[level-membership-for-critical-care-medicine-category]
83 Myocarditis and Acute Myopathies
 Myocarditis in the Intensive Care Unit
Myocarditis in the Intensive Care Unit
Myocarditis is defined as inflammation of heart muscle.1 Many different etiologic agents have been implicated in this disease, but viral infections are the most common cause. Myocarditis is also associated with autoimmune and other systemic diseases.2 The clinical picture of myocarditis varies widely, from asymptomatic patients who recover without specific therapy and suffer no long-term sequelae to critically ill patients with heart failure and cardiogenic shock. There are no standardized, specific, and widely agreed-upon criteria for making the diagnosis of myocarditis or for determining a cause in many patients.3 Lastly, there has been controversy regarding the most appropriate medical therapy for this condition.
On pathologic examination of myocardial biopsy specimens or on autopsy series, myocarditis is usually apparent as infiltration of myocardium with lymphocytes and fibroblasts, accompanied by myocyte necrosis (myocytolysis).3 It is this type of myocarditis, often termed lymphocytic myocarditis, that will be referred to in this chapter unless otherwise specified. Other types of inflammatory reactions can be seen less frequently in myocarditis, involving giant cells, eosinophils, or granulomas, which can be associated with specific clinical conditions.
In most patients with myocarditis, a specific cause is not found.4 It is presumed that in North America and Europe, the most common etiologic agent is viral.1 Coxsackie B enterovirus was felt to be the most common cause up to the 1990s, but adenoviruses and parvovirus 19 have been implicated as causative agents more frequently over the past 20 years. Other viral causes include hepatitis C, cytomegalovirus, and human herpesvirus 6.2 Myocarditis is a common finding in patients infected with human immunodeficiency virus (HIV). However, the causative agent responsible in these cases may be a secondary viral infection such as cytomegalovirus or other opportunistic infection such as mycobacteria, fungi, or parasites, rather than HIV itself.1,5,6 Infectious illnesses such as Lyme disease, acute rheumatic fever, and diphtheria often have myocarditis as a prominent feature. In Central and South America, the most common cause of myocarditis is the protozoan, Trypanosoma cruzi, the cause of Chagas’ disease (Table 83-1). Systemic and autoimmune diseases such as systemic lupus erythematosus, polymyositis, scleroderma, sprue, Whipple’s disease, and sarcoidosis can be complicated by myocarditis, and myocarditis can be a feature of the infiltrative cardiomyopathies seen in hemochromatosis or amyloidosis. Idiopathic specific forms of myocarditis include hypersensitivity or eosinophilic myocarditis, which has also been reported after smallpox vaccination,7 and giant cell myocarditis.8 Lastly, myocarditis can be associated with doxorubicin cardiomyopathy or with peripartum cardiomyopathy, or it can be a manifestation of a hypersensitivity reaction to medications9,10 (Table 83-2).
| Infectious | Immune-Mediated | Toxic Myocarditis |
|---|---|---|
| Bacterial: Brucella, Corynebacterium diphtheriae, gonococcus, Haemophilus influenzae, meningococcus, Mycobacterium, Mycoplasma pneumoniae, pneumococcus, salmonella, Serratia marcescens, staphylococcus, Streptococcus pneumoniae, Streptococcus pyogenes, Treponema pallidum, Tropheryma whippelii, and Vibrio cholerae Spirochetal: Borrelia and Leptospira Fungal: actinomyces, aspergillus, blastomyces, Candida, Coccidioides, Cryptococcus, Histoplasma, mucormycoses, Nocardia, and Sporothrix Protozoal: Toxoplasma gondii and Trypanosoma cruzi Parasitic: ascaris, Echinococcus granulosus, Paragonimus westermani, Schistosoma, Taenia solium, Trichinella spiralis, visceral larva migrans, and Wuchereria bancrofti Rickettsial: Coxiella burnetii, Rickettsia rickettsii, and Rickettsia tsutsugamushi Viral: coxsackievirus, cytomegalovirus, dengue virus, echovirus, encephalomyocarditis, Epstein-Barr virus, hepatitis A virus, hepatitis C virus, herpes simplex virus, herpes zoster, human immunodeficiency virus, influenza A virus, influenza B virus, Junin virus, lymphocytic choriomeningitis, measles virus, mumps virus, parvovirus, poliovirus, rabies virus, respiratory syncytial virus, rubella virus, rubeola, vaccinia virus, varicella-zoster virus, variola virus, and yellow fever virus |
Allergens: acetazolamide, amitriptyline, cefaclor, colchicine, furosemide, isoniazid, lidocaine, methyldopa, penicillin, phenylbutazone, phenytoin, reserpine, streptomycin, tetanus toxoid, tetracycline, and thiazides Alloantigens: heart transplant rejection Autoantigens: Chagas’ disease, Chlamydia pneumoniae, Churg-Strauss syndrome, inflammatory bowel disease, giant cell myocarditis, insulin-dependent diabetes mellitus, Kawasaki’s disease, myasthenia gravis, polymyositis, sarcoidosis, scleroderma, systemic lupus erythematosus, thyrotoxicosis, and Wegener’s granulomatosis |
Drugs: amphetamines, anthracyclines, catecholamines, cocaine, cyclophosphamide, ethanol, fluorouracil, hematin, interleukin-2, lithium, and trastuzumab Heavy metals: copper, iron, and lead Physical agents: electric shock, hyperpyrexia, and radiation Miscellaneous: arsenic, azides, bee and wasp stings, carbon monoxide, inhalants, phosphorus, scorpion bites, snake bites, and spider bites |
* The most common causes are shown in boldface type.
From Feldman A, McNamara D. Myocarditis. N Engl J Med 2000;343:1388-98.
TABLE 83-2 Distinct Forms of Myocarditis
From Haas G. Etiology, evaluation, and management of acute myocarditis. Cardiol Rev 2001;9:88-95.
Unfortunately, it is difficult to make a clinical diagnosis of a specific viral cause of myocarditis. This usually requires measurement of antiviral antibody titers in acute and convalescent-phase sera. Viral cultures of tissue specimens are unreliable.4 Identification of viral genomes incorporated in myocyte DNA suggests but does not specifically prove that the virus is the cause.
 Pathogenesis
Pathogenesis
Based on observations of human myocarditis, as well as murine models of the disease caused by coxsackie B3, the pathogenesis of viral myocarditis can be described in three stages.2,11 The first stage is initiated by viral infection and replication within myocytes. Viral proteases and activation of cytokines may produce myocyte damage and apoptosis.12 The presence of this viral replication phase is difficult to detect clinically because patients may be asymptomatic during this phase or only have nonspecific viremic symptoms. In addition, there is no rapid screening test to confirm viral infection.
The second stage involves host immune activation. Stimulation of cellular immunity and humoral responses attenuates viral proliferation and can result in recovery from the illness. However, unabated immune activation can result in activated T cells targeting myocardial antigens that cross-react with viral peptides. This leads to release of cytokines such as tumor necrosis factor (TNF), interleukin (IL)-1, and IL-6, resulting in further myocyte damage.1,12 Activation of CD4 cells and antibody production plays a less important pathogenetic role. It is believed that this secondary immune response to viral infection plays a greater role in disease pathogenesis than the primary infection.12
Evidence supporting these mechanisms includes several key observations. Myocardial biopsy with recombinant DNA techniques can detect viral genomes in 20% to 35% of patients. Tissue-specific autoantibodies have been detected in 25% to 73% of patients with evidence of myocarditis on biopsy, with antibodies directed against contractile, structural, and mitochondrial myocyte proteins. Inappropriate expression of the major histocompatibility complex can frequently be demonstrated on biopsy specimens.1 Elevated levels of inflammatory cytokines are detected in patients with active myocarditis.
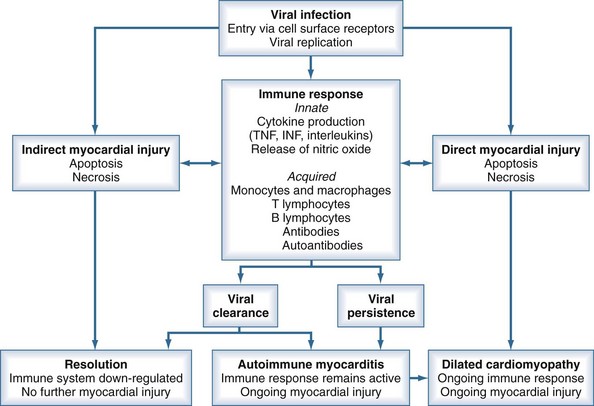
Either persistent overactivation of cellular immune activity or incomplete clearing with persistent or recurrent viral replication can lead to the third stage, during which significant myocardial damage occurs. This leads to left ventricular (LV) dilatation and remodeling, LV systolic dysfunction, and manifestations of heart failure.12 These processes can then abate, with reduction in LV size and improvement of LV function, or can continue to progress with development of dilated cardiomyopathy, worsening ventricular function, and chronic heart failure. Chronic dilated cardiomyopathy is the major long-term sequela of acute myocarditis (Figure 83-1).
 Clinical Presentation and Diagnosis
Clinical Presentation and Diagnosis
The incidence of myocarditis is difficult to determine; many cases are mild with subclinical disease. Myocarditis is diagnosed on clinical grounds, as there are no specific clinical diagnostic criteria. The presentation of myocarditis varies widely. Patients can be asymptomatic insofar as myocarditis has been found in 1% to 10% of autopsy specimens of young adults who had no history of cardiac illness. Myocarditis can be found at autopsy in up to 20% of cases of young, apparently healthy adults who die suddenly and unexpectedly.1,4,10
Patients ill with myocarditis present with nonspecific symptoms of dyspnea (72%), chest pain (32%), and symptoms of arrhythmia (18%).13 The presentation may be indistinguishable from acute coronary syndromes due to coronary artery disease. There may have been a preceding viral prodrome with fever, malaise, and arthralgias. Physical examination can show fever, tachycardia, S3 and S4 gallop sounds, and a pericardial rub if myopericarditis is present. Signs of heart failure can be present, including pulmonary rales and wheezes, elevated jugular venous pulse, and peripheral edema. Murmurs of mitral regurgitation and tricuspid regurgitation may be heard. Infrequently, the presentation is fulminant and severe, with acute heart failure, pulmonary edema, and cardiogenic shock.4
Laboratory findings can include leukocytosis, eosinophilia, and an elevated erythrocyte sedimentation rate. Cardiac biomarkers such as creatine kinase, troponin T, and troponin I may be elevated, with sensitivity of troponin I reported at 34% and specificity of 89%.14 Rheumatologic serologic markers and HIV status should be evaluated.
The 12-lead electrocardiogram (ECG) is an insensitive test for the diagnosis of myocarditis. It shows sinus tachycardia and nonspecific ST-segment depression and T-wave inversion most often. Patients may present with chest pain and ST-segment elevation, with a picture mimicking AMI. More severe cases can be associated with supraventricular or ventricular arrhythmias, conduction disturbances, and heart block.1
Echocardiography is essential to diagnose and quantitate regional or global LV wall-motion abnormalities, left ventricular and right ventricular size and function, the presence of pericardial effusion, and valvular regurgitation. Fulminant myocarditis is characterized by a nondilated left ventricle, with severe systolic dysfunction and increased wall thickness reflecting myocardial edema.15 Findings on myocardial nuclear scintigraphy are frequently abnormal, but this test is not useful in the diagnosis of myocarditis. Cardiac catheterization and coronary angiography are often necessary to exclude acute ischemia as the cause of chest pain or acute heart failure.
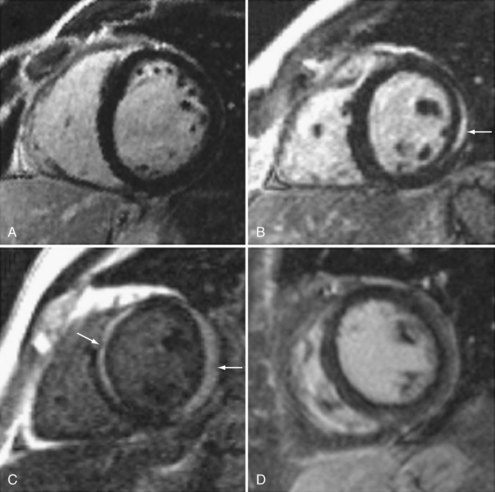
There is increasing use of cardiac magnetic resonance imaging (CMR) in the diagnosis of myocarditis.16,17,18 This technique has the potential to offer a noninvasive means to make this diagnosis. CMR should be considered in symptomatic patients with a high clinical suspicion of disease when the results are likely to affect management decisions. Diagnostic criteria include: (1) focal or diffuse myocardial edema in T2-weighted images, (2) early gadolinium enhancement indicating inflammation, and (3) late gadolinium enhancement in subepicardial or mid-myocardial areas indicating necrosis and fibrosis. Abnormalities may be diffuse or patchy, often confined to the lateral free wall of the left ventricle or the base of the interventricular septum (Figure 83-2). Diagnostic accuracy of CMR is reported at 78% when 2 or 3 criteria are present and 68% when only late gadolinium enhancement is present. CMR is more likely to be abnormal when performed more than 7 days after onset of symptoms. CMR may also detect pericardial effusion (seen in 32%-57% of patients) and gives information regarding LV function. CMR can also be used to direct myocardial biopsy in patients with patchy uptake. The value of CMR for assessing prognosis is unknown, and this presently represents a major limitation of this diagnostic technique.19,20,21
 Endomyocardial Biopsy
Endomyocardial Biopsy
Percutaneous endomyocardial biopsy (EMB) is currently used to aid in the diagnosis of myocarditis and is considered the definitive diagnostic technique. The Dallas criteria have been accepted as the standard for histopathologic diagnosis. These criteria define active myocarditis as the presence of an inflammatory myocardial infiltrate (more than five lymphocytes per high-power field) accompanied by myocyte necrosis. Borderline myocarditis is defined as inflammation without myocyte necrosis. However, there is no difference in prognosis in patients with either of these biopsy results.9 Thus, lymphocyte infiltration (with or without myocyte necrosis) is the most important diagnostic criterion.
Although EMB is useful for diagnostic purposes, there are a number of significant limitations. A high frequency of interobserver variation has been noted among pathologists in applying the Dallas criteria. Biopsies are not sensitive in diagnosing myocarditis; various series have reported positive right ventricular biopsy results in only 10% to 67% of patients with myocarditis suspected on clinical grounds or with recent-onset idiopathic dilated cardiomyopathy. This variability may relate to the timing of biopsies in respect to the stage or chronicity of the patient’s illness. In addition, the myocardial inflammation may not be diffuse and may be patchy, or may predominantly involve the left ventricle, so random right ventricular biopsies may miss affected myocardium.22 Thus, performing a biopsy earlier in a patient’s clinical course, taking multiple biopsy specimens, and performing LV biopsies are ways of improving diagnostic yield. In addition, immunohistochemical staining for human leukocyte antigens can improve diagnostic sensitivity.11,23 EMB should be performed in centers with a high-volume experience, with proven safety and availability of appropriate pathologic techniques.24 However, it is important to emphasize that a negative biopsy finding does not preclude the diagnosis of myocarditis.
Although EMB is an insensitive test with a number of problems, a positive biopsy finding has a high positive predictive value.9 Some authors question the benefits of performing biopsy with standard staining techniques as a routine in suspected myocarditis cases, but this remains the best diagnostic test currently available. Other analyses such as examining specimens for viral genomes utilizing polymerase chain reaction (PCR) or using immunohistochemistry technology to identify up-regulated HLA proteins may offer improved diagnostic yield.22
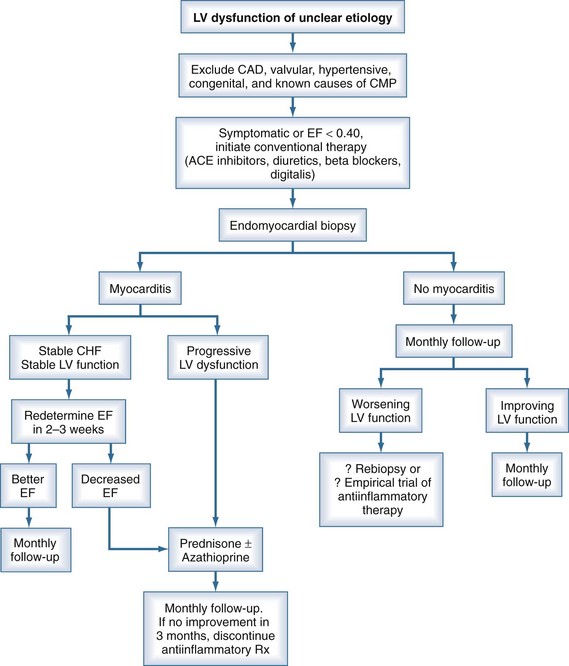
Endomyocardial biopsy should be strongly considered in cases of suspected myocarditis when pathology results will affect management decisions. A recent American Heart Association/American College of Cardiology/European Society of Cardiology (AHA/ACC/ESC) scientific statement offered recommendations concerning the appropriate use of EMB based on patients’ clinical presentations.25 EMB was deemed useful, beneficial. and effective (class I indication) in patients with acute heart failure with hemodynamic compromise, after causes such as coronary artery disease are excluded. EMB is this setting is necessary to differentiate giant cell myocarditis and eosinophilic myocarditis from lymphocytic myocarditis, since immunosuppressive therapy is mandated in the first two conditions (see later). A class I indication for EMB was also recommended for patients with new-onset subacute heart failure, with duration of illness of 2 weeks to 3 months, who fail to improve with medical therapy for heart failure or who demonstrate severe ventricular arrhythmia or advanced heart block. EMB should be considered if causes such as sarcoidosis or collagen vascular disease are suspected and should be performed to diagnose giant cell myocarditis or eosinophilic myocarditis.26 Endomyocardial biopsy should always be performed prior to initiating immunosuppressive therapy (Table 83-3).
Adapted with permission from Wu L, Lapeyre A, Cooper L. Current role of endomyocardial biopsy in the management of dilated cardiomyopathy and myocarditis. Mayo Clin Proc 2001;76:1030-8.
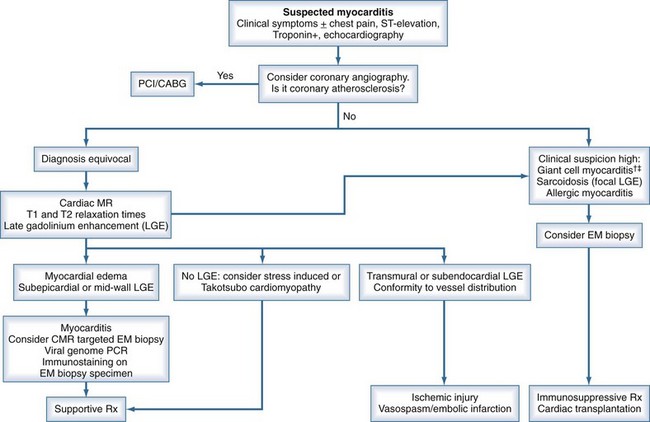
An algorithm has been proposed outlining the steps in evaluating patients suspected of having acute myocarditis (Figure 83-3).
 Clinical Course and Prognosis
Clinical Course and Prognosis
The clinical course and prognosis of acute myocarditis is variable. The majority of patients diagnosed with myocarditis will improve. Patients with mild symptoms most often recover without complications. Eight to 12% of young, apparently healthy adults who die suddenly from a cardiac cause are found to have myocarditis at autopsy, suggesting that patients even with apparently mild illness can suffer fatal arrhythmias.11 Some patients with myocarditis will progress to chronic dilated cardiomyopathy with manifestations of systolic heart failure,3 although a precise incidence is not known. Fifteen to 25% of patients who present with new-onset dilated cardiomyopathy have evidence for antecedent myocarditis.3 Patients with heart failure and LV dysfunction will experience spontaneous resolution of their illness within 12 months in up to 40% of cases, without long-term sequelae. Roughly one-quarter of patients with acute myocarditis and ejection fraction less than 35% will improve, half will develop chronic cardiomyopathy and heart failure, and one-quarter will deteriorate and may be candidates for cardiac transplantation.27
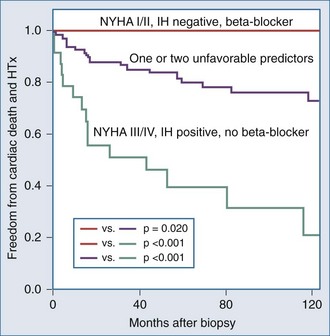
It is important to examine the patient population under study and the criteria used for diagnosing myocarditis in any series assessing prognosis and mortality. No clinical markers reliably predict which patients with myocarditis will recover or worsen.9 In the Myocarditis Treatment Trial, 1-year mortality rate was 20% and 5-year mortality was 56% in patients with biopsy-confirmed lymphocytic myocarditis.28 A series of 21 patients with active myocarditis on biopsy was analyzed for predictors of disease course. Variables assessed included baseline hemodynamics, use of ventilatory and circulatory support, and serum cardiac biomarkers. Overall, there was a 37% mortality rate (8 of 21), with death occurring at 27.6 ± 6.9 days. Factors predicting a worse prognosis included hypotension (mean 84/49 mm Hg), higher pulmonary capillary wedge pressure (mean of 24 mm Hg), and use of mechanical ventilation. Factors that were not predictive of mortality included sex, age, heart rate, cardiac index, peak creatine kinase, or the use of intraaortic balloon counterpulsation for circulatory support.29 Another trial reported 181 patients with myocarditis confirmed by EMB utilizing the Dallas criteria, immunohistochemical staining and PCR, which assesses for viral genome. LV biopsy was performed in 90% of patients. Patients were followed for an average of 59 months, and 22% died or received cardiac transplantation. Multivariate analysis concluded that functional class III and IV heart failure and a positive immunohistochemical result were the only predictors of poor outcome, and treatment with beta-blockers was associated with better outcomes23 (Figure 83-4). Other series have reported that LV ejection fraction (LVEF) less than 40% and right ventricular dysfunction also predict a poorer prognosis.11
Fulminant Myocarditis
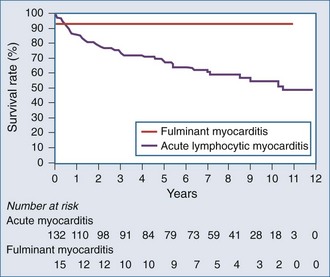
In a study of 147 patients presenting with heart failure due to biopsy-positive active myocarditis with ejection fraction less than 40%, 10% of patients were diagnosed with fulminant myocarditis and 90% with acute lymphocytic myocarditis.8 The patients with fulminant myocarditis needed hemodynamic support with high-dose vasopressors or left ventricular assist devices (LVADs). The acute myocarditis patients had more stable hemodynamics and did not require vasopressors or received them at low doses. Patients with fulminant myocarditis tended to be younger and had higher heart rates and lower systemic blood pressure. There was no difference between the groups in mean pulmonary capillary wedge pressure or cardiac index.
In summary, fulminant myocarditis has a distinct clinical course, with critical illness at presentation but with excellent long-term survival once patients recover from the acute phase of their illness. Healing of myocardial injury and significant improvement of LV systolic function can be expected. Therefore, an aggressive approach to therapy, including the use of ventricular assist devices or other mechanical assist devices, without resorting to early cardiac transplantation, is warranted (Figure 83-5).9
Giant Cell Myocarditis
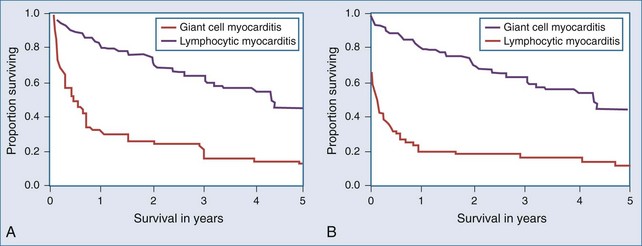
Giant cell myocarditis is a distinct form of myocarditis, generally with a rapidly progressive course without significant likelihood of spontaneous resolution. On endomyocardial biopsy, infiltration with inflammatory giant cells is seen. Although the pathogenesis is not clear, it is believed to be an autoimmune disorder, and CD4 T lymphocytes are thought to play an important role. A total of 63 patients with biopsy-confirmed giant cell myocarditis were studied retrospectively.30 Heart failure was the presentation in 75% of cases; 14% presented with ventricular arrhythmias, and 11% presented with chest pain, an abnormal ECG, or heart block. There was an association with inflammatory bowel disease in 8% of cases. Survival was poor, with a median time of 5.5 months to death or cardiac transplantation (Figure 83-6). In this uncontrolled series, immunosuppressive therapy was associated with prolonged survival from 3 months in 30 patients not given immunosuppressive drugs and 3.8 months in patients treated with prednisone, to 11.5 months in patients given prednisone plus azathioprine and 12.6 months in patients who were given cyclosporine as part of their regimen. Prognosis after cardiac transplantation was also worse when compared with other forms of heart disease, with a 30-day mortality rate of 15% and a 26% mortality rate during the 3.7-year posttransplant follow-up period. Twenty-six percent of patients had giant cell infiltrates seen in their transplanted heart at an average time of 3 years after transplant.
Eosinophilic Myocarditis
Eosinophilic myocarditis, also termed hypersensitivity myocarditis, is a rare form of myocarditis characterized by eosinophilic infiltration and degranulation seen on endomyocardial biopsy. It is believed that pathogenesis involves a direct role of eosinophil-mediated myocyte damage. There can be associated arteritis. This entity is distinct from eosinophilic endocarditis (Löffler endocarditis). The clinical manifestations are not specific, aside from a high incidence of eosinophilia in peripheral blood. Patients usually present with heart failure due to LV systolic dysfunction. Fever and rash may be present. Untreated, the disease is often rapidly fatal.7
The cause is believed to be a hypersensitivity reaction, usually to medication or rarely in association with parasitic infections. Drugs most often implicated are sulfonamides, diuretics, angiotensin-converting enzyme (ACE) inhibitors, cephalosporins, digoxin, or dobutamine. Eosinophilic myocarditis has been reported to occur weeks after smallpox vaccination, with an incidence of 1 in 16,000 vaccinated.31 The clinical course is unfavorable, often with rapidly worsening heart failure and sudden death due to ventricular arrhythmia. Treatment involves the discontinuation of all potentially offending medication and the use of high-dose corticosteroids. Excellent responses to corticosteroids, as well as some spontaneously resolving illness, have been reported.32,33
Eosinophilic myocardial infiltration has been reported in 2% to 7% of myocardial biopsy specimens of patients awaiting cardiac transplantation, or in the explanted heart after transplant. The cause is unclear, but dobutamine therapy, sodium bisulfite used as a preservative in dobutamine solutions, and use of LVADs have been implicated. The presence of eosinophilic myocarditis in this setting did not have an adverse affect on posttransplant survival and did not recur in the transplanted heart.34,35
 Therapy
Therapy
General Management of Heart Failure
Treatment of myocarditis is based on the clinical presentation. Patients with mild disease can be treated expectantly, with dietary sodium restriction and avoidance of strenuous exercise for several weeks or months.3 Animal models indicate that strenuous exercise can worsen myocarditis. Elimination of unnecessary medications is important in patients with eosinophilia.
Nonsteroidal antiinflammatory drugs should be avoided because they may worsen myocarditis.4 The routine use of anticoagulants for prophylaxis of systemic emboli is not recommended. Patients who present with symptoms of arrhythmia or heart failure should be hospitalized, with continuous cardiac rhythm monitoring performed for evaluation of potential life-threatening arrhythmias or conduction abnormalities. If these are diagnosed, they are treated in a similar matter as in patients with other causes of heart disease, utilizing antiarrhythmic drugs or pacemakers. However, a period of observation is recommended to assess for improvement of cardiac function prior to implantation of an implantable cardioverter-defibrillator (ICD).
There are data in murine models of myocarditis supporting the use of ACE inhibitors, angiotensin blockers, and beta-blockers. These drugs reduce inflammation and lessen necrosis and fibrosis.2,3,11,19 There are convincing data in humans supporting the use of these medications, as well as aldosterone antagonists in patients with dilated cardiomyopathy. Therefore, in patients with myocarditis and heart failure, the use of standard multidrug medical therapy for heart failure and LV systolic dysfunction is indicated.3,10 These medications have been shown to improve symptoms, prolong life, and regress the adverse LV remodeling in patients with dilated cardiomyopathy of various causes.36,37,38
As described earlier, β-adrenergic blockade was associated with improved survival in a multivariate analysis of patients with acute myocarditis.23 Large randomized controlled clinical trials, which included patients with idiopathic dilated cardiomyopathy, have unequivocally shown benefit from beta-blockers in patients with LV systolic dysfunction,39–43 and these agents should also be used in patients with heart failure due to myocarditis. Beta-blockers should be initiated after patients are on a stable dose of ACE inhibitors and when signs of fluid overload have resolved. Contraindications to beta-blocker therapy include bronchospastic disease or severe chronic obstructive lung disease, heart block, or significant underlying bradycardia. Hypotension should be corrected prior to initiating beta-blocker therapy.
Digoxin has been shown in animal models to decrease levels of cytokines, but digoxin was associated with adverse outcomes in one murine model of myocarditis. Digoxin can be useful in helping to control ventricular rates in patients with atrial fibrillation. After ACE inhibitors and beta-blockers have been initiated, the use of digoxin should be considered in patients with significant LV systolic dysfunction. However, no survival benefit for digoxin has ever been shown in patients with heart failure due to dilated cardiomyopathy.44 Contraindications to the use of digoxin include renal failure or heart block.
Use of the aldosterone antagonist, spironolactone, has been shown to have symptomatic and survival benefit in patients with class III-IV chronic systolic heart failure.45 In experimental models, these agents can reverse the progressive myocardial fibrosis that occurs in the remodeling process of dilated cardiomyopathy. These agents have not been studied in patients with myocarditis, but their use should be strongly considered in patients with severe LV dysfunction (ejection fraction less than 35%) and symptomatic heart failure.2 Contraindications to the use of aldosterone antagonists include renal insufficiency, serum creatinine levels above 2.0 mg%, or hyperkalemia. Serum potassium levels must be carefully monitored during initiation and dose titration.
In critically ill patients with severe heart failure and low cardiac index, parenteral vasodilators should be used. Intravenous (IV) nitroprusside is a powerful venous and arterial dilator which significantly reduces systemic vascular resistance, mean systemic arterial pressure, and pulmonary capillary wedge pressure, raising cardiac index. It must be administered in the intensive care unit (ICU), with invasive hemodynamic monitoring with a pulmonary artery catheter, to best gauge the appropriate dose of medication and accurately assess response to therapy. Prolonged use of nitroprusside is associated with accumulation of the toxic metabolites thiocyanate and cyanide, and serum levels of these compounds must be monitored. Intravenous nitroglycerin is also an effective venodilator and coronary vasodilator, with less arterial dilating property than nitroprusside. The use of nitroglycerin in cases of myocarditis has not been studied. Patients often develop tolerance to this drug.46–48
Patients with severe myocarditis may develop cardiogenic shock, with hypotension, respiratory failure, and signs of end-organ hypoperfusion. In these instances, initial treatment with inotropic agents or vasopressors is indicated. Dobutamine is a potent β1-agonist with less β2– and α-agonist properties. Dobutamine has favorable short-term hemodynamic effects with increasing myocardial contractility, reducing systemic vascular resistance and reducing pulmonary capillary wedge pressure. However, dobutamine can be proarrhythmic, and patients can develop tolerance to the drug. Routine use of dobutamine in patients with exacerbations of chronic systolic heart failure was associated with increased mortality rates when compared with placebo.49
Milrinone is another parenteral inotropic agent that works by inhibiting phosphodiesterase. This drug leads to increased inotropy and decreased systemic vascular resistance and pulmonary capillary wedge pressure, with resultant increased stroke volume and cardiac index. Milrinone may cause hypotension. It is less proarrhythmic than dobutamine, and it does not induce tolerance.50,51
In patients with fulminant myocarditis or cardiogenic shock not responding to pharmacologic therapy, intraaortic balloon counterpulsation should be utilized. Mechanical ventricular assist devices (VADs) are used for patients requiring greater hemodynamic support. These devices are mechanical pumps which provide physiologic cardiac output and LV afterload reduction and may provide time for spontaneous improvement or recovery of normal LV function. VADs are usually univentricular but can be biventricular, supporting both right and LV function. With improved technology, these devices are smaller and can be implanted through smaller incisions or percutaneously. VADs are connected to an external power pack via a driveline through the skin. The power pack is now small and portable, so patients have freedom of movement and can participate in rehabilitation efforts during VAD use. Routine anticoagulation therapy is not required. Complications of VADs include local site infection, sepsis, thromboemboli, and device failure.52,53
In patients with myocarditis, VADs can be used to provide circulatory needs and improve coronary flow during the time necessary for spontaneous resolution of myocarditis to occur. Beneficial reverse remodeling may occur while patients are on VAD support, resulting in improved myocyte structure and function. VADs can provide support for months or even years. Some authors believe that patients with fulminant myocarditis should be given every opportunity to recover ventricular function with VAD use, and that cardiac transplantation should be used only as a last resort when severe heart damage is irreversible.54
Cardiac transplantation is the final option for treating critically ill patients with myocarditis. However, these patients have a higher rate of transplant rejection and a lower survival rate when compared with patients transplanted for ischemic or other causes of cardiomyopathy. Myocarditis has been reported to recur in the transplanted heart (Figure 83-7).10
Immunosuppressive Therapy
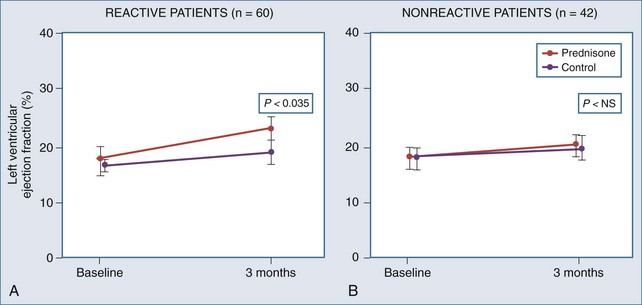
High-dose daily prednisone therapy was used for a 3-month course in 102 patients with dilated cardiomyopathy, 59% of whom were classified as having “reactive” myocarditis on endomyocardial biopsy.55 The authors found a significant improvement in LVEF at 3 months in treated patients with reactive myocarditis (Figure 83-8), but this improvement was not sustained at 9 months. Improvement did not occur in patients with nonreactive biopsies treated with prednisone. No significant mortality benefit from immunosuppressive treatment was noted, although this was not a prespecified primary endpoint.
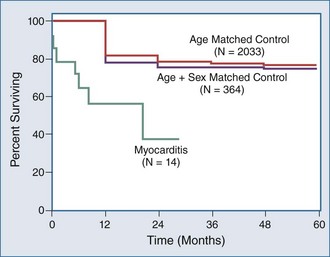
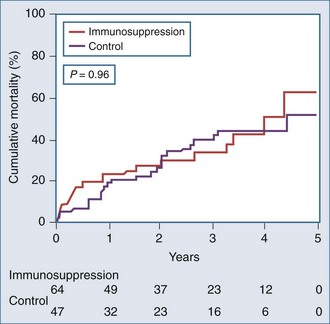
The Myocarditis Treatment Trial enrolled 111 patients with a positive endomyocardial biopsy finding and LVEF less than 45%, with a duration of illness of less than 2 years.28 Three treatment groups were compared: daily prednisone plus azathioprine, prednisone plus cyclosporine, and placebo. Mortality was 20% at 1 year and 56% at 3 years. These investigators found no difference in ejection fraction at week 28 or week 52, no change in LV size at week 28, and no difference in 1-year mortality between treated and untreated groups. Their conclusion was that these immunosuppressive strategies were not beneficial. Significant limitations of this study include a 30% dropout rate and significant interobserver variability among pathologists’ diagnoses of biopsy specimens, despite utilizing the Dallas criteria (Figure 83-9).
In view of the limitations of histopathologic diagnosis using the Dallas criteria, another group of investigators used immunohistologic markers of inflammation, up-regulation of HLA, to diagnose active myocarditis as an indication for immunosuppressive therapy.56 This criterion has the advantage of indicating that autoimmunity is playing a role in pathogenesis. Also, since HLA is distributed throughout the entire myocardium, biopsy sampling error is eliminated as a confounding variable in assessing response to therapy. In this study, 84 of 202 patients with chronic (>6 months) idiopathic dilated cardiomyopathy (ejection fraction < 40%) were found to have strong expression of HLA in biopsy specimens and were randomized to receive placebo or prednisone plus azathioprine for 3 months. At 3 months’ follow-up, a significant improvement in the prespecified secondary endpoints of LVEF, LV volumes, and functional capacity was seen in the treated group, and this improvement was maintained at 2 years (71.8% improvement in the treated group versus 30.8% in the untreated group). However, there was no improvement in the prespecified composite primary endpoint of death, cardiac transplant, or hospital readmission. This study was limited by a 31% dropout rate.
In another study, patients with positive endomyocardial biopsy specimens and progressive heart failure who responded to 6 months of therapy with prednisone and azathioprine were more likely to have circulating cardiac autoantibodies and no viral genome in their myocardium as compared with nonresponders.57
Studies have suggested that in patients with heart failure and low ejection fraction, IV immunoglobulin has a pronounced antiinflammatory effect, as measured by circulating levels of inflammatory markers.58 Uncontrolled studies suggested benefit in patients with myocarditis from treatment with IV immunoglobulin.59,60 However, a placebo-controlled double-blind trial of IV immunoglobulin in patients with myocarditis or idiopathic dilated cardiomyopathy of less than 6 months’ duration showed no significant improvement with therapy, as assessed by ejection fraction or functional capacity at 6 and 12 months.50 In this study, average LVEF improved from 25% ± 8% at baseline to 41% ± 17% at 6 months in both treated and untreated groups. One-year event-free survival rate was 91.9% in both groups, indicating a favorable prognosis.
In summary, there is no evidence that patients with lymphocytic myocarditis or idiopathic dilated cardiomyopathy benefit from routine use of immunosuppressive therapy. However, this treatment approach should be considered in patients with myocarditis and positive endomyocardial biopsy findings, those who develop early signs of severe heart failure, and those who are shown to experience progressive worsening of LV function. Lastly, immunosuppressive therapy should be used in patients with myocarditis associated with connective tissue diseases such as systemic lupus erythematosus (SLE), eosinophilic or granulomatous forms of the disease, and in giant cell myocarditis (Figure 83-10).
 Transient Apical Ballooning Syndrome
Transient Apical Ballooning Syndrome
A distinctive cardiomyopathy with acute onset, frequently precipitated by emotional or physical stress, is termed transient apical ballooning syndrome (TABS) owing to a distinctive LV wall-motion abnormality. This cardiomyopathy was first described in patients in Japan in 1991,61 and the syndrome has subsequently been described in the United States and Europe.62,63 It is characterized by the sudden onset of chest pain and/or dyspnea, ECG changes mimicking AMI, and mild elevation of serum myocardial biomarkers. The syndrome is precipitated by extreme emotional or physical stress in over 70% of cases.64 The characteristic LV wall-motion abnormality is akinesis or dyskinesis of a large area of the LV apex (Figure 83-11 and 83-12). Coronary artery stenosis is not present. TABS is also known as stress cardiomyopathy or takotsubo cardiomyopathy, so named because the takotsubo pot used by Japanese fishermen to trap octopus has a shape similar to the left ventricle in this condition (“short neck, round flask”).63–66
Echocardiography and left ventriculography show moderate to severe LV dysfunction in these patients, with characteristic hyperkinesis of inferior-basal and basal-septal segments, with severe hypokinesis or dyskinesis involving mid-anteroseptal, apical, and inferior-apical wall segments. Acutely, LVEF is reduced to 20% to 40%.64,65 Up to 20% of patients may demonstrate a LV outflow tract gradient due to basal septal hyperkinesis and transient systolic anterior motion of the anterior leaflet of the mitral valve.61,62,66
Patients with TABS often present critically ill, with pulmonary edema, hypotension, and shock. Cardiogenic shock develops secondary to marked LV systolic dysfunction and decreased stroke volume. Shock can also be exacerbated by the development of an LV outflow tract gradient.68 Cardiogenic shock has been reported in 5% of patients at presentation and has occurred during the course of the illness in 6% to 46% of patients in different series.63–6669
Suspicion of TABS and urgent diagnosis are important, since therapy and prognosis differ substantially from AMI. TABS should not be treated with thrombolytic therapy, as coronary occlusion is not involved in the pathogenesis. If cardiogenic shock develops, treatment with intraaortic balloon pump (IABP) counterpulsation is indicated. Inotropic therapy should be used judiciously or not at all. Dobutamine and other β-agonists may worsen cardiogenic shock by increasing hyperkinesis of the basal portion of the heart and causing or aggravating an LV outflow tract gradient. There have been several case reports of patients with TABS with hypotension who develop frank cardiogenic shock after initiation of inotropic therapy. Since a hyperadrenergic state has been proposed to be a major pathogenic mechanism, empirical use of beta-blockers while patients are being supported with IABP counterpulsation has been used successfully. Echocardiography can be useful to guide therapy. For those with extensive wall-motion abnormalities but no outflow obstruction, IABP support without beta-blockers is recommended. Administration of the α-agonist, phenylephrine, can also be considered in cases with a high LV outflow tract gradient, because this drug increases afterload, causing LV dilatation and a decrease in mitral valve systolic anterior motion and lowering of intraventricular gradients.67
TABS is associated with a good prognosis; therefore aggressive therapy of hemodynamic compromise and cardiogenic shock is indicated. In almost all patients, the marked apical wall-motion abnormalities begin to improve within days, and LV function can be expected to recover to normal during the ensuing weeks or months. Follow-up in various series has shown recovery of LVEF to normal in most instances. In-hospital mortality in larger series has been reported at 0% to 4%.62,64–66,69,70 The large majority of survivors will recover completely, with normal functional status. The long-term prognosis is good. In one series, only 2 out of 72 patients had recurrence of TABS within 13 months.63 In another series, the recurrence of TABS was calculated at 2.9% per year. Over a 4-year follow-up, long-term survival of patients who recovered from TABS was equivalent to sex- and age-matched control groups without a history of TABS.70
The pathogenesis of TABS is unknown. Transient multivessel coronary spasm has been proposed, but this has not been demonstrated at the time of acute coronary angiography in the vast majority of patients. In most patients, the extent of LV wall-motion abnormality is larger than the distribution of a single coronary artery.62,63 Cardiac MRI has not shown evidence of infarction or myocarditis.71 In our judgment, a hyperadrenergic state precipitated by acute stress and causing myocardial stunning is the most attractive hypothesis. One study documented supraphysiologic levels of serum catecholamines and stress neuropeptides in patients during the acute phase of TABS, likely due to adrenal and sympathoneuronal hyperactivity. The apex of the left ventricle may be more sensitive than other LV wall segments to the deleterious effects of adrenergic hyperstimulation.65
TABS has been reported to occur in approximately 1.7% to 2.2% of admissions for acute coronary syndrome in Japan and 2% of cases of acute heart failure due to acute coronary syndrome.69,71 TABS may be more common than currently recognized. Correct diagnosis is more likely to be made in centers where emergency coronary angiography and primary percutaneous coronary intervention are used in the treatment of acute coronary syndrome and ST-segment elevation myocardial infarction (STEMI).
 Tachycardia-Induced Cardiomyopathy
Tachycardia-Induced Cardiomyopathy
A sustained rapid heart rate can lead to the acute development of LV dilation and dysfunction with symptoms of heart failure. This is termed tachycardia-induced cardiomyopathy (TICMP) and can occur in otherwise normal hearts or can exacerbate heart failure in patients with preexisting cardiomyopathy. Supraventricular or ventricular arrhythmias of any type can lead to this syndrome. Arrhythmias which may be responsible for TICMP include atrial fibrillation, atrial flutter, automatic atrial tachycardia, AV node reentry tachycardia, supraventricular tachycardia involving accessory pathways, accelerated junctional tachycardia, ventricular tachycardia (from RV and LV sites) and even prolonged, sustained ventricular bigeminy.72 It is not known how long the tachycardia needs to be present in order to cause LV dysfunction, but sustained arrhythmia for days to weeks is likely necessary. The presence of an underlying predisposing substrate has been postulated, as not all patients with sustained tachycardia will develop cardiomyopathy.73
Animal models of TICMP have been established and studied to elucidate pathophysiologic mechanisms and clinical correlates. In these models, sustained, rapid atrial or ventricular pacing leads to severe biventricular systolic and diastolic dysfunction with four-chamber dilation. Within 24 hours of initiating rapid pacing, there is a fall in cardiac output and an increase in ventricular filling pressures. Neurohormonal activation occurs, typical for dilated cardiomyopathy. Cardiac output, ejection fraction, and ventricular volume continue to deteriorate over 3 to 5 weeks. When pacing is discontinued, cardiac output improves to near normal in 48 hours, and hemodynamics are normal within 4 weeks. Ejection fraction recovers to normal in 1 to 2 weeks, although end-diastolic volume remains high at 12 weeks, suggesting persistent remodeling. Structural cardiac changes seen include myocyte hypertrophy and apoptosis and altered extracellular matrix. Proposed pathophysiologic mechanisms include myocardial energy depletion, ischemia, and altered myocyte handling of calcium.73–75
There currently are no data in humans regarding the time course, mechanisms, or cellular biochemical alterations. TICMP can occur at any age, from infants to the elderly. TICMP has been reported to occur in fetuses with sustained supraventricular tachycardia, which resolved with correction of the arrhythmia.74 It is not known if there is a minimal heart rate necessary to induce cardiomyopathy. The longer the duration of arrhythmia, the more likely cardiomyopathy is to occur and the more severe it will tend to be. The incidence and prevalence of TICMP are not known.
In patients with TICMP who have received appropriate arrhythmia therapy, heart failure symptoms improve rapidly. LV systolic function will generally recover to normal within 4 weeks if there is no other underlying heart disease. Cardiac rhythm monitoring for 24 to 48 hours is often necessary to ensure that heart rate is controlled during activity as well as at rest.74 In a report of 11 patients with atrial flutter and abnormal systolic function who underwent atrial flutter ablation, ejection fraction improved from an average of 31% at baseline to 41% within 7 months of ablation. Lack of resolution of cardiomyopathy was predicted by a lower baseline ejection fraction.76 A series of 24 patients with TICMP was reported whose cardiomyopathy initially resolved with arrhythmia control, but who experienced repeated rapid decline in LV function and recurrent heart failure when their arrhythmias reoccurred. These patients again had improvement or normalization of ejection fraction following repeated arrhythmia control within 6 months. However, three of the patients died suddenly and unexpectedly, emphasizing that structural and electrical abnormalities may persist on a chronic basis.77
Key Points
Cooper LT, Baughman K, Feldman AM, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology and the European Society of Cardiology. Circulation. 2007;116:2216-2233.
Cooper L, Berry G, Shabetai R. Idiopathic giant cell myocarditis—natural history and treatment. N Engl J Med. 1997;336:1860-1866.
Gianni M, Dentali F, Grandi AM, Sumner G, Hiralal R, Lonn E. Apical ballooning syndrome or takotsubo cardiomyopathy: a systematic review. Eur Heart J. 2006;27:1523-1529.
A comprehensive review of the reported series of patients with this condition.
Magnani JW, Dec GW. Myocarditis. Current trends in diagnosis and treatment. Circulation. 2006;113:876-890.
An excellent overview of the etiology, pathogenesis, diagnosis, and treatment of myocarditis.
Mason J, O’Connell J, Herskowitz A, et al. A clinical trial of immunosuppressive therapy for myocarditis. N Engl J Med. 1995;333:269-275.
McCarthy R, Boehmer J, Hruban R, et al. Long-term outcome of fulminant myocarditis as compared with acute (nonfulminant) myocarditis. N Engl J Med. 2000;342:690-695.
Parrillo JE, Cunnion RE, Epstein SE, Parker MM, Suffredini AF, Brenner M, et al. A prospective randomized controlled trial of prednisone for dilated cardiomyopathy. N Engl J Med. 1989;321:1061-1068.
1 Feldman A, McNamara D. Myocarditis. N Engl J Med. 2000;343:1388-1398.
2 Cooper LT. Myocarditis. N Engl J. Med 2009;360:1526-1538.
3 Winkel E, Costanzo M, Parrillo JE. Myocarditis. Curr Treat Options Cardiovasc Med. 2000;2:407-419.
4 Kavinsky CJ, Parrillo JE. Rheumatic fever and cardiovascular diseases. Samter’s Immunol Dis. 1995;5:823-840.
5 Barbaro G, Fisher SD, Lipshultz SE. Pathogenesis of HIV-associated cardiovascular complications. Lancet Infect Dis. 2001;1:115-124.
6 Anderson DW, Virmani R, Reilly JM, O’Leary T, Cunnion RE, Robinowitz M, et al. Prevalent myocarditis at necropsy in the acquired immunodeficiency syndrome. J. Am Coll. Cardiol. 1988;11:792-799.
7 Ginsberg FL, Parrillo JE. Eosinophilic Myocarditis. In: Dec GW, editor. Heart Failure Clinics, Myocarditis, vol.1. Philadelphia, PA: W.B. Saunders Co; 2005:419-429. Issue 3
8 Cooper L, Berry G, Shabetai R. Idiopathic giant cell myocarditis-natural history and treatment. N Engl J Med. 1997;336:1860-1866.
9 McCarthy R, Boehmer J, Hruban F, et al. Long-term outcome of fulminant myocarditis as compared with acute (nonfulminant) myocarditis. N Engl J Med. 2000;342:690-695.
10 Haas G. Etiology, evaluation, and management of acute myocarditis. Cardiol Rev. 2001;9:88-95.
11 Magnani JW, Dec GW. Myocarditis. Current trends in diagnosis and treatment. Circulation. 2006;113:876-890.
12 Liu P, Mason J. Advances in the understanding of myocarditis. Circulation. 2001;104:1076-1082.
13 Hufnagel G, Pankuweit S, Richter A, et al. The European Study of Epidemiology and Treatment of Cardiac Inflammatory Disease (ESETCID). First epidemiological results. Herz. 2000;25:279-285.
14 Smith SC, Ladenson JH, Mason JW, Jaffe AS. Elevations of cardiac troponin I associated with myocarditis; experimental and clinical correlates. Circulation. 1997;30:1354-1359.
15 Schultz JC, Hilliard AA, Cooper LT, Rihal CS. Diagnosis and treatment of viral myocarditis. Mayo Clin Proc. 2009;84(11):1001-1009.
16 Liu PP, Yan AT. Cardiovascular magnetic resonance for the diagnosis of acute myocarditis; prospects for detecting myocardial inflammation. J Am Coll Cardiol. 2005;45:1923-1925.
17 Abdel-Aty H, Boye P, Zagrosek A, et al. Diagnostic performance of cardiovascular magnetic resonance in patients with suspected acute myocarditis. J Am Coll Cardiol. 2005;45:1815-1822.
18 Marholdt H, Goedecke C, Wagner A, et al. Cardiovascular magnetic resonance assessment of human myocarditis. Circulation. 2004;109:1250-1258.
19 Elllis CR, DiSalvo T. Myocarditis. basic and clinical aspects. Cardiol Rev. 2007;15:170-177.
20 Nelson KH, Li T, Afonso L. Diagnostic approach and role of MRI in the assessment of acute myocarditis. Cardiol Rev. 2009;17:24-30.
21 Friedrich MG, Sechtem U, Schulz-Menger J, et al. Cardiovascular magnetic resonance in myocarditis: A JACC White Paper. J Am Coll Cardiol. 2009;53(17):1475-1487.
22 Parrillo JE. Inflammatory cardiomyopathy (myocarditis): Which patients should be treated with anti-inflammatory therapy? Circulation. 2001;104:4-6.
23 Kindermann I, Kindermann M, Kandolf R, et al. Predictors of outcome in patients with suspected myocarditis. Circulation. 2008;118:639-648.
24 McKenna W, Davies M. Immunosuppression for myocarditis. N Engl J Med. 1995;333:312-313.
25 Cooper LT, Baughman K, Feldman AM, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology and the European Society of Cardiology. Circulation. 2007;116:2216-2233.
26 Wu L, Lapeyre A, Cooper L. Current role of endomyocardial biopsy in the management of dilated cardiomyopathy and myocarditis. Mayo Clin Proc. 2001;76:1030-1038.
27 Dec GW. Introduction to clinical myocarditis. In: Cooper LT, editor. Myocarditis: from bench to bedside. Totowa: NJ, Humana Press; 2003:257-281.
28 Mason J, O’Connell J, Herskowitz A, et al. A clinical trial of immunosuppressive therapy for myocarditis. N Engl J Med. 1995;333:269-275.
29 Fuse K, Kodama M, Okura Y, et al. Predictors of disease course in patients with acute myocarditis. Circulation. 2000;102:2829-2835.
30 Cooper L, Berry G, Shabetai R. Idiopathic giant cell myocarditis-natural history and treatment. N Engl J Med. 1997;336:1860-1866.
31 Murphy J, Wright S, Bruce K. Eosinophilic lymphocytic myocarditis after smallpox vaccination. Lancet. 2003;362:1378-1380.
32 Galiuto L, Enriquez-Sarano M, Reeder G, et al. Eosinophilic myocarditis manifesting as myocardial infarction: Early diagnosis and successful treatment. Mayo Clin Proc. 1997;72:603-610.
33 Kazama R, Okura Y, Hoyano M. Therapeutic role of pericardiocentesis for acute necrotizing eosinophilic myocarditis with cardiac tamponade. Mayo Clin Proc. 2003;78:901-907.
34 Takkenberg J, Czer L, Fishbein M. Eosinophilic myocarditis in patients awaiting heart transplantation. Crit Care Med. 2004;32:714-721.
35 Johnson M. Eosinophilic myocarditis in the explanted hearts of cardiac transplant recipients: interesting pathologic finding or pathophysiologic entity of clinical significance? Crit Care Med. 2004;32:888-890.
36 Pfeffer M, Braunwald E, Moye L, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 1992;327:669-677.
37 The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;25:293-302.
38 Garg R, Yusuf S. Overview of randomized trials of angiotensin-converting enzyme inhibitors on mortality and morbidity in patients with heart failure. JAMA. 1995;273:1450-1456.
39 Packer M. Current role of beta-adrenergic blockers in the management of chronic heart failure. Am J Med. 2001;110:81S-84S.
40 CIBIS II Investigators. The Cardiac Insufficiency Bisoprolol Study II: A randomized trial. Lancet. 1999;353:9-13.
41 The MERIT-HF Study Group. Effects of controlled release metoprolol on total mortality, hospitalizations and well-being in patients with heart failure. JAMA. 2000;283:1295-1302.
42 Bristow M, Gilbert E, Abraham W, et al. Carvedilol produces dose related improvements in left ventricular function and survival in subjects with chronic heart failure. Circulation. 1996;94:2807-2816.
43 Packer M, Coats A, Fowler M, et al. Effects of carvedilol on survival in severe chronic heart failure. N Engl J Med. 2001;344:1651-1658.
44 The Digitals Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med. 1997;336:525-533.
45 Pitt B, Zannad F, Remme W, et al. The effects of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999;341:709-717.
46 Steimle A, Stevenson L, Chelimsky-Fallick C, et al. Sustained hemodynamic efficacy of therapy tailored to reduce the filling pressures in survivors with advanced heart failure. Circulation. 1997;96:1165-1172.
47 Stevenson L. Tailored therapy for hemodynamic goals for advanced heart failure. Eur J Heart Fail. 1999;1:251-527.
48 Stevenson L, Tillisch J. Maintenance of cardiac output with normal filling pressures in patients with dilated heart failure. Circulation. 1986;74:1303-1308.
49 Jain P, Massie B, Gattis W, et al. Current medical treatment for the exacerbation of chronic heart failure resulting in hospitalization. Am Heart J. 2003;145:S3-17.
50 Jaski B, Fifer M, Wright R, et al. Positive inotropic and vasodilator actions of milrinone in patients with severe congestive heart failure. J Clin Invest. 1985;75:643-649.
51 Cuffe M, Califf R, Adams K, et al. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: A randomized controlled trial. JAMA. 2002;287:1541-1547.
52 Goldstein G, Oz M, Rose E. Medical progress: Implantable left ventricular assist devices. N Engl J Med. 1998;339:1522-1533.
53 Nemeh H, Smedira N. Mechanical treatment of heart failure: the growing role of LVADs and artificial hearts. Cleve Clin J Med. 2003;70:223-234.
54 Farrar D, Holman W, McBride L, et al. Long-term follow-up of Thoratec ventricular assist device bridge-to-recovery patients successfully removed from support after recovery of ventricular function. J Heart Lung Transplant. 2002;21:516-521.
55 Parrillo JE, Cunnion R, Epstein S, et al. A prospective randomized controlled trial of prednisone for dilated cardiomyopathy. N Engl J Med. 1989;321:1061-1068.
56 Wojnicz R, Nowalany-Kozielska E, Wojciechowska C, et al. Randomized placebo controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: two year follow-up results. Circulation. 2001;104:39-45.
57 Frustaci A, Chimenti C, Calabrese F, et al. Immunosuppressive therapy for active lymphocytic myocarditis: Virological and immunologic profile of responders versus non-responders. Circulation. 2003;107:857-863.
58 Gullestad L, Aass H, Field J, et al. Immunomodulating therapy with intravenous immunoglobulin in patients with chronic heart failure. Circulation. 2001;103:220-225.
59 McNamara D, Rosenblum W, Janosko K, et al. Intravenous immune globulin in the therapy of myocarditis and acute cardiomyopathy. Circulation. 1997;95:2476-2478.
60 Patel P, Lenihan D. The current status of immune modulating therapy for myocarditis: A case of acute parvovirus myocarditis treated with intravenous immunoglobulin. Am J Med Sci. 2003;26:369-374.
61 Dote I, Sato H, Tateishi H, et al. Myocardial stunning due to simultaneous multivessel spasm: a review of five cases. J Cardiol. 1991;21:203-214.
62 Desmet WJR, Adriaenssens BFM, Dens JAY. Apical ballooning of the left ventricle: first series in white patients. Heart. 2003;89:1027-1031.
63 Sharkey SW, Lesser JR, Zenovich AG, et al. Acute and reversible cardiomyopathy provoked by stress in women from the United States. Circulation. 2005;111:472-479.
64 Tsuchihashi K, Ueshima K, Uchida T, et al. Transient left ventricular apical ballooning without coronary artery stenosis: a novel heart syndrome mimicking acute myocardial infarction. J Am Coll Cardiol. 2001;38(1):11-18.
65 Wittstein IS, Thiemann DR, Lima JAC, et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. 2005;352(6):539-548.
66 Donohue D, Movahed M. Clinical characteristics, demographics and prognosis of transient left ventricular apical ballooning syndrome. Heart Fail Rev. 2005;10:311-316.
67 Villareal RP, Achari A, Wilansky S, Wilson JM. Anteroapical stunning and left ventricular outflow tract obstruction. Mayo Clin Proc. 2001;76(1):79-83.
68 Prasad A. Apical ballooning syndrome. An important differential diagnosis of acute myocardial infarction. Circulation. 2007;115:e56-e59.
69 Gianni M, Dentali F, Grandi AM, Sumner G, Hiralal R, Lonn E. Apical ballooning syndrome or takotsubo cardiomyopathy: a systematic review. Eur Heart J. 2006;27:1523-1529.
70 Elesber AA, Prasad A, Lennon RJ, Wright RS, Lerman A, Rihal CS. Four-year recurrence rate and prognosis of the apical ballooning syndrome. J Am Coll Cardiol. 2007;50(5):448-452.
71 Dec GW. Recognition of the apical ballooning syndrome in the United States. Circulation. 2005;111:388-390.
72 Shinbane JS, Wood MA, Jensen DN, et al. Tachycardia-induced cardiomyopathy: a Review of Animal Models and Clinical Studies. J Am Coll Cardiol. 1997;29:709-715.
73 Dandamuli G, Rampurwala AY, Mahenthiran J, et al. Persistent LV dilatation in tachycardia-induced cardiomyopathy patients with appropriate treatment and normalization of ejection fraction. Heart Rhythm. 2008;5:1111-1114.
74 Umana E, Solares A, Alpert MA. Tachycardia-induced cardiomyopathy. Am J Med. 2003;114:51-55.
75 Khasnis A, Jongnarangsin K, Abela G, et al. Tachycardia-induced cardiomyopathy: a Review of Literature. Pace. 2005;28(7):710-721.
76 Luchsiger JA, Steinberg JS. Resolution of cardiomyopathy with ablation of atrial flutter. J Am Coll Cardiol. 1998;32:205-210.
77 Nerheim P, Birger-Botkin S, Piracha L, Olshansky B. Heart failure and sudden death in patients with tachycardia-induced cardiomyopathy and recurrent tachycardia. Circulation. 2004;110:247-252.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
83 Myocarditis and Acute Myopathies
Myocarditis in the Intensive Care Unit
Myocarditis is defined as inflammation of heart muscle.1 Many different etiologic agents have been implicated in this disease, but viral infections are the most common cause. Myocarditis is also associated with autoimmune and other systemic diseases.2 The clinical picture of myocarditis varies widely, from asymptomatic patients who recover without specific therapy and suffer no long-term sequelae to critically ill patients with heart failure and cardiogenic shock. There are no standardized, specific, and widely agreed-upon criteria for making the diagnosis of myocarditis or for determining a cause in many patients.3 Lastly, there has been controversy regarding the most appropriate medical therapy for this condition.
On pathologic examination of myocardial biopsy specimens or on autopsy series, myocarditis is usually apparent as infiltration of myocardium with lymphocytes and fibroblasts, accompanied by myocyte necrosis (myocytolysis).3 It is this type of myocarditis, often termed lymphocytic myocarditis, that will be referred to in this chapter unless otherwise specified. Other types of inflammatory reactions can be seen less frequently in myocarditis, involving giant cells, eosinophils, or granulomas, which can be associated with specific clinical conditions.
In most patients with myocarditis, a specific cause is not found.4 It is presumed that in North America and Europe, the most common etiologic agent is viral.1 Coxsackie B enterovirus was felt to be the most common cause up to the 1990s, but adenoviruses and parvovirus 19 have been implicated as causative agents more frequently over the past 20 years. Other viral causes include hepatitis C, cytomegalovirus, and human herpesvirus 6.2 Myocarditis is a common finding in patients infected with human immunodeficiency virus (HIV). However, the causative agent responsible in these cases may be a secondary viral infection such as cytomegalovirus or other opportunistic infection such as mycobacteria, fungi, or parasites, rather than HIV itself.1,5,6 Infectious illnesses such as Lyme disease, acute rheumatic fever, and diphtheria often have myocarditis as a prominent feature. In Central and South America, the most common cause of myocarditis is the protozoan, Trypanosoma cruzi, the cause of Chagas’ disease (Table 83-1). Systemic and autoimmune diseases such as systemic lupus erythematosus, polymyositis, scleroderma, sprue, Whipple’s disease, and sarcoidosis can be complicated by myocarditis, and myocarditis can be a feature of the infiltrative cardiomyopathies seen in hemochromatosis or amyloidosis. Idiopathic specific forms of myocarditis include hypersensitivity or eosinophilic myocarditis, which has also been reported after smallpox vaccination,7 and giant cell myocarditis.8 Lastly, myocarditis can be associated with doxorubicin cardiomyopathy or with peripartum cardiomyopathy, or it can be a manifestation of a hypersensitivity reaction to medications9,10 (Table 83-2).
| Infectious | Immune-Mediated | Toxic Myocarditis |
|---|---|---|
| Bacterial: Brucella, Corynebacterium diphtheriae, gonococcus, Haemophilus influenzae, meningococcus, Mycobacterium, Mycoplasma pneumoniae, pneumococcus, salmonella, Serratia marcescens, staphylococcus, Streptococcus pneumoniae, Streptococcus pyogenes, Treponema pallidum, Tropheryma whippelii, and Vibrio cholerae Spirochetal: Borrelia and Leptospira Fungal: actinomyces, aspergillus, blastomyces, Candida, Coccidioides, Cryptococcus, Histoplasma, mucormycoses, Nocardia, and Sporothrix Protozoal: Toxoplasma gondii and Trypanosoma cruzi Parasitic: ascaris, Echinococcus granulosus, Paragonimus westermani, Schistosoma, Taenia solium, Trichinella spiralis, visceral larva migrans, and Wuchereria bancrofti Rickettsial: Coxiella burnetii, Rickettsia rickettsii, and Rickettsia tsutsugamushi Viral: coxsackievirus, cytomegalovirus, dengue virus, echovirus, encephalomyocarditis, Epstein-Barr virus, hepatitis A virus, hepatitis C virus, herpes simplex virus, herpes zoster, human immunodeficiency virus, influenza A virus, influenza B virus, Junin virus, lymphocytic choriomeningitis, measles virus, mumps virus, parvovirus, poliovirus, rabies virus, respiratory syncytial virus, rubella virus, rubeola, vaccinia virus, varicella-zoster virus, variola virus, and yellow fever virus |
Allergens: acetazolamide, amitriptyline, cefaclor, colchicine, furosemide, isoniazid, lidocaine, methyldopa, penicillin, phenylbutazone, phenytoin, reserpine, streptomycin, tetanus toxoid, tetracycline, and thiazides Alloantigens: heart transplant rejection Autoantigens: Chagas’ disease, Chlamydia pneumoniae, Churg-Strauss syndrome, inflammatory bowel disease, giant cell myocarditis, insulin-dependent diabetes mellitus, Kawasaki’s disease, myasthenia gravis, polymyositis, sarcoidosis, scleroderma, systemic lupus erythematosus, thyrotoxicosis, and Wegener’s granulomatosis |
Drugs: amphetamines, anthracyclines, catecholamines, cocaine, cyclophosphamide, ethanol, fluorouracil, hematin, interleukin-2, lithium, and trastuzumab Heavy metals: copper, iron, and lead Physical agents: electric shock, hyperpyrexia, and radiation Miscellaneous: arsenic, azides, bee and wasp stings, carbon monoxide, inhalants, phosphorus, scorpion bites, snake bites, and spider bites |
* The most common causes are shown in boldface type.
From Feldman A, McNamara D. Myocarditis. N Engl J Med 2000;343:1388-98.
TABLE 83-2 Distinct Forms of Myocarditis
From Haas G. Etiology, evaluation, and management of acute myocarditis. Cardiol Rev 2001;9:88-95.
Unfortunately, it is difficult to make a clinical diagnosis of a specific viral cause of myocarditis. This usually requires measurement of antiviral antibody titers in acute and convalescent-phase sera. Viral cultures of tissue specimens are unreliable.4 Identification of viral genomes incorporated in myocyte DNA suggests but does not specifically prove that the virus is the cause.
Pathogenesis
Based on observations of human myocarditis, as well as murine models of the disease caused by coxsackie B3, the pathogenesis of viral myocarditis can be described in three stages.2,11 The first stage is initiated by viral infection and replication within myocytes. Viral proteases and activation of cytokines may produce myocyte damage and apoptosis.12 The presence of this viral replication phase is difficult to detect clinically because patients may be asymptomatic during this phase or only have nonspecific viremic symptoms. In addition, there is no rapid screening test to confirm viral infection.
The second stage involves host immune activation. Stimulation of cellular immunity and humoral responses attenuates viral proliferation and can result in recovery from the illness. However, unabated immune activation can result in activated T cells targeting myocardial antigens that cross-react with viral peptides. This leads to release of cytokines such as tumor necrosis factor (TNF), interleukin (IL)-1, and IL-6, resulting in further myocyte damage.1,12 Activation of CD4 cells and antibody production plays a less important pathogenetic role. It is believed that this secondary immune response to viral infection plays a greater role in disease pathogenesis than the primary infection.12
Evidence supporting these mechanisms includes several key observations. Myocardial biopsy with recombinant DNA techniques can detect viral genomes in 20% to 35% of patients. Tissue-specific autoantibodies have been detected in 25% to 73% of patients with evidence of myocarditis on biopsy, with antibodies directed against contractile, structural, and mitochondrial myocyte proteins. Inappropriate expression of the major histocompatibility complex can frequently be demonstrated on biopsy specimens.1 Elevated levels of inflammatory cytokines are detected in patients with active myocarditis.
Either persistent overactivation of cellular immune activity or incomplete clearing with persistent or recurrent viral replication can lead to the third stage, during which significant myocardial damage occurs. This leads to left ventricular (LV) dilatation and remodeling, LV systolic dysfunction, and manifestations of heart failure.12 These processes can then abate, with reduction in LV size and improvement of LV function, or can continue to progress with development of dilated cardiomyopathy, worsening ventricular function, and chronic heart failure. Chronic dilated cardiomyopathy is the major long-term sequela of acute myocarditis (Figure 83-1).
Clinical Presentation and Diagnosis
The incidence of myocarditis is difficult to determine; many cases are mild with subclinical disease. Myocarditis is diagnosed on clinical grounds, as there are no specific clinical diagnostic criteria. The presentation of myocarditis varies widely. Patients can be asymptomatic insofar as myocarditis has been found in 1% to 10% of autopsy specimens of young adults who had no history of cardiac illness. Myocarditis can be found at autopsy in up to 20% of cases of young, apparently healthy adults who die suddenly and unexpectedly.1,4,10
Patients ill with myocarditis present with nonspecific symptoms of dyspnea (72%), chest pain (32%), and symptoms of arrhythmia (18%).13 The presentation may be indistinguishable from acute coronary syndromes due to coronary artery disease. There may have been a preceding viral prodrome with fever, malaise, and arthralgias. Physical examination can show fever, tachycardia, S3 and S4 gallop sounds, and a pericardial rub if myopericarditis is present. Signs of heart failure can be present, including pulmonary rales and wheezes, elevated jugular venous pulse, and peripheral edema. Murmurs of mitral regurgitation and tricuspid regurgitation may be heard. Infrequently, the presentation is fulminant and severe, with acute heart failure, pulmonary edema, and cardiogenic shock.4
Laboratory findings can include leukocytosis, eosinophilia, and an elevated erythrocyte sedimentation rate. Cardiac biomarkers such as creatine kinase, troponin T, and troponin I may be elevated, with sensitivity of troponin I reported at 34% and specificity of 89%.14 Rheumatologic serologic markers and HIV status should be evaluated.
The 12-lead electrocardiogram (ECG) is an insensitive test for the diagnosis of myocarditis. It shows sinus tachycardia and nonspecific ST-segment depression and T-wave inversion most often. Patients may present with chest pain and ST-segment elevation, with a picture mimicking AMI. More severe cases can be associated with supraventricular or ventricular arrhythmias, conduction disturbances, and heart block.1
Echocardiography is essential to diagnose and quantitate regional or global LV wall-motion abnormalities, left ventricular and right ventricular size and function, the presence of pericardial effusion, and valvular regurgitation. Fulminant myocarditis is characterized by a nondilated left ventricle, with severe systolic dysfunction and increased wall thickness reflecting myocardial edema.15 Findings on myocardial nuclear scintigraphy are frequently abnormal, but this test is not useful in the diagnosis of myocarditis. Cardiac catheterization and coronary angiography are often necessary to exclude acute ischemia as the cause of chest pain or acute heart failure.
There is increasing use of cardiac magnetic resonance imaging (CMR) in the diagnosis of myocarditis.16,17,18 This technique has the potential to offer a noninvasive means to make this diagnosis. CMR should be considered in symptomatic patients with a high clinical suspicion of disease when the results are likely to affect management decisions. Diagnostic criteria include: (1) focal or diffuse myocardial edema in T2-weighted images, (2) early gadolinium enhancement indicating inflammation, and (3) late gadolinium enhancement in subepicardial or mid-myocardial areas indicating necrosis and fibrosis. Abnormalities may be diffuse or patchy, often confined to the lateral free wall of the left ventricle or the base of the interventricular septum (Figure 83-2). Diagnostic accuracy of CMR is reported at 78% when 2 or 3 criteria are present and 68% when only late gadolinium enhancement is present. CMR is more likely to be abnormal when performed more than 7 days after onset of symptoms. CMR may also detect pericardial effusion (seen in 32%-57% of patients) and gives information regarding LV function. CMR can also be used to direct myocardial biopsy in patients with patchy uptake. The value of CMR for assessing prognosis is unknown, and this presently represents a major limitation of this diagnostic technique.19,20,21
Endomyocardial Biopsy
Percutaneous endomyocardial biopsy (EMB) is currently used to aid in the diagnosis of myocarditis and is considered the definitive diagnostic technique. The Dallas criteria have been accepted as the standard for histopathologic diagnosis. These criteria define active myocarditis as the presence of an inflammatory myocardial infiltrate (more than five lymphocytes per high-power field) accompanied by myocyte necrosis. Borderline myocarditis is defined as inflammation without myocyte necrosis. However, there is no difference in prognosis in patients with either of these biopsy results.9 Thus, lymphocyte infiltration (with or without myocyte necrosis) is the most important diagnostic criterion.
Although EMB is useful for diagnostic purposes, there are a number of significant limitations. A high frequency of interobserver variation has been noted among pathologists in applying the Dallas criteria. Biopsies are not sensitive in diagnosing myocarditis; various series have reported positive right ventricular biopsy results in only 10% to 67% of patients with myocarditis suspected on clinical grounds or with recent-onset idiopathic dilated cardiomyopathy. This variability may relate to the timing of biopsies in respect to the stage or chronicity of the patient’s illness. In addition, the myocardial inflammation may not be diffuse and may be patchy, or may predominantly involve the left ventricle, so random right ventricular biopsies may miss affected myocardium.22 Thus, performing a biopsy earlier in a patient’s clinical course, taking multiple biopsy specimens, and performing LV biopsies are ways of improving diagnostic yield. In addition, immunohistochemical staining for human leukocyte antigens can improve diagnostic sensitivity.11,23 EMB should be performed in centers with a high-volume experience, with proven safety and availability of appropriate pathologic techniques.24 However, it is important to emphasize that a negative biopsy finding does not preclude the diagnosis of myocarditis.
Although EMB is an insensitive test with a number of problems, a positive biopsy finding has a high positive predictive value.9 Some authors question the benefits of performing biopsy with standard staining techniques as a routine in suspected myocarditis cases, but this remains the best diagnostic test currently available. Other analyses such as examining specimens for viral genomes utilizing polymerase chain reaction (PCR) or using immunohistochemistry technology to identify up-regulated HLA proteins may offer improved diagnostic yield.22
Endomyocardial biopsy should be strongly considered in cases of suspected myocarditis when pathology results will affect management decisions. A recent American Heart Association/American College of Cardiology/European Society of Cardiology (AHA/ACC/ESC) scientific statement offered recommendations concerning the appropriate use of EMB based on patients’ clinical presentations.25 EMB was deemed useful, beneficial. and effective (class I indication) in patients with acute heart failure with hemodynamic compromise, after causes such as coronary artery disease are excluded. EMB is this setting is necessary to differentiate giant cell myocarditis and eosinophilic myocarditis from lymphocytic myocarditis, since immunosuppressive therapy is mandated in the first two conditions (see later). A class I indication for EMB was also recommended for patients with new-onset subacute heart failure, with duration of illness of 2 weeks to 3 months, who fail to improve with medical therapy for heart failure or who demonstrate severe ventricular arrhythmia or advanced heart block. EMB should be considered if causes such as sarcoidosis or collagen vascular disease are suspected and should be performed to diagnose giant cell myocarditis or eosinophilic myocarditis.26 Endomyocardial biopsy should always be performed prior to initiating immunosuppressive therapy (Table 83-3).
Adapted with permission from Wu L, Lapeyre A, Cooper L. Current role of endomyocardial biopsy in the management of dilated cardiomyopathy and myocarditis. Mayo Clin Proc 2001;76:1030-8.
An algorithm has been proposed outlining the steps in evaluating patients suspected of having acute myocarditis (Figure 83-3).
Clinical Course and Prognosis
The clinical course and prognosis of acute myocarditis is variable. The majority of patients diagnosed with myocarditis will improve. Patients with mild symptoms most often recover without complications. Eight to 12% of young, apparently healthy adults who die suddenly from a cardiac cause are found to have myocarditis at autopsy, suggesting that patients even with apparently mild illness can suffer fatal arrhythmias.11 Some patients with myocarditis will progress to chronic dilated cardiomyopathy with manifestations of systolic heart failure,3 although a precise incidence is not known. Fifteen to 25% of patients who present with new-onset dilated cardiomyopathy have evidence for antecedent myocarditis.3 Patients with heart failure and LV dysfunction will experience spontaneous resolution of their illness within 12 months in up to 40% of cases, without long-term sequelae. Roughly one-quarter of patients with acute myocarditis and ejection fraction less than 35% will improve, half will develop chronic cardiomyopathy and heart failure, and one-quarter will deteriorate and may be candidates for cardiac transplantation.27
It is important to examine the patient population under study and the criteria used for diagnosing myocarditis in any series assessing prognosis and mortality. No clinical markers reliably predict which patients with myocarditis will recover or worsen.9 In the Myocarditis Treatment Trial, 1-year mortality rate was 20% and 5-year mortality was 56% in patients with biopsy-confirmed lymphocytic myocarditis.28 A series of 21 patients with active myocarditis on biopsy was analyzed for predictors of disease course. Variables assessed included baseline hemodynamics, use of ventilatory and circulatory support, and serum cardiac biomarkers. Overall, there was a 37% mortality rate (8 of 21), with death occurring at 27.6 ± 6.9 days. Factors predicting a worse prognosis included hypotension (mean 84/49 mm Hg), higher pulmonary capillary wedge pressure (mean of 24 mm Hg), and use of mechanical ventilation. Factors that were not predictive of mortality included sex, age, heart rate, cardiac index, peak creatine kinase, or the use of intraaortic balloon counterpulsation for circulatory support.29 Another trial reported 181 patients with myocarditis confirmed by EMB utilizing the Dallas criteria, immunohistochemical staining and PCR, which assesses for viral genome. LV biopsy was performed in 90% of patients. Patients were followed for an average of 59 months, and 22% died or received cardiac transplantation. Multivariate analysis concluded that functional class III and IV heart failure and a positive immunohistochemical result were the only predictors of poor outcome, and treatment with beta-blockers was associated with better outcomes23 (Figure 83-4). Other series have reported that LV ejection fraction (LVEF) less than 40% and right ventricular dysfunction also predict a poorer prognosis.11
Fulminant Myocarditis
In a study of 147 patients presenting with heart failure due to biopsy-positive active myocarditis with ejection fraction less than 40%, 10% of patients were diagnosed with fulminant myocarditis and 90% with acute lymphocytic myocarditis.8 The patients with fulminant myocarditis needed hemodynamic support with high-dose vasopressors or left ventricular assist devices (LVADs). The acute myocarditis patients had more stable hemodynamics and did not require vasopressors or received them at low doses. Patients with fulminant myocarditis tended to be younger and had higher heart rates and lower systemic blood pressure. There was no difference between the groups in mean pulmonary capillary wedge pressure or cardiac index.
In summary, fulminant myocarditis has a distinct clinical course, with critical illness at presentation but with excellent long-term survival once patients recover from the acute phase of their illness. Healing of myocardial injury and significant improvement of LV systolic function can be expected. Therefore, an aggressive approach to therapy, including the use of ventricular assist devices or other mechanical assist devices, without resorting to early cardiac transplantation, is warranted (Figure 83-5).9
