Chapter 33 Myocardial Blood Flow Measurement
Evaluating Coronary Pathophysiology and Monitoring Therapy
INTRODUCTION
Positron emission tomography (PET) combined with tracer kinetic modeling allows the assessment of regional myocardial blood flow (MBF) of the left ventricle in mL/g/min and adds a new dimension to the noninvasive evaluation and understanding of coronary pathophysiology. Measuring coronary flow responses to sympathetic stimulation with cold pressor testing and/or hyperemic flow increases due to pharmacologic vasodilation enable the noninvasive identification and characterization of coronary circulatory function. These flow responses can identify both functional and structural abnormalities of the coronary circulation, reflecting early stages of developing coronary artery disease (CAD) long before the development of macroscopic morphologic alterations of the arterial wall and the clinical manifestation of CAD. PET measurements of MBF combined with various forms of vasomotor stress, therefore, extend the scope of conventional scintigraphic imaging in the noninvasive delineation of coronary circulatory dysfunction in early subclinical stages of CAD. The identification of the CAD process does not depend on demonstrating regional perfusion defects at rest or during stress on conventional myocardial perfusion imaging. Rather, the extent of coronary flow responses to vasomotor stress or the coronary flow reserve may help identify coronary functional abnormalities that may precede or accompany CAD-related structural alterations of the arterial wall.1
METHODOLOGY OF BLOOD FLOW MEASUREMENT
PET approaches for the measurement of regional MBF in mL/g/min entail intravenous administration of positron-emitting tracers of MBF such as nitrogen-13-labeled (13N) ammonia, oxygen-15-labeled (15O) water, or rubidium-82 (82Rb) (Table 33-1) and imaging of the radiotracer’s transit time through the central circulatory system and its extraction and retention in the myocardium. 13N-ammonia and 15O-water, the most commonly used flow tracers with PET, are short-lived (physical half-life 9.8 and 2.4 minutes, respectively) and therefore necessitate on-site cyclotron production. Rubidium-82, with an ultrashort physical half-life of 78 seconds, is available through a strontium-82/rubidium-82 generator system with a 4- to 5-week shelf life and thus does not require an on-site cyclotron.
| Tracer | Half-Life | Method |
|---|---|---|
| O-15-water | 2.4 min | Cyclotron |
| N-13-ammonia | 9.8 min | Cyclotron |
| Rubidium-82 | 78 sec | Generator |
Cyclotron, in-house cyclotron needed; Generator, available through generator; Half-Life, physical half-life.
Beginning with the intravenous injection of the radiotracer of blood flow, serial images are acquired to capture the initial transit of the flow tracer through the central circulation and its extraction into the myocardium (see Table 33-1). The serially acquired image data sets are then reformatted into short-axis and long-axis myocardial slices and assembled into polar maps.2,3 Regions of interest (ROIs) are assigned by the analysis software program to the territories of the three major coronary arteries; a 25 mm2 ROI is positioned in the left ventricular blood pool on a short-axis slice. The ROIs are then copied to all serially acquired image data sets, and time-activity curves are generated that reflect the arterial radiotracer input function and the myocardial response to it. The time-activity curves are then fitted with operational equations derived from tracer kinetic models, which describe the exchange of radiotracer between tissue compartments and the volume of tracer distribution in each compartment. The operational equations then yield regional MBF in mL/g/min. Tracer kinetic models also correct for physical decay of the radioisotope, partial volume–related underestimations of the true myocardial tissue concentrations (by assuming a uniform myocardial wall thickness of 1 cm),4 and spillover of radioactivity between the left ventricular blood pool and myocardium.5
PET TRACERS OF MYOCARDIAL BLOOD FLOW (See Chapter 19)
Estimates of MBF obtained with 13N-ammonia and 15O-water have been widely validated against independent microsphere blood flow measurements in animals and yielded highly reproducible values of MBF (correlation coefficient for 13N-ammonia: r = 0.99 and standard error of the estimate (SEE): 0.17; correlation coefficient for 15O-water: r = 0.95).6,7 Similarly, measurements of MBF with 13N-ammonia and 15O-water in humans yield comparable estimates of MBF,8,9 but these tracers differ in several aspects in their use to measure MBF.
13N-Ammonia
Used as a myocardial flow tracer, 13N-ammonia is selectively retained by the myocardial cells and has a longer physical half-life (9.8 minutes). This allows acquisition of statistically high-count 13N-ammonia images of the myocardium, and thus the relative distribution of the myocardial flow can be denoted with high diagnostic quality in the visual and semiquantitative assessment of myocardial perfusion defects during stress underlying hemodynamically obstructive CAD.10–15 Although 13N-ammonia becomes metabolically trapped in myocardium, mainly in the form of glutamate and mostly glutamine, alterations in cardiac work or myocardial metabolism do not significantly affect the association between tracer tissue concentrations and MBF.10 Acute ischemia may moderately reduce the retention fraction of 13N-ammonia in the myocardium, but it does not significantly affect its use as a flow tracer. The longer physical half-life of 13N-ammonia, however, necessitates longer time intervals (≈45 minutes) between repeat assessments of MBF, restraining the numbers of interventions that can be undertaken during a single PET flow study.
Rubidium-82
As a myocardial flow tracer, 82Rb is increasingly used for the assessment of myocardial perfusion or its relative radiotracer uptake of the left ventricle during stress and rest in the evaluation of flow-limiting epicardial stenosis.12,16–22 Recent investigations also suggest the utility of 82Rb for quantifying MBF in mL/g/min.23–27 The ultrashort physical half-life of 82Rb (only 76 seconds) allows serial investigations of myocardial perfusion at short time intervals (e.g., 10 minutes). Yet the ultrashort physical half-life necessitates the delivery of high 82Rb activity doses (i.e., 60 mCi) for adequate visualization of the myocardial tracer uptake after 1 to 2 physical half-lives for tracer clearance from the blood pool. Clinical investigations report the feasibility and utility of MBF quantification with 82Rb PET combined with one- or two-compartment tracer kinetic models.24–27 Initial animal validation studies24,28 using a two-compartment tracer kinetic model yielded precise MBF values at rest, while the hyperemic flow was generally underestimated and highly variable. Applying 82Rb with a one-compartment tracer kinetic model, however, appears to be more promising in the quantifications of MBF.26,27 In 14 healthy volunteers, for example, estimates of MBF both at rest and during dipyridamole-stimulated hyperemia correlated well with flow values obtained with 13N-ammonia (r = 0.85). Mean hyperemic MBF values were comparable between both flow tracers (13N-ammonia: 2.71 mL/g/min versus 82Rb: 2.83 mL/g/min); however, the corresponding standard deviations (SD), as an index for the individual measurement variability between both flow studies, was higher for rubidium-82 with 0.81 mL/g/min when compared to 13N-ammonia with an SD of 0.50 mL/g/min.27 In another investigation,25 PET measurements of myocardial blood flow with 82Rb using a wavelet-based noise-reduction protocol29 and a two-compartment model were compared to myocardial flows as determined with 15O-water in eleven healthy individuals. For these two approaches to quantify myocardial flow, the authors found a close association of resting and hyperemic MBF (r = 0.94; P < 0.001) ranging between 0.45mL/g/min to 2.75 mL/g/min, emphasizing the potential role of 82Rb in the quantification of myocardial blood flows by PET. Further studies on the interstudy and interobserver variability of 82Rb-estimated MBF are needed to determine the exact range of measurement-related errors of this approach in the individual person and, thereby, its utility in the assessment of subclinical and clinically manifest CAD.
15O-Water
As a myocardial flow tracer, 15O-water distributes into the water spaces of both the myocardium and the blood pool. Thus, 15O-water does not selectively accumulate in the myocardial cells, and corrections for the blood-pool activity are needed. These corrections are commonly performed by blood-pool imaging with 15O water or 11C-labeled red blood cells and subtraction of the blood pool from the 15O water images. Applying a one-compartment tracer kinetic model, estimates of MBF are then determined from the rate of clearance of 15O-water from the left ventricular myocardium.7,30 The subtraction of the blood pool from the 15O water images, in concert with the rapid clearance of 15O-water and its short half-life, may produce statistically low-count images of the myocardium. Thus, the visual evaluation and semiquantitative analysis of the relative distribution of the myocardial 15O-water uptake (“static” image) may be of limited value in the diagnostic evaluation of flow-limiting epicardial lesions. On the other hand, using 15O-water, with its short physical half-life (2.4 minutes), affords repeat MBF measurements at short intervals of 10 to 15 minutes. This allows a convenient assessment of MBF at rest as well as the response to vasomotor stress before and after intervention during the same study session.
DETERMINANTS OF MYOCARDIAL BLOOD FLOW AND FLOW RESERVE
Coronary Circulatory Function: Definitions
The investigation of coronary vasomotor (circulatory) function is usually confined to patients with chest pain syndromes undergoing coronary angiography. Although the identification of coronary vasomotor dysfunction during coronary angiography entails important diagnostic and prognostic information,31–36 the invasive nature and time-consuming process to determine this abnormality in vasomotor function pose major limitations. Invasive approaches to assess abnormalities in coronary vasomotor function, therefore, cannot be applied for widespread clinical use.37–43 A variety of invasive methods for evaluating coronary vasomotor function exist.37,44,45 Such methods commonly necessitate computer-based measurements of coronary vessel diameter (QCA; quantitative coronary angiography) and intracoronary flow-velocity probes for determining coronary flow alterations in response to infused acetylcholine, bradykinin, or substance P–stimulated release of endothelium-derived nitric oxide (NO), or flow-mediated (and thus endothelium-dependent) alterations of the lumen of the epicardial artery.37,38,42,44,45 As regards the latter approach, increases in coronary flow can be stimulated either by intracoronary infusion of papaverine or adenosine or by a more physiologic stress test such as sympathetic stimulation with cold pressor testing (CPT).36–38,40,46
In more detail, endothelium-dependent vasodilators such as acetylcholine specifically stimulate the muscarinic receptor–mediated release of endothelium-derived NO that induces the epicardial artery to dilate through relaxation of the vascular smooth muscle cells. It is worth noting that such vasodilation in response to acetylcholine can only be partially prevented by inhibitors of endothelial NO synthase (eNOS).37 Thus there appears to also be a concomitant release of other endothelium-derived vasodilators, such as prostacyclin and/or endothelium-derived hyperpolarizing factors (EDHF) that contribute to the acetylcholine-stimulated normal coronary vasodilation.37,45 The specific intracoronary infusion of acetylcholine, for example, through an infusion catheter defines normal endothelial function when there is a vasodilation of the epicardial artery. In the presence of dysfunctional epicardial artery endothelium, however, the concurrent smooth muscle cell constrictor effects of acetylcholine overcome the endothelium-mediated vasodilation,37 and a lack of vasodilation or, more commonly, a paradoxical vasoconstriction ensues.37,47 For the determination of coronary blood flow to evaluate the responses of the coronary microcirculatory system, an intracoronary Doppler catheter and the placement of a flow wire are necessary.37,45 Endothelium-dependent vasodilators such as acetylcholine also increase coronary blood flow, paralleling their concurrent vasodilator effects on the coronary arteriolar resistance vessels. Increases in coronary flows to acetylcholine stimulation as measured with the Doppler flow wire, therefore, identify normal endothelium-dependent coronary arteriolar vasomotor function, whereas an impairment or absence of coronary flow is appreciated as endothelial dysfunction of the arteriolar vessels.37,48 Notably, a similar phenomenon has been described for sympathetically induced coronary flow increases during CPT with immersion of the left hand into ice water.40,46,48,49 Normally, CPT-induced and metabolically induced vasodilation of the coronary arteriolar vessels induces an increase in flow in the coronary circulatory system. This increase in coronary flow results in an elevation of shear forces on the vascular wall, with a concomitant release of endothelial-derived NO50,51 and thereby induces a flow-mediated vasodilation of the upstream coronary vessels.37,38,46 If the endothelium is dysfunctional, however, the sympathetically mediated vasoconstrictor response of the vascular smooth muscle cells during CPT prevails and causes an impairment, absence, or even decrease in coronary flow.46,52–54 Of note, the CPT-induced coronary vasomotor response closely correlates with the specific determination of coronary vasomotion due to acetylcholine stimulation at the site of the epicardial conduit artery and the coronary microcirculation.40,48,52,55
Substances such as nitroglycerin or sodium nitroprusside provide NO directly to the vascular smooth muscle cell layer, causing epicardial vasodilation. Thus, the latter vasomotor response to nitroglycerin or sodium nitroprusside is independent of the functional state of the vascular endothelium and thereby provides specific information on vascular smooth muscle cell function of the epicardial artery.37 Similarly, adenosine or papaverine relax vascular smooth muscle cells of the coronary vessels and are commonly applied intracoronarily to gain information on the predominantly endothelium-independent hyperemic coronary flows or so-called total coronary vasodilatory capacity. At the same time, determination of flow-mediated epicardial vasodilation by QCA during hyperemic flow increases with adenosine or papaverine may serve as another important and more physiologic index of endothelium-dependent epicardial vasomotion.37,40,56 Applying such a protocol, a flow-related and NO-mediated epicardial vasodilation during a hyperemic coronary flow increase defines normal endothelial function, whereas an impairment or absence of flow-related epicardial vasodilation is indicative of a dysfunctional state of the coronary endothelium.
NONINVASIVE ASSESSMENT OF CORONARY CIRCULATORY FUNCTION
Myocardial Blood Flow at Rest
PET-measured MBF at rest in healthy volunteers has been reported to range between 0.4 and 1.2 mL/g/min.23,57–61 Apart from some interstudy variations of PET-measured MBF related to methodologic differences, including radiotracers, tracer kinetic models, and image analysis, the variability in these individual resting blood flows is likely attributable to differences in left ventricular myocardial workload at the time of assessment.57,62 As observed in several clinical investigations,57,58,63 there is a close linear correlation between resting MBF and the rate-pressure product (RPP; defined as the product of systolic blood pressure and heart rate) as an index of cardiac work and therefore metabolic oxygen demand. These observations indicate that increases in myocardial work are closely accompanied by commensurate flow increases to adequately meet increases in oxygen demand. Accordingly, MBF is closely coupled to myocardial oxygen demand and myocardial work, both at rest and during physical stress (e.g., bicycle exercise, treadmill exercise, dobutamine stimulation).14,63 Age-related increase in resting MBF has also been related to increases in cardiac work due to higher systolic blood pressures.58,61 More controversial are possible differences in MBF at rest between males and females. Some but not all investigations observed higher values of resting MBF,57,61,64 that are thought to be related to gender-dependent differences in plasma lipid profiles.
Assessment of Coronary Reactivity
Approaches to assessing coronary circulatory function include measurements of MBF with PET at rest and its responses to physiologically or pharmacologically stimulated coronary flow increases, including bicycle exercise, dobutamine stress, sympathetic stimulation with CPT, as well as vascular smooth muscle relaxation with vasodilator agents.14,46,63,65–71 An additional indicator of coronary function is a heterogeneous perfusion response to pharmacologic or sympathetic stimulation, with a progressive decrease in perfusion from the base to the apex of the left ventricle. This abnormal perfusion response might prove useful as a noninvasive indicator of epicardial coronary dysfunction.72,73
Total Integrated Vasodilator Capacity and Coronary Flow Reserve
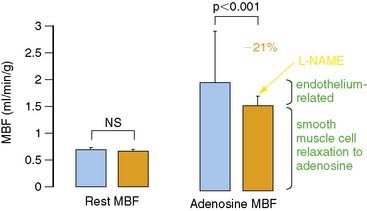
The most widely and clinically used approach for the assessment of coronary circulatory function is the pharmacologically induced hyperemic MBF.62,65,70,74,75 Vascular smooth muscle–relaxing substances such as dipyridamole, adenosine, adenosine triphosphate (ATP), or adenosine receptor agonists76 lower the resistance to flow at the site of the coronary arterioles and produce maximum or submaximum hyperemic MBF. Hyperemic flow is considered a measure of a predominantly endothelium-independent flow response. However, inhibition of eNOS by intravenous infusion of NG-nitro-l-arginine methyl ester (L-NAME) significantly reduces adenosine-induced MBF increases by 21% to 25%, as measured with PET (Fig. 33-1).77,78 This attenuation in hyperemic flow responses most likely reflects an impairment of flow-mediated vasodilation during higher coronary flows. Thus, shear-sensitive components of the coronary endothelium contribute, in part, through flow-mediated coronary vasodilation to the overall hyperemic flow during pharmacologic vasodilation.37,77,78 Since pharmacologically induced hyperemic MBF increases reflect smooth muscle cell and, in part, endothelium-related vasodilatory effects, it is also defined as the “total integrated coronary circulatory function.”44,65,70
The coronary vasodilatory capacity reflects the ability of the coronary circulatory system to increase flows from baseline in order to meet increases in myocardial metabolic demand. This capacity to increase coronary flow to a maximum from rest is known as coronary flow reserve and first described by Coffman and Gregg.79 A more physiologic framework for the coronary flow reserve was provided by Mosher et al.80 by adding the concept of coronary autoregulation. Within the latter concept, resting coronary blood flow is determined by several factors, while myocardial oxygen demand—as a function of heart rate, blood pressure, myocardial contractility, and ventricular preload—is appreciated as the predominant factor in the regulation of resting coronary flows. Given that the metabolic myocardial oxygen demand is widely constant, coronary flow within the range of its autoregulation is widely independent of the coronary perfusion pressure. Consequently, within the range of coronary autoregulation, so-called plateau coronary flow changes little, despite alterations in perfusion pressures. Conversely, a pharmacologically induced dilation of the coronary arteriolar resistance vessels leads to a hyperemic coronary inflow that no longer is governed by autoregulatory mechanisms and that changes linearly with changes in intracoronary perfusion pressure. In this consideration, the ratio of hyperemic to resting coronary flow is defined as the coronary flow reserve. Values for the flow reserve should be interpreted with caution, however. Under certain conditions, the coronary flow reserve does not necessarily indicate the true coronary vasodilator capacity. The coronary flow reserve can decline due to increase in resting flow or decrease in maximum hyperemic flow. Factors that increase the demand for myocardial oxygen, such as arterial hypertension, increased myocardial contractility, increased left ventricular wall stress, and tachycardia, cause an increase in resting flow. On the other hand, maximum hyperemic coronary flow may decline in the presence of a focal flow-limiting epicardial lesion, in the presence of microvascular disease in patients with hypertension or diabetes, or as a consequence of increases in extravascular resistive forces paralleled by increases in left ventricular pressures in patients with congestive heart failure or hypertension. Thus, there are several limitations when interpreting the coronary flow reserve. Nevertheless, the concept of the coronary flow reserve remains an important and useful index for assessing the functional significance or downstream effects of focal epicardial arterial lesions, functional improvement after coronary revascularization, and coronary circulatory function in individuals with subclinical or clinically manifest CAD.65,81,82
Sympathetic Stimulation with Cold Pressor Testing
PET measurements of alterations in MBF from rest to sympathetic stimulation by CPT entail specific information on coronary endothelial function.39,46,48,53,83,84 CPT is performed with immersion of a hand into ice water, which in turn induces a sympathetically mediated increase in heart rate and blood pressure and thus an increase in myocardial workload. The RPP can be used as an index of myocardial workload and oxygen demand. Increases in myocardial workload during CPT are associated with vasodilation of the coronary arteriolar resistance vessels through the release of (presumably) adenosine as a metabolic vasodilator.85 As a consequence, there is a decrease in coronary vascular resistance that causes an increase in coronary inflow. This increase in coronary inflow causes in turn a flow-mediated, endothelium-dependent dilation of the upstream coronary vessel segments. As demonstrated in Figure 33-2, an increase in cardiac work is normally accompanied by commensurate flow-mediated coronary vasodilation, and an increase in MBF is determined with PET.46,53,54,61 In the presence of coronary endothelial dysfunction, however, the sympathetically induced increase in coronary inflow does not mediate a flow-related vasodilation. Under such conditions, the sympathetically mediated vasoconstrictor effects of the vascular smooth muscle cells predominate and are not overcome by normal flow-related coronary vasodilation (Fig. 33-3).37,46,86 Myocardial blood flows during CPT are then attenuated, absent, or even paradoxically decreased, which denotes a dysfunction of the coronary endothelium.46,53,54,84
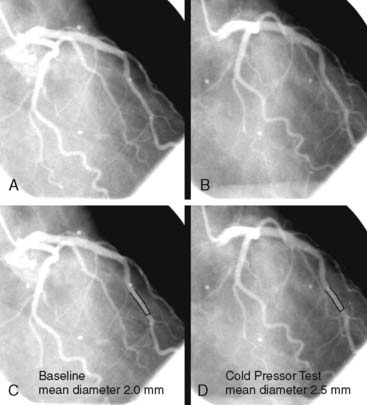
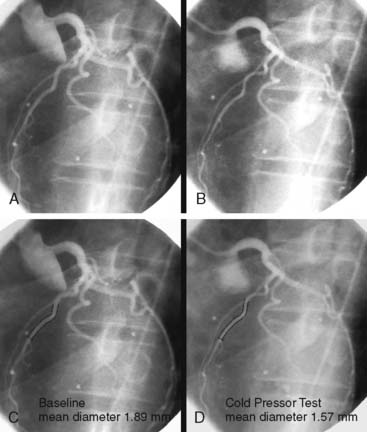
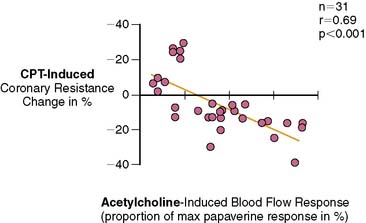
Several clinical investigations in the assessment of coronary vasomotor function emphasize the validity and value of PET-measured changes in MBF during CPT from rest as a noninvasive index of coronary endothelial function. For example, coronary flow increases during CPT, as assessed invasively with Doppler wire during coronary angiography, closely correlate with the flow response to acetylcholine stimulation, which is considered a specific probe of endothelial function (Fig. 33-4).37,48 Coronary flows during sympathetic stimulation with CPT, therefore, may probe endothelium-related vasomotor function and thereby delineate the functional integrity of the vascular wall.37,46 In particular, it was observed that the epicardial vasomotor response to CPT was closely paralleled by the acetylcholine-stimulated response, suggesting the epicardial vasomotor response to CPT may be intimately related to the integrity of endothelial function.37,52 Further, CPT-induced changes in epicardial luminal diameter, as determined by QCA, and PET-measured responses of MBF to CPT, are closely correlated.46,53,54 More direct evidence for the involvement of the endothelium in CPT-mediated MBF responses has been provided by Campisi et al.84 In chronic smokers exhibiting an impairment of MBF responses to CPT, intravenous infusion of l-arginine as a substrate of nitric oxide synthase restored the MBF increase to CPT, most likely due to increases in the bioavailability of endothelium-derived NO.
Perfusion Heterogeneity and Base-to-Apex Myocardial Perfusion Gradient
In the last 2 decades, the interrelation of structural and functional determinants of clinically manifest CAD has been extensively studied.87–92 As shown by previous fundamental work of Gould et al.,87 focal coronary artery lesions between 60% and 85% diameter stenosis commonly do not affect the resting MBF, as the result of a compensatory vasodilation of the downstream arteriolar resistance vessels.87 This adaptive vasodilation under resting conditions may fail to compensate for greater increases in epicardial resistance, that is, for cases of severe coronary artery stenoses (greater than 85%), so stress-induced myocardial ischemia may manifest.69,93 In light of several investigations, it has been widely accepted that hyperemic coronary flows during pharmacologic vasodilation commonly begin to decline when focal epicardial artery narrowing reaches 50% diameter stenosis.87,88,90,92
Whether structural changes of the arterial wall in early stages of CAD may also affect the coronary flow is an area of ongoing debate and interest. PET flow studies1,82,94–96 add evidence that hyperemic flow increases may be mildly diminished in the presence of diffuse CAD with coronary lesions less than 50% and/or coronary circulatory dysfunction. Such observations have raised a new concept that even subclinical CAD-related structural alterations of the arterial wall may exert downstream fluid dynamic effects that, as reported recently, may manifest as mild myocardial perfusion heterogeneity at rest,97,98 and during vasomotor stress98 or as longitudinal base-to-apex perfusion gradient during hemodynamic stress.72,73,94,99 Gould et al.94 were the first to describe heterogeneity in longitudinal myocardial perfusion during pharmacologically induced hyperemia in patients with diffuse CAD. Other investigators found that PET-determined quantitative estimates of MBF during pharmacologically induced hyperemia also denoted a relative decrease in longitudinal myocardial flow from the mid to the mid-distal part of the left ventricular myocardium in individuals with coronary risk factors but without clinically manifest CAD.72,73 It has been argued that fluid dynamic consequences of CAD- related vessel stiffness and/or functional abnormalities of the epicardial conduit vessels may account for the longitudinal heterogeneity in myocardial perfusion. Based on the Hagen-Poiseuille equation, intracoronary resistance relates to the velocity of the blood flow and inversely to the fourth power of the vessel diameter.91,100,101 Normal function of the vascular endothelium ascertains that increases in flow velocity during exercise or pharmacologic vasodilation are associated with a flow-dependent and nitric-oxide-mediated dilation of the coronary artery, which balances the velocity-induced increase in coronary resistance, so that the resistance is kept low or does not increase.38,42 Structural and/or functional disturbances of coronary arterial walls in the early development of CAD, however, commonly impair a flow-mediated and thus endothelial-derived NO–mediated dilation of the epicardial artery. In this concept, the absence of a flow-mediated epicardial vasodilation causes an increase in intracoronary resistance during higher coronary flows that leads to a progressive proximal-to-distal decline in intracoronary pressure along the epicardial artery.100 Such a decrease in intracoronary pressure has been put forth as cause for a gradual base-to-apex, relative decline or heterogeneity in myocardial perfusion or MBF.72,73,94,100
There is some direct evidence of a cause-and-effect relationship of structural and/or functional changes of the epicardial artery and downstream hemodynamic effects. As previous investigations have demonstrated, there is a close association between heterogeneity in myocardial perfusion during dipyridamole stimulation and the presence of diffuse CAD.94 A more recently performed comparative study of patients with coronary risk factors and angina pectoris symptoms provided further insight.102 Comparisons were made between the invasive assessment of epicardial endothelial function in response to acetylcholine stimulation, and 99mTc tetrofosmin SPECT-measured myocardial perfusion during both bicycle exercise and dipyridamole stimulation. In these patients without flow-limiting epicardial coronary lesions, a moderate but significant association (r = 0.49; P < 0.002) between acetylcholine-induced alterations in epicardial artery diameter and the degree of exercise-induced myocardial perfusion defects was observed, suggesting that functional abnormalities of the epicardial artery may indeed account, at least in part, for stress-induced regional myocardial perfusion defects.102,103 A perfusion heterogeneity in longitudinal (base-to-apex) MBF during sympathetic stimulation with CPT, as measured with PET, was observed to be related to a functional decrease in epicardial luminal diameter as determined with quantitative coronary angiography (r = 0.77; P < 0.0001).99 Apart from structural alterations of the coronary arterial wall, an impairment of flow-mediated, endothelium-dependent epicardial coronary vasomotor function may contribute to the manifestation of a longitudinal heterogeneity in MBF during vasomotor stress.44,103 The assessment of heterogeneity in longitudinal myocardial perfusion or quantitative blood flow with PET could be a promising noninvasive index of early structural and/or functional alterations of the CAD process, predominantly at the site of the epicardial artery. As another noninvasive index of the early stages of the development of CAD, it may carry important predictive information on future cardiovascular events95,104 but awaits further confirmation through clinical endpoint investigations.
Reproducibility of Measurements of Myocardial Blood Flow and Responses to Stressors
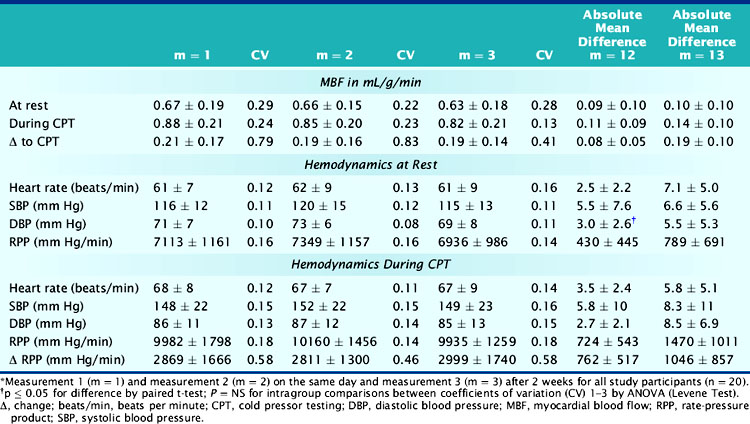
As outlined before, an impairment of coronary circulatory function in individuals with subclinical stages of the CAD process has been widely appreciated to carry important diagnostic and prognostic information.31,32,105–107 If this holds true, then a restoration of abnormalities in coronary circulatory function by lifestyle modifications or therapeutic interventions should lead to an improved clinical outcome, as some preliminary data of the peripheral circulation may indeed suggest.108–110 PET measurements of MBF responses to cold exposure and pharmacologic vasodilation are increasingly applied to determine and monitor the effects of lifestyle modifications or therapeutic interventions on coronary circulatory function.53,67,111–117 Such serial PET flow studies raise the need for establishing the reproducibility of repeat PET measurements of MBF.118 This allows determination of the methodologic measurement error of PET-determined flows in repeat assessments62,74,118 and also the biological and hemodynamic variability of these flow measurements.46,58,118–120 By using the mean difference of repeat MBF measurements, the sample size of the study populations needed for adequately powering clinical investigations with serial PET blood-flow measurements can be determined.
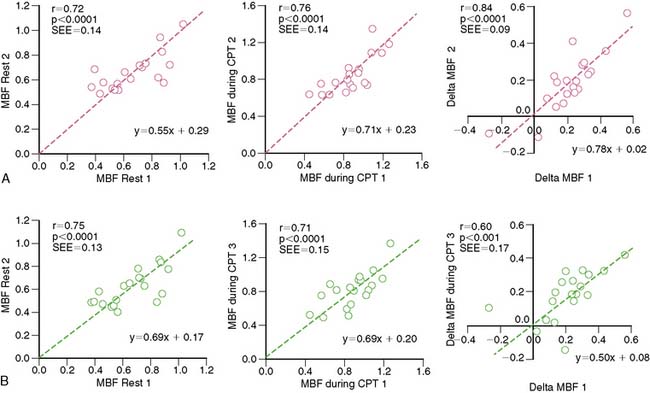
A reasonable reproducibility of hyperemic MBF during pharmacologic vasodilation with 13N-ammonia or 15O-labeled water and PET has been demonstrated previously.66,74,121 As recently shown, CPT-related MBF measurements were also reproducible when determined with 15O-labeled water on a 1-day protocol.122 Further, in a more detailed and extended investigation,118 the hemodynamic and endothelium-related MBF responses to CPT or its change from rest (ΔMBF), as measured with 13N-ammonia and PET, were demonstrated to be not only highly reproducible in short-term (1-day protocol) but also in long-term (2 to 3 weeks protocol) measurements (Table 33-2). There are important considerations in interpreting the reproducibility data of PET-measured MBF. For example, the Pearson’s correlation coefficient (r) of the least-square regression analysis can be used to denote the strength of agreement between repeat MBF measurements. Notably, the standard error of the estimate (SEE) indicates the tightness of the linear fit of the data and is considered to represent the actual range of method-related measurement error (or its variability). For example, the range of measurement errors, as denoted by the SEE for the endothelium-related change of MBF from rest to CPT, was found to be 0.09 mL/g/min for short-term and 0.17 mL/g/min for long-term repeat measurements (Fig. 33-5). According to the latter values of the SEE, alterations in the change in MBF in serial pharmaceutical studies that are above this range of SEE are likely to be related to beneficial effects of pharmaceutical interventions on coronary endothelial function.118 In addition, the mean difference and the corresponding standard deviation (SD) of repeat MBF measurements are used to calculate the sample size of a study population needed to sufficiently power a serial flow study. For example, using the longitudinal mean difference and the corresponding SD of the endothelium-related change in MBF from rest to CPT of 0.08 ± 0.05 mL/g/min in the short-term and of 0.19 ± 0.10 mL/g/min in the long-term (see Table 33-2), at a 5% significance level with a power of 87%, a sample size of 14 and 22 individuals would be needed for serial PET flow studies to identify possible intervention-related, statistically significant alterations in MBF responses to CPT.118 Another important statistical parameter used in the interpretation of reproducibility data is the repeatability coefficient (RPC) as proposed by Bland and Altman.123 The RPC serves as a useful index to denote the agreement between repeat measurements. Given a normal Gaussian distribution of MBF measurements,123 the RPC denotes the expected range of measurement error between repeat assessments of myocardial blood flows. It is critical, however, to keep in mind that most PET flow studies have investigated relatively small sample sizes, between 11 and 25 study participants.66,74,118,124 Given the relatively small sample sizes of these PET flow studies, they may not have fully met the assumption of a normal Gaussian distribution of flow values. Further, the RPC can also be applied as an index of precision of MBF measurements between different studies (Table 33-3). For example, in the aforementioned investigation,118 the RPC for the endothelium-related change in MBF from rest to CPT was 0.18 mL/g/min for the short-term and 0.27 mL/g/min for the long-term reproducibility measurements with 13N-ammonia and PET. Both RPCs indicate less measurement error in the assessment of endothelium-related MBF responses to CPT with 13N-ammonia PET than was observed with the short-term RPC of CPT-related flows and 15O-water measurements (1-day study protocol)122 and also for hyperemic flow increases reported in previous studies,66,74,124 which have been reported between 0.49 and 1.34 mL/g/min (see Table 33-3).

ALTERED CORONARY CIRCULATORY FUNCTION AND CARDIOVASCULAR EVENTS
Normal endothelium-dependent vasomotor function has been widely appreciated to play an active and central role in the modulation of the integrity and metabolism of the vascular wall, vasomotor tone, and hemostasis.125,126 The coronary vasomotor tone underlies the endothelium-dependent production and release of vasoactive mediators such as prostacyclin, endothelin-1, endothelium-derived hyperpolarizing factor (EDHF), and in particular, NO. Given normal function of the coronary endothelium, increases in coronary flow with physical exercise lead to a flow-mediated and thus endothelium-dependent release of NO, prompting a NO-mediated relaxation of the vascular smooth muscle cells and vasodilation. This NO-mediated mechanism offsets possible vasoconstrictor effects of elevated endothelin-1 and angiotensin-II levels, and/or sympathetic activation.37,44,127 Coronary risk factors, however, not only cause a reduction in the bioavailability of endothelial-derived NO and/or prostacyclin but are usually associated with increased activity of vasoconstrictors such as endothelin-1, angiotensin II, and reactive oxygen species (ROS), most likely accounting for the diminished endothelium-dependent coronary vasodilator capacity. Coronary vasomotor function, therefore, constitutes a tenuous balance between vasodilator and vasoconstrictor mechanisms, where the endothelium-mediated vasodilation normally prevails.
Importantly, the flow-mediated release of endothelial-derived NO not only leads to coronary vasodilation during higher flow velocities but also exerts antiatherosclerotic and antithrombotic effects.37,126,127 For example, one atheroprotective mechanism of endothelial-derived NO is to act in concert with prostacyclin to prevent platelet aggregation, while another is to reduce expression of cellular adhesion molecules, thereby preventing the attachment of neutrophils to the endothelium and their migration into the subintimal space.37,126,127 NO further inhibits vascular smooth muscle cell proliferation and exerts antiinflammatory and antioxidative effects within the arterial wall. Reductions in the bioavailability of atheroprotective and endothelial-derived NO contribute to the initiation or acceleration of the atherosclerotic process. Risk factors for CAD as diverse as smoking, hypercholesterolemia, hypertension, hyperglycemia, obesity, and a family history of premature atherosclerotic disease have all been shown to be associated with an attenuation or loss of endothelium-dependent vasodilation.37,105,126 The causes of coronary circulatory dysfunction in patients with coronary risk factors are certainly multifactorial. Increased vascular production of ROS derived from superoxide-producing endothelial enzymes, such as NAD(P)H oxidase, xanthine oxidase, or uncoupled NO synthase, reduces the bioavailability of endothelium-derived NO, which has been implicated as a leading cause and common final pathway of abnormal coronary vasodilatory capacity.128
An impairment of endothelium-dependent coronary circulatory function is commonly associated with other active processes such as inflammation, proliferation or apoptosis, and the expression of vascular cellular adhesion molecules (ICAM). This so-called endothelial activation reflects an initial injury of the vascular wall that may initiate and contribute to the development and progression of the atherosclerotic process. In particular, this “endothelial activation” plays an important role in the pathogenesis of acute coronary syndromes characterized by coronary plaque vulnerability and paradoxical vasoconstriction, paralleled by endothelial dysfunction, which is likely to contribute to plaque rupture,129,130 and increased thrombogenicity due to loss of a potent antithrombotic endothelial surface.131 An impairment of coronary circulatory function, therefore, is likely to reflect the vulnerability of plaques, which may explain the independent predictive value of coronary circulatory dysfunction for future cardiovascular events.31,32,105,106 Alterations of coronary circulatory function appear to reflect active processes that modify the functional and structural state of the arterial wall and may serve as an explanation for the independent predictive value of coronary circulatory dysfunction for future cardiovascular events.32,126,127
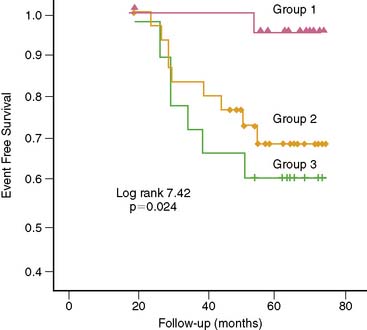
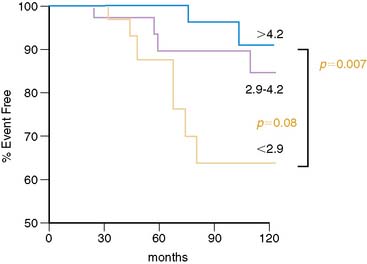
Individuals with traditional and novel coronary risk factors and those with clinically manifest CAD commonly present an impairment of endothelium-dependent vasomotion.35 The degree of the dysfunctional endothelium probably reflects the cumulative effect of several risk factors on the arterial wall. Nevertheless, there is considerable variability in the extent of endothelial dysfunction in individuals with similar coronary risk factor profiles.36 It is worth noting that the assessment of peripheral and coronary vasomotor dysfunction has been considered an important marker of the inherent atherosclerotic risk in an individual, reflecting the cumulative risk of various coronary risk factors as well as of yet-unknown variables and genetic predispositions.132,133 Of particular interest is that PET-measured flow responses to sympathetic stimulation with CPT, indicative of coronary endothelial dysfunction in patients with angiographically normal coronary vessels but with coronary risk factors, appear to be associated with an increased risk for future cardiovascular events (Fig. 33-6).106 This appears to be similar to observations by Britten et al.34 They found, in patients with normal or mildly diseased epicardial coronary arteries, an inverse relationship between the attenuation of the flow response to intracoronary papaverine and a higher risk of future cardiovascular events (Fig. 33-7).
These observations on coronary circulatory dysfunction or of an impairment of the total coronary vasodilator capacity as a predictor of cardiovascular events in patients with normal coronary angiograms or with only mild or subclinical CAD are, at first sight, in contrast to the more favorable outcome of a normal SPECT or PET stress/rest perfusion imaging study.134–138 Cardiovascular events predicted by the presence of stress-induced perfusion defects on SPECT or PET normally occur within 2 to 3 years, whereas cardiovascular events predicted by PET-determined coronary vasomotor abnormality or by invasively determined measures of endothelial function in patients without epicardial flow-limiting lesions appeared in some studies to have occurred after longer time periods. Thus, it is possible that assessment of coronary circulatory dysfunction by PET may identify individuals without clinically manifest CAD but who may be at risk of developing CAD and its atherothrombotic sequelae. Such functional alterations of the coronary circulation also appear to be superior to CAD-related structural alterations of the epicardial wall in the prediction of future cardiovascular clinical outcome.139–143
Of further interest is that impairment in coronary circulatory function may be predictive of future cardiovascular outcome not only in individuals with early stages of the CAD process but also in patients with hypertrophic or idiopathic cardiomyopathy.107,144 For example, Cecchi et al.107 stratified the extent of diminished vasodilatory capacity during dipyridamole-stimulated and PET-determined flow increases in patients with hypertrophic cardiomyopathy, which reflected the severity of microvascular dysfunction to functional and/or structural abnormalities of the coronary arteriolar vessels. In 51 patients with hypertrophic cardiomyopathy, a clinical follow-up of 8.1 ± 2.1 years was performed.107 The authors observed that the extent of reduced hyperemic coronary flow in these patients was independently predictive of death from cardiovascular causes, progression toward severe congestive heart failure, or sustained ventricular arrhythmias requiring the implantation of a cardioverter-defibrillator. Similarly, Neglia et al.144 observed that in 67 patients with idiopathic cardiomyopathy, severe reductions in hyperemic myocardial blood flows during dipyridamole stimulation of ≤ 1.36 mL/g/min were independently associated with a 3.5 and 3.3 relative risk of death or development or progression of heart failure during a mean follow-up of 45 ± 37 months. These observations clearly suggest that in patients with hypertrophic or idiopathic cardiomyopathy, dysfunction of the coronary arteriolar vessels may precede clinical deterioration,70,107 although its diagnostic value and its impact on medical treatment remain uncertain.
Insights Into Mechanisms of Coronary Circulatory Dysfunction by PET
Vascular Inflammatory States
The CAD process is commonly characterized by low-grade inflammation affecting coronary vasomotor function and is associated with increases in C-reactive protein (CRP) serum levels, levels of soluble endothelial cell adhesion molecules, and procoagulant activity.126,130 The exact mechanism underlying abnormal coronary vasomotion is certainly multifactorial, but it has been attributed in part to an imbalance of the redox equilibrium between endothelial-derived NO and ROS toward oxidative stress. Increases in oxidative stress in turn, apart from reducing the bioavailability of endothelial-derived NO associated with an impairment of endothelium-dependent coronary vasodilatory function, may lead to activation of a whole array of inflammatory genes such as nuclear factor κB (NF-κB), activator protein 1 (AP-1), or peroxisome proliferator, activated receptors (PPARs) involved in the pathogenesis of the CAD process. The inflammatory activation at the site of the arterial system is associated with the production of interleukin 6 (IL-6), which increases systemic markers of inflammation such as CRP.130,145 Whether elevated CRP levels reflect an alternate mediator to CAD or an epiphenomenon of no pathologic consequence is a matter of ongoing experimental and clinical research. Several studies have demonstrated that increases in CRP serum levels correlated with the degree of endothelial dysfunction in both the peripheral and the coronary circulation.31,54,146 Such findings suggest that inflammatory mechanisms might indicate, at least in part, the presence of endothelial dysfunction, providing an important link between inflammation and the development of atherosclerosis. Notably, PET flow measurements not only unmask the pathophysiologic consequences of elevated CRP serum levels on the epicardial conduit vessels31 but also extend into the coronary microcirculation and, in particular, appear to contribute to the effects of coronary risk factors in the initiation and development of CAD.54 The latter conclusion may also be supported by the independent value of elevated CRP serum levels in the predicting of future cardiovascular events.147,148
Smoking, Vascular Oxidation-Reduction State, and Hypertension
Noninvasive PET measurements of MBF have contributed to elucidating the complex relationship between the oxidation-reduction state of the arterial wall and coronary circulatory function.44,65 For example, Kaufmann et al.114 demonstrated that acute intravenous antioxidant intervention with vitamin C, given to reduce the oxidative stress burden in smokers, significantly increased hyperemic coronary flows during pharmacologic vasodilation, implicating ROS as a dominant cause of the attenuated total coronary vasodilator function in chronic smokers. In regard to the effects of hypercholesterolemia on coronary circulatory function, hyperemic flows during pharmacologic vasodilation were found to be reduced in individuals with familial hypercholesterolemia or with secondary hypercholesterolemia in comparison to age-matched controls.149–151 Importantly, the hyperemic flow increases were inversely related to the severity of abnormal plasma lipid levels. The detrimental effects of elevated total plasma cholesterol levels on the coronary circulation are well known, but it appears that LDL cholesterol is also a major determinant of reduced coronary vasodilatory capacity, as observed with PET.149 Another interesting observation is that a restoration of tetrahydrobiopterin (BH4) deficiency in hypercholesterolemic individuals restored the total hyperemic vasodilatory capacity of the coronary circulation.112 BH4 deficiency may thus contribute to coronary circulatory dysfunction, most likely through an uncoupling of eNOS,152 with further increases of ROS in hypercholesterolemia.
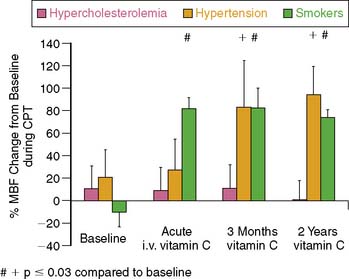
Of further interest is that the assessment of coronary vasomotor function by PET53 unmasked distinct differences in responses of coronary endothelial dysfunction to short-term and long-term antioxidant intervention with vitamin C in patients with different coronary risk factors such as smoking, arterial hypertension, and hypercholesterolemia (Fig. 33-8). These in vivo findings by PET53 differed from those in other investigations, that proposed increases in ROS as a primary common pathway underlying endothelial dysfunction,128,153 and suggested complex mechanisms accounting for abnormalities in coronary circulatory function in humans.37 In chronic smokers, short-term and long-term antioxidant vitamin C challenges improved abnormalities in endothelium-related MBF responses to CPT, while no such beneficial effect was observed in individuals with hypercholesterolemia. Such observation may indicate that abnormalities in endothelium-related coronary vasomotion in smokers are predominantly mediated by the release of ROS, while other mechanisms appeared to prevail in hypercholesterolemia-related coronary endothelial dysfunction. For example, selective targeting of G protein–dependent signal transduction by oxidized LDL results in a diminished receptor-mediated stimulation of endothelial NO production154 not affected by antioxidant intervention. Interestingly, although short-term vitamin C challenges did not lead to an improvement in coronary endothelial dysfunction in hypertensive patients, a normalization of the endothelium-related vasodilation was observed after long-term application of vitamin C for 3 months, which was sustained after a 2-year follow-up. The reason for the delayed onset of the beneficial effect of vitamin C challenges on coronary endothelial dysfunction remains uncertain; it may be related to an improvement of the endothelial redox equilibrium, resulting in an increased expression of eNOS or prevention of eNOS uncoupling through enhanced bioavailability of BH4.152,155,156 Another PET flow study investigated the effects of deferoxamine, an iron chelator that inhibits the generation of hydroxyl radicals, on endothelium-related flow responses to CPT in type 1 diabetes mellitus.116 Acute intravenous challenges of deferoxamine normalized the abnormality in endothelium-related flow responses to CPT, suggesting that ROS within the endothelium or in the subendothelial space plays a central role in mediating coronary flow abnormalities in type 1 diabetes mellitus.
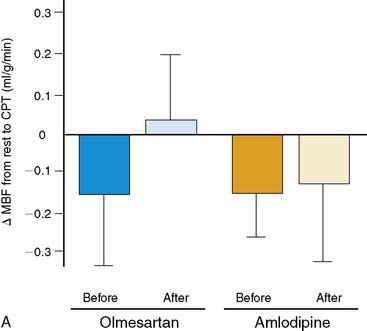
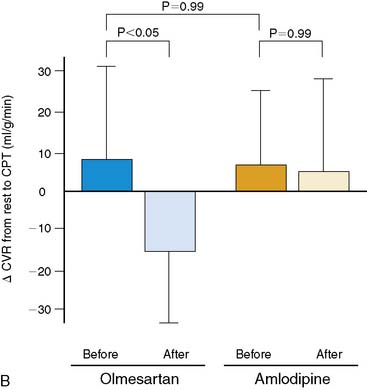
More recently, PET flow studies also contributed to unmasking heterogeneous effects of drug interventions on endothelium-related coronary vasomotion. Naya et al.157 studied 26 patients with untreated essential hypertension at baseline and after medical control of blood pressures with the angiotensin II receptor blocker (ARB) olmesartan or with the calcium antagonist amlodipine. The investigators demonstrated that only ARB inhibition improved endothelium-related MBF responses to sympathetic stimulation, while no such effect was observed for the calcium antagonist amlodipine (Fig. 33-9). The study further demonstrated that the elevation of superoxide dismutase (SOD) by ARB inhibition significantly correlated with the improvement of endothelium-dependent vasomotion in essential hypertension (r = 0.61; P < 0.05). These observations suggest that specific antioxidative effects of ARB inhibition, possibly mediated by inhibition of endothelial NADPH oxidase activation associated with a decrease in ROS or increases in antioxidative superoxide dismutase concentrations,128 account for the improvement in coronary endothelial function in these hypertensive patients.
Obesity, Insulin-Resistance, and Adipocytokines
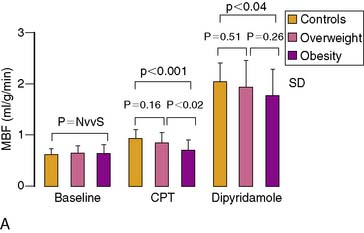
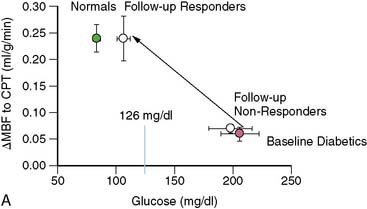
Obesity, frequently associated with the insulin-resistance syndrome and systemic microinflammation, has been recognized as a risk factor of cardiovascular morbidity and mortality.158 The exact mechanism by which obesity initiates and accelerates coronary vascular disease is still poorly understood. Coronary flow studies with PET demonstrated that insulin-resistant patients may present abnormalities in coronary circulatory function in the absence of traditional coronary risk factors.113 In these individuals, the endothelium-related MBF response to CPT was diminished, while hyperemic flows during dipyridamole stimulation were preserved. Thus, abnormalities of coronary circulatory function in insulin-resistant individuals appear to be confined to the coronary vascular endothelium. It should be noted that initial stages of the vascular injury may only involve the endothelium,54,84,111,159 whereas more advanced stages of coronary risk-factor states, such as increases in oxidative stress burden, may also lead to an impairment in smooth muscle cell vasodilator function.152 For example, functional alterations of the coronary circulation in individuals with increasing body weight progress from an impairment in endothelium-dependent coronary flow response to CPT, to an impairment of the predominantly endothelium-independent hyperemic flows during dipyridamole stimulation in obesity (Fig. 33-10).2 Similarly, Prior et al.3 observed a progressive worsening of functional abnormalities of endothelium-dependent vasomotion with increasing severity of insulin resistance and carbohydrate intolerance, while an attenuation of the total vasodilator capacity only became apparent in clinical type 2 diabetes. The observations are in agreement with earlier findings in type 1 and type 2 diabetes mellitus patients, with comparable reductions in hyperemic coronary flows.160–165 These reductions in hyperemic coronary flows were also related to euglycemic control and were correlated inversely with plasma glucose concentrations averaged over several months.
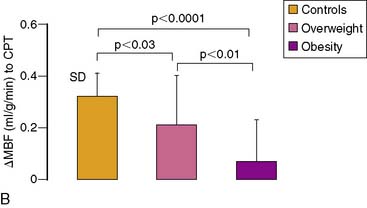
PET flow studies combined with various plasma or metabolic markers of coronary risk factors and evaluated by univariate and multivariate analysis appear to be promising in teasing out adverse or beneficial effects of various factors on coronary circulatory function in complex in vivo conditions in humans.2,44,166 Assessing coronary circulatory function in overweight individuals with PET demonstrated that an increase in body weight paralleled by an increase in plasma markers of insulin-resistance syndrome, and chronic inflammation was independently associated with abnormal endothelium-related coronary circulatory function.2 These findings provided first evidence that obesity is indeed an alternate mediator of coronary vascular disease rather than an epiphenomenon related to other traditional coronary risks.2,167 Of further interest is the role of various adipocytokines such as leptin, adiponectin, or ghrelin affecting the coronary circulation in humans.168,169 For example, in in vitro studies, leptin released from adipose tissue may stimulate pro-atherosclerotic effects such as increases in ROS in cultured human endothelial cells, acceleration of vascular cell calcification, and smooth muscle cell proliferation and migration.170 This is in contrast to more recent findings of a leptin-induced improvement in both endothelium-dependent and independent vasodilation.171–173 Notably, intracoronary infusion of leptin in humans with angiographically normal coronary arteries also may directly cause a coronary vasodilation of the conduit and arteriolar vessels.173 In view of these contradictory observations, PET coronary flow investigations in obese individuals unraveled a significant and positive association between leptin plasma levels and endothelium-related flow responses to CPT (r = 0.37; P < 0.036). Thus, increases in leptin plasma levels in obesity were associated with relatively higher endothelium-related flow increases to CPT. These observations in humans appear to be in agreement with the reported beneficial effect of leptin substitution on endothelium-dependent and thus nitric-oxide-mediated vasoreactivity in obese leptin knockout mice.174 The observed positive association between leptin plasma levels in obesity and relatively higher endothelium-related MBF responses to CPT in humans may indeed indicate a beneficial effect of leptin—and/or leptin-related but still undetermined factors—on the coronary endothelium to counteract the adverse effects of increases in body weight on coronary circulatory function.2 Thus, in vivo imaging with PET may contribute to delineating important associations between coronary risk factors and coronary vascular states that may add to or even contrast with experimental findings to date. In this way, PET flow studies of human coronary circulatory function may also provide information that could aid in defining investigations of more specific direct cause/effect relationships.
DELINEATION OF CORONARY RISK AND MONITORING OF THERAPEUTIC STRATEGIES
According to the Framingham Heart Study,175 individual persons with a high coronary risk profile (with diabetes or multiple coronary risk factors) are at risk for future cardiovascular events and can substantially benefit from medical intervention in primary and secondary prevention of coronary artery disease.176–178 The National Health and Nutrition Examination (NHANES) put forth, however, that there is a wide range of individuals (about 40% to 50%) who are estimated to be at intermediate risk for cardiovascular events and yet are not necessarily considered for preventive medical intervention and/or lifestyle modifications.179 These intermediate-risk individuals could benefit from further risk stratification with surrogates for subclinical CAD such as increases in carotid intima media thickness (IMT) by vascular ultrasound,180 MDCT-determined coronary artery calcification (CAC),69,93,181–183 and assessment of peripheral or coronary vasomotor function.44,65,70,184 Functional measures of vasomotor abnormalities have the advantage that they precede early CAD-related structural alterations of the arterial wall1 and appear to be more accurate in predicting future cardiovascular outcomes than structural alterations of the arterial wall.139,140,184 Use of noninvasive methods for identifying and characterizing subclinical stages of the CAD process44,69,93,185 could in fact lead to more effective primary and secondary prevention. Identification and characterization of functional and/or structural abnormalities of the arterial wall in subclinical stages of the atherosclerotic process in asymptomatic individuals at low or moderate risk could identify those individuals who are in fact at higher cardiac risk.45,69,93,126,127,179 Whether these individuals would benefit from initiation of or intensified medical preventive therapy remains to be studied.44,126,186
Based on the central role of functional abnormalities of the coronary circulation in the development and progression of atherosclerosis, improvement of endothelium-dependent coronary vasomotion by a variety of interventions, such as angiotensin-converting enzyme-inhibitors,187 β-hydroxymethylglutaryl-coenzyme A reductase inhibitors,115,188 hormone replacement therapy in postmenopausal women,111 euglycemic control in diabetes, and physical exercise,189,190 has become a primary therapeutic strategy for prevention of the atherosclerotic process. Notably, Fichtlscherer et al.109 reported that normalization of endothelial function of the forearm circulation in patients after an acute coronary syndrome was paralleled by an event-free survival. Similar findings were reported by Modena et al.108 in hypertensive postmenopausal women. An improvement in brachial artery flow–mediated, endothelium-dependent vasodilation after institution of aggressive antihypertensive therapy resulted in an improved clinical outcome. Although low in numbers, these preliminary results support the evolving concept that improvement in vasomotor function in peripheral and coronary circulation in patients with CAD or at risk of developing CAD may indeed lead to an improved clinical outcome. If this holds true in future clinical studies, then the assessment of coronary circulatory function by PET imaging could be a promising and unique tool to successfully guide preventive therapeutic strategies.
Hormone Replacement Therapy in Postmenopausal Women
Postmenopausal women are commonly at increased risk of developing cardiovascular disease.191,192 The exact mechanisms underlying the increased risk in developing cardiovascular disease in postmenopausal women have been related to changes in cardiovascular risk profile, such as the metabolic syndrome, insulin resistance, dyslipidemia, arterial hypertension, and deprivation of endogenous estroge, that commonly accompany menopause. Conceptually, insofar as abnormalities in coronary vasomotor function reflect an increased risk for future cardiovascular events, hormone replacement therapy (HRT) in postmenopausal women aiming to maintain or normalize coronary vasomotion should lead to an improved clinical outcome. PET measurements of MBF combined with CPT and pharmacologic stress, therefore, investigated the effects of HRT on coronary vasomotion in postmenopausal women. Long-term estrogen replacement in postmenopausal women without traditional coronary risk factors improved endothelium-related MBF responses to CPT. This beneficial effect, however, was not observed for short-term estrogen administration111,193,194 or in postmenopausal women with traditional coronary risk factors.111 Thus it appears that the presence of other coronary risk factors apart from the postmenopausal state may offset potential beneficial effects of HRT on endothelium-dependent coronary vasomotor function. On the other hand, it is intriguing to hypothesize that HRT may exert a beneficial effect on the coronary endothelium when coronary risk factors in postmenopausal women are controlled by medical intervention. Indeed, preliminary results in young postmenopausal women195 suggest that HRT, in addition to standard management of traditional risk factors, may contribute to maintaining the functional integrity of vascular endothelium.
Regarding the effects of HRT on attenuated hyperemic flows during pharmacologic vasodilation, there is some evidence of a beneficial effect of short-term oral estrogen medication on hyperemic flows in asymptomatic healthy postmenopausal women as well as in symptomatic postmenopausal women, of whom the majority had coronary risk factors.194 More recently, the effects of 17β-estradiol combined with the progestin, drospirenone on hyperemic flows in symptomatic postmenopausal women, and in part with other cardiovascular pharmaceuticals, were studied.196 HRT with 17β-estradiol and drospirenone administered for 6 weeks was associated with an improvement of hyperemic MBF when compared to the placebo group. The latter observation194,196 differs, however, from findings with other PET flow studies and HRT.111,193,194 The reason for the contradictory observations of a beneficial effect of HRT on hyperemic myocardial blood flows in postmenopausal women remains uncertain but is likely to be multifactorial. For example, differences in the clinical characteristics of postmenopausal women studied, elapsed time after menopause, the state of altered vasomotor function, and/or differences in the use of progestins combined with estrogens could account for differences in the observed effects of HRT on hyperemic flow increases in postmenopausal women.
Responses to Lipid-Lowering Treatments
Multicenter trials have provided convincing evidence of primary and secondary prevention of cardiac events by long-term treatment with reductase inhibitors.147,197,198 Notably, the beneficial effect in secondary prevention trials was observed despite minimal effects on the anatomic severity of coronary stenosis. Improvement in coronary endothelial dysfunction and stabilization in coronary plaques most likely account for the reported improvement in clinical outcome in hypercholesterolemic patients after long-term treatment with HMG-CoA reductase inhibitors. Several PET flow studies also investigated effects of HMG-CoA reductase inhibitors on diminished vasodilatory coronary capacity as a surrogate marker for early functional stages of developing CAD in hypercholesterolemic patients.
In one study, for example, repeat measurements of MBF were obtained in 51 asymptomatic young men with moderately elevated plasma cholesterol levels who were randomized with placebo or treatment. No significant effect on adenosine hyperemic blood flow was seen after 6 months of treatment with pravastatin (4 mg/day) (Table 33-4).199 However, average values of hyperemic blood flow and flow reserve were normal at baseline. Post hoc analysis of 15 study participants with diminished hyperemic MBF at baseline (<4 mL/min/g), showed a significant 27% increase in hyperemic MBF and a similar increase in myocardial flow reserve. Similar improvements were reported in asymptomatic patients with familial hypercholesterolemia after 9 to 15 months of treatment with simvastatin (5 to 10 mg/day),200 as well as in patients with normal or minimally diseased (<30% diameter narrowing or irregular luminal surface) coronary arteries after 6 months of simvastatin (20 mg/day) treatment.188
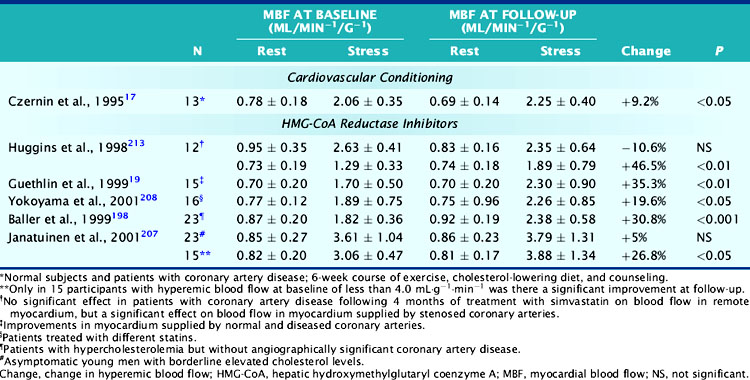
In these studies, the total cholesterol and LDL plasma cholesterol concentrations in asymptomatic individuals declined by 20% to 30% and 30% to 40%, respectively. Whether the improvement in coronary vasodilator capacity relates directly and quantitatively to reductions in total cholesterol or LDL plasma cholesterol concentrations is uncertain. The improvement in dipyridamole hyperemic MBF within 18 to 20 hours after plasma LDL apheresis suggests such a direct effect.201 Plasma LDL concentrations in this study were high at baseline (194 ± 38 mg/dL−1) and had decreased by 58%. Hyperemic MBF had increased by 31%. This observation of a significant improvement in coronary vasodilator capacity differs from findings of a delayed improvement in coronary vasoreactivity in patients with hypercholesterolemia who underwent 6 months of fluvastatin treatment (see Table 33-4).202 After 2 months of treatment, total and LDL plasma cholesterol concentrations had decreased by 28% and 37%, respectively, but hyperemic MBF remained unchanged. When these patients were reevaluated after 6 months, no further changes in plasma lipid levels were noted, but hyperemic MBF had improved by 35%. Unlike the study with LDL plasma apheresis, the delayed increase in coronary vasodilator capacity in the fluvastatin study argues against a direct effect of plasma cholesterol lowering and suggests that the beneficial vasomotor effects of HMG-CoA reductase inhibitors may be independent of plasma cholesterol concentrations. Further, these findings suggest that the effects of reductions in plasma cholesterol levels are mediated by other structural and functional mechanisms. Because of the striking reductions in plasma LDL cholesterol concentrations in the LDL apheresis study, rheologic factors, rather than a direct effect on coronary vasomotor function, may account for the higher postapheresis hyperemic MBF.201 Consistent with this possibility is a statistically significant 6.3% decrease in plasma viscosity that would reduce resistance to flow and thus result in higher hyperemic blood flow. It is also possible that the delay in improvement seen in the fluvastatin study was associated with structural improvements of the coronary vessels that required time, e.g., decrease in intima media thickness, and improvement in the bioactivity of or (as postulated by others) down-regulation of angiotensin-II receptors on endothelial cells.203–205
Measurements of MBF in other studies also show improvements in hyperemic flow and flow reserve in patients with clinically and angiographically documented CAD.202,206,207 These studies showed a beneficial effect on blood flow in myocardium subtended by angiographically normal and angiographically diseased coronary arteries. In only one study was the beneficial effect of HMG-CoA reductase inhibitors on hyperemic coronary flow increases confined to myocardial areas supplied by angiographically stenosed coronary arteries.206 Because the improvement in hyperemic flow was restricted to myocardium subtended by obstructive CAD, an effect on microvascular capacity appears unlikely. Rather, an increase in flow-mediated coronary vasodilation at the site of coronary stenosis may be a possible explanation. Reasons for the lack of improvement in remote myocardial areas include the shorter duration of treatment with HMG-CoA reductase inhibitors (only 4 months compared with 6 months or longer in the other studies), and hyperemic MBF and flow reserves that were essentially normal in the remote myocardium at baseline.
Insulin Resistance, Impaired Glucose Tolerance, and Diabetes Mellitus
More recently, the effects of preventive therapeutic intervention with insulin-sensitizing thiazolidinedione on coronary circulatory function in insulin-resistant patients were studied.113 The observed abnormalities in coronary endothelial dysfunction normalized with insulin-sensitizing thiazolidinedione treatment in 25 individuals with insulin resistance but without other coronary risk factors. These findings strongly suggest that insulin resistance possibly still has undetermined factors that affect coronary endothelial function. In view of these recent data, insulin-sensitizing pioglitazone application, added to conventional lipid-lowering therapy, did not only lead to an improvement in myocardial glucose utilization but resulted in an increase in resting MBF in nondiabetic patients with combined hyperlipidemia.166 The pioglitazone-mediated improvements in myocardial glucose utilization and resting MBF in these study participants were associated with a significant decrease in plasma insulin levels; glucose levels, however, were not altered.166 This might suggest that the insulin-sensitizing effects of pioglitazone accounted for the observed beneficial effects on myocardial metabolism and vascular parameter that go beyond those effects appreciated with conventional lipid-lowering therapy. As observed in the latter study166 and also in type 2 diabetic patients, pioglitazone medication did not beneficially alter adenosine-induced hyperemic coronary flows.208 A reasonable explanation for these inconsistent findings is still lacking113,166,208 but may be related to differences in patient characteristics, differences in the duration of insulin resistance, co-medication, and/or differences in angiopathy and vascular smooth muscle involvement. Another explanation is that in insulin-resistant study participants, alterations of coronary vasomotor function were largely limited to the endothelium and had not yet altered vascular smooth muscle cell function, as was reported for forearm blood flow measurements in individuals with CAD, new onset type 2 diabetes mellitus, and for the coronary circulation in individuals with increasing body weight or insulin resistance.2,3,209 In more advanced stages of diabetes, a higher burden of ROS and/or greater abnormalities in LDL subfractions and LDL oxidation within the vascular wall may also directly impair vascular smooth muscle cell function and thereby cause an impairment of the predominantly endothelium-independent hyperemic flow increase.208,210 The latter possibility may be supported by a recently observed association between PET-measured reductions in hyperemic coronary flows and structural alterations of the retinal arteries in more advanced stages of diabetes mellitus, while the hyperemic flow increase was still preserved in diabetic patients without background retinopathy.211 The restoration of coronary and peripheral vasomotor function by insulin-sensitizing thiazolidinedione therapy in insulin-resistant and diabetic individuals,113,166,209 however, may indeed reflect an important mechanistic link between increases in insulin sensitivity and an improvement in cardiovascular outcome in clinically manifest insulin resistance.178,212
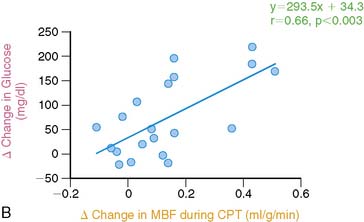
Effects of glucose lowering therapy on coronary circulatory function in type 2 diabetic patients were investigated as well.189 Three months of glucose-lowering treatment with glyburide and metformin significantly improved the coronary endothelium-mediated vasomotor function (Fig. 33-11A). The decrease in plasma glucose levels significantly correlated with the improvement in endothelium-related myocardial flow responses to CPT and thus an improvement in coronary vasomotor (dys)function (r = 0.67; P < 0.01) (see Fig. 33-11B). This association suggests a direct adverse effect of elevated plasma glucose, apart from the adverse effects of the insulin resistance syndrome, on diabetes-related coronary vascular disease in a preclinical state of CAD.
ACKNOWLEDGMENTS
Some sections of the manuscript are similar to sections of an extensive review of cardiac PET by Schelbert213 and Schindler et al.44
1. Reddy K.G., Nair R.N., Sheehan H.M., et al. Evidence that selective endothelial dysfunction may occur in the absence of angiographic or ultrasound atherosclerosis in patients with risk factors for atherosclerosis. J Am Coll Cardiol. 1994;23(4):833-843.
2. Schindler T.H., Cardenas J., Prior J.O., et al. Relationship between increasing body weight, insulin resistance, inflammation, adipocytokine leptin, and coronary circulatory function. J Am Coll Cardiol. 2006;47(6):1188-1195.
3. Prior J.O., Quinones M.J., Hernandez-Pampaloni M., et al. Coronary circulatory dysfunction in insulin resistance, impaired glucose tolerance, and type 2 diabetes mellitus. Circulation. 2005;111(18):2291-2298.
4. Gambhir S.S., Schwaiger M., Huang S.C., et al. Simple noninvasive quantification method for measuring myocardial glucose utilization in humans employing positron emission tomography and fluorine-18 deoxyglucose. J Nucl Med. 1989;30(3):359-366.
5. Weinberg I.N., Huang S.C., Hoffman E.J., et al. Validation of PET-acquired input functions for cardiac studies. J Nucl Med. 1988;29(2):241-247.
6. Kuhle W.G., Porenta G., Huang S.C., et al. Quantification of regional myocardial blood flow using 13N-ammonia and reoriented dynamic positron emission tomographic imaging. Circulation. 1992;86(3):1004-1017.
7. Bergmann S.R., Fox K.A., Rand A.L., et al. Quantification of regional myocardial blood flow in vivo with H215O. Circulation. 1984;70(4):724-733.
8. Nitzsche E.U., Choi Y., Czernin J., et al. Noninvasive quantification of myocardial blood flow in humans. A direct comparison of the [13N]ammonia and the [15O]water techniques. Circulation. 1996;93(11):2000-2006.
9. Bol A., Melin J.A., Vanoverschelde J.L., et al. Direct comparison of [13N]ammonia and [15O]water estimates of perfusion with quantification of regional myocardial blood flow by microspheres. Circulation. 1993;87(2):512-525.
10. Schelbert H.R., Phelps M.E., Huang S.C., et al. N-13 ammonia as an indicator of myocardial blood flow. Circulation. 1981;63(6):1259-1272.
11. Schelbert H.R., Wisenberg G., Phelps M.E., et al. Noninvasive assessment of coronary stenoses by myocardial imaging during pharmacologic coronary vasodilation. VI. Detection of coronary artery disease in human beings with intravenous N-13 ammonia and positron computed tomography. Am J Cardiol. 1982;49(5):1197-1207.
12. Gould K.L., Goldstein R.A., Mullani N.A., et al. Noninvasive assessment of coronary stenoses by myocardial perfusion imaging during pharmacologic coronary vasodilation. VIII. Clinical feasibility of positron cardiac imaging without a cyclotron using generator-produced rubidium-82. J Am Coll Cardiol. 1986;7(4):775-789.
13. Gould K.L., Schelbert H.R., Phelps M.E., et al. Noninvasive assessment of coronary stenoses with myocardial perfusion imaging during pharmacologic coronary vasodilatation. V. Detection of 47 percent diameter coronary stenosis with intravenous nitrogen-13 ammonia and emission-computed tomography in intact dogs. Am J Cardiol. 1979;43(2):200-208.
14. Krivokapich J., Czernin J., Schelbert H.R. Dobutamine positron emission tomography: absolute quantitation of rest and dobutamine myocardial blood flow and correlation with cardiac work and percent diameter stenosis in patients with and without coronary artery disease. J Am Coll Cardiol. 1996;28(3):565-572.
15. Chow B.J., Beanlands R.S., Lee A., et al. Treadmill exercise produces larger perfusion defects than dipyridamole stress N-13 ammonia positron emission tomography. J Am Coll Cardiol. 2006;47(2):411-416.
16. Go R.T., Marwick T.H., MacIntyre W.J., et al. A prospective comparison of rubidium-82 PET and thallium-201 SPECT myocardial perfusion imaging utilizing a single dipyridamole stress in the diagnosis of coronary artery disease. J Nucl Med. 1990;31(12):1899-1905.
17. Marwick T.H., Shan K., Patel S., et al. Incremental value of rubidium-82 positron emission tomography for prognostic assessment of known or suspected coronary artery disease. Am J Cardiol. 1997;80(7):865-870.
18. Sampson U.K., Dorbala S., Limaye A., et al. Diagnostic accuracy of rubidium-82 myocardial perfusion imaging with hybrid positron emission tomography/computed tomography in the detection of coronary artery disease. J Am Coll Cardiol. 2007;49(10):1052-1058.
19. Yoshinaga K., Chow B.J., Williams K., et al. What is the prognostic value of myocardial perfusion imaging using rubidium-82 positron emission tomography? J Am Coll Cardiol. 2006;48(5):1029-1039.
20. Chow B.J., Ananthasubramaniam K., dekemp R.A., et al. Comparison of treadmill exercise versus dipyridamole stress with myocardial perfusion imaging using rubidium-82 positron emission tomography. J Am Coll Cardiol. 2005;45(8):1227-1234.
21. Bateman T.M., Heller G.V., McGhie A.I., et al. Diagnostic accuracy of rest/stress ECG-gated Rb-82 myocardial perfusion PET: comparison with ECG-gated Tc-99m sestamibi SPECT. J Nucl Cardiol. 2006;13(1):24-33.
22. Groves A.M., Speechly-Dick M.E., Dickson J.C., et al. Cardiac (82)rubidium PET/CT: initial European experience. Eur J Nucl Med Mol Imaging. 2007;34(12):1965-1972.
23. Bergmann S.R., Herrero P., Markham J., et al. Noninvasive quantitation of myocardial blood flow in human subjects with oxygen-15-labeled water and positron emission tomography. J Am Coll Cardiol. 1989;14(3):639-652.
24. Herrero P., Markham J., Shelton M.E., et al. Noninvasive quantification of regional myocardial perfusion with rubidium-82 and positron emission tomography. Exploration of a mathematical model. Circulation. 1990;82(4):1377-1386.
25. Lin J.W., Sciacca R.R., Chou R.L., et al. Quantification of myocardial perfusion in human subjects using 82Rb and wavelet-based noise reduction. J Nucl Med. 2001;42(2):201-208.
26. El Fakhri G., Sitek A., Guerin B., et al. Quantitative dynamic cardiac 82Rb PET using generalized factor and compartment analyses. J Nucl Med. 2005;46(8):1264-1271.
27. Lortie M., Beanlands R.S., Yoshinaga K., et al. Quantification of myocardial blood flow with 82Rb dynamic PET imaging. Eur J Nucl Med Mol Imaging. 2007;34(11):1765-1774.
28. Huang S.C., Williams B.A., Krivokapich J., et al. Rabbit myocardial 82Rb kinetics and a compartmental model for blood flow estimation. Am J Physiol. 1989;256(4 Pt 2):H1156-H1164.
29. Lin J.W., Laine A.F., Akinboboye O., et al. Use of wavelet transforms in analysis of time-activity data from cardiac PET. J Nucl Med. 2001;42(2):194-200.
30. Iida H., Kanno I., Takahashi A., et al. Measurement of absolute myocardial blood flow with H215O and dynamic positron-emission tomography. Strategy for quantification in relation to the partial-volume effect. Circulation. 1988;78(1):104-115.
31. Schindler T.H., Hornig B., Buser P.T., Olschewski M., Magosaki N., Pfisterer M., Nitzsche E.U., Solzbach U., Just H. Prognostic value of abnormal vasoreactivity of epicardial coronary arteries to sympathetic stimulation in patients with normal coronary angiograms. Arterioscler Thromb Vasc Biol. 2003;23(3):495-501.
32. Lerman A., Zeiher A.M. Endothelial function: cardiac events. Circulation. 2005;111(3):363-368.
33. Suwaidi J.A., Hamasaki S., Higano S.T., et al. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. 2000;101(9):948-954.
34. Britten M.B., Zeiher A.M., Schachinger V. Microvascular dysfunction in angiographically normal or mildly diseased coronary arteries predicts adverse cardiovascular long-term outcome. Coron Artery Dis. 2004;15(5):259-264.
35. Schachinger V., Britten M.B., Zeiher A.M. Prognostic impact of coronary vasodilator dysfunction on adverse long-term outcome of coronary heart disease. Circulation. 2000;101(16):1899-1906.
36. Halcox J.P., Schenke W.H., Zalos G., et al. Prognostic value of coronary vascular endothelial dysfunction. Circulation. 2002;106(6):653-658.
37. Drexler H. Endothelial dysfunction: clinical implications. Prog Cardiovasc Dis. 1997;39(4):287-324.
38. Drexler H., Zeiher A.M., Wollschlager H., et al. Flow-dependent coronary artery dilatation in humans. Circulation. 1989;80(3):466-474.
39. Zeiher A.M., Drexler H., Saurbier B., et al. Endothelium-mediated coronary blood flow modulation in humans. Effects of age, atherosclerosis, hypercholesterolemia, and hypertension. J Clin Invest. 1993;92(2):652-662.
40. Zeiher A.M., Drexler H., Wollschlager H., et al: Modulation of coronary vasomotor tone in humans. Progressive endothelial dysfunction with different early stages of coronary atherosclerosis, Circulation 83(2):391–401
41. Zeiher A.M., Krause T., Schachinger V., et al. Impaired endothelium-dependent vasodilation of coronary resistance vessels is associated with exercise-induced myocardial ischemia. Circulation. 1995;91(9):2345-2352.
42. Cox D.A., Vita J.A., Treasure C.B., et al. Atherosclerosis impairs flow-mediated dilation of coronary arteries in humans. Circulation. 1989;80(3):458-465.
43. Gordon J.B., Ganz P., Nabel E.G., et al. Atherosclerosis influences the vasomotor response of epicardial coronary arteries to exercise. J Clin Invest. 1989;83(6):1946-1952.
44. Schindler T.H., Zhang X.L., Vincenti G., et al. Role of PET in the evaluation and understanding of coronary physiology. J Nucl Cardiol. 2007;14(4):589-603.
45. Cohn J.N., Quyyumi A.A., Hollenberg N.K., et al. Surrogate markers for cardiovascular disease: functional markers. Circulation. 2004;109(25 Suppl 1):IV31-IV46.
46. Schindler T.H., Nitzsche E.U., Olschewski M., et al. PET-Measured Responses of MBF to Cold Pressor Testing Correlate with Indices of coronary vasomotion on quantitative coronary angiography. J Nucl Med. 2004;45(3):419-428.
47. Ludmer P.L., Selwyn A.P., Shook T.L., et al. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315(17):1046-1051.
48. Zeiher A.M., Drexler H., Wollschlager H., et al. Endothelial dysfunction of the coronary microvasculature is associated with coronary blood flow regulation in patients with early atherosclerosis. Circulation. 1991;84(5):1984-1992.
49. Nitenberg A., Ledoux S., Valensi P., et al. Impairment of coronary microvascular dilation in response to cold pressor–induced sympathetic stimulation in type 2 diabetic patients with abnormal stress thallium imaging. Diabetes. 2001;50(5):1180-1185.
50. Dimmeler S., Fleming I., Fisslthaler B., et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399(6736):601-605.
51. Dimmeler S., Zeiher A.M. Exercise and cardiovascular health: get active to “AKTivate” your endothelial nitric oxide synthase. Circulation. 2003;107(25):3118-3120.
52. Zeiher A.M., Drexler H., Wollschlaeger H., et al. Coronary vasomotion in response to sympathetic stimulation in humans: importance of the functional integrity of the endothelium. J Am Coll Cardiol. 1989;14(5):1181-1190.
53. Schindler T.H., Nitzsche E.U., Munzel T., et al. Coronary vasoregulation in patients with various risk factors in response to cold pressor testing: contrasting myocardial blood flow responses to short- and long-term vitamin C administration. J Am Coll Cardiol. 2003;42(5):814-822.
54. Schindler T.H., Nitzsche E.U., Olschewski M., et al. Chronic inflammation and impaired coronary vasoreactivity in patients with coronary risk factors. Circulation. 2004;110(9):1069-1075.
55. Zeiher A.M., Drexler H. Coronary hemodynamic determinants of epicardial artery vasomotor responses during sympathetic stimulation in humans. Basic Res Cardiol. 1991;86(Suppl 2):203-213.
56. Rossen J.D., Quillen J.E., Lopez A.G., et al. Comparison of coronary vasodilation with intravenous dipyridamole and adenosine. J Am Coll Cardiol. 1991;18(2):485-491.
57. Chareonthaitawee P., Kaufmann P.A., Rimoldi O., et al. Heterogeneity of resting and hyperemic myocardial blood flow in healthy humans. Cardiovasc Res. 2001;50(1):151-161.
58. Czernin J., Muller P., Chan S., et al. Influence of age and hemodynamics on myocardial blood flow and flow reserve. Circulation. 1993;88(1):62-69.
59. Tamaki N., Yonekura Y., Senda M., et al. Myocardial positron computed tomography with 13N-ammonia at rest and during exercise. Eur J Nucl Med. 1985;11(6–7):246-251.
60. Senneff M.J., Geltman E.M., Bergmann S.R. Noninvasive delineation of the effects of moderate aging on myocardial perfusion. J Nucl Med. 1991;32(11):2037-2042.
61. Prior J.O., Schindler T.H., Facta A.D., et al. Determinants of myocardial blood flow response to cold pressor testing and pharmacologic vasodilation in healthy humans. Eur J Nucl Med Mol Imaging. 2007;34(1):20-27.
62. Sawada S., Muzik O., Beanlands R.S., et al. Interobserver and interstudy variability of myocardial blood flow and flow-reserve measurements with nitrogen 13 ammonia-labeled positron emission tomography. J Nucl Cardiol. 1995;2(5):413-422.
63. Krivokapich J., Smith G.T., Huang S.C., et al. 13N ammonia myocardial imaging at rest and with exercise in normal volunteers. Quantification of absolute myocardial perfusion with dynamic positron emission tomography. Circulation. 1989;80(5):1328-1337.
64. Duvernoy C.S., Meyer C., Seifert-Klauss V., et al. Gender differences in myocardial blood flow dynamics: lipid profile and hemodynamic effects. J Am Coll Cardiol. 1999;33(2):463-470.
65. Kaufmann P.A., Camici P.G. Myocardial blood flow measurement by PET: technical aspects and clinical applications. J Nucl Med. 2005;46(1):75-88.
66. Wyss C.A., Koepfli P., Mikolajczyk K., et al. Bicycle exercise stress in PET for assessment of coronary flow reserve: repeatability and comparison with adenosine stress. J Nucl Med. 2003;44(2):146-154.
67. Namdar M., Koepfli P., Grathwohl R., et al. Caffeine decreases exercise-induced myocardial flow reserve. J Am Coll Cardiol. 2006;47(2):405-410.
68. Jagathesan R., Barnes E., Rosen S.D., et al. Comparison of myocardial blood flow and coronary flow reserve during dobutamine and adenosine stress: Implications for pharmacologic stress testing in coronary artery disease. J Nucl Cardiol. 2006;13(3):324-332.
69. Di Carli M.F., Hachamovitch R. New technology for noninvasive evaluation of coronary artery disease. Circulation. 2007;115(11):1464-1480.
70. Camici P.G., Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356(8):830-840.
71. Alexanderson E., Cruz P., Vargas A., et al. Endothelial dysfunction in patients with antiphospholipid syndrome assessed with positron emission tomography. J Nucl Cardiol. 2007;14(4):566-572.
72. Hernandez-Pampaloni M., Keng F.Y., Kudo T., et al. Abnormal longitudinal, base-to-apex myocardial perfusion gradient by quantitative blood flow measurements in patients with coronary risk factors. Circulation. 2001;104(5):527-532.
73. Schindler T.H., Facta A.D., Prior J.O., et al. PET-measured heterogeneity in longitudinal myocardial blood flow in response to sympathetic and pharmacologic stress as a non-invasive probe of epicardial vasomotor dysfunction. Eur J Nucl Med Mol Imaging. 2006;33(10):1140-1149.
74. Kaufmann P.A., Gnecchi-Ruscone T., Yap J.T., et al. Assessment of the reproducibility of baseline and hyperemic myocardial blood flow measurements with 15O-labeled water and PET. J Nucl Med. 1999;40(11):1848-1856.
75. Go V., Bhatt M.R., Hendel R.C. The diagnostic and prognostic value of ECG-gated SPECT myocardial perfusion imaging. J Nucl Med. 2004;45(5):912-921.
76. Kubo S., Tadamura E., Toyoda H., et al. Effect of caffeine intake on myocardial hyperemic flow induced by adenosine triphosphate and dipyridamole. J Nucl Med. 2004;45(5):730-738.
77. Buus N.H., Bottcher M., Hermansen F., et al. Influence of nitric oxide synthase and adrenergic inhibition on adenosine-induced myocardial hyperemia. Circulation. 2001;104(19):2305-2310.
78. Tawakol A., Forgione M.A., Stuehlinger M., et al. Homocysteine impairs coronary microvascular dilator function in humans. J Am Coll Cardiol. 2002;40(6):1051-1058.
79. Coffman J.D., Gregg D.E. Reactive hyperemia characteristics of the myocardium. Am J Physiol. 1960;199:1143-1149.
80. Mosher P., Ross J.Jr, McFate P.A., et al. Control of coronary blood flow by an autoregulatory mechanism. Circ Res. 1964;14:250-259.
81. Morita K., Tsukamoto T., Naya M., et al. Smoking cessation normalizes coronary endothelial vasomotor response assessed with 15O-water and PET in healthy young smokers. J Nucl Med. 2006;47(12):1914-1920.
82. Tsukamoto T., Morita K., Naya M., et al. Myocardial flow reserve is influenced by both coronary artery stenosis severity and coronary risk factors in patients with suspected coronary artery disease. Eur J Nucl Med Mol Imaging. 2006;33(10):1150-1156.
83. Nabel E.G., Ganz P., Gordon J.B., et al. Dilation of normal and constriction of atherosclerotic coronary arteries caused by the cold pressor test. Circulation. 1988;77(1):43-52.
84. Campisi R., Czernin J., Schoder H., et al. L-Arginine normalizes coronary vasomotion in long-term smokers. Circulation. 1999;99(4):491-497.
85. Sato A., Terata K., Miura H., et al. Mechanism of vasodilation to adenosine in coronary arterioles from patients with heart disease. Am J Physiol Heart Circ Physiol. 2005;288(4):H1633-H1640.
86. Schindler T.H., Schelbert H.R. Measurements of myocardial blood flow and monitoring therapy. In: Zaret B.L., Beller G.A., editors. Nuclear Cardiology: State of the Art and Future Directions. ed 3. Philadelphia: Mosby; 2005:399-412.
87. Gould K.L., Lipscomb K., Calvert C. Compensatory changes of the distal coronary vascular bed during progressive coronary constriction. Circulation. 1975;51(6):1085-1094.
88. Di Carli M., Czernin J., Hoh C.K., et al. Relation among stenosis severity, myocardial blood flow, and flow reserve in patients with coronary artery disease. Circulation. 1995;91(7):1944-1951.
89. Beanlands R.S., Muzik O., Melon P., et al. Noninvasive quantification of regional myocardial flow reserve in patients with coronary atherosclerosis using nitrogen-13 ammonia positron emission tomography. Determination of extent of altered vascular reactivity. J Am Coll Cardiol. 1995;26(6):1465-1475.
90. Uren N.G., Melin J.A., De Bruyne B., et al. Relation between myocardial blood flow and the severity of coronary-artery stenosis. N Engl J Med. 1994;330(25):1782-1788.
91. Kern M.J. Coronary physiology revisited: practical insights from the cardiac catheterization laboratory. Circulation. 2000;101(11):1344-1351.
92. Gould K.L., Lipscomb K. Effects of coronary stenoses on coronary flow reserve and resistance. Am J Cardiol. 1974;34(1):48-55.
93. Di Carli M.F., Dorbala S., Hachamovitch R. Integrated cardiac PET-CT for the diagnosis and management of CAD. J Nucl Cardiol. 2006;13(2):139-144.
94. Gould K.L., Nakagawa Y., Nakagawa K., et al. Frequency and clinical implications of fluid dynamically significant diffuse coronary artery disease manifest as graded, longitudinal, base-to-apex myocardial perfusion abnormalities by noninvasive positron emission tomography. Circulation. 2000;101(16):1931-1939.
95. Gould K.L. Assessing progression or regression of CAD: the role of perfusion imaging. J Nucl Cardiol. 2005;12(6):625-638.
96. Graf S., Khorsand A., Gwechenberger M., et al. Typical chest pain and normal coronary angiogram: cardiac risk factor analysis versus PET for detection of microvascular disease. J Nucl Med. 2007;48(2):175-181.
97. Johnson N.P., Gould K.L. Clinical evaluation of a new concept: resting myocardial perfusion heterogeneity quantified by Markovian analysis of PET identifies coronary microvascular dysfunction and early atherosclerosis in 1,034 subjects. J Nucl Med. 2005;46(9):1427-1437.
98. Schindler T.H., Facta A.D., Prior J.O., et al. Structural alterations of the coronary arterial wall are associated with myocardial flow heterogeneity in type 2 diabetes mellitus. Eur J Nucl Med Mol Imaging. 2009;36(2):219-229. (Epub 2008)
99. Schindler T.H., Zhang X.L., Vincenti G., et al. Diagnostic value of PET-measured heterogeneity in myocardial blood flows during cold pressor testing for the identification of coronary vasomotor dysfunction. J Nucl Cardiol. 2007;14(5):688-697.
100. De Bruyne B., Hersbach F., Pijls N.H., et al. Abnormal epicardial coronary resistance in patients with diffuse atherosclerosis but “normal” coronary angiography. Circulation. 2001;104(20):2401-2406.
101. Lim M.J., Kern M.J. Coronary pathophysiology in the cardiac catheterization laboratory. Curr Probl Cardiol. 2006;31(8):493-550.
102. Verna E., Ceriani L., Provasoli S., et al. Larger perfusion defects with exercise compared with dipyridamole SPECT (exercise-dipyridamole mismatch) may reflect differences in epicardial and microvascular coronary dysfunction: when the stressor matters. J Nucl Cardiol. 2007;14(6):818-826.
103. Schindler T.H., Schelbert H.R. “Mismatch” in regional myocardial perfusion defects during exercise and pharmacologic vasodilation: a noninvasive marker of epicardial vasomotor dysfunction. J Nucl Cardiol. 2007;14(6):769-774.
104. Sdringola S., Loghin C., Boccalandro F., et al. Mechanisms of progression and regression of coronary artery disease by PET related to treatment intensity and clinical events at long-term follow-up. J Nucl Med. 2006;47(1):59-67.
105. Bonetti P.O., Lerman L.O., et al. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003;23:168-175.
106. Schindler T.H., Nitzsche E.U., Schelbert H.R., et al. Positron emission tomography-measured abnormal responses of myocardial blood flow to sympathetic stimulation are associated with the risk of developing cardiovascular events. J Am Coll Cardiol. 2005;45(9):1505-1512.
107. Cecchi F., Olivotto I., Gistri R., et al. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med. 2003;349(11):1027-1035.
108. Modena M.G., Bonetti L., Coppi F., et al. Prognostic role of reversible endothelial dysfunction in hypertensive postmenopausal women. J Am Coll Cardiol. 2002;40(3):505-510.
109. Fichtlscherer S., Breuer S., Zeiher A.M. Prognostic value of systemic endothelial dysfunction in patients with acute coronary syndromes: further evidence for the existence of the “vulnerable” patient. Circulation. 2004;110(14):1926-1932.
110. Papaioannou G.I., Kasapis C., Seip R.L., et al. Value of peripheral vascular endothelial function in the detection of relative myocardial ischemia in asymptomatic type 2 diabetic patients who underwent myocardial perfusion imaging. J Nucl Cardiol. 2006;13(3):362-368.
111. Campisi R., Nathan L., Pampaloni M.H., et al. Noninvasive assessment of coronary microcirculatory function in postmenopausal women and effects of short-term and long-term estrogen administration. Circulation. 2002;105(4):425-430.
112. Wyss C.A., Koepfli P., Namdar M., et al. Tetrahydrobiopterin restores impaired coronary microvascular dysfunction in hypercholesterolaemia. Eur J Nucl Med Mol Imaging. 2005;32(1):84-91.
113. Quinones M.J., Hernandez-Pampaloni M., Schelbert H., et al. Coronary vasomotor abnormalities in insulin-resistant individuals. Ann Intern Med. 2004;140(9):700-708.
114. Kaufmann P.A., Gnecchi-Ruscone T., di Terlizzi M., et al. Coronary heart disease in smokers: vitamin C restores coronary microcirculatory function. Circulation. 2002;102(11):1233-1238.
115. Wielepp P., Baller D., Gleichmann U., et al. Beneficial effects of atorvastatin on myocardial regions with initially low vasodilatory capacity at various stages of coronary artery disease. Eur J Nucl Med Mol Imaging. 2005;32(12):1371-1377.
116. Hattori N., Schnell O., Bengel F.M., et al. Deferoxamine improves coronary vascular responses to sympathetic stimulation in patients with type 1 diabetes mellitus. Eur J Nucl Med Mol Imaging. 2002;29(7):891-898.
117. Bengel F.M., Abletshauser C., Neverve J., et al. Effects of nateglinide on myocardial microvascular reactivity in Type 2 diabetes mellitus–a randomized study using positron emission tomography. Diabet Med. 2005;22(2):158-163.
118. Schindler T.H., Zhang X.L., Prior J.O., et al. Assessment of intra- and interobserver reproducibility of rest and cold pressor test-stimulated myocardial blood flow with (13) N-ammonia and PET. Eur J Nucl Med Mol Imaging. 2007;34:1178-1188.
119. Panza J.A., Quyyumi A.A., Diodati J.G., et al. Long-term variation in myocardial ischemia during daily life in patients with stable coronary artery disease: its relation to changes in the ischemic threshold. J Am Coll Cardiol. 1992;19(3):500-506.
120. el-Tamimi H., Mansour M., Pepine C.J., et al. Circadian variation in coronary tone in patients with stable angina. Protective role of the endothelium. Circulation. 1995;92(11):3201-3205.
121. Nagamachi S., Czernin J., Kim A.S., et al. Reproducibility of measurements of regional resting and hyperemic myocardial blood flow assessed with PET. J Nucl Med. 1996;37(10):1626-1631.
122. Siegrist P.T., Gaemperli O., Koepfli P., et al. Repeatability of cold pressor test-induced flow increase assessed with H(2)(15)O and PET. J Nucl Med. 2006;47(9):1420-1426.
123. Bland J.M., Altman D.G. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet. 1986;1(8476):307-310.
124. Jagathesan R., Kaufmann P.A., Rosen S.D., et al. Assessment of the long-term reproducibility of baseline and dobutamine-induced myocardial blood flow in patients with stable coronary artery disease. J Nucl Med. 2005;46(2):212-219.
125. Celermajer D.S. Endothelial dysfunction: does it matter? Is it reversible. J Am Coll Cardiol. 1997;30(2):325-333.
126. Ganz P., Vita J.A. Testing endothelial vasomotor function: nitric oxide, a multipotent molecule. Circulation. 2003;108(17):2049-2053.
127. Widlansky M.E., Gokce N., Keaney J.F., et al. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42(7):1149-1160.
128. Cai H., Harrison D.G. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87(10):840-844.
129. Tomai F., Crea F., Gaspardone A., et al. Unstable angina and elevated C-reactive protein levels predict enhanced vasoreactivity of the culprit lesion. Circulation. 2001;104(13):1471-1476.
130. Faxon D.P., Fuster V., Libby P., et al. Atherosclerotic vascular disease conference: Writing group III: pathophysiology. Circulation. 2004;109(21):2617-2625.
131. Topol E.J., Yadav J.S. Recognition of the importance of embolization in atherosclerotic vascular disease. Circulation. 2000;101(5):570-580.
132. Berliner J.A., Navab M., Fogelman A.M., Frank J.S., Demer L.L., Edwards P.A., Watson A.D., Lusis A.J. Atherosclerosis: basic mechanisms. Oxidation, inflammation, and genetics. Circulation. 1995;91(9):2488-2496.
133. Brull D.J., Serrano N., Zito F., et al. Human CRP gene polymorphism influences CRP levels: implications for the prediction and pathogenesis of coronary heart disease. Arterioscler Thromb Vasc Biol. 2003;23(11):2063-2069.
134. Hachamovitch R., Hayes S.W., Friedman J.D., et al. Stress myocardial perfusion single-photon emission computed tomography is clinically effective and cost effective in risk stratification of patients with a high likelihood of coronary artery disease (CAD) but no known CAD. J Am Coll Cardiol. 2004;43(2):200-208.
135. Hachamovitch R., Hayes S.W., Friedman J.D., et al. A prognostic score for prediction of cardiac mortality risk after adenosine stress myocardial perfusion scintigraphy. J Am Coll Cardiol. 2005;45(5):722-729.
136. Beller G.A. Cardiac imaging: value, cost, and appropriateness. J Nucl Cardiol. 2007;14(6):763-764.
137. Beller G.A. Nuclear cardiology: where are we and where are we going? J Nucl Cardiol. 2006;13(5):601-602.
138. Berman D.S., Wong N.D., Gransar H., et al. Relationship between stress-induced myocardial ischemia and atherosclerosis measured by coronary calcium tomography. J Am Coll Cardiol. 2004;44(4):923-930.
139. Rozanski A., Gransar H., Wong N.D., et al. Clinical outcomes after both coronary calcium scanning and exercise myocardial perfusion scintigraphy. J Am Coll Cardiol. 2007;49(12):1352-1361.
140. Nishimura R.A., Lerman A., Chesebro J.H., et al. Epicardial vasomotor responses to acetylcholine are not predicted by coronary atherosclerosis as assessed by intracoronary ultrasound. J Am Coll Cardiol. 1995;26(1):41-49.
141. Schuijf J.D., Wijns W., Jukema J.W., et al: A comparative regional analysis of coronary atherosclerosis and calcium score on multislice CT versus myocardial perfusion on SPECT, J Nucl Med 47(11): 1749–1755
142. Madjid M., Toutouzas K., Stefanadis C., et al. Coronary thermography for detection of vulnerable plaques. J Nucl Cardiol. 2007;14(2):244-249.
143. Cury R.C., Nieman K., Shapiro M.D., et al. Comprehensive cardiac CT study: evaluation of coronary arteries, left ventricular function, and myocardial perfusion–is it possible? J Nucl Cardiol. 2007;14(2):229-243.
144. Neglia D., Michelassi C., Trivieri M.G., et al. Prognostic role of myocardial blood flow impairment in idiopathic left ventricular dysfunction. Circulation. 2002;105(2):186-193.
145. Szmitko P.E., Wang C.H., Weisel R.D., et al. New markers of inflammation and endothelial cell activation: Part I. Circulation. 2003;108(16):1917-1923.
146. Fichtlscherer S., Rosenberger G., Walter D.H., et al. Elevated C-reactive protein levels and impaired endothelial vasoreactivity in patients with coronary artery disease. Circulation. 2000;102(9):1000-1006.
147. Blake G.J., Ridker P.M. C-reactive protein, subclinical atherosclerosis, and risk of cardiovascular events. Arterioscler Thromb Vasc Biol. 2002;22(10):1512-1513.
148. Tracy R.P., Lemaitre R.N., Psaty B.M., et al. Relationship of C-reactive protein to risk of cardiovascular disease in the elderly. Results from the cardiovascular health study and the rural health promotion project. Arterioscler Thromb Vasc Biol. 1997;17(6):1121-1127.
149. Kaufmann P.A., Gnecchi-Ruscone T., Schafers K.P., et al. Low density lipoprotein cholesterol and coronary microvascular dysfunction in hypercholesterolemia. J Am Coll Cardiol. 2000;36(1):103-109.
150. Yokoyama I., Ohtake T., Momomura S., et al. Reduced coronary flow reserve in hypercholesterolemic patients without overt coronary stenosis. Circulation. 1996;94(12):3232-3238.
151. Pitkanen O.P., Nuutila P., Raitakari O.T., et al. Coronary flow reserve in young men with familial combined hyperlipidemia. Circulation. 1999;99(13):1678-1684.
152. Munzel T., Daiber A., Ullrich V., et al. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler Thromb Vasc Biol. 2005;25(8):1551-1557.
153. Munzel T., Keaney J.F.Jr. Are ACE inhibitors a “magic bullet” against oxidative stress? Circulation. 2001;104(13):1571-1574.
154. Ohgushi M., Kugiyama K., Fukunaga K., et al. Protein kinase C inhibitors prevent impairment of endothelium-dependent relaxation by oxidatively modified LDL. Arterioscler Thromb Vasc Biol. 1993;13(10):1525-1532.
155. May J.M. How does ascorbic acid prevent endothelial dysfunction? Free Radic Biol Med. 2000;28(9):1421-1429.
156. Heller R., Unbehaun A., Schellenberg B., et al. L-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. J Biol Chem. 2001;276(1):40-47.
157. Naya M., Tsukamoto T., Morita K., et al. Olmesartan, but not amlodipine, improves endothelium-dependent coronary dilation in hypertensive patients. J Am Coll Cardiol. 2007;50(12):1144-1149.
158. Eckel R.H., Daniels S.R., Jacobs A.K., et al. America’s children: a critical time for prevention. Circulation. 2005;111(15):1866-1868.
159. Campisi R., Czernin J., Schoder H., et al. Effects of long-term smoking on myocardial blood flow, coronary vasomotion, and vasodilator capacity. Circulation. 1998;98(2):119-125.
160. Yokoyama I., Momomura S., Ohtake T., et al. Reduced myocardial flow reserve in non-insulin-dependent diabetes mellitus. J Am Coll Cardiol. 1997;30(6):1472-1477.
161. Pitkanen O.P., Nuutila P., Raitakari O.T., et al. Coronary flow reserve is reduced in young men with IDDM. Diabetes. 1998;47(2):248-254.
162. Di Carli M.F., Bianco-Batlles D., Landa M.E., et al. Effects of autonomic neuropathy on coronary blood flow in patients with diabetes mellitus. Circulation. 1999;100(8):813-819.
163. Di Carli M.F., Janisse J., Grunberger G., et al. Role of chronic hyperglycemia in the pathogenesis of coronary microvascular dysfunction in diabetes. J Am Coll Cardiol. 2003;41(8):1387-1393.
164. Srinivasan M., Herrero P., McGill J.B., et al. The effects of plasma insulin and glucose on myocardial blood flow in patients with type 1 diabetes mellitus. J Am Coll Cardiol. 2005;46(1):42-48.
165. Herrero P., Peterson L.R., McGill J.B., et al. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol. 2006;47(3):598-604.
166. Naoumova R.P., Kindler H., Leccisotti L., et al. Pioglitazone improves myocardial blood flow and glucose utilization in nondiabetic patients with combined hyperlipidemia: a randomized, double-blind, placebo-controlled study. J Am Coll Cardiol. 2007;50(21):2051-2058.
167. Al Suwaidi J., Higano S.T., Holmes D.R.Jr, et al. Obesity is independently associated with coronary endothelial dysfunction in patients with normal or mildly diseased coronary arteries. J Am Coll Cardiol. 2001;37(6):1523-1528.
168. Matsuzawa Y. Therapy Insight: adipocytokines in metabolic syndrome and related cardiovascular disease. Nat Clin Pract Cardiovasc Med. 2006;3(1):35-42.
169. Avogaro A., de Kreutzenberg S.V. Mechanisms of endothelial dysfunction in obesity. Clin Chim Acta. 2005;360(1–2):9-26.
170. Bouloumie A., Marumo T., Lafontan M., et al. Leptin induces oxidative stress in human endothelial cells. Faseb J. 1999;13(10):1231-1238.
171. Vecchione C., Maffei A., Colella S., et al. Leptin effect on endothelial nitric oxide is mediated through Akt-endothelial nitric oxide synthase phosphorylation pathway. Diabetes. 2002;51(1):168-173.
172. Lembo G., Vecchione C., Fratta L., et al. Leptin induces direct vasodilation through distinct endothelial mechanisms. Diabetes. 2000;49(2):293-297.
173. Matsuda K., Teragawa H., Fukuda Y., et al. Leptin causes nitric-oxide independent coronary artery vasodilation in humans. Hypertens Res. 2003;26(2):147-152.
174. Winters B., Mo Z., Brooks-Asplund E., et al. Reduction of obesity, as induced by leptin, reverses endothelial dysfunction in obese (Lep(ob)) mice. J Appl Physiol. 2000;89(6):2382-2390.
175. Wilson P.W., D’Agostino R.B., Levy D., et al. Prediction of coronary heart disease using risk factor categories. Circulation. 1998;97(18):1837-1847.
176. Yusuf S., Sleight P., Pogue J., et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342(3):145-153.
177. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994;344(8934):1383-1389.
178. Dormandy J.A., Charbonnel B., Eckland D.J., et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366(9493):1279-1289.
179. Verma S., Buchanan M.R., Anderson T.J. Endothelial function testing as a biomarker of vascular disease. Circulation. 2003;108(17):2054-2059.
180. Graner M., Varpula M., Kahri J., et al. Association of carotid intima-media thickness with angiographic severity and extent of coronary artery disease. Am J Cardiol. 2006;97(5):624-629.
181. Hoffmann U., Ferencik M., Cury R.C., et al. Coronary CT angiography. J Nucl Med. 2006;47(5):797-806.
182. Hoffmann U., Nagurney J.T., Moselewski F., et al. Coronary multidetector computed tomography in the assessment of patients with acute chest pain. Circulation. 2006;114(21):2251-2260.
183. Schepis T., Gaemperli O., Koepfli P., et al: Added value of coronary artery calcium score as an adjunct to gated SPECT for the evaluation of coronary artery disease in an intermediate-risk population, J Nucl Med 48(9):1424–1430.
184. Chan S.Y., Mancini G.B., Kuramoto L., et al. The prognostic importance of endothelial dysfunction and carotid atheroma burden in patients with coronary artery disease. J Am Coll Cardiol. 2003;42(6):1037-1043.
185. Husmann L., Valenta I., Gaemperli O., et al. Feasibility of low-dose coronary CT angiography: first experience with prospective ECG-gating. Eur Heart J. 2008;29(2):191-197.
186. Quyyumi A.A. Prognostic value of endothelial function. Am J Cardiol. 2003;91(12A):19H-24H.
187. Mancini G.B., Henry G.C., Macaya C., et al. Angiotensin-converting enzyme inhibition with quinapril improves endothelial vasomotor dysfunction in patients with coronary artery disease. The TREND (Trial on Reversing Endothelial Dysfunction) Study. Circulation. 1996;94(3):258-265.
188. Baller D., Notohamiprodjo G., Gleichmann U., et al. Improvement in coronary flow reserve determined by positron emission tomography after 6 months of cholesterol-lowering therapy in patients with early stages of coronary atherosclerosis. Circulation. 1999;99(22):2871-2875.
189. Schindler T.H., Facta A.D., Prior J.O., et al. Improvement in coronary vascular dysfunction produced with euglycaemic control in patients with type 2 diabetes. Heart. 2007;93(3):345-349.
190. Czernin J., Barnard R.J., Sun K.T., et al. Effect of short-term cardiovascular conditioning and low-fat diet on myocardial blood flow and flow reserve. Circulation. 1995;92(2):197-204.
191. Gordon T., Kannel W.B., Hjortland M.C., et al. Menopause and coronary heart disease. The Framingham Study. Ann Intern Med. 1978;89(2):157-161.
192. Rossi R., Nuzzo A., Origliani G., et al. Prognostic role of flow-mediated dilation and cardiac risk factors in post-menopausal women. J Am Coll Cardiol. 2008;51(10):997-1002.
193. Peterson L.R., Eyster D., Davila-Roman V.G., et al. Short-term oral estrogen replacement therapy does not augment endothelium-independent myocardial perfusion in postmenopausal women. Am Heart J. 2001;142(4):641-647.
194. Duvernoy C.S., Rattenhuber J., Seifert-Klauss V., et al. Myocardial blood flow and flow reserve in response to short-term cyclical hormone replacement therapy in postmenopausal women. J Gend Specif Med. 2001;4(3):21-27. 47
195. Schindler T.H., Campisi R., Dorsey D., et al. Effect of hormone replacement therapy on vasomotor function of the coronary microcirculation in postmenopausal women with medically treated cardiovascular risk factors. Eur Heart J. 2009;30(8):978-986.
196. Knuuti J., Kalliokoski R., Janatuinen T., et al. Effect of estradiol-drospirenone hormone treatment on myocardial perfusion reserve in postmenopausal women with angina pectoris. Am J Cardiol. 2007;99(12):1648-1652.
197. Collins R., Armitage J., Parish S., et al. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361(9374):2005-2016.
198. Collins R., Armitage J., Parish S., et al. Effects of cholesterol-lowering with simvastatin on stroke and other major vascular events in 20536 people with cerebrovascular disease or other high-risk conditions. Lancet. 2004;363(9411):757-767.
199. Janatuinen T., Laaksonen R., Vesalainen R., et al. Effect of lipid-lowering therapy with pravastatin on myocardial blood flow in young mildly hypercholesterolemic adults. J Cardiovasc Pharmacol. 2001;38(4):561-568.
200. Yokoyama I., Yonekura K., Inoue Y., et al. Long-term effect of simvastatin on the improvement of impaired myocardial flow reserve in patients with familial hypercholesterolemia without gender variance. J Nucl Cardiol. 2001;8(4):445-451.
201. Mellwig K.P., Baller D., Gleichmann U., et al. Improvement of coronary vasodilatation capacity through single LDL apheresis. Atherosclerosis. 1998;139(1):173-178.
202. Guethlin M., Kasel A.M., Coppenrath K., et al. Delayed response of myocardial flow reserve to lipid-lowering therapy with fluvastatin. Circulation. 1999;99(4):475-481.
203. Kaesemeyer W.H., Caldwell R.B., Huang J., et al. Pravastatin sodium activates endothelial nitric oxide synthase independent of its cholesterol-lowering actions. J Am Coll Cardiol. 1999;33(1):234-241.
204. Laufs U., La Fata V., Plutzky J., et al. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97(12):1129-1135.
205. Nickenig G., Baumer A.T., Temur Y., et al. Statin-sensitive dysregulated AT1 receptor function and density in hypercholesterolemic men. Circulation. 1999;100(21):2131-2134.
206. Huggins G.S., Pasternak R.C., Alpert N.M., et al. Effects of short-term treatment of hyperlipidemia on coronary vasodilator function and myocardial perfusion in regions having substantial impairment of baseline dilator reverse. Circulation. 1998;98(13):1291-1296.
207. Yokoyama I., Momomura S., Ohtake T., et al. Improvement of impaired myocardial vasodilatation due to diffuse coronary atherosclerosis in hypercholesterolemics after lipid-lowering therapy. Circulation. 1999;100(2):117-122.
208. McMahon G.T., Plutzky J., Daher E., et al. Effect of a peroxisome proliferator-activated receptor-gamma agonist on myocardial blood flow in type 2 diabetes. Diabetes Care. 2005;28(5):1145-1150.
209. Sourij H., Zweiker R., Wascher T.C. Effects of pioglitazone on endothelial function, insulin sensitivity, and glucose control in subjects with coronary artery disease and new-onset type 2 diabetes. Diabetes Care. 2006;29(5):1039-1045.
210. Tan K.C., Ai V.H., Chow W.S., et al. Influence of low density lipoprotein (LDL) subfraction profile and LDL oxidation on endothelium-dependent and independent vasodilation in patients with type 2 diabetes. J Clin Endocrinol Metab. 1999;84(9):3212-3216.
211. Sundell J., Janatuinen T., Ronnemaa T., et al. Diabetic background retinopathy is associated with impaired coronary vasoreactivity in people with Type 1 diabetes. Diabetologia. 2004;47(4):725-731.
212. Erdmann E., Dormandy J.A., Charbonnel B., et al. The effect of pioglitazone on recurrent myocardial infarction in 2,445 patients with type 2 diabetes and previous myocardial infarction: results from the PROactive (PROactive 05) Study. J Am Coll Cardiol. 2007;49(17):1772-1780.
213. Schelbert H.R. Positron emission tomography of the heart: Methodology, findings in the normal and the diseased heart, and clinical applications. In: Phelps M., editor. PET: Molecular imaging and its biological applications. New York: Springer-Verlag; 2005:389-508.