Chapter 108 Naturally Occurring Antioxidants
 Free Radicals and Nonradical Oxidants
Free Radicals and Nonradical Oxidants
Normal Metabolism Generates Reactive Oxygen Species
Cyclooxygenase and Lipoxygenase
Inflammation activates the arachidonic acid cascade, which converts arachidonate to eicosanoids that mediate inflammation and activate NADPH oxidase to increase production of ROS.1
Redox Cycling
Several xenobiotics, such as paraquat and alloxan, catalyze the formation of superoxide through cyclic reactions, promoting auto-oxidation.
Reactive Oxygen Species Generated Directly from Oxygen
In the presence of reduced iron or copper ions, oxygen can produce H2O2 and hydroxyl radicals. Oxygen can also react spontaneously with heme proteins such as myoglobin, hemoglobin, and cytochrome c to generate superoxide. Excessive iron and iron overload may cause hydroxyl radical production. Consequently, release of iron from storage sites during inflammation and injury may promote the spontaneous production of free radicals.2
The Free Radical Theory of Aging
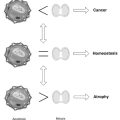
According to this seminal proposal, free radicals from normal metabolism or external sources gradually overcome antioxidant mechanisms, leading to cumulative oxidative damage to essential cellular elements: proteins, DNA, and lipids.3 Accordingly, increased prevalence of chronic degenerative diseases can be viewed in terms of irreversible cellular deficits. The consequences of oxidative stress are often subtle: increased membrane fluidity and damage to membrane receptor proteins may alter cellular regulatory mechanisms such as signal transduction, inactivation of proteins required for ATP production, or calcium homeostasis. The more or less continuous production of ROS by activated phagocytes during chronic, possibly low-level, inflammation may eventually deplete antioxidant defenses, allowing ROS to attack cells with ensuing tissue injury.
More than 100 conditions provide circumstantial evidence linking oxidative damage to disease processes, although correlation cannot distinguish cause or consequence. This list includes several forms of cancer,4,5 atherosclerosis,6,7 hypertension, diabetes mellitus,8 cataracts,9 inflammation and autoimmune disease,10,11 lung disease,12 neurologic disorders including Alzheimer’s disease and Parkinson’s diease,13,14 hepatitis,15 obesity,16 fibromyalgia and chronic fatigue syndrome,17 in addition to cell death18 and attributes of aging.19,20 Psychological stress and responses to social stressors can affect antioxidant enzymes.21
Mitochondrial Deterioration
Mitochondrial deterioration poses a test of the free radical theory. An estimated 3% of oxygen molecules passing through mitochondria is converted superoxide, due to leakage of the electron transport chain.22 Mitochondria also produce H2O2 and lipid peroxides, which can damage highly susceptible mitochondrial DNA and alter mitochondrial membrane permeability to release apoptogenic substances such as cytochrome c. Paradoxically, mice lacking the mitochondrial defensive enzyme, manganese superoxide dismutase (MnSOD), have increased pathology and increased DNA damage, yet life span is unchanged.23
Hydroxyl Radicals and Peroxynitrite
Peroxynitrite from Superoxide and Nitric Oxide
High, sustained levels of NO and superoxide are associated with tissue toxicity, cancer, and inflammatory conditions, such as arthritis, juvenile diabetes, and ulcerative colitis.24 Oxidative stress can result from increased production of superoxide, which reacts spontaneously with NO to produce peroxynitrite (ONOO−). Excessive superoxide can come from upregulated xanthine oxidase or cytochrome P450. ROS uncouple nitric oxide synthase (NOS) to produce superoxide, not NO. Thus, exposure of human endothelial cells to lysophosphatidylcholine leads to downregulation of endothelial NOS and SOD, resulting in a superoxide overload.25,26 Under inflammatory conditions, simultaneous production of superoxide and NO can increase 1000-fold, and production of peroxynitrite can increase by a million-fold.27
Reactive Oxygen Species: Function as Broad-Spectrum Antibiotics
Infection, toxic exposure, ischemia, and trauma activate phagocytic cells—macrophages, monocytes, neutrophils, and eosinophils to create ROS.28 The binding of immune complexes, bacterial endotoxins, or other inflammatory agents to cell surface receptors triggers a respiratory burst, a localized production of ROS able to oxidize viruses and bacteria. Excessive superoxide from NADPH oxidase undergoes dismutation to H2O2 via SOD. Myeloperoxidase in lysosomes then converts H2O2 and the chloride ion to hypochlorite, which spontaneously produces highly reactive chloramines from amines.
Oxidative Stress and Cell Signaling Cascades
Physiologic levels of ROS influence cell regulatory processes as diverse as apoptosis, cell growth, and chemotaxis.29,30 Many signal cascades are sensitive to redox balance and can be modulated by ROS and antioxidants. Essential to these pathways are multiple protein kinases, whose phosphorylation products are often other kinases or kinase inhibitors. Related protein phosphatases reverse those effects.
Transcriptions Factors
Nuclear Factor-κB
Proinflammatory cytokines and lipopolysaccharide (LPS) promote NF-κB activation. NF-κB upregulation may contribute to chronic oxidative stress and diseases, such as rheumatoid arthritis, hypertension, and even overweightness and obesity.31 In contrast, transcription factors can boost defenses against ROS, depending upon which signaling cascade is activated, to increase transcription of glutathione-S transferase (GSH S-transferase), metallothionein-1, and manganese-dependent SOD.
Nuclear Factor Erythroid Related Factor 2
Nuclear factor erythroid related factor acts as a master redox switch to activate cell-protective proteins and detoxifying enzymes, which include NADPH:quinone reductase, glutamate cysteine ligase, GSH S-transferase, GSH peroxidase (GPx), and thioredoxin. Release from its inactive cytoplasmic complex is redox-dependent.32
AP-1
This transcription factor controls additional processes impacting cell proliferation and apoptosis suppression, including cyclooxygenase-2 (COX-2) expression. As an example of AP-1 activation, epidermal growth factor and platelet-derived growth factor bind to their receptors, activating phosphatidylinositol kinase, followed by Rac (guanosine triphosphatase [GTPase]) activation, in turn stimulating NADPH oxidase to produce superoxide.33
Protein Kinases and Protein Phosphatases
The kinase family includes the protein kinase C (PKC) group, which includes serine/threonine kinases that are dependent on calcium and phospholipids; the mitogen-activated protein kinase (MAPK) family (extracellular signal-regulated kinases, Jun N-terminal kinase [JNK], and p38 kinases); and tyrosine protein kinases. Related phosphatases reverse their actions. Members of these broad families can participate in post-transcriptional control due to their redox-sensitive cysteinyl domains.34 As an example, vasocontraction occurs when angiotensin II binds to its receptor and activates PKC, in turn activating NADPH oxidase to produce superoxide, which destroys NO. As another example, H2O2 from SOD can oxidize catalytic cysteinyl moieties of tyrosine phosphatases, thereby preventing inactivation of tyrosine kinases, including Src, to stimulate AP-1.33
Inflammatory Eicosanoids
The arachidonate cascade features proinflammatory hydroperoxides and endoperoxide eicosanoids: prostaglandins such as PGG2, PGH2, and PGE2 (via COX); leukotrienes; and hydroxyeicosatetraenoic acid (via lipoxygenase). COX-2 can be induced by superoxide, H2O2, and inflammatory cytokines such as IL-1 and TNF-α. Stimulated lipoxygenase can trigger ROS production through sequential activation of GTPase (Rac), PKC, phosphorylation of the NOx subunit of NADPH oxidase, leading to activation of plasma membrane NADPH oxidase and ultimately superoxide synthesis.35
Calcium Homeostasis
Transcription factors may be regulated through calcium signaling mechanisms, and H2O2 can stimulate calcium release from mitochondria.33 H2O2 can increase L-type calcium channels and calcium influx by vascular smooth muscle cells linked to hypertension.36
Apoptosis and Cell Cycle Regulation
Programmed cell death is regulated by complex pathways involving transcription factors; therefore, it too relies on the cellular redox balance. Oxidative stress triggers apoptosis in several model systems, and apoptosis may be regulated by antioxidants.37,38 Conditions ranging from diabetes mellitus and heart failure to HIV infection may entail altered apoptosis.39
Regulation of Xenobiotic, Phytochemical, and Carcinogen Metabolism
ROS modulate components of the cytochrome P450 system to activate potential carcinogens. Phase II detoxification enzymes (sulfotransferases, quinone reductase, GSH S-transferase, uridine diphosphate glucuronyl transferase) can block carcinogenic effects and modulate detoxification.40
Specific Reactive Oxygen Species as Secondary Messengers for Signaling Cascades
Nitric Oxide
NO is synthesized by NOS, and physiologic NO levels regulate multiple processes; thus, NO produced by the endothelial isoform (eNOS) regulates vasodilation. In contrast, large amounts of NO are produced during inflammation by phagocytic cells by an inducible isoform (iNOS) in response to proinflammatory cytokines.41 NO is involved in S-nitrosylation of proteins by reacting with cysteinyl residues to form S-nitrothiols, which possess vasodilation activity and resist superoxide in plasma. As an example, ROS such as H2O2 can inhibit protein tyrosine phosphatase and its downstream actions via activation of NOS, with increased NO production and reversible active site cysteinyl S-nitrosylation.42 High levels of NO induce apoptosis by stimulating release of cytochrome c from mitochondria, activating the AKDK1, cyclic dependent kinase 1 (CDK1) and JNK pathway, leading to active caspases that trigger cell death.43
Hydrogen Peroxide
The primary source of H2O2 is superoxide, constitutively produced by mitochondria and NADPH oxidases. To control H2O2, catalase and peroxidases specifically degrade H2O2, whereas SOD limits superoxide to curtail H2O2 accumulation. A high steady-state level of H2O2, associated with inflammation, increases production of NF-κB and AP-1. In post-transcriptional mechanisms, H2O2 oxidizes cysteinyl moieties in redox switching enzymes. This process activates tyrosine kinases and inhibits related phosphatases,44 leading to nuclear factor translocation and increased expression of adhesion molecules, vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), to promote neutrophil binding. ROS, like H2O2, can oxidize GSH peroxidase, peroxiredoxins (Prx), and inactive transcription factors directly by the formation of protein disulfides or sulfenic acid derivatives.45
Regulatory Roles for Superoxide and Peroxynitrite
The importance of superoxide in regulatory cascade mechanisms is restricted by its short half life and limited diffusion. Studies with SOD inhibitors suggest that superoxide may mediate sympathetic excitation in rabbit brain and regulate cardiovascular responses to stress.46 Some authors propose that peroxynitrite participates in control mechanisms.47 At low concentration in vitro, peroxynitrite may modulate signaling pathways by nitrating tyrosine residues in target enzymes, such as tyrosine kinases. The inhibition of phosphotyrosine-dependent signaling to block cell adhesion molecule production suggests a role in modulating signal transduction. Furthermore, peroxynitrite can promote tyrosine phosphorylation (activation) in various cell types, for example, by activating MAPKs or by inhibiting phosphotyrosine phosphatases. This upregulation can be transient and reversible, and achievable at relatively low peroxynitrite concentrations in vitro.48
Laboratory Assessment of Oxidative Stress
Currently used methods measure a small fraction of potential oxidation products, given the wide range of ROS and possible cell targets. These markers include urinary and/or plasma products of lipid peroxidation, such as F2 isoprostanes,49 and aldehydes, such as malondialdehyde from lipid fragmentation; products of protein degradation as protein carbonyls and nitrosylated adducts; and products of purine oxidation, such as 8-oxo-2-deoxyguanosine.19,50,51 Inflammation and oxidative stress are related mechanistically. The C-reactive protein serves as a marker for systemic inflammation, as well as liver dysfunction,52 and can be reduced by antioxidant supplementation.53 Gamma-glutamyltransferase (γ-glutamyltranspeptidase [GTT]) is linked to the redox state and to GSH/ oxidized GSH (GSSH) balance, therefore it promotes GSH production. GTT may be useful in assessing oxidative stress associated with type 2 diabetes, the metabolic syndrome,54 and Alzheimer’s disease,55 in addition to coronary heart disease and stroke.56 The ferric ion reducing ability (FRAP) assay has been used to correlate serum total antioxidant capacity (TAC) with metabolic risk factors. Ingestion of foods with high TAC values reduced markers of systemic inflammation and liver dysfunction.52,57
 Antioxidants
Antioxidants
Antioxidants inhibit the oxidation of target molecules by radicals and ROS.58 There is an apparent “pecking order” among antioxidants; some are more readily oxidized than others and will be consumed rapidly unless replenished or recycled.59 Certain antioxidants are preventive inhibitors that block the initiation of free radical attack. Preventive inhibitors include defensive enzymes such as catalase, SOD, and peroxidases (GPx), as well low-molecular-weight compounds, including reduced GSH. Beta-carotene, chelating agents such as organic acids, and plant polyphenols represent preventive antioxidants when they quench singlet oxygen or sequester metal ion catalysts. Antioxidants can function as chain breakers, which convert free radicals to stable products and thus block free radical chain reactions. Vitamin E and ascorbic acid are chain-breaking antioxidants.
Laboratory Assessment of Antioxidant Activity
Studies of ROS and radical quenching by antioxidants frequently employ single time points (end-point assays). A more reliable approach evaluates the IC50, the concentration of antioxidant yielding 50% inhibition of a given oxidant or radical. It also is important to compare antioxidant activities in several assay systems. Studies showed that procyanidolic oligomers (PCOs) found in numerous foods and used in supplements (e.g., pine bark and grape seed extract) effectively quench diphenyl picrylhydrazyl radicals.60 Galloyl catechins were shown to be more effective than simple catechins in inhibition of NADPH-dependent lipid peroxidation of rat liver microsomes.61
The oxygen radical absorbance capacity (ORAC) is often used to compare antioxidant activity of foods and complex mixtures. This assay measures the decay in fluorescence (possibly from peroxyl radicals), and results are expressed as “trolox equivalents.”62 ORAC values for foods have been published by the U.S. Food and Drug Administration (FDA).63 In addition the FRAP assay is sometimes used. This method assesses ferric to ferrous reduction to evaluate TAC. The TAC content of more than 3000 foods and supplements has been published.64 To establish cell functionality, preliminary studies explored the feasibility of measuring antioxidant protection of erythrocytes and chemoattractant activity of polymorphs after oral supplementation.65 Cellular antioxidant activity (CAA), as measured by the ability to quench peroxyl radical-induced fluorescence of a fluorescein probe in cultured hepatoma cells, was developed to account for update and metabolism of antioxidants.66 It is worth noting that CAA values of flavonoids do not correlate with ORAC values.
Enzymes as Antioxidants
Superoxide Dismutase: Antioxidant Role for Manganese, Copper, and Zinc
SODs rapidly convert superoxide to H2O2. The mitochondrial enzyme (SOD2) requires manganese, whereas the cytoplasmic (SOD1) and extracellular (SOD3) enzymes require both copper and zinc. SOD2 is induced during acute inflammation,67 whereas SOD3 in vessel walls plays a major role in regulating vascular ROS.68 Knockout mice lacking SOD2 die several days after birth due to massive oxidative damage. Mice lacking SOD1 develop ROS-related pathologies with reduced lifespan. SOD may be effective in treating experimental ulcerative colitis.69 High risk, premature infants treated with prophylactic recombinant human SOD1 had reduced early pulmonary injury at 1 year.70 Preliminary evidence suggests that SOD can reduce free radical damage to skin after radiation treatment for breast cancer.71
Several lines of evidence suggest an involvement of SOD in neurologic abnormalities. Mutations in the SOD1 gene account for 20% of patients with a familial dominant form of amyotrophic lateral sclerosis, although the mechanism is unknown.72 Over-expression of SOD2 in a mouse model of Alzheimer’s disease was shown to reduce brain superoxide and prevent memory losses.73
Catalase
This iron-dependent enzyme converts H2O2 to diatomic oxygen extremely efficiently. It occurs widely in cells and is a component of peroxisomes. Animal studies with catalase, usually in conjunction with SOD, suggest protection against ischemic injury to the lung,74 intestinal ROS damage,75 and radiation.76 Examination of human atherosclerotic coronary arteries revealed that vascular antioxidant enzymes, including catalase, were selectively elevated in smooth muscle cells and macrophages in atherosclerotic lesions.77 In contrast, knockout mice lacking catalase were phenotypically normal, possibly due to effective scavenging by peroxidases.78
The Peroxidase Family
Glutathione Peroxidases: Antioxidant Role for Selenium
GPxs reduce H2O2 as well as lipid peroxides in the presence of reduced GSH. Unlike catalase and SOD, GPxs require selenocysteine. GPxs exist as several isoforms. GPx1 is a cytoplasmic isoform that prefers H2O2 as a substrate. GPx4 has a high affinity for lipid hydroperoxides, reducing membrane lipid peroxides to nontoxic fatty acid alcohols.79 Antioxidant enzymes, including extracellular GSH peroxidase, are induced by oxidative stress associated with lung diseases.80 Mice lacking GPx1 have a normal life span; however, they develop early cataracts. In contrast, GPx4 knockout mice do not survive early embryonic development.81 Curiously, reduced levels of GPx4 may increase the mouse life span.82
Peroxiredoxin
This group of six thiol peroxidases reduces H2O2, lipid peroxide, and peroxynitrite via an active site cysteinyl, –SH. The PrxII isoform is one of the most abundant proteins in erythrocytes. Prxs control intracellular levels of H2O2 to regulate cell signaling in various cell types. Mice lacking PrxI or PrxII develop severe hemolytic anemia and are prone to cancer due to elevated H2O2. Prx1 knockout mice exhibit atherosclerotic lesions due to endothelial cell dysfunction.83 NO may protect macrophages against oxidative and nitrosative stress by inducing Prx.84
Storage and Transport Proteins
Ferritin, Transferrin, and Ceruloplasmin
Free iron and copper ions catalyze the conversion of H2O2 to hydroxyl radicals; therefore, proteins that bind these ions help protect tissues against ROS. Iron stored in ferritin does not participate in generation of free radicals. Under normal circumstances, little unbound iron is present in cells. However, with chronic inflammation, unbound iron may be released from ferritin, posing a potential hazard. Iron storage disease is linked to oxidative damage. Transferrin (which has a high affinity for iron) and ceruloplasmin (which binds copper) can be considered part of the antioxidant defenses.85
Metallothionein
This cysteine-rich protein binds many metals. It is induced by cytokines, toxic metals, and oxidative stress. Mitochondrial-specific ROS generators can increase the production of metallothionein in the liver by 3.7- to 11.8-fold in mice, far more than SOD or GSH peroxidase.86 Similarly acute ethanol-induced hepatotoxicity and associated oxidative stress were curtailed in mice that had been genetically manipulated to overexpress metallothionein, compared with wild-type mice.87
Redox Regulation: The Glutathione System
Glutathione
This sulfhydryl-reducing agent is a tripeptide containing cysteine that occurs in millimolar concentrations in most cells, where it acts as a detoxifying agent, assists amino acid transport, and quenches free radicals, in addition to regulating the internal redox environment of cells. Together with ascorbate, GSH participates in the regeneration of vitamin E, which emphasizes the cooperation of antioxidants. Reduced GSH reacts directly with singlet oxygen, hydroxyl radicals, and superoxide radicals to form oxidized glutathione (GSSG). With aging, there is a general decline in GSH levels, and low GSH levels are associated with a variety of chronic conditions, such as diabetes, age-related macular degeneration, gastrointestinal disorders, and neurodegenerative diseases.89
GSH reductase reduces GSSG with NADPH, linking thiol regulation to a robust glucose metabolism. GSH reductase helps maintain a high GSH/GSSG ratio. Oxidative stress reduces this ratio, activates transcription factors, and increases production of IL-1 and TNF.90 NADPH and GSH are cofactors for other reductases that help regenerate tocopherol and ascorbate, demonstrating the principle that overall metabolic balance is a prerequisite for adequate antioxidant defense.
Redox Regulation: The Thioredoxin System: Antioxidant Role for Selenium
Thioredoxin (TRX) is a small sulfhydryl protein that acts as a reducing agent for Prxs and ribonucleotide reductase, and TRX also promotes the activation of several transcription factors.91 TRX reductase is a selenocysteine-containing enzyme that specifically catalyzes the reduction of TRX. Together, TRX and TRX reductase represent a widespread endogenous redox-regulating system with multiple roles in intracellular signaling and resistance to oxidative stress. Thus, thioredoxin activates apoptosis signal-regulating kinase-1. It reduces redox factor-1 (REF-1), which then reduces oxidized (inactive) transcription factors such as NF-κB and Nref-2. Apurinic/apyrimidinic endonuclease 1 (APE-1), a dual functional DNA repair enzyme that can also reduce transcription factors (such as AP-1 and p53) is regenerated by thioredoxin, linking DNA repair to cellular redox balance.92
Antioxidant Enzymes in Response to Oxidative Stress
Antioxidant enzyme levels tend to decrease with age-related conditions.93,94 In contrast, several antioxidant enzymes are often induced during oxidative stress. In human atherosclerotic coronary arteries, catalase, GSH peroxidase, and SOD were upregulated in atherosclerotic lesions, possibly as the result of oxidative stress.77 These enzymes are part of a battery of proteins regulated by defensive genes. Nuclear transcription factors such as Nfr2 can be regarded as a cell-sensing system for oxidative and electrophilic stress, which is responsive within 15 minutes after exposure to a stressor.95 Transactivation of AREs is regulated by redox balance, in which nuclear factor Nrf2 migrates to the nucleus in response to ROS, where it activates genes responsible for a battery of antioxidant enzymes, including Prx-6, GPx, GSH transferase, TRX, as well as phase II detoxifying enzymes.32
Micronutrients as Antioxidants
Vitamin C
Ascorbic acid can react with a wide range of ROS, including superoxide, singlet oxygen, hypochlorite, and sulfur radicals.96 Ascorbic acid is an efficient chain-breaking antioxidant in human plasma,97 where it protects lipids and membranes by scavenging peroxyl and hydroxyl radicals.98 The recommended dietary allowance (90 mg for men >19 years old; 75 mg for women >19 years old) is probably an underestimate. Healthy young women may require at least 400 mg/day of ascorbic acid according to tissue saturation studies.99 Vitamin C can reduce heavy metal toxicity.100 Dehydroascorbate is reduced back to ascorbate via GSH and NADPH. In animal models, high levels of ascorbate compensated for low GSH production and vice versa. A combination of ascorbate with vitamin E is possibly more effective than ascorbate alone for older adults.101 Ascorbic acid functions with GSH and lipoic acid to regenerate α-tocopherol.
Cardiovascular Disease
Observational studies suggest that ascorbic acid has, at best, modest effects on the risk of coronary heart disease. Although a meta-analysis found no benefit with vitamin C supplementation on survival and cardiovascular disease (CVD) risk,102 others reported benefits with high supplemental vitamin C.103 The Nurses’ Health Study suggested that vitamin C intake of more than 359 mg/day (diet plus supplements) reduced the risk of CVD.104 For diabetic postmenopausal women, high vitamin C supplementation correlated with an increased risk of CVD mortality.105 The Physicians’ II study found that vitamin C supplementation at 500 mg/day was not cardioprotective for middle-aged men.106
Hypertension
In young healthy women, a higher level of plasma vitamin C was associated with decreased blood pressure.107 A randomized controlled study of hypertensives supplemented with ascorbic acid 500 mg/day found a significant reduction of blood pressure only during the first month of treatment, suggesting a short-term benefit.108
Cancer
High ascorbic acid intake has been linked to a reduced risk of cancers of the oral cavity and esophagus,109 ovaries,110 stomach,111 and colon. However, pooled analysis of eight prospective studies found vitamin C intake unrelated to lung cancer.112 Treatment of male physicians aged equal to or more than 50 years with 500 mg ascorbic acid daily for 8 years found no reduced risk of prostate cancer or total cancer.113 Several studies suggested that extracellular ascorbate at millimolar concentrations (achieved via intravenous or intraperitoneal administration) acted as an antitumor agent in animals and humans via a pro-oxidant mechanism. A plausible mechanism involves ascorbate reduction of transition metals (cupric to cuprous, ferric to ferrous), which react with oxygen, yielding superoxide and H2O2, to which tumor cells are susceptible.114
Neurodegeneration
The Rotterdam Study suggested a high intake of ascorbic acid and vitamin E was associated with a reduced risk of Alzheimer’s disease, especially among cigarette smokers.115 Although ascorbate in plasma or cerebrospinal fluid of 32 patients with Alzheimer’s disease did not correlate with cognitive decline over 1 year, the ratio of cerebrospinal fluid ascorbate/plasma ascorbate increased, which was possibly related to an impaired ability of the brain to concentrate neuroprotective nutrients.116
Cataract
Although some investigations reported the risk of cataracts and retinal damage with lowered vitamin C status, a prospective study employing 500 mg of vitamin C, 400 IU of vitamin E, and 15 mg of β-carotene found no effect on the development or progression of cataracts.117
Diabetes
A 12-year prospective study of nondiabetic participants in the European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk Study noted those with the highest 20% of plasma vitamin C had a 62% lower risk of developing type 2 diabetes compared with those in the lowest 20%.118
Vitamin C: A Cell Response Modifier?
Vitamin C was able to increase the efficiency of generating mouse and human pluripotent stem cells from somatic cells, in part by attenuating cell senescence.119 In other research, expression of five functional protein groups related to signaling, apoptosis, and activated transcription, among others, was detected in vitamin C–treated T cells.120 Generally, vitamin C appears to function primarily as an antioxidant in modulating redox balance in a variety of situations, such as attenuating cytochrome P450 detoxification or plasma C-reactive protein levels.
Vitamin E (Tocopherols)
Vitamin E refers to four tocopherols (α, β, γ, δ) and tocotrienols (α, β, γ, δ) that possess an unsaturated isoprenoid side chain. Vitamin E represents a primary chain-breaking antioxidant of lipids, lipoproteins, and membranes, where it acts as a peroxyl radical scavenger, creating a tocopheryl radical. This radical will decompose unless converted back to tocopherol by ascorbic acid, GSH, and coenzyme Q10 (CoQ10), illustrating that antioxidant defenses complement one another.97,121 Vitamin E can exacerbate hypertension in susceptible people. High levels may antagonize other fat-soluble vitamins, thus decreasing bone mineralization. This vitamin may be contraindicated in patients receiving anticoagulants or in those with a vitamin K deficiency.
Immunity
Supplementing healthy elderly people (consuming an otherwise typical diet) with 60 to 800 mg vitamin E improved several aspects of cell-mediated immunity within 6 to 12 months.122 In other research, ingestion of 268 mg/day of natural vitamin E for 8 months significantly reduced serum immunoglobulin-E, with apparent normalization of most lesions in patients with atopic dermatitis.123
Cardiovascular Disease
Hypothetically, the oxidation of low-density lipoprotein (LDL) and other lipoproteins can initiate atherosclerosis. Increasing dietary vitamin E intake to more than 20 times the usual intake in a Western-type diet was suggested to reduce the risk of heart disease and its manifestations, such as myocardial infarction, among low-risk populations.124 However, randomized, placebo-controlled clinical trials with α-tocopherol (200 to 800 IU/day) in Western populations have been mainly negative.125
Only 5 of 12 clinical studies on vitamin antioxidants and CVD reported reduced cardiovascular death and nonfatal myocardial infarction. These include the Cambridge Heart and Antioxidant Study (CHAOS), the Womens’ Health Study (reduced risk of sudden death from CVD), and the Secondary Prevention with Antioxidants of CVD in End Stage Renal Disease (SPACE) study of hemodialysis patients, who were at high risk of oxidative stress. Inconsistent results with vitamin E may reflect compounding variables including the heterogeneity of risk factors for coronary heart disease, treatment for late course of disease versus treatment for early-onset oxidative stress,126 and genotypic differences among subgroups, for example, the haptoglobin 2-2 genotype in type 2 diabetic patients.127 More generally, it is unrealistic to expect that dosing with one or two antioxidants can substitute for multiple synergistic factors that support interlocking regulatory pathways in reducing risks of chronic degenerative disease.
Neurodegenerative Disease
A high intake of vitamin E and vitamin C was found to be associated with a reduced risk of Alzheimer’s disease according to the Rotterdam study.115 In a controlled trial of patients with moderately severe Alzheimer’s disease, α-tocopherol administered at a daily level of 2000 IU delayed the onset of severe dementia or death in comparison with controls.128 However, a controlled clinical study of patients with mild cognitive impairment found that treatment with 2000 IU vitamin E failed to slow mental decline.129
Cancer
Effects of antioxidant supplementation often depend on the subject’s nutritional status. Clinical studies with vitamin E illustrate this principle. The Shanghai Breast Cancer Study observed a 20% reduction in breast cancer risk with vitamin E supplementation among women with low dietary intake.130 The Linxian General Population Nutrition Intervention Trial, which included adults deficient in several micronutrients, examined the effects of 50 mcg/day of selenium, 30 mg/day of vitamin E, and 15 mg/day of β-carotene. Supplementation led to decreased all-cause mortality, including overall cancer risk.131 In contrast, U.S. studies of vitamin E on cancer rates reported negative results for breast cancer,132 endometrial cancer,133 lung cancer (pooled analysis of eight studies),134 or cancer incidence and cancer mortality.135 Although earlier studies suggested an inverse association between vitamin E intake and risk of prostate cancer, the Physician’s Health Study II of men aged fifty years or older found no reduction in risk with vitamin E supplementation at 400 IU after eight years.113 Supplementation of men equal to or greater than fifty-five years with vitamin E 400 IU per day for up to twelve years increased the risk of prostate cancer by 17%. Men who took vitamin E together with selenomethionine (200 mcg per day) had no higher risk than those taking a placebo.136 Compounding variables in such studies included status of vitamin D, ω-6 fatty acids, and γ-tocopherol, testosterone status in addition to polymorphisms for α-tocopherol–associated protein.137
Diabetes
Whether α-tocopherol can benefit diabetes has not been established through well-controlled clinical studies. Treatment of elderly patients with impaired glucose regulation with α-tocopherol 1000 IU and vitamin C 1000 mg/day reduced markers of oxidative stress and inflammation while improving insulin sensitivity.138
Gamma-Tocopherol
Although the typical U.S. diet supplies twice as much γ-tocopherol as the α form or tocotrienols, α-tocopherol predominates in the body, due to selection by liver tocopherol-transfer protein. Gamma-tocopherol is also rapidly degraded. It selectively blocks reactive nitrogen species and complements antioxidant actions of α-tocopherol.139 Gamma-tocopherol can protect pancreatic β-cells against NO inhibition in vitro.140 Mice supplementation with a mixture of α- and γ-tocopherol (not α-tocopherol alone) reduced age-related transcriptional changes in the brain.141 Clinical findings using γ-tocopherol are limited. Patients with metabolic syndrome treated with γ-tocopherol 800 mg and α-tocopherol 800 mg/day for 6 weeks had significantly decreased markers of inflammation and oxidative stress (high-sensitivity C-reactive protein and nitrotyrosine) compared with placebo.142 The EPIC Study found no correlation between plasma levels of either α- or γ-tocopherol and risk of prostate cancer.143
Tocotrienols
Tissue distributions of tocotrienols and tocopherols differ, which is perhaps indicative of selective mechanisms in various tissues.144 Tocotrienols can induce apopotosis in prostate cancer and breast cancer cells.145 Tocotrienols can suppress inflammatory markers, such as IL-6, TNF-α, and NO, by stimulated macrophages.146 Clinical studies are in their infancy. Hypercholesterolemic subjects treated with mixtures of tocotrienols daily for 28 days did not have improved serum lipid or glucose concentrations or lipid peroxidation (urinary 8-iso-prostaglandin F2a).147 In contrast, treatment of healthy adults with tocotrienols 160 mg/day for 6 months according to a randomized, double-blind protocol resulted in reduced DNA damage.148
Vitamin E: A Specific Cellular Response Modifier
Several studies suggest that vitamin E administration can increase expression of genes related to Th1/Th2 ratios in older mice.149 Alpha-tocopherol can block upregulation of NF-κB, PKC, and p38 MAPK pathways to prevent inflammatory cytokine production in stimulated human monocytes and in human dendritic cells.150 Variation in cytokine responses to vitamin E in elderly populations seems to reflect polymorphisms in cytokine genes.151 Nonetheless, such observations reflect the altered redox state of cells and tissues. Because other antioxidants yield similar effects as α-tocopherol, some authors contend that vitamin E protects membrane domains and polyunsaturated fatty acid pathways from ROS as its underlying mechanism of action, rather than involvement in specific signaling actions.152
Carotenoids
These plant pigments are conveniently divided into carotenes and xanthophylls (oxygenated carotenes). The best-known carotenoid, β-carotene, represents only 25% to 33% of plasma carotenoids. In general, carotenoids are versatile antioxidants. Beta-carotene is especially effective at low oxygen tension, as found in tissues.153 Increased carotenoid levels have been associated with decreased LDL oxidation.154 They can quench ROS produced during inflammation and modulate levels of proinflammatory prostaglandins and leukotrienes.155,156
Lung Cancer
Although retrospective studies suggested an inverse correlation, analysis of pooled results from six prospective cohort studies found no relationship between dietary β-carotene and lung cancer risk reduction.157 A recent systematic review of prospective studies concluded that dietary intake of total carotenoids did not significantly reduce the risk of lung cancer.158 Supplementation was studied in three large randomized controlled trials (RCTs). The two studies (Alpha-Tocopherol, beta Carotene Cancer Prevention Trial and the beta Carotene and Retinol Efficacy Trial) reported an increased risk of lung cancer in high-risk groups supplemented with β-carotene. In contrast, the Physicians’ Health Study (only 11% smokers) found no effect with β-carotene supplements.
Prostate Cancer
A meta-analysis of 11 case–control studies and 10 prospective studies found that the highest intake of lycopene or tomatoes accounted for a 11% to 19% reduction in prostate cancer risk.159 Dietary lycopene intake was not related to prostate cancer risk in a more recent large prospective study.143
Breast Cancer
The inverse relationship between consumption of carotenoid-rich fruits and vegetables for the risk of breast cancer was strongest among premenopausal women.160 For women who were smokers and did not use supplements, dietary α-carotene and β-carotene intake was inversely associated with the risk of breast cancer.161 In contrast, the Women’s Health Study found no link between lycopene intake and reduced risk of breast cancer.
Cardiovascular Disease
Although several prospective studies found that higher carotenoid-rich foods correlated with reduced risk of CVD, others did not.103 Four RCTs found no effect of oral β-carotene, ranging from 20 to 50 mg/day, in preventing CVD.
Adult Macular Degeneration
The xanthophylls lutein and zeaxanthin are the only carotenoids in the retina and macula, where they filter blue light. Cross-section and retrospective case–control studies suggested that increased consumption of carotenoids, especially lutein and zeaxanthin, correlated with a lower risk of advanced, age-related macular degeneration.162 Individuals with intermediate risk of adult macular degeneration (AMD) or with advanced AMD in one eye should consider using a combination of vitamin C, vitamin E, β-carotene, and zinc, as recommended by the Age-Related Eye Disease Study (AREDS). A large scale clinical trial of lutein, zeaxanthin, and ω-3 fatty acids is in progress (AREDS-2).163
Carotenoids: Cell Response Modifiers?
Interpretation of effects of β-carotene and other provitamin A carotenoids is complicated by their potential formation of retinoids, which are well-known regulatory species. Although they are effective antioxidants, non-provitamin A carotenoids (lycopene, lutein, canthaxanthin, and astaxanthin) do not present this complication. Much of lycopene’s activity relates to its antioxidant actions; however, its metabolites could provide regulatory effects.164 The non-provitamin A carotenoids may affect cancer cells by increased apoptosis, decreased cell cycle progression, or decreased production of cytokines. Alternatively, lycopene’s inhibition of cancer cell proliferation may involve increased gap junction-mediated intercellular communication.165
Coenzyme Q10 (Ubiquinone)
Ubiquinones contain side chains with isoprene units, and the predominant form in humans is ubiquinone 10 (CoQ10). It functions as an essential electron carrier in mitochondrial ATP production. As the only lipophilic antioxidant synthesized in the body, CoQ10 also stabilizes membranes and functions as an important antioxidant that can recycle α-tocopherol.166 Ubiquinol, the reduced form of CoQ10, protects LDL against lipid peroxidation.167 CoQ10 synthesis requires vitamins B2, B6, B12, and folate, and synthesis may not be optimal in people with a low intake of these important vitamins.
Immunity
CoQ10 may enhance the immune system. Supplementation of healthy volunteers with CoQ10 3 mg/kg daily for 12 weeks was found to increase white cell levels and decrease lymphocyte DNA damage.168
Cardiovascular Disease
In a mouse model for atherosclerosis, large doses of CoQ10 reduced plaque formation.169 In a RCT of patients at risk for CVD, 6 month treatment with a combination of vitamins C and E, CoQ10, and selenium increased arterial elasticity, lowered glycosylated hemoglobin, and increased high-density lipoprotein cholesterol compared with controls.170 CoQ10 has been used with conventional therapy in treatment of congestive heart failure. Studies employing doses ranging 100 to 200 mg/day for up to 3 months reported minor improvements.171 Drugs such as β-blockers and 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase inhibitors lower serum levels of CoQ10.172 However, the necessity of CoQ10 supplementation in these situations remains uncertain. A meta-analysis of 12 clinical trials of various protocols suggested a reduction in elevated systolic and diastolic blood pressure among hypertensive patients supplemented with CoQ10.173 Large, well-controlled trials of CoQ10 are needed. Results of a multinational RCT of patients with cardiac failure (New York Heart Association Classes III & IV) receiving standard therapy and 300 mg of CoQ10 daily can be expected in the near future.174
Thiols as Antioxidants
Alpha-Lipoic Acid
Diabetes
Meta-analysis of four RCTs with intravenous ALA indicated significant improvement in diabetic neuropathy.175 Whether oral ALA supplementation offers benefits is less clear. The highest tissue level likely attainable from oral doses is less than 10% of other intracellular antioxidants such as GSH.176 Using a rat model of diabetes, investigators showed that month long ALA intraperitoneal treatment worsened energy balance and increased fat accumulation.177
Aging
Animal studies suggested that high-dose ALA and/or acetyl-L-carnitine can improve brain function,178 reduce brain lipid peroxidation, and increase brain antioxidant enzyme activities in the brains of aged rats,179 especially in the hippocampus.180 Aged mice supplemented orally with ALA had increased glucose tolerance, energy expenditure, and skeletal muscle mitochondria biogenesis, but also had increased loss of lean body mass.181 It is not clear that these results can be extrapolated clinically.
Cardiovascular Disease
Feeding high-dose ALA also reduced myocardial oxidant production to levels observed in healthy young rats.182 ALA supplementation was used in mouse models of ischemia reperfusion and atherosclerosis, where it slowed the formation of atherosclerotic lesions.183 In a rat model of diabetes, intraperitoneal ALA reduced cardiac apoptosis, caspase expression, and enhanced MnSOD activity and GSH levels.184
Obesity
Preliminary data are suggestive. A study of 1127 of obese and pre-obese subjects supplemented for 4 months with ALA 800 mg/day reported significant reduction in weight, blood pressure, body mass index, and abdominal circumference.185
Neurologic Disease
Clinical results are preliminary. Patients with varying stages of Alzheimer’s disease administered 600 mg/day of ALA for 48 months showed slowed disease progression in patients with mild, not moderate, dementia.186
Alpha-Lipoic Acid: A Cell Response Modifier?
ALA can induce several phase 2 antioxidant and thiol-protective enzymes, including enzymes needed for GSH synthesis. Old rats treated with ALA intraperitoneally were found to have increased γ-glutamylcysteine ligase (GGL) activity, reflecting induced Nrf2 binding to ARE and increased transcriptional levels of GGL subunits, thus reversing the age-related losses.187 ALA activated protein kinase B (Akt) to increase survival of cultured neuronal cells exposed to oxidative stress.188
N-Acetylcysteine and Glutathione
GSH synthesis is regulated in part by cysteine availability. As a supplement, N-acetylcysteine is an effective precursor for GSH. It is deacetylated in the gut to free cysteine, and oral supplementation can efficiently raise intracellular GSH189 in model systems and in healthy individuals.190 N-acetylcysteine would be expected to play a supportive role to improve GSH status and maintain cell redox balance, counter ROS, and attenuate drug toxicity as a conjugating agent in drug metabolism.191 Orally administered GSH may increase intestinal GSH levels via a GSH transport system.192
Oxidative Stress
Supplementation of healthy, trained men with N-acetylcysteine 1800 mg/day for 3 days before exhaustive resistance exercise significantly increased plasma thiols and TAC and reduced lipid peroxidation and protein oxidation.193 A RCT of hypertensive patients with type 2 diabetes reported that 6 month treatment with N-acetylcysteine and arginine significantly reduced systolic blood pressure and lowered levels of oxidized LDL, C-reactive protein, VCAM, and protein oxidation (plasma nitrotyrosine), suggesting improved endothelial function along with reduced oxidative stress.194
Minerals and Oxidative Stress
Magnesium
As a cofactor for nearly all ATP-requiring reactions, magnesium is integral to intermediary metabolism. Low magnesium status is linked to inflammation and activation of the systemic stress response. Subclinical magnesium deficiency is common in the United States, and magnesium deficiency is correlated with chronic diseases associated with aging.195
In rodents, low magnesium status caused release of substance P, leading to increased circulating inflammatory cells, cytokines, and ROS, with depletion of antioxidants associated with cardiomyopathy.196 Furthermore, magnesium deficiency promoted senescence in primary human fibroblasts.197 Experimental magnesium deficiency created systemic inflammation with leukocyte and macrophage activation, inflammatory cytokine release, formation of acute phase proteins, and oxidative stress. Possible mechanisms included opening calcium channels and activation of NF-κB.198 High fructose intake, for example, from excessive high fructose corn syrup intake, coupled with low magnesium status, could set the stage for oxidative stress, insulin resistance, and metabolic syndrome in susceptible individuals.199
Zinc
Beyond its role in SOD, zinc is required by many transcription factors, including tumor suppression protein p53. “Zinc fingers”—domains that bind multiple zinc atoms—stabilize these proteins. In addition to stabilizing membranes, zinc is involved in apoptosis, hormone release, and nerve transmission.200 Subclinical zinc deficiency manifests as inflammation and oxidative stress, and it often accompanies aging.201 That zinc deficiency diminishes immune function, especially Th1 activity, is well established. Zinc is essential for IL-2 mediated T-cell activation. The mechanism seems to rely on activation of NF-κB, resulting in increased expression of TNF-α, IL-1B, and IL-8 cytokines.202 Although a RCT of subjects over 65 years old found that supplementation with zinc 25 mg/day for 3 months increased levels of cytotoxic T cells and CD4 T cells, other clinical studies reported negative results.203,204
Metabolites as Secondary Antioxidants
L-Carnitine
This metabolite is synthesized mainly by the liver and kidney, then transported to tissues using fatty acids and carbohydrate as fuel. Fatty acid transport into mitochondria relies on a carnitine shuttle for β-oxidation. A decline in carnitine correlates with the age-related decline in mitochondrial energy production and increased ROS.205 After demonstrations that supplementation with acetyl-L-carnitine reversed age-related decline in tissue carnitine level in rats, later experiments showed that animals supplemented with high doses of ALA and acetyl-L-carnitine for 1 month exhibited improved substrate binding of brain carnitine acetyltransferase,178,206 improved memory,207 and decreased DNA damage.208 Whether such improvements with high doses of acetyl-L-carnitine translate into long-term benefits for patients remains to be investigated.
Uric Acid (Urate)
Uric acid is a waste product of a purine metabolism that occurs in high levels in plasma. Urate is a broad-spectrum antioxidant capable of scavenging free radicals, and it can chelate transition metals.209 Uric acid is responsible for 21% to 34% of the total plasma antioxidant activity, in which it appears to protect α-tocopherol from peroxyl radicals.210 Also, assays of TAC indicated that 49% of the TAC of human plasma is due to uric acid.211 Although a ubiquitous antioxidant, elevated urate is not necessarily beneficial.
Does a Bilirubin–Biliverdin Cycle Protect Against Reactive Oxygen Species?
Bilirubin bound to albumin can act as an antioxidant in vitro, although the in vivo mechanism is unclear. Earlier cell culture studies suggested a protective role of bilirubin–biliverdin cycling via heme oxygenase and biliverdin reductase. However, more recent data indicated that bilirubin is degraded by ROS, rather than being converted to biliverdin and recycled.212
Melatonin: A Hormone with Antioxidant Potential
In addition to helping set the circadian rhythm and sleep patterns, this hormone also acts as an antioxidant in vitro and in vivo.213 In senescence-accelerated mice, long-term administration of physiologic levels of melatonin corrected hepatic mitochondrial dysfunction, suggesting that melatonin may reduce oxidative damage associated with aging.214 Melatonin can act by inhibiting the expression of eNOS in laboratory animals.215 It can also upregulate SOD and catalase in the brain and liver of rats exposed to carcinogens.216 In an experimental model of diabetes, melatonin was shown to reduce lipid peroxidation and upregulate GSH reductase, GPx, and γ-glutamylcysteine synthetase.217
Non-Nutritive Antioxidants
Flavonoids
Flavonoids (bioflavonoids) represent one of the largest classes of polyphenols, and more than 4000 different flavonoids have been identified. Their antioxidant properties, physiologic activities, and bioavailability vary according to major subclasses.218
Dietary Patterns
For typical Western diets, the estimated daily consumption of total flavonoids is in the range of 100 to 200 mg,219 although intake reflects cultural and even regional preferences. As examples, simple phenolic acids (caffeic acid, ferulic acid, gallic acid, and coumaric acids) accounted for 75% of total phenolic intake among Finnish adults, whereas flavonoids accounted for 24% of the total.220 In an adult Spanish population, the mean intake of total flavonoids was shown to be 313 mg/day, with proanthocyanidins accounting for 60% of the total.221
Flavonoid Bioavailability/Biotransformation
The uptake of individual flavonoids is generally quite limited. Most flavonoids have very low water solubility. Phosphatidylcholine complexes of poorly soluble flavonoids may improve uptake.222 Absorbed aglycone flavonoids are rapidly conjugated (sulfates, glucuronides) or methyated by intestinal and liver systems, then excreted. Such flavonoid derivatives may have different biological effects than parent compounds. Additionally, gut bacteria can degrade polyphenols by ring session, and absorbed monophenols may act differently than parent compounds. Enterohepatic and enteric recycling of flavonoid metabolites is another complication.223 Analysis of plasma and urine of healthy volunteers 2 hours after ingesting 200 mg quercetin or green flavonoids failed to detect increases in any of the 71 aromatic metabolites observable at 5 hours after ingestion, suggesting that flavonoid metabolites are not responsible for the rapid responses to ingested flavonoid-rich foods or beverages.224
Are Flavonoids Important Antioxidants In Vivo?
In vitro, various flavonoids scavenge a variety of radicals225,226 and block LDL oxidation.227 As chelators, flavonoids can inhibit spontaneous generation of hydroxyl radicals228 catalyzed by copper and iron ions. Although data are conflicting, it appears that flavonoids and their derivatives probably make small contributions as direct antioxidants in vivo.229 The observed increased antioxidant capacity in plasma after ingestion of fruits, fruit juice, and flavonoid-rich foods may be due to increased production of the antioxidant uric acid, rather than due to flavonoids.230,231 Peak plasma concentrations of flavonoids and their metabolites after acute ingestion are often less than 1 µmol/L, several orders of magnitude lower than plasma or intracellular concentrations of ascorbic acid, uric acid, and GSH.
Flavonoids in Cancer Prevention
Evidence for a relationship between flavonoids and cancer incidence is conflicting. Among healthy postmenopausal women, there was an inverse association between the incidence of lung cancer intake of flavanones and proanthocyanidins for current or past smokers232; the highest intake of quercetin had a lower incidence of lung cancer, and those with the highest intake of myricetin had a reduced risk of prostate cancer. In contrast, among elderly Dutchmen, dietary catechins mainly from tea were not associated with a reduced risk of lung cancer or epithelial cancers.233 In earlier studies flavonoid consumption was not associated with reduced cancer risk in this population234 although it was inversely related to mortality due to CHD.235
Soy
Soy isoflavones can suppress tumors in animal models. Genistein can induce apoptosis via downregulation of Bcl-2 and Bcl-XL, antiapoptotic proteins in human hepatoma cell lines,236 as well as in breast, prostate, pancreatic, and non-small lung cancer cells. Genistein also inhibits activation of NF-κB and Akt signaling pathways that normally balance cell survival and apoptosis.237 A recent meta-analysis suggested a significant association between consumption of soy-based foods and reduction of the risk of prostate cancer.238 Epidemiologic studies on the relationship between a reduced risk of breast cancer and soy intake have yet to provide definitive evidence for taking soy supplements. RCTs are in progress.
Tea
Green tea contains epigallocatechin, epigallocatechin-3 gallate (EGCG), and epicatechin-3 gallate. These flavonoids may block signal transduction pathways and induce apoptosis in various tumor cells, including skin, prostate, colon, breast, and lung.239 EGCG blocked the growth of human cervical cancer cells via cell cycle arrest and induced apoptosis.240 It did so by inhibiting tyrosine kinases, thus blocking MAPK signaling pathways and NF-κB and AP-1.241 Green tea catechins can limit DNA oxidation, lipid peroxidation, and free radical production in smokers.242
Analysis of nearly 200 epidemiologic studies examined associations between green tea consumption and risks of cancer at different sites and found that the majority of reports did not show a preventive effect. Inhibition with green tea consumption was more frequent among Asian populations according to case–control studies.239,243 Tea consumption may help specific subgroups, rather than a general population, such as lowering the risk of breast cancer only among women with low catechol-O-methyl transferase.244 Intervention studies are a more effective way to establish protective effects of tea on cancer risk. In a double-blind study, patients at risk for prostate cancer treated with green tea catechins 600 mg/day for 1 year had a cancer rate of 3% compared with a 30% rate for the placebo group.245 A prospective study of patients after resection for colon cancer or polypectomy employed a daily dose of EGCG 20 mg and apigenin 20 mg for up to 4 years. The recurrence rate of neoplasia was 7% for treated patients versus 47% for untreated patients.246
Flavonoids in Cardiovascular Protection
Whether increased consumption of flavonoid-rich foods can reduce the risk of CVD is not clear. For example, a study of postmenopausal American women found significant reductions in the risk of CVD and all-cause mortality with increased flavonoid intake.247 Results of the Cancer Prevention Study II Nutrition Cohort suggested an inverse relationship between total dietary flavonoid intake and the risk of stroke and total CVD, especially for men.248 In contrast, a study of women reported no significant relationship between consumption of flavanols and flavones and coronary heart disease,249 and there was no strong relationship between flavonoid intake and total coronary heart disease among American male health professionals.250
Antioxidant-Rich Beverages
Wine and Grape Juice
Wine contains abundant polyphenols, and drinking red wine can increase the antioxidant capacity of serum.251,252 A meta-analysis of 133 RCTs studying effects of flavonoids and flavonoid-rich foods suggested that red wine consumption did not improve flow-mediated dilation, alter blood pressure, or change LDL levels.253 Results for grape juice consumption were also mixed. Overweight volunteers who consumed 480 mL/day grape juice for 12 weeks did not have improved plasma antioxidant capacity or lipid profiles.254
Cocoa
A double blind study of postmenopausal, hypercholesterolemic women found that high cocoa flavonol consumption daily for 6 weeks improved endothelial functions.255 Overweight adults who ingested cocoa beverages experienced significantly improved flow-mediated dilation in a randomized trial.256 Furthermore, administration of 40 g/day cocoa powder for 4 weeks significantly reduced ICAM-1 and P-selectin in high-risk patients with CVD.257
Tea
In laboratory animals, green tea prevented loss of liver SOD, GSH peroxidase, and catalase activities due to alcohol intoxication, while protecting membrane lipids from peroxidation.258 In apolipoprotein-E gene knockout mice, an animal model of atherosclerosis, theaflavin with quercetin reduced formation of atherosclerotic lesions and vascular superoxide and ILB-4, while increasing vascular endothelial NOS activity, NO production, and vasodilation.259 A meta-analysis of seven cohort and case–control studies reported an 11% decrease in the risk of myocardial infarction with daily consumption of three cups of tea.260 A later meta-analysis of nine observational studies suggested that consumption of the equivalent of three cups of green or black tea daily was associated with a significantly reduced risk of ischemic stroke.261
Soy
Studies with vascular cells suggest that soy isoflavones can increase eNOS activity262 while activating the redox regulated Nfr2-Keap1 signal pathway to increase transcription of phase 2 enzymes and antioxidant enzymes.263 Some epidemiologic studies implicated soy, especially soy protein, with reduced risk of coronary heart disease.253 Several RCTs reported cardiovascular benefits with improved systemic arterial function after a meal enriched with soy isoflavones.264 In a RCT, supplementation of postmenopausal women with genistein for up to 36 months decreased homocysteine and fasting glucose.265 However, results were inconsistent.
Quercetin
Inflammation
Experiments with guinea pig epidermal cells suggest that flavonoids, such as quercetin, can inhibit COX-2 and 5-lipoxygenase pathways to curtail production of inflammatory eicosanoids.266 Flavonoids may reduce expression of adhesion molecules needed to recruit white cells to arterial walls, a property unrelated to their antioxidant potential. Quercetin and kaempferol inhibited adhesion molecule expression induced by TNF-α in human aortic endothelial cells. Pre-exposure of hepatocytes to flavonoids blocked this inhibitory effect, suggesting that liver biotransformation reduces the ability of flavonoids to influence endothelial cells.267 Quercetin supplementation of healthy volunteers with quercetin (500 or 1000 mg) together with vitamin C (125 mg or 250 mg) daily over 12 weeks significantly increased plasma quercetin without changing plasma oxidant capacity (FRAP and ORAC).268
Oxidative Stress
Stress induced by strenuous exercise increased plasma cortisone and lipid peroxides and decreased SOD, GPx, and catalase in rat brains. Treatment with quercetin significantly increased these activities, while lowering plasma peroxides.269 In contrast, long-term quercetin supplementation (1000 mg/day) in athletes did not protect against exercise-induced oxidative stress, although supplementation might lead to a modest improvement in maximal oxygen uptake in untrained individuals.270,271
Cardiovascular Disease
Short-term consumption of flavonoid-rich foods and beverages can improve CVD risk factors, including endothelial function in healthy volunteers and patients with coronary artery disease. Interest was stimulated by the Zutphen study, which reported an inverse relationship between the incidence of stroke and coronary artery disease and consumption of flavonoids, particularly quercetin.235
Using a RCT with crossover design, healthy men who ingested 200 mg of quercetin experienced lowered plasma endothelin-1 and increased plasma nitrite and S-nitrothiols, which are biomarkers of NO production.272
Chemoprevention
Quercetin can induce apoptosis and stimulate production of antioxidant enzymes such as NADPH quinone oxidoreductase activity in human breast cancer cells.273,274 With induced lung carcinogenesis in mice, quercetin pretreatment effectively prevented losses of SOD, catalase, GPx, GSH S-transferase, GSH reductase, while limiting tumor formation.275 A case–control study found an inverse relationship between the intake of quercetin-rich foods and lung cancer risk, especially for heavy smokers. This effect corresponded to upregulation of GSH transferase isozymes and downregulation of cytochrome P450 isoforms.276
Proanthocyanidins (Oligomeric Proanthocyanidins)
Pine Bark Extract
Pine bark extract (PBE) represents a complex mixture of polyphenols enriched in procyanidolic oligomers (PCOs), containing two to seven monomers of catechin and epicatechin. PCOs are effective antioxidants in vitro and can protect and/or regenerate α-tocopherol and vitamin C. PBE can also upregulate antioxidative enzymes in cultured cells.277
Inflammation
PBE can inhibit production of ROS by macrophages blocking the expression of proinflammatory cytokines, such as IL-1β.278 Plasma from subjects treated with PBE can block NF-κB activation and production of matrix metalloproteinases in human monocytes.279 Rodents fed diets supplemented with PBE had increased plasma antioxidant capacity and reduced lipid peroxidation (thiobarbituric acid reactive substances).280 Plasma taken from healthy subjects 30 minutes after ingestion of a single dose of 300 mg PBE had inhibited COX-1 and COX-2 in human monocytes.281 These observations suggest that PCOs may be useful in reducing inflammation; standardized PBE has been used to treat capillary dysfunction in patients with diabetes, venous abnormalities,270 and knee osteoarthritis.282
Grape Seed Extract
Oxidative Stress
Wine flavonoids including PCOs are effective antioxidants in vitro. They can reduce lipid peroxides and decrease susceptibility of isolated LDL to oxidation. Grape seed extract (GSE) treatment led to significant postprandial reduction in plasma lipid hydroperoxides and increased antioxidant capacity in healthy volunteers.283 Male smokers administered GSE had significantly decreased platelet reactivity compared with control subjects given a placebo.284 Type 2 diabetic patients received GSE 600 mg/day according to a controlled double-blind protocol. After 4 weeks, significant decreases were observed for plasma fructosamine and high-sensitivity C-reactive protein, and significant increases were seen for whole blood GSH compared with placebo.285
Neurologic Disease
After animal experiments indicating that GSEs can limit lipid peroxidation in the brain,286 GSE polyphenols were found to inhibit protofibril formation and protofiber oligomerization in a mouse model of Alzheimer’s disease.287
Chemoprevention
GSE is able to inhibit proliferation of a variety of cancer cell lines involving regulatory targets such as MAPKs, NF-κB, phosphoinositide 3-kinases (PI 3-kinases), and angiogenesis.288 Furthermore, pretreatment with GSE protected rodents against experimentally induced hepatotoxicity, pulmonary toxicity, and doxorubicin-induced cardiotoxicity by suppressing oxidative stress.286,289
Other Potential Phytochemical Antioxidants
Hepatoprotection
Silymarin reduced oxidative stress and prevented liver necrosis in mice exposed to toxic agents.290 Silymarin blocked production of TNF-α, interferon-γ, and IL-2 by stimulated peripheral blood mononuclear cells and T cells isolated from hepatitis C virus infected and noninfected subjects.291 Phase I to II trials have not demonstrated consistent reduction in liver function tests, viral load, or improved liver histology in patients with hepatitis C.292
Diabetes
An open trial with cirrhotic diabetic patients treated with 600 mg/day silymarin for 12 months reported significant reduction in glucosuria, glycosylated hemoglobin, and malondialdehye levels compared with controls.293 Similar positive results were found in a RCT of treatment of patients with type 2 diabetes with 600 mg/day silymarin for 4 months.294
Cancer
Androgen-responsive prostate cancer cells can be inhibited by silymarin. Using a transgenic mouse prostate cancer model, dietary phosphatidylcholine-silibinin complex inhibited tumor growth and metastasis by reducing expression of metalloproteinases and vascular endothelial growth factor and its receptor, while blocking epithelial to mesenchymal associated with metastasis.295
Resveratrol
Grapes, blueberries, and peanuts contain resveratrol and methylated resveratrol belonging to the stilbene family of polyphenols. Resveratrol can act as an antioxidant to protect human erythrocytes against H2O2–induced lipid peroxidation in vitro.296 Stilbenes exhibit low solubility and rapid clearance, and ingestion of acute doses of resveratrol by healthy subjects achieved only nanomolar plasma concentrations, concentrations several orders of magnitude less than those used for in vitro or animal studies.
Inflammation
Possible mechanisms relating to anti-inflammatory effects include inhibition of COX and lipoxygenase and blocking the action of NK-κB and AP-1 in cultured cells.297
Cardiovascular Protection
Exposure to resveratrol correlated with decreased myocardial damage associated with ischemia reperfusion and inhibition of LDL oxidation.298 Proteomic and immunoblot study revealed upregulation of five different redox-regulated proteins, including MAPKs.299 Resveratrol may also promote vasodilation. Resveratrol activated human platelet eNOS, increased NO production, and reduced NOx activity, thus lowering superoxide production.300
Diabetes
Using experimentally-induced diabetes in rats, resveratrol reduced glucose levels compared with untreated animals. Cardioprotection of diabetic rats involved induction of eNOS, vascular endothelial growth factor, and heme oxygenase.301
Chemoprevention
In various cancer cell lines, resveratrol exposure can stimulate apoptosis and inhibit cell proliferation.302 A study of rats fed resveratrol demonstrated that high-dose resveratrol suppressed a transgenic rat model of spontaneous prostate cancer.303
Aging
Resveratrol can increase longevity in a variety of organisms, including mice.304 It may modulate transcription of the sirtuin gene family implicated in senescence, endothelial function, and angiogenesis. Sirt1, a deacetylase that silences transcription, can be induced by resveratrol in vitro. It is worth noting that low doses of resveratrol can improve cell survival by upregulating antiapoptotic and redox regulating proteins, whereas high doses induce cell death by downregulating redox proteins and inducing production of apoptotic proteins.305 Questions of bioavailability, long-term safety, effective dosage, and optimal duration of treatment have not yet been established.306
Ginkgo biloba
Oxidative Stress
In part, beneficial effects appear related to antioxidant and anti-inflammatory actions, and it is likely that active constituents, ginkgolides, and related flavonoids reduce oxidative stress.307,308 Cell culture studies suggest that EGb 761 can raise GSH levels and induce γ-glutamylcysteinyl synthetase, the rate-limiting enzyme in GSH synthesis.309 Damage to human endothelial cells exposed to oxidized LDL could be prevented by EGb 761.310 Rats treated with EGb 761 had increased catalase and SOD activity and reduced lipid peroxidation in the hippocampus, striatum, and substantia nigra compared with controls.311
Aging
Mice supplemented with EGb 761 also had less ROS-induced apoptosis in lymphocytes than untreated mice; this result was greater in old mice than in young animals.312 In clinical studies, type 2 diabetic patients treated with EGb 761 for 3 months were found to have reduced markers of lipid peroxidation in platelets, potentially through inhibition of the COX-1 mediated cascade.313 A RCT of elderly subjects administered EGb 761 long-term found no benefit in reducing the risk of CVD.314
Curcumin
Curcuminoids are bright yellow pigments found in turmeric, a family of lipophilic diketones. Curcumin, the principal curcuminoid, is poorly absorbed with serum concentrations in the nanomolar to micromolar range after oral ingestion of up to 8 g.315 Extending the observation that curcumin scavenges ROS and RNS in vitro, consumption of 200 mg/day of curcuminoids decreased serum lipid peroxide levels and LDL lipid peroxidation in healthy subjects.316
Inflammation
In cultured colonic cells, curcumin inhibited iNOS and blocked lipoxygenase, COX-2, and phospholipase A, and reduced production of proinflammatory prostaglandins, thromboxanes, and leukotrienes by inhibiting NF-κB activation.317 Clinical studies are preliminary. For example, a RCT of patients experiencing remission of ulcerative colitis examined high-dose curcumin supplementation in addition to standard treatment. After 6 months, 2 of 43 patients relapsed compared with 8 of 39 patients in a placebo group.318
Cancer
Curcumin can induce apoptosis and inhibit angiogenesis and production of matrix metalloproteinases in several cancer cell lines. Oral curcumin administration inhibited a mouse model of familial adenomatous polypsis,319 and it inhibited the growth of human tumors in xenotransplant animal models. Clinical studies are limited. Two of 21 patients with advanced pancreatic cancer administered 8 g/day curcumin until cancer regressed experienced some anticancer activity.320 Several Phase II and III clinical trials are near completion (Curcuminoids in colon cancer prevention, University of Pennsylvania; curcumin and capecitabine in rectal cancer, MD Anderson Cancer Center; curcumin for pancreatic cancer, MD Anderson Cancer Center; and for pediatric oral mucositis, Hadassah Medical Organization).
Neurodegeneration
Curcumin can bind to amyloid deposits. A mouse model of Alzheimer’s disease treated with curcumin 7 days reduced existing plaques and structural irregularities in dystrophic dendrites.321 Of four clinical trials of curcumin supplementation for Alzheimer’s disease, the two that have been reported found no significant differences in cognitive performance between treated and placebo groups.322
Garlic
Oxidative Stress
Rabbits fed an atherogenic diet and garlic extract had significantly better antioxidant status and reduced plaque area compared with animals not fed garlic.323 S-allyl cysteine inhibited oxidative damage and blocked the formation of H2O2 in cultured endothelial cells.324 Using an animal model of type 2 diabetes, investigators found that aged garlic extract lowered lipid peroxidation and increased SOD and GPx activity compared with controls.325
A pilot study of smokers and nonsmokers found that daily supplementation with aged garlic extract for 2 weeks reduced lipid peroxidation (8-iso-prostaglandin F2).326 A study of hypertensive patients supplemented 2 months with garlic observed significant reduction of DNA oxidation (8-hydroxyguanosine), NO production, and lipid peroxidation compared with controls.327 However, using a RCT and crossover protocol, investigators observed that supplementation of men with coronary artery disease with aged garlic extract for 1 month increased arterial flow-mediated dilation without lowering markers of oxidative stress (oxidized LDL) inflammation (C-reactive protein).328
Inflammation and Detoxification
Garlic can attenuate inflammatory cytokine production in stimulated macrophages by suppressing NO production.329 Garlic also upregulates phase II detoxification enzymes in vitro. Human hepatoma cells exposed to diallyl sulfides activated the AREs and Nerf2 family of detoxification enzymes, possibly through calcium-dependent signaling.330
Specific Antioxidant Mechanisms for Polyphenols In Vivo?
Mechanism
Depending upon the experimental design, polyphenols can inhibit COX, lipooxygenase, telomerase, xanthine oxidase, cytochrome P450, and protein glycation, as well as modulating drug transport systems and platelet function. Flavonoids may influence multiple signaling pathways, making it difficult to assign specific actions. As an example, red wine polyphenols administered to rats for 16 weeks inhibited chemically induced colon cancer.331 In the process, responses to oxidative stress were downregulated. Microarray analysis of cellular changes after polyphenol treatment for 14 days revealed the following frequencies of downregulated genes: receptors and signal transduction, 22%; transport and binding proteins, 14%; inflammatory and immune response, 11%; metabolic enzymes, 4%; gene expression/control, 4%; xenobiotic metabolism, 3%; and cell cycle regulation, 3%. In the final analysis, observed protective effects of flavonoid-rich foods in humans may be independent of flavonoids.
Gastrointestinal Tract as Primary Target for Polyphenols
The GI tract is continuously exposed to ROS and RNS from multiple sources. The gut epithelium may not adapt to long-term oxidative stress, and because of low initial levels of defensive enzymes, it may be susceptible to oxidative damage with even moderate inflammation.332 After ingestion of flavonoid-rich foods, phenolic compounds are present in the stomach and intestinal lumen at much higher concentrations than those found in plasma.333 Of particular interest are inflammatory bowel disease, gastric ulcers, and liver disease.334,335 ROS are present at high levels in the colons of patients with ulcerative colitis.336 Green tea polyphenols can reduce inflammation in models of inflammatory bowel disease.337 Polyphenolic by-products of intestinal metabolism, such as hydroxybenzoic acid and protocatechuic acid, exhibit important properties. As an example, they can induce apoptosis in gastric cancer cells.338
 Therapeutic Use of Antioxidants
Therapeutic Use of Antioxidants
Immunodeficiency, Oxidative Stress, and Antioxidants
Supplementation with antioxidants, including vitamins C and E, and carotenoids, along with selenium, copper, and zinc, has been reported to improve aspects of innate and humoral immunity. Used alone or in combination, these nutrients may improve lymphocyte proliferation, delayed hypersensitivity responses, and immune cell functions. These include Th1 cytokine-mediated responses. Adequate levels of proinflammatory cytokines help balance anti-inflammatory Th2-mediated responses.339,340 However, a small RCT of allergic individuals found no change in immune responses, serum antioxidant capacity, or markers of oxidative stress after 4-week supplementation with vitamins C and E, β-carotene, selenium, and zinc at levels several-fold higher than recommended dietary allowances.341
The growing literature suggests that HIV infection is associated with oxidative stress. Sero-positive HIV patients were shown to have significantly reduced plasma TAC compared with healthy controls, a finding that correlated with significantly elevated lipid peroxidation and reduced levels of vitamins C and E, SOD342, and oxidative stress, which may relate to HIV dementia.342,343 A cross-sectional study demonstrated increased plasma lipid peroxide levels along with depleted GSH levels in HIV-infected individuals compared with healthy controls.344 Low levels of reduced GSH could activate transcription factor NF-κB to increase transcription of the HIV genome.345
It is logical to propose that antioxidant treatment could benefit HIV-positive individuals. Polyphenols can inhibit HIV infectivity of cells in vitro.346 However, results of clinical studies are mixed. A review of 6 RCTs concluded that β-carotene/vitamin A did not change overall mortality, morbidity, or CD4 and CD8 counts, whereas 4 RCTs of other micronutrients did not improve overall mortality.347 Benefits of micronutrient supplementation in improving mortality among HIV-positive patients appears to involve correcting nutrient deficiencies.348 High vitamin C or high vitamin E levels can potentially interfere with indinavir therapy.349
Exercise, Oxidative Stress, and Antioxidants
The health benefits of regular physical activity are well established, but the exact mechanisms are uncertain. Some data suggest that long-term exercise increases antioxidant defenses as an adaptation. Clearly, strenuous physical exercise increases ROS production and muscle fiber damage. Although vitamin C, vitamin E, ALA, L-carnitine, and flavonoids can attenuate certain of the signs and symptoms due to resistance training, they do not eliminate tissue injury. Green tea extracts appear to have marginal effects on metabolism and performance in trained athletes.350 Supplementing with antioxidant vitamins such as E, C, and CoQ10 may decrease associated oxidative damage in untrained individuals, especially in older people.351 Nonetheless, optimal doses of putative beneficial antioxidants and pretreatment intervals are unknown.352 The field is confusing because a variety of markers of antioxidant status and different training levels have been used. Furthermore, many studies use blood levels of antioxidant and ROS-induced by-products as end points to assess antioxidant status after exercise, contributing to variable test results. It is questionable whether these endpoints represent tissue level changes. Benefits of supplementation are most often seen in untrained populations rather than in athletes, especially when there are nutrient deficiencies. For example, in resistance-trained men, supplementation with 1000 mg/day of vitamin C and 378 mg/day of mixed tocopherols or placebo for 14 days did not change markers of muscle injury or oxidative stress (blood protein carbonyls and peroxides).353 Currently, there does not appear to be enough evidence for recommending antioxidant supplements for well-trained and well-nourished athletes.354,355 Antioxidant supplementation may actually be counterproductive. A RCT of kayakers given standard antioxidants (vitamin C 400 mg, α-tocopherol 272 mg, β-carotene 30 mg, lutein 2 mg, selenium 400 mcg, zinc 30 mg, magnesium 600 mg) daily for 4 weeks found that supplementation failed to lower lipid peroxidation induced by strenuous exercise, and it reduced muscle recovery times.356
Do Antioxidants Explain the Health Benefits of Plant-Based Foods?
Consumption of plant-based foods rich in antioxidants was reported to decrease oxidative damage in humans by some authors,333 although others found little effect.357 Recent analysis of data from the EPIC study found that eating five portions of fruits and vegetables daily lowered the overall risk of cancer by 9%, far less than expected.358 Nonetheless, previous studies concluded that five servings could reduce the risk of stroke by 26%; reduce the risk of cancers of the oral cavity, bowel and lung; and reduce the risk of obesity. Do antioxidants account for such benefits? Results of assays of antioxidants and assessments of oxidative stress are suggestive, but as yet there is no firm demonstration that in vitro measurements translate into specific health benefits. The U.S. FDA and the European Food Safety Authority concluded that claims cannot be made linking foods and food constituents to protection of body cells and molecules, such as DNA, proteins, and lipids from oxidative damage.359
Supplementation with Antioxidants
Specific Goals
A primary goal is to adjust supplementation to correct any imbalances in an individual’s nutritional status. Overall, studies suggest that the recommended dietary allowances for antioxidants are inadequate for optimal immune function for certain groups. Table 108-1 provides a summary of a recommended intake of antioxidants for maintenance as compiled by the European Federation of Health Product Manufacturers.301 For most adults, intakes of vitamin C up to 2000 mg/day are believed to be safe.360 The levels of vitamins and minerals often prescribed in treatment protocols may be considerably higher than those listed in Table 108-1.
TABLE 108-1 Recommended Safe Upper Intake Levels for Antioxidant Nutrients
| Vitamin E | 1000 mg* |
| Vitamin C | 2000 mg* |
| Vitamin A | 3000 mcg* |
| Beta carotene | None established (5 or more servings of fruits and vegetables would provide an estimated 3-6 mg β-carotene daily.) |
| Copper | 10 mg* |
| Magnesium | 250 mg† |
| Selenium | 300 mcg* |
| Zinc | 40 mg* |
* U.S. Food and Nutrition Board, Institute of Medicine Tolerable Upper Intake Levels.
† European Commission Scientific Committee on Food, Tolerable Upper Intake Level.
Data from Flynn A, Moreiras O, Stehle P, et al. Vitamins and minerals: a model for safe addition to foods. Eur J Nutr 2003;42:118-130.
A second goal is to support the body’s defense systems. Antioxidant requirements should be balanced against oxidant burden. Multiple, complementary antioxidants are far more effective than large amounts of a single antioxidant, because antioxidants often work synergistically.361 Animal studies suggest that consumption of a greater diversity of antioxidants is more effective than use of single antioxidants. Daily supplementation with vitamin C (500 mg), vitamin E (400 IU), β-carotene (15 mg), zinc (80 mg), and copper (2 mg) significantly slowed the progression of advanced AMD, although this regimen did not affect the risk of cataracts or loss of visual acuity.362 In other research, the combination of CoQ10, vitamin E, and ALA were shown to act synergistically in protecting LDL against lipid peroxidation in vitro.363
How Common Are Antioxidant Deficiencies?
The prevalence of subclinical deficiencies of nutrients with antioxidant roles may be quite high—magnesium (56%), zinc (12%), vitamin E (93%), and vitamin C (31%) for general populations, as judged by intakes less than an estimated average requirement.19 More than 90% of Americans do not consume the dietary reference intake (DRI) for vitamin E of 15 mg, which is judged as adequate to prevent myopathy and neuropathy.364 Success with antioxidant supplementation is most notable when there are deficiencies, overt or not. According to a metabolic “triage” hypothesis, low micronutrient intake can increase the risk of chronic degenerative diseases without overt deficiencies, because the body’s homeostatic mechanisms direct essential nutrients to support the most critical functions.19
Limits to Antioxidant Supplementation
The use of antioxidant supplementation to completely quench free radicals is counterproductive. As discussed earlier, physiologic levels of ROS are essential in maintaining homeostasis for multiple physiologic systems, for example, superoxide and NO play essential roles in maintaining the body’s defenses and homeostatic mechanisms. Antioxidant supplementation may be counterproductive, as examples, acute supplementation with vitamins C, E, and α-lipid acid negated the reduction of blood pressure and the improvement in flow-mediated vasodilation due to exercise in elderly hypertensive men.365 Transient oxidative stress, induced by short-term physical activity, can ameliorate insulin resistance. However, pretreatment with α-tocopherol 400 IU/day and vitamin C 1000 mg/day for 4 weeks prevented exercise-induced insulin sensitivity and blocked induction of endogenous antioxidant enzymes in healthy volunteers.366 Quercetin and myricetin efficiently inhibited TRX reductase to induce apoptosis and cell cycle arrest in tumor cell lines by direct binding and by semiquinone radical oxidation.367 However, the TRX system broadly affects redox balance in normal cells and tissues, and its inhibition may be detrimental to health.
What is the most effective way to increase intake of a broad spectrum of defined and undefined antioxidants? The increased consumption of a variety of antioxidant-rich foods—especially colored fruits, vegetables and legumes, and beverages such as green tea—remains a prudent strategy. Because short-term interventions with vitamins C or E, β-carotene, or flavonoids have not demonstrated consistent health benefits, the data suggest that other plant food factors may account for such protection.368 As an example, flavonoids seem to account for only a small percentage of the oxidant capacity of apple extracts.230
Additional Considerations for Antioxidant Supplementation
Does Vitamin E Increase Mortality Risk
Meta-analysis of 68 randomized trials found that supplementation up to 800 IU/day did not change the risk of all-cause mortality.102 A more recent meta-regression analysis of 29 clinical trials representing a total of 138,462 participants and vitamin E dosages ranging from 16.5 to 5000 IU/day found no causal relationship between high-dose vitamin E and mortality.369 Indiscriminant use of high doses of vitamin E is not recommended.136,370 Vitamin E can interfere with vitamin K-dependent clotting factors, and high doses increase the risk of bleeding in at-risk individuals.
Does Excessive Beta-Carotene Contribute to Disease?
Beta-carotene may act as a pro-oxidant for those at risk for oxidative stress (smoking, alcohol consumption); β-carotene may also raise the risk of lung cancer in high-risk populations unless protected by antioxidants like vitamin E.371 When β-carotene is supplemented during oxidative stress, cleavage products are produced, which are genotoxic, and these may account for the carcinogenic effects observed in the Alpha Tocopherol, Beta Carotene Cancer Prevention Study (ATBC) and Beta Carotene and Retinol Efficacy Trial (CARET).372 High levels of β-carotene may be contraindicated in smokers with myocardial infarctions. In one study, the rate of fatal coronary heart disease rose in patients receiving 20 mg/day β-carotene (with or without α-tocopherol).373 Carotenoids generally lose their effectiveness at high concentrations and at high partial pressures of oxygen. This feature may account for the possible benefits of consumption of carotenoid-rich foods, although therapeutic doses of isolated carotenoids may be deleterious.374
Can Antioxidants Contribute to Oxidative Stress?
The potential pro-oxidant activity of polyphenolic radicals is well documented.375 Flavonoids are metabolized and detoxified by liver enzymes, and pharmacologic doses can induce phase I detoxification enzymes, increasing the ability to transform toxins. The tradeoff lies in the possibly greater sensitivity to mutagens. Vitamins C and E exhibit pro-oxidant activity in vitro under certain conditions. When exposed to catalytic levels of iron or copper ions, ascorbate promotes the formation of H2O2 and hydroxyl radicals.376,377 In vitro studies suggest that ascorbic acid can decompose lipid peroxides to genotoxins.378 In vivo, these effects would likely require high ascorbate concentrations coupled with the depletion of tocopherol.379 Excessive dietary iron or iron accumulation could promote ascorbate-induced oxidation, and ascorbate could act as an oxidant in the presence of iron or copper released during inflammation or injury. In a RCT, hypertensive patients were treated with 500 mg ascorbic acid and/or 1000 mg grape seed polyphenols daily for 6 weeks. Although ascorbic acid alone reduced elevated systolic pressure, the combination significantly increased both systolic and diastolic blood pressure.380
Antioxidants as Adjuvant Cancer Therapy
A systematic review found that 24 of 33 randomized, controlled studies reported decreased toxicity of chemotherapy with concomitant use of antioxidants, including vitamin E and vitamin C, with one study of vitamin A reporting increased toxicity.381 As a recent example, a study of pediatric patients with acute lymphoblastoma being treated with chemotherapy and concurrent supplementation with 400 IU vitamin E and 600 mg N-acetylcysteine daily for 28 days found that children taking the supplements experienced less liver toxicity, fewer blood abnormalities, and fewer blood transfusions.382 The use of α-tocopherol succinate to inhibit tumor growth in vivo remains unproven. Because this derivative possesses no antioxidant activity, presumably its inhibition of cancer cell proliferation and induction of apoptosis in tumor cells relies on other mechanisms.
Caveats Regarding Potential Nutrient–Drug Interactions and Phytochemical–Drug Interactions
The popularity of antioxidant supplementation comes with additional concerns regarding interactions with over-the-counter and prescription medications. Literature in these areas is steadily growing, and several useful compendia are available.383–386 In addition, the editors of the Pharmacist’s Letter publish the Natural Medicines Comprehensive Database, which is updated frequently.
 Future Directions
Future Directions
The broad outlines for antioxidant defenses against oxidative stress are established. However, our understanding of the inner workings of these mechanisms remains incomplete, and details of the body’s response to oxidative stress remain poorly understood. Use of newer research tools, such as genetically manipulated laboratory animals (e.g., SOD−/− or GPx1−/− animals) and analysis of global changes in gene expression induced by oxidative stress, offer powerful approaches to understanding in vivo responses.387 Incorporation of a systems biology analysis can also provide powerful insights.388
In general, the consumption of a broad spectrum of antioxidants in amounts geared to meet an individual’s oxidant burden and nutritional status may minimize the effects of genetic predisposition that compromise defenses against aging and degenerative disease and promote optimal health. Yet for most antioxidants, used singly or in combinations, the dosage, duration, efficacy, safety, and levels needed to sustain optimal health remain unknown. This is especially true for polyphenols.389 In addition, a therapeutic window for antioxidant action is often inferred, although not defined. Target dietary reference intakes for phytochemicals may eventually reflect composites beyond the currently recommended five daily servings for fruits and vegetables.390
Nonetheless, essential data needed to support the therapeutic use of antioxidants cannot be obtained from in vitro or from animal studies. Future claims for the impact of antioxidants on health must await additional studies, including intervention clinical trials demonstrating better function with supplementation and prospective studies exploring risk reduction for degenerative diseases.391 A “nutrigenomics” strategy embraces the large-scale analysis of gene expression, individual proteins, and lipid markers, as well as metabolic pathways integrated using functional analysis software.392 This approach demonstrated that a diet enriched in putative antioxidants can modulate low grade oxidative stress and inflammation in overweight men. The potential of herbs and botanicals in the prevention and treatment of conditions involving free radical pathology further enriches the future of antioxidant research.393
1. Cho K.J., Seo J.M., Kim J.H. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol Cells. 2011;32:1–5.
2. Li L., Frei B. Prolonged exposure to LPS increases iron, heme, and p22phox levels and NADPH oxidase activity in human aortic endothelial cells: inhibition by desferrioxamine. Arterioscler Thromb Vasc Biol. 2009;29:732–738.
3. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1957;2:298–300.
4. Kovacic P., Jacintho J.D. Mechanisms of carcinogenesis: focus on oxidative stress and electron transfer. Curr Med Chem. 2001;8:773–796.
5. Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95.
6. Ross J.S., Stagliano N.E., Donovan M.J., et al. Atherosclerosis and cancer: common molecular pathways of disease development and progression. Ann N Y Acad Sci. 2001;947:271–292.
7. Iuliano L. The oxidant stress hypothesis of atherogenesis. Lipids. 2001;36:S41–S44.
8. Wei W., Liu Q., Tan Y., et al. Oxidative stress, diabetes and diabetic complications. Hemoglobin. 2009;33:370–377.
9. Ottonello S., Foroni C., Carta A., et al. Oxidative stress and age-related cataract. Ophthalmologica. 2000;214:78–85.
10. D’Odorico A., Bortolan S., Cardin R., et al. Reduced plasma antioxidant concentrations and increased oxidative DNA damage in inflammatory bowel disease. Scand J Gastroenterol. 2001;36:1289–1294.
11. Tak P.P., Zvaifler N.J., Green D.R., et al. Rheumatoid arthritis and p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today. 2000;21:78–82.
12. Rahman I. Oxidative stress, transcription factors and chromatin remodeling in lung inflammation. Biochem Pharmacol. 2002;64:935–942.
13. Esposito E., Rotilio D., Di Matteo V., et al. A review of specific dietary antioxidants and the effects on biochemical mechanisms related to neurodegenerative processes. Neurobiol Aging. 2002;23:719–735.
14. Moosmann B., Behl C. Antioxidants as treatment for neurodegenerative disorders. Expert Opin Investig Drugs. 2002;11:1407–1435.
15. Jain S.K., Pemberton P.W., Smith A., et al. Oxidative stress in chronic hepatitis C: not just a feature of late stage disease. J Hepatol. 2002;36:805–811.
16. Vincent H.K., Taylor A.G. Biomarkers and potential mechanisms of obesity-induced oxidative stress in humans. Int J Obes (Lond). 2006;30:400–418.
17. Maes M., Twisk F.N. Why myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) may kill you: disorders in the inflammatory and oxidative and nitrosative stress (IO&NS) pathways may explain cardiovascular disorder in ME/CFS. Neuro Endocrinol Lett. 2009;30:677–693.
18. Curtin J.F., Donovan M., Cotter T.G. Regulation and measurement of oxidative stress in apoptosis. J Immunol Methods. 2002;265:49–72.
19. Ames B.N. Low micronutrient intake may accelerate the degenerative diseases of aging through allocation of scarce micronutrients by triage. Proc Natl Acad Sci USA. 2006;103:17589–17594.
20. Wei Y.H., Lee H.C. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med. 2002;227:671–682.
21. Goncalves L., Dafre A.L., Carobrez S.G., et al. A temporal analysis of the relationships between social stress, humoral immune response and glutathione-related antioxidant defenses. Behav Brain Res. 2008;192:226–231.
22. Lanza I.R., Nair K.S. Mitochondrial function as a determinant of life span. Pflugers Arch. 2010;459:277–289.
23. Van Remmen H., Jones D.P. Current thoughts on the role of mitochondria and free radicals in the biology of aging. J Gerontol A Biol Sci Med Sci. 2009;64A:171–174.
24. Keshavarzian A., Sedghi S., Kanotsky J., et al. Excessive production of reactive oxygen metabolites by inflamed colon: analysis by chemiluminescence probe. Gastroenterol. 1992;103:177–185.
25. Droge W. Free radicals in the physiological control of cell function. Phys Rev. 2002;82:47–95.
26. Choi S., Park S., Laing G.H., et al. Superoxide generated by lysophosphatiylcholine induces endothelial nitric oxide synthase downregulation in human endothelial cells. Cell Physiol Biochem. 2010;25:233–240.
27. Pacher P., Beckman J.S., Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424.
28. Roos D. The involvement of oxygen radicals in microbicidal mechanisms of leukocytes and macrophages. Klin Wochenschr. 1991;69:975–980.
29. Suzuki Y.J., Forman H.J., Sevanian A. Oxidants as stimulators of signal transduction. Free Radic Biol Med. 1997;22:269–285.
30. Lam G.Y., Huang J., Brumell J.H. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin Immunopathol. 2010;32:415–430.
31. Pierce G.L., Lesniewski L.A., Lawson B.R., et al. Nuclear factor kappaB activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation. 119, 2009. 1294–1292
32. Suhr Y.J., Kundu J.K., Na H.K. Nfr2 as a master redox switch in turning on the cellular signaling involved in the induction of cytoprotective genes by some chemoprotective phytochemicals. Planta Med. 2008;74:1526–1539.
33. Fruefhauf J.P., Meyskens F.L., Jr. Reactive oxygen species: a breath of life or death? Clin Cancer Res. 2007;13:789–794.
34. Giorgi C., Agnoletto C., Baldini C., et al. Redox control of protein kinase C: cell and disease specific aspects. Antioxid Redox Signal. 2010;13:1051–1058.
35. Edderkoui M., Hong P., Vaquero E.C., et al. Extracellular matrix stimulates reactive oxygen specific production and increases pancreatic cell survival through 5-lipoxygenase and NADPH oxidase. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1137–G1147.
36. Tabet F., Savoia C., Schiffrin E.L., et al. Differential calcium regulation by hydrogen peroxide and superoxide in vascular smooth muscle cells from spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2004;44:200–208.
37. Vissers M.C., Lee W.G., Hampton M.B. Regulation of apoptosis by vitamin C. Specific protection of the apoptotic machinery against exposure to chlorinated oxidants. J Biol Chem. 2001;276:46835–46840.
38. Roberts R.A., Soames A.R., James N.H., et al. Dosing-induced stress causes hepatocyte apoptosis in rats primed by the rodent nongenotoxic hepatocarcinogen cyproterone acetate. Toxicol Appl Pharmacol. 1995;135:192–199.
39. Narula J., Haider N., Virmani R., et al. Apoptosis in myocytes in end stage heart failure. N Engl J Med. 1996;335:1182–1189.
40. Moon Y.J., Wang X., Morris M.E. Dietary flavonoids: effects on xenobiotic and carcinogen metabolism. Toxicol In Vitro. 2006;20:187–210.
41. Pfeilschifter J., Eberhardt W., Huwiler A. Nitric oxide and mechanisms of redox signaling. J Am Soc Nephrol. 2003;14:S237–S240.
42. Barrett D.M., Black S.M., Todor H., et al. Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on S-nitrosylation. J Biol Chem. 2005;280:14453–14461.
43. Snyder C.M., Shroff E.H., Jing L., et al. Nitric oxide induces cell death by regulating anti-apoptotic BCL-2 family members. PloS ONE. 2009;4:e7059.
44. Cho S.H., Lee C.H., Ahn Y., et al. Redox regulation of PTEN and protein tyrosine phosphatases in H2O2 mediated cell signaling. FEBS Lett. 2004;560:7–13.
45. Forman H.J., Maiorino M., Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835–842.
46. Mayorov D.M. Brain superoxide as a key regulator of the cardiovascular response to emotional stress in rabbits. Exp Physiol. 2007;92:471–479.
47. Liaudet L., Vassalli G., Pacher P. Role of peroxynitrite in the redox regulation of cell signal transduction pathways. Front Biosci. 2009;14:4809–4814.
48. Kohr M.J., Traynham C.J., Roof S.R., et al. cAMP-independent activation of protein kinase A by the peroxynitrite generator SIN-1 elicits positive inotropic effects in cardiomyocytes. J Mol Cell Cardiol. 2010;48:645–648.
49. Roberts L.J., 2nd., Morrow J.D. Products of isoprostane pathway: unique bioactive compounds and markers of lipid peroxidation. Cell Mol Life Sci. 2002;59:808–820.
50. Chevion M., Berenshtein E., Stadtman E.R. Human studies related to protein oxidation: protein carbonyl content as a marker of damage. Free Radic Res. 2000;33:S99–S108.
51. Halliwell B., Lee C.Y. Using isoprostanes as biomarkers of oxidative stress: Some rarely considered issues. Antioxid Redox Signal. 2010;13:145–156.
52. Valtuena S., Pellegrini N., Franzini L., et al. Food selection based on total antioxidant capacity can modify antioxidant intake, systemic inflammation, and liver function without altering markers of oxidative stress. Am J Clin Nutr. 2008;87:1290–1297.
53. Block G., Jensen C., Dietrich M., et al. Plasma C-reactive protein concentrations in active and passive smokers: influence of antioxidant supplementation. J Am Coll Nutr. 2004;23:141–147.
54. Forlani G., Di Bonito P., Mannucci E., et al. Prevalence of elevated liver enzymes in Type 2 diabetes mellitus and its association with the metabolic syndrome. J Endocrinol Invest. 2008;31:146–152.
55. Yavus B.B., Yavuz B., Halil M., et al. Serum elevated gamma glutamyltransferase levels may be a marker for oxidative stress in Alzheimer’s disease. Int Psychogeriatr. 2008;20:815–823.
56. Fraser A., Harris R., Sattar N., et al. Gamma-glutamyltransferase is associated with incident vascular events independently of alcohol content: analysis of the British Women’s Heart and Health Study and meta-analysis. Arterioscler Thromb Vasc Biol. 2007:2729–2735.
57. Kwak H.K., Yoon S. Relation of serum total antioxidant status with metabolic risk factors in Korean adults. Nutr Res Pract. 2007;1:335–340.
58. Smith C.V. Free radical mechanisms in tissue injury. In: Moslen M.Y., Smith C.V. Free Radical Mechanisms of Tissue Injury. Boca Raton: CRC Press; 1992:2–22.
59. Buettner G.R. The pecking order of free radicals and antioxidants: lipid peroxidation, a-tocopherol, and ascorbate. Arch Biochem Biophys. 1993;300:535–543.
60. Sandoval M., Ronzio R.A., Muanza D.N., et al. Peroxynitrite-induced apoptosis in epithelial (T84) and macrophage (RAW 264.7) cell lines: effect of legume-derived polyphenols (phytolens). Nitric Oxide. 1997;1:476–483.
61. Yang B., Kotani A., Arai K., et al. Relationship of electrochemical oxidation of catechins on their antioxidant activity in microsomal lipid peroxidation. Chem Pharm Bull. 2001;49:747–751.
62. Garrett A.R., Murray B.K., Robinson R.A., et al. Measuring antioxidant capacity using the ORAC and TOSC assays. Methods Mol Biol. 2010;594:251–262.
63. Nutrient Data Laboratory, Agricultural Research Service. USDA Oxygen radical absorbance capacity (ORAC) of selected foods. Washington, DC: USDA; 2007.
64. Carlsen M.H., Halvorsen B.L., Holte K., et al. The total antioxidant content of more than 3100 foods, herbs, beverages, spices, and herbs and supplements used worldwide. Nutr J. 2010;9:3. PMCID: (PMC2841576)
65. Jensen G.S., Wu X., Patterson K.M., et al. In vitro and in vivo antioxidant capacities of an antioxidant-rich fruit and berry juice blend. Results of a pilot and randomized, double blinded, placebo-controlled crossover study. J Agric Food Chem. 2008;56:8326–8333.
66. Wofe K.L., Liu R.H. Cellular antioxidant activity (CAA) assay for assessing antioxidants, foods, and dietary supplements. J Agric Food Chem. 2007;55:8896–8907.
67. Visner G.A., Dougall W.C., Wilson J.M., et al. Regulation of manganese superoxide dismutase by lipopolysaccharide, interleukin-1, and tumor necrosis factor. J Biol Chem. 1990;265:2856–2864.
68. Fukai T., Folz R.J., Landmesser U., et al. Extracellular superoxide dismutase and cardiovascular disease. Cardiovasc Res. 2002;55:239–249.
69. Segui J., Gironella M., Sans M., et al. Superoxide dismutase ameliorates TNBS-induced colitis by reducing oxidative stress, adhesion molecule expression, and leukocyte recruitment into the inflamed intestine. Leukoc Biol. 2004;76:537–544.
70. Davis J.M., Parad R.B., Michele T., et al. Pulmonary outcome at 1 year corrected age in premature infants treated at birth with recombinant human CuZn superoxide dismutase. Pediatrics. 2003;111:469–476.
71. Campana F., Zevoudis S., Perdereau B., et al. Topical superoxide dismutase reduces post-irradiation breast cancer fibrosis. J Cell Mol Med. 2004;8:109–116.
72. Deng H.X., Hentati A., Tainer J.A., et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science. 1993;261:1047–1051.
73. Massaad C.A., Washington T.M., Paulter R.G., et al. Overexpression of SOD2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:13576–13581.
74. Muzykantov V.R. Delivery of antioxidant enzyme proteins to the lung. Antioxid Redox Signal. 2001;3:39–62.
75. Sanders L.M., Henderson C.E., Hong M.Y., et al. Pro-oxidant environment of the colon compared to the small intestine may contribute to greater cancer susceptibility. Cancer Lett. 2004;208:155–161.
76. Jones J.B., Cramer H.M., Inch W.R., et al. Radioprotective effect of free radical scavenging enzymes. J Otolaryngol. 1990;19:299–306.
77. Kobayashi S., Inoue N., Azumi H., et al. Expressional changes of the vascular antioxidant system in atherosclerotic coronary arteries. J Atheroscler Thromb. 2002;9:184–190.
78. Ho Y.S., Xiong Y., Ma W., et al. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem. 2004;279:32804–32812.
79. Lu J., Holmgren A. Selenoproteins. J Biol Chem. 2009;284:723–727.
80. Comhair S.A., Erzurum S.C. Antioxidant responses to oxidant-mediated lung diseases. Am J Physiol Lung Cell Mol Physiol. 2002;283:L246–L255.
81. Muller F.L., Lustgarten M.S., Jang Y., et al. Trends in oxidative aging theories. Free Radic Biol Med. 2007;43:477–503.
82. Ran Q., Liang H., Ikeno Y. Reduction in glutathione peroxidase 4 increases lifespan through increased sensitivity to apoptosis. J Gerontol A Biol Sci Med Sci. 2007;62:932–942.
83. Kisucka J., Chauhan A.K., Patten I.S., et al. Peroxiredoxin-1 prevents excessive activation and early atherosclerosis. Circ Res. 2008;103:598–605.
84. Diet A., Abbas K., Bouton C., et al. Regulation of peroxiredoxins by nitric oxide in immunostimulated macrophages. J Biol Chem. 2007;282:36199–36205.
85. Krsek-Staples J.A., Webster R.O. Ceruloplasmin inhibits carbonyl formation in endogenous cell proteins. Free Radic Biol Med. 1993;14:115–125.
86. Kondoh M., Inoue Y., Atagi S., et al. Specific induction of metallothionein synthesis by mitochondrial oxidative stress. Life Sci. 2001;69:2137–2146.
87. Zhou Z., Sun X., Kang Y.J. Metallothionein protection against alcoholic liver injury through inhibition of oxidative stress. Exp Biol Med. 2002;227:214–222.
88. Moschos M.P. Selenoprotein P. Cell Mol Life Sci. 2000;57:1836–1845.
89. Ballatori N., Krance S.M., Notenboom S., et al. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. 2009;390:191–214.
90. Peristeris P., Clark B.D., Gatti S., et al. N-acetylcysteine and glutathione as inhibitors of tumor necrosis factor production. Cell Immunol. 1992;140:390–399.
91. Nordberg J., Arner E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–1312.
92. Luo M., He H., Kelley M.R., et al. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxid Redox Signal. 2010;12:1247–1269.
93. Wei Y.H., Lee H.C. Oxidative stress, mitochondrial DNA mutation, and impairment of antioxidant enzymes in aging. Exp Biol Med. 2002;227:671–682.
94. Ansari M.A., Scheff S.W. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69:155–167.
95. Kaspar J.W., Niture S.K., Jaiswal A.K. Nrf2:Inrf2(keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309.
96. Krinsky N.I. Mechanism of action of biological antioxidants. Proc Soc Exp Biol Med. 1992;200:248–254.
97. Frei B., England L., Ames B.N. Ascorbate is an outstanding antioxidant in human blood plasma. Proc Natl Acad Sci U S A. 1989;86:6377–6381.
98. Chakraborty S., Nandi A., Mukhopadhyay M., et al. Ascorbate protects guinea pig tissues against lipid peroxidation. Free Radic Biol Med. 1994;16:417–426.
99. Levine M., Wang Y., Padayatty S.J., et al. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci U S A. 2001;98:9842–9846.
100. Calabrese E.J., Stoddard A., Leonard D.A., et al. The effect of vitamin C supplementation on blood and hair levels of cadmium, lead, and mercury. Ann N Y Acad Sci. 1987;498:347–353.
101. Losonczy K.G., Harris T.B., Havlik R.J. Vitamin E and vitamin C supplement use and risk of all-cause and coronary heart disease mortality in older persons: the Established Populations for Epidemiologic Studies of the Elderly. Am J Clin Nutr. 1996;64:190–196.
102. Bjelakovic G., Nikolova D., Gluud L.L., et al. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systemic review and meta-analysis. JAMA. 2007;297:842–857.
103. Knekt P., Ritz J., Pereiara M.A., et al. Antioxidant vitamins and coronary heart disease risk: a pooled analysis of 9 cohorts. Am J Clin Nutr. 2004;80:1508–1520.
104. Osganian S.K., Stampfer M.J., Rimm E., et al. Vitamin C and risk of coronary heart disease in women. J Am Coll Cardiol. 2003;42:246–252.
105. Lee D.H., Folsom A.R., Harnack L., et al. Does supplemental vitamin C increase cardiovascular disease risk in women with diabetes? Am J Clin Nutr. 2004;80:1194–1200.
106. Sesso H.D., Buring J.E., Christen W.G., et al. Vitamins E and C in the prevention of cardiovascular disease and cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2008;300:2123–2133.
107. Block G., Jensen C.D., Norkus E.P., et al. Vitamin C in plasma is inversely related to blood pressure and change in blood pressure during the previous year in young Black and White women. Nutr J. 2008;7:35. PMCID: PMC2621233
108. Hajjar I.M., George V., Sasse E.A., et al. A randomized, double-blind, controlled trial of vitamin C in the management of hypertension and lipids. Am J Ther. 2002;9:289–293.
109. Negri E., Franceschi S., Bosetti C., et al. Selected micronutrients and oral and pharyngeal cancer. Int J Cancer. 2000;86:122–127.
110. Fleischauer A.T., Olson S.H., Mignone L., et al. Dietary antioxidants, supplements, and risk of epithelial ovarian cancer. Nutr Cancer. 2002;40:92–98.
111. You W.C., Zhang L., Gail M.H., et al. Gastric dysplasia and gastric cancer: Helicobacter pylori, serum vitamin C, and other risk factors. J Natl Cancer Inst. 2000;92:1607–1612.
112. Cho E., Hunter D.J., Spiegelman D., et al. Intakes of vitamin A, C and E and folate and multivitamins and lung cancer: a pooled analysis of 8 prospective studies. Int J Cancer. 2006;118:970–978.
113. Gaziaono J.M., Glynn R.J., Christen W.G., et al. Vitamins E and C in the prevention of prostate and total cancer in men: The Physicians’ Health Study II randomized controlled trial. JAMA. 2009;301:52–62.
114. Chen Q., Espey M.G., Sun A.Y., et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A. 2008;105:11105–11109.
115. Engelhart M.J., Geerlings M.I., Ruitenberg A., et al. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002;287:3223–3229.
116. Bowman G.L., Dodge H., Frei B., et al. Ascorbic acid and rates of cognitive decline in Alzheimer’s disease. J Alzheimers Dis. 2009;16:93–98.
117. Age-Related Eye Disease Study Research Group. AREDS report no. 9. A randomized, placebo-controlled trial of high dose supplementation with vitamins C, E and beta carotene for age-related cataract and vision loss. Arch Ophthalmol. 2001;119:1439–1452.
118. Harding A.H., Wareham N.J., Bingham S.A., et al. Plasma vitamin C level, fruit and vegetable consumption, and the risk of new onset type 2 diabetes mellitus: European prospective investigation of cancer – Norfolk Prospective Study. Arch Intern Med. 2008;168:1493–1499.
119. Estaban M.A., Wang I., Qin B., et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell. 2010;6:71–79.
120. Grant M.M., Mistry N., Lunec J., et al. Dose-dependent modulation of the T cell proteome by ascorbic acid. Br J Nutr. 2007;97:19–26.
121. Chan A.C. Partners in defense, vitamin E and vitamin C. Can J Physiol Pharmacol. 1993;71:725–731.
122. Meydani S.N., Meydani M., Blumberg J.B., et al. Vitamin E supplementation and in vivo immune response in healthy elderly subjects. JAMA. 1997;277:1380–1386.
123. Tsoureli-Nikita E., Hercogova J., Lotti T., et al. Evaluation of dietary intake of vitamin E in the treatment of atopic dermatitis: a study of the clinical course and evaluation of the immunoglobulin E serum levels. Int J Dermatol. 2002;41:146–150.
124. Stampfer M.J., Hennekens C.H., Manson J.E., et al. Vitamin E consumption and the risk of coronary disease in women. N Engl J Med. 1993;328:1444–1449.
125. Sesso H.D., Buring J.E., Christen W.G., et al. Vitamins E and C in the prevention of cardiovascular disease and cancer in men: the Physicians’ Health Study II randomized controlled trial. JAMA. 2008;300:2123–2133.
126. Frei B. Efficacy of dietary antioxidants to prevent oxidative damage and inhibit chronic disease. J Nutr. 2004;134:S3196–S3198.
127. Milman U., Blum S., Shapira C., et al. Vitamin E supplementation reduces cardiovascular events in a sub group of middle-aged individuals with both type 2 diabetes mellitus and the haptoglobin G2-2 genotype. Arterioscler Thromb Vasc Biol. 2008;28:341–347.
128. Sano M., Ernesto C., Thomas R.G., et al. A controlled trial of selegiline, alpha tocopherol, or both as treatment for Alzheimer’s disease. N Engl J Med. 1997;336:1216–1222.
129. Petersen R.C., Thomas R.G., Grundman M., et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352:2379–2388.
130. Dorjgochoo T., Shrubsole M.J., Suh X.O., et al. Vitamin supplement use and risk of breast cancer: the Shanghai Breast Cancer Study. Breast Cancer Res Treat. 2008;111:269–278.
131. Qiao Y.-L., Dawsey S.M., Kamangar F., et al. Total and cancer mortality after supplementation with vitamins and minerals: follow-up of the Linxian General Population Intervention Trial. J Natl Cancer Inst. 2009;101:507–518.
132. Hunter D.J., Manson J.E., Colditz G.A., et al. A prospective study of the intake of vitamins C, E and A and the risk of breast cancer. N Engl J Med. 1993;329:234–240.
133. Cui X., Rosner B., Wilett W.C., et al. Antioxidant intake and risk of endometrial cancer: results from the Nurses’ Health Study. Int J Cancer. 2011;128:1169–1178.
134. Slatore C.G., Littman A.J., Au D.H., et al. Long term use of supplemental multivitamins vitamin C, vitamin E, and folate does not reduce the risk of lung cancer. Am J Respir Crit Care Med. 2008;177:524–530.
135. Lin J., Cook N.R., Albert C., et al. Vitamins C and E and beta carotene supplementation and cancer risk: a randomized, controlled trial. J Natl Cancer Inst. 2009;101:14–23.
136. Klein E.A., Thompson I.M., Tangen I.M., Jr., et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011;306:1549–1556.
137. Wright M.E., Peters U., Gunter M.J., et al. Association of variants of two vitamin E transport genes with circulating vitamin E concentrations and prostate cancer risk. Cancer Res. 2009;69:1429–1438.
138. Rizzo M.R., Abbatecola A.M., Barbieri M., et al. Evidence for anti-inflammatory effects of combined administration of vitamin E and C in older persons with impaired fasting glucose: impact on insulin action. J Am Coll Nutr. 2008;27:505–511.
139. Christen S., Woodall A.A., Shigenaga M.K., et al. Gamma tocopherol traps mutagenic electrophiles such as Nox and complements alpha tocopherol. Physiological implications. Proc Natl Acad Sci U S A. 1997;94:3217–3222.
140. Sjoholm A., Berggren P.O., Coney R.V. Gamma tocopherol partially protects insulin-secreting cell against functional inhibition by nitric oxide. Biochem Biophy Res Commun. 2000;277:334–340.
141. Park S.K., Page G.P., Kim K., et al. Differential effect of alpha- and gamma-tocopherol supplementation in age-related transcriptional alterations in heart and brain of B6/C3H mice. J Nutr. 2008;138:1010–1018.
142. Devaraj S., Leonard S., Traber M.G., et al. Gamma-tocopherol supplementation alone or in combination with alpha-tocopherol alters biomarkers of oxidative stress and inflammation in subjects with metabolic syndrome. Free Radica Biol Med. 2008;44:1203–1208.
143. Key T.J., Appleby P.N., Allen N.E., et al. Plasma carotenoids, retinol, and tocopherols and the risk of prostate cancer in the European Prospective Investigation into Cancer and Nutrition Study. Am J Clin Nutr. 2007;86:672–681.
144. Podda M., Weber C., Traber M.G., et al. Simultaneous determination of tissue tocopherols, tocotrienols, ubiquinols and ubiquinones. J Lipid Res. 1996;37:893–901.
145. Bachawal S.V., Wali V.B., Sylvester P.W. Enhanced antiproliferative and apoptotic response to combined treatment of gamma-tocotrienol with erlotinib or gefitinib in mammary tumor cells. BMC Cancer. 2010;10:84–97.
146. Yam M.L., Abdul Haifid S.R., Cheng H.M., et al. Tocotrienols suppress proinflammatory markers and cyclooxygenase-2 expression in RAW264.7 macrophages. Lipids. 2009;44:787–797.
147. Mustad V.A., Smith C.A., Ruey P.P., et al. Supplementation with 3 compositionally different tocotrienol supplements does not improve cardiovascular disease risk factors in men and women with hypercholesterolemia. Am J Clin Nutr. 2002;76:1237–1243.
148. Chin S.F., Hamid N.A., Latiff A.A., et al. Reduction of DNA damage in older healthy adults by Tri E Tocotrienol supplementation. Nutrition. 2008;24:1–10.
149. Han S.N., Adolfsson O., Lee C.K., et al. Age- and vitamin E-induced changes in gene expression profiles of T cells. J Immunol. 2006;177:6052–6061.
150. Tan B.H., Sagoo P., Chan C., et al. Inhibition of NF-kB and oxidative pathways in human dendritic cells by antioxidant vitamins generates regulatory T cells. J Immunol. 2005;174:7633–7644.
151. Belisle S.E., Leka L.S., Delgado-Lista J., et al. Polymorphisms at cytokine genes may determine the effect of vitamin E on cytokine production in the elderly. J Nutr. 2009;139:1855–1860.
152. Traber M.G., Atkinson J. Vitamin E, antioxidant and nothing more. Free Radic Biol Med. 2007;43:4–15.
153. Vile G.F., Winterbourn C.C. Inhibition of adriamycin-promoted microsomal lipid peroxidation by beta-carotene, alpha-tocopherol and retinal at high and low oxygen partial pressures. FEBS Lett. 1988;238:353–356.
154. Jialal I., Norkus E.P., Cristol L., et al. Beta carotene inhibits the oxidative modification of low-density lipoprotein. Biochim Biophys Acta. 1991;1086:134–138.
155. Rock C.L. Carotenoids: biology and treatment. Pharmacol Ther. 1997;75:185–197.
156. Bendich A. Carotenoids and the immune response. J Nutr. 1989;119:112–115.
157. Mannisto S., Smith-Warner S.A., Spiegelman D., et al. Dietary carotenoids and risk of lung cancer in a pooled analysis of seven cohort studies. Cancer Epidemiol Biomarkers Prev. 2004;13:40–48.
158. Gallichio L., Boyd K., Matanoski G., et al. Carotenoids and the risk of developing lung cancer: a systematic review. Am J Clin Nutr. 2008;88:372–383.
159. Etminan M., Takkouche B., Caamano-Isora F. The role of tomato product and lycopene in the prevention of prostate cancer: a meta-analysis of observational studies. Cancer Epidemiol Biomarkers Prev. 2004;13:340–345.
160. Zhang S., Hunter D.J., Forman M.R., et al. Dietary carotenoids and vitamins A, C, and E and risk of breast cancer. J Natl Cancer Inst. 91, 1999. 547:556
161. Larsson S.C., Bergkvist L., Wolk A. Dietary carotenoids and risk of hormone-receptor defined breast cancer in a prospective cohort of Swedish women. Eur J Cancer. 2010;46:1079–1085.
162. Seddon J.M., Ajani U.A., Sperduto R.D., et al. Dietary carotenoids, vitamins A, C and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group. JAMA. 1994;272:1413–1420.
163. Coleman H., Chew E. Nutritional supplementation in age-related macular degeneration. Curr Opin Opthalmol. 2007;18:220–223.
164. Erdman J.W., Jr., Ford N.A., Lindshield B.L. Are the health attributes of lycopene related to its antioxidant function? Arch Biochem Biophysics. 2009;483:229–235.
165. Chalab N., Delort L., Satih S., et al. Immunohistochemical expression of RARalpha, RARbeta and CX43 in breast tumor cell lines after treatment with lycopene and correlation with RT-QPCR. J Histochem Cytochem. 2007;55:877–883.
166. Maguire J.J., Kagan V., Ackrell B.A., et al. Succinate-ubiquinone reductase linked recycling of alpha-tocopherol in reconstituted systems and mitochondria: requirement for reduced ubiquinone. Arch Biochem Biophys. 1992;292:47–53.
167. Stocker R., Bowry V.W., Frei B. Ubiquinol-10 protects human low density lipoproteins more efficiently against lipid peroxidation than does alpha tocopherol. Proc Natl Acad Sci U S A. 1991;88:1646–1650.
168. Niklowitz P., Sonnenshein A., Janetzky B., et al. Enrichment of coenzyme Q10 in plasma and blood cells. Defense against oxidative damage. Int J Biol Sci. 2007;3:257–262.
169. Witting P.K., Pettersson K., Letters J., et al. Anti-atherogenic effect of coenzyme Q10 in apolipoprotein E gene knockout mice. Free Radic Biol Med. 2000;29:295–305.
170. Shargorodsky M., Debbi O., Matas Z., et al. Effect of long term treatment with antioxidants (vitamin C, vitamin E, coenzyme Q10 and selenium) on arterial compliance, humoral factors and inflammatory markers in patients with multiple cardiovascular risk factors. Nutr Metab. 2010;7:55. PMCID: PMC2911454
171. Molyneux S.L., Florkowski C.M., Richards A.M., et al. Coenzyme Q10: an adjunctive therapy for congestive heart failure? N Z Med J. 2009;122:74–79.
172. De Pinieux G., Chariot P., Ammi-Said M., et al. Lipid-lowering drugs and mitochondrial function: effects of HMG-CoA reductase inhibitors on serum ubiquinone and blood lactate/pyruvate ratio. Br J Clin Pharmacol. 1996;42:333–337.
173. Rosenfeldt F.L., Haas S.J., Krum H., et al. Coenzyme Q10 in the treatment of hypertension. A meta-analysis of the clinical trials. J Hum Hypertens. 2007;21:297–306.
174. Mortensen S.A. Overview on coenzyme Q10 as adjunctive therapy in chronic heart failure. Rationale, design and endpoints of “Q-symbio” – a multinational trial. Biofactors. 2003;18:79–89.
175. Ziegler D., Nowak H., Kempler P., et al. Treatment of symptomatic diabetic neuropathy with the antioxidant alpha lipoic acid: a meta-analysis. Diabetic Med. 2004;21:114–121.
176. Smith A.R., Shenvi S.V., Widlansky M., et al. Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr Med Chem. 2004;11:1135–1146.
177. Luz J., Zemdengs J.C.S., Amaral L.S.G. Chronic lipoic acid treatment worsens energy imbalances in streptozotocin-induced diabetic rats. Diabetic Metab. 2009;35:137–142.
178. Liu J., Killilea D.W., Ames B.N. Age-associated mitochondrial oxidative decay: improvement of carnitine acetyltransferase substrate binding affinity and activity in brain by feeding old rats acetyl-L-carnitine and/or R-alpha-lipoic acid. Proc Natl Acad Sci U S A. 2002;99:1876–1881.
179. Arivazhagan P., Shila S., Kumaran S., et al. Effect of DL-alpha-lipoic acid on the status of lipid peroxidation and antioxidant enzymes in various brain regions of aged rats. Exp Gerontol. 2002;37:803–811.
180. Aliev G., Liu J., Shenk J.C., et al. Neuronal mitochondrial amelioration by feeding acetyl-L-carnitine and lipoic acid to aged rats. J Cell Mol Med. 2009;13:320–333.
181. Wang Y., Li X., Guo Y., et al. Alpha lipoic acid increases energy expenditure by enhancing adenosine monophosphate-activated protein kinase-peroxisome proliferator-activated receptor gamma coactivator-1-alpha signaling in the skeletal muscle of aged rats. Metabolism. 2010;59:967–976.
182. Suh J.H., Shigeno E.T., Morrow J.D., et al. Oxidative stress in the aging rat heart is reversed by dietary supplementation with (R)-(alpha)-lipoic acid. FASEB J. 2001;15:700–706.
183. Zhang W.J., Bird K.E., McMillen T.S., et al. Dietary lipoic acid supplementation inhibits atherosclerotic lesion development in Apolipoprotein E deficient and Apolipoprotein E/low density lipoprotein receptor deficient mice. Circulation. 2008;117:421–428.
184. Li C.-J., Zhang Q.M., Zhang J., et al. Attenuation of myocardial apoptosis by alpha lipoic acid through suppression of oxidative stress to reduce diabetic cardiomyopathy. Chinese Med J. 2009;122:2580–2586.
185. Carbonelli M.G., Di Renzo L., Bigioni M., et al. Alpha lipoic acid supplementation: a tool for obesity therapy? Curr Pharm Des. 2010:16840–16846.
186. Maczurek A., Hager K., Kenlies M., et al. Lipoic acid as an anti-inflammatory and neuroprotective treatment for Alzheimer’s disease. Adv Drug Deliv Rev. 2008;60:1463–1470.
187. Suh J.H., Shenvi S.V., Dixon B.M., et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible by lipoic acid. Proc Natl Acad Sci USA. 2004;101:3381–3386.
188. Zhang L., Xing G.Q., Barker J.L., et al. Alpha lipoic acid protects rat cortical neurons against cell death induced by amyloid and hydrogen peroxide through the Akt signaling pathway. Neurosci Lett. 2001;312:125–128.
189. Yim C.Y., Hibbs J.B., Jr., McGregor J.R., et al. Use of N-acetyl cysteine to increase intracellular glutathione during induction of antitumor responses by IL-2. J Immunol. 1994;152:5796–5805.
190. Roes E.M., Raijmakers M.T., Peters W.H., et al. Effects of oral N-acetylcysteine on plasma homocysteine and whole blood glutathione levels in healthy, non-pregnant women. Clin Chem Lab Med. 2002;40:496–498.
191. Wu G., Fang Y.Z., Yang S., et al. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492.
192. Iantomasi T., Favilli F., Marraccini P., et al. Glutathione transport system in human small intestine epithelial cells. Biochim Biophys Acta. 1997;1330:274–283.
193. Zembron-Lacny A., Ostapiuk J., Szyszka K. Effects of sulfur-containing compounds on plasma redox status in muscle damaging exercise. Chin J Physiol. 2009;52:289–294.
194. Martina V., Masha A., Gigliardi V.R., et al. Long-term N-acetylcysteine and L-arginine administration reduces endothelial activation and systolic blood pressure in hypertensive patients with Type 2 diabetes. Diabetes Care. 2008;31:940–943.
195. Ames B.N. Low micronutrient intake may accelerate the degenerative diseases of aging though allocation of scarce micronutrients by triage. Proc Natl Acad Sci U S A. 2006;103:17589–17594.
196. Tejero-Taldo M.I., Kramer J.H., Mak I., et al. The nerve-heart connection in the pro-oxidant response to Mg-deficiency. Heart Fail Rev. 2006;11:35–44.
197. Killilea D.W., Ames B.N. Magnesium deficiency accelerates cellular senescence in culture human fibroblasts. Proc Natl Acad Sci U S A. 2008;105:5768–5773.
198. Mazur A., Maier J.A., Rock E., et al. Magnesium and the inflammatory response: potential physiopathological implications. Arch Biochem Biophys. 2007;458:48–56.
199. Rayssiguier Y., Gueux E., Nowacki W., et al. High fructose consumption combined with low dietary magnesium intake may increase the incidence of the metabolic syndrome by inducing inflammation. Magnes Res. 2006;19:237–243.
200. Truong-Tan A.Q., Ho L.H., Chai F., et al. Cellular zinc fluxes and the regulation of apoptosis/gene-directed cell death. J Nutr. 2000;130(suppl 5):S1459–S1466.
201. Prasad A.S. Zinc: mechanisms of host defense. J Nutr. 2007;137:1345–1349.
202. Shen H., Oesterling E., Stromberg A., et al. Zinc deficiency induces vascular pro-inflammatory parameters associated with NF-kappaB and PAR signaling. J Am Coll Nutr. 2008;27:577–587.
203. Fortes C., Forastiere F., Agabiti N., et al. The effect of zinc and vitamin A supplementation on immune response in an older population. J Am Geriatr Soc. 1998;46:19–26.
204. Prasad A.S. Clinical, immunological, anti-inflammatory and antioxidant roles of zinc. Exp Gerontol. 2008;43:370–377.
205. Ames B.N., Liu J. Delaying the mitochondrial decay of aging with acetylcarnitine. Ann NY Acad Sci. 2004;1033:108–116.
206. Savitha S., Sivarajan K., Haripriya D., et al. Efficacy of levo-carnitine and alpha lipoic acid in ameliorating the decline in mitochondrial enzymes during aging. Clin Nutr. 2005;24:794–800.
207. Liu J., Head E., Gharib A.M., et al. Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: partial reversal by feeding acetyl-L-carnitine and/or alpha-lipoic acid. Proc Natl Acad Sci U S A. 2002;99:2356–2361.
208. Savitha S., Panneerselvan C. Mitigation of age-dependent oxidative damage to DNA in rat heart by carnitine and lipoic acid. Mech Ageing Dev. 2007;128:206–212.
209. Ames B.N., Cathcart R., Schwiers E., et al. Uric acid provides an antioxidant defense in humans against oxidant and radical caused aging and cancer: a hypothesis. Proc Natl Acad Sci U S A. 1981;78:6858–6862.
210. Wayner D.D., Burton G.W., Ingold K.U., et al. Quantitative measurement of the total peroxyl radical-trapping antioxidant capability of human blood plasma by controlled peroxidation. FEBS Lett. 1985;187:33–37.
211. Kampa M., Nistikaki A., Tsaousis V., et al. A new automated method for the determination of the Total Antioxidant Capacity (TAC) of human plasma, based on the crocin bleaching assay. BMC Clin Pathol. 2002;2:3. PMCID: PMC128814
212. McDonagh A.F. The biliverdin-bilirubin antioxidant cycle of cellular protection: missing a wheel? Free Radic Biol Med. 2010;49:814–820.
213. Stetinova V., Smetanova L., Grossmann V., et al. In vitro and in vivo assessment of the antioxidant activity of melatonin and related indole derivatives. Gen Physiol Biophys. 2002;21:153–162.
214. Okatani Y., Wakatsuki A., Reiter R.J., et al. Hepatic mitochondrial dysfunction in senescence-accelerated mice: correction by long-term, orally administered physiological levels of melatonin. J Pineal Res. 2002;33:127–133.
215. Aydogan S., Yerer M.B., Goktas A. Melatonin and nitric oxide. J Endocrinol Invest. 2006;29:281–287.
216. Murawska-Cialowicz E., Jethon Z., Magdalen J., et al. Effects of melatonin on lipid peroxidation and antioxidative enzyme activities in the liver, kidneys and brain of rats administered with benzo(a)pyrene. Exp Toxicol Pathol. 2009;63:97–103.
217. Winiarska K., Fraczyk T., Malinska D., et al. Melatonin attenuates diabetes-induced oxidative stress in rabbits. J Pineal Res. 2006;40:168–176.
218. Tapiero H., Tew K.D., Ba G.N., et al. Polyphenols: do they play a role in the prevention of human pathologies? Biomed Pharmacother. 2002;56:200–207.
219. Manach C., Williamson G., Morand C., et al. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr. 2005;81(suppl):S230–S242.
220. Ovaskainen M.L., Torronen R., Koponen J.M., et al. Dietary intake and major food sources of polyphenols in Finnish adults. J Nutr. 2008;138:562–566.
221. Zamora-Ros R., Andres-Lacueva C., Lamuela-Raventos R.M., et al. Estimation of dietary source of flavonoid intake in Spanish adult population (EPIC-Spain). J Am Diet Assoc. 2010;110:390–398.
222. Kidd P.M. Bioavailability and activity of phytosome complexes from botanical polyphenols: the silymarin, curcumin, green tea and grape seed extracts. Alt Med Rev. 2009;14:226–246.
223. Liu Z., Hu M. Natural polyphenol disposition via coupled metabolic pathways. Expert Opin Drug Metab Toxicol. 2007;3:389–406.
224. Loke W.M., Jenner A.M., Proudfoot J.M., et al. A metabolic profiling approach to identify biomarkers of flavonoid intake in humans. J Nutr. 2009;139:2309–2314.
225. Rice-Evans C.A., Miller N.J., Pagangu G. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Bio Med. 1996;20:933–956.
226. Roy M.K., Coide M., Rao T.P., et al. ORAC & DPPH assay comparison to assess antioxidant capacity of tea infusions: relationship between total polyphenol and individual catechin content. Int J Food Sci Nutr. 2010;61:109–124.
227. De Whalley C.V., Rankin S.M., Hoult J.R., et al. Flavonoids inhibit the oxidative modification of low density lipoproteins by macrophages. Biochem Pharmacol. 1990;39:1743–1750.
228. Afanas’ev I.B., Dorozhko A.I., Brodskii A.V., et al. Chelating and free radical scavenging mechanisms of inhibitory action of rutin and quercetin in lipid peroxidation. Biochem Pharmacol. 1989;38:1763–1769.
229. Lotito S.B., Frei B. Consumption of flavonoid-rich foods and increased plasma antioxidant capacity in humans: cause, consequence or epiphenomenon? Free Radic Biol Med. 2006;41:1727–1746.
230. Lotito S.B., Frei B. Relevance of apple polyphenols as antioxidants in human plasma: contrasting in vitro and in vivo effects. Free Radic Biol Med. 2004;36:201–211.
231. Lotito S.B., Frei B. The increase in plasma antioxidant capacity after apple consumption is due to the metabolic effect of fructose on urate, not apple-derived antioxidant flavonoids. Free Radic Biol Med. 2004;37:251–258.
232. Cutler G.J., Nettleton J.A., Ross J.A., et al. Dietary flavonoid intake and risk of cancer in postmenopausal women: the Iowa Women’s Health Study. Int J Cancer. 2008;123:664–671.
233. Arts I.C., Hollman P.C., Bueno De Mesquita H.B., et al. Dietary catechins and epithelial cancer incidence: the Zutphen elderly study. Int J Cancer. 2001;92:298–302.
234. Hertog M.G., Feskens E.J., Hollman P.C., et al. Dietary flavonoids and cancer risk in the Zutphen elderly study. Nutr Cancer. 1994;22:175–184.
235. Hertog M.G., Feskens E.J., Hollman P.C., et al. Dietary antioxidant flavonoids and risk of coronary heart disease: the Zutphen elderly study. Lancet. 1993;342:1007–1011.
236. Su S.J., Chow N.H., Kung M.L., et al. Effects of soy isoflavones on apoptosis induction and G2-M arrest in human hepatoma cells involvement of capsase-3 activation, Bcl-2 and Bcl-XL downregulation and Cdc2 activity. Nutr Cancer. 2003;45:113–123.
237. Sarkar F.H., Li Y. Soy isoflavones and cancer prevention. Cancer Invest. 2003;21:744–757.
238. Yan L., Spitznagel E.L. Soy consumption and prostate cancer risk in men: a revisit of a meta-analysis. Am J Clin Nutr. 2009;89:1156–1163.
239. Ju J., Lu G., Lambert J.D., et al. Inhibition of carcinogenesis by tea constituents. Semin Cancer Biol. 2007;17:395–402.
240. Qiao Y., Cao J., Xie L., et al. Cell growth inhibition and gene expression regulation by (-)-epigallocatechin-3-gallate in human cervical cancer cells. Arch Pharm Res. 2009;32:1309–1315.
241. Shimizu M., Shirakami Y., Moriwaki H. Targeting receptor tyrosine kinases for chemoprevention by green tea catechin, EGCG. Int J Mol Sci. 2008;9:1034–1049.
242. Klaunig J.E., Xu Y., Han C., et al. The effect of tea consumption on oxidative stress in smokers and nonsmokers. Proc Soc Exp Biol Med. 1999;220:249–254.
243. Yang C.S., Wang X., Lu G., et al. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat Rev Cancer. 2009;9:429–439.
244. Wu A.H., Tseng C.C., Van Den Berg D., et al. Tea intake, COMT genotype, and breast cancer in Asian-American women. Cancer Res. 2003;63:7526–7529.
245. Bettuzzi S., Brausi M., Rizzi F., et al. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high grade prostate interepithelial neoplasia: a preliminary report from a one year proof of principle study. Cancer Res. 2006;66:1234–1240.
246. Hoensch H., Groh B., Edler L, Kirch W. Prospective cohort comparison of flavonoid treatment in patients with resected colorectal cancer to prevent recurrence. World J Gastroenterol. 2008;14:2187–2193.
247. Mink P.J., Scrafford C.G., Barraj L.M., et al. Flavonoid intake and the risk of cardiovascular disease in postmenopausal women. Am J Clin Nutr. 2007;85:895–909.
248. McCullough M.L., Peterson J.J., Patel R., et al. Flavonoid intake and cardiovascular disease mortality in a prospective cohort of US adults. AM J Clin Nutr. 2012;95:454–464.
249. Lin J., Rexrode K.M., Hu F., et al. Dietary intakes of flavanols and flavones and coronary heart disease in US women. Am J Epidemiol. 2007;165:1305–1313.
250. Rimm E.B., Katan M.B., Ascherio A., et al. Relation between intake of flavonoids and risk of coronary heart disease in male health professionals. Ann Intern Med. 1996;125:384–389.
251. Whitehead T.P., Robinson D., Allaway S., et al. Effect of red wine ingestion on the antioxidant capacity of serum. Clin Chem. 1995;41:32–35.
252. Simonetti P., Gardana C., Pietta P. Plasma levels of caffeic acid and antioxidant status after red wine intake. J Agric Food Chem. 2001;49:5964–5968.
253. Hooper L., Kroon P.A., Rimm E.B., et al. Flavonoids, flavonoid-rich foods, and cardiovascular risk: a meta-analysis of randomized controlled trials. Am J Clin Nutr. 2008;88:38–50.
254. Hollis J.H., Houchins J.A., Blumberg J.B., et al. Effects of concord grape juice on appetite, diet, body weight, lipid profile and antioxidant status in adults. J Am Coll Nutr. 2009;289:574–582.
255. Wang-Polagruto J.F., Villablanca A.C., Polagruto J.A., et al. Chronic consumption of flavanol-rich cocoa improves endothelial function and decreases vascular adhesion molecule in hypercholesterolemic postmenopausal women. J Cardiovasc Pharmacol. 2006;47(suppl 2):S177–S186.
256. Njike V.Y., Faridi Z., Shuval K., et al. Effect of sugar-sweetened and sugar-free cocoa on endothelial function in overweight adults. Int J Cardiol. 2011;149:83–88.
257. Monagas M., Kahan N., Andres-Lacueve C., et al. Effect of cocoa powder on the modulation of inflammatory biomarkers in patients of cardiovascular disease. Am J Clin Nutr. 2009;90:1144–1150.
258. Skrzydlewska E., Ostrowska J., Stankiewicz A., et al. Green tea as a potent antioxidant in alcohol intoxication. Addict Biol. 2002;7:307–314.
259. Loke W.M., Proudfoot J.M., Hodgson J.M., et al. Specific dietary polyphenols attenuate atherosclerosis in apolipoprotein E-knockout mice by alleviating inflammation and endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2010;30:749–757.
260. Peters U., Poole C., Arab L. Does tea affect cardiovascular disease? A meta-analysis. Am J Epidemiol. 2001;154:495–503.
261. Arab L., Liu W., Elashoff D. Green and black tea consumption and risk of stroke: a meta-analysis. Stroke. 2009;40:1786–1792.
262. Joy S., Siow R.C., Rowlands D.J., et al. The isoflavone equol mediates rapid vascular relaxation: Ca-independent activation of endothelial nitric oxide synthase involving ERK1/2 and Akt phosphorylation in human endothelial cells. J Biol Chem. 2006;281:27335–27345.
263. Mann G.E., Bonacasa B., Ishi T.Y., et al. Targeting the redox sensitive Nrf2-Keap1 pathway in cardiovascular disease: protection afforded by dietary isoflavones. Curr Opin Pharmacol. 2009;9:139–145.
264. Hall W.L., Formanuik N.L., Harnparich D.M., et al. A meal enriched with soy isoflavones increases nitric oxide mediated vasodilation in healthy postmenopausal women. J Nutr. 2008;138:1288–1292.
265. Marini H., Bitto A., Altavilla D., et al. Efficacy of genistein aglycone on some cardiovascular risk factors and homocysteine levels: a follow-up study. Nutr Metab Cardiovasc Dis. 2010;20:332–340.
266. Kim H.P., Mani I., Iversen L., et al. Effects of naturally-occurring flavonoids and bioflavonoids on epidermal cyclooxygenase and lipoxygenase from guinea pigs. Prostaglandins Leukot Essent Fatty Acids. 1998;58:17–24.
267. Lotito S.B., Frei B. Dietary flavonoids attenuate tumor necrosis factor-alpha induced adhesion molecule expression in human aortic endothelial cells. J Biol Chem. 2006;281:37102–37110.
268. Shanely R.A., Knab A.M., Nieman D.C., et al. Quercetin supplementation does not alter antioxidant status in humans. Free Radica Res. 2010;44:224–231.
269. Haleagrahara N., Radhakrishnan A., Lee N., et al. Flavonoid quercetin protects against swimming stress-induced changes in oxidative biomarkers in the hypothalamus of rats. Eur J Pharmacol. 2009;621:46–52.
270. McAnulty S.R., McAnulty L.S., Nieman D.C., et al. Chronic quercetin ingestion and exercise-induced oxidative damage and inflammation. Appl Physiol Nutr Metab. 2008;33:254–262.
271. Davis J.M., Carlstedt C.J., Chen S., et al. The dietary flavonoid quercetin increases VO(2max) and endurance capacity. Int J Sport Nutr Exerc Metab. 2010;20:56–62.
272. Loke W.M., Hodgson J.M., Proudfoot J.M., et al. Pure dietary flavonoids quercetin and (-)-epicatechin augment nitric oxide products and reduce endothelin-1 acutely in healthy men. Am J Clin Nutr. 2008;88:1018–1025.
273. Chien S.Y., Wu Y.C., Chung J.G., et al. Quercetin-induced apoptosis acts through mitochondrial and caspase-3 dependent pathways in human breast cancer MDA-MB-231 cells. Hum Exp Toxicol. 2009;28:493–503.
274. Valerio L.G., Jr., Kepa J.K., Pickwell G.V., et al. Induction of human NADPH quinone oxidoreductase (NQO1) gene expression by the flavonol quercetin. Toxicol Lett. 2001;119:49–57.
275. Kamaraj S., Vinodhkumar R., Anandakumar R., et al. The effects of quercetin on antioxidant status and tumor markers in lung and serum of mice treated with benzo(a)pyrene. Biol Pharmac Bull. 2007;30:2268–2273.
276. Lam T.K., Rotunno M., Lubin J.H., et al. Dietary quercetin, quercetin-gene interaction, metabolic gene expression in lung tissue and lung cancer risk. Carcinogenesis. 2010;31:634–642.
277. Rohdewald P. A review of the French maritime pine bark extract (Pycnogenol), a herbal medication with a diverse pharmacology. Int J Clin Pharmacol Ther. 2002;40:158–168.
278. Cho K.J., Yun C.H., Packer L., et al. Inhibition mechanisms of bioflavonoids extracted from the bark of Pinus maritima on the expression of proinflammatory cytokines. Ann N Y Acad Sci. 2001;928:141–156.
279. Grimm T., Chovanova Z., Muchova J., et al. Inhibition of NF-kappaB and MM-9 secretion by plasma of human volunteers after ingestion of maritime pine bark extract (Pycnogenol). J Inflamm. 2006;3:1. PMCID: PMC1413525
280. Busserolles J., Geux E., Balasinska B., et al. In vivo antioxidant capacity of procyanidin-rich extracts from grape seed and pine (Pinus maritima) bark in rats. Int J Vitamin Nutr Res. 2006;76:22–27.
281. Schafer A., Chovanova Z., Muchova J., et al. Inhibition of COX-1 and COX-2 activity by plasma of human volunteers after ingestion of French maritime pine bark extract (Pycnogenol). Biomed Pharmacother. 2006;60:5–9.
282. Cisar P., Jany R., Waczulikova I., et al. Effect of pine bark extract (Pycnogenol) on symptoms of knee osteoarthritis. Phytother Res. 2008;22:1087–1092.
283. Natella F., Belelli F., Gentili V., et al. Grape seed proanthocyanidins prevent plasma postprandial oxidative stress in humans. J Agric Food Chem. 2002;50:7720–7725.
284. Polagruto J.A., Gross H.B., Kamangar F., et al. Platelet reactivity in male smokers following acute consumption of flavonol-rich grapeseed extract. J Med Food. 2007;10:725–730.
285. Kar P., Laight D., Rooprai H.K., et al. Effects of grape seed extract in Type 2 diabetic subjects at high cardiac risk: a double blind randomized placebo controlled trial examining metabolic markers, vascular tone, inflammation, oxidative stress and insulin sensitivity. Diabet Med. 2009;26:526–531.
286. Bagchi D., Bagchi M., Stohs S.J., et al. Free radicals and grape seed proanthocyanidin extract: importance in human health and disease prevention. Toxicology. 2000;148:187–197.
287. Ono K., Condron M.M., Ho L., et al. Effects of grape seed-derived polyphenols on amyloid beta-protein self-assembly and cytotoxicity. J Biol Chem. 2008;283:32176–32187.
288. Nandakumar V., Singh T., Katiyar S.K. Multi-targeted prevention and therapy by proanthocyanidins. Cancer Lett. 2008;269:378–387.
289. Li W., Zu B., Xu J., et al. Procyanidins produce significant attenuation of doxorubicine-induced cardiotoxicity via suppression of oxidative stress. Basic Clin Pharmacol Toxicol. 2009;104:192–197.
290. Patel J., Joseph C., Corcoran G.B., et al. Silymarin modulates doxorubicine-induced oxidative stress, Bcl-xL and p35 expression while preventing apopotic and necrotic cell death in the liver. Toxicol Appl Pharmacol. 2010;245:143–152.
291. Mroishima C., Shuhart M.C., Wang C.C., et al. Silymarin inhibits in vitro T-cell proliferation and cytokine production in hepatitis C virus infection. Gastroenterol. 2010;138:671–681.
292. Hawke R.L., Schrieber S.J., Soule T.A., et al. Silymarin ascending multiple oral dosing phase I studies in noncirrhotic patients with chronic hepatitis C. J Clin Pharmcol. 2010;50:434–449.
293. Velussi M., Cerrigoi A.M., De Monte A., et al. Long term (12 months) treatment with an antioxidant drug, silymarin, is effective on hyperinsulinemia, exogenous insulin need and malondialdehyde levels in cirrhotic diabetic patients. J Hepatol. 1997;26:871–879.
294. Huseini H.F., Larijani B., Heshmat R., et al. The efficacy of silybum marianum (L) Gaertn. (silymarin) in the treatment of type II diabetes: a randomized, double-blind, placebo-controlled clinical trial. Phytother Res. 2006;20:1036–1039.
295. Singh R.P., Raina K., Sharma G., et al. Silibinin inhibits established prostate tumor growth, progression, invasion, and metastasis and suppresses tumor angiogenesis and epithelial-mesenchymal transition in transgenic adenocarcinoma of the mouse prostate model. Clin Cancer Res. 2008;14:7773–7780.
296. Mijkstacka R., Rimando A.M., Ignatowicz E. Antioxidant effect of trans resveratrol, pterostilbene, quercetin and their combinations in human erythrocytes in vitro. Plant Foods Hum Nutr. 2010;65:57–63.
297. Das S., Das D.K. Anti-inflammatory responses of resveratrol. Inflamm Allergy Drug Targets. 2007;6:168–173.
298. Wu J.M., Wang Z.R., Hsieh T.C., et al. Mechanism of cardioprotection by resveratrol, a phenolic antioxidant present in red wine. Int J Mol Med. 2001;8:3–17.
299. Bezstarosti K., Das S., Lamers J.M., et al. Differential proteomic profiling to study the mechanism of cardiac pharmacological preconditioning by resveratrol. J Cell Mol Med. 2006;10:896–907.
300. Gresele P., Pignatelli P., Guglielmini G., et al. Resveratrol at concentrations attainable with moderate wine consumption stimulates human platelet nitric oxide production. J Nutr. 2008;138:1602–1608.
301. Thirunavukkarasu M., Penumathsa S.V., Koneru S., et al. Resveratrol alleviates cardiac dysfunction in streptozotocin-induced diabetes: role of nitric oxide, thioredoxin and heme oxygenase. Free Radic Biol Med. 2007;43:720–729.
302. Udenigwe C.C., Ramprasath V.R., Aluko R.E., et al. Potential of resveratrol in anticancer and anti-inflammatory therapy. Nutr Rev. 2008;66:445–454.
303. Harper C.E., Cook L.M., Patel B.B., et al. Genistein and resveratrol, alone and in combination, suppress cancer in SV-40 tag rats. Prostate. 2009;69:1668–1682.
304. Baur J.A., Pearson K.J., Price N.L., et al. Resveratrol improves health and survival of mice on a high calorie diet. Nature. 2006;444:337–342.
305. Dudley J., Das S., Mukherjee S., et al. Resveratrol, a unique phytoalexin present in red wine, delivers either survival signal or death signal to the ischemic myocardium, depending on dose. J Nutr Biochem. 2009;20:443–452.
306. Cottart C.H., Nivet-Antoine V., Laguillier-Morizot C., et al. Resveratrol bioavailability and toxicity in humans. Mol Nutr Food Res. 2010;54:7–16.
307. Rong Y., Geng Z., Lau B.H. Ginkgo biloba attenuates oxidative stress in macrophages and endothelial cells. Free Radic Biol Med. 1996;20:121–127.
308. Otamiri T., Tagesson C. Ginkgo biloba extract prevents mucosal damage associated with small intestinal ischemia. Scand J Gastroenterol. 1989;24:666–670.
309. Rimbach G., Gohil K., Matsugo S., et al. Induction of glutathione synthesis in human keratinocytes by Ginkgo biloba extract (EGb 761). Biofactors. 2001;15:39–52.
310. Pierre S.V., Lesnik P., Bonello L., et al. The standardized Ginkgo biloba extract Egb-761 protects vascular endothelium exposed to oxidized low density lipoproteins. Cell Mol Biol. 2008;54(suppl):1032–1042.
311. Bridi R., Crossetti F.P., Steffen V.M., et al. The antioxidant activity of standardized extract of Ginkgo biloba (EGb 761) in rats. Phytother Res. 2001;15:449–451.
312. Schindowski K., Leutner S., Kressmann S., et al. Age-related increase of oxidative stress-induced apoptosis in mice prevention by Ginkgo biloba extract (EGb 761). J Neural Transm. 2001;108:969–978.
313. Kudolo G.R., Delaney D., Blodgett J. Short-term ingestion of Ginkgo biloba extract (EGb 761) reduces malondialdehyde levels in washed platelets of type 2 diabetic patients. Diabetes Res Clin Pract. 2005;68:29–38.
314. Kuller L.H., Ives D.G., Fitzpatrick A.L., et al. Does Ginkgo biloba reduce the risk of cardiovascular events? Circ Cardiovasc Qual Outcomes. 2010;3:41–47.
315. Cheng A.L., Ch Hsu, Lin J.K., et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high risk or premalignant lesions. Anticancer Res. 2001;21:2895–2900.
316. Miquel J., Bernd A., Sempere J.M., et al. The curcuma antioxidants: pharmacological effects and prospects for future chemical use. A review. Arch Gerontol Geriatr. 2002;34:37–46.
317. Plummer S.M., Holloway K.A., Manson M.M., et al. Inhibition of cyclooxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-kappa activation via the NIK/IKK signaling complex. Oncogene. 1999;18:6013–6020.
318. Hanai H., Iida T., Takeuchi K., et al. Curcumin maintenance therapy for ulcerative colitis: randomized, multicenter, double-blind placebo controlled trial. Clin Gastroenterol Hepatol. 2006;4:1502–1506.
319. Perkins K., Vershoyle R.D., Hill K., et al. Chemopreventive efficacy and pharmacokinetics of curcumin in the min/+mouse, a model of familial adenomatous polyposis. Cancer Epidemiol Biomarkers Prev. 2002;11:535–540.
320. Dhillon N., Aggarwal B.B., Newman R.A., et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14:4491–4499.
321. Garcia-Alloza M., Borrelli L.A., Rozkalne A., et al. Curcumin labels amyloid pathology in vivo, disrupts existing plaques and partially restores distorted neurite in an Alzheimer mouse model. J Neurochem. 2007;102:1095–1104.
322. Hamaguchi T., Ono K., Yamada M. Curcumin and Alzheimer’s disease. CNS Neurosci Ther. 2010;16:285–297.
323. Durak I., Ozturk H.S., Olcay E., et al. Effects of garlic extract on oxidant/antioxidant status and atherosclerotic plaque formation on rabbit aorta. Nutr Metab Cardiovasc Dis. 2002;12:141–147.
324. Ho S.E., Ide N., Lau B.H. S-allyl cysteine reduces oxidant load in cells involved in the atherogenic process. Phytomedicine. 2001;8:39–46.
325. Lee Y.M., Gweon O.C., Se Y.J., et al. Antioxidant effect of garlic and aged black garlic in animal model of type 2 diabetes mellitus. Nutr Res Pract. 2009;3:156–161.
326. Dillon S.A., Lowe G.M., Billington D., et al. Dietary supplementation with aged garlic extract reduces plasma and urine concentrations of 8-isoprostaglandin F(2alpha) in smoking and nonsmoking men and women. J Nutr. 2002;132:168–171.
327. Dhawan V., Jain S. Garlic supplementation prevents oxidative DNA damage in essential hypertension. Mol Cell Biochem. 2005;275:85–94.
328. Williams M.J., Sutherland W.H., McCormick M.P., et al. Aged garlic extract improves endothelial function in men with coronary artery disease. Phytother Res. 2005;19:314–319.
329. Chang H.P., Huang S.Y., Chen Y.H. Modulation of cytokine secretion by garlic oil derivatives is associated with suppressed nitric oxide production in stimulated macrophages. J Agri Food Chem. 2005;53:2530–2534.
330. Chen C., Pung D., Loeng V., et al. Induction of detoxifying enzymes by garlic organosulfur compounds through transcription factor Nrf2: effect of chemical structure and stress signals. Free Radic Biol Med. 2004;37:1570–1580.
331. Dolara P., Luceri C., De Filippo C., et al. Red wine polyphenols influence carcinogenesis, intestinal microflora, oxidative damage and gene expression profiles of colonic mucosa in F344 rats. Mutat Res. 2005;591:237–246.
332. Grisham M.B., MacDermott R.P., Deitch E.A. Oxidant defense mechanisms in the human colon. Inflammation. 1990;14:669–680.
333. Halliwell B., Rafter J., Jenner A. Health promotion by flavonoids, tocopherols, tocotrienols, and other phenols: direct or indirect effects? Antioxidant or not? Am J Clin Nutr. 2005;81(suppl):S268–S276.
334. Dryden G.W., Song M., McClain C. Polyphenols and gastrointestinal disease. Curr Opin Gastroenterol. 2006;22:165–170.
335. Romier B., Schneider Y.J., Larondelle Y., et al. Dietary polyphenols can modulate the intestinal inflammatory response. Nutr Rev. 2009;67:363–378.
336. Keshavarzian A., Sedghi S., Kanotsky J., et al. Excessive production of reactive oxygen metabolites by inflamed colon: analysis by chemiluminescence probe. Gastroenterol. 1992;103:177–185.
337. Mota K.S., Dias M.E., Luiz-Ferreira A., et al. Flavonoids with gastroprotective activity. Molecules. 2009;14:979–1012.
338. Hoensch H.P., Krich W. Potential role of flavonoids in the prevention of intestinal neoplasia: a review of their mode of action and their primary clinical perspectives. Int J Gastroenterol Cancer. 2005;35:187–195.
339. Webb A.L., Villamor E. Update: effects of antioxidant and non-antioxidant supplementation on immune function. Nutr Rev. 2007;65:181–217.
340. Wintergerst E.S., Maggini S., Horrig D.H. Contribution of selected vitamins and trace minerals to immune function. Ann Nutr Metab. 2007;51:301–323.
341. Dunstan J.A., Breckler L., Hale J., et al. Supplementation with vitamins C, E, beta-carotene and selenium has no effect on anti-oxidant status and immune responses in allergic individuals. A randomized controlled trial. Clin Exp Allergy. 2007;37:180–187.
342. Suresh D.R., Annam V., Pratibha K., et al. Total antioxidant capacity – a novel early biochemical marker of oxidative stress in HIV infected individuals. J Biomed Sci. 2009;16:61. PMCID: PMC2714592
343. Steiner J., Haugey N., Li W., et al. Oxidative stress and therapeutic approaches in HIV dementia. Antiox Redox Signal. 2006;8:2089–2100.
344. Wanchu A., Rana S.V., Pallikkuth S., et al. Short communication: oxidative stress in HIV-infected individuals: a cross-sectional study. AIDS Res Hum Retroviruses. 2009;25:1307–1311.
345. Greenspan H.C. The role of reactive oxygen species, antioxidants and phytopharmaceuticals in human immunodeficiency virus activity. Med Hypotheses. 1993;40:85–92.
346. Nance C.L., Siwak E.B., Shearer W.T. Preclinical development of the green tea catechin epigallocatechin gallate as an HIV-1 therapy. J Allergy Clin Immunol. 2009;123:459–465.
347. Irlam J.H., Visser M.E., Rollins N., et al. Micronutrient supplementation in children and adults with HIV infection. Cochrane Database Syst Rev. 2005;19:CD003650.
348. Austin J., Singhal N., Voight R., et al. A community randomized controlled clinical trial of mixed carotenoids and micronutrient supplementation of patients with acquired immunodeficiency syndrome. Eur J Clin Nutr. 2006;60:1266–1276.
349. Slain D., Amsden J.R., Khakoo R.A., et al. Effect of high dose vitamin C on the steady state pharmacokinetics of the protease inhibitor indinavir in healthy volunteers. Pharmacotherapy. 2005;25:165–170.
350. Eichenberger P., Colomban P.C., Mettler S. Effects of 3 week consumption of green tea extracts on whole body metabolism during cycling exercise in endurance trained men. Int J Vitam Nutr Res. 2009;79:24–33.
351. Goldfarb A.H. Antioxidants: role of supplementation to prevent exercise induced oxidative stress. Med Sci Sports Exerc. 1993;25:232–236.
352. Bloomer R.J. The role of nutritional supplements in the prevention of resistance-induced skeletal muscle injury. Sports Med. 2007;37:519–532.
353. Bloomer R.J., Falvo M.J., Schilling B.K., et al. Prior exercise and antioxidant supplementation: effect on oxidative stress and muscle injury. J Int Soc Sports Nutr. 2007;4:9. PMCID: PMC2131751
354. Williams S.L., Strobel N.A., Lexis L.A., et al. Antioxidant requirements of endurance athletes: implications for health. Nutr Rev. 2006;64:93–108.
355. Margaritis I., Rousseau A.S. Does physical exercise modify antioxidant requirements? Nutr Res Rev. 2008;21:2–12.
356. Teixeria V.H., Valente H.F., Casal S.I., et al. Antioxidants do not prevent post-exercise peroxidation and may delay muscle recovery. Med Sci Sports Exerc. 2009;49:1752–1760.
357. McAnulty S.R., McAnulty L.S., Morrow J.D., et al. Effect of daily fruit ingestion on angiotensin converting enzyme activity, blood pressure, and oxidative stress in chronic smokers. Free Radic Res. 2005;39:1241–1248.
358. Boffetta P., Couto E., Wichmann J., et al. Fruit and vegetable intake and overall cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC). J Natl Cancer Inst. 2010;102:529–537.
359. European Food Safety Authority Scientific Opinion. EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). EFSA. 2011;9:2474–2486.
360. Hathcock J.N., Azzi A., Blumberg J., et al. Vitamins E and C are safe across a broad range of intakes. Am J Clin Nutr. 2005;81:736–745.
361. May J.M., Qu Z.C., Whitesell R.R., et al. Ascorbate recycling in human erythrocytes: role of GSH in reducing dehydroascorbate. Free Radic Biol Med. 1996;20:543–551.
362. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled clinical trial of high-dose supplementation with vitamins C and E, beta carotene and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119:1417–1436.
363. Schneider D., Elstner E.F. Coenzyme Q10, vitamin E, and dihydrothioctic acid cooperatively prevent diene conjugation in isolated low density lipoprotein. Antioxid Redox Signal. 2000;2:327–333.
364. Maras J.E., Bermudez O.I., Qiano N. Intake of alpha-tocopherol is limited among US adults. J Am Diet Assoc. 2004;104:567–575.
365. Wray D.W., Uberoi A., Lawrenson L., et al. Oral antioxidants and cardiovascular health in exercise trained and untrained elderly: a radically different outcome. Clin Sci (Lond.). 2009;116:433–441.
366. Ristow M., Zarse K., Oberback A., et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci U S A. 2009;106:8665–8670.
367. Lu J., Papp L.V., Fang J., et al. Inhibition of mammalian thioredoxin reductase by some flavonoids: implication for myricetin and quercetin. Cancer Res. 2006;66:4410–4418.
368. Halliwell B. Effect of diet on cancer development: is oxidative DNA damage a biomarker? Free Radic Biol Med. 2002;32:968–974.
369. Gerss J., Kopcke W. The questionable association of vitamin E supplementation and mortality – inconsistent results from different meta-analytic approaches. Cell Mol Biol. 2009;55(suppl O):L1111–L1120.
370. Dolan Y., Lichtenberg D., Pincuk I. No evidence supports vitamin E indiscriminate supplementation. Biofactors. 2009;35:469–473.
371. Paiva S.A., Russell R.M. Beta carotene and other carotenoids as antioxidants. J Am Coll Nutr. 1999;18:426–433.
372. Alija A.J., Bresgen N., Sommerburg O., et al. Cyto- and genotoxic potential of beta carotene and cleavage products under oxidative stress. Biofactors. 2005;24:159–163.
373. Rimm E.B., Stampfer M.J., Ascherio A., et al. Vitamin E consumption and the risk of coronary heart disease. N Engl J Med. 1993;328:1450–1456.
374. Young A.J., Lowe G.M. Antioxidant and prooxidant properties of carotenoids. Arch Biochem Biophys. 2001;385:20–27.
375. Galati G., Sabzevari O., Wilson J.X., et al. Prooxidant activity and cellular effects of the phenoxyl radicals of dietary flavonoids and other polyphenolics. Toxicology. 2002;177:91–104.
376. Ames B.N., Shigenaga M.K., Hagen T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci U S A. 1993;90:7915–7922.
377. Chen L.H. Interaction of vitamin E and ascorbic acid. In Vivo. 1989;3:199–209.
378. Lee S.H., Oe T., Blair I.A. Vitamin C-induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science. 2001;292:2083–2086.
379. Childs A., Jacobs C., Kaminski T., et al. Supplementation with vitamin C and N-acetylcysteine increases oxidative stress in humans after an acute muscle injury induced by eccentric exercise. Free Radic Biol Med. 2001;31:745–753.
380. Ward N.C., Hodgson J.M., Croft K.D., et al. The combination of vitamin C and grape seed polyphenols increases blood pressure: a randomized, double blind, placebo-controlled trial. J Hypertens. 2005;23:427–434.
381. Block K.I., Koch A.C., Mead M.N., et al. Impact of antioxidant supplementation on chemotherapeutic toxicity: a systematic review of evidence from randomized controlled trials. Int J Cancer. 2008;123:1227–1239.
382. Al-Tonbary Y., Al-Haggar M., El-Ashry R., et al. Vitamin E and N-acetylcysteine as antioxidant adjuvant therapy, in children with acute lymphoblastic leukemia. Adv Hematol. 2009:689639. PMCID: PMC2778172
383. Cassileth B.R., Yeung K.S., Gubili J. Herb-Drug Interactions in Oncology, 2nd ed. China: Sloan Kettering Cancer Center, Integrative Medicine Service. People’s Medical Publishing House; 2010.
384. Harkness R. Mosby’s Handbook of Drug-Herb and Drug-Supplement Interactions. St. Louis: Mosby; 2002.
385. Stargrove M.B., Treasure J., McKee D.L. Herb, Nutrient, and Drug Interactions: Clinical Implications and Therapeutic Strategies. St. Louis: Mosby; 2007.
386. PDR for Herbal Medicines. Physicians’ Desk Reference, 3rd ed. Montvale, NJ: Thompson PDR; 2004.
387. Han E.S., Muller F.L., Perez V., et al. The in vivo gene expression signature of oxidative stress. Physiol Genomics. 2008;34:112–126.
388. Zhang Q., Pi J., Woods C.G., et al. A systems biology perspective on Nrf2-mediated antioxidant response. Toxicol Appl Pharmacol. 2010;244:84–97.
389. Canada A.T., Watkins W.D., Nguyen T.D. The toxicity of flavonoids to guinea pig enterocytes. Toxicol Appl Pharmacol. 1989;99:357–361.
390. Williamson G., Holst B. Dietary reference intake (DRI) value for dietary polyphenols: are we heading in the right direction? Br J Nutr. 2008;99(suppl 3):S55–S58.
391. Van’t Veer P., Kok F.J. Human studies to substantiate health effects of antioxidants: what is needed? Free Radic Res. 2000;33:S109–S115.
392. Bakker G.C., van Erk M.J., Pellis L., et al. An anti-inflammatory dietary mix modulates inflammation and oxidative and metabolic stress in overweight men: a nutrigenomics approach. Am J Clin Nutr. 2010;91:1044–1059.
393. Hasani-Ranibar S., Larijani B., Abdollahi M. 2009. A systematic review of the potential herbal sources of future drugs effective in oxidant-induced diseases. Inflamm Allergy Drug Targets. 2009;8:2–10.