Chapter 54 Myelomeningocele and Other Spinal Dysraphisms
Myelomeningocele (MMC) is the most common congenital anomaly of the central nervous system. It exists in a spectrum of neural tube defects (NTDs), ranging from cranioschisis, or complete failure of neurulation, at one end to spina bifida occulta, with minimal or no neurologic involvement, on the other end.92 Although the most severe NTDs result in stillbirth or death shortly after birth, the majority of patients with a spinal dysraphism survive. MMC, which composes 90% of open spinal dysraphic states, is the most complex congenital anomaly compatible with life and is the second most common disabling condition in childhood after cerebral palsy.20,119,169 The varying degrees of organ involvement seen with these conditions have major implications for long-term health and physical function, as well as the psychologic and social well-being of the affected individual. Survival and quality of life for those with MMC have improved because of advances in medical, surgical, and rehabilitative care during the past 50 years. Many challenges remain, however, regarding advancing prevention, understanding etiology, maximizing health-related outcomes, transitioning to adult-based health care systems, and improving activity and participation at the societal level. These challenges are best met by a team of specialists working with patients, their families, and the community.
Terminology and Historical Background
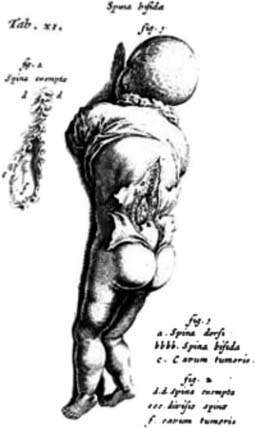
The first clear description of spinal dysraphism was by Casper Bauhin, a Swiss physician, anatomist, and botanist, in his publication Theatrum Anatomicum in 1592. The term spina bifida, however, is often historically associated with Nicholas Tulp, a Dutch physician who published a sketch (Figure 54-1) and description of several patients with the condition in 1641. In 1875, Rudolf Virchow described spina bifida occulta, which refers to a hidden bony defect, as well as other potential hidden anomalies.165 Spina bifida has been further delineated with the term spina bifida aperta, a midline defect that communicates with the external environment and includes MMC and meningocele. The term spina bifida cystica is also sometimes used, and simply refers to a sac filled with cerebrospinal fluid protruding from the spinal column but can also refer to MMC and meningocele. MMC at the level of the spine refers to protrusion of the meninges through a defect in the posterior elements of the spine, with involvement of the spinal cord or nerve roots. Meningocele refers only to protrusion of the meninges and cerebrospinal fluid through a defect in the posterior elements of the spine into the tissue beneath the skin, without involvement of functional neural elements. These distinctions are important and will be further explained below.
Because this terminology is often confusing and minimally descriptive, an alternative system based on advances in clinical recognition and imaging of spinal lesions now allows for more useful classification, especially as it relates to prognosis. This point is exemplified by studies reporting a 17% incidence of bony spina bifida occulta in the general population and in 30% of normal individuals aged 1 to 10 years with no neurologic involvement or anatomic abnormality other than incomplete closure of the posterior elements of the spine.16,91 In contrast, occult spinal dysraphism, including diastematomyelia, lipomyelomeningocele, and tight filum terminale, among others, can present with neurologic compromise or orthopedic deformity, with no further outward anatomic abnormalities other than possible cutaneous anomalies. Modern imaging studies are able to reveal these underlying defects and provide a system of classification based on neuroradiologic and clinical findings.178
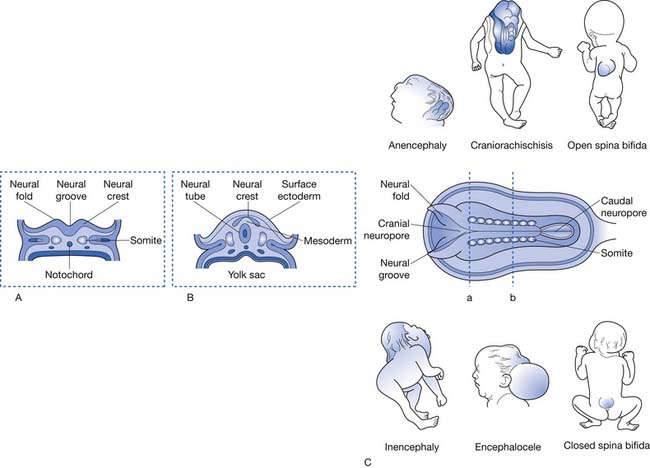
This system of classification simply divides the various forms of spinal dysraphism into open spinal dysraphisms (i.e., open spina bifida) and closed spinal dysraphisms (i.e., closed spina bifida) (Figure 54-2). It is useful from a prognostication standpoint because it is based on the observation that those with open defects generally have lesions with visible neural elements, often leaking cerebrospinal fluid, which are associated with malformations that involve the entire central nervous system, including Chiari II malformations, midline defects, and hydrocephalus. In contrast, closed spinal dysraphisms (meningocele and occult spinal dysraphism) are lesions that are fully epithelialized with no neural tissue exposed, and generally have the malformation limited to the spine and spinal cord, with only rare involvement of the brain.3 Box 54-1 displays a complete listing of spinal dysraphic states.178 Although most of the conditions listed in Box 54-1 can result in a patient having rehabilitation needs, the most severely affected and vast majority of affected individuals will have a history of MMC. For this reason, the remainder of this chapter refers to rehabilitation concepts as they apply to MMC, although they can be applied to the various other spinal dysraphisms as appropriate.
BOX 54-1 Cliniconeuroradiologic Classification of Spinal Dysraphism
From Tortori-Donati P, Rossi A, Cama A: Spinal dysraphism: a review of neuroradiological features with embryological correlations and proposal for a new classification, Neuroradiology 42(7):471–491, 2000, with permission.
Epidemiology
Incidence and Prevalence
Epidemiologic studies of spinal dysraphisms typically include both MMC and meningocele, which are collectively referred to as spina bifida.18 One clear observation from these studies is that the prevalence of MMC has decreased worldwide during the past century.3 In the United States, rates have varied from as high as 2.31/1000 births in Boston during the 1930s to as low as 0.51/1000 births in Atlanta in the early 1990s.127,144 The most recent U.S. data indicate a 6.9% decrease in prevalence, from 2.04/10,000 live births from 1999 to 2000, to 1.90/10,000 live births from 2003 to 2005.34 Rates in other parts of the world have been as high as 3.4/1000 live births in Dublin, Ireland in 1953, although these rates have generally decreased in a fashion similar to that in the United States.38
The reasons for the decreasing prevalence of MMC are multifactorial and not completely understood. A portion of the decrease can be attributed to the advent of prenatal screening and elective termination of pregnancy.42,114 This is only partially responsible, however, and other factors are known to play a role. The most influential factor has been the increased consumption of folic acid among women of childbearing age. Folate supplementation was first shown to decrease rates of NTDs in studies performed in Wales in the early 1980s.93 Multiple observational and controlled trials followed, leading up to the Medical Research Council study, which involved seven countries and 3012 women. The study was ended early after supplementation with folic acid (4 mg/day) was shown to prevent 72% of NTDs in women with a previously affected pregnancy.115 A subsequent study proved the efficacy of folic acid in preventing the first occurrence of an NTD.43 Both recommendations by the U.S. Public Health Service beginning in 1992 for daily folic acid intake for women of childbearing age and mandatory supplementation of all enriched cereal grain products in the United States since 1998, however, have not resulted in the 48% reduction in NTDs predicted by these studies.44 A decrease of approximately 26% has been observed, illustrating the need to further educate all women of childbearing age on the importance of daily folic acid intake of 0.4 mg for those who could become pregnant and 4 mg for those with a previously affected pregnancy or a history of MMC themselves. These statistics also place emphasis on the importance of health care provider participation in this effort.35 A recent study showed that women aged 18 to 24, who account for almost one third of U.S. births, had the least awareness regarding the need for folic acid consumption, appropriate timing of consumption, and the lowest daily use of supplements.188 Also of note, folic acid supplementation was ineffective in decreasing the overall incidence of lipomyelomeningocele between 1995 and 2001 in Nova Scotia in one study, providing evidence that the embryogenesis of various spinal dysraphisms could be fundamentally different.114 Subsequent studies have shown a more substantial reduction in cranial, cervical, and thoracic defects related to folic acid consumption compared with lumbar and sacral defects, further pointing to the etiologic heterogeneity of NTDs.46 A recent U.S. study conducted after the implementation of fortification of the food supply found little evidence of a link between maternal folic acid intake and NTDs. The authors theorize that folic acid fortification has reduced the folic acid–sensitive NTDs, with those that remain resulting from additional risk factors or underlying mechanisms.121
Geographic variation in birth prevalence of NTDs occurs both between and within countries.86 Across Europe, rates have been noted to be higher in Germany and Hungary than in Scandinavian countries. In the United States, there has been an observed trend of decreasing rates of NTD from the east coast to the west coast.64 Ethnicity has been shown to play a role in MMC as well, with those of Celtic descent (Irish, Welsh, and Scottish) having higher rates than those of Anglo-Saxon or Norman origin. These findings have been correlated with the high rates of MMC in Boston in the 1930s, which were highest among those who had mothers of Irish descent.30,128 In addition, those of Hispanic, Chinese, and Sikh ethnicity are at a greater risk for MMC.34 (1) Rates among black and Asian populations have been observed to be low, with similar rates among populations living in different areas.86 Female gender has also consistently been shown to be a risk factor for MMC, with a female preponderance observed in both still and live births.84,86 Additionally, the prevalence of spina bifida in Texas along the Mexico border was recently reported to be 3.52/10,000 live births, well above the most recent Centers for Disease Control and Prevention data for the entire country.28
Embryology and Etiology
Embryology
NTDs are known to occur as a result of failure of neurulation between the seventeenth and thirtieth days of gestation.74 Primary neurulation refers to the development of the neural tube, which forms the brain and spinal cord. Secondary neurulation refers to formation of the remainder of the neural tube from a cell mass caudal to the posterior neuropore, which forms the lower sacral and coccygeal segments. The caudal neuropore closes around the twenty-sixth day of gestation, and, as a result, teratogenic events that take place after this closure cannot cause thoracic or lumbosacral MMC.94,161 Failure of primary neurulation can lead to an open NTD, consisting not only of a spinal anomaly but also other defects, including a Chiari II malformation and hydrocephalus, possibly resulting from cerebrospinal fluid loss during early development.82,112 Most posterior lumbar and sacral meningoceles are thought to occur during secondary neurulation, with those higher on the spinal axis resulting from defects in primary neurulation that do not cause an open NTD.3
Etiology
Although the mechanisms are not well understood, up to 80% of all NTDs are thought to be due to multifactorial influences (i.e., genetic and environmental factors). Certain other NTDs are associated with NTD syndromes, single-gene disorders, and chromosomal disorders and are relatively well defined.5 Recurrence rates of NTDs are influenced by family history, geography, and severity of anomaly. These rates have been reported to vary from 2.4% to 5% after the birth of one affected child, with the risk doubling after two affected children.40 A Hungarian study reported that recurrence risk among those with spina bifida was greatest for MMC associated with hydrocephalus, at 4.79%.137 In addition, studies in the early 1990s showed that at least 70% of NTDs are “folic acid–sensitive” or “folic acid–dependent,” with the remaining 30% being “folic acid–resistant.”6 Recent studies from Ireland have confirmed that both homozygosity and heterozygosity for the T allele of the C677T polymorphism of the gene encoding the folate-dependent enzyme 5,10-MTHFR are risk factors for NTDs. This polymorphism is associated with lower tissue folate concentrations, higher homocysteine concentrations, and lower enzyme activity than in the wild-type genotype. This single genetic variant could account for up to half of the folate-related NTDs in Ireland.88 These combined at-risk phenotypes are present in approximately 59% of the European population and 53% of the North American population.19 Recent studies have also investigated links between genetic dysregulation of platelet-derived growth factor, myoinositol, and nitric oxide synthase, among others, as contributors to the formation of NTDs.25,66,207 Two large trials are currently ongoing to further elucidate the genetics of spina bifida and other NTDs.118
A number of environmental factors have been implicated in the development of NTDs, including low socioeconomic status130,196; maternal diabetes mellitus190; maternal hyperthermia153; folate deficiency117; hyperzincemia113; maternal obesity195; and certain drug exposures, including to carbamazepine,146 valproic acid,135 diuretics, antihistamines, and sulfonamides.125 Of the medications mentioned, carbamazepine and valproic acid have the strongest correlation with spina bifida, with an estimated risk of 1% and 1% to 2%, respectively.135,146 Seasonal variation, with peaks in midspring conception for spina bifida, has been reported in some regions but has not been confirmed in others.33,60 The factors for which there is more convincing evidence of an association to NTDs, however, include maternal obesity, maternal hyperthermia, and lack of maternal periconceptional folic acid supplementation.118 As well, a recent Canadian study suggests that vitamin B12 supplementation in combination might be more effective in preventing NTDs than folic acid alone.177
Prenatal Counseling (Diagnosis and Management)
Prenatal screening now allows for diagnosis of the majority of cases of MMC before birth. Initial screening of women who are at high risk, including those with a positive family history, a previous child with spina bifida, or exposure to teratogenic agents, should include measurement of serum α-fetoprotein and acetylcholinesterase levels at 16 to 18 weeks postconception. Based on these results, a patient-specific risk can be calculated and repeat testing of serum levels performed. Subsequently, high-resolution ultrasound can be performed, which is sensitive in 95% of cases in which good images are retrieved. If the images are of poor quality because of maternal obesity or other factors, amniocentesis can be performed to obtain amniotic fluid α-fetoprotein and acetylcholinesterase levels. These tests can be repeated based on previous equivocal results; however, after diagnosis, genetic counseling along with a discussion of management options should occur. A recent study showed the diversity of physician views regarding prenatal screening, selective termination, and disability, suggesting a need to better understand how these differences affect reproductive technology, health care policy, and medical practice.199
Prenatal detection of MMC is important to educate the patient and family regarding the diagnosis and management options. Early detection also allows time to prepare for a safe delivery in a medical center that offers neurologic closure. Functional motor outcome can also be predicted by high-resolution ultrasound before delivery.39 Previous consideration has been given to performing cesarean section for all mothers with an affected pregnancy to avoid further damage to neural structures. One study reported a less severe lower extremity paralysis in infants born by cesarean section before the onset of labor but showed no difference in those who received cesarean section after the onset of labor.102 Subsequent studies, however, have found no difference in motor outcome with cesarean section.74,97
Intrauterine Surgical Procedures
It is estimated that more than 400 fetuses have received in utero closure of MMC by open maternal–fetal surgery worldwide.173 Studies involving these patients have suggested that the incidence of shunt-dependent hydrocephalus is significantly reduced, that the brain stem and cerebellum are restored to a more normal anatomic configuration, and leg function might be improved in some patients receiving intrauterine MMC repair.45,173,174,186 Criticism exists, however, regarding methods of data collection and study design, among other issues.203 A multicenter, randomized, open-label trial is currently underway to further evaluate this procedure and should be completed in 2010. As well, a percutaneous fetoscopic approach to closure has been reported in a few cases with the purported benefit of reduced maternal risk compared with open maternal–fetal surgery.90
Neonatal Management
Back Defect
After an infant is delivered with an open NTD, a sequence of events is set in place to preserve neurologic function, prevent infection, and stabilize cerebrospinal fluid flow. Early closure (within 72 hours of delivery) reduces the risk of infection in the central nervous system.37 Before closure, the open defect must be protected to prevent contamination or further damage from trauma. Closure occurs in three stages:
After closure of the open spinal defect, hydrocephalus often develops.3 Some centers advocate simultaneous back closure and insertion of a ventriculoperitoneal (VP) shunt (Figure 54-3).103,117
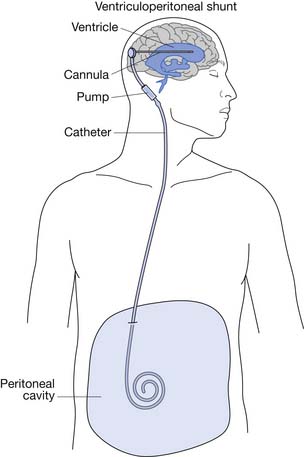
Hydrocephalus
Most infants with MMC require VP shunting.106 Approximately 15% are born with severe hydrocephalus and require immediate shunting.111 The 85% who do not require immediate shunting should be watched closely after their back closure for signs of increased intracranial pressure. The leaking cerebrospinal fluid serves as a decompression before closure, and once the defect is closed, cerebrospinal fluid can accumulate in the ventricular system. The white matter of a neonate is relatively compliant, and therefore the ventricles can become enlarged before the head circumference changes.13 The presence of hydrocephalus correlates well with the level of the spinal defect, with thoracic lesions having a higher incidence than lumbar or sacral lesions.143
Early Bladder Management
More than 90% of infants with MMC will have a neurogenic bladder. Management decisions made in infancy can affect renal health and the eventual development of urinary continence.166 The importance of aggressive urinary management should be stressed to the family before the child leaves the hospital. Early goals include avoiding infections, preventing upper tract damage, and identifying anatomic abnormalities in the genitourinary system.166 Baseline investigations should include renal–bladder sonography and a voiding cystourethrogram. Hydronephrosis is found in 7% to 30% of infants, and reflux occurs in approximately 20% of infants.77,205 Infants with hydronephrosis or reflux should be started on prophylactic antibiotics. Infants who are unable to void begin intermittent catheterization programs. If the infant is able to void, he or she should be checked for complete emptying by checking a postvoid residual volume, either by catheterization or bladder scan. Incomplete emptying can lead to urinary tract infections because the retained urine serves as a culture medium.
Assessment of the Neurologic Level
Even if not spoken out loud, the first question parents of a newborn with MMC often ask is, “Will my child be able to walk?” A careful neurologic examination can give them an idea even within the first few days of life. The best predictor of motor function is the actual motor examination. Information regarding the best motor examination can be obtained by observation, palpation, and postural changes. Motor examinations can improve after the initial examination, which might be related to a period of spinal shock associated with the delivery or the closure.56,160 This can make a newborn’s motor function appear worse than it might eventually be.
Therapy
The goal of any MMC team should be to develop and implement a comprehensive plan that enables the child to attain a maximal level of function in all areas.119 Therapists play a key role in this endeavor and often develop a very good working relationship with families of children with MMC. Therapists are invaluable in providing education and anticipatory guidance for the family. For children born with contractures at the hips, knees, ankles, or feet, a program of passive range of motion can be taught to the family even before patient discharge. Splints are often fabricated soon after birth by the therapists. Throughout life, the MMC clinic therapist will help to educate and coordinate with community therapists regarding the plan developed by the MMC team.
Childhood Management
Shunts
Almost all children with MMC require placement of a VP shunt for management of hydrocephalus. The two most common shunt complications are infection and obstruction.185 Presenting signs and symptoms of shunt malfunction vary with the age of the child. Mechanical obstructions tend to present more acutely with signs and symptoms related to increased intracranial pressure, and infections tend to present more insidiously (Box 54-2).
Infections have a greater long-term morbidity than malfunctions. The overall risk of shunt infection is 12% per child. Staphylococcus epidermidis is the organism that causes most shunt infections.55 Symptoms do not usually develop until several weeks after the shunt is placed or revised. Epidemiologic factors seem to influence the incidence of shunt infections more than surgical factors. Aside from skin contamination during shunt placement, shunts can also become infected with gram-negative rods if the distal end of the shunt erodes into an intraabdominal organ. Gram-negative infections have a much poorer prognosis.57
In the first year of life half of all children with a VP shunt develop obstruction requiring revision. Of those children who require a revision in the first year, 31% will require a second revision in the second year, and then have a risk recurrence rate of 12% per year thereafter.170 Endoscopic third ventriculostomies are performed in carefully selected patients and can become an alternative to chronic VP shunts.23
Arnold–Chiari II Malformations
The Chiari II malformation is characterized by variable displacement of cerebellar tissue into the spinal canal, accompanied by caudal dislocation of the lower brain stem and fourth ventricle (Figure 54-4). Although these posterior fossa abnormalities are the most often described, the Chiari II malformation is also associated with a wide range of abnormalities throughout the neuraxis.3
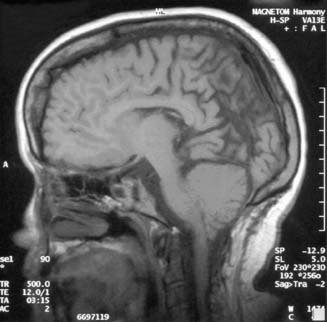
Although the operative mortality for closure of the spinal defect in children with MMC is very low, the operative mortality for symptomatic Arnold–Chiari malformations is relatively high (34% to 38%).3,150 A symptomatic Chiari II malformation remains the leading cause of death for infants with MMC.171 Signs and symptoms of symptomatic Chiari II malformations include intermittent obstructive or central apnea, cyanosis, bradycardia, dysphagia, nystagmus, stridor, vocal cord paralysis, torticollis, opisthotonos, hypotonia, upper extremity weakness, and spasticity. The constellation of stridor, central apnea, and aspiration is sometimes referred to as central ventilatory dysfunction.71
Before hindbrain decompression for a symptomatic Chiari II malformation, the child’s shunt system should be evaluated carefully because shunt malfunctions can cause Chiari malformations to become symptomatic. Hindbrain decompressions should be performed early to minimize the progression of symptoms of the Chiari malformation. Poor preoperative prognostic signs include bilateral vocal cord paralysis, severe neurogenic dysphagia, and prolonged apnea.36
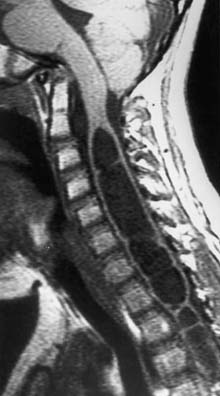
Hydromyelia
Hydromyelia is the dilatation of the central canal of the spinal cord (Figure 54-5). It is analogous to dilatation of the ventricles in the brain and is a relatively common occurrence in children with MMC. Hydromyelia is probably much more common than we are aware because it often does not cause obvious symptoms in patients with MMC.27 When symptomatic, it usually presents with rapidly progressive scoliosis, a change in strength or coordination of the upper or lower extremities, and spasticity. Magnetic resonance imaging is the best study to demonstrate this spinal cord abnormality.26 When suspected, the entire neuraxis should be imaged because untreated or subclinical hydrocephalus can produce hydromyelia. Hydrocephalus should be treated before surgical treatment for hydromyelia.
Tethered Cord Syndrome
In children with MMC, the spinal cord can be fixed or “tethered” at one point, causing traction, which can lead to progressive urologic, orthopedic, or neurologic decline. It was first described in 1857, and the first known detethering of the spinal cord was performed on a previously healthy 17-year-old subject who had progressive loss of lower extremity function in 1891.111
The spinal cord usually terminates at the level of L1–L2. However, MMC repair invariably is followed by the development of arachnoiditis, fibrosis, and adhesions between the intraspinal neural structures, the meninges, and the surrounding vertebral structures.206 These adhesions can tether the cord to the low lumbar or sacral region.
Most children with MMC will show signs of tethering on magnetic resonance imaging. Therefore symptoms should develop before surgical correction is pursued. Typical signs and symptoms in children include increased weakness (54%), worsening gait (54%), scoliosis (51%), pain (32%), orthopedic deformity (11%), and urologic dysfunction (6%).78 Surgical correction should be considered early because most cases will improve or stabilize if treated early.78 Delayed correction can result in irreversible loss of function because the natural history is for symptoms to worsen with time.138
At least three other lesions can lead to tethering of the spinal cord: diastematomyelia, lipomyelomeningocele, and tight filum terminale. Diastematomyelia refers to divisions (not duplications) of the spinal cord. It is usually associated with a bony spur. Even if asymptomatic, the natural history is for symptoms to develop that can be irreversible.5,69 Lipomyelomeningocele refers to a subcutaneous lipoma, continuous with the cauda equina, which also has a meningocele with neural elements enclosed extending outside the dura. A tight filum terminale is another congenital malformation in which the filum terminale does not elongate. Prophylactic surgery is usually recommended for these three lesions.
Neurogenic Bladder
Urologic involvement in MMC and other spinal dysraphisms varies, and is not necessarily correlated with the level of the lesion as in traumatic spinal cord injury. In MMC, more than 90% of patients have partial or complete denervation of the bladder, with poor compliance and contractibility resulting in unacceptable residual urine volumes.166 The urethral sphincter is incompetent in 86% of patients, so that incontinence occurs with increases in intravesical pressure. About one third of patients have detrusor–sphincter dyssynergia, resulting in high intraluminal pressures.109 The external sphincter is usually partially functional and can improve in the first year after birth.168 Patients should be observed at least annually because deterioration or improvement can occur in the first year of life, and tethering of the spinal cord with a change in bladder function can occur over the years. Although the majority of individuals with MMC have normal renal function at birth, 40% to 90% will experience a decline by age 10 if left unattended.164
Prevention of damage to the urinary tract and continence are the primary goals of neurogenic bladder management. Urodynamic evaluation is generally performed in infants with MMC, with 75% showing a normal upper urinary tract. The remaining infants show some degree of hydronephrosis resulting from vesicoureteral reflux, detrusor–sphincter dyssynergia, an enlarged bladder, or other structural abnormality. Infants with normal anatomy should receive a renal ultrasound biannually. Those with incomplete emptying and no outlet resistance can be taught the Credé maneuver. Those with detrusor–sphincter dyssynergia, or who have already developed hydronephrosis, should be treated with anticholinergic medications and clean intermittent catheterization to prevent the development or worsening of hydronephrosis. Children with vesicoureteral reflux, which develops when detrusor pressure exceeds 40 mm Hg, are often prescribed prophylactic antibiotics. If they have persistent febrile urinary tract infection or persistent hydronephrosis, surgical intervention is often necessary. Cutaneous vesicostomy can be performed, with reversal done at a later time when the patient is capable of effective clean intermittent catheterization.110,158
As fewer than 10% of children with MMC have normal urinary control, continence of urine is a prominent issue.96 Although there are no effective external collection devices for girls, excluding diapers, condom catheters are an option for boys with reflex emptying who do not have vesicoureteral reflux or large residual volumes. Appropriate sizing can be a difficult issue for some, and impaired sensation can lead to skin breakdown.
The high prevalence of small bladder capacity and low outlet resistance in many children results in only about one fourth of children being continent with clean intermittent catheterization alone.187 The addition of anticholinergic medications, α-adrenergic agonists, and antibiotic instillations still only resulted in complete continence in 49% of patients in one study.198 In general, frequent catheterization less than every 4 hours is required to achieve continence.96
Multiple surgical options are available for those who do not achieve continence with clean intermittent catheterization and medications. Although a full description of these techniques is beyond the scope of this chapter, bladder augmentation along with artificial sphincter placement can be used individually or in combination. Success for long-term continence after artificial sphincter placement is more than 60%.17 In addition, for patients who have difficulty performing urethral clean intermittent catheterization, continent diversion, with the appendix used as a conduit to the bladder to create an abdominal stoma, can create easier access for many patients.158
Independence with toileting in children with MMC is delayed more than all other self-care tasks, regardless of intelligence. Although most children achieve independent control of bowel and bladder function by the age of 4, those with MMC might not achieve this until age 10 to 15.134 The cause of this is multifactorial and includes level of paralysis, intelligence, difficulty with visuospatial tasks, kyphoscoliosis, parental support, sensation, sphincter control, and bladder perception. Children can be taught to perform clean intermittent catheterization as early as age 5, although they will still need assistance with maintaining a schedule. Parents need to be trained not only in their child’s bladder program but also instructed in the importance of allowing the child to accept responsibility once she or he is able. A recent study of young adults revealed that up to 60% reported urinary incontinence, with approximately 70% perceiving this as a problem.167,195
Latex Allergy
Although the incidence of latex allergy in the general population is estimated to be less than 1% to 2%, the prevalence among children with MMC ranges from 20% to 65%.176 This IgE-mediated response to natural rubber latex is related to repeated mucosal exposure during surgical, therapeutic, and diagnostic procedures as well as atopic predisposition. The allergic response can range from dermatitis, allergic rhinitis, asthma, and angioedema to anaphylaxis.163,173 Patients with spina bifida have been determined to have a 500-fold greater risk for anaphylaxis in the operating room compared with control groups.176 Because latex sensitization takes place over time, a previous lack of sensitivity or negative allergy test does not preclude the possibility of a life-threatening reaction. Parents should be educated regarding the presence or risk of latex allergy. It is generally recommended that all patients with MMC use nonlatex catheters and avoid all other latex-containing products, whether medical or nonmedical.
Neurogenic Bowel
Patterns of neurogenic bowel involvement in children with MMC vary from normal bowel control in about 20% to incontinence caused by impaired rectal sensation, impaired sphincter function, and altered colonic motility in the remainder.96 Those without control of the external anal sphincter become incontinent when the pressure inside the rectum is sufficient to produce reflex relaxation of the internal anal sphincter. In those with lesions above L3, the internal anal sphincter has low tone, which further contributes to incontinence. As well, lesions above this level generally result in absent sensation, although sensation can be present but impaired in lower-level lesions.1 Presence of the bulbocavernous or anocutaneous reflex has been associated with a greater likelihood of achieving bowel continence.87
The goal of a bowel management program is to achieve efficient, regular, and predictable emptying before the rectum becomes full enough to stimulate reflex relaxation of the internal anal sphincter (in those in whom innervation is present). This can be achieved through the use of stool softeners, bulking agents, suppositories, digital stimulation, manual removal, or enemas. However, many patients and families prefer dietary manipulation. Clinicians often recommend performing bowel programs after a meal to take advantage of the gastrocolic reflex, although it is not clear that this is intact in patients with MMC.50
In patients who cannot become continent, surgical options are available. Although a full description of these techniques is beyond the scope of this chapter, options include the antegrade continence enema (ACE) procedure72 and colostomy. In an ACE procedure the appendix is used as a conduit to the bowel, through which a catheter can then be inserted into the cecum and saline or tap water infused, with bowel emptying achieved within 15 to 45 minutes. Colostomy is another option if standard treatment and antegrade continence enema have failed.
The importance of achieving bowel continence at an early age is important to provide a smooth transition into preschool and kindergarten, where children can be severely criticized by peers. Encouraging children to assume increasing responsibility for their bowel program is again emphasized. Unfortunately, one study showed that as many as 86% of teenagers aged 13 to 18 with MMC needed assistance from a caregiver for their bowel program.96 One study of young adults revealed that approximately 34% reported fecal incontinence regardless of their means of management, and 77% perceived this as a problem.194 The reasons for this are, again, believed to be multifactorial.
Endocrine Disorders
Individuals with MMC are known to have disturbed growth and development. Although spinal cord lesions, vertebral anomalies, and other skeletal deformities reduce growth of the lower limbs and spine, complex central nervous system abnormalities and hydrocephalus put the patient with MMC at risk for hypothalamic–pituitary dysfunction, including central precocious puberty and growth hormone deficiency.∗ Precocious puberty has been reported in 12% to 16% of patients with MMC.54,55 The mechanism of this is thought to be related to increased pressure on the hypothalamus, resulting in premature activation of the hypothalamic–pituitary–gonadal axis. Treatment with gonadotropin-releasing hormone analogs has proven beneficial in several studies, halting the progression of puberty, stopping menses, and decreasing hormone levels.182 It is more common for girls with MMC to experience precocious puberty, and it is unclear whether patients without hydrocephalus are at risk. Currently, only limited data show that treatment of growth hormone deficiency with growth hormone provides significant improvement in growth velocity, height, muscle strength, and mobility, as well as a reduction in obesity.147,180 Ongoing debate continues as to whether to treat patients with MMC with growth hormone. Clarification of the goals of treatment is required because efficacy with regard to functional improvement can be influenced by the level of the lesion and the presence of complicating factors such as syringomyelia, tethered cord syndrome, scoliosis, vertebral anomalies, contracture, and advanced pubertal development. Further studies are required to assess the ultimate role of growth hormone treatment in individuals with MMC.184
Musculoskeletal Considerations
Motor Innervation
It is important for practitioners and parents to appreciate that the level of motor function does not necessarily correspond to the anatomic vertebral level on radiographic studies.143 Spinal defects are generally described as cervical-, upper thoracic-, midthoracic-, low thoracic-, upper lumbar-, midlumbar-, low lumbar-, lumbosacral-, or sacral-level lesions. Many writers comment that cervical and upper thoracic dysraphisms might be an entirely different entity because these children tend to have a better prognosis and less neurologic deficit than children with lower thoracic and lumbar lesions.151 The majority of children with MMC have lumbosacral-level vertebral lesions. Very few have cervical or upper thoracic vertebral lesions. Approximately one fourth of patients have midlumbar-level lesions, and one fifth present with sacral-level involvement.
It is important to recognize that these children do not present simply with flaccid paraplegia below their anatomic lesion. Only one third present with flaccid paralysis. Most have a combination of upper and lower motor neuron signs. Many are asymmetric on motor and sensory testing. Some have voluntary motor control below other segments of paralysis and sensory loss.108
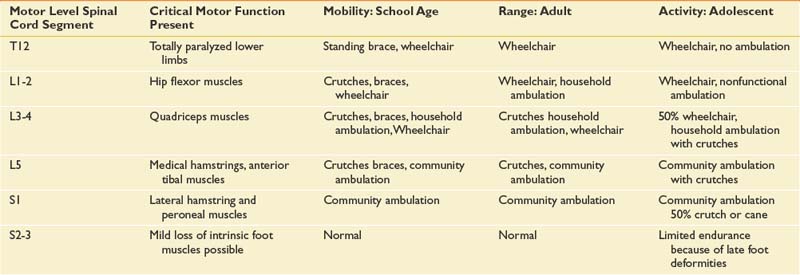
The level of neurologic impairment influences medical providers’ expectations for functional outcome, as well as musculoskeletal deformities and complications to anticipate (Table 54-1).
Hips
Most patients with MMC will have some hip deformity that interferes with ambulation, seating, or bracing. The proper management of hip deformity remains a topic of debate among orthopedic surgeons.72 Muscle imbalance at the hip accounts for most of the hip flexion deformity, but sitting at 90 degrees and hip flexor spasticity can also contribute to hip flexion deformities. Patients with thoracic-level lesions develop hip flexion deformities related to positioning or hip flexor spasticity. Patients with upper- and midlumbar-level lesions develop hip flexion deformities related to unopposed hip flexors and adductors. Hip flexion contractures create anterior pelvic tilt, which increases lumbar lordosis and can interfere with ambulation. Hip flexor lengthening procedures can preserve ambulation in the ambulatory MMC patient but should not be considered in the nonambulatory patient unless skin integrity, pain, or seating is compromised.
Hips that slowly migrate laterally (subluxation) over time are usually related to muscle imbalance around the hip. In a natural history study, Broughton et al.24 found that hip dislocation had occurred by the age of 11 years in 28% of thoracic-level lesion patients, in 30% of upper lumbar-level lesion patients, in 36% of patients with L4 function, in 7% of patients with L5 function, and in 1% of patients with sacral function. In the ambulatory child, aggressive treatment including femoral and acetabular osteotomy should be considered. A recent study by Gabrieli et al.,63 however, found that gait characteristics are not significantly different with regard to hip subluxation, and therefore concluded that surgery for the dislocated hip in the ambulatory child is not indicated. Little debate exists that, in the nonambulatory child, a dislocated hip probably does not cause much morbidity and therefore does not require surgical treatment. Debate continues regarding unilateral hip dislocation leading to pelvic obliquity that eventually contributes to scoliosis.
Knees
Children with MMC can develop knee flexion or extension contractures.55 Knee flexion contractures are generally seen in patients with thoracic-level MMC but can also be seen in patients with lumbar and sacral lesions.204 Knee flexion contractures can interfere with upright mobility, transfers, and, if severe, hygiene in the popliteal space. Knee flexion contractures of 20 degrees or less are generally well tolerated in the ambulatory patient. Nonambulatory patients might tolerate even more severe contractures without loss of function. The surgical treatment for knee flexion contractures is a radical knee flexor release. This procedure is well tolerated, with a low rate of recurrence in the ambulatory child.107 Knee extension contractures are much less common. They can be related to unopposed knee extensors, congenital dislocation of the knee, immobilization for fractures, or after treatment for flexion contractures. Knee extension contractures can interfere with seating and sit-to-stand transfers in the nonambulatory patient but generally do not impede ambulation in the ambulatory child.72 In the nonambulatory child, adequate flexion can usually be obtained by transection of the patellar ligament.154
Feet
Foot deformities are present in almost all children with MMC.132 Foot deformities can be present at birth, related to lack of intrauterine movement or muscle imbalance across the ankle. Foot deformities also develop over time from imbalanced muscle pull around the ankle and weight-bearing forces. Lack of sensation and autonomic instability can lead to secondary skin problems and poor wound healing.
The common foot deformities in children with MMC are equinus, equinovarus, calcaneus, cavus, and rocker-bottom deformity. The goal in treating these children is to achieve a plantigrade foot with stable skin. Splinting and passive manipulation can be helpful in infancy, but many of these children will require corrective surgical procedures.
Calcaneus deformities can be seen at birth or develop postnatally as a response to unopposed dorsiflexion in patients with midlumbar-level paralysis. Calcaneus deformity makes bracing difficult and often ineffective. This deformity predisposes the patient to pressure sores over the heel, which can lead to osteomyelitis. Patients with progressive deformities or a propensity toward pressure ulceration should be treated aggressively because delay in treatment can result in a greatly increased risk of pressure ulceration.61 Solid ankle–foot orthoses are the most appropriate orthosis for this deformity.
Spine
Deformities of the spine in children with MMC can be congenital, paralytic, or a combination of the two. Congenital spinal anomalies include scoliosis secondary to vertebral malformations, congenital kyphosis related to posterior element dysplasia, and intrathecal anomalies such as diastematomyelia.72 Predictors of the development of scoliosis include ambulatory status, clinical motor level, and last intact laminar arch.179 Spinal deformities are more likely to occur in those with thoracic lesions (>90% of patients with thoracic lesions develop scoliosis), but they can also be seen in those with sacral lesions, although they are relatively rare.123,179 Kyphotic deformities can cause severe seating and skin problems. Surgical treatment of kyphotic deformities carries significant risk of complications but usually has a good outcome.131 Kyphotic deformities seen at birth are related to congenital malformations. Lordosis is usually related to hip flexion contractures. Orthotic management of scoliosis does not provide a complete or permanent correction but might delay the need for surgical correction until the child is closer to skeletal maturity. Rapid progression of otherwise stable curves can be seen in patients with hydromyelia or tethered cord syndromes.
Fractures
Children with MMC are susceptible to pathologic fractures in the lower limbs.139 One study reported a 20% incidence of fracture in its record review.99 Neonates with contractures in the lower limbs are more likely to have fractures with mobilization.21 Risk factors include osteopenia, insensate extremities, contractures, and immobilization. Children with thoracic lesions are more likely to have femur fractures, and children with lumbar lesions are more likely to have tibial fractures.99 A recent study documented fractures to be most common in the early adolescent period, with most fractures seen in community ambulators.51 Fractures manifest with localized erythema, heat, and swelling. Crepitus and deformity occur only with displaced fractures. Often no complaint is made of pain, and no trauma is reported. Fractures are often confused with cellulitis or osteomyelitis. Fractures tend to heal well, with exuberant callus formation, in children with MMC.85 Prolonged immobilization should be avoided because individuals can become more osteopenic during a period of immobilization. One study has demonstrated the use of oral bisphosphonates to treat osteopenia in nonambulatory children, including those with spina bifida.159
Mobility
Almost all children with MMC are able to achieve some degree of independent mobility. This might not be in an upright position, although upright mobility is the goal of almost all parents of a child with MMC. Delay in achievement of ambulation can be expected in all children with MMC, regardless of level.197 Maintaining ambulation is often sought after, even when it is highly inefficient and time-consuming. Many teenagers with MMC will choose wheelchair mobility when given the opportunity, even after a decade of being ambulatory. The ability for a child to become ambulatory and to maintain ambulatory function is determined by a number of factors, including lesion level, cognitive ability, motivation, musculoskeletal complications, growth, age, and obesity.152
Ambulatory function can be divided into four groups: community ambulation, household ambulation, nonfunctional ambulation, and no ambulation. Generally, most patients with sacral-level involvement are community ambulators, and those with thoracic-level involvement are nonambulators. Most patients with lumbar lesions will achieve some level of ambulation but can lose it as adolescence and growth progress.197
Orthoses
The typical four goals or objectives in prescribing an orthotic device for a child with MMC are:
Three bracing systems are used that are relatively unique to the child with MMC. The parapodium is a device that allows even the child with thoracic-level involvement an opportunity for upright mobility. It offers structural support all the way from the trunk to the floor. A swing-to type of gait is used for ambulation. A swivel walker is a modification of the parapodium that translates trunk rotation into forward movement of a dual footplate mechanism. Finally, a reciprocating gait orthosis (RGO) combines bilateral hip–knee–ankle–foot orthoses with a cable system to coordinate hip flexion with hip extension at the opposite hip. Active hip flexion is required to use this type of orthosis. An energy consumption study was performed on three children comparing the use of an RGO and a swing-through type of gait pattern. The swing-through gait was more efficient, but all three children preferred the RGO.68 None of these orthoses allow for efficient gait, but they are often used by smaller children for upright mobility (see Chapter 15).
Skin Breakdown
Skin breakdown is a very common occurrence in children and adults with MMC. Countless dollars are spent treating what should be a preventable complication. One clinic reported that approximately 50% of its patients had skin breakdown. Forty-two percent of these cases were attributed to excessive pressure. In this group, the prevalence steadily increased between infancy and 10 years of age.133 Other factors contributing to the development of pressure sores include mental retardation, chronic soiling, and parental involvement. Morbidity associated with pressure sores can also be severe, including risk of amputation of feet and limbs.
The best treatment for pressure sores is to relieve the pressure. Almost any local approach will work if pressure is relieved. Occasionally, surgical procedures are required to close open wounds. Primary closures or skin flaps are preferred over myocutaneous procedures for initial wounds (see Chapter 32).
Obesity
Obesity has achieved national attention in the able-bodied population. Obesity rates for children and adolescents with spina bifida are similar to the general population; however, obesity rates are higher among adults with spina bifida (38%), particularly women.52 Children with MMC have a lower metabolic rate and lower energy expenditure, which predisposes them to obesity.98 Once they have become obese, exercise is more difficult, which exacerbates the problem. Daily physical activity should be encouraged, beginning at the toddler stage.67 Dietary management should begin in infancy to prevent obesity because outcomes from weight reduction programs are poor. In addition to the obvious problems associated with obesity, individuals with spina bifida or spinal cord injury are at increased risk for developing metabolic syndrome associated with the development of cardiovascular disease and diabetes.129
Psychologic and Social Issues
Cognitive Function
Parents of children with MMC rate their medical support much higher than they do the support received for psychosocial problems.165 Children with MMC have specific behavioral and cognitive issues that need to be addressed or at least recognized by parents, health care providers, and school personnel. As a group, children with MMC have lower IQ scores compared with their able-bodied peers. Their verbal IQ is usually higher than their performance IQ.31 On the Wechsler Intelligence Scale for Children, only 6% of children with MMC scored in the high average to extremely high range compared with 27% of control subjects. Seventy-five percent of children with MMC score in the low average to extremely low average range compared with 25% of matched peers.7 These children also have more difficulty with math and visual perceptual tasks than their able-bodied peers.62,198 IQ scores correlate with the level of lesion. Children with thoracic-level lesions tend to have lower IQ scores than children with lumbar- or sacral-level lesions. IQ scores are also adversely affected by central nervous system infections80 but not necessarily by recurrent shunt revisions.140
Behavior
The temperament characteristics of children with MMC and shunted hydrocephalus were recently described. Children with MMC were found to be less adaptable, more withdrawn, more distractible, less attentive, and less predictable than those in the control group.189 Stimulant medications are often prescribed for these children.
Myelomeningocele in Adults
Transition to Adult Health Care
Because of advances in medical care, 75% to 85% of individuals with MMC now survive into early adulthood.20,122 These individuals can experience secondary complications from accelerated impairment as a result of aging and progression of their underlying disease process.89 Although children are generally observed closely by their pediatric physicians, adults with MMC often do not have regular medical follow-up that is comprehensive in nature. A lack of data exists regarding the best approaches to transition that improve medium and long-term outcomes, and there is a need for evaluation of transitional models using controlled trials.14 In our state, patients who receive care at The Children’s Hospital of Alabama in conjunction with Children’s Rehabilitation Service, a state-funded agency, are generally followed until the age of 21. Although some of these patients continue to be followed by their pediatric providers into adulthood, a well-organized transition to adult health care is optimal for appropriate medical surveillance and presents an ongoing challenge to all who care for this patient population.12,120
Late Neurologic Changes
Adults with MMC remain at risk of developing neurologic complications of their disease. Complications seen in adults include VP shunt infections and malfunctions, syringomyelia, symptomatic tethered cord, and symptomatic type II Chiari malformation. Adolescents and adults with MMC appear not to outgrow their need for shunting.101 As well, up to 10% of adults with VP shunts can develop chronic idiopathic headaches requiring specialized pain management, although at presentation headaches should always receive a comprehensive evaluation to rule out life-threatening causes.53 Likewise, adults with MMC can develop syringomyelia, presenting with complaints of pain, paresthesias, and weakness in the upper extremities.41 Symptomatic tethering of the spinal cord in adults has been associated with herniated disks, pregnancy, and traumatic injuries.70,105 Entrapment syndromes, overuse syndromes, and herniated disks need to be considered in the differential diagnosis of upper limb symptoms in adults more so than in children. An incidence of seizures ranging from 3% to 23% in the adult population has also been reported and is thought to be multifactorial in origin.20,192
Late Musculoskeletal Considerations
A recent review summarizes musculoskeletal considerations in adults with spina bifida.49 Because there is little information regarding this population specifically, much of the information is gleaned from the spinal cord injury population. Shoulder pain is very common in wheelchair users,4,145 although it might be less common in patients who began using a wheelchair earlier in life.156 Rotator cuff disorders and bicipital tendonitis are the most common injuries in chronic wheelchair users.15,59 Physiatrists and therapists can play a key role in prevention and treatment of chronic shoulder problems.
Charcot joints can develop in the lower limbs as a result of lack of sensation and demineralization.22,83 They are most common in the foot and ankle followed by the hip and knee.125 Although more typical in older patients, we recently reported an unusual case of a 5-year-old patient with MMC who developed a Charcot arthropathy of the elbow.208 Charcot joints can lead to significant decline in functional status. Appropriate bracing is necessary to limit movement in affected joints.
The ambulation status of adults with spina bifida has been shown to deteriorate.49,175 Factors explaining this loss of function can include spasticity, knee and hip flexion contractures, low back pain, lack of motivation, or medical complications.11 Physiatrists are particularly well equipped to diagnose and treat these patients to help maintain mobility.
Renal and Urologic Damage
Renal damage remains one of the most common causes of morbidity and mortality among individuals with MMC.124,162 Although rates of bladder cancer appear to be the same as in the general population, age at presentation can be younger, with a more invasive process and poor survival among individuals with MMC.8 Affected adults should receive ongoing urologic care to achieve or maintain social continence and normal renal function. It has been reported that up to 80% of adults with MMC can achieve social continence with proper management.20 Further studies are needed to properly evaluate the effectiveness of both proactive and observational approaches to management, but it is clear that adults with MMC require regular urologic assessment to optimize outcomes.2
Fertility, Sexuality, and Reproductive Issues
Few studies elucidate issues related to fertility, sexuality, and satisfaction with sexual function among individuals with MMC. Despite this, there are some facts that can guide clinicians in caring for and advising patients. Fertility among women with MMC is thought to be normal, and affected individuals are capable of becoming pregnant.201 One study showed that 95% of female patients with MMC menstruated regularly.75 Complications can be experienced during pregnancy related to recurrent urinary tract infection, worsening kyphoscoliosis, VP shunt malfunction, and failure of genitourinary diversions. Vaginal delivery is generally indicated, especially in those with a VP shunt, and cesarean section should be performed only for obstetric reasons such as a contracted, underdeveloped pelvis.141 Many men with MMC are infertile, with one study showing 10 of 10 men with MMC to be azoospermic, exhibiting primary testicular failure on biopsy.207 Another study showed, however, that six of nine men with MMC appeared capable of reproducing with assisted reproductive technologies, although their overall semen quality remained poor.79 In addition, boys display a 15% to 25% incidence of cryptorchidism, further contributing to infertility.58,65,116 Regardless, male and female patients with MMC should receive both basic and specialized sex education, particularly as it pertains to their increased risk of having a child with an NTD. It is recommended that women with MMC of childbearing age take 4 mg of folic acid daily to prevent having an affected child.
The degree of satisfaction with sexual function among individuals with MMC is unknown. Both men and women generally have decreased sensation in the perineum, which can impair the ability to reach orgasm. In addition, nonverbal learning disorders and societal attitudes toward individuals with disabilities can affect self-esteem, social interactions, and ultimately psychosexual development. The reported incidence of the ability to achieve erection ranges from 14% in those with higher-level lesions to 85% in those with lower-level lesions, although many of these are achieved reflexively.47,155 The ability to sustain these erections during intercourse is uncertain, and many patients report dissatisfaction with the degree of rigidity.75 Lesions above the level of sympathetic outflow, as well as a negative anocutaneous reflex, are correlated with increased difficulty achieving erection.47 Although one study reports that most adults with MMC have satisfactory sexual function, another showed that only 18% of the men and 33% of the women with MMC had sexual intercourse activities.32,75 The latter study also reported that orgasm was achieved in 67% of men but only 19% of women. More recent studies by Verhoef et al.193 and Cardenas et al.24 reported women with spina bifida to be more than twice as sexually active as men with spina bifida. Treatment of erectile dysfunction in men with MMC with sildenafil (Viagra) has been successful.147 Retrograde ejaculation is common among men as well, but the exact incidence is unknown. Both men and women can experience skin breakdown during sexual intercourse and should be educated in this regard. Further research is needed to more properly address this topic (see also Chapter 31).
Educational Issues, Vocational Issues, and Independent Living
Most adolescents with MMC complete high school, and about half move on to further education. Although little is published regarding the educational levels achieved, employment status, or living situation of persons with MMC, one long-term follow-up survey reported that 85% of children who survived to adulthood either attended or graduated from high school and/or college, with 36% requiring special education services. Forty-five percent of participants were employed, and 15% lived independently.20 Another study reported more specifically that the college graduation rate was only 14.6% for their cohort.81 A recent Canadian study of individuals with spina bifida and cerebral palsy revealed that transportation was a major barrier to employment, fewer females were employed, and employment was inversely related to IQ.104 Other studies have reported rates of independent living ranging from 14% to 41%.100,191 Rates of employment reported range from 25% to 62.5%, depending on the criteria being considered, with variables considered including intelligence, academic qualifications, behavior, continence, and severity of physical disability.5,10,191 A recent study of young adults from The Netherlands reported that only 16% of those with spina bifida were living independently, 53% of those who finished secondary education were employed, and 71% did not have a partner.10
Conclusion
For more information about MMC, contact the Spina Bifida Association of America through their website at http://www.sbaa.org; by e-mail at sbaa@sbaa.org; by mail at 4590 MacArthur Boulevard NW, Suite 250, Washington, DC 20007-4226; or by telephone at (800) 621-3141.
1. Agnarsson U., Warde C., McCarthy G., et al. Anorectal function of children with neurological problems. I. Spina bifida. Dev Med Child Neurol. 1993;35(10):893-902.
2. Ahmad I., Granitsiotis P. Urological follow-up of adult spina bifida patients. Neurourol Urodyn. 2007;26(7):978-980.
3. Albright A.L., Pollack I.F., Adelson P.D., editors. Principles and practice of pediatric neurosurgery. New York: Thieme, 1999.
4. Alm M., Saraste H., Norrbrink C. Shoulder pain in persons with thoracic spinal cord injury: prevalence and characteristics. J Rehabil Med. 2008;40(4):277-283.
5. Andar U.B., Harkness W.F., Hayward R.D. Split cord malformations of the lumbar region. A model for the neurosurgical management of all types of ‘occult’ spinal dysraphism? Pediatr Neurosurg. 1997;26(1):17-24.
6. [Anonymous]. Mental retardation and developmental disabilities research reviews,, Vol 4. New York: Wiley, 1998.
7. Appleton P.L., Minchom P.E., Ellis N.C., et al. The self-concept of young people with spina bifida: a population-based study. Dev Med Child Neurol. 1994;36(3):198-215.
8. Austin J.C., Elliott S., Cooper C.S. Patients with spina bifida and bladder cancer: atypical presentation, advanced stage and poor survival. J Urol. 2007;178(3 Pt 1):798-801.
9. Barf H.A., Post M.W., Verhoef M., et al. Life satisfaction of young adults with spina bifida. Dev Med Child Neurol. 2007;49(6):458-463.
10. Barf H.A., Post M.W., Verhoef M., et al: Restrictions in social participation of young adults with spina bifida, Disabil Rehabil 31: 921-927,2009
11. Bartonek A., Saraste H., Samuelsson L., et al. Ambulation in patients with myelomeningocele: a 12-year follow-up. J Pediatr Orthop. 1999;19(2):202-206.
12. Begeer I.H., Staal-Schreinemachers A.L. The benefits of team treatment and control of adult patients with spinal dysraphism. Eur J Pediatr Surg. 1996;6(suppl 1):15-16.
13. Bell W.O., Sumner T.E., Volberg F.M. The significance of ventriculomegaly in the newborn with myelodysplasia. Childs Nerv Syst. 1987;3(4):239-241.
14. Binks J.A., Barden W.S., Burke T.A., et al. What do we really know about the transition to adult-centered health care? A focus on cerebral palsy and spina bifida. Arch Phys Med Rehabil. 2007;88(8):1064-1073.
15. Boninger M.L., Towers J.D., Cooper R.A., et al. Shoulder imaging abnormalities in individuals with paraplegia. J Rehabil Res Dev.. 2001;38(4):401-408.
16. Boone D., Parsons D., Lachmann S.M., et al. Spina bifida occulta: lesion or anomaly? Clin Radiol. 1985;36(2):159-161.
17. Bosco P.J., Bauer S.B., Colodny A.H., et al. The long-term results of artificial sphincters in children. J Urol. 1991;146(2):396-399.
18. Botto L.D., Moore C.A., Khoury M.J., et al. Neural-tube defects. N Engl J Med. 1999;341(20):1509-1519.
19. Botto L.D., Yang Q. 5,10-Methylenetetrahydrofolate reductase gene variants and congenital anomalies: a HuGE review. Am J Epidemiol. 2000;151(9):862-877.
20. Bowman R.M., McLone D.G., Grant J.A., et al. Spina bifida outcome: a 25-year prospective. Pediatr Neurosurg. 2001;34(3):114-120.
21. Boytim M.J., Davidson R.S., Charney E., et al. Neonatal fractures in myelomeningocele patients. J Pediatr Orthop. 1991;11(1):28-30.
22. Brinker M.R., Rosenfeld S.R., Feiwell E., et al. Myelomeningocele at the sacral level. Long-term outcomes in adults. J Bone Joint Surg Am. 1994;76(9):1293-1300.
23. Brockmeyer D., Abtin K., Carey L., et al. Endoscopic third ventriculostomy: an outcome analysis. Pediatr Neurosurg. 1998;28(5):236-240.
24. Broughton N.S., Menelaus M.B., Cole W.G., et al. The natural history of hip deformity in myelomeningocele. J Bone Joint Surg Br. 1993;75(5):760-763.
25. Brown K.S., Cook M., Hoess K., et al. Evidence that the risk of spina bifida is influenced by genetic variation at the NOS3 locus. Birth Defects Res Part A Clin Mol Teratol. 2004;70(3):101-106.
26. Caldarelli M., Di Rocco C., Colosimo C.Jr., et al. Surgical treatment of late neurological deterioration in children with myelodysplasia. Acta Neurochir (Wien). 1995;137(3–4):199-206.
27. Caldarelli M., Di Rocco C., La Marca F. Treatment of hydromyelia in spina bifida. Surg Neurol. 1998;50(5):411-420.
28. Canfield M.A., Marengo L., Ramadhani T.A., et al. The prevalence and predictors of anencephaly and spina bifida in Texas. Paediatr Perinat Epidemiol. 2009;23(1):41-50.
29. Cardenas D.D., Topolski T.D., White C.J., et al. Sexual functioning in adolescents and young adults with spina bifida. Arch Phys Med Rehabil. 2008;89(1):31-35.
30. Carter C.O., Evans K. Spina bifida and anencephalus in greater London. J Med Genet. 1973;10(3):209-234.
31. Casari E.F., Fantino A.G. A longitudinal study of cognitive abilities and achievement status of children with myelomeningocele and their relationship with clinical types. Eur J Pediatr Surg. 1998;8(suppl 1):52-54.
32. Cass A.S., Bloom B.A., Luxenberg M. Sexual function in adults with myelomeningocele. J Urol. 1986;136(2):425-426.
33. Castilla E.E., Orioli I.M., Lugarinho R., et al. Monthly and seasonal variations in the frequency of congenital anomalies. Int J Epidemiol. 1990;19(2):399-404.
34. Centers for Disease Control and Prevention (CDC). Racial/ethnic differences in the birth prevalence of spina bifida—United States, 1995-2005. MMWR Morb Mortal Wkly Rep. 2009;57(53):1409-1413.
35. Centers for Disease Control and Prevention. Spina bifida and anencephaly before and after folic acid mandate—United States, 1995–1996 and 1999–2000. MMWR Morb Mortal Wkly Rep. 2004;53(17):362-365.
36. Charney E.B., Rorke L.B., Sutton L.N., et al. Management of Chiari II complications in infants with myelomeningocele. J Pediatr. 1987;111(3):364-371.
37. Charney E.B., Weller S.C., Sutton L.N., et al. Management of the newborn with myelomeningocele: time for a decision-making process. Pediatrics. 1985;75(1):58-64.
38. Coffey V.P. Neural tube defects in Dublin 1953–1954 and 1961–1982. Ir Med J. 1983;76(10):411-413.
39. Coniglio S.J., Anderson S.M., Ferguson J.E.II. Functional motor outcome in children with myelomeningocele: correlation with anatomic level on prenatal ultrasound. Dev Med Child Neurol. 1996;38(8):675-680.
40. Cowchock S., Ainbender E., Prescott G., et al. The recurrence risk for neural tube defects in the United States: a collaborative study. Am J Med Genet. 1980;5(3):309-314.
41. Craig J.J., Gray W.J., McCann J.P. The Chiari/hydrosyringomyelia complex presenting in adults with myelomeningocoele: an indication for early intervention. Spinal Cord. 1999;37(4):275-278.
42. Cuckle H., Wald N. The impact of screening for open neural tube defects in England and Wales. Prenat Diagn. 1987;7(2):91-99.
43. Czeizel A.E., Dudas I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327(26):1832-1835.
44. Daly L.E., Kirke P.N., Molloy A., et al. Folate levels and neural tube defects. Implications for prevention. JAMA. 1995;274(21):1698-1702.
45. Danzer E., Gerdes M., Bebbington M.W., et al. Lower extremity neuromotor function and short-term ambulatory potential following in utero myelomeningocele surgery. Fetal Diagn Ther. 2009;25(1):47-53.
46. De Wals P., Tairou F., Van Allen M.I., et al. Spina bifida before and after folic acid fortification in Canada. Birth Defects Res A Clin Mol Teratol. 2008;82(9):622-626.
47. Diamond D.A., Rickwood A.M., Thomas D.G. Penile erections in myelomeningocele patients. Br J Urol. 1986;58(4):434-435.
48. Dias L.S. Surgical management of knee contractures in myelomeningocele. J Pediatr Orthop. 1982;2(2):127-131.
49. Dicianno B.E., Kurowski B.G., Yang J.M., et al. Rehabilitation and medical management of the adult with spina bifida. Am J Phys Med Rehabil. 2008;87(12):1027-1050.
50. Dietrich S., Okamoto G. Bowel training for children with neurogenic dysfunction: a follow-up. Arch Phys Med Rehabil. 1982;63(4):166-170.
51. Dosa N.P., Eckrich M., Katz D.A., et al. Incidence, prevalence, and characteristics of fractures in children, adolescents, and adults with spina bifida. J Spinal Cord Med. 2007;30(suppl 1):S5-S9.
52. Dosa N.P., Foley J.T., Eckrich M., et al: Obesity across the lifespan among persons with spina bifida, Disabil Rehabil 31:914-920,2009
53. Edwards R.J., Witchell C., Pople I.K. Chronic headaches in adults with spina bifida and associated hydrocephalus. Eur J Pediatr Surg. 2003;13(Suppl 1):S13-S17.
54. Elias E.R., Sadeghi-Nejad A. Precocious puberty in girls with myelodysplasia. Pediatrics. 1994;93(3):521-522.
55. Enger P.O., Svendsen F., Wester K. CSF shunt infections in children: experiences from a population-based study. Acta Neurochir (Wien). 2003;145(4):243-248. discussion 248
56. Erickson D., Bartholomew T., Marlin A. Sonographic evaluation and conservative management of newborns with myelomeningocele and hydronephrosis. J Urol. 1989;142(2 part 2):592-594. discussion 603–605
57. Ersahin Y., Mutluer S., Guzelbag E. Cerebrospinal fluid shunt infections. J Neurosurg Sci. 1994;38(3):161-165.
58. Ferrara P., Rossodivita A., Ruggiero A., et al. Cryptorchidism associated with meningomyelocele. J Paediatr Child Health. 1998;34(1):44-46.
59. Finley M.A., Rodgers M.M. Prevalence and identification of shoulder pathology in athletic and nonathletic wheelchair users with shoulder pain: A pilot study. J Rehabil Res Dev. 2004;41(3B):395-402.
60. Fraser F.C., Frecker M., Allderdice P. Seasonal variation of neural tube defects in Newfoundland and elsewhere. Teratology. 1986;33(3):299-303.
61. Fraser R.K., Hoffman E.B. Calcaneus deformity in the ambulant patient with myelomeningocele. J Bone Joint Surg Br. 1991;73(6):994-997.
62. Friedrich W.N., Lovejoy M.C., Shaffer J., et al. Cognitive abilities and achievement status of children with myelomeningocele: a contemporary sample. J Pediatr Psychol. 1991;16(4):423-428.
63. Gabrieli A.P., Vankoski S.J., Dias L.S., et al. Gait analysis in low lumbar myelomeningocele patients with unilateral hip dislocation or subluxation. J Pediatr Orthop. 2003;23(3):330-334.
64. Greenberg F., James L.M., Oakley G.P.Jr. Estimates of birth prevalence rates of spina bifida in the United States from computer-generated maps. Am J Obstet Gynecol. 1983;145(5):570-573.
65. Greene S.A., Frank M., Zachmann M., et al. Growth and sexual development in children with meningomyelocele. Eur J Pediatr. 1985;144(2):146-148.
66. Groenen P.M., Klootwijk R., Schijvenaars M.M., et al. Spina bifida and genetic factors related to myo-inositol, glucose, and zinc. Mol Genet Metab. 2004;82(2):154-161.
67. Grogan C.B., Ekvall S.M. Body composition of children with myelomeningocele, determined by 40K, urinary creatinine and anthropometric measures. J Am Coll Nutr. 1999;18(4):316-323.
68. Guidera K.J., Smith S., Raney E., et al. Use of the reciprocating gait orthosis in myelodysplasia. J Pediatr Orthop. 1993;13(3):341-348.
69. Guthkelch A.N. Diastematomyelia with median septum. Brain. 1974;97(4):729-742.
70. Harashima S., Taira T., Hori T. [Adult type tethered cord syndrome with chronic attackwise pain in the bilateral feet]. No Shinkei Geka. 2004;32(5):481-485.
71. Hays R.M., Jordan R.A., McLaughlin J.F., et al. Central ventilatory dysfunction in myelodysplasia: an independent determinant of survival. Dev Med Child Neurol. 1989;31(3):366-370.
72. Herndon C.D., Rink R.C., Cain M.P., et al. In situ Malone antegrade continence enema in 127 patients: a 6-year experience. J Urol. 2004;172(4 part 2):1689-1691.
73. Herring J.A., Tachdjian M.O. Texas Scottish Rite Hospital for Children. Tachdjian’s pediatric orthopaedics, ed 3. Philadelphia: Saunders; 2002.
74. Hill A.E., Beattie F. Does caesarean section delivery improve neurological outcome in open spina bifida? Eur J Pediatr Surg. 1994;4(suppl 1):32-34.
75. Hirayama A., Yamada K., Tanaka Y., et al. [Evaluation of sexual function in adults with myelomeningocele]. Hinyokika Kiyo. 1995;41(12):985-989.
76. Hochhaus F., Butenandt O., Schwarz H.P., et al. Auxological and endocrinological evaluation of children with hydrocephalus and/or meningomyelocele. Eur J Pediatr. 1997;156(8):597-601.
77. Hopps C.V., Kropp K.A. Preservation of renal function in children with myelomeningocele managed with basic newborn evaluation and close followup. J Urol. 2003;169(1):305-308.
78. Hudgins R.J., Gilreath C.L. Tethered spinal cord following repair of myelomeningocele. Neurosurg Focus. 2004;16(2):E7.
79. Hultling C., Levi R., Amark S.P., et al. Semen retrieval and analysis in men with myelomeningocele. Dev Med Child Neurol. 2000;42(10):681-684.
80. Hunt G.M., Holmes A.E. Factors relating to intelligence in treated cases of spina bifida cystica. Am J Dis Child. 1976;130(8):823-827.
81. Hurley A.D., Bell S. Educational and vocational outcome of adults with spina bifida in relationship to neuropsychological testing. Eur J Pediatr Surg. 1994;(4 Suppl 1):17-18.
82. Inagaki T., Schoenwolf G.C., Walker M.L. Experimental model: change in the posterior fossa with surgically induced spina bifida aperta in mouse. Pediatr Neurosurg. 1997;26(4):185-189.
83. Jones E.A., Manaster B.J., May D.A., et al. Neuropathic osteoarthropathy: diagnostic dilemmas and differential diagnosis. Radiographics. 2000;20(special number):S279-S293.
84. Kallen B., Cocchi G., Knudsen L.B., et al. International study of sex ratio and twinning of neural tube defects. Teratology. 1994;50(5):322-331.
85. Khoury J.G., Morcuende J.A. Dramatic subperiosteal bone formation following physeal injury in patients with myelomeningocele. Iowa Orthop J. 2002;22:94-98.
86. Kiely M. Reproductive and perinatal epidemiology. Boca Raton: CRC Press; 1991.
87. King J.C., Currie D.M., Wright E. Bowel training in spina bifida: importance of education, patient compliance, age, and anal reflexes. Arch Phys Med Rehabil. 1994;75(3):243-247.
88. Kirke P.N., Mills J.L., Molloy A.M., et al. Impact of the MTHFR C677T polymorphism on risk of neural tube defects: case-control study. BMJ. 2004;328(7455):1535-1536.
89. Klingbeil H., Baer H.R., Wilson P.E. Aging with a disability. Arch Phys Med Rehabil. 2004;85(7 suppl 3):S68-S73. quiz S74–S75
90. Kohl T., Tchatcheva K., Merz W., et al. Percutaneous fetoscopic patch closure of human spina bifida aperta: advances in fetal surgical techniques may obviate the need for early postnatal neurosurgical intervention. Surg Endosc. 2009;23:890-895.
91. Kriss V.M., Desai N.S. Occult spinal dysraphism in neonates: assessment of high-risk cutaneous stigmata on sonography. AJR Am J Roentgenol. 1998;171(6):1687-1692.
92. Lary J.M., Edmonds L.D. Prevalence of spina bifida at birth—United States, 1983–1990: a comparison of two surveillance systems. MMWR CDC Surveill Summ. 1996;45(2):15-26.
93. Laurence K.M., James N., Miller M.H., et al. Double-blind randomised controlled trial of folate treatment before conception to prevent recurrence of neural-tube defects. Br Med J (Clin Res Ed). 1981;282(6275):1509-1511.
94. Lemire R.J. Neural tube defects. JAMA. 1988;259(4):558-562.
95. Leveratto L., Picco P., Cama A., et al. Insulin like growth factor I (IGF1) in children with neural tube closure defects: a preliminary report. Eur J Pediatr Surg. 1993;3(suppl 1):19-20.
96. Lie H.R., Lagergren J., Rasmussen F., et al. Bowel and bladder control of children with myelomeningocele: a Nordic study. Dev Med Child Neurol. 1991;33(12):1053-1061.
97. Lingman G. Management of pregnancy and labour in cases diagnosed with major fetal malformation. Curr Opin Obstet Gynecol. 2005;17(2):143-146.
98. Littlewood R.A., Trocki O., Shepherd R.W., et al. Resting energy expenditure and body composition in children with myelomeningocele. Pediatr Rehabil. 2003;6(1):31-37.
99. Lock T.R., Aronson D.D. Fractures in patients who have myelomeningocele. J Bone Joint Surg Am. 1989;71(8):1153-1157.
100. Lonton A.P., O’Sullivan A.M., Loughlin A.M. Spina bifida adults. Z Kinderchir. 1983;38(suppl 2):110-112.
101. Lorber J., Pucholt V. When is a shunt no longer necessary? An investigation of 300 patients with hydrocephalus and myelomeningocele: 11–22 year follow up. Z Kinderchir. 1981;34(4):327-329.
102. Luthy D.A., Wardinsky T., Shurtleff D.B., et al. Cesarean section before the onset of labor and subsequent motor function in infants with meningomyelocele diagnosed antenatally. N Engl J Med. 1991;324(10):662-666.
103. Machado H.R., Santos de Oliveira R. Simultaneous repair of myelomeningocele and shunt insertion. Childs Nerv Syst. 2004;20(2):107-109.
104. Magill-Evans J., Galambos N., Darrah J., et al. Predictors of employment for young adults with developmental motor disabilities. Work. 2008;31(4):433-442.
105. Maliszewski M., Ladzinski P., Majchrzak H. [Tethered cord syndrome in adults]. Neurol Neurochir Pol. 2000;34(6):1269-1279.
106. Marlin A.E. Management of hydrocephalus in the patient with myelomeningocele: an argument against third ventriculostomy. Neurosurg Focus. 2004;16(2):E4.
107. Marshall P.D., Broughton N.S., Menelaus M.B., et al. Surgical release of knee flexion contractures in myelomeningocele. J Bone Joint Surg Br. 1996;78(6):912-916.
108. Mazur J.M., Stillwell A., Menelaus M. The significance of spasticity in the upper and lower limbs in myelomeningocele. J Bone Joint Surg Br. 1986;68(2):213-217.
109. McGuire E: Spina bifida: a multidisciplinary approach. In Mclaren RI: Neuroanatomy and neurophysiology, New York, 1986, praeger
110. McGuire E.J., Woodside J.R., Borden T.A., et al. Prognostic value of urodynamic testing in myelodysplastic patients. J Urol. 1981;126(2):205-209.
111. McLone D.G. American Society of Pediatric Neurosurgeons, American Association of Neurological Surgeons, Section of Pediatric Neurosurgery: Pediatric neurosurgery: surgery of the developing nervous system, ed 4. Philadelphia: Saunders; 2001.
112. McLone D.G., Knepper P.A. The cause of Chiari II malformation: a unified theory. Pediatr Neurosci. 1989;15(1):1-12.
113. McMichael A.J., Dreosti I.E., Ryan P., et al. Neural tube defects and maternal serum zinc and copper concentrations in mid-pregnancy: a case-control study. Med J Aust. 1994;161(8):478-482.
114. McNeely P.D., Howes W.J. Ineffectiveness of dietary folic acid supplementation on the incidence of lipomyelomeningocele: pathogenetic implications. J Neurosurg Spine. 2004;100(2):98-100.
115. Medical Research Council Vitamin Study Research Group. Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet. 1991;338(8760):131-137.
116. Meyer S., Landau H. Precocious puberty in myelomeningocele patients. J Pediatr Orthop. 1984;4(1):28-31.
117. Miller P.D., Pollack I.F., Pang D., et al. Comparison of simultaneous versus delayed ventriculoperitoneal shunt insertion in children undergoing myelomeningocele repair. J Child Neurol. 1996;11(5):370-372.
118. Mitchell L.E. Spina Bifida Research Resource: study design and participant characteristics. Birth Defects Res A Clin Mol Teratol. 2008;82(10):684-691.
119. Molnar G.E., Alexander M.A., editors. Pediatric rehabilitation, ed 3, Philadelphia: Hanley & Belfus, 1999.
120. Morgan D.J., Blackburn M., Bax M. Adults with spina bifida and/or hydrocephalus. Postgrad Med J. 1995;71(831):17-21.
121. Mosley B.S., Cleves M.A., Siega-Riz A.M., et al. Neural tube defects and maternal folate intake among pregnancies conceived after folic acid fortification in the United States. Am J Epidemiol. 2009;169(1):9-17.
122. Mukherjee S. Transition to adulthood in spina bifida: changing roles and expectations. Sci World J. 2007;7:1890-1895.
123. Muller E.B., Nordwall A. Prevalence of scoliosis in children with myelomeningocele in western Sweden. Spine. 1992;17(9):1097-1102.
124. Muller T., Arbeiter K., Aufricht C. Renal function in meningomyelocele: risk factors, chronic renal failure, renal replacement therapy and transplantation. Curr Opin Urol. 2002;12(6):479-484.
125. Myrianthopoulos N.C., Melnick M. Studies in neural tube defects. I. Epidemiologic and etiologic aspects. Am J Med Genet. 1987;26(4):783-796.
126. Nagarkatti D.G., Banta J.V., Thomson J.D. Charcot arthropathy in spina bifida. J Pediatr Orthop. 2000;20(1):82-87.
127. Naggan L. The recent decline in prevalence of anencephaly and spina bifida. Am J Epidemiol. 1969;89(2):154-160.
128. Naggan L., MacMahon B. Ethnic differences in the prevalence of anencephaly and spina bifida in Boston, MA. N Engl J Med. 1967;277(21):1119-1123.
129. Nelson M.D., Widman L.M., Abresch R.T., et al. Metabolic syndrome in adolescents with spinal cord dysfunction. J Spinal Cord Med. 2007;30(Suppl 1):S127-S139.
130. Nevin N.C., Johnston W.P., Merrett J.D. Influence of social class on the risk of recurrence of anencephalus and spina bifida. Dev Med Child Neurol. 1981;23(2):155-159.
131. Niall D.M., Dowling F.E., Fogarty E.E., et al. Kyphectomy in children with myelomeningocele: a long-term outcome study. J Pediatr Orthop. 2004;24(1):37-44.
132. Noonan K.J., Didelot W.P., Lindseth R.E. Care of the pediatric foot in myelodysplasia. Foot Ankle Clin. 2000;5(2):281-304. vi
133. Okamoto G.A., Lamers J.V., Shurtleff D.B. Skin breakdown in patients with myelomeningocele. Arch Phys Med Rehabil. 1983;64(1):20-23.
134. Okamoto G.A., Sousa J., Telzrow R.W., et al. Toileting skills in children with myelomeningocele: rates of learning. Arch Phys Med Rehabil. 1984;65(4):182-185.
135. Omtzigt J.G., Los F.J., Grobbee D.E., et al. The risk of spina bifida aperta after first-trimester exposure to valproate in a prenatal cohort. Neurology. 1992;42(4 suppl 5):119-125.
136. Palmer J.S., Kaplan W.E., Firlit C.F. Erectile dysfunction in patients with spina bifida is a treatable condition. J Urol. 2000;164(3 part 2):958-961.
137. Papp C., Adám Z., Tóth-Pál E., et al. Risk of recurrence of craniospinal anomalies. J Matern Fetal Med. 1997;6(1):53-57.
138. Phuong L.K., Schoeberl K.A., Raffel C. Natural history of tethered cord in patients with meningomyelocele. Neurosurgery. 2002;50(5):989-993. discussion 993–995
139. Quan A., Adams R., Ekmark E., et al. Bone mineral density in children with myelomeningocele. Pediatrics. 1998;102(3):E34.
140. Ralph K., Moylan P., Canady A., et al. The effects of multiple shunt revisions on neuropsychological functioning and memory. Neurol Res. 2000;22(1):131-136.
141. Rietberg C.C., Lindhout D. Adult patients with spina bifida cystica: genetic counselling, pregnancy and delivery. Eur J Obstet Gynecol Reprod Biol. 1993;52(1):63-70.
142. Rinck C., Berg J., Hafeman C. The adolescent with myelomeningocele: a review of parent experiences and expectations. Adolescence. 1989;24(95):699-710.
143. Rintoul N.E., Sutton L.N., Hubbard A.M., et al. A new look at myelomeningoceles: functional level, vertebral level, shunting, and the implications for fetal intervention. Pediatrics. 2002;109(3):409-413.
144. Roberts H.E., Moore C.A., Cragan J.D., et al. Impact of prenatal diagnosis on the birth prevalence of neural tube defects, Atlanta, 1990–1991. Pediatrics. 1995;96(5 part 1):880-883.
145. Roehrig S., Like G. Factors affecting shoulder pain in adolescents and young adults with spina bifida. Pediatr Phys Ther. 2008;20(3):224-232.
146. Rosa F.W. Spina bifida in infants of women treated with carbamazepine during pregnancy. N Engl J Med. 1991;324(10):674-677.
147. Rotenstein D., Bass A.N. Treatment to near adult stature of patients with myelomeningocele with recombinant human growth hormone. J Pediatr Endocrinol Metab. 2004;17(9):1195-1200.
148. Rotenstein D., Reigel D.H. Growth hormone treatment of children with neural tube defects: results from 6 months to 6 years. J Pediatr. 1996;128(2):184-189.
149. Rotenstein D., Reigel D.H., Flom L.L. Growth hormone treatment accelerates growth of short children with neural tube defects. J Pediatr. 1989;115(3):417-420.
150. Salomao J.F., Bellas A.R., Leibinger R.D., et al. [Symptomatic Chiari type II malformation]. Arq Neuropsiquiatr. 1998;56(1):98-106.
151. Salomao J.F., Cavalheiro S., Matushita H., et al. Cystic spinal dysraphism of the cervical and upper thoracic region. Childs Nerv Syst. 2006;22(3):234-242.
152. Samuelsson L., Skoog M. Ambulation in patients with myelomeningocele: a multivariate statistical analysis. J Pediatr Orthop. 1988;8(5):569-575.
153. Sandford M.K., Kissling G.E., Joubert P.E. Neural tube defect etiology: new evidence concerning maternal hyperthermia, health and diet. Dev Med Child Neurol. 1992;34(8):661-675.
154. Sandhu P.S., Broughton N.S., Menelaus M.B. Tenotomy of the ligamentum patellae in spina bifida: management of limited flexion range at the knee. J Bone Joint Surg Br. 1995;77(5):832-833.
155. Satin-Smith M.S., Katz L.L., Thornton P., et al. Arm span as measurement of response to growth hormone (GH) treatment in a group of children with meningomyelocele and GH deficiency. J Clin Endocrinol Metab. 1996;81(4):1654-1656.
156. Sawatzky B.J., Slobogean G.P., Reilly C.W., et al. Prevalence of shoulder pain in adult- versus childhood-onset wheelchair users: a pilot study. J Rehabil Res Dev. 2005;42(3 suppl 1):1-8.
157. Sawyer S.M., Roberts K.V. Sexual and reproductive health in young people with spina bifida. Dev Med Child Neurol. 1999;41(10):671-675.
158. Selzman A.A., Elder J.S., Mapstone T.B. Urologic consequences of myelodysplasia and other congenital abnormalities of the spinal cord. Urol Clin North Am. 1993;20(3):485-504.
159 Sholas M.G., Tann B., Gaebler-Spira D. Oral bisphosphonates to treat disuse osteopenia in children with disabilities: a case series. J Pediatr Orthop. 2005;25(3):326-331.
160. Shurtleff D.B., editor. Myelodysplasias and exstrophies: significance, prevention, and treatment. Orlando: Grune & Stratton, 1986.
161. Shurtleff D.B., Lemire R.J. Epidemiology, etiologic factors, and prenatal diagnosis of open spinal dysraphism. Neurosurg Clin North Am. 1995;6(2):183-193.
162. Singhal B., Mathew K.M. Factors affecting mortality and morbidity in adult spina bifida. Eur J Pediatr Surg. 1999;9(suppl 1):31-32.
163. Slater J.E. Rubber anaphylaxis. N Engl J Med. 1989;320(17):1126-1130.
164. Smith E.D. Urinary prognosis in spina bifida. J Urol. 1972;108(5):815-817.
165. Smith G.K. The history of spina bifida, hydrocephalus, paraplegia, and incontinence. Pediatr Surg Int. 2001;17(5–6):424-432.
166. Snodgrass W.T., Adams R. Initial urologic management of myelomeningocele. Urol Clin North Am. 2004;31(3):427-434. viii
167. Sousa J.C., Gordon L.H., Shurtleff D.B. Assessing the development of daily living skills in patients with spina bifida. Dev Med Child Neurol Suppl. 1976;37:134-142.
168. Spindel M.R., Bauer S.B., Dyro F.M., et al. The changing neurourologic lesion in myelodysplasia. JAMA. 1987;258(12):1630-1633.
169. Stark G. Spina bifida: problems and management. Oxford: Blackwell Scientific Publications; 1977.
170. Steinbok P., Irvine B., Cochrane D.D., et al. Long-term outcome and complications of children born with meningomyelocele. Childs Nerv Syst. 1992;8(2):92-96.
171. Stevenson K.L. Chiari type II malformation: past, present, and future. Neurosurg Focus. 2004;16(2):E5.
172. Sussman G.L., Tarlo S., Dolovich J. The spectrum of IgE-mediated responses to latex. JAMA. 1991;265(21):2844-2847.
173. Sutton L.N. Fetal surgery for neural tube defects. Best Pract Res Clin Obstet Gynaecol. 2008;22(1):175-188.
174. Sutton L.N., Adzick N.S., Bilaniuk L.T., et al. Improvement in hindbrain herniation demonstrated by serial fetal magnetic resonance imaging following fetal surgery for myelomeningocele. JAMA. 1999;282(19):1826-1831.
175. Taylor A., McNamara A. Ambulation status of adults with myelomeningocoele. Z Kinderchir. 1990;45(suppl 1):32-33.
176. Taylor J.S., Erkek E. Latex allergy: diagnosis and management. Dermatol Ther. 2004;17(4):289-301.
177. Thompson M.D., Cole D.E., Ray J.G. Vitamin B-12 and neural tube defects: the Canadian experience. Am J Clin Nutr. 2009;89(2):697S-701S.
178. Tortori-Donati P., Rossi A., Cama A. Spinal dysraphism: a review of neuroradiological features with embryological correlations and proposal for a new classification. Neuroradiology. 2000;42(7):471-491.
179. Trivedi J., Thomson J.D., Slakey J.B., et al. Clinical and radiographic predictors of scoliosis in patients with myelomeningocele. J Bone Joint Surg Am. 2002;84(8):1389-1394.
180. Trollmann R., Bakker B., Lundberg M., et al. Growth in pre-pubertal children with myelomeningocele (MMC) on growth hormone (GH): the KIGS experience. Pediatr Rehabil. 2006;9(2):144-148.
181. Trollmann R., Dörr H.G., Strehl E., et al. Growth and pubertal development in patients with meningomyelocele: a retrospective analysis. Acta Paediatr. 1996;85(1):76-80.
182. Trollmann R., Strehl E., Dorr H.G. Precocious puberty in children with myelomeningocele: treatment with gonadotropin-releasing hormone analogues. Dev Med Child Neurol. 1998;40(1):38-43.
183. Trollmann R., Strehl E., Wenzel D., et al. Arm span, serum IGF-1 and IGFBP-3 levels as screening parameters for the diagnosis of growth hormone deficiency in patients with myelomeningocele: preliminary data. Eur J Pediatr. 1998;157(6):451-455.
184. Trollmann R., Strehl E., Wenzel D., et al. Does growth hormone (GH) enhance growth in GH-deficient children with myelomeningocele? J Clin Endocrinol Metab. 2000;85(8):2740-2743.
185. Tuli S., Drake J., Lamberti-Pasculli M. Long-term outcome of hydrocephalus management in myelomeningoceles. Childs Nerv Syst. 2003;19(5–6):286-291.
186. Tulipan N., Sutton L.N., Bruner J.P., et al. The effect of intrauterine myelomeningocele repair on the incidence of shunt-dependent hydrocephalus. Pediatr Neurosurg. 2003;38(1):27-33.
187. Uehling D.T., Smith J., Meyer J., et al. Impact of an intermittent catheterization program on children with myelomeningocele. Pediatrics. 1985;76(6):892-895.
188. Use of supplements containing folic acid among women of childbearing age—United States 2007. MMWR Morb Mortal Wkly Rep. 2008;57(01):5-8.
189. Vachha B., Adams R. Myelomeningocele, temperament patterns, and parental perceptions. Pediatrics. 2005;115(1):e58-e63.
190. Van Allen M.I., Kalousek D.K., Chernoff G.F., et al. Evidence for multi-site closure of the neural tube in humans. Am J Med Genet. 1993;47(5):723-743.
191. van Mechelen M.C., Verhoef M., van Asbeck F.W., et al. Work participation among young adults with spina bifida in the Netherlands. Dev Med Child Neurol. 2008;50(10):772-777.
192. Verhoef M., Barf H.A., Post M.W., et al. Secondary impairments in young adults with spina bifida. Dev Med Child Neurol. 2004;46(6):420-427.
193. Verhoef M., Barf H.A., Vroege J.A., et al. Sex education, relationships, and sexuality in young adults with spina bifida. Arch Phys Med Rehabil. 2005;86(5):979-987.
194. Verhoef M., Lurvink M., Barf H.A., et al. High prevalence of incontinence among young adults with spina bifida: description, prediction and problem perception. Spinal Cord. 2005;43(6):331-340.
195. Waller D.K., Shaw G.M., Rasmussen S.A., et al. Prepregnancy obesity as a risk factor for structural birth defects. Arch Pediatr Adolesc Med. 2007;161(8):745-750.
196. Watkins M.L., Rasmussen S.A., Honein M.A., et al. Maternal obesity and risk for birth defects. Pediatrics. 2003;111(5 part 2):1152-1158.
197. Williams E.N., Broughton N.S., Menelaus M.B. Age-related walking in children with spina bifida. Dev Med Child Neurol. 1999;41(7):446-449.
198. Wills K.E., Holmbeck G.N., Dillon K., et al. Intelligence and achievement in children with myelomeningocele. J Pediatr Psychol. 1990;15(2):161-176.
199. Winquist B., Ogle K., Muhajarine N. Exploring physicians’ views and values in relation to maternal serum screening. J Obstet Gynaecol Can. 2008;30(7):564-572.
200. Wolraich M.L., Hawtrey C., Mapel J., et al. Results of clean intermittent catheterization for children with neurogenic bladders. Urology. 1983;22(5):479-482.
201. Woodhouse C.R. Prospects for fertility in patients born with genitourinary anomalies. J Urol. 2001;165(6 part 2):2354-23560.
202. Woodhouse C.R. Sexual function in boys born with exstrophy, myelomeningocele, and micropenis. Urology. 1998;52(1):3-11.
203. Worley G. A new look at meningomyeloceles. Pediatrics. 2003;111(6 part 1):1494-1495. author reply 1494–1495
204. Wright J.G., Menelaus M.B., Broughton N.S., et al. Natural history of knee contractures in myelomeningocele. J Pediatr Orthop. 1991;11(6):725-730.
205. Wu H.Y., Baskin L.S., Kogan B.A. Neurogenic bladder dysfunction due to myelomeningocele: neonatal versus childhood treatment. J Urol. 1997;157(6):2295-2297.
206. Yamada S., Won D.J., Yamada S.M. Pathophysiology of tethered cord syndrome: correlation with symptomatology. Neurosurg Focus. 2004;16(2):E6.
207. Zhu H., Wicker N.J., Volcik K., et al. Promoter haplotype combinations for the human PDGFRA gene are associated with risk of neural tube defects. Mol Genet Metab. 2004;81(2):127-132.
208. Zimmermann A., Law C., Blount J., et al. Neuropathic arthropathy of the elbow in a paediatric patient with myelomeningocele. Dev Neurorehabil. 2007;10(3):261-264.