CHAPTER 214 Myelomeningocele and Myelocystocele
Myelomeningocele is a manifestation of a generalized malformation of the central nervous system (CNS) that occurs in 2500 to 6000 newborns per year in the United States.1,2 Its initial management will affect not only the neonates’ survival but also the handicaps that they will have to cope with throughout their lives. Myelocystoceles are uncommon, embryologically unrelated lesions of the distal end of the spinal cord. Although these lesions do not require urgent surgical repair, affected children require lifelong neurosurgical care.
Terminology
Myelomeningocele most likely results from failure of proper neural tube closure during primary neurulation, and such failure leaves a flat plate of neural tissue called the neural placode (Fig. 214-1). As a result, the overlying mesodermal and ectodermal elements fail to form, and an open spinal defect is produced that is almost invariably associated with a Chiari II malformation and its cranial manifestations (Fig. 214-2). A terminal myelocystocele is a skin-covered midline mass composed of a low-lying conus medullaris with a cystic trumpet-like dilation of the caudal central canal, a surrounding meningocele, and a lipoma (Fig. 214-3). These lesions are believed to result from defective cellular differentiation during secondary neurulation. Myelomeningocele and myelocystocele are discussed separately.
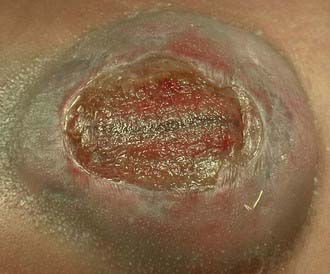

Myelomeningocele
Anatomic Considerations
Although failure of closure of the neural tube is thought to be the cause of myelomeningocele, it is likely that other processes, such as errors in gastrulation, may also disrupt neurulation and ultimately produce an open neural tube defect (NTD).3 In all cases of myelomeningocele, failure of neural tube closure results in an exposed neural placode (Fig. 214-4). The groove in the center of the placode is the remnant of the central canal. The spinal roots exit from the anterior surface of the placode such that the ventral roots lie medially and the dorsal roots lie laterally. The dura fuses with the defect in the fascia laterally. Functional neural tissue may be present caudal to the placode or in the nerve roots exiting from the placode.
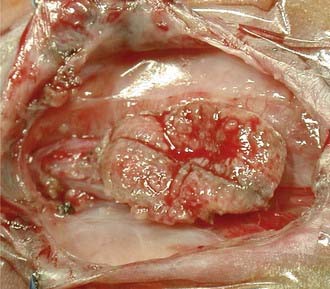
Most myelomeningoceles (85%) are located in the caudal thoracolumbar spine or more distally. Ten percent are in the thorax, and the rest are cervical. Cervical myelomeningoceles are often very similar to meningoceles but without the associated Chiari II malformation and hydrocephalus.4
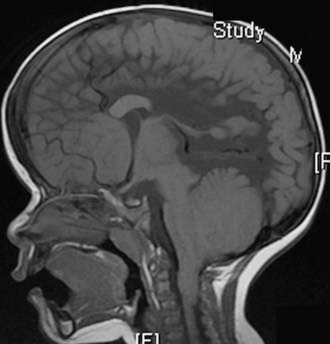
Almost all patients with myelomeningocele also have the Chiari II hindbrain malformation, a constellation of abnormalities that affect the entire CNS (see Fig. 214-2). Associated brainstem defects include medullary kinking, tectal beaking, and intrinsic nuclei abnormalities.5 Supratentorial abnormalities include partial or complete dysgenesis of the corpus callosum, polymicrogyria, a large massa intermedia, and gray matter heterotopia. Mesodermal development of the skull is also affected and leads to a small posterior fossa, short clivus, low-lying tentorium and torcular Herophili, wide incisura, and enlarged foramen magnum. Lückenschädel, or craniolacunia (scalloping of the skull bones noted on computed tomography [CT] and plain radiographs of the skull in these infants), is also a result of this mesodermal dysplastic process; these skull lacunae are not caused by increased intracranial pressure and usually resolve by 1 year of age.
The majority (80% to 90%) of patients with myelomeningocele have hydrocephalus that requires treatment. The hydrocephalus may result from both obstructive and communicating components. Syringomyelia occurs in 40% to 80% of patients with spina bifida and is usually nonprogressive.6 In patients with spina bifida, neurological deterioration can result from symptomatic hydrocephalus, Chiari II malformation, syringomyelia, or retethering. Most often, the cause of neurological deterioration is hydrocephalus from malfunction of a shunt.6
History
There is evidence that spina bifida existed in ancient civilizations.7 Peter Van Forest first recorded a child with spina bifida in 1587,8 and in 1610 he performed the first reported surgical resection of the myelomeningocele sac.9 The first anatomic illustration was drawn by Tulp in 1641.10 In 1761, Morgagni was the first to associate the clinical changes observed in spina bifida patients with the myelomeningocele.11 The first theory to explain spina bifida was advanced by Lebedeff in 1881.12 He attributed myelomeningocele to failure of the neural tube to close. In 1886, von Recklinghausen described the types of spina bifida and reviewed the surgical treatment.13 In Fraser’s report of the first series of spina bifida patients treated surgically, two thirds of the patients operated on between 1898 and 1923 survived until hospital discharge. Six years later, nearly a quarter (23%) of the patients were still alive.14
More aggressive surgical treatment for children with spina bifida was undertaken after the development of ventriculoperitoneal shunting for hydrocephalus in the 1950s. Because delayed complications developed in many patients, some physicians suggested selective surgical treatment of neonates with spina bifida.15,16 The ethical debate about selective treatment ended in the early 1980s when reports demonstrated that patients in nonselected series did as well as or better than those in series with selection.17 In North America, unless there is an anomaly incompatible with life, almost all newborns with myelomeningocele are treated aggressively to optimize their quality of life.
Epidemiology
In the United States, the prevalence of myelomeningocele has declined because of both prenatal folate supplementation and termination of pregnancy. Before 1980, the prevalence of myelomeningocele in the United States was 1 to 2 per 1000 live births. More recently, the prevalence has declined to 0.44 per 1000 live births.18 Twenty percent to 30% of the decline can be attributed to pregnancy termination after prenatal diagnosis.19–21
Geographic variation in the prevalence of spina bifida has been recognized for many years. The United Kingdom, particularly Ireland, has a higher prevalence of NTDs than do continental Europe and the United States. The prevalence of spina bifida in the United Kingdom after 1980 was 0.74 to 2.5 per 1000 live births; the prevalence in the United States during the same period was 0.41 to 1.43 per 1000 live births.22
Racial and ethnic variation in the prevalence of spina bifida exists and persists after immigration. In the United States, the prevalence of NTDs is highest in Hispanics, followed by whites and then African Americans and Asians. The overall U.S. prevalence was 0.46 per 1000 live births, but Hispanics had a prevalence rate of 0.6 per 1000 and Asians 0.23 per 1000.18 Internationally, the prevalence of spina bifida varies from a low of 0.1 per 1000 live births in native Africans to a high of 12.5 per 1000 in Celtics.22 Patterns of variation in the severity of the defect also exist among ethnic and racial groups. With regard to gender differences, a slight (0.57% to 0.71%) female preponderance exists.23
Pathogenesis
Closure of the posterior neuropore occurs during human embryonic stage 12, at approximately 26 days of gestation. Although the etiology of some types of open NTDs may be related to errors in gastrulation,3 the most widely supported theory of spina bifida formation proposes that the posterior neuropore fails to close completely during neurulation and a myelomeningocele, Chiari II malformation, and hydrocephalus result. The nonclosure theory has been substantiated by recent experimental studies using toxic agents and animal mutants. Toxic agents include cytochalasin, vinblastine, calcium-channel antagonists, phospholipase C, concanavalin A, retinoic acid, hydroxyurea, and mitomycin C. Folate and its pathophysiology have been the focus of the majority of recent investigations. Mutant mice, such as the splotch and curly tail/loop tail mouse,24 have been used to study the pathogenesis of NTDs.
A unified hypothesis to explain the sequential development of myelomeningocele and Chiari II hindbrain malformation has been proposed.25 In normal development, during the period of rapid brain enlargement that occurs after closure of the posterior neuropore, the neurocele (primitive central canal) becomes transiently occluded. If the posterior neuropore has failed to close, thereby causing a myelomeningocele, the neurocele fails to occlude. Without appropriate neurocele occlusion, cerebrospinal fluid (CSF) flows out through the defect in the open posterior neuropore. With the lack of distention of the brain by CSF, formation of the cranium and its contents is disrupted. The posterior fossa is small, and both upward and downward herniation of the cerebellum occurs and results in the Chiari II malformation.25 The unified hypothesis has been supported by experiments with chick embryos and with the homozygote splotch mouse.
Etiology
Folate
The coenzyme folate is required for hematopoiesis, metabolism, and normal gastrointestinal and neurological function. The association between folate insufficiency and NTDs was suspected by Hibbard in 1964. A randomized, double-blind study in 1991 demonstrated that couples in the United States with a previous history of an infant with an NTD have a 2% to 3% chance of having a second child born with an NTD. If 4 mg of folic acid was consumed during the critical period (before and during pregnancy), the risk of having a second child born with spina bifida dropped 71%,26 thus suggesting that some, but not all NTDs are related to abnormal folate supplementation (Table 214-1).27 Current recommendations state that women who could potentially become pregnant should consume at least 0.4 mg/day of folic acid (Table 214-2).28 The cause of the NTDs that are not prevented by folate supplementation is not known.
| RISK FACTORS | RISK (%) |
|---|---|
| Medical | |
| History of pregnancy with an NTD | 2-3 |
| Partner with an NTD | 2-3 |
| Diabetes mellitus type 1 | 1 |
| Seizure disorder (valproic acid or carbamazepine) | 1 |
| Close relative with an NTD | 0.3-1 |
| Prepregnancy obesity (>110 kg) | 0.2 |
| Nonmedical | |
| Agricultural pesticides and chemicals | |
| Cleaning solvents and disinfectants | |
| Nursing | |
| Radiation exposure | |
| Anesthetic agents | |
| Hot tubs, saunas, and fever (hyperthermia) | |
| Lead | |
| Tobacco smoke | |
NTD, neural tube defect.
Adapted from Cohen AR, Robinson S. Myelomeningocele: Early management. In: McLone DG, ed. Pediatric Neurosurgery, 4th ed. Philadelphia: WB Saunders; 2000:241-259.
TABLE 214-2 Recommendations for Daily Folate Supplementation
| CIRCUMSTANCE | DOSE (mg) |
|---|---|
| Before Pregnancy | |
| Women with no known risk factor | 0.4 |
| Women at high risk | 4 |
| During Pregnancy | |
| Women with no known risk factor | 0.6 |
| Women at high risk | 4 |
| Post Partum While Breast-Feeding | |
| Women with no known risk factor | 0.5 |
| Women at high risk | 4 |
Adapted from Cohen AR, Robinson S. Myelomeningocele: Early management. In: McLone DG, ed. Pediatric Neurosurgery, 4th ed. Philadelphia: WB Saunders; 2000:241-259.
Anticonvulsants
NTDs are associated with antiepileptic medications taken during pregnancy. The risk of a mother treated with carbamazepine or valproic acid having a child with spina bifida or anencephaly is 1% or 1% to 2%, respectively.29
Prenatal Diagnosis
Diagnostic Studies
Maternal Serum Alpha Fetoprotein
Determination of maternal serum alpha fetoprotein (MSAFP) levels in the early part of the second trimester is the initial screen for NTDs. In amniotic fluid, the AFP concentration is 100-fold less than in fetal CSF.32 The optimal time for sampling MSAFP is at 16 to 18 weeks, although MSAFP levels can be determined between 14 and 21 weeks’ gestation. Seventy-nine percent of pregnancies with open NTDs and 3% of normal singleton pregnancies have an MSAFP level that is 2.5 multiples of the median (MoM) at 16 to 18 weeks’ gestation.33 Based on the adjusted MoM MSAFP level for the patient’s age and population, the MSAFP level at a specific gestational age provides a patient-specific risk for an NTD. For example, an MSAFP level of 1.5 MoM predicts a risk of 1 in 2317 for an open NTD, and an MSAFP level of 2.5 MoM predicts a risk of 1 in 98. The estimated risk is adjusted according to the prevalence of NTDs in the population. The diagnostic accuracy of a single MSAFP level is limited to 60% to 70%. If the risk for spina bifida predicted by the MSAFP concentration is greater than 1 in 500, further testing with high-resolution ultrasonography is recommended.
If the screening MSAFP level is elevated, the test can be repeated. A constant or decreasing MSAFP level is not consistent with a pregnancy with an NTD. In 40% of cases, the repeated level is normal.34 Instead of repeating the test, high-resolution fetal ultrasonography can be performed. Ultrasonography can identify false-positive MSAFP levels caused by incorrect estimated gestational age, multiple pregnancy, or fetal demise. If the initial and repeated MSAFP levels are elevated more than 3 MoM and the first fetal ultrasound study is normal, a second ultrasound study is recommended.
Ultrasonography
The sensitivity of high-resolution fetal ultrasonography is nearly 100% in prenatal screening for NTDs. An accurate prediction of the anatomic level of the spinal cord defect can be obtained in 64% of cases and is within one level of the defect in 79%.35 High-resolution ultrasonography can also visualize two cranial abnormalities that are characteristic of hydrocephalus and the Chiari II malformation associated with spina bifida. The first, consisting of scalloping of the frontal bones on a biparietal view, is called the lemon sign.36,37 The second, consisting of an abnormally shaped midbrain, elongated cerebellum, and obliteration of the cisterna magna characteristic of the Chiari II malformation, is referred to as the banana sign. The lemon sign was present in 80% of fetuses with myelomeningocele, and the banana sign was present in 93%.36,37
The correlation among the findings of prenatal ultrasonography, level of spina bifida, degree of posterior fossa deformity, and degree of hydrocephalus has been examined.38 No correlation was found between the ventricular diameter and the level of the spinal defect.39,40 The posterior fossa deformity did not progress on serial ultrasound examinations.38 If ultrasonography is nondiagnostic, magnetic resonance imaging (MRI) or amniocentesis may be indicated.
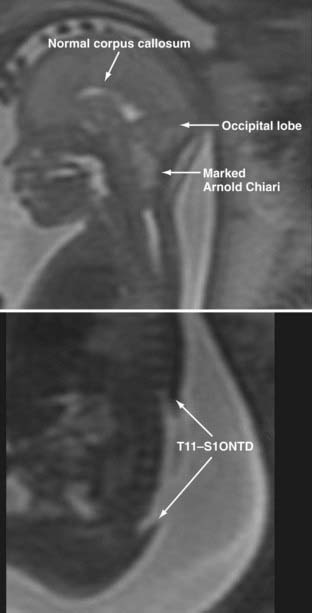
Magnetic Resonance Imaging
Prenatal MRI is increasingly being used to examine potential fetal neurological anomalies (Fig. 214-5), and it provides greater resolution than ultrasonography.41 Because the recent development of fast spin echo techniques has largely overcome the challenges associated with fetal movement, MRI has become an excellent noninvasive second-line imaging modality for cases in which screening high-resolution ultrasonography is nondiagnostic. Data regarding rates of diagnostic accuracy in detecting myelomeningocele are not currently available.
Amniocentesis
Amniocentesis may be indicated if the MSAFP level and imaging studies suggest the presence of an NTD. The amniotic acetylcholinesterase (AChE) level is used to increase the diagnostic accuracy of the amniotic AFP level because the latter can have a high false-positive rate in a population at low risk for NTDs.42 If an NTD is present, neural AChE from CSF leaks into the amniotic fluid. Other anomalies such as an omphalocele or Turner’s syndrome can elevate the amniotic AChE level.43 Amniotic fluid AFP and AChE levels had an accuracy of 99% and a false-positive rate of 0.34% in a large study of singleton pregnancies.44
Differential Diagnosis
At least 22 other fetal abnormalities besides myelomeningocele increase MSAFP levels.32 Abdominal abnormalities such as omphalocele, cloacal exstrophy, esophageal atresia, annular pancreas, duodenal atresia, and gastroschisis and urologic abnormalities such as congenital nephrosis, polycystic kidneys, urinary tract obstruction, and renal agenesis can also elevate MSAFP levels.33,42 Elevated MSAFP and elevated amniotic fluid AFP and AChE can be caused by a sacrococcygeal cystic teratoma, but the teratoma can usually be distinguished from a myelomeningocele by fetal ultrasonography.
Prenatal Counseling
Prognosis
The present 2-year survival rate of neonates born with myelomeningocele is greater than 95%. Ten percent to 15% of children with spina bifida die before 6 years of age, even with aggressive treatment.1,45 Hydrocephalus that requires treatment occurs in most (90% to 98%) of the patients.1,45
The functional motor level determines the ambulatory status of a patient. The motor level may or may not correspond to the anatomic level of the myelomeningocele. The prenatal anatomic level determined by ultrasonography accurately predicts the motor function level at the age of 3 to 4 years better than does determination of the neonatal motor level by examination.46 L3 function allows one to stand erect, and L4 and L5 function allows ambulation.47 During the first decade, approximately 60% of children with spina bifida are community ambulators, without or with assistive devices (including wheelchairs); 15% are household ambulators and 26% are nonambulatory.48 The percentage of community ambulators decreases by about 17% during adolescence. As teenagers, many patients with spina bifida have to increase the level of assistance provided by devices. Normal urinary continence is present in only a small portion (6% to 17%) of these children.47 Most (85%) are socially continent by using a combination of clean intermittent catheterization and medications, and most (86%) have social fecal continence.49
The majority (75% to 80%) of children with spina bifida can have normal intelligence with aggressive management of hydrocephalus and infections. A correlation between a higher myelomeningocele level or severe ventricular enlargement on prenatal ultrasonography and lower intelligence has been found in some studies.17,50,51 The intelligence quotient (IQ) remained normal if the CNS was not infected, but only about a third (31%) of the patients with a history of CNS infection had a normal IQ.52 It remains controversial whether the number of shunt revisions affects IQ.50,53 Although complex learning disabilities are not unusual in children with spina bifida, more than half (58%) of children with spina bifida perform at their grade level.49 Because of severe cognitive impairment, 10% to 15% require custodial care.1 Only a small portion (<10%) of patients with spina bifida become economically independent. The majority (>80%) of patients with spina bifida need psychiatric care or counseling to help them cope with their disabilities.
Risk in Siblings
The parents of a child with a myelomeningocele may seek counseling about the chance of having another child with an NTD. The risk of a sibling having an NTD is estimated to be 2.8% in the United States.54 If the parents have had two previous pregnancies with NTDs, the risk increases to 4.8%.55 If a parent has an NTD, the risk of a child having spina bifida is 3%.56 Monozygotic twins have a low concordance rate.57 Autosomal dominant, autosomal recessive, and X-linked inheritance patterns have been described.
Fetal Management of Spina Bifida and Hydrocephalus
A role for intrauterine surgery to minimize the neurological deficits of patients with myelomeningoceles has been suggested but remains highly controversial. According to some proponents of intrauterine myelomeningocele repair (IUMR), a portion of the neurological deficit present in children with myelomeningoceles is acquired from persistent exposure of the dysplastic spinal cord to amniotic fluid.58 In experimental animal models, intrauterine repair to cover the exposed spinal cord restored normal development.59–62 Other experiments in rodents have shown that the dysplasia of the dorsal spinal cord results from the lack of normal afferent input from aberrant dorsal roots rather than from deterioration of normal tissue as a result of exposure to amniotic fluid.63
Recent studies have shown that IUMR does not improve the motor deficit, but it may minimize the degree of cerebellar herniation in the Chiari II malformation. In a nonrandomized observational study, patients who underwent IUMR were compared with patients who underwent standard neonatal repair.64 The infants undergoing intrauterine repair were less likely than the control infants to require a shunt for symptomatic hydrocephalus during the first 6 months (59% versus 90%), and they demonstrated less hindbrain herniation (38% versus 95%). It is, however, important to note that this was an uncontrolled, observational study. The infants who underwent intrauterine repair were born significantly earlier than were the control infants (33 versus 37 weeks’ gestation). Further studies have suggested that greater rates of prematurity translate into an increased risk for fetal morbidity and mortality.65 The long-term effects on the cognitive development of children who have undergone IUMR have yet to be assessed, although studies of short-term neurodevelopmental outcomes indicate that those who do not require shunting may have improved outcomes.66 Data with regard to urologic outcomes after IUMR are also inconclusive at this time.67 To determine whether IUMR is beneficial when compared with conventional postnatal closure of myelomeningocele, the Management of Myelomeningocele Study (MOMS) was initiated in 2003. MOMS is a prospective, randomized trial that is being completed at three U.S. centers. The primary end points include death and shunt placement. Secondary outcomes include leg function, bowel and bladder function, and intelligence, among others. MOMS was scheduled for completion in 2009.
Perinatal Management
Labor and Delivery
No difference in outcome between infants delivered vaginally or by cesarean section, with or without labor, was found in two retrospective studies.68,69 In contrast, a prospective study found that outcome differed depending on the type of delivery and the presence or absence of labor. The risk for neurological injury rose 2.2-fold if labor occurred.70 The evidence from these and other studies is not conclusive, but it suggests that the exposed neural placode may suffer trauma during labor. Neurological outcome may be improved by performing an elective cesarean section after lung maturity but before the onset of labor. The exceptions are infants who are not expected to survive because of other anomalies or chromosomal abnormalities.
Preoperative Evaluation and Management
Neurosurgical
During the initial neurological examination, the spinal level of the defect and the severity of hydrocephalus, if present, are determined. Care in interpreting abnormal reflexes is important because these reflexes may not reflect volitional function. For example, perineal stimulation may induce a contraction and spreading response that should not be mistaken for voluntary anal contraction. About two thirds of affected neonates show signs of myelopathy, including hyperreflexia and clonus, because they often have an incomplete functional spinal cord transection.71 To determine the sensory level, a stimulus is applied to the lower extremities distally to proximally until the infant grimaces. To determine the motor level, a stimulus is applied above the sensory level, and the distal-most voluntary motion is noted. With an L1-3 level, the infant has hip flexion with extended knees and clubfeet. The presence of intact hip adduction, hip flexion, and knee extension with inverted feet is indicative of an L2-4 level. With an L5-S2 level, the infant has hip adduction, knee extension, and knee flexion with dorsiflexed feet. Infants with a sacral level may appear intact except for weakness of plantar flexion and rocker-bottom feet. A flaccid pelvic floor and patulous anus are often present. Future anal sphincter function is not accurately predicted by the presence of an anal wink or sphincter tone.47 An underlying split cord malformation may cause kyphoscoliosis or significant orthopedic or neurological (more than one level) asymmetry between the lower extremities.72 Other stigmata of occult spinal bifida such as hypertrichosis, hemangioma, or hyperpigmented areas may suggest the presence of a tandem lesion.
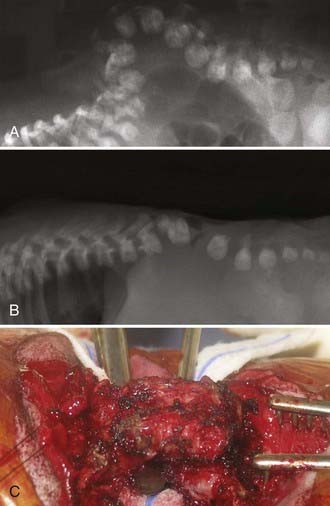
To determine ventricular size, a cranial ultrasound study is performed. Until the spinal defect is closed, relative microcephaly with small ventricles may be present. CT or MRI is not usually necessary before myelomeningocele repair. If severe kyphosis is present, plain radiographs of the spine are indicated (Fig. 214-6).
Although neonates are not generally symptomatic from the Chiari II malformation, the patient should be observed for central or obstructive apnea, stridor, and opisthotonos. If the infant has stridor, inspection of the vocal cords by direct laryngoscopy is necessary to document their function because bilateral vocal cord paralysis is predictive of a poor outcome.66
Myelomeningocele Repair
Timing
Myelomeningocele repair can be performed safely up to 72 hours after birth. This allows time for evaluation of the cardiopulmonary and genitourinary systems to be initiated and for cranial ultrasonography to be performed.1 Morbidity increases if repair is delayed beyond 72 hours. Ventriculitis occurs 5 times more frequently in infants who undergo delayed closure.1 In patients with delayed repair, shunt infection developed in about 75%, and the mortality was 13%.73 Cultures from the neural placode should be obtained and shown to have no growth before repair of the myelomeningocele if it is delayed beyond 72 hours after birth. If the cultures confirm infection, the neonate is treated by external ventricular drainage and appropriate antibiotics until the infection clears. Patients who undergo shunt placement before a delayed repair have a very high risk for shunt infection. Intellectual development is negatively affected by meningitis in the neonatal period.52
Hydrocephalus
Immediate CSF diversion is required in a small proportion (15%) of patients with spina bifida and severe hydrocephalus. If the patient has evidence of significant hydrocephalus at birth, CSF diversion before closure of the myelomeningocele should be considered to minimize pressure on the myelomeningocele dural closure. Insertion of a temporary drain or ventriculoperitoneal shunt is performed with the patient supine and the myelomeningocele protected in a doughnut-shaped sponge support. During the same anesthesia, the patient is repositioned prone for repair of the myelomeningocele. The risk for shunt infection with simultaneous shunt placement rises significantly if repair of the myelomeningocele is delayed. No significant difference in the shunt infection rate between a single-stage repair with shunt insertion (5 of 17, 29%) and a two-stage repair with shunt insertion (3 of 14, 21%) was found in a retrospective study.73 Nevertheless, concern for shunt infection may lead some neurosurgeons to perform the procedures at separate sittings.
Operative Technique
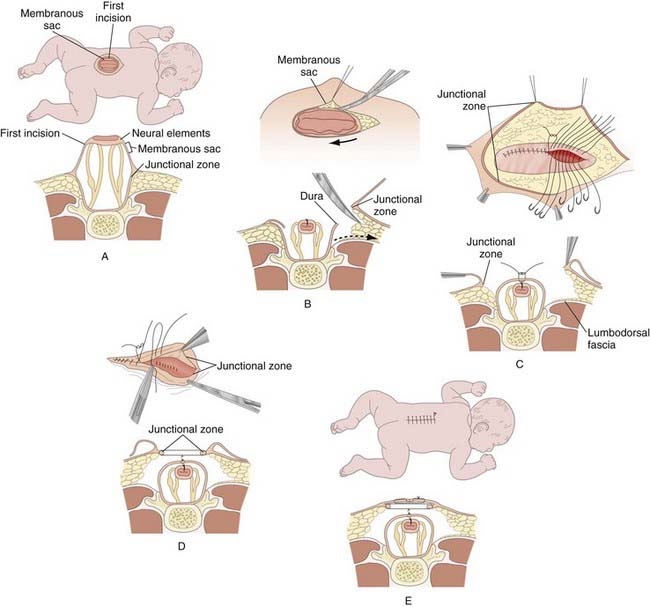
The purposes of myelomeningocele repair are to protect the functional spinal cord tissue, prevent loss of CSF, and minimize the risk for meningitis by reconstructing the neural tube and its coverings. The margin between the arachnoid of the neural placode and the dystrophic epidermis, or the junctional zone, is the site of the initial incision (Fig. 214-7). The goal is to free the neural placode from the surrounding junctional zone circumferentially. Effort is made to preserve all functional neural tissue. If indicated, any associated congenital lesions such as dermoid, lipoma, neurenteric cyst, myelomeningocele manqué (dorsal roots exiting from the cord or neural placode and aberrantly entering the dura), or lesions associated with split notochord syndrome are addressed. To minimize the risk for later development of a dermoid or lipoma, any residual epidermal or dermal elements are resected from the periphery of the neural placode.1 Using monofilament suture, the pia is closed if possible to re-form the neural placode into a tube because pial closure is thought to lower the incidence of retethering. The dura is mobilized and closed by identifying normal dura below the last intact lamina. It is then dissected free from surrounding tissue by developing a plane in the epidural space in a rostral-to-caudal direction bilaterally. To reconstruct the thecal sac, the dura is closed in the midline with nonabsorbable suture. Duraplasty with thoracolumbar fascia or another dural substitute is performed when necessary to prevent leakage of CSF. The thoracolumbar fascia is reapproximated in the midline. To obtain a closure without tension, releasing incisions in the fascia laterally away from the defect may be necessary. Absorbable sutures are used to close subcutaneous tissue in the midline; when these sutures are placed through the so-called junctional layer—the area where the dura had fused with the subcutaneous tissue—the surgeon can often use traction sufficient to draw the skin edges together. Although it is the authors’ preference to use nonabsorbable suture on the skin, many neurosurgeons prefer absorbable suture.
Kyphosis
Patients with thoracic spina bifida have a propensity for the development of kyphosis. About 15% of spina bifida patients have kyphosis at birth. If possible, spinal reconstruction for the kyphosis is delayed until the infant is older. In the presence of severe kyphosis (or gibbus deformity), kyphectomy may be necessary to allow successful wound closure (see Fig. 214-6).74 After kyphectomy, an orthotic device should be considered.75
Complications
Early Complications of Myelomeningocele Repair
The most common complication after repair of a myelomeningocele is superficial wound dehiscence.75 Particularly if the infant does not yet have a shunt, daily examination of head circumference, fontanelle fullness, and the back incision for CSF leakage should be performed. An external ventricular drain should be placed or periodic ventricular puncture should continue until CSF cultures show no growth if the repair has already developed a leak before shunt placement.75 Superficial wound infections are usually accompanied by systemic symptoms such as mild fever, ileus, and mild leukocytosis. Dressing changes and intravenous antibiotics are the recommended treatment.
Hydrocephalus
Shunt malfunctions develop frequently in children with spina bifida. At least one shunt revision occurs during the first year in almost half the shunted patients with spina bifida.76 More shunt complications occur in children with higher spinal lesions and more severe hydrocephalus. An alternative to CSF diversion with a shunt in some spina bifida patients is endoscopic third ventriculostomy.77 Endoscopic third ventriculostomy is not generally recommended for infants with spina bifida because the outcome is poor, but it can be considered as an alternative to a shunt revision if a child with spina bifida has a shunt malfunction later in life. In children with spina bifida who are evaluated for shunt malfunction and have appropriate ventricular anatomy, endoscopic third ventriculostomy is successful in the majority of patients, although some authors favor ventricular shunting because malfunctions may be more readily diagnosed.78
Chiari II Malformation
Although a symptomatic Chiari II malformation rarely occurs in infants, it is associated with relatively high (34% to 38%) mortality.79 The degree of caudal herniation of the cerebellum does not correlate well with the severity of the symptoms and signs of brainstem dysfunction.75 Intermittent obstructive or central apnea, cyanosis, bradycardia, dysphagia, nystagmus, stridor, vocal cord paralysis, torticollis, opisthotonos, hypotonia, upper extremity weakness, and spasticity are signs of a symptomatic Chiari II malformation. Twenty-three percent of children with spina bifida had symptoms from the Chiari II malformation at some point, but only 17% required hindbrain decompression.45 The average age at decompression was 14 months.
The manifestations of a symptomatic Chiari II malformation may vary with the child’s age. Within the first 3 months of life, a symptomatic Chiari II malformation develops in less than 20% of patients with spina bifida.75,80 Infants with a symptomatic Chiari II malformation can have swallowing difficulties (69%), apnea (58%), stridor (56%), aspiration (40%), arm weakness (27%), and opisthotonos (18%).79 Although neonatal symptoms of brainstem dysfunction may be a harbinger of irreversible neurological dysfunction, with rapid diagnosis and decompression, up to 69% of infants may become asymptomatic within 2 days.80
Hindbrain decompression should be performed early for a symptomatic Chiari II malformation to achieve the best outcome.80 Because the symptoms can be due to increased intracranial pressure, proper function of the shunt should be confirmed. If there is no shunt, an external ventriculostomy or shunt should be placed. If the symptoms are not relieved after 48 hours, hindbrain decompression should be performed.75 Urgent hindbrain decompression is indicated if a child has a functioning shunt and life-threatening symptoms. A poor prognosis is associated with preoperative bilateral vocal cord paralysis, severe neurogenic dysphagia, and prolonged apnea.
For planning the extent of the hindbrain decompression, MRI of the craniovertebral junction and cervical spine is helpful. Intraoperative ultrasound and determination of the extent of compression during exposure also aid in this decision (see Fig. 214-2). Cervical laminectomies may be all that is required to decompress the malformation. The foramen magnum is generally enlarged in these patients, and because the tentorium is so low, removal of occipital bone rarely creates significant additional room in these children. Narrow laminectomies, performed at levels that overlie the compressed cerebellum, may minimize the likelihood of future cervical kyphosis. Because of the low-lying torcular Herophili and the putative risk for hemorrhage associated with a complete dural opening in these infants, some surgeons have advocated only lysis of the compressive dural band and no duraplasty.
Latex Allergy
Many spina bifida patients acquire an allergy to latex, which can have life-threatening consequences if they experience a severe anaphylactic reaction. A 3.4% mortality rate has been reported in patients who have severe intraoperative complications from a type I hypersensitivity response (anaphylaxis) to latex.81 Latex antibodies can be detected in the serum in up to half (36% to 51%) of spina bifida patients.82,83 A correlation has been found between the number of surgeries that a patient has experienced and the incidence of latex allergy.75 In particular, a higher incidence of latex allergy was found in patients who had undergone more ventriculoperitoneal shunt revisions. More than a third (36%) of patients with a ventriculoperitoneal shunt were sensitized to latex in one study.82
Latex precautions are recommended for all patients with spina bifida. The first allergic reaction can be anaphylaxis. The incidence of latex reactions can be lowered to almost zero (0% to 0.3%) with appropriate precautions.84 Food allergies are also common in patients with latex allergy. Foods that can induce an allergic reaction, presumably by cross-reaction with the latex antibodies, include avocados, bananas, celery, chestnuts, kiwi, papaya, peaches, pears, and tomatoes.
Long-Term Follow-up
Children with spina bifida are born with a neurological deficit that should remain stable after the neonatal period. If there is progression of neurological, orthopedic, or urologic deficits, an evaluation of possible causes is recommended. For example, a tethered cord may be manifested as subtle changes, such as an increase in the frequency of urinary tract infections or progression of a foot deformity. In some infants, urologic deficits do not appear initially. In one study, half the children with myelomeningocele and normal bladder function near birth demonstrated abnormal function at 4 months.85 Continual urologic follow-up is recommended for almost all patients with spina bifida, even those with no perceptible neurological deficit in the lower extremities. Abnormal urination occurs in the vast majority (>90%) of patients with normal neurological function.86
In addition to addressing the neurological, orthopedic, and urologic problems, a spina bifida clinic can screen for other problems, such as pulmonary and sleep disorders, endocrine abnormalities, and neuropsychological issues. The importance of latex precautions can be reinforced. Learning disabilities, behavioral problems, and excessive weight gain can be addressed promptly to maximize the child’s cognitive, psychological, and social development. Kaufman and colleagues reported that disbanding of a multidisciplinary clinic caring for patients with myelomeningocele resulted in 45% to 66% of patients failing to continue with regular follow-up despite the availability of the same specialists in the same locations. They reported a concomitant increase in surgical procedures that could probably have been prevented with more structured outpatient care.87
Myelocystocele
Terminal myelocystoceles are skin-covered midline masses that occur in the lumbosacral area and are distinct from myelomeningoceles (Fig. 214-8). Myelocystoceles account for 5% of skin-covered masses and represent one of a spectrum of lesions that result from errors in secondary neurulation. Alternative terms for myelocystocele are syringocele, syringomyelocele, and lipomeningomyelocystocele. A terminal myelocystocele is composed of a low-lying conus medullaris with cystic dilation of the caudal central canal, a surrounding meningocele, and a lipoma that extends from the conus to a subcutaneous fat collection. The terminal intraconus cyst is lined with ependyma and dysplastic glia and does not communicate with the subarachnoid space. The meningocele communicates with the subarachnoid space and may lie dorsal or ventral to or circumferentially around the terminal cyst. The overlying skin may have a cutaneous signature typical of occult spina bifida, such as hemangioma, nevus, hypertrichosis, or dystrophic epithelium (see Figs. 214-3 and 214-8).
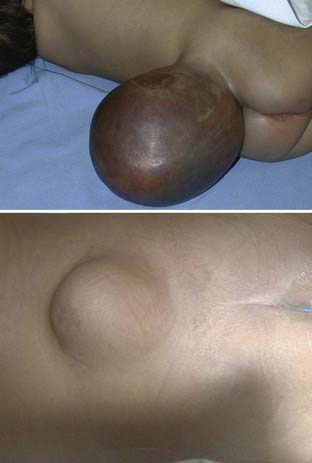
Myelocystoceles are often associated with cloacal exstrophy or other severe genitourinary anomalies. Cloacal exstrophy, also called vesicointestinal fissure, is a severe malformation of the abdomen and pelvis consisting of an omphalocele, bladder exstrophy, ambiguous genitalia, imperforate anus, and pelvic abnormalities. When all these abnormalities are present, the defect is called the OEIS complex (for omphalocele, bladder exstrophy, imperforate anus, and spinal defect). Almost two thirds (62%) of patients with cloacal exstrophy have some form of myelodysplasia.88
History
Although cloacal exstrophy with an associated myelocystocele was first described in 1709 by Littre, the first successful closure was not reported until 1960 by Rickham.89 One of four patients survived. A literature review in 1976 showed that the survival rate remained at 22%. With recent advances in surgical technique, most children survive but require extensive reconstructive surgery involving the sphincters and genitalia.88,90
Epidemiology
Myelocystoceles are extremely rare lesions. Cloacal exstrophy occurs sporadically, with an incidence of 1 in 200,000 to 400,000 live births.88 The change in prevalence that occurred with myelomeningoceles has not been noted with myelocystoceles, most likely because of the different causes of these lesions. No risk in siblings has been identified,91 although one case of two siblings with OEIS complex has been reported.92
Pathogenesis
As noted previously, myelocystoceles are often associated with cloacal exstrophy. During embryogenesis, the cloaca is an endodermally lined chamber that receives the hindgut and allantois. The cloacal membrane, a bilaminar veil of ectoderm and endoderm, divides the cloaca. Cloacal exstrophy occurs as a result of rupture of the cloacal membrane and failure of the lateral abdominal wall to close. According to one theory, herniation of the lower part of the abdomen and pelvis disrupts notochord secretion of inductive factors that regulate development of the neural tube and retrogressive differentiation.93 An alternative theory suggests that CSF is unable to exit from the early neural tube as it should and distends the distal terminal ventricle after canalization. The terminal ventricle distends the surrounding dorsal membrane and arachnoid and causes the surrounding meningocele.94 Neither of these theories have been verified experimentally. Teratogens that cause spina bifida aperta in hamsters, such as retinoic acid, also cause lesions resembling myelocystoceles. Most likely, the pathogenesis reflects both mechanical disruption of normal development and a lack of appropriate induction of differentiation factors.
Etiology
In contrast to the strong association between folate and myelomeningoceles, no such association with myelocystoceles is known, but this may reflect the rarity of these lesions. Loperamide hydrochloride, diphenylhydantoin, and retinoic acid have been associated with myelocystoceles. The influenza vaccine has been associated with OEIS complex. No genetic association has been identified.91
Prenatal Diagnosis
The AFP level is not usually elevated with myelocystocele because it is a skin-covered lesion, but if there is a concurrent omphalocele, AFP is frequently elevated. A myelocystocele can be diagnosed with high-resolution fetal ultrasonography or prenatal MRI.95 The primary lesion in the differential diagnosis is sacrococcygeal teratoma, which is characterized by the presence of polyhydramnios and intact vertebrae. By contrast, with a myelocystocele associated with OEIS complex, other abnormalities are often visualized, including hemivertebrae, sacral agenesis, a small pelvic cavity, a flared iliac bone, and no bladder.91 Prenatal diagnosis allows the family to prepare, if necessary, for a scheduled cesarean section and for the multiple issues that must be addressed if cloacal exstrophy is present.
Operative Repair
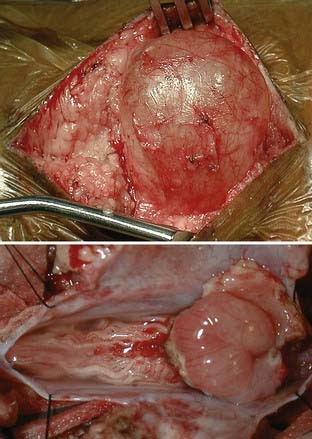
The same general principles are used for repair of myelocystocele as for repair of other myelodysplasias. The patient is positioned prone, and electrodes for electromyography are placed if intraoperative monitoring is desired. Either of two similar strategies may be used when approaching the lesion, and extensive understanding of the dysraphic anatomy is required. Through a midline incision, the most caudal intact lamina is identified and removed to localize the normal thecal sac. The myelocystocele sac and dura are then circumferentially dissected away from the surrounding soft tissue (Fig. 214-9). This usually requires release of the dorsal fibrous band that connects the most cranial bifid lamina across the midline. The normal dura can then be visualized as it continues caudally over the meningocele. The dura and arachnoid are opened at the level of the laminectomy, and the incision proceeds caudally over the myelocystocele sac to allow visualization of the dilated spinal cord, terminal lipoma, and associated nerve roots that traverse the meningocele and reenter the spinal canal. Careful dissection through the fat caudal to the meningocele will reveal the terminal cyst, which can then be opened in the midline, caudal to the emerging nerve roots. At this point, the terminal spinal cord is inspected caudally and dorsally for tethering elements, which are released if encountered. The wall of the cyst and associated lipoma can then be resected distal to the caudal arachnoid reflection. The free edges of the remaining sac may be approximated and the residual dura closed primarily to reestablish the thecal sac. The soft tissues are then closed in layers. Although a significant fascial defect may be present, there is sufficient overlying skin to complete a primary closure.
Bruner J, Tulipan N, Paschall R, et al. Fetal surgery for myelomeningocele and the incidence of shunt-dependent hydrocephalus. JAMA. 1999;282:1819-1825.
Dias Dias MS, Partington M. Embryology of myelomeningocele and anencephaly. Neurosurg Focus. 2004;16(2):E1.
Kaufman BA, Terbrock A, Winters N, et al. Disbanding a multidisciplinary clinic: effects on the health care of myelomeningocele patients. Pediatr Neurosurg. 1994;21:36-44.
Lewis D, Van Dyke D, Stumbo P, et al. Drug and environmental factors associated with adverse pregnancy outcomes. Part I. Anti-epileptic drugs, contraceptives, smoking, and folate. Ann Pharmacother. 1998;32:802-817.
McLone D. The cause of Chiari II malformation: a unified theory. Pediatr Neurosci. 1989;15:1-12.
McLone D, Czyzewsky D, Raimondi A. Central nervous system infections as a limiting factor in the intelligence of children with myelomeningocele. Pediatrics. 1980;70:338-342.
Tulipan N. Intrauterine closure of myelomeningocele: an update. Neurosurg Focus. 2004;16(2):E2.
Worley G, Schuster J, Oakes W. Survival at 5 years of a cohort of newborn infants with myelomeningocele. Dev Med Child Neurol. 1996;38:816-822.
1 McLone D. Care of the neonate with a myelomeningocele. Neurosurg Clin N Am. 1998;9:111-120.
2 American Academy of Pediatrics Committee on Genetics. Folic acid for the prevention of neural tube defects. Pediatrics. 1999;104:325-327.
3 Dias MS, Partington M. Embryology of myelomeningocele and anencephaly. Neurosurg Focus. 2004;16(2):E1.
4 Dias M, Walker M. The embryogenesis of complex dysraphic malformations: a disorder of gastrulation. Pediatr Neurosurg. 1992;18:229-253.
5 Gilbert J, Jones K, Rorke L, et al. Central nervous system anomalies associated with meningomyelocele, hydrocephalus, and the Arnold-Chiari malformation: reappraisal of theories regarding the pathogenesis of posterior neural tube closure defects. Neurosurgery. 1986;18:559-564.
6 LaMarca F, Hermen M, Grant SA, et al. Presentation and management of hydromyelia in children with Chiari type-II malformation. Pediatr Neurosurg. 1997;26:57-67.
7 Kozma C. Skeletal dysplasia in ancient Egypt. Am J Med Genet A. 2008;146A:3104-3112.
8 Forestus P. De capitis et cerebre morbis ac symptomatis. Observationum Medicinalium, libri III. Leiden: Ex Off Plantiniana; 1587.
9 Forestus P. Observation chir, libri V, lib III, obs VII, 1610.
10 Tulpius N. Cum atieis figuris. Observationum Medicinalium, libra III, vol cap XXIX, XXX. Amsterdam: Apud Ludovican Elzevirium; 1641.
11 Morgagni J-B. De sedubis et causis morborum per anatomen indigatis, libri V. Venice: Ex Typographia Remondiona; 1761.
12 Lebedeff A. Uber che Enstehurg der Anencephalie und Spina Bifida dei Vogelin und Menschen. Virchows Arch Pathol Anat Physiol. 1881;86:263.
13 Von Recklinghausen F. Untersuchungen uber che Spina bifida. Arch Pathol Anat. 1886;105:243.
14 Fraser J. Spina bifida. Edinb Med J. 1929;36:284.
15 Lorber J. Results of treatment of myelomeningocele: An analysis of 524 unselected cases with special references to possible selection of treatment. Dev Med Child Neurol. 1971;13:279-303.
16 Lorber J, Salfield S. Results of selective treatment of spina bifida cystica. Arch Dis Child. 1981;56:822-830.
17 McLone D. Results of treatment of children born with a myelomeningocele. Clin Neurosurg. 1983;30:407-412.
18 Lary JM, Edmonds LD. Prevalence of spina bifida at birth—United States, 1983-1990: A comparison of two surveillance systems. MMWR CDC Surveill Summ. 1996;45(2):15-26.
19 Cragan J, Roberts H, Edmonds L, et al. Surveillance for anencephaly and spina bifida and the impact of prenatal diagnosis—United States, 1985-1994. MMWR Morb Mortal Wkly Rep. 1995;44(SS-4):1-13.
20 Roberts H, Moore C, Cragan J, et al. Impact of prenatal diagnosis on the birth prevalence of neural tube defects. Atlanta, 1990-1991. Pediatrics. 1995;96:880-883.
21 VanDorsten J, Hulsey T, Newman R, et al. Fetal anomaly detection by second-trimester ultrasonography in a tertiary center. Am J Obstet Gynecol. 1998;178:742-749.
22 Shurtleff D, Lemire R. Epidemiology, etiologic factors, and prenatal diagnosis of open spinal dysraphism. Neurosurg Clin N Am. 1995;6:183-193.
23 Greene W, Terry R, DeMasi R, et al. Effect of race and gender on neurological level in myelomeningocele. Dev Med Child Neurol. 1991;33:110.
24 Reis JL, Correia-Pinto J, Monteiro MP, et al. Vascular and apoptotic changes in the placode of myelomeningocele mice during the final stages of in utero development. J Neurosurg Pediatr. 2008;2:150-157.
25 McLone D. The cause of Chiari II malformation: a unified theory. Pediatr Neurosci. 1989;15:1-12.
26 Centers for Disease Control and Prevention (CDC). Use of folic acid for prevention of spina bifida and other neural tube defects—1983-1991. MMWR Morb Mortal Wkly Rep. 1991;40(30):513-516.
27 Lewis D, Van Dyke D, Stumbo P, et al. Drug and environmental factors associated with adverse pregnancy outcomes. Part III. Folic acid: pharmacology, therapeutic recommendations, and economics. Ann Pharmacother. 1998;32:1087-1095.
28 Recommendations for the use of folic acid to reduce the number of cases of spina bifida and other neural tube defects. MMWR Recomm Rep. 1992;41(RR-14):1-7.
29 Lewis D, Van Dyke D, Stumbo P, et al. Drug and environmental factors associated with adverse pregnancy outcomes. Part I. Anti-epileptic drugs, contraceptives, smoking, and folate. Ann Pharmacother. 1998;32:802-817.
30 Shaw G, Velie E, Schaffer S. Risk of neural tube defect—affected pregnancies among obese women. JAMA. 1996;275:1093-1096.
31 Werler M, Louik C, Shapiro S, et al. Prepregnant weight in relation to risk of neural tube defects. JAMA. 1996;275:1089-1092.
32 Main D, Mennuti M. Neural tube defects: issues in prenatal diagnosis and counseling. Obstet Gynecol. 1986;67:1-16.
33 Wald N, Cuckle H, Brock J, et al. Maternal serum-alpha-fetoprotein measurement in antenatal screening for anencephaly and spina bifida in early pregnancy: report of UK collaborative study on alpha-fetoprotein in relation to neural-tube defects. Lancet. 1977;1:1323-1332.
34 Park T. Myelomeningocele. In: Albright L, Pollack I, Adelson D, editors. Principles and Practice of Pediatric Neurosurgery. New York: Thieme Medical; 1999:291-320.
35 Kollias S, Goldstein R, Cogen P, et al. Prenatally detected myelo-meningoceles: sonographic accuracy in estimation of the spinal level. Radiology. 1992;185:109-112.
36 Nicolaides K, Campbell S, Gabbe S, et al. Ultrasound screening for spina bifida: cranial and cerebellar signs. Lancet. 1986;2:72-74.
37 Nyberg D, Mack L, Hirsch J, et al. Abnormalities of fetal contour in sonographic detection of spina bifida: evaluation of the “lemon sign.”. Radiology. 1988;167:387-392.
38 Babook C, Goldstein R, Barth R, et al. Prevalence of ventriculomegaly in association with myelomeningocele: correlation with gestational age and severity of posterior fossa deformity. Radiology. 1994;190:703-707.
39 Babook C, Drake C, Goldstein R. Spinal level of fetal myelomeningocele: does it influence ventricular size. AJR Am J Roentgenol. 1997;169:207-210.
40 Lee CS, Hong SH, Wang KC, et al. Fetal ventriculomegaly: prognosis in cases in which prenatal neurosurgical consultation was sought. J Neurosurg. 2006;105:265-270.
41 Glenn OA, Barkovich AJ. Magnetic resonance imaging of the fetal brain and spine: an increasingly important tool in prenatal diagnosis, part 1. AJNR Am J Neuroradiol. 2006;27:1604-1611.
42 Loft A, Hogdall E, Larsen S, et al. A comparison of amniotic fluid alpha-fetoprotein and acetylcholinesterase in the prenatal diagnosis of open neural tube defects and anterior abdominal wall defects. Prenat Diagn. 1993;13:93-109.
43 Aitken D, Morrison N, Ferguson-Smith M. Predictive value of amniotic acetylcholinesterase analysis in the diagnosis of fetal abnormality in 3700 pregnancies. Prenat Diagn. 1984;4:329-340.
44 Wald N, Cuckle H, Nanchahal K. Amniotic fluid acetylcholinesterase measurement in the prenatal diagnosis of open neural tube defects: second report of the Collaborative Acetylcholinesterase Study. Prenat Diagn. 1989;9:813-829.
45 Worley G, Schuster J, Oakes W. Survival at 5 years of a cohort of newborn infants with myelomeningocele. Dev Med Child Neurol. 1996;38:816-822.
46 Coniglio S, Anderson S, Ferguson J. Functional motor outcome in children with myelomeningocele: Correlation with anatomic level on prenatal ultrasound. Dev Med Child Neurol. 1996;38:675-680.
47 Hahn Y. Open myelomeningocele. Neurosurg Clin N Am. 1995;6:231-241.
48 Swank M, Dias L. Myelomeningocele: a review of the orthopaedic aspects of 206 patients treated from birth with no selection criteria. Dev Med Child Neurol. 1992;34:1047-1052.
49 Steinbok P, Irvine B, Cochrane D, et al. Long-term outcome and complications of children born with meningomyelocele. Childs Nerv Syst. 1992;8:92-96.
50 Bier J, Morales Y, Liebling J, et al. Medical and social factors associated with cognitive outcome in individuals with myelomeningocele. Dev Med Child Neurol. 1997;39:263-266.
51 Coniglio S, Anderson S, Ferguson J. Developmental outcomes of children with myelomeningocele: prenatal predictors. Am J Obstet Gynecol. 1997;177:319-326.
52 McLone D, Czyzewsky D, Raimondi A. Central nervous system infections as a limiting factor in the intelligence of children with myelomeningocele. Pediatrics. 1980;70:338-342.
53 Jensen P. Psychological aspects of myelomeningocele—a longitudinal study. Scand J Psychol. 1987;28:313-321.
54 Toriello H, Higgins J. Occurrence of neural tube defects among first-, second-, and third-degree relatives of probands: results of a United States study. Am J Med Genet. 1983;15:601-606.
55 McBride M. Sib risk of anencephaly and spina bifida in British Columbia. Am J Med Genet. 1979;3:377-387.
56 Carter C, Evans K. Children of adult survivors with spina bifida cystica. Lancet. 1973;2:924-926.
57 Myriathopolous N, Melnick M. Studies in neural tube defects. I. Epidemiologic and etiologic aspects. Am J Med Genet. 1987;26:783-796.
58 Meuli M, Meuli-Simmen C, Hutchins G, et al. The spinal cord defect in human fetuses with myelomeningocele: implications for fetal surgery. J Pediatr Surg. 1997;32:448-452.
59 Michejda M. Intrauterine treatment of spina bifida: primate model. Z Kinderchir. 1984;39:259-261.
60 Heffez D, Aryanpur J, Hutchins G, et al. The paralysis associated with myelomeningocele: clinical and experimental data implicating a preventable spinal cord injury. Neurosurgery. 1990;26:987-992.
61 Heffez D, Aryanpur J, Cuello-Rotellini N, et al. Intrauterine repair of experimental surgically created dysraphism. Neurosurgery. 1993;32:1005-1010.
62 Meuli M, Meuli-Simmen C, Yingling C, et al. Creation of myelo-meningocele in utero: a model of functional damage from spinal cord exposure in fetal sheep. J Pediatr Surg. 1995;30:1028-1033.
63 McLone D, Dias M, Goosens W, et al. Pathological changes in exposed neural tissue of fetal delayed splotch (Spd) mice. Childs Nerv Syst. 1997;13:1-7.
64 Bruner J, Tulipan N, Paschall R, et al. Fetal surgery for myelomeningocele and the incidence of shunt-dependent hydrocephalus. JAMA. 1999;282:1819-1825.
65 Tulipan N. Intrauterine closure of myelomeningocele: an update. Neurosurg Focus. 2004;16(2):E2.
66 Johnson MP, Gerdes M, Rintoul N, et al. Maternal-fetal surgery for myelomeningocele: neurodevelopmental outcomes at 2 years of age. Am J Obstet Gynecol. 2006;194(4):1145-1150.
67 Carr MC. Fetal myelomeningocele repair: urologic aspects. Curr Opin Urol. 2007;17:257-262.
68 Bensen J, Dillard R, Burton B. Open spina bifida: does cesarean section delivery improve prognosis? Obstet Gynecol. 1988;71:532-534.
69 Cochrane D, Aronyk K, Sawatzky B, et al. The effects of labor and delivery on spinal cord function and ambulation in patients with meningomyelocele. Childs Nerv Syst. 1991;7:312-315.
70 Luthy D, Wardinsky T, Shurtleff D, et al. Cesarean section before the onset of labor and subsequent motor function in infants with meningomyelocele diagnosed antenatally. N Engl J Med. 1991;324:662-666.
71 Stark G. Neonatal assessment of the child with a myelomeningocele. Arch Dis Child. 1971;46:539-548.
72 Hankinson TC, Klimo P, Feldstein NA, et al. Chiari malformations, syringomyelia and scoliosis. Neurosurg Clin N Am. 2007;18:549-568.
73 Gamache F. Treatment of hydrocephalus in patients with meningomyelocele or encephalocele: a recent series. Childs Nerv Syst. 1995;11:487-488.
74 Reigel D. Kyphectomy and myelomeningocele repair. In: Ransohoff J, editor. Modern Techniques in Surgery, vol 13, Neurosurgery. Mount Kisco, NY: Futura; 1979:1-9.
75 Pang D. Surgical complications of open spinal dysraphism. Neurosurg Clin N Am. 1995;6:243-257.
76 Caldarelli M, DiRocco C, LaMarca F. Shunt complications in the first postoperative year in children with meningomyelocele. Childs Nerv Syst. 1996;12:748-754.
77 Cohen AR, Perneczky A. Endoscopy and the management of third ventricular lesions. In: Apuzzo M, editor. Surgery of the Third Ventricle. Baltimore: Williams & Wilkins; 1998:889-936.
78 Marlin AE. Management of hydrocephalus in the patient with myelomeningocele: an argument against third ventriculostomy. Neurosurg Focus. 2004;16(2):E4.
79 Park T, Hoffman H, Hendrick E, et al. Experience with surgical decompression of the Arnold-Chiari malformation in young infants with myelomeningocele. Neurosurgery. 1983;13:147-152.
80 Pollack I, Kinnunen D, Albright A. The effect of early craniocervical decompression on functional outcome in neonates and young infants with myelodysplasia and symptomatic Chiari II malformations: results from a prospective series. Neurosurgery. 1996;38:703-710.
81 Mazagri R, Ventureyra E. Latex allergy in neurosurgical practice. Childs Nerv Syst. 1999;15:404-407.
82 Nieto A, Estornell F, Mazon A, et al. Allergy to latex in spina bifida: a multivariate study of associated risk factors in 100 consecutive patients. J Allergy Clin Immunol. 1996;98:501-507.
83 Cremer R. The role of shunted hydrocephalus in the development of allergy to latex in patients with spina bifida [letter]. J Allergy Clin Immunol. 1997;100:719.
84 Holzman R. Clinical management of latex-allergic children. Anesth Analg. 1997;85:529-533.
85 Sillen U, Hansson E, Hermansson G, et al. Development of the urodynamic pattern in infants with myelomeningocele. Br J Urol. 1996;78:596-601.
86 Mevorach R, Bogaert G, Baskin L, et al. Lower urinary tract function in ambulatory children with spina bifida. Br J Urol. 1996;77:593-596.
87 Kaufman BA, Terbrock A, Winters N, et al. Disbanding a multidisciplinary clinic: effects on the health care of myelomeningocele patients. Pediatr Neurosurg. 1994;21:36-44.
88 Davidoff A, Hebra A, Balmer D, et al. Management of the gastro-intestinal tract and nutrition in patients with cloacal exstrophy. J Pediatr Surg. 1996;31:771-773.
89 Rickham P. Vesico-intestinal fissure. Arch Dis Child. 1960;35:97-102.
90 Muthukumar N. Terminal and nonterminal myelocystoceles. J Neurosurg. 2007;107(2 suppl):87-97.
91 Chen C-P, Shih S-L, Liu F-F, et al. Perinatal features of omphalocele-exstrophy–imperforate anus–spinal defects (OEIS complex) associated with large meningomyeloceles and severe limb defects. Am J Perinatol. 1997;14:275-279.
92 Smith N, Chambers H, Furness M, et al. The OEIS complex (omphalocele-exstrophy–imperforate anus–spinal defects): recurrence in sibs. J Med Genet. 1992;29:730-732.
93 Cohen A. The mermaid malformation: cloacal exstrophy and occult spinal dysraphism. Neurosurgery. 1991;28:834-843.
94 McLone D, Naidich T. Terminal myelocystocele. Neurosurgery. 1985;16:36-43.
95 Yu JA, Sohaey R, Kennedy AM, et al. Terminal myelocystocele and sacrococcygeal teratoma: a comparison of fetal ultrasound presentation and perinatal risk. AJNR Am J Neuroradiol. 2007;28:1058-1060.






