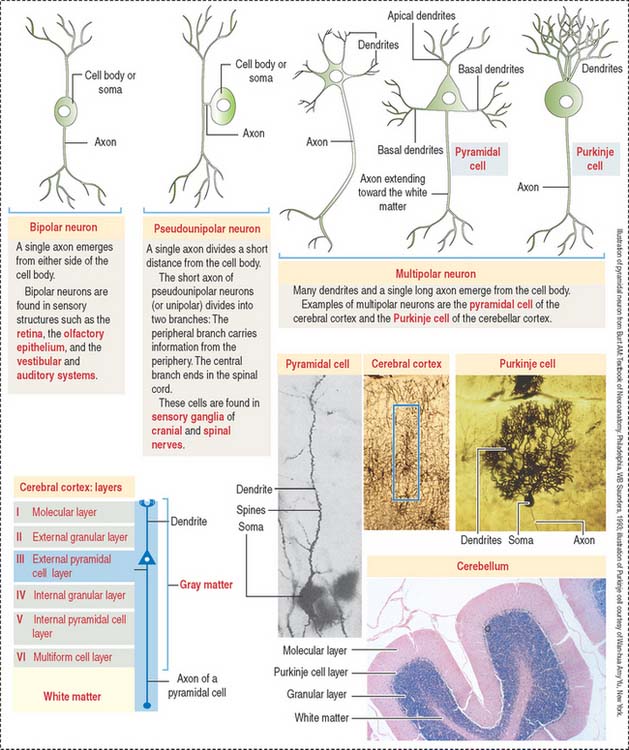
Types of neurons
Different types of neurons can be identified on the basis of the number and length of processes emerging from the soma (Figure 8-5):
According to the number of processes, neurons can be classified as:
Based on the length of the axon relative to the dendritic tree, multipolar neurons can be subclassified into (1) Golgi type I neurons, when the axon extends beyond the limits of the dendritic tree and (2) Golgi type II neurons, when an axon terminates in the immediate area of the cell body and does not extend beyond the limits of the dendritic tree. Small stellate cells of the cerebral cortex are Golgi type II cells.
Designation of groups of neurons and axons
Clusters of neurons arranged in a layer form a stratum or lamina (cerebral cortex). When neurons form longitudinal groups, these groups are designated columns.
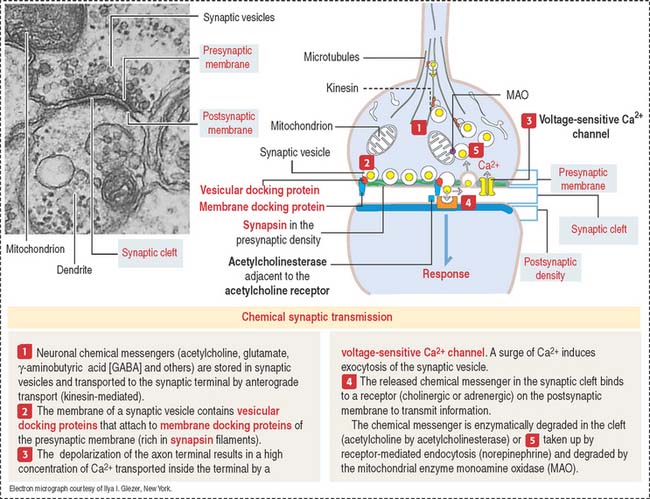
Synaptic terminals and synapses
The synaptic terminal (Figure 8-6) is specialized for the transmission of a chemical message in response to an action potential. The synapse is the junction between the presynaptic terminal of an axon and a postsynaptic membrane receptor surface, generally a dendrite.
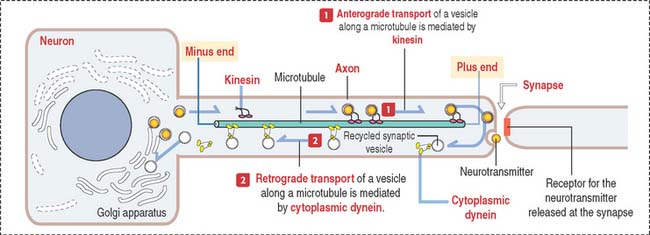
Presynaptic terminals contain a large number of membrane-bound vesicles (40 to 100 nm in diameter), the synaptic vesicles. Synaptic vesicles originate in the neuronal soma and are transported by molecular motor proteins along the axon (axonal transport) (Figure 8-7). Each vesicle contains a neurotransmitter. Presynaptic terminals contain mitochondria, components of the smooth endoplasmic reticulum, microtubules, and a few neurofilaments.
Synapses are classified by their location on the postsynaptic neuron (Figure 8-8) as follows:
Clinical significance: Axonal transport of rabies virus
The role of the axonal cytoskeleton and motor proteins (kinesin and cytoplasmic dynein; see Figure 8-7) was discussed in the Cytoskeleton section of Chapter 1, Epithelium. We emphasize once more the bidirectional transport of molecules along the axon: kinesin-mediated anterograde axonal transport of neurotransmitters—from the cell body toward the axon terminals, and the cytoplasmic dynein-mediated retrograde axonal transport of growth factors and recycling of axon terminal components—from the axon terminals to the cell body (see Box 8-D).
Box 8-D Neurotransmitters
Axonal transport is important in the pathogenesis of neurologic infectious diseases. For example, the rabies virus introduced by the bite of a rabid animal replicates in the muscle tissue from as little as 2 to 16 weeks or longer. After binding to the acetylcholine receptor, the viral particles are mobilized by retrograde axonal transport to the cell body of neurons supplying the affected muscle. The rabies virus continues to replicate within infected neurons and after the shedding of the virions by budding, they are internalized by the terminals of adjacent neurons. Further dissemination of the rabies virus occurs in the CNS. From the CNS, the rabies virus is transported by anterograde axonal transport by the peripheral nerves to the salivary glands. The virus enters the saliva to be transmitted by the bite. Painful spasm of the throat muscles on swallowing accounts for hydrophobia (aversion to swallowing water).
Astrocytes
Fibrous astrocytes are found predominantly in white matter and have long thin processes with few branches. Protoplasmic astrocytes reside predominantly in gray matter and have shorter processes with many short branches. Astrocytic processes end in expansions called end-feet (Figure 8-9).
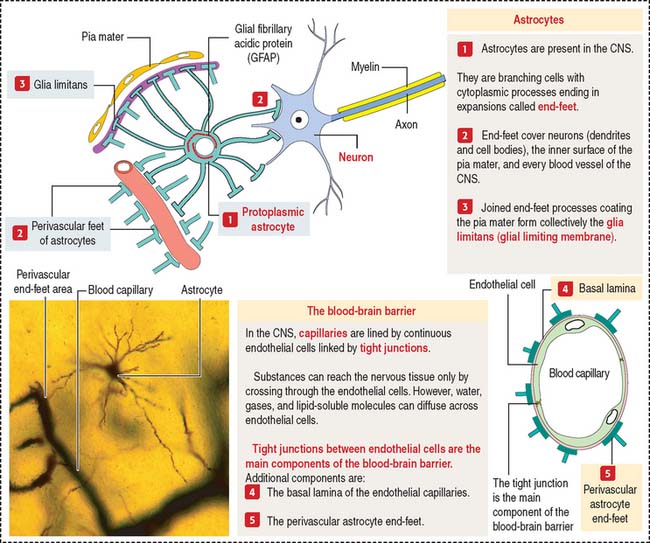
One of the distinctive features of astrocytes is the presence of a large number of glial filaments (glial fibrillary acidic protein, a class of intermediate filament studied in Chapter 1, Epithelium). Glial fibrillary acidic protein is a valuable marker for the identification of astrocytes by immunohistochemistry. Nuclei of astrocytes are large, ovoid, and lightly stained.
Most brain capillaries and the inner surface of the pia mater are completely surrounded by astrocytic end-feet (see Figure 8-9) forming the glia limitans (also called the glial limiting membrane). The close association of astrocytes and brain capillaries suggests a role in the regulation of brain metabolism.
Astrocytes surround neurons and neuronal processes in areas devoid of myelin sheaths and form the structural matrix for the nervous system.
Oligodendrocytes and Schwann cells: Myelinization
Processes of oligodendrocytes envelop axons and form a sheathlike covering (Figure 8-10). The formation of this sheath is similar to that of Schwann cells in peripheral nerves.
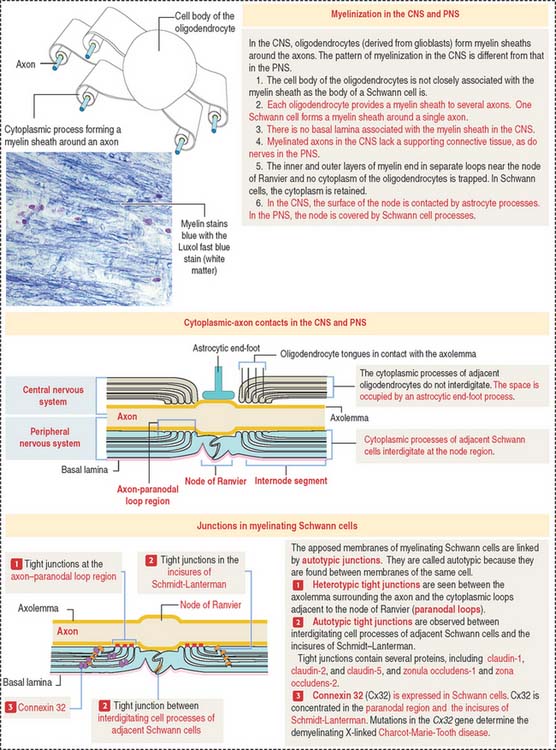
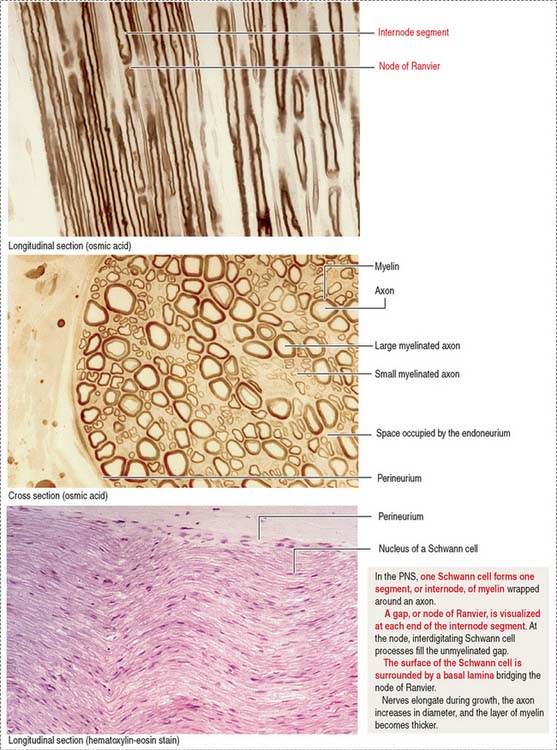
Myelin sheaths extend from the initial segments of axons to their terminal branches. The segments of myelin formed by individual oligodendrocyte processes are internodes. The periodic gaps between the internodes are the nodes of Ranvier.
During the formation of the myelin sheath, a cytoplasmic process of the oligodendrocyte wraps around the axon and, after one full turn, the external surface of the glial membrane makes contact with itself, forming the inner mesaxon (Figure 8-11).
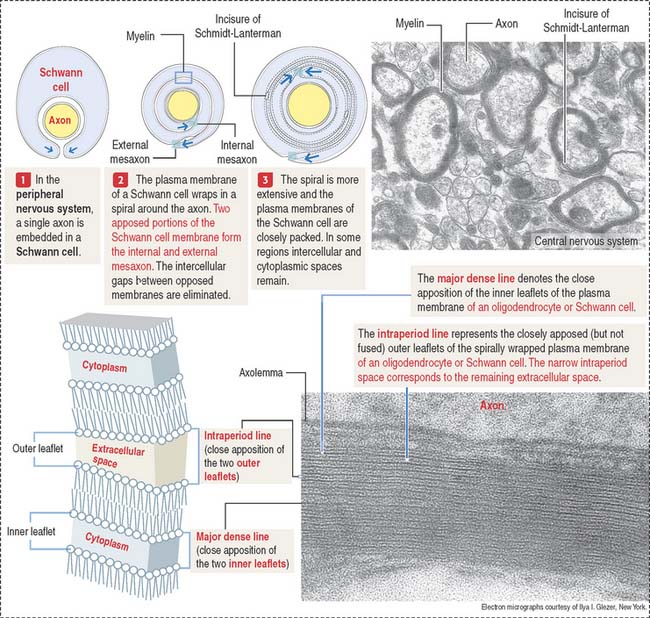
Spiraling continues until the axon is invested with a number of wrappings. The alternate fusion of both the cytoplasmic and external surfaces of the membrane results in an interdigitated double spiral (see Figure 8-11), one of intraperiod lines (fused external surfaces with remnant extracellular space), and one of major dense lines (fused cytoplasmic surfaces).
The apposed interdigitating processes of myelinating Schwann cells and the incisures of Schmidt-Lanterman are linked by tight junctions. They are called autotypic tight junctions because they link plasma membranes of the same cell. Heterotypic tight junctions are seen between the axolemma (surrounding the axon) and the Schwann cell paranodal cytoplasmic loops adjacent to the node of Ranvier.
Tight junctions contain claudins (claudin-1, claudin-2, and claudin-5) and zonula occludens (ZO) proteins (ZO-1 and ZO-2) (see Figure 8-10). Tight junctions (1) stabilize newly formed wraps of myelin during nerve development, (2) act as a selective permeability barrier, and (3) restrict the movement of lipids and proteins from specific membrane domains.
Connexin 32 (Cx32) is found in Schwann cells. Cx32 does not form gap junctions with other Schwann cells. Instead, Cx32 predominates in the para-nodal membranes and incisures of Schmidt-Lanterman and forms intercellular channels linking portions of the same cell. Mutations in the Cx32 gene causes X-linked Charcot-Marie-Tooth disease, a demyelinating disorder of the PNS characterized by the progressive loss of both motor and sensory functions of the distal legs (see Box 8-E).
Box 8-E Charcot-Marie-Tooth disease
Myelin: Protein and lipid components
Myelin in the CNS and PNS is similar in overall protein and lipid composition, except that myelin in the PNS contains more sphingomyelin and glycoproteins. Three proteins are particularly relevant (Figure 8-12): myelin basic protein (MBP), proteolipid protein (PLP), and myelin protein zero (MPZ).
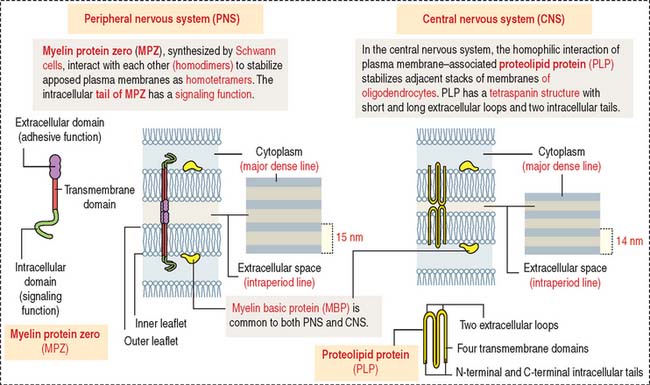
The predominant protein in myelin of the PNS is MPZ, a functional equivalent to PLP in the CNS. The extracellular domain of two MPZ proteins extends into the extracellular space to establish homophilic interaction with a similar pair of MPZ molecules on an opposite membrane. The homotetrameric structure provides intermembrane adhesion essential for the compactation of myelin (see Figure 8-13). The intracellular domain of MPZ participates in a signaling cascade that regulates myelinogenesis. In the CNS, plasma membrane–associated PLPs interact with each other and have a similar stabilizing function.
Proteins of myelin are strong antigens with a role in autoimmune diseases such as multiple sclerosis in the CNS and Guillain-Barré syndrome in the PNS.
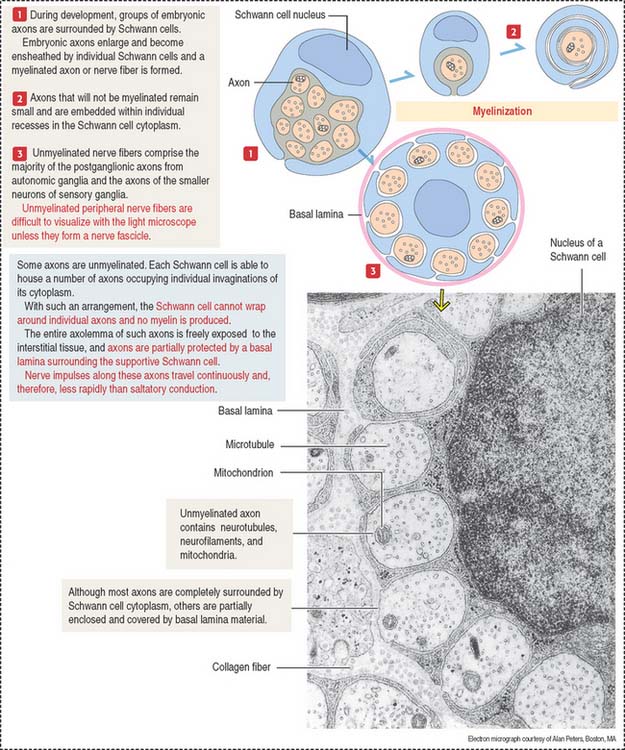
Some axons of the PNS are unmyelinated (see Figure 8-13). A Schwann cell can accommodate several axons in individual cytoplasmic invaginations and no myelin is produced.
Clinical significance: Demyelinating diseases
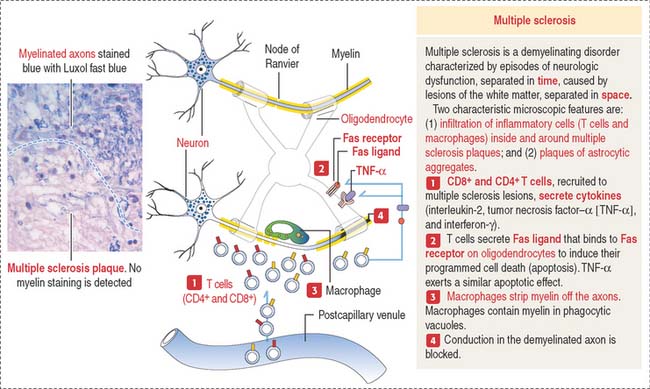
Multiple sclerosis (Figure 8-14) is characterized by clinically recurrent or chronically progressive neurologic dysfunction caused by multiple areas of demyelination in the CNS, in particular the brain, optic nerves, and spinal cord. An immune-mediated origin of multiple sclerosis is supported by an increase of immunoglobulin G (IgG) in the cerebrospinal fluid (CSF), and abnormalities of T cell function. A characteristic pathologic finding is the multiple sclerosis plaque, a demyelination lesion of the white matter, where the primary target is the myelin sheath and oligodendrocytes.
An inherited demyelination disorder is adrenoleukodystrophy, in which progressive demyelination is associated with dysfunction of the adrenal cortex. The X-linked form of this disease is caused by a mutation of a gene encoding a membrane protein of peroxisomes. A defect in this gene leads to the accumulation of very-long-chain fatty acids (VLCFAs) in serum (discussed under Peroxisomes in Chapter 2, Epithelial Glands).
Metabolic demyelination disorders include central pontine myelinolysis, a syndrome in which neurologic dysfunction is observed following rapid correction of hyponatremia in individuals with alcohol abuse or malnutrition. A typical pathologic finding is the presence of symmetrical demyelinated lesions in the central pons.
Clinical significance: Neurodegenerative diseases
Plaques and tangles lead to neuronal and white matter loss. Figure 8-16 and Box 8-F summarize and highlight the major molecular events observed in the brains of patients with Alzheimer’s disease, in particular the formation of amyloid plaques. A disproportion between production and clearance, and accumulation of Aβ peptides may be the initiation factor in Alzheimer’s disease.
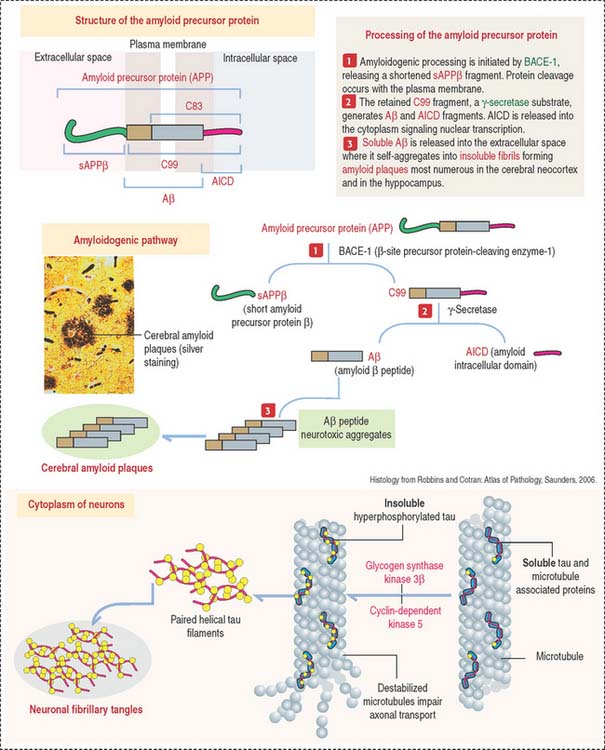
Box 8-F Amyloid precursor protein: Secretases and ADAM family proteins are sheddases
Neurofibrillary tangles in pyramidal neurons are typical of Alzheimer’s disease and other neurodegenerative disorders called tauopathies. Alterations in the stabilizing function of tau, a microtubule-associated protein, result in the accumulation of twisted pairs of tau in neurons. In normal neurons, soluble tau promotes the assembly and stability of microtubules and axonal vesicle transport. Hyperphosphorylated tau is insoluble, lacks affinity for microtubules, and self-associates into paired helical filaments (see Figure 8-16). Figure 8-17 stresses the role of microtubules in axonal transport, a function affected by abnormal tau.
Autosomal recessive juvenile parkinsonism is a relatively rare syndrome characterized by a mutation in a gene encoding the protein parkin. Parkin is detected primarily in the nervous system and is a member of the family of E3 ubiquitin ligases. Ubiquitin ligases attach ubiquitin protein chains to proteins, a process called ubiquination, thereby targeting them for degradation by the 26S proteasome (see Figure 3-14, Chapter 3, Cell Signaling). The ubiquination by parkin may be important for the normal turnover of α-synuclein. Aggregation of abnormal proteins and defective ubiquination may be important in the pathogenesis of Parkinson’s disease. A routine clinical test of the α-synuclein and parkin genes is currently not available. Essentially, the common neurodegenerative diseases are idiopathic (unknown cause) disorders of undefined pathogenesis.
Microglial cells
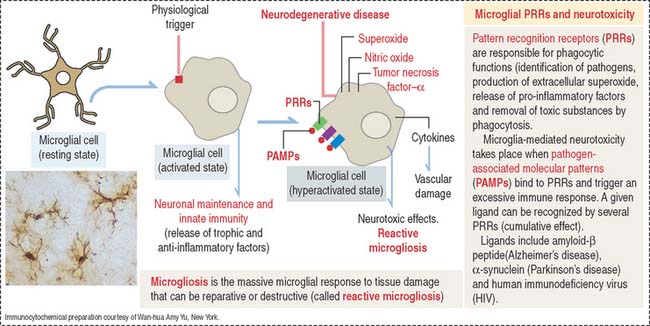
Activated microglial cells participate in brain development by supporting the clearance of neural cells undergoing apoptosis, eliminating toxic debris and enhancing neuronal survival through the release of trophic and anti-inflammatory factors. In the mature brain, microglia facilitate repair by steering the migration of stem cells to the site of inflammation and injury.
Microglial cells may become overactivated and exert neurotoxic effects by the excessive production of cytotoxic substances such as superoxide, nitric oxide, and tumor necrosis factor–α (TNF-α). Activated microglial cells are present in large numbers in neurodegenerative diseases (Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, amyotrophic lateral sclerosis, Huntington’s disease), causing a generalized microglial hyperactivity, a condition called reactive microgliosis. Figure 8-17 provides a summary of the structural and functional aspects of resting, activated and hyperactivated microglial cells.
EPENDYMA
Ependyma designates the simple cuboidal epithelium covering the surface of the ventricles of the brain and the central canal of the spinal cord. The ependyma consists of two cell types (Figure 8-18): (1) ependymal cells and (2) tanycytes.
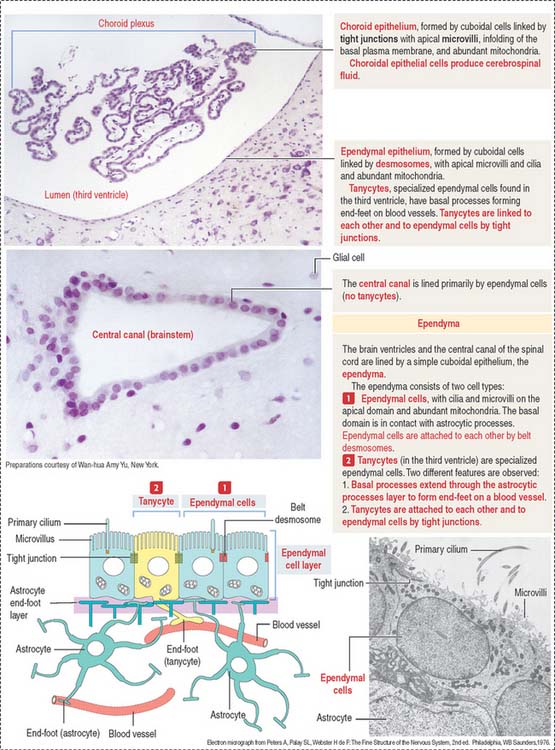
CHOROID PLEXUS
During development, the ependymal cell layer comes in contact with the highly vascularized meninges, forming the tela choroidea in the roof of the third and fourth ventricles and along the choroid fissure of the lateral ventricles. These cells differentiate into secretory cells, which in combination with the meningeal blood vessels form the choroid plexus.
The cells of the choroid plexus are highly polarized (Figure 8-19). The apical domain contains microvilli, and tight junctions connect adjacent cells. The basolateral domain forms interdigitating folds, and the cell rests on a basal lamina.
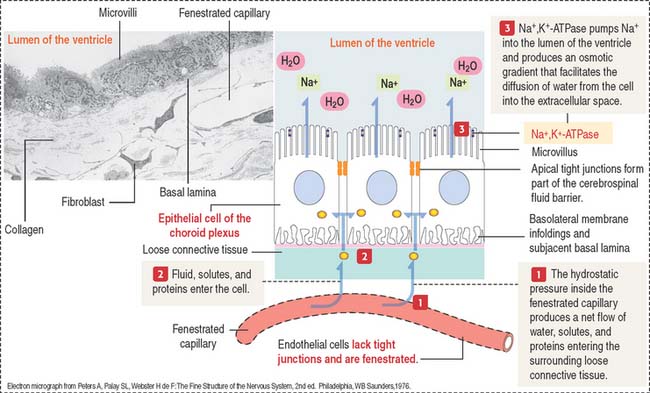
Cerebrospinal fluid
The choroid plexuses of the lateral, third, and fourth ventricles produce CSF.
CSF flows from the fourth ventricle into the brain and spinal subarachnoid space through median and lateral apertures. After entering the subarachnoid space, CSF flows outside the CNS into the blood, at the superior sagittal sinus (see Figure 8-18). The epithelium of the choroid plexus represents a barrier between the blood and the CSF. Several substances can leave the capillaries of the choroid plexus but cannot enter the CSF.
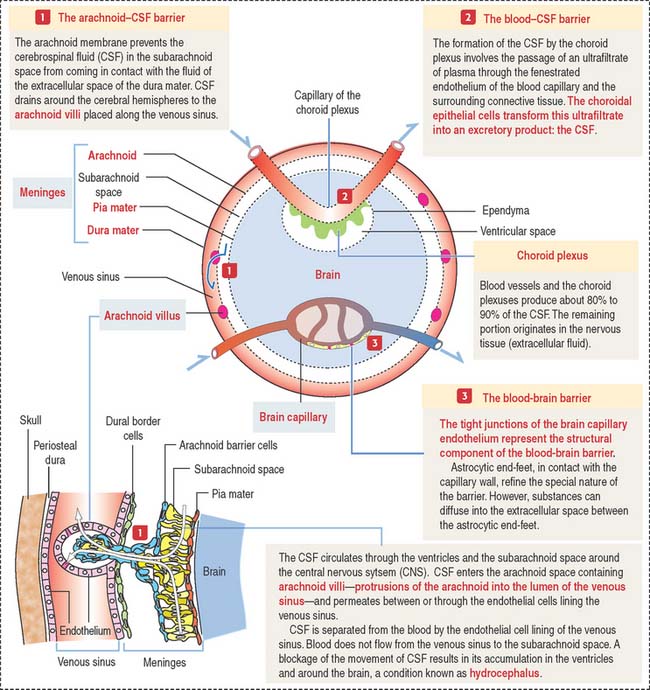
Clinical significance: Brain permeability barriers
Tight junctions represent the structural basis of the blood-brain barrier. This barrier offers free passage to glucose and other selected molecules but excludes most substances, in particular potent drugs required for the treatment of an infection or tumor. If the blood-brain barrier breaks down, tissue fluid accumulates in the nervous tissue, a condition known as cerebral edema.
Figure 8-20 illustrates details of three brain permeability barriers: (1) the arachnoid-CSF barrier, represented by arachnoid villi distributed along the venous sinus, in particular the arachnoid barrier cells linked by tight junctions. Arachnoid villi transfer CSF to the venous system (superior sagittal sinus). Fluid in the subarachnoid space operates like a shock absorber, which prevents the mass of the brain from compressing nerve roots and blood vessels. (2) The blood-CSF barrier. It involves tight junctions in the choroidal epithelium, responsible for the production of the CSF. (3) The blood-brain barrier, represented by tight junctions sealing the endothelial intercellular space.
PERIPHERAL NERVOUS SYSTEM
Individual nerve fibers of the PNS are ensheathed by Schwann cells (Figure 8-21). In myelinated fibers, individual Schwann cells wrap around the axon, forming a myelin sheath analogous to that of the oligodendrocytes of the CNS (see Figure 8-11). In unmyelinated fibers, a single Schwann cell envelops several axons (see Figure 8-13).
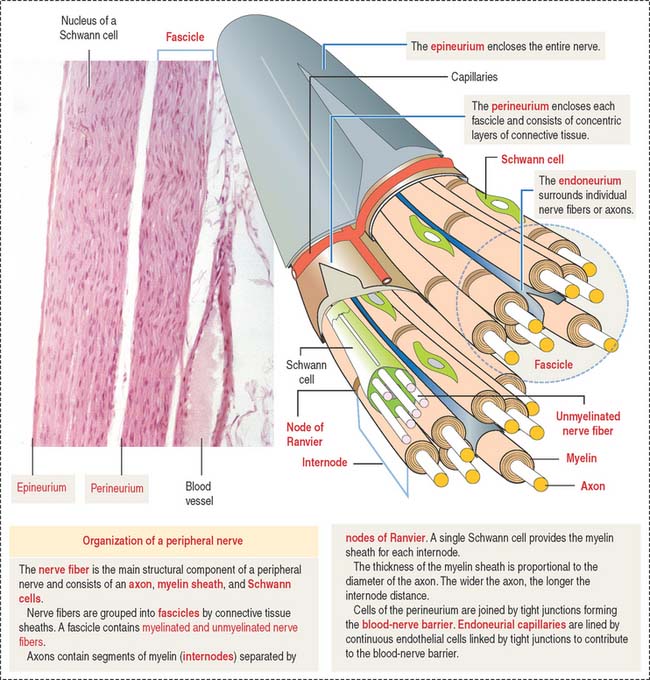
Structure of a peripheral nerve
In addition to Schwann cells, peripheral nerves have three additional connective tissue coverings (see Figures 8-21 and 8-22): (1) the epineurium, (2) the perineurium, and (3) the endoneurium.
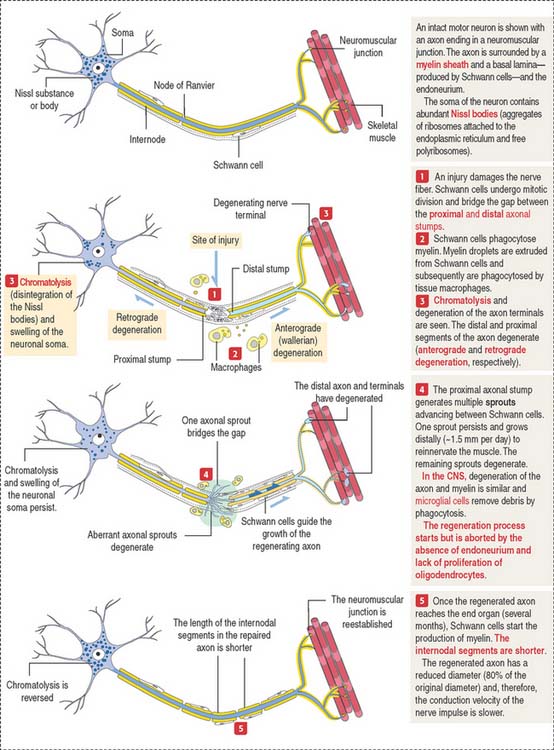
Clinical significance: Segmental demyelination and axonal degeneration
Axonal degeneration (Figure 8-23) may be followed by axonal regeneration. Recall from our discussion in Chapter 7, Muscle Tissue, that the motor unit is the functional unit of the neuromuscular system. Therefore, segmental demyelination and axonal degeneration affect the motor unit and cause muscle paralysis and atrophy. Physiotherapy for the paralyzed muscles is necessary to prevent muscle degeneration before regenerating motor axons can reach the motor unit. Neurotrophins play a significant role in the survival of neurons uncoupled from a peripheral target (see Box 8-G).
Box 8-G Neurotrophins
Segmental demyelination occurs when the function of the Schwann cell is abnormal or there is damage to the myelin sheath, for example, a crush nerve injury. If the nerve fiber is completely severed, the chances of recovery decrease unless a nerve segment is grafted. The presence of the endoneurium is essential for the proliferation of Schwann cells. Schwann cells guide an axonal sprout, derived from the proximal axonal stump, to reach the end organ (for example, a muscle).
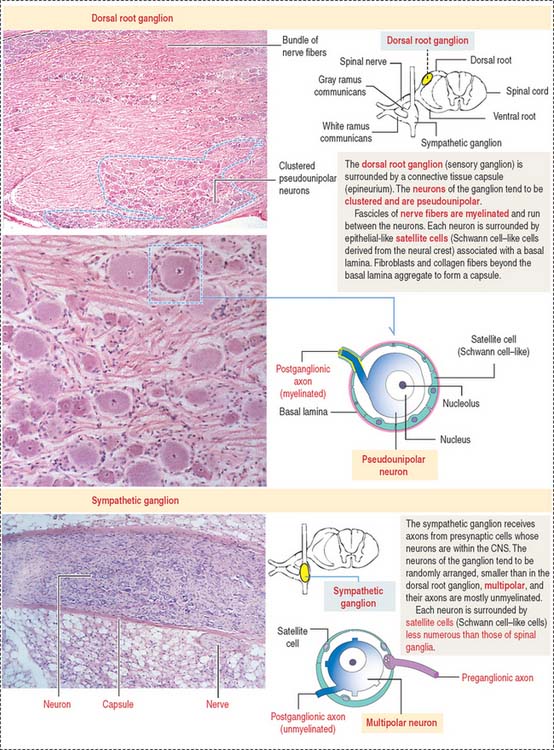
SENSORY GANGLIA
Sensory ganglia of the posterior spinal nerve roots and the trunks of the trigeminal, facial, glossopharyngeal, and vagal cranial nerves have a similar organization (Figure 8-24).
AUTONOMIC NERVOUS SYSTEM
The enteric nervous system consists of two interconnected plexuses—the myenteric plexus of Auerbach and the submucosal plexus of Meissner—within the walls of the alimentary tube. Each plexus consists of neurons and associated cells, and bundles of nerve fibers passing between plexuses. We discuss the enteric nervous system in Chapter 15, Upper Digestive Segment, and Chapter 16, Lower Digestive Segment.
Similar to the sensory ganglion, a layer of connective tissue continuous with the epineurium and perineurium of the peripheral nerve fiber (see Figure 8-24) surrounds each autonomic ganglion. The neurons of the autonomic ganglia are multipolar. The dendrites are contacted by myelinated axons of preganglionic neurons (white rami). The axons have a small diameter and are unmyelinated (grey rami). Each neuronal cell body is surrounded by Schwann cell–like satellite cells.
Neurohistochemistry
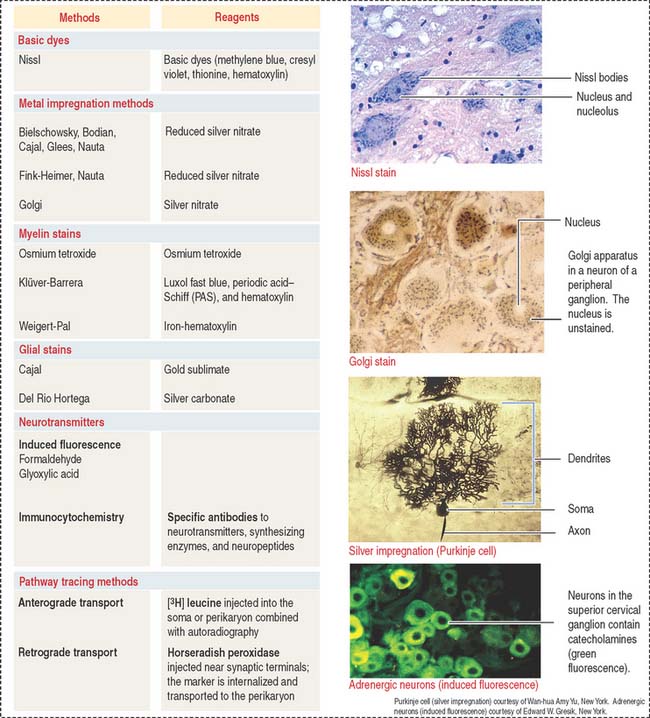
The nervous tissue has specialized features not observed in other basic tissues stained with routine staining methods such as hematoxylin-eosin. For example, basic dyes can demonstrate the cytoplasmic Nissl substance (ribonucleoproteins) in the cytoplasm of neurons (Figure 8-25).
Essential concepts
Microglial cells are phagocytic cells and immunoprotect the brain and spinal cord.
Myelin is separated from the axon by the axolemma, the surface membrane of the axon. Tight junctions (represented by claudins and zonula occludens proteins) are found linking the plasma membranes of the same Schwann cell and adjacent Schwann cell at the level of the node of Ranvier. Gap junctions, containing connexin 32 (Cx32), are present in the region of the incisures of Schmidt-Lanterman. Mutations in the Cx32 gene determine the X-linked Charcot-Marie-Tooth disease, a demyelinating disorder of the PNS.