CHAPTER 118 Neoplasms of the Neck
Radiologic Studies
Similar to CT, MRI should be performed with contrast enhancement using gadolinium agents. Fat suppression is helpful for postcontrast and T2-weighted sequences; however, nonenhanced T1-weighted images without fat suppression should always be obtained because they frequently provide the best delineation of the normal anatomic structures and the extent of pathologic processes.1,2 Positron emission tomography (PET) with fluorine-18 fluorodeoxyglucose (FDG), primarily PET-CT scanning, has evolved as a well-established modality in the evaluation of the primary site, regional nodal disease, and the presence of distant tumor for staging information, particularly for squamous cell carcinoma and the detection of synchronous primary tumors such as lung and esophagus. PET-CT scans may allow for precise localization of the lesion and may decrease the number of false-positive and false-negative findings.2,3 A controversial use of PET/CT imaging is in the clinical decision making of neck treatment following definitive chemoradiation therapy for advanced head and neck cancer. Several studies have addressed the use of PET scanning in the postirradiated neck. Porceddu and colleagues have shown that in patients with N2 and N3 neck disease a negative PET scan at 8 weeks allowed the observation of those patients with residual neck mass without compromising regional control. Of 27 patients observed by Porceddu with negative PET scans there were no failures in the neck. Yao and associates obtained similar results in a similar patient population. Optimal timing for post-therapy PET scanning appears to be at least 8 weeks, because scanning at 8 to 12 weeks significantly improved the negative predictive value over those who scanned earlier.4–6 On the basis of these studies it may be a reasonable strategy to obtain a PET-CT scan 8 to 12 weeks following therapy to evaluate for residual disease and performing neck dissection on those with positive scans while observing those with negative scans whether a neck mass is present or not. It should be mentioned that the spatial resolution of PET (and PET-CT) allows for detection of lesions that are at least 8 mm in size.
Ultrasound imaging has become a valuable tool in the head and neck surgeons’ diagnostic repertoire. It has the ability to allow the addition of imaging to the in-office examination of the neck as an adjunct to the physical examination. In addition, for the evaluation of the N0 neck it allows not only the characterization of lymph nodes in the neck but allows guided FNA biopsy of nodes of interest.7 The ability for ultrasound to be used as a primary prognostic tool in the neck to help dictate whether elective neck dissection is necessary has been investigated but has not changed the treatment paradigm in the N0 head and neck patient.
Benign Neoplasms of the Neck
Vascular Neoplasms
Pathology
Normal paraganglia contain two types of cells: type 1, chief cells or granular cells; and type 2, the supporting or sustentacular cells. Type 1 cells contain dense-core granules filled with catecholamines, a property that places them in the amine precursor and uptake decarboxylase (APUD) system. Type 2, or sustentacular cells, are elongated cells that closely resemble Schwann cells. Their function is not entirely clear. Tumors of paraganglia such as carotid body tumors contain both type 1 and 2 cells. Type 1 cells predominate and are arranged in an organized-nested pattern, known as Zellballen, surrounded by sustentacular cells in a fibrous stoma. These nests of Zellballen are illustrated in Figure 118-1. Type 1 chief cells tend to be polygonal shaped with abundant granular eosinophilic cytoplasm. They are peripherally surrounded by type 2 sustentacular cells that are difficult to identify by light microscopy and appear as spindle-shaped basophilic cells. Nuclear pleomorphism and cellular hyperchromatism are common in paragangliomas and should not be considered evidence of malignancy. Immunohistochemistry aids the diagnosis and differential diagnosis of these neoplasms. Type 1 cells stain positively with neuron-specific enolase, chromogranin A, and synaptophysin. Type 2 cells stain with S-100 and focally with glial fibrillary acidic protein.
Nomenclature
Other terms such as chemodectoma, glomus tumor, and nonchromaffin tumor are less accurate terms and should be avoided. Chemodectoma is an inaccurate term to describe all paragangliomas of the head and neck because the carotid body is the only known paraganglia of the head and neck that behaves as a chemoreceptor. The term glomus tumor more accurately describes benign cutaneous tumors arising from neuromyoarterial cells surrounding arteriovenous anastomoses. Designation as nonchromaffin tumor relates to histologic staining characteristics. An early histologic staining technique employing the chromaffin reaction failed to show the presence of catecholamines; paragangliomas were therefore described as nonchromaffin tumors. Newer techniques however have detected catecholamines in small quantities.8 The chromaffin reaction is a highly insensitive method on which to classify these tumors.9
Epidemiology
The most common paraganglioma of the head and neck is the carotid body tumor, followed by jugulotympanic paragangliomas and vagal paragangliomas. Other sites include the larynx, nasal cavity, orbit, trachea, aortic body, lung, and mediastinum. It has been estimated that paragangliomas comprise 1 in 30,000 head and neck tumors.10 However, the true incidence of paragangliomas may be unknown because previous reports have confused paragangliomas with neuroendocrine tumors.11 Further complicating an accurate estimate is the multicentricity of these tumors, particularly in familial paragangliomas.
Carotid Paragangliomas
History
The anatomist von Haller first described the carotid body in 1743; its function, however, was unknown at the time. Histologic studies of the carotid body revealed glandular acini, so the carotid body was renamed the carotid gland. Von Luschka first described a tumor of the carotid body in 1862. In 1880 Reigner performed the first resection of a carotid body tumor, but the patient did not survive. Six years later Maydl resected a carotid body tumor, and the patient survived but had postoperative hemiplegia and aphasia. In 1889 Albert was the first surgeon to resect successfully a carotid body without ligating the carotid vessels. The first successful removal of a carotid body tumor in the United States was reported by Scudder in 1903.12 The term paraganglion was first used by histologist Kohn in 1903 to describe the carotid body.13 This term was most appropriate because cells of the carotid body originate from the neural crest and migrate in close association with autonomic ganglion cells, hence the name “paraganglionic.” In 1950 Mulligan described in the dog neoplastic degeneration of the carotid body as chemodectoma because of the chemoreceptor function of the carotid body.14
Anatomy and Physiology
The carotid body is located in the adventitia of the posteromedial aspect of the bifurcation of the common carotid artery. The normal carotid body measures 3 to 5 mm in diameter but is often larger in persons living at higher altitudes. The average weight of the normal adult gland is 12 mg, with a wide range previously reported as 1 to 47 mg.15 During surgical removal, the typical finding is a small, reddish-brown to tan, ovoid structure attached to the carotid vessels at the bifurcation by Mayer’s ligament, through which the feeding vessels run, primarily from the external carotid artery. Blood flow and oxygen consumption of the carotid body, gram for gram, exceed those of the brain or thyroid gland.16 Sensory innervation is from Hering’s nerve, a branch of the glossopharyngeal nerve that originates approximately 1.5 cm distal to the jugular foramen.
Etiology
The etiology of paragangliomas appears to be multifactorial. Most paragangliomas are solitary. Multiple pheochromocytomas and paragangliomas are seen in familial syndromes, mainly multiple endocrine neoplasia types 2A and 2B. Other syndromes associated with paragangliomas are neurofibromatosis type 1 and von Hippel-Lindau disease, which is characterized by retinal angiomas and cerebellar hemangioblastomas. Carney’s triad demonstrates the association of paraganglioma, pulmonary chondroma, and gastric leiomyosarcoma.17
In addition to these associations, a syndrome of familial paragangliomas characterized by multiple paragangliomas, especially in the head and neck region, has been described and occurs in at least 10% of cases. The familial nature of carotid body tumors was first suggested by Chase in 1933 in his description of two sisters with carotid body tumors.18 Many developments have occurred recently in the characterization of the familial paraganglioma (PGL) syndromes including the genes and mutations involved and recommendations for the head and neck surgeon in managing these patients and their at-risk family members (Table 118-1). Genetic mutations responsible for the hereditary form of PGL have been identified in genes coding for succinate-dehydrogenase subunit D (SDHD), B (SDHB) and C (SDHC) genes, which map to chromosome 11, 1 and 1, respectively. Hereditary PGL syndrome has been classified genetically into four entities: PGL1, PGL2, PGL3, and PGL4. Germline mutations in SDHD, SDHB, and SDHC have been identified in PGL1, PGL4, and PGL3, respectively. The gene for PGL2 has not been identified.19 Individuals with hereditary paraganglioma syndrome have early onset of tumors and a higher frequency of bilateral and/or multiple tumors than do those with sporadic disease. Past reports suggested familial paraganglioma to be rare, although recent literature challenges this assertion and suggests the association of paragangliomas with germline genetic mutations is likely to be significantly higher, representing 17% of sporadic patients in one study19 and up to 28% to 40% in other studies.20–22
Table 118-1 Screening Recommendations for Carriers of SDHB, SDHD, or SDHC Gene Mutations*
| Annual physical examination and measurement of blood pressure |
| Annual levels of urinary catecholamines and metanephrines |
| Imaging of neck, thorax, abdomen and pelvis every 6-12 mo by computed tomography and/or magnetic resonance imaging |
* Starts in early teens. Succinate-dehydrogenase subunits D (SDHD), B (SDHB), and C (SDHC).
From Drucker AM, Houlden RL. A case of familial paraganglioma syndrome type 4 caused by a mutation in the SDHB gene. Nat Clin Pract Endocrinol Metab. 2006;2(12):706-712 and Isik C, Erem C, et al. Familial paraganglioma. Eur Arch Otorhinolaryngol. 2006;263(1):23-31.
PGL1 is caused by mutations in the D subunit of the succinate dehydrogenase (SDH) gene and is the most common inherited genetic abnormality in families with a history of paragangliomas. All three syndromes follow an autosomal pattern of inheritance, but the inheritance pattern for PGL1 is autosomal dominant modified by genomic imprinting. Genomic imprinting in paragangliomas was described by van der Mey and colleagues after reviewing data from 15 large Dutch pedigrees.23 The imprintable gene is transmitted in a mendelian manner, but the expression of the gene is determined by the sex of the transmitting parent. With paragangliomas, the gene results in the development of a tumor when it is paternally inherited. Offspring of male carriers were observed to demonstrate a 50% incidence of tumors, whereas children of female carriers never developed tumors. PGL1 is associated most commonly with head and neck tumors but also confers a risk for pheochromocytoma. It is estimated that 86% of individuals with a gene mutation will develop a tumor by age 50.24 Gene mutations in SDHC are rare, with few families identified to date.
Of further interest is the development of a paraganglioma syndrome in head and neck patients due to the inheritance of mutations in the SDHB gene. Although the PGLs associated with mutations in SDHB are less common than their D subunit counterpart, mutations in this gene confer a higher risk for the development of extra-adrenal, catecholamine-secreting, and often malignant pheochromocytomas, making identification of these individuals crucial in management of the at-risk family members to ensure that proper screening for pheochromocytoma and other paragangliomas takes place. Several cases of renal cell carcinoma in carriers of these mutations have also been reported, and one case of papillary thyroid cancer has been documented in the literature.24 The role of genetic counseling and testing in this population of patients cannot be overemphasized. Due to the apparent high rate of mutations in patients with a history consistent with sporadic paragangliomas, consideration of genetic counseling and testing should be given to every patient with a carotid body tumor or other paragangliomas.
Clinical Presentation and Diagnosis
The characteristic feature of carotid body tumors is slow growth rate, which is reflected clinically by the delay between the first symptoms and the diagnosis, which averages between 4 and 7 years. A carotid body tumor usually presents as a lateral cervical mass, which is mobile laterally but less mobile in the cranio-caudal direction because of its adherence to the carotid arteries. This physical finding has been called a positive Fontaine sign.25 Alternatively, a carotid body tumor may present as a parapharyngeal mass. Many carotid body tumors are pulsatile by transmission from the carotid vessels or, less commonly, expand themselves, reflecting their extreme intrinsic vascularity. Sometimes a bruit may be heard by auscultation, but it can disappear with carotid compression. The consistency varies from soft and elastic to firm and these tumors are generally nontender. As they enlarge, progressive symptoms of dysphagia, odynophagia, hoarseness, and other cranial nerve (IX-XII) deficits appear. Carotid sinus syndrome syncope has been described in association with carotid body tumors.26 The syndrome refers to a loss of consciousness accompanied with a reflex bradycardia and hypertension. Inciting stimuli include spontaneous movement of the head or following digital pressure applied to the tumor. Rarely, paraganglioma of the head and neck may present as a functional neuropeptide secreting tumor.
The capacity for catecholamine synthesis in head and neck paragangliomas, however, does not translate immediately to clinical findings. Although all paragangliomas have neurosecretory granules, only 1% to 3% are considered functional.27 Glenner and coworkers first described a functional carotid body tumor secreting norepinephrine in 1962.9 Patients should be asked about signs and symptoms indicating elevated catecholamines. Complaints of headaches, palpitations, flushing, and perspiration should be evaluated. In these patients a 24-hour urine collection is examined for norepinephrine and its metabolites including vanillylmandelic acid (VMA) and normetanephrine. Alternatively, plasma metanephrine may be assessed. Excess epinephrine or metanephrine should prompt suspicion of an adrenal pheochromocytoma because head and neck paraganglioma lack the enzyme to convert norepinephrine to epinephrine (phenylethanolamine-N-methyltransferase). An abdominal CT scan should be performed to rule out a concomitant adrenal pheochromocytoma. α- and β-Adrenergic blocking is undertaken if a tumor is found to be functional preoperatively. This decreases the risk from sudden catecholamine release that may occur with tumor manipulation in surgery. Routine screening for urinary metanephrines and VMA and serum catecholamines is only indicated for multiple or familial paragangliomas or in the presence of catecholamine-related symptoms.28
Malignancy
Cellular criteria for malignancy have not been established. Harrington and Dockerty29 attempted to classify malignant tumors of the carotid body. Criteria for malignancy included mitoses with giant cells, nuclear pleomorphism, and capsular invasion. Using these criteria, 50% of the 20 tumors studied would be considered malignant. Batsakis30 concurred that increased mitotic rate and capsular invasion should not be considered as determinants of malignancy. Other authors have hypothesized that all carotid body tumors demonstrate some degree of capsular invasion.31 Malignancy is determined by metastasis, which must be proven with biopsy, because paragangliomas may exhibit multicentricity. There are no histologic criteria for malignancy. In fact, previous reports have described metastatic carotid body tumors without mitoses.32 The diagnosis of malignancy should be made by evidence of spread to regional lymph nodes or distant sites33,34 (most common being lung and bones).
Malignant paragangliomas have been reported in 6% of carotid body paragangliomas by Batsakis.30 Accurate 5-year survival rates are not available because of the low malignancy rate of an uncommon tumor. Data from the National Cancer Data Base35 suggest an overall 5-year survival rate of 60%. Distant metastases had a worse prognosis with a 5-year survival rate of 11.8%, while those with regional spread of disease fared much better with a 5-year survival rate of 78%.
Imaging Studies
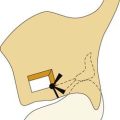
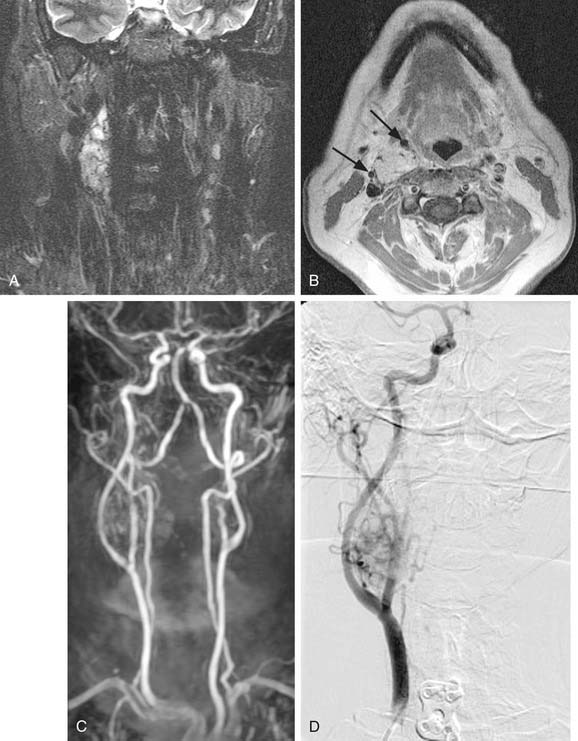
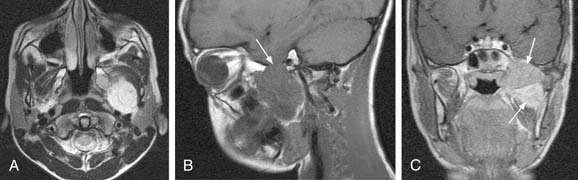
MRI with gadolinium may be the most useful imaging study for evaluating carotid body tumors (see Fig. 118-1A and B) because it offers superior soft tissue contrast without the need for ionizing radiation compared with CT scanning. MRI is sensitive for tumors as small as 0.8 cm.36 Paragangliomas larger than 2 cm in diameter typically demonstrate on T2-weighted images internal flow voids, dark lines, and dots that correspond to vascular structures. This is, however, not always present with carotid paragangliomas. Carotid body tumors demonstrate a characteristic lyre sign characterized as a bowing and displacing of the internal and external carotid arteries as shown in Figure 118-1B and C. Radiographic evaluation should be sufficient to make the diagnosis of carotid body tumor.
Carotid angiography has been replaced by MRI (including MR angiography, see Fig. 118-1C), but catheter angiography remains useful when preoperative embolization is necessary (see Fig. 118-1D). The use of preoperative embolization is controversial, but many authors have recommended its use for large tumors to decrease blood loss.37 Preoperative embolization may also be used in those rare instances where a malignant carotid body may be suspected. If preoperative embolization is planned, surgery should be performed 24 to 48 hours following in order to avoid revascularization, edema, or local inflammation. Additionally, angiography with temporary balloon occlusion using clinical and electroencephalographic monitoring, combined with brain CT perfusion scanning, can provide specificity as to the tolerance of collateral cerebral circulation across the circle of Willis in select cases.
The high density of somatostatin receptors in paragangliomas provides for newer functional nuclear medicine imaging techniques including metaiodobenzylguanidine (MIBG) scanning and octreotide scanning. MIBG scanning uses I-131 labeled tracer, which is concentrated in intracellular storage vesicles of paragangliomas.38 Octreotide scanning uses Indium-111 labeled somatostatin analog octreotide to diagnose primary tumors of the APUD system, as well as their metastases.39 These functional imaging studies have been recommended as a possible screening test for familial paragangliomas for patients at risk.40 They also allow the detection of additional tumors when malignant paraganglioma is suspected.41
Classification
Although not universally adopted in the literature on carotid body tumors, a classification system has been previously proposed for carotid body tumors. In 1971, while a surgery resident at the Mayo Clinic, Shamblin and coworkers described a classification system used to grade the difficulty of resection in carotid body tumors.42 Group I tumors were defined as localized, relatively small, and minimally attached to the carotid vessels. Surgical excision was described as carried out without difficulty in this group. Group II encompassed tumors adherent to or partially surrounding the vessels, with moderate arterial attachment. These tumors were described as amenable to careful surgical removal. Group III carotid body tumors completely encased the carotids. Shamblin and his colleagues recommended approaching these tumors with great care and with consideration for vessel replacement.
Surgery
In a report from Memorial Sloan-Kettering Cancer Center, Lack and colleagues discussed 39 of 43 patients with carotid body tumors treated surgically; one patient received definitive radiation therapy; and 3 others were observed but not treated.43 In this cohort of patients, 24 of 39 patients were free of disease following surgery, at an average follow-up interval of 12 years (6 months to 38 years). Local recurrence occurred in 4 of 39 patients (10%). Regional or distant metastases occurred in 4 of 39 patients (10%). All four of these patients were dead of disease within 6 years.
The incidence of permanent cranial nerve impairment as a complication of surgery has been reported in the literature to occur in approximately 20% of cases. In this report by Lack and colleagues,43 cranial nerve sacrifice of the vagus or hypoglossal nerve was necessary in 15% (6 of 39) of patients. An additional patient developed Horner’s syndrome postoperatively. Although not quantified, the superior laryngeal nerve supplying sensory innervation to the larynx and motor innervation to the cricothyroid muscle may be the most frequently injured nerve during carotid body resection. Paralysis of one superior laryngeal nerve may result in some degree of aspiration, although isolated palsy of one superior laryngeal nerve typically requires no additional rehabilitation. Furthermore, denervation of one cricothyroid muscle may result in pitch changes in singers, but changes in voice may not be perceptible. Injury to the cervical sympathetic chain will result in Horner’s syndrome, with ipsilateral ptosis, miosis, and anhidrosis. Netterville and colleagues have described first bite syndrome resulting from injury to the cervical sympathetic chain, resulting in loss of sympathetic input to the parotid gland.36 Patients with first bite syndrome complain of severe cramping in the parotid area when they take the first bite of food, particularly with the first meal of the day. The pain generally subsides over the next several bites but is more intense with strong sialogogues such as tart or bitter foods. The physiologic mechanism behind first bite syndrome is likely due to denervation supersensitivity of the sympathetic receptors that control the myoepithelial cells in the parotid gland. With oral intake, parasympathetic neurotransmitters are released and cross-stimulation of the sympathetic receptors causes a supramaximal response of the myoepithelial cells. Treatment includes restriction to bland foods and oral carbamazepine (100 to 200 mg twice daily) for those with severe pain.
Anand and colleagues44 reviewed 1181 published cases of carotid body tumors treated with surgical resection. Internal carotid artery injury was identified in 23% of cases (275 cases), with an overall occurrence of central nervous system (CNS) complications of 26%. This subcategory of internal carotid artery injury was further examined. In 23% of cases (62 cases), internal carotid artery repair was accomplished with simply suture or patch repair, with a CNS complication rate of 3%. The internal carotid artery was reconstructed in 45% (125 cases), with a CNS complication rate of 10% and a mortality rate of 2%. It was necessary to ligate the internal carotid artery in 32% (89 cases), resulting in a CNS complication rate of 66% and a mortality rate of 46%.
Controversy over Surgery for Multicentric Tumors
Another recently described and rarely recognized problem following bilateral carotid body excision is baroreflex failure syndrome.36 The clinical manifestations are caused by bilateral denervation of the carotid sinus. The carotid sinus is situated in the adventitia of the carotid bulb and serves as a baroreceptor to decrease systemic blood pressure. Bilateral baroreceptor dysfunction causes unopposed sympathetic outflow, resulting in marked fluctuations in blood pressure and a sustained tachycardia postoperatively. Over time, compensation occurs, but it is variable and unpredictable. Compensation may occur by baroreceptor fibers in the aorta or through neural regrowth at the carotid sinus. Therapy consists of sodium nitroprusside in the early recovery phase to prevent excessive hypertension. Long-term control may be accomplished using oral antihypertensives such as clonidine or phenoxybenzamine. Clonidine is an α-2 agonist, resulting in decreased release of norepinephrine into synaptic clefts, and stimulation of parasympathetic outflow, slowing the heart rate. Phenoxybenzamine is an α-1 and α-2 antagonist that decreases peripheral resistance and increases cardiac output.
Radiation Therapy
Radiation oncologists at the University of Florida have described effective local control of 23 carotid and vagal paragangliomas using definitive radiotherapy.45 In their study, 15 patients with 23 carotid or vagal paragangliomas were treated with radiotherapy between 1981 and 1995. Nineteen tumors were treated with radiation therapy alone, and four were treated with surgery and postoperative radiation therapy. For benign tumors, total doses ranged from 35 to 48.5 Gy. The two malignant tumors received 64.8 and 70 Gy, respectively. Follow-up ranged from 1.5 to 10 years. Local control was achieved in 96% of tumors at 5 years. Five-year disease-specific survival was 89%, with one patient dead of locally recurrent disease 5 years after radiation therapy. This patient had been previously treated with surgery and radiation therapy before treatment by the authors. Another patient died of atherosclerotic disease 13.5 years after radiation therapy. There were no patients with regional or distant failure following treatment. Complications reported by these authors included one patient experiencing a delayed transient CNS syndrome. No other complications were reported.
Valdagni and Amichetti reported 7 patients with 13 carotid body tumors treated with radiation therapy between 1968 and 1987.46 Treatment consisted of radiotherapy alone for 10 tumors and surgery plus radiation therapy for 3 tumors. Total doses ranged from 46 to 60 Gy at 1.8 to 2.5 Gy per fraction. Follow-up ranged from 1 to 19 years. Local control was achieved in all patients. Acute side effects were minimal. No short or long-term toxicities were reported.
Even proponents of radiation therapy for carotid body tumors concur with surgical resection as the preferred modality of treatment for most lesions.47 Definitive radiation therapy may be reserved for patients who are poor surgical candidates due to a debilitated medical condition and for locally advanced tumors where anticipated postoperative morbidity may preclude consideration for surgical resection. Adjuvant radiation therapy may be considered following surgery for malignant carotid body tumors for locoregional control.
Observation
Using sequential radiologic imaging, Jansen and colleagues48 have estimated the median tumor doubling time for 20 carotid body tumors to be 7.1 years including 12 cases without detectable growth. Farr has estimated a growth rate for carotid body tumors, plotting tumor size against years of symptom duration, of 2 cm in 5 years.49 A scan-and-wait policy may be considered for those patients who are not suitable candidates for surgery or radiation therapy. This highly select group of patients includes those individuals whose medical condition is so poor that both surgery and radiation therapy are contraindicated, or those patients of such advanced age that the carotid body tumor may have minimal impact on their life expectancy or quality of life. This group may also include those patients with malignant carotid body tumors with distant metastases where locoregional treatment would be only palliative in intent.
Vagal Paragangliomas
Vagal paragangliomas are tumors derived from paraganglionic tissue associated with one of the ganglia of the vagus nerve.50 Vagal paragangliomas most commonly arise from the inferior vagal ganglion, also referred to as the nodose ganglion. Tumors arising from the superior vagal ganglion, or jugular ganglion, may be dumbbell-shaped and may extend from the neck intracranially through the jugular foramen.
Clinical Presentation and Diagnosis
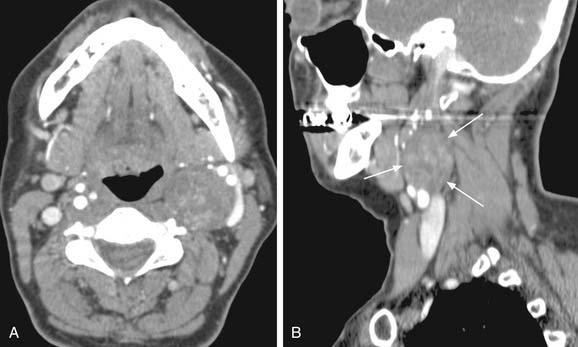
Vagal paragangliomas, like carotid body tumors, may present as a palpable neck mass that is more mobile in a lateral direction than a craniocaudal orientation. Paralysis of the ipsilateral true vocal cord or Horner’s syndrome, from involvement of the ipsilateral sympathetic chain, may be present as the tumor grows in size. True vocal cord paralysis may result in hoarseness with or without aspiration of liquids from glottic incompetence. Large tumors arising from the jugular ganglion may be associated with cranial neuropathies of IX, XI, and XII. Diagnostic imaging studies described previously in the chapter may demonstrate anterior displacement of the carotid artery from the tumor present in the posterior carotid sheath. Unlike carotid body tumors, the internal and external carotid arteries do not manifest a splayed configuration in vagal paragangliomas (Fig. 118-2).
Management
Arteriovenous Malformations of the Head and Neck
Arteriovenous malformations (AVMs) represent another form of benign vascular neoplasms that present in the head and neck. The most common site of head and neck AVMs are the cheek, ear, nose, forehead, and upper aerodigestive tract. These can be difficult neoplasms to manage. Both excision and interventional radiologic procedures have been used with some success. Kohout and colleagues51 presented a large series of 81 patients with head and neck arteriovenous malformations in which patients were separated on the basis of the Schobinger classification and outcomes over a 20-year period were discussed. These authors found that lesions in stage I responded well to therapy and had a higher cure rate than those in stage II or III, although this did not meet statistical significance. They also found that the largest percentage of patients who did not undergo therapy were in stage I, leading to the conclusion that more potentially curable patients were left untreated. Their recommendations for intervention include early-stage lesions and rapidly progressive and painful lesions. Treatment options include both resection and embolization or a combination. Reconstructive considerations must also play a role in decision making with surgical intervention.
Schwannomas
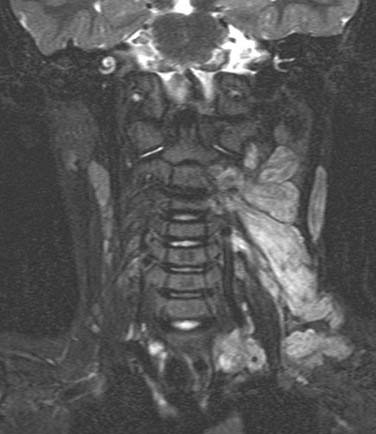
Schwannomas, also called neurilemomas, are typically well-encapsulated, slowly growing tumors that arise from Schwann cells of peripheral nerves. These tumors are typically solitary but may occur in multiples, and up to half occur in the head and neck region.52 Schwannomas may arise from cranial nerves such as VIII (acoustic neuromas) or X, the sympathetic chain, cervical nerve roots, or the brachial plexus. Clinically, schwannomas of the lateral neck may present as a painless neck mass. The clinical presentation of acoustic neuromas is covered elsewhere in this text (see Chapter 177). Radiologically, schwannomas typically present as a well-circumscribed mass that enhances on contrasted studies. They are usually heterogeneous and contain bright areas on T2-weighted images (Fig. 118-3A and B). Histologically, schwannomas demonstrate a characteristic cellular pattern of alternating regions containing compact spindle cells called Antoni type A areas and more loosely arranged, hypocellular zones called Antoni type B areas. Rows of nuclear palisading may be observed, and such arrangements are referred to as Verocay bodies. The treatment of choice of schwannomas of the neck typically involves surgical resection. Cranial neuropathies following surgical resection of head and neck schwannomas are common, and preoperative counseling of the patient as to predicted deficits is crucial in management. Preoperative speech and swallowing evaluation, as well as postoperative management, are critically important in voice and swallowing rehabilitation in these patients. Malignant transformation of schwannomas is rare.
Neurofibromas
Neurofibromas (Fig. 118-4) are benign nerve sheath tumors that may present either as a solitary neck mass or as multiple tumor nodules in association with the autosomal dominant disorder neurofibromatosis. In contrast to schwannomas, neurofibromas are unencapsulated and histologically demonstrate an interlacing bundle of spindle cells. Like schwannomas, solitary neurofibromas undergo malignant transformation uncommonly and are best treated by complete surgical resection. Neurofibromas associated with neurofibromatosis are more difficult to treat, in light of the multiple numbers of infiltrative tumors that are not well-defined. Surgery for neurofibromatosis is typically reserved for those lesions that are painful, that may cause compressive symptoms from their large size, or for lesions that are malignant. Malignant transformation of neurofibromatosis occurs more commonly than solitary neurofibromas.
Benign Lesions of the Parapharyngeal Space
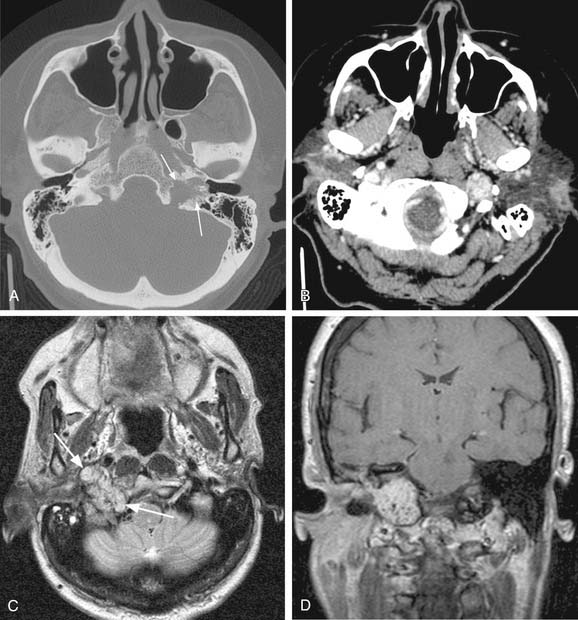
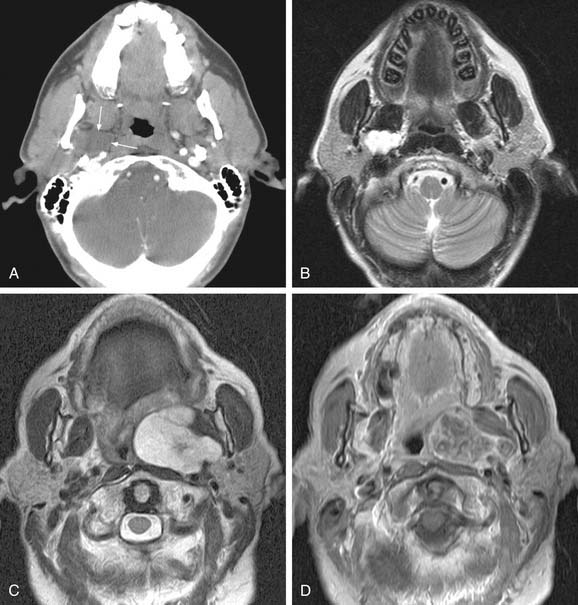
The parapharyngeal space is a pyramidal area bounded by the skull base superiorly, the hyoid bone inferiorly, the pterygomandibular raphe anteriorly, the vertebral fascia posteriorly, the medial pterygoid laterally, and the superior constrictor muscle medially. The space contains both the prestyloid and poststyloid compartments, and pathology in this area is based on these anatomic structures. Some tumors presented earlier in the chapter including paragangliomas and benign peripheral nerve tumors are also neoplasms of the parapharyngeal space. Salivary gland tumors are the most common tumors in the parapharyngeal space and are the main consideration in the differential diagnosis. A full description of both benign and malignant tumors of the salivary glands can be found elsewhere in this volume, in Chapters 87 and 88. The most common benign neoplasm of the parapharyngeal space is the pleomorphic adenoma followed by paragangliomas. Parapharyngeal space neoplasms can arise de novo or extend from surrounding structures. In the case of benign salivary gland tumors, it is hypothesized that they arise de novo from rests of salivary tissue in the parapharyngeal space. Both MRI and CT scanning are useful in assessing masses of the parapharyngeal space; however, MRI is superior in detailing the complicated neurovascular anatomy of the parapharyngeal space as illustrated in Figure 118-5A through D. Successful resection of the lesions can be accomplished by both a transparotid/transcervical approach or a strictly transcervical approach. Mandibulotomy is generally not necessary except in large tumors where wide exposure is necessary for the control of vessels. Warthin’s tumors and other neurogenic tumors have been described in the parapharyngeal space but are much less common, as are salivary malignancies.53,54
Malignant Neoplasms of the Neck
Primary malignant neoplasms of the neck are rare entities. Few studies have elucidated the true incidence of primary malignancies of the neck because they are often described in case reports or small series or not reported at all. Thus our knowledge and data are based on these limited publications and reports on non–head- and neck-related soft tissue tumors. In the larger series of reports of these tumors of the entire head and neck region, only approximately 4 to 20 cases are listed per year.55,56 The following discussion provides the reader with an overview of the differential diagnosis, evaluation, and management of malignant tumors of the neck.
Metastatic Lesions to the Neck
Unknown Primary
The location of the neck mass signals a clue regarding the primary site because of the well-known lymphatic drainage routes in the head and neck region and the updated classification system of neck lymph node levels. The location of the metastatic regional lymph node may direct the physician to the most common site of the primary lesion. Regional metastatic lesions from primary lesions of the oral cavity typically are found in levels 1, 2, and 3; from the oropharynx and hypopharynx, and larynx levels 2, 3, and 4; while level 5 metastases are more commonly associated with nasopharyngeal primaries. When the mass presents in the supraclavicular region, esophageal and pulmonary primary sites should be considered in addition to abdominal and pelvic locations.57,58
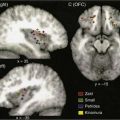
Usually, the primary site is identified during the head and neck examination or during operative endoscopy, at which time biopsies confirm the location and histology of the primary site. When the primary site cannot be identified, imaging techniques including CT, MRI, and/or PET scan have been used with varying degrees of success et al.59,60 Fused PET/CT imaging has been helpful in determining the site of disease in unknown primary tumors of the neck. Fencl and associates found a sensitivity of 62% and a specificity of 81.9% in 190 patients with unknown primary lesions.61 Figure 118-6 demonstrates the utility of this modality in detection of the primary site in a case of unknown primary in level 2 with detection of the primary site following PET/CT imaging. The treatment of unknown primary cancers of the neck continues to evolve, particularly related to consideration of directed biopsies, tonsillectomy, and extent of radiation therapy and neck dissection.
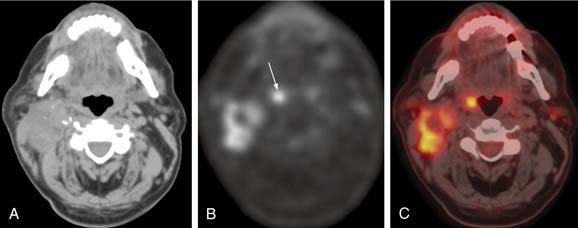
Figure 118-6. Unknown primary. A, Nonenhanced axial computed tomography (CT) image from positron emission tomography (PET)-CT study shows large heterogeneous level 2 lymph nodes. The palatine tonsils appear symmetric. B, Corresponding FDG (18fluorodeoxyglucose)-PET image reveals increased metabolic activity in the nodes, as well as another focus of high FDG uptake (arrow). C, Fused PET-CT image clearly demonstrates high metabolic activity in the right palatine tonsil and metastatic nodes.
Metastatic Neoplasms to the Neck—Regional
Aside from the unknown primary and the far more common squamous cell carcinomas of the aerodigestive tract, it is important to not overlook the possibility of metastatic disease from other sites in the head and neck region. Common primary sites of metastatic disease include the skin of the ear, face, scalp, and neck, which often present with an enlarged lymph node in the parotid, cheek, submandibular, or cervical region. The major and minor salivary glands may present with a primary neoplasm that initially appears to be a metastatic lesion to the neck manifesting as a level 1 or level 2 neck mass. The surprising diagnosis of adenocarcinoma may lead one to an exhausting search for a primary site in the lower digestive tract only to surmise eventually that this is a primary salivary gland tumor. Another common primary site that may be diagnosed first as a primary neck neoplasm is the thyroid gland, which not uncommonly initially presents as a neck mass.62
Metastatic Neoplasms to the Neck—Distant
Any neck mass determined to be malignant should also be considered as originating at a distant site. Although hundreds of neoplasms have been described to metastasize to the neck, the more common distant sites include pulmonary, esophageal, renal, ovarian, cervical, and prostate.63
Parapharyngeal Space Neoplasms
Numerous histologic types of primary malignancies of the PPS have been reported including malignant salivary gland tumors (mucoepidermoid carcinoma, adenocarcinoma, adenoid cystic carcinoma, carcinoma ex-pleomorphic adenoma, acinic cell carcinoma); malignant neurogenic tumors; lymphoma; liposarcoma; fibrosarcoma; and malignant meningioma.53,64–67
Sarcomas
The neck and parotid have been described as the most common head and neck sites involved by sarcomas, although they represent less than 1% of all head and neck malignancies.68 In the United States fewer than 5000 cases of sarcomas of all sites are reported annually with approximately 80% diagnosed in adults. Of these, only 15% to 20% are identified in the head and neck region with soft tissues of the neck and paranasal sinus region the most common sites identified. Although the etiology has not been determined, these neoplasms arise from mesenchymal cells, which may include endothelial cells, muscle, cartilage, and supporting connective tissue. More than 80% of sarcomas are derived from soft tissue, and approximately 20% arise within bone.
Classification and Staging
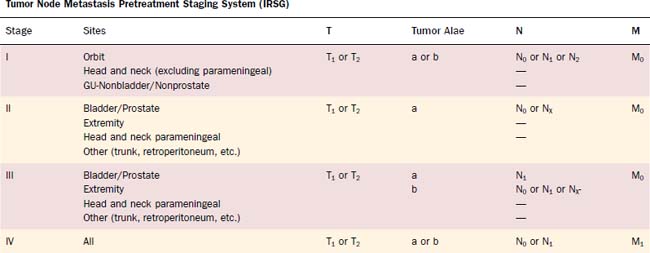
Sarcomas have generally been classified and named according to the tissue of origin rather than “site” of origin. Many “soft tissue” sarcomas such as malignant fibrous histiocytoma may be diagnosed within bone, but the diagnosis depends on the histologic material for confirmation. The newer American Joint Commission for Cancer (AJCC) staging system now takes into account differing histologies and separates bone and soft tissue. The listing of types of soft tissue sarcomas is provided in Table 118-2, and the approved 2003 AJCC staging system is shown in Table 118-3 for the soft tissue histologies. The listing of bone histologies can be found in Table 118-4, and the AJCC staging system in Table 118-5.
Table 118-3 American Joint Committee on Cancer Staging System for Soft Tissue Sarcomas (2003)
| Stage | Definition |
|---|---|
| Primary Tumor | |
| Tx | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor < 5 cm in greatest dimension (T1a, superficial; T1b, deep) |
| T2 | Tumor > 5 cm in greatest dimension (T2a, superficial; T2b, deep) |
| Regional Lymph Nodes | |
| Nx | Lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1* | Regional lymph node metastasis |
| Distant Metastases | |
| Mx | Distant metastasis cannot be assessed |
| M0 | No distant metastasis |
| M1 | Distant metastasis present |
| Histopathologic Grade | |
| Gx | Grade cannot be assessed |
| G1 | Well differentiated |
| G2 | Moderately differentiated |
| G3 | Poorly differentiated |
| G4 | Poorly differentiated or undifferentiated |
| Combined | |
| IA—G1-2, T1a-b, N0, M0 | Low-grade, small, and superficial or deep tumor |
| IB—G1-2, T2a, N0, M0 | Low-grade, large, and superficial tumor |
| IIA—G1-2, T2b, N0, M0 | Low-grade, large, and deep tumor |
| IIB—G3-4, T1a-b, N0, M0 | High-grade, small, and superficial or deep tumor |
| IIC—G3-4, T2a, N0, M0 | High-grade, large, and superficial tumor |
| III—G3-4, T2b, N0, M0 | High-grade, large, and deep tumor |
| IV—any G, any T, N1, M0 | Any metastasis |
Table 118-5 American Joint Committee on Cancer (AJCC) Staging System for Bone Sarcomas (2003)
| Stage | Definition |
|---|---|
| Primary Tumor | |
| Tx | Primary tumor cannot be assessed |
| T0 | No evidence of primary tumor |
| T1 | Tumor confined within the cortex |
| T2 | Tumor invades beyond the cortex |
| Regional Lymph Nodes | |
| Nx | Lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1 | Regional lymph node metastasis |
| Distant Metastases | |
| Mx | Distant metastasis cannot be assessed |
| M0 | No distant metastasis |
| M1 | Distant metastasis present |
| Histopathologic Grade | |
| Gx | Grade cannot be assessed |
| G1 | Well differentiated (low grade) |
| G2 | Moderately differentiated (low grade) |
| G3 | Poorly differentiated (high grade) |
| G4* | Poorly differentiated or undifferentiated (high grade) |
* Ewing’s sarcoma, considered G4.
The AJCC staging system for soft tissue sarcoma is shown in Table 118-3, although other staging systems have been described, particularly for various types of tumors. In addition, fibrosarcoma grade I (fibromatosis or desmoid tumor) and dermatofibrosarcoma are not listed under sarcoma staging. Other changes effective in 2003 include the addition of subdivisions of tumor (T) category into superficial (Ta) and deep (Tb) lesions. In general, the tumor should be described pathologically by grade as G1-G4 with tumor size less than 5 cm (T1) or greater than 5 cm (T2).
The AJCC staging system for bone sarcoma is shown in Table 118-5, although other staging systems have been described, particularly for various types of tumors. In addition, malignant lymphoma, leukemia, and multiple myeloma are not listed under bone sarcoma staging. Unlike the soft tissue sarcomas, the bone sites do not include subdivisions by depth of tumor as superficial or deep. Instead, T1 tumors are confined to within the cortex, whereas T2 tumors invade beyond the cortex.
Treatment
Malignant Fibrous Histiocytoma
Malignant fibrous histiocytoma (MFH) is the most common soft tissue sarcoma in adults. Rarely, it involves the head and neck region including the soft tissues of the paranasal sinuses, neck, skull base, and parotid gland. Of 88 fibrous histiocytomas (benign and malignant) of the head and neck analyzed, the neck was the second most common site behind the sinonasal region.69 Etiologic factors include prior radiation therapy and historical use of silica as injection material. The cell of origin has received much discussion, although consideration of fibroblastic or primitive mesenchymal cell has emerged as the leading theory.70 Microscopically, these tumors tend to reveal histiocytes, fibroblasts, giant cells, spindle cells, and collagen with the storiform-pleomorphic form the most common. MFH is considered a high-grade sarcoma and is classified into numerous subtypes providing for a separation from the standard staging for sarcomas. There is evidence that the survival and course of the disease is related to the size and depth of these tumors.71 The treatment is surgical with wide margins. Although elective treatment of the neck is not indicated due to the low incidence of cervical metastases, a possible exception may be the oral cavity where MFH has a higher metastatic potential to regional lymph nodes.72,73 Local recurrence rates approach 30% with overall lymph node metastases near 10% and distant metastases approximately 35%, most of which occur within the first 2 years.71 Recurrence seems to result in a lower surgical salvage rate for head and neck MFH than in extremities and overall survival is poorer for head and neck sites. Survival approaches 75% for patients without local recurrence following surgery and drops to 38% with local recurrence for an overall 5-year survival of approximately 51%.74
Rhabdomyosarcoma
RMS is a malignancy that is derived from mesenchymal cells associated with skeletal muscle differentiation. It represents the most common soft tissue sarcoma in children and accounts for 20% of sarcomas in all age groups. More than 45% of RMSs arise in the head and neck region with the highest incidence in the first decade and another peak occurring in the second and third decades. The most common sites in the head and neck from a recent series of 50 cases include the face, orbit, nasal cavity, neck, paranasal sinuses, and parameningeal sites.75 MRI imaging provides detailed evaluation of the location and extension of disease, allowing a more precise approach to these tumors (Fig. 118-7A through D). Metastatic disease was present in 33% of cases with the most common sites being bone marrow, cerebrospinal fluid, peritoneal fluid, and lung.75 Other reports reveal the neck soft tissue to be involved in almost 14% of adult head and neck RMSs.76 These tumors are categorized by the Intergroup Rhabdomyosarcoma Study (IRS) into the following subtypes: embryonal, embryonal-botryoid variant, embryonal-spindle cell variant, alveolar-classic and solid variants, undifferentiated, and anaplastic. These are also commonly classified as embryonal, alveolar, pleomorphic, and mixed types. The embryonal type represents the most common RMS in both children and adults and microscopically, reveals small spindle cells with a central nucleus, although round cells often resembling lymphocytes may be seen. The botryoid variant grows in a polypoid, grapelike fashion and differs microscopically from the classic embryonal by a subepithelial condensation of tumor cells. The alveolar type occurs primarily in the teenage and young adult population and is identified histologically by an alveolar pattern of loosely arranged cells with hyperchromatic nuclei. The pleomorphic type represents approximately 17% of the adult RMS and less than 5% of pediatric cases. These lesions are found to have large, pleomorphic cells with eosinophilic cytoplasm. Immunohistochemistry has provided valuable diagnostic techniques to the histologic diagnosis of these lesions with antidesmin staining in 94%; 77% positive for desmin, 78% positive for muscle specific actin, and 30% for myoglobin.77
Treatment of RMS has continued to improve since the development of the IRS and now often involves combined modality treatment to include surgery, chemotherapy, and radiation therapy. Therapy has been based on the site category, which the IRS had delineated as (1) orbit, (2) parameningeal, and (3) other head and neck. Primary treatment often includes induction chemotherapy followed by radiation therapy, although concomitant therapy may be used. Surgery is typically reserved for debulking or for tumors that can be resected entirely without functional or cosmetic deformity. Neck dissection is warranted in obvious neck involvement or in clinically enlarged adenopathy. The survival rates for each of these sites were 92%, 69%, and 81%, respectively.78–81 IRS III and IV analysis of nonorbital nonparameningeal patients does show an overall 83% five-year survival and a better prognosis in N0 patients than those with N1 disease.82 Prognosis correlates with patient age, site of disease, histologic type, size of tumor, and metastases with botryoid and spindle associated with a better prognosis than the embryonal type, which is better than alveolar and pleomorphic.83 Long-term follow-up has identified significant morbidity related to these treatments and long-term functional outcomes should be considered in addition to locoregional control and survival.84 Lymphatic metastases are found to occur in 3% to 20% of RMS, although hematogenous spread may also occur.85,86 Staging of RMS is commonly based on the IRS, which incorporates extent of disease with metastases and surgical results. It was also recommended by IRS that staging systems for this disease require ongoing analysis to confirm prognostic correlation with stage77,85 (Table 118-6).
Osteosarcoma
Osteosarcoma of the head and neck primarily involves the mandible and maxilla with the mandible having a slightly higher incidence. The tumor rarely involves the soft tissues of the neck, although isolated regional metastases have been reported in addition to several reports involving the hyoid and larynx.87,88 The treatment of these lesions has primarily included surgical resection with or without radiation therapy and chemotherapy. The incidence of cervical metastases is reported to be less than 10%, making routine neck dissection unwarranted.89 The poor prognosis of soft tissue osteosarcomas of the larynx may encourage multimodality treatment for these sites in the future.
Fibrosarcoma
The neck is the second most common site of presentation of head and neck fibrosarcoma, following the paranasal sinus region. Although it can occur at any age, it is more common in adults between 40 and 70 years of age. There also exists a subset of children diagnosed before age 2. This neoplasm originates from the fibroblast and usually arises spontaneously but is known to arise in areas of prior burn scars and radiation therapy.90,91 Histologically, they are identified by a malignant fibroblastic proliferation with variable amounts of collagen and reticulin forming a “herringbone” pattern. These lesions have a broad differential diagnosis and the well-differentiated types are commonly confused with fibromatosis and other benign processes. They typically present in the neck as a painless enlarging firm mass and have a low rate of lymphatic metastasis, thus making routine neck dissection unwarranted. There tends to be a high local recurrence rate up to 50% despite radical surgical excision with a survival of 50% to 75% and possibly higher in young children.90,92–95 Adjuvant treatment should be based on size of the tumor, tumor grade, and status of surgical margins.94 Review of the literature on these tumors should proceed with caution because of changes in histochemical diagnosis that may alter the inclusion of certain tumors previously classified as fibrosarcoma in earlier series.
Alveolar Soft Part Sarcoma
Alveolar soft part sarcoma is a rare tumor that is stated to involve the head and neck region in approximately 25% of cases, although it represents less than 1% of all sarcomas. The exact differentiation of this tumor remains elusive; however, muscular and neural derivations are hypothesized. Recent work has failed to recognize the cell of origin in ASPS, but identification of the unbalanced translocation der(17)t(X:17)(p11;p25) leading to the fusion gene ASPL-TFE3 has generated two important realizations. First, the presence of this fusion protein is causative in ASPS, and second, specific immunohistochemical stain for this fusion protein has led to improved accuracy in diagnosis.96 The common sites affected in the head and neck include the tongue and orbit with the orbit having the best prognosis. ASPS rarely involves the neck and is reported to metastasize to the neck from head and neck primary sites in less than 10% of cases making elective neck dissection unwarranted. Distant metastatic disease does occur and may not present for years or decades after the primary site was treated. Surgery remains the mainstay of treatment, although local recurrence is common. More recent reports have shown success with multimodality treatment including chemotherapy. Overall survival is approximately 65% at 5 years but drops to 50% at 10 years.97–103
Angiosarcoma
Treatment is primarily surgical using wide margins due to the multicentric nature of these tumors and local recurrence rate nearing 50%. Postoperative adjuvant radiation therapy is also generally recommended. There is limited experience using primary or adjuvant chemotherapy in these neoplasms. Metastatic disease commonly occurs in the lung and liver, whereas regional metastatic disease is common in lesions of the scalp. Elective neck dissection is recommended for clinically and radiographically evident disease and/or primary lesion involving the scalp. The 5-year survival remains low with most reporting less than 25%.104–110
Epithelioid Hemangioendothelioma
This tumor is extremely rare and is described to involve the head and neck region in only approximately 10% to 15% of the cases. These lesions are found to be derived from an epithelioid or histiocytoid type of endothelial cell.111 It does manifest a wide spectrum of biologic behavior ranging from a benign form to an extremely aggressive form of the disease, although all are vascular in nature. Aggressiveness and eventual mortality seem to be related to location of the lesion with liver lesions having a worse prognosis than other sites including the head and neck.112 There has recently been described a more “benign” variant without distant metastases.113 Another form has been described as spindle cell hemangioendothelioma but is now thought to be a benign process. This variant is referred to as spindle cell hemangioma for solitary lesions.114 Variants that are more aggressive and similar to angiosarcoma most often arise in the thyroid, submandibular, and neck soft tissues, although mucosal and skin sites have been described including the paranasal sinuses, larynx, and temporal bone.115–117 Demonstration of endothelially derived cells is important in arriving at a correct diagnosis, and immunohistochemical studies for endothelial markers such as FVIII-Rag (factor VIII related antigen) and CD 34 have been found to be positive in a majority of cases.112,118 Treatment has typically included surgical excision with possible radiation therapy. Recurrence and metastatic potential correlates with biologic aggressiveness with the more epithelioid lesions having a better prognosis, whereas the sarcomatous lesions have a higher metastatic potential and poorer prognosis.119,120
Chondrosarcoma
Although chondrosarcoma is typically found in the maxillary and mandibular region of the head and neck, presentation in the neck or origin in soft tissues can occur.121,122 Histologically, evidence of cartilage formation exists with varying degrees of differentiation and grade. These tumors are typically classified as osseous or extraosseous and may be subtyped into conventional, myxoid, and mesenchymal with mesenchymal being much more common in children and young adults. Prognosis does seem to be related to the subtype with myxoid having the worst prognosis followed by mesenchymal and conventional. There has been some controversy surrounding the histologic separation of osteoblastic chondrosarcoma from osteosarcoma. From the National Cancer Database (NCDB) report, the average age of patients with chondrosarcoma of the head and neck is 51, although more than 32% were younger than age 40. There is a slight male predominance and ethnicity reveals that non-Hispanic white constitutes more than 86% of cases. Only a small percentage of cases with regional or distant metastases at diagnosis with 5.6% and 6.7%, respectively, are found in the NCDB report.122 Treatment includes wide surgical resection, although consideration of postoperative radiation may be entertained, particularly in high-grade tumors. Survival from the NCDB report reveals a surprisingly high survival of head and neck chondrosarcomas at 87.2% five-year and 70.6% ten-year survival, with 59.5% undergoing surgery alone and 21.0% having adjuvant radiation therapy.
Leiomyosarcoma
This is a neoplasm that generally affects older adults, although it can occur at any age. It represents 6% of all sarcomas, with 3% involving the head and neck region. The oral cavity followed by the sinonasal region and subcutaneous areas are the most common sites.76 Although the scalp and face are the most common sites of occurrence of the subcutaneous lesions, there are reports involving the superficial and deep tissues of the cervical region.123,124 The neoplasm develops from smooth muscle origin and histologically has a typical appearance of fascicles arranged in a perpendicular fashion with the cigar-shaped nuclei, eosinophilic cytoplasm, and paranuclear vacuoles. Most leiomyosarcomas also expresse muscle-specific actin, smooth muscle actin, and desmin.125 These can be differentiated from fibrosarcoma by “cigar-shaped” nuclei rather than the “pointy” nuclei of fibrosarcoma. The most common presentation is a nodular dark blue or black lesion involving the dermis and epidermis, which may be tender to palpation. Those that arise in the subcutaneous tissues have a higher local recurrence, metastatic rate, and worse prognosis. Of those originating in the oral cavity, there is a high rate of local recurrence in addition to metastatic disease to the cervical nodes, lungs, and subcutaneous tissues.76 Deep neck masses should also be considered in the differential even without skin involvement. The treatment includes wide resection with negative margins. Neck dissection may be indicated due to the potential for regional and distant metastases.126,127 The prognosis varies greatly with site of origin and histologic variations, thus making accurate estimates of survival of each site difficult.
Liposarcoma
Although considered the most common soft tissue sarcoma of adults, constituting 12% to 18% of cases, involvement in the head and neck region is rare, occurring in an estimated 3% to 6% of cases.76 A relationship to previous lipoma and traumatic events has been considered, yet there is insufficient evidence to confirm the relationship to the development of liposarcoma. In a review of head and neck liposarcomas, Barnes identified the larynx and hypopharynx to be the most common sites, followed closely by the neck. Others report the neck to be more commonly involved.76,128 The prognosis appears to be dependent on site and classification with the well differentiated and myxoid having a better prognosis (75% to 100%) than the round cell and pleomorphic varieties (12% to 30%).76,128–130 The liposarcoma is considered to occur primarily in deeper soft tissue locations than the lipoma or atypical lipoma. Although cervical metastases are rare, distant metastases have been reported primarily to the lung and liver.
Atypical Lipoma
Despite its benign behavior, the atypical lipoma or pleomorphic lipoma may be misdiagnosed as liposarcoma because of the histologic similarity. These are typically more superficial and radical treatment is not necessary if wide surgical margins are obtained. Similarly, the spindle cell lipoma has also been described and behaves in a similar fashion to the atypical lipoma. Both of these processes are more common in adult males.131
Malignant Hemangiopericytoma
Hemangiopericytoma (HPC) arises from the cells of Zimmerman, which occur around capillaries and postcapillary venules. The majority of HPCs of the head and neck are found in the paranasal sinuses; however, because of their cells of origin, nearly any tissue could be involved, including the neck.132,133 The tumor primarily affects adults, although there is a subset of children from birth to 5 years of age who can be affected. The treatment is surgical because HPC has been shown to be relatively radioresistant, and the highly vascular nature of these tumors may require preoperative embolization. Adjuvant radiation therapy has been recommended for those with high-grade features and/or positive margins. Neck dissection is not necessary because lymphatic spread is rare. Varying reports of distant metastases seem to correlate HPCs with histologic pattern, mitotic figures, and proliferation indices.134–136 The 5-year survival is near 70% and distant metastases usually portend recurrence at the primary site.
Malignant Peripheral Nerve Sheath Tumor
The malignant peripheral nerve sheath tumor (MPNST) refers to a type of neurosarcoma that represents nearly 10% of all sarcomas and behaves in an aggressive fashion with a poor prognosis. The tumor appears to have varying distribution by gender and is typically a disease of adults.137,138 The tumor typically arises in the neck in up to half of the head and neck cases, although the sinonasal region, parapharyngeal space, parotid, and thyroid have been involved. These tumors are generally considered to occur in two settings: (1) spontaneous and (2) within the setting of a neurofibroma, particularly neurofibromatosis I (NF-1). Those arising in the latter typically occur at a younger age (fourth decade) and have a worse prognosis.139 It is stated that for patients diagnosed with NF-1, the risk of developing an MPNST is 2%.140 The typical presentation is of a progressive swelling with many presenting with pain in the region. Those with a history of NF-1 may describe a long history of a mass with a recent rapid enlargement. Associated neurogenic symptoms of weakness or paresthesias may be associated. Microscopically, they typically reveal atypical spindle cells similar to Schwann cells that are closely associated with a peripheral nerve. There remains significant controversy and variability in the histopathologic diagnostic criteria. The treatment includes wide resection with clear margins and postoperative radiation with margin status and tumor size correlating with survival.139,141,142 The prognosis is poor despite aggressive treatment with more than 40% developing local recurrence. The presence of lymphatic metastases is rare.138,141,143,144
Synovial Sarcoma
Synovial sarcoma (SS) comprises 6% to 10% of all soft tissue sarcomas and 3% to 10% of all head and neck sarcomas. This neoplasm has been described to arise in the periarticular regions of the body, although the sites of the head and neck are not usually in these areas. This tumor typically arises in people aged 20 to 40 with the hypopharyngeal and retropharyngeal regions the most likely sites of the head and neck. It is thought to be derived from a pluripotential mesenchymal cell with both epithelioid and spindle differentiation. Microscopically, the tumor is found to have a predominant spindle cell component with cuboidal and columnar cells surrounding glandular areas and may have calcifications in up to 30% of cases.145,146 Prognosis appears related to tumor size, mitotic indices, high grade, local recurrence, and tumor necrosis, although absence of calcifications and ploidy may also be related.145,147 The symptoms on presentation are usually related to mass effect, although a painful mass may be identified. The treatment requires wide surgical resection. Chemotherapy may also be beneficial when used preoperatively.148,149 Neck dissection is not necessary due to the absence of cervical metastases. The 5-year survival is 47% to 58% with up to 40% incidence of local recurrence.76,146,150
Malignant Giant Cell Tumor
The malignant giant cell tumor (MGCT) of the head and neck region is extremely rare and may be considered to be radiation induced after patients are treated for a benign giant cell tumor. MGCTs account for less than 10% of all GCTs. The sinonasal region and mandible are the most common sites in the head and neck.151,152 The secondary MGCT appears more common than primary MGCTs, which arise de novo without prior evidence of a benign GCT. Overall 5-year survival of secondary MGCTs was 32%.153 Metastatic lesions of GCT are usually identified in the lungs.154
Ewing’s Sarcoma
Ewing’s sarcoma represents a malignancy derived from primitive neuroectoderm and is the second most common bone tumor in children. Of 70 cases described at a single institution, only 5 (7.1%) occurred in the head and neck region.155 These are separated into osseous and extraosseous types with approximately 75% occurring in the first 2 decades. Primitive neuroectodermal tumor (PNET) is a diagnosis that has many overlapping features with extraosseous Ewing’s sarcoma and may be related.89 PNET occurs in the paraspinous regions in approximately 50% of cases. The most common head and neck sites for Ewing’s sarcoma include the mandible, maxilla, skull, and sinonasal region, although soft tissue sites have been described.156 The incidence of lymphatic spread to the cervical nodes is uncommon. Treatment involves multimodality therapy including chemotherapy, whereas surgery may be necessary for complete control of the primary site and for reconstructive considerations. Radiation may be beneficial in combination with these other modalities.
Solitary Fibrous Tumor
There exists a wide range of benign and malignant fibrous tumors with varying degrees of local, regional, and metastatic growth potential. The one that deserves mention because of its frequency in the head and neck, particularly in children, is desmoid fibromatosis. These tumors have a wide range of behavior and very low mortality rate. Treatment includes wide resection because of the high local recurrence rate of 21% to 47%.69,157,158
Lymphoma
Lymphoma deserves mention because of the common presentation in enlarged cervical lymph nodes but is presented in detail in Chapter 119.
Melanoma
Although melanoma may arise or metastasize to the neck without a known primary site, a thorough evaluation should be undertaken to identify the primary site. A review of 300 cases of melanoma by Balm159 revealed that 17 (5.7%) presented with a cervical lymph node without known primary. The treatment included surgery, and the 5-year disease-specific survival was 48% with a median of 36 months, which correlated with other patients with a stage II cutaneous melanoma.159
In a large series of head and neck melanomas, elective neck dissection and therapeutic neck dissection did not appear to improve survival over patients undergoing delayed neck dissection for regional metastases developing later than 3 months following primary treatment. There was, however, a higher incidence of distant metastatic disease in patients who did develop regional metastatic disease.160
Squamous Cell Carcinoma Arising in Branchial Cleft Cyst
Rarely, squamous cell carcinomas have been documented to arise within branchial cleft cysts of the neck. Cytopathologic diagnosis is difficult but should be considered in the differential diagnosis of a cystic neck mass. Final confirmation of the diagnosis should follow the criteria proposed in differentiating these tumors from cystic squamous cell carcinoma of cervical lymph nodes.161
Carcinoma Arising within Thyroglossal Duct Cyst
This extremely rare occurrence has been described, providing evidence for cytologic examination of thyroglossal duct cysts in suspicious cases.162–165
Badenhop RF, Jansen JC, Fagan PA, et al. The prevalence of SDHB, SDHC, and SDHD mutations in patients with head and neck paraganglioma and association of mutations with clinical features. J Med Genet. 2004;41(7):e99.
Crist W, Gehan EA, Ragab AH, et al. The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol. 1995;13(3):610-630.
DeSanto LW, Neel HB3rd. Squamous cell carcinoma. Metastasis to the neck from an unknown or undiscovered primary. Otolaryngol Clin North Am. 1985;18(3):505-513.
Ducatman BS, Scheithaur BW, Piepgras DG, et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006-2021.
Fencl P, Belohlavek O, Skopalova M, et al. Prognostic and diagnostic accuracy of [(18)F]FDG-PET/CT in 190 patients with carcinoma of unknown primary. Eur J Nucl Med Mol Imaging. 2007;34(11):1783-1792.
Gordin A, Golz A, Keidar Z, et al. The role of FDG-PET/CT imaging in head and neck malignant conditions: impact on diagnostic accuracy and patient care. Otolaryngol Head Neck Surg. 2007;137(1):130-137.
Hughes KV3rd, Olsen KD, McCaffrey TV. Parapharyngeal space neoplasms. Head Neck. 1995;17(2):124-130.
Koch BB, Karnell LH, Hoffman HT, et al. National cancer database report on chondrosarcoma of the head and neck. Head Neck. 2000;22(4):408-425.
Lee JH, Barich F, Karnell LH, et al. National Cancer Data Base report on malignant paragangliomas of the head and neck. Cancer. 2002;94(3):730-737.
Lydiatt WM, Shaha AR, Shah JP. Angiosarcoma of the head and neck. Am J Surg. 1994;168(5):451-454.
Netterville JL, Reilly KM, Robertson D, et al. Carotid body tumors: a review of 30 patients with 46 tumors. Laryngoscope. 1995;105(2):115-126.
Neumann HP, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292(8):943-951.
Pappo AS, Meza JL, Donaldson SS, et al. Treatment of localized nonorbital, nonparameningeal head and neck rhabdomyosarcoma: lessons learned from intergroup rhabdomyosarcoma studies III and IV. J Clin Oncol. 2003;21(4):638-645.
Porceddu SV, Jarmolowski E, Hicks RJ, et al. Utility of positron emission tomography for the detection of disease in residual neck nodes after (chemo)radiotherapy in head and neck cancer. Head Neck. 2005;27(3):175-181.
Raney RB, Asmar L, Vassilopoulou-Sellin R, et al. Late complications of therapy in 213 children with localized, nonorbital soft-tissue sarcoma of the head and neck: a descriptive report from the Intergroup Rhabdomyosarcoma Studies (IRS)-II and-III. IRS Group of the Children’s Cancer Group and the Pediatric Oncology Group. Med Pediatr Oncol. 1999;33(4):362-371.
Rumboldt Z, Gordon L, Gordon L, et al. Imaging in head and neck cancer. Curr Treat Options Oncol. 2006;7(1):23-34.
Wang SJ, Wang MB, Barauskas TM, Calcaterra TC. Surgical management of carotid body tumors. Otolaryngol Head Neck Surg. 2000;123(3):202-206.
Weiss SW, Enzinger FM. Malignant fibrous histiocytoma: an analysis of 200 cases. Cancer. 1978;41(6):2250-2266.
1. Curtin HD. Detection of perineural spread: fat suppression versus no fat suppression. AJNR. 2004;25(1):1-3.
2. Rumboldt Z, Gordon L, Gordon L, et al. Imaging in head and neck cancer. Curr Treat Options Oncol. 2006;7(1):23-34.
3. Gordin A, Golz A, Keidar Z, et al. The role of FDG-PET/CT imaging in head and neck malignant conditions: impact on diagnostic accuracy and patient care. Otolaryngol Head Neck Surg. 2007;137(1):130-137.
4. Porceddu SV, Jarmolowski E, Hicks RJ, et al. Utility of positron emission tomography for the detection of disease in residual neck nodes after (chemo)radiotherapy in head and neck cancer. Head Neck. 2005;27(3):175-181.
5. Yao M, Luo P, Hoffman HT, et al. Pathology and FDG PET correlation of residual lymph nodes in head and neck cancer after radiation treatment. Am J Clin Oncol. 2007;30(3):264-270.
6. Rogers JW, Greven KM, McGuirt WF, et al. Can post-RT neck dissection be omitted for patients with head-and-neck cancer who have a negative PET scan after definitive radiation therapy? Int J Radiat Oncol Biol Phys. 2004;58(3):694-697.
7. Richards PS, Peacock TE. The role of ultrasound in the detection of cervical lymph node metastases in clinically N0 squamous cell carcinoma of the head and neck. Cancer Imaging. 2007;7:167-178.
8. Kersing W. Demonstration of hormonal activity of a glomus juglare tumour by catecholamine determination. Arch Otorhinolaryngol. 1977;217(4):463-473.
9. Glenner GG, Crout JR, Roberts WC. A functional carotid-body-like tumor. Secreting levarterenol. Arch Pathol. 1962;73:230-240.
10. Mariman EC, van Beersum SE, Cremers CW, et al. Fine mapping of a putatively imprinted gene for familial non-chromaffin paragangliomas to chromosome 11q13.1: evidence for genetic heterogeneity. Hum Genet. 1995;95(1):56-62.
11. Barnes L. Paraganglioma of the larynx. A critical review of the literature. ORL J Otorhinolaryngol Relat Spec. 1991;53(4):220-234.
12. Scudder C. Tumor of the intercarotid body: a report of one case, together with all cases in the literature. Am J Med Sci. 1903;126:384-389.
13. Kohn A. Kie paraganglien. Arch Mikr Anat. 1903:62.
14. Mulligan R. Chemodectoma in the dog. Am J Pathol. 1950;26:680.
15. Heath D, Edwards C, Harris P. Post-mortem size and structure of the human carotid body. Thorax. 1970;25(2):129-140.
16. Frey CF, Karoll RP. Management of chemodectomas. Am J Surg. 1966;111(4):536-542.
17. Carney JA, Sheps SG, Go VL, et al. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N Engl J Med. 1977;296(26):1517-1518.
18. Chase W. Familial and bilateral tumors of the carotid body. J Pathol Bacteriol. 1933;36:1-12.
19. Badenhop RF, Jansen JC, Fagan PA, et al. The prevalence of SDHB, SDHC, and SDHD mutations in patients with head and neck paraganglioma and association of mutations with clinical features. J Med Genet. 2004;41(7):e99.
20. Bayley JP, van Minderhout I, Weiss MM, et al. Mutation analysis of SDHB and SDHC: novel germline mutations in sporadic head and neck paraganglioma and familial paraganglioma and/or pheochromocytoma. BMC Med Genet. 2006;7:1.
21. Dannenberg H, Dinjens WN, Abbou M, et al. Frequent germ-line succinate dehydrogenase subunit D gene mutations in patients with apparently sporadic parasympathetic paraganglioma. Clin Cancer Res. 2002;8(7):2061-2066.
22. Taschner PE, Jansen JC, Baysal BE. Nearly all hereditary paragangliomas in the Netherlands are caused by two founder mutations in the SDHD gene. Genes Chromosomes Cancer. 2001;31:274-281.
23. van der Mey AG, Maaswinkel-Mooy PD, Cornelisse CJ, et al. Genomic imprinting in hereditary glomus tumours: evidence for new genetic theory. Lancet. 1989;2(8675):1291-1294.
24. Neumann HP, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292(8):943-951.
25. Wang SJ, Wang MB, Barauskas TM, et al. Surgical management of carotid body tumors. Otolaryngol Head Neck Surg. 2000;123(3):202-206.
26. Rosenkranz L, Schell AR. Carotid body tumor as reversible cause of recurrent syncope. NY State. J Med. 1984;84(1):38-39.
27. Manolidis S, Shohet JA, Jackson CG, et al. Malignant glomus tumors. Laryngoscope. 1999;109(1):30-34.
28. Johnson JT. Parapharyngeal space masses: diagnosis and management. In: Paparella M, editor. Otolaryngology. Philadelphia: WB Saunders; 1991:2584.
29. Harrington SW, Dockerty MB. Tumors of the carotid body: clinical and pathological considerations of 20 tumors affecting 19 patients. Ann Surg. 1941;114:820-833.
30. Batsakis JG. Paragangliomas of the head and neck. 2nd ed. Tumors of the Head and Neck: Clinical and Pathologic Considerations. Baltimore: Williams and Wilkins. 1979:369-380.
31. Bestler JM, Toomey JM. Malignant carotid body tumor. Report of a case. Arch Otolaryngol. 1969;89(5):752-755.
32. Zbaren P, Lehmann W. Carotid body paraganglioma with metastases. Laryngoscope. 1985;95(4):450-454.
33. Conley JJ. The management of carotid body tumors. Surg Gynecol Obstet. 1963;117:722-732.
34. Staats EF, Brown RL, Smith RR. Carotid body tumors, benign and malignant. Laryngoscope. 1966;76(5):907-916.
35. Lee JH, Barich F, Karnell LH, et al. National Cancer Data Base report on malignant paragangliomas of the head and neck. Cancer. 2002;94(3):730-737.
36. Netterville JL, Reilly KM, Robertson D, et al. Carotid body tumors: a review of 30 patients with 46 tumors. Laryngoscope. 1995;105(2):115-126.
37. Ward PH, Liu C, Vinuela F, et al. Embolization: an adjunctive measure for removal of carotid body tumors. Laryngoscope. 1988;98(12):1287-1291.
38. Von Moll L, McEwan AJ, Shapiro B, et al. Iodine-131 MIBG scintigraphy of neuroendocrine tumors other than pheochromocytoma and neuroblastoma. J Nucl Med. 1987;28(6):979-988.
39. Kau R, Arnold W. Somatostatin receptor scintigraphy and therapy of neuroendocrine (APUD) tumors of the head and neck. Acta Otolaryngol. 1996;116(2):345-349.
40. Myssiorek D, Palestro CJ. 111Indium pentetreotide scan detection of familial paragangliomas. Laryngoscope. 1998;108(2):228-231.
41. Inoue T, Yoshinaga K, Morita K, et al. Whole-body iodine-131 metaiodobenzylguanidine imaging for detection of bone metastases in patients with paraganglioma: comparison with bone scintigraphy. Ann Nucl Med. 2007;21(5):307-310.
42. Shamblin WR, ReMine WH, Sheps SG, et al. Carotid body tumor (chemodectoma). Clinicopathologic analysis of ninety cases. Am J Surg. 1971;122(6):732-739.
43. Lack EE, Cubilla AL, Woodruff JM, et al. Paragangliomas of the head and neck region: a clinical study of 69 patients. Cancer. 1977;39(2):397-409.
44. Anand VK, Alemar GO, Sanders TS. Management of the internal carotid artery during carotid body tumor surgery. Laryngoscope. 1995;105(3 Pt 1):231-235.
45. Evenson LJ, Mendenhall WM, Parsons JT, et al. Radiotherapy in the management of chemodectomas of the carotid body and glomus vagale. Head Neck. 1998;20(7):609-613.
46. Valdagni R, Amichetti M. Radiation therapy of carotid body tumors. Am J Clin Oncol. 1990;13(1):45-48.
47. Mendenhall WM, Hinerman RW, Amdur RJ, et al. Treatment of paragangliomas with radiation therapy. Otolaryngol Clin North Am. 2001;34(5):1007-1020. vii-viii
48. Jansen JC, van den Berg R, Kuiper A, et al. Estimation of growth rate in patients with head and neck paragangliomas influences the treatment proposal. Cancer. 2000;88(12):2811-2816.
49. Farr HW. Carotid body tumors: a 40-year study. CA Cancer J Clin. 1980;30(5):260-265.
50. Walsh RM, Leen EJ, Gleeson MJ, et al. Malignant vagal paraganglioma. J Laryngol Otol. 1997;111(1):83-88.
51. Kohout MP, Hansen M, Pribaz JJ, et al. Arteriovenous malformations of the head and neck: natural history and management. Plast Reconstr Surg. 1998;102(3):643-654.
52. Das Gupta TK, Brasfield RD, Strong EW, et al. Benign solitary schwannomas (neurilemomas). Cancer. 1969;24(2):355-366.
53. Hughes KV3rd, Olsen KD, McCaffrey TV. Parapharyngeal space neoplasms. Head Neck. 1995;17(2):124-130.
54. Khafif A, Segev Y, Kaplan DM, et al. Surgical management of parapharyngeal space tumors: a 10-year review. Otolaryngol Head Neck Surg. 2005;132(3):401-406.
55. Farr HW. Soft part sarcomas of the head and neck. Semin Oncol. 1981;8(2):185-189.
56. Jaffe BF. Pediatric head and neck tumors: a study of 178 cases. Laryngoscope. 1973;83(10):1644-1651.
57. DeSanto LW, Neel HB3rd. Squamous cell carcinoma. Metastasis to the neck from an unknown or undiscovered primary. Otolaryngol Clin North Am. 1985;18(3):505-513.
58. Shah JP. Patterns of cervical lymph node metastasis from squamous carcinomas of the upper aerodigestive tract. Am J Surg. 1990;160:405-409.
59. Greven KM, Keyes JWJr, Williams DW3rd, et al. Occult primary tumors of the head and neck: lack of benefit from positron emission tomography imaging with 2-[F-18]fluoro-2-deoxy-D-glucose. Cancer. 1999;86(1):114-118.
60. Jungehulsing M, Scheidhauer K, Damm M, et al. 2[F]-fluoro-2-deoxy-D-glucose positron emission tomography is a sensitive tool for the detection of occult primary cancer (carcinoma of unknown primary syndrome) with head and neck lymph node manifestation. Otolaryngol Head Neck Surg. 2000;123(3):294-301.
61. Fencl P, Belohlavek O, Skopalova M, et al. Prognostic and diagnostic accuracy of [(18)F]FDG-PET/CT in 190 patients with carcinoma of unknown primary. Eur J Nucl Med Molecular Imaging. 2007;34(11):1783-1792.
62. Coleman SC, Smith JC, Burkey BB, et al. Long-standing lateral neck mass as the initial manifestation of well-differentiated thyroid carcinoma. Laryngoscope. 2000;110(2 Pt 1):204-209.
63. Batsakis JG, McBurney TA. Metastatic neoplasms to the head and neck. Surg Gynecol Obstet. 1971;133(4):673-677.
64. Carrau RL, Johnson JT, Myers EN. Management of tumors of the parapharyngeal space. Oncology (Williston Park. NY. 1997;11(5):633-640.
65. Olsen KD. Tumors and surgery of the parapharyngeal space. Laryngoscope. 1994;104(5 Pt 2 Suppl 63):1-28.
66. Pang KP, Goh CH, Tan HM. Parapharyngeal space tumours: an 18 year review. J Laryngol Otol. 2002;116(3):170-175.
67. Pensak ML, Gluckman JL, Shumrick KA. Parapharyngeal space tumors: an algorithm for evaluation and management. Laryngoscope. 1994;104(9):1170-1173.
68. Wanebo HJ, Koness RJ, MacFarlane JK, et al. Head and neck sarcoma: report of the Head and Neck Sarcoma Registry. Society of Head and Neck Surgeons Committee on Research. Head Neck. 1992;14(1):1-7.
69. Barnes L. Tumors and tumorlike lesions of the soft tissues. In: Barnes L, editor. Surgical Pathology of the Head and Neck. New York: Marcel Dekker, 1985.
70. Wood GS, Beckstead JH, Turner RR, et al. Malignant fibrous histiocytoma tumor cells resemble fibroblasts. Am J Surg Pathol. 1986;10(5):323-335.
71. Weiss SW, Enzinger FM. Malignant fibrous histiocytoma: an analysis of 200 cases. Cancer. 1978;41(6):2250-2266.
72. Barnes L, Kanbour A. Malignant fibrous histiocytoma of the head and neck. A report of 12 cases. Arch Otolaryngol Head Neck Surg. 1988;114(10):1149-1156.
73. Bras J, Batsakis JG, Luna MA. Malignant fibrous histiocytoma of the oral soft tissues. Oral Surg Oral Med Oral Pathol. 1987;64(1):57-67.
74. Wu X, Qi Y, Tang P. [A comparison of malignant fibrous histiocytoma of head, neck and extremities]. Chinese Med J. 2000;113(6):532-535.
75. Hicks J, Flaitz C. Rhabdomyosarcoma of the head and neck in children. Oral Oncol. 2002;38(5):450-459.
76. Barnes L. Tumors and tumorlike lesions of the soft tissues. In: Barnes L, editor. Surgical Pathology of the Head and Neck. New York: Marcel Dekker; 2001:889-1048.
77. Parham DM, Webber B, Holt H, et al. Immunohistochemical study of childhood rhabdomyosarcomas and related neoplasms. Results of an Intergroup Rhabdomyosarcoma study project. Cancer. 1991;67(12):3072-3080.
78. Crist W, Gehan EA, Ragab AH, et al. The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol. 1995;13(3):610-630.
79. Maurer HM, Beltangady M, Gehan EA, et al. The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer. 1988;61(2):209-220.
80. Maurer HM, Gehan EA, Beltangady M, et al. The Intergroup Rhabdomyosarcoma Study-II. Cancer. 1993;71(5):1904-1922.
81. Rodary C, Gehan EA, Flamant F, et al. Prognostic factors in 951 nonmetastatic rhabdomyosarcoma in children: a report from the International Rhabdomyosarcoma Workshop. Med Pediatr Oncol. 1991;19(2):89-95.
82. Pappo AS, Meza JL, Donaldson SS, et al. Treatment of localized nonorbital, nonparameningeal head and neck rhabdomyosarcoma: lessons learned from intergroup rhabdomyosarcoma studies III and IV. J Clin Oncol. 2003;21(4):638-645.
83. Newton WAJr, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification—an Intergroup Rhabdomyosarcoma Study. Cancer. 1995;76(6):1073-1085.
84. Raney RB, Asmar L, Vassilopoulou-Sellin R, et al. Late complications of therapy in 213 children with localized, nonorbital soft-tissue sarcoma of the head and neck: a descriptive report from the Intergroup Rhabdomyosarcoma Studies (IRS)-II and-III. IRS Group of the Children’s Cancer Group and the Pediatric Oncology Group. Med Pediatr Oncol. 1999;33(4):362-371.
85. Lawrence WJr, Hays DM, Moon TE. Lymphatic metastasis with childhood rhabdomyosarcoma. Cancer. 1977;39(2):556-559.
86. Shimada H, Newton WAJr, Soule EH, et al. Pathology of fatal rhabdomyosarcoma. Report from Intergroup Rhabdomyosarcoma Study (IRS-I and IRS-II). Cancer. 1987;59(3):459-465.
87. Anderson TD, Kearney JJ. Osteosarcoma of the hyoid bone. Otolaryngol Head Neck Surg. 2002;126(1):81-82.
88. Pinsolle J, LeCluse I, Demeaux H, et al. Osteosarcoma of the soft tissue of the larynx: report of a case with electron microscopic studies. Otolaryngol Head Neck Surg. 1990;102(3):276-280.
89. Barnes L. Diseases of the bones and joints. In: Barnes L, editor. Surgical Pathology of the Head and Neck. New York: Marcel Dekker; 2001:1049-1232.
90. Greager JA, Reichard K, Campana JP, et al. Fibrosarcoma of the head and neck. Am J Surg. 1994;167(4):437-439.
91. Mahmoud NA. Radiation-induced fibrosarcoma of the head and neck. J Laryngol Otol. 1980;94(2):231-242.
92. Conley J, Stout AP, Healey WV. Clinicopathologic analysis of eighty-four patients with an original diagnosis of fibrosarcoma of the head and neck. Am J Surg. 1967;114(4):564-569.
93. Frankenthaler R, Ayala AG, Hartwick RW, et al. Fibrosarcoma of the head and neck. Laryngoscope. 1990;100(8):799-802.
94. Mark RJ, Sercarz JA, Tran L, et al. Fibrosarcoma of the head and neck. The UCLA experience. Arch Otolaryngol Head Neck Surg. 1991;117(4):396-401.
95. Swain RE, Sessions DG, Ogura JH. Fibrosarcoma of the head and neck in children. Laryngoscope. 1976;86(1):113-116.
96. Folpe AL, Deyrup AT. Alveolar soft-part sarcoma: a review and update. J Clin Pathol. 2006;59(11):1127-1132.
97. Batsakis JG. Alveolar soft-part sarcoma. Ann Otol Rhinol Laryngol. 1988;97(3 Pt 1):328-329.
98. Batsakis JG, Regezi JA, Rice DH. The pathology of head and neck tumors: fibroadipose tissue and skeletal muscle, Part 8. Head Neck Surg. 1980;3(2):145-168.
99. Hunter BC, Devaney KO, Ferlito A, et al. Alveolar soft part sarcoma of the head and neck region. Ann Otol Rhinol Laryngol. 1998;107(9 Pt 1):810-814.
100. Ordonez NG. Alveolar soft part sarcoma: a review and update. Adv Anat Pathol. 1999;6(3):125-139.
101. Portera CAJr, Ho V, Patel SR, et al. Alveolar soft part sarcoma: clinical course and patterns of metastasis in 70 patients treated at a single institution. Cancer. 2001;91(3):585-591.
102. Simmons WB, Haggerty HS, Ngan B, et al. Alveolar soft part sarcoma of the head and neck. A disease of children and young adults. Int J Pediatr Otorhinolaryngol. 1989;17(2):139-153.
103. Spector RA, Travis LW, Smith J. Alveolar soft part sarcoma of the head and neck. Laryngoscope. 1979;89(8):1301-1306.
104. Cochran JHJr, Fee WEJr. Angiosarcoma of the head and neck. Otolaryngol Head Neck Surg. 1979;87(4):409-416.
105. el-Sharkawi S. Angiosarcoma of the head and neck. J Laryngol Otol. 1997;111(2):175-176.
106. Hodgkinson DJ, Soule EH, Woods JE. Cutaneous angiosarcoma of the head and neck. Cancer. 1979;44(3):1106-1113.
107. Lydiatt WM, Shaha AR, Shah JP. Angiosarcoma of the head and neck. Am J Surg. 1994;168(5):451-454.
108. Mark RJ, Tran LM, Sercarz J, et al. Angiosarcoma of the head and neck. The UCLA experience 1955 through 1990. Arch Otolaryngol Head Neck Surg. 1993;119(9):973-978.
109. Morrison WH, Byers RM, Garden AS, et al. Cutaneous angiosarcoma of the head and neck. A therapeutic dilemma. Cancer. 1995;76(2):319-327.
110. Panje WR, Moran WJ, Bostwick DG, et al. Angiosarcoma of the head and neck: review of 11 cases. Laryngoscope. 1986;96(12):1381-1384.
111. Weiss SW, Ishak KG, Dail DH, et al. Epithelioid hemangioendothelioma and related lesions. Semin Diagnost Pathol. 1986;3(4):259-287.
112. Sun ZJ, Zhang L, Zhang WF, et al. Epithelioid hemangioendothelioma of the oral cavity. Oral Dis. 2007;13(2):244-250.
113. Billings SD, Folpe AL, Weiss SW. Epithelioid sarcoma-like hemangioendothelioma. Am J Surg Pathol. 2003;27(1):48-57.
114. Perkins P, Weiss SW. Spindle cell hemangioendothelioma. An analysis of 78 cases with reassessment of its pathogenesis and biologic behavior. Am J Surg Pathol. 1996;20(10):1196-1204.
115. Goldstein WS, Bowen BC, Balkany T. Malignant hemangioendothelioma of the temporal bone masquerading as glomus tympanicum. Ann Otol Rhinol Laryngol. 1994;103(2):156-159.
116. Wesley RK, Mintz SM, Wertheimer FW. Primary malignant hemangioendothelioma of the gingiva. Report of a case and review of the literature. Oral Surg Oral Med Oral Pathol. 1975;39(1):103-112.
117. Yasuoka T, Okumura Y, Okuda T, et al. Hemangioma and malignant hemangioendothelioma of the maxillary sinus: case reports and clinical consideration. J Oral Maxillofac Surg. 1990;48(8):877-881.
118. Makhlouf HR, Ishak KG, Goodman ZD. Epithelioid hemangioendothelioma of the liver: a clinicopathologic study of 137 cases. Cancer. 1999;85(3):562-582.
119. Mentzel T, Beham A, Calonje E, et al. Epithelioid hemangioendothelioma of skin and soft tissues: clinicopathologic and immunohistochemical study of 30 cases. Am J Surg Pathol. 1997;21(4):363-374.
120. Weiss SW, Enzinger FM. Epithelioid hemangioendothelioma: a vascular tumor often mistaken for a carcinoma. Cancer. 1982;50(5):970-981.
121. Gadwal SR, Fanburg-Smith JC, Gannon FH, et al. Primary chondrosarcoma of the head and neck in pediatric patients: a clinicopathologic study of 14 cases with a review of the literature. Cancer. 2000;88(9):2181-2188.
122. Koch BB, Karnell LH, Hoffman HT, et al. National cancer database report on chondrosarcoma of the head and neck. Head Neck. 2000;22(4):408-425.
123. de Saint Aubain Somerhausen N, Fletcher CD. Leiomyosarcoma of soft tissue in children: clinicopathologic analysis of 20 cases. Am J Surgical Pathol. 1999;23(7):755-763.
124. Snowden RT, Osborn FD, Wong FS, et al. Superficial leiomyosarcoma of the head and neck: case report and review of the literature. Ear Nose Throat J. 2001;80(7):449-453.
125. Montgomery E, Goldblum JR, Fisher C. Leiomyosarcoma of the head and neck: a clinicopathological study. Histopathology. 2002;40(6):518-525.
126. Izumi K, Maeda T, Cheng J, et al. Primary leiomyosarcoma of the maxilla with regional lymph node metastasis. Report of a case and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1995;80(3):310-319.
127. Mindell RS, Calcaterra TC, Ward PH. Leiomyosarcoma of the head and neck: a review of the literature and report of two cases. Laryngoscope. 1975;85(5):904-910.
128. Golledge J, Fisher C, Rhys-Evans PH. Head and neck liposarcoma. Cancer. 1995;76(6):1051-1058.
129. McCulloch TM, Makielski KH, McNutt MA. Head and neck liposarcoma. A histopathologic reevaluation of reported cases. Arch Otolaryngol Head Neck Surg. 1992;118(10):1045-1049.
130. Otte T, Kleinsasser O. Liposarcoma of the head and neck. Arch Otorhinolaryngol. 1981;232(3):285-291.
131. Enzinger FM, Harvey DA. Spindle cell lipoma. Cancer. 1975;36(5):1852-1859.
132. DelGaudio JM, Garetz SL, Bradford CR, et al. Hemangiopericytoma of the oral cavity. Otolaryngol Head Neck Surg. 1996;114(2):339-340.
133. Walike JW, Bailey BJ. Head and neck hemangiopericytoma. Arch Otolaryngol. 1971;93(4):345-353.
134. Billings KR, Fu YS, Calcaterra TC, et al. Hemangiopericytoma of the head and neck. Am J Otolaryngol. 2000;21(4):238-243.
135. Carew JF, Singh B, Kraus DH. Hemangiopericytoma of the head and neck. Laryngoscope. 1999;109(9):1409-1411.
136. Kowalski PJ, Paulino AF. Proliferation index as a prognositic marker in hemangiopericytoma of the head and neck. Head Neck. 2001;23:492-496.
137. Ducatman BS, Scheithauer BW, Piepgras DG, et al. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57(10):2006-2021.
138. Vege DS, Chinoy RF, Ganesh B, et al. Malignant peripheral nerve sheath tumors of the head and neck: a clinicopathological study. J Surg Oncol. 1994;55(2):100-103.
139. Loree TR, North JHJr, Werness BA, et al. Malignant peripheral nerve sheath tumors of the head and neck: analysis of prognostic factors. Otolaryngol Head Neck Surg. 2000;122(5):667-672.
140. Kapadia SB. Tumors of the nervous system. In: Barnes L, editor. Surgical Pathology of the Head and Neck. New York: Marcel Dekker; 2001:787-888.
141. Bailet JW, Abemayor E, Andrews JC, et al. Malignant nerve sheath tumors of the head and neck: a combined experience from two university hospitals. Laryngoscope. 1991;101(10):1044-1049.
142. Hoffmann DF, Everts EC, Smith JD, et al. Malignant nerve sheath tumors of the head and neck. Otolaryngol Head Neck Surg. 1988;99(3):309-314.
143. Guccion JG, Enzinger FM. Malignant schwannoma associated with von Recklinghausen’s neurofibromatosis. Virchows Archiv. 1979;383(1):43-57.
144. Sordillo PP, Helson L, Hajdu SI, et al. Malignant schwannoma—clinical characteristics, survival, and response to therapy. Cancer. 1981;47(10):2503-2509.
145. Hasegawa T, Yokoyama R, Matsuno Y, et al. Prognostic significance of histologic grade and nuclear expression of beta-catenin in synovial sarcoma. Hum Pathol. 2001;32(3):257-263.
146. Roth JA, Enzinger FM, Tannenbaum M. Synovial sarcoma of the neck: a followup study of 24 cases. Cancer. 1975;35(4):1243-1253.
147. el-Naggar AK, Ayala AG, Abdul-Karim FW, et al. Synovial sarcoma. A DNA flow cytometric study. Cancer. 1990;65(10):2295-2300.
148. Kampe CE, Rosen G, Eilber F, et al. Synovial sarcoma. A study of intensive chemotherapy in 14 patients with localized disease. Cancer. 1993;72(7):2161-2169.
149. Rosen G, Forscher C, Lowenbraun S, et al. Synovial sarcoma. Uniform response of metastases to high dose ifosfamide. Cancer. 1994;73(10):2506-2511.
150. Shmookler BM, Enzinger FM, Brannon RB. Orofacial synovial sarcoma: a clinicopathologic study of 11 new cases and review of the literature. Cancer. 1982;50(2):269-276.
151. Mintz GA, Abrams AM, Carlsen GD, et al. Primary malignant giant cell tumor of the mandible. Report of a case and review of the literature. Oral Surg Oral Med Oral Pathol. 1981;51(2):164-171.
152. Potter GD, McClennan BL. Malignant giant cell tumor of the sphenoid bone and its differential diagnosis. Cancer. 1970;25(1):167-170.
153. Rock MG, Sim FH, Unni KK, et al. Secondary malignant giant-cell tumor of bone. Clinicopathological assessment of nineteen patients. J Bone Joint Surg. 1986;68(7):1073-1079.
154. Rock MG, Pritchard DJ, Unni KK. Metastases from histologically benign giant-cell tumor of bone. J Bone Joint Surg. 1984;66(2):269-274.
155. Vaccani JP, Forte V, de Jong AL, et al. Ewing’s sarcoma of the head and neck in children. Int J Pediatr Otorhinolaryngol. 1999;48(3):209-216.
156. Jones JE, McGill T. Peripheral primitive neuroectodermal tumors of the head and neck. Arch Otolaryngol Head Neck Surg. 1995;121(12):1392-1395.
157. Conley J, Healey WV, Stout AP. Fibromatosis of the head and neck. Am J Surg. 1966;112(4):609-614.
158. Dehner LP, Askin FB. Tumors of fibrous tissue origin in childhood. A clinicopathologic study of cutaneous and soft tissue neoplasms in 66 children. Cancer. 1976;38(2):888-900.
159. Balm AJ. Lymph node metastasis in the neckand parotid gland from an unknown primary melanoma. Clin Otolaryngol. 1994;19:161-165.
160. Fisher SR. Elective, therapeutic, and delayed lymph node dissection for malignant melanoma of the head and neck: analysis of 1444 patients from 1970 to 1998. Laryngoscope. 2002;112(1):99-110.
161. Singh B, Balwally AN, Sundaram K, et al. Branchial cleft cyst carcinoma: myth or reality? Ann Otol Rhinol Laryngol. 1998;107(6):519-524.
162. Deshpande A, Bobhate SK. Squamous cell carcinoma in thyroglossal duct cyst. J Laryngol Otol. 1995;109(10):1001-1004.
163. Hanna E. Squamous cell carcinoma in a thyroglossal duct cyst (TGDC): clinical presentation, diagnosis, and management. Am J Otolaryngol. 1996;17(5):353-357.
164. Kwan WB, Liu FF, Banerjee D, et al. Concurrent papillary and squamous carcinoma in a thyroglossal duct cyst: a case report. Can J Surg. 1996;39(4):328-332.
165. Ranieri E, DAndrea MR, Vecchione A. Fine needle aspiration cytology of squamous cell carcinoma arising in a thyroglossal duct cyst. A case report. Acta Cytol. 1996;40(4):747-750.