[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 28 Myeloma and Leukemia
Myeloma
Epidemiology and Risk Factors
Multiple myeloma is a malignant disorder that results from a clonal proliferation of marrow plasma cells that usually results in the production of a monoclonal immunoglobulin in the serum or urine. Approximately 20,180 patients are diagnosed, and 10,650 die, annually in the U.S. with this disorder, making it the second most common hematologic malignancy in the United States.1 The disease most commonly occurs during the seventh and eighth decades of life, is more frequent in men (1.6:1), and has an incidence in African Americans over twice as high as that noted in whites.1
There is no undisputable risk factor for myeloma, but ionizing radiation or work in the paper, pulp, and leather tanning industries and other chemical exposures have been implicated.2 Numerous reports in people with chronic exposure to low-dose ionizing radiation exist, including radiology technicians/physicians prior to routine shielding and radium watch dial painters; perhaps the best evidence was among atomic bomb survivors at Hiroshima in an initial report, but the follow-up of this report failed to confirm those initial findings.3 Similarly, benzene has been implicated, but a thorough retrospective analysis of available reports eliminated this factor as well.4 Hereditary factors have not previously played a prominent role as risk factors for multiple myeloma, but recent reports suggest that at least among certain populations the incidence of monoclonal gammopathy of unknown significance (MGUS)/myeloma may have a familial/genetic association.5
Pathology
Myeloma begins with the clonal expansion of a malignant plasma cell that is usually positive for CD38, CD138, and monoclonal cytoplasmic immunoglobulin with a clonal light chain of either kappa or lambda type. These cells typically have an eccentric nucleus within a large cytoplasm and often have an area of central clearing that corresponds to intracytoplasmic immunoglobulin. Cells of plasmacytic origin are usually negative for other markers of the B-cell lineage such as CD19 and CD20. Recent studies suggest different categories of myeloma, which may separate disease into different levels of outcome.6 Although a heterogeneous array of cytogenetic abnormalities have been described in patients, deletions of chromosomes 13 or 17 p, certain translocations involving chromosome 14, on which the immunoglobulin H (IgH) locus resides [t(4;14), t(14;16)], and chromosome 1 abnormalities have predicted shortened survival; [t(11;14)] has usually been associated with either an average or an improved prognosis and rarely noted to have a subgroup with poor prognosis.6
The abnormal clonal proliferation usually results in the production of monoclonal immunoglobulin in the serum and/or urine; IgG is secreted in approximately 60%, IgA in 20%, IgD in 2%, and IgE in less than 1%; biclonal secretion is rare. Secretion of light chain as the sole monoclonal protein is noted in 18%.7 Previously, 3% of patients were noted to have nonsecretory disease; this number has declined with the advent of serum free light chain (kappa, lambda free kappa:lambda) detection.
Clinical Presentation
Bone disease occurs in nearly 70% of newly diagnosed patients with multiple myeloma and results, in part, from RANKL overexpression and OPG inhibition resulting in unbridled activation of osteoclasts. Other cytokines such as interleukin-1beta (IL-1β), IL-6, and tumor necrosis factor-alpha (TNF-α) also have a role in lytic bone disease.8,9 Progressive bone destruction results in hypercalcemia in approximately 20% of patients with newly diagnosed myeloma.
Marrow infiltration with plasma cells results in a normocytic, normochromic anemia in the majority of patients with previously untreated myeloma. Recently, up-regulation of hepcidin mRNA in myeloma patients has been noted and may play a causative role in this complication.10 In patients with high levels of circulating monoclonal serum protein, stacking of red blood cells on the peripheral blood smear, known as rouleaux formation, may occur.
Nearly one half of patients will develop renal failure at some time during the course of their disease. Cast nephropathy of the distal tubule is the most common cause of this complication, but hypercalcemia, dehydration, hyperuricemia, and concomitant conditions such as amyloidosis and light or heavy chain deposition diseases may also be contributing factors.11
Frequent bacterial infections may also be noted in patients with myeloma due to suppression of uninvolved immunoglobulins, decreased antibody response, and impaired opsonization among other abnormalities reflective of the impaired immune response in patients with multiple myeloma.12,13
Although the diagnosis of myeloma by fixed criteria may be difficult, the International Myeloma Working Group (IMWG) proposed a simple system based on the presence of 10% or greater clonal marrow plasma cells and/or the presence of a serum monoclonal protein 3.0 g/dL or greater.14 The same group developed the “CRAB” (HyperCalcemia, Renal Failure, Anemia, Bone lesions) criteria to determine whether there has been end-organ damage that would classify a patient as having symptomatic disease that would require initiation of treatment.14 These criteria include elevated serum Calcium (≥11.5 mg/dL), Renal insufficiency (creatinine > 2 mg/dL), Anemia (>2 g/dL below the lower limit of normal or < 10 g/dL), Bone lesions (lytic disease or osteoporosis with compression fractures), or other complications (hyperviscosity, amyloidosis, and light chain deposition diseases). Patients without any of these complications are considered asymptomatic and may be observed, without treatment, with frequent follow-up until progression to symptomatic disease.
It is important to distinguish multiple myeloma from related plasma cell dyscrasias such as MGUS and solitary bone plasmacytoma (SBP) or extramedullary plasmacytoma (EMP). The diagnosis of MGUS relies on the presence of fewer than 10% marrow clonal plasma cells and a monoclonal serum protein of less than 3 g/dL in a patient who has no evidence of end-organ/tissue damage, including anemia, hypercalcemia, renal failure, or lytic bone lesions attributable to myeloma and no evidence of a B-cell proliferative disorder.14 SBP or EBP is diagnosed when a single lesion of biopsy-proven clonal plasma cells is detected without evidence of other disease including negative magnetic resonance imaging (MRI) of the spine and bone marrow examination; it is particularly important to identify these patients because 35% to 65% may be curable when given radiation therapy with curative intent.15
Key Points Monoclonal gammopathy of unknown significance
<10% clonal marrow plasma cells
AND serum monoclonal protein less than 3.0 g/dL
• Single lesion of biopsy-proven clonal plasma cells without other evidence of myeloma including negative MRI of the spine and negative bone marrow aspiration.
Staging
Numerous factors have been suggested to determine the prognosis of patients with myeloma. In the 1970s, Durie and Salmon16 proposed a staging system based on the degree of anemia, level of paraprotein, presence or absence of hypercalcemia, lytic lesions, and renal insufficiency that subsequently became the standard staging system for myeloma for several decades (Table 28-1). Although this system was predictive of the degree of myeloma tumor mass, several components were imprecise, such as extent of bone lesions and the inclusion of factors such as anemia, hypercalcemia, and renal function, which may be affected by factors other than myeloma infiltration. Subsequently, multiple new prognostic factors emerged including plasma cell hypodiploidy, C-reactive protein, lactate dehydrogenase, labeling index, cytogenetic abnormalities, and beta-2 microglobulin (β2M).
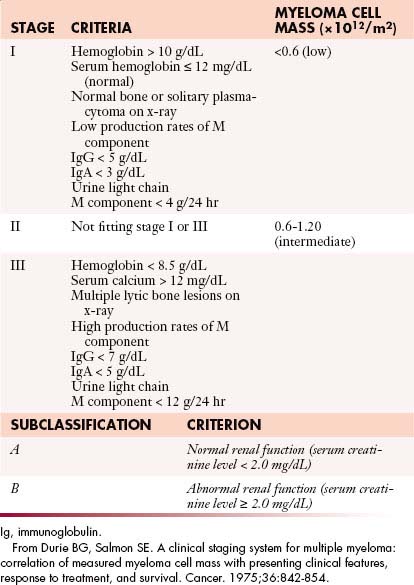
In 2005, Greipp and coworkers17 proposed the International Staging System (ISS) that separated patients into three stages based on the level of serum albumin and β2M. The data from 10,750 patients were analyzed by univariate and multivariate analysis; the system was designed using half the data set and validated in the other half of the population (Table 28-2). This system effectively identified a group of patients with a relatively short median survival of 29 months (β2M ≥ 5.5 mg/L, stage III), another with longer median survival of 62 months (β2M < 3.5 mg/L and albumin > 3.5 g/dL, stage III), as well as a group with intermediate survival (stage II). Although some patients in the data set may have received thalidomide, the impact on survival of bortezomib, lenalidomide, and multiple combinations utilizing both of these drugs will likely lead to improved median survivals for one or more stages defined by the ISS; the Greek Myeloma Study Group also confirmed the validity of the ISS in the era of novel agents.18 The ISS has subsequently become a standard staging system for patients with myeloma.
Table 28-2 International Staging System for Multiple Myeloma
| STAGE | CRITERIA |
|---|---|
| 1 | β2M < 3.5 mg/L Albumin ≥ 3.5 g/dL |
| 2 | β2M 3.5-5.5 mg/L or β2M < 3.5 mg/L and Albumin < 3.5 g/dL |
| 3 | β2M > 5.5 mg/L |
β2M, beta-2 microglobulin.
From Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23:3412-3420.
Although the Durie-Salmon system and ISS effectively separate patients into different survival categories, the impact of chromosomal abnormalities on prognosis is significant, and some have even suggested a molecular classification of myeloma.6 Certain translocations involving chromosome 14 [t(4;14), t(14;16)] and deletion of chromosome 13 have previously predicted a poor prognosis, but treatment with bortezomib appears to overcome the prognostic impact of these abnormalities; the same data with lenalidomide are premature and need to be validated over time.6,19,20 Deletion of chromosome 17p remains prognostic even in the era of novel agents; other considerations include the poor prognosis of chromosome 1 abnormalities and the possibility of an improved prognosis with t(11;14).6 Although no staging system based on chromosomal abnormalities has been specifically agreed upon, the aforementioned considerations are essential for the determination of prognosis and appropriate treatment decisions.
Imaging
Goals
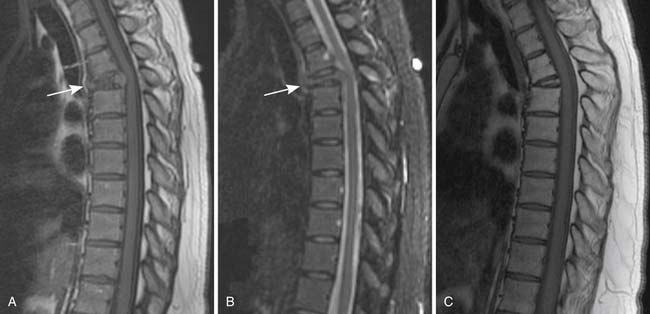
When a patient is suspected of having an isolated plasmacytoma, imaging initially serves two roles. The first is similar to its role in any focal solid tumor—to define the anatomy of the tumor and its relationship to other structures in order to assist with local treatment planning, which will usually be radiation therapy with curative intent. The second is to determine whether the seemingly isolated plasmacytoma is the only site of disease or whether it is merely the tip of the iceberg in a patient who should properly be classified as having multiple myeloma. To that end, one may weigh and choose between the various forms of systemic imaging, to be discussed later. If no other tumor is found elsewhere, and the patient remains classified as having solitary plasmacytoma, then follow-up imaging also serves two purposes. The first is again similar to its role in other solid tumors—to evaluate the site of the primary disease for evidence of healing or, alternatively, recurrence or progression (Figure 28-1). The second is surveillance for development of multiple myeloma, again by use of systemic imaging along with other criteria.
For patients with multiple myeloma, imaging helps at initial evaluation to distinguish those with asymptomatic or smoldering myeloma, who have no visible bone disease, from those with symptomatic myeloma. Once the patient has been categorized into the symptomatic or asymptomatic group, imaging is used to assess stability versus progression of disease.
Key Points Radiology report
• Identify and characterize focal bone lesions: Number, location, and size when practical. (For multiple lesions, it is often impractical to discuss each separately.)
• Clearly indicate when a bone lesion is thought due to disease other than myeloma, such as a geode due to degenerative joint disease.
• Discuss specific lesions with significantly elevated risk of pathologic fracture.
• Assess subjective bone mineralization—normal, decreased absolutely, decreased allowing for age and gender.
• Look for and discuss compression fractures in the spine.
• Follow-up examinations: Indicate whether the disease is stable or progressive or shows signs of healing to include resolution of fluoro-2-deoxy-D-glucose (FDG) avidity at positron-emission tomography/computed tomography (PET/CT) or decrease in contrast enhancement at MRI.
Techniques
Conventional Radiography
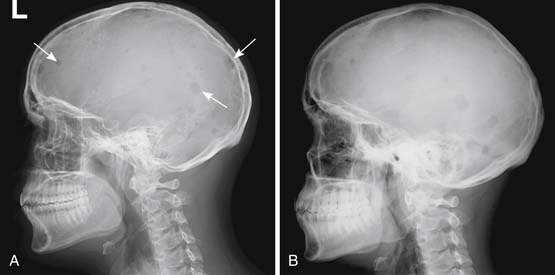
Skeletal surveys have for decades been a mainstay of imaging in multiple myeloma and related diseases and are included in the original Durie and Salmon staging system.16 They consist of a series of images intended to include all the bones that have a reasonable likelihood of developing visible signs of multiple myeloma. There is no generally agreed-upon grouping of images to be included in a skeletal survey, and so there will be variation from one institution to another as to exactly what is included. Based on observation not only of our own practice pattern but also of the assortment of images obtained at outside institutions and then reviewed within our institution, the minimum number and type of views might reasonably be lateral skull, lateral cervical spine, frontal and lateral thoracic and lumbar spine, frontal chest or ribs, frontal pelvis, and frontal views of each humerus and femur. That would be 12 views. The assortment of images suggested by the IMWG21 is listed in a key point text box later. Whole body radiography has been suggested as an alternative to the individual views obtained with skeletal surveys.22
Skeletal surveys have the advantage of being relatively cheap and easy to perform using widely available equipment. They are adequate for finding lytic bone lesions that have reached a sufficient size. Generally that means loss of 30% to 50% of the bone mineralization.23,24 For an individual lesion, the size needed for it to be visible will depend greatly on which bone is involved and where the lesion is in the bone. For example, if a femoral lesion is located along the lateral or medial margin of the bone, it will erode the endosteal surface of the cortex and create a scalloped border that will be noticeable earlier than a similarly sized lesion located either anteriorly or posteriorly, assuming that the bone is being imaged in the frontal projection. Lesions in the pelvis and sacrum often have to attain a size of several centimeters before they are noticed because overlying bowel gas may mask them. In the skull, the presence of normal lucencies due to venous lakes can make it difficult to decide whether a particular lucent area is normal or abnormal.
Skeletal surveys are obtained as part of the initial diagnostic workup of patients with MGUS or suspected SBP because the presence of unexpected lytic bone lesions would necessitate recategorization, probably to multiple myeloma. Skeletal surveys are also obtained in patients with myeloma to help determine the stage of disease when diagnosed. If the skeletal survey shows lytic lesions, more advanced imaging may be unnecessary. The skeletal survey is also useful for searching for progression after treatment.25
Skeletal surveys and other conventional radiographs can reveal disease progression but are of limited utility in assessing response to therapy because lytic bone lesions heal very slowly, if at all24 (Figures 28-2 and 28-3).
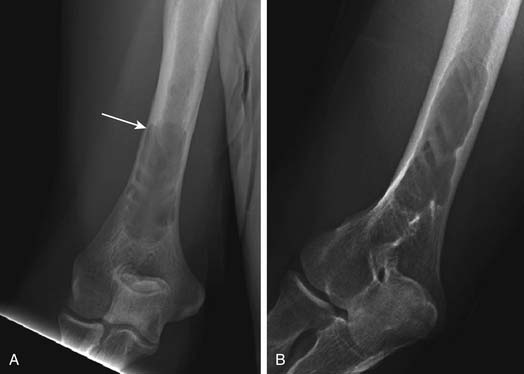
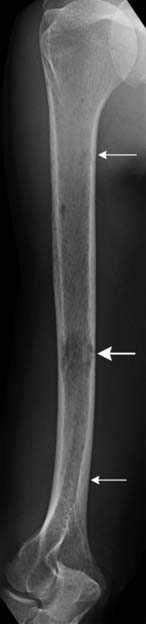
Another use of conventional radiography is in assessing the risk of pathologic fracture once a lytic lesion has been identified by any means. For this one usually wants both frontal and lateral views of the area in question (Figure 28-4).
Computed Tomography
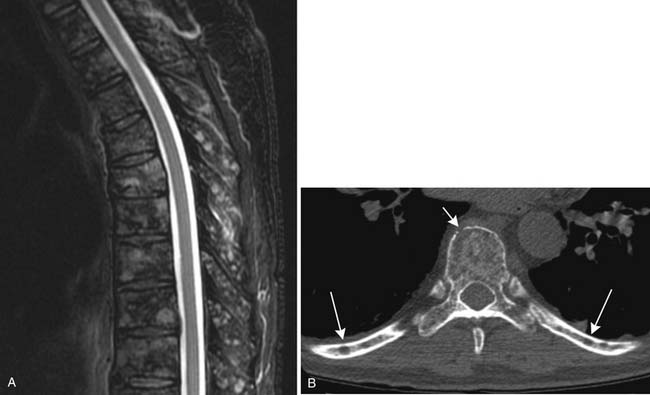
CT provides cross-sectional radiographic images of the body and can serve many purposes. Myeloma’s most common anatomic manifestation is in the bones. CT can demonstrate smaller lytic lesions than would be apparent by conventional radiography and can, like conventional radiography, help estimate the risk of pathologic fracture21–23 (Figure 28-5). With attention to the difference in attenuation between tumor (water density) and marrow (fat density), CT can also demonstrate marrow involvement. Despite these capabilities, CT is not commonly used for evaluation of osseous myeloma, although low-dose whole body CT has been suggested as an alternative to the skeletal survey.26 CT is also quite useful for detection and characterization of the uncommon soft tissue manifestations of myeloma or complications of its treatment.
Magnetic Resonance Imaging
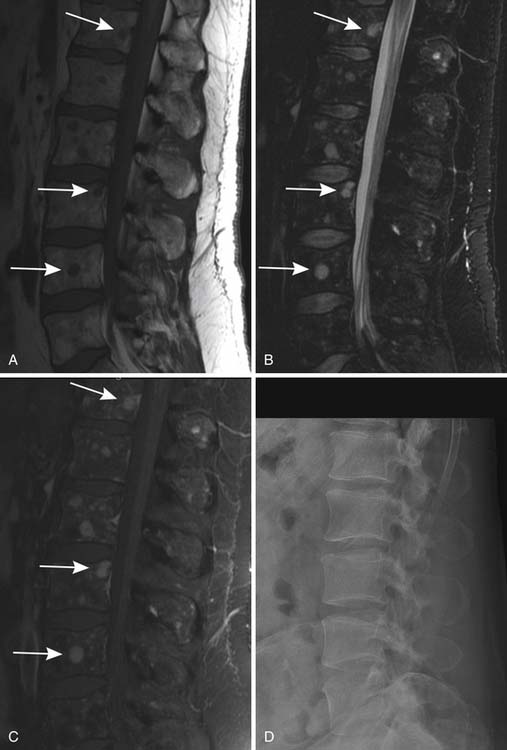
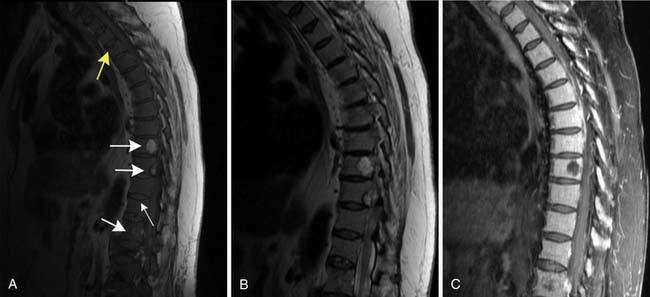
MRI is also a cross-sectional imaging technique that differs from CT in having inherently lower spatial resolution but greater contrast resolution, an advantage in imaging the bone marrow. Because of this, its most typical use in myeloma is in staging assessment of bone marrow involvement. Myeloma can involve any bone, but for assessing a large amount of bone marrow in one MRI examination, spine MRI has been traditionally used for myeloma assessment. With the advancement of new techniques using coils adapted to image large areas, imaging from the skull base to the proximal thighs can be done, including assessment of the entire body in one scan. This whole body MRI examination gives a very broad view of the marrow at the cost of spatial resolution. Whole body MRI has recently been tested as a staging method for plasma cell neoplasms. Ghanem and colleagues27 compared whole body MRI and skeletal surveys in patients with either myeloma or MGUS. Among 54 patients, whole body MRI found bone marrow involvement in 10 who had negative skeletal surveys. Walker and associates28 also found MRI to be more sensitive to focal bone lesions than skeletal survey. Evaluating 100 patients with MGUS or myeloma, Bäuerle and coworkers29 compared spinal MRI with whole body MRI and found 9 patients with isolated extra-axial disease that was identified only with whole body MRI.
Patterns of myeloma involvement seen on MRI include normal marrow (which does not exclude low-level infiltration of myelomatous cells), focal involvement (Figure 28-6), diffuse homogeneous involvement (Figure 28-7), and heterogeneous involvement, with combinations of fatty marrow and tiny areas of myeloma creating a salt-and-pepper appearance.30–32 Shorter time to progression and worse response to therapy have been found with diffuse and focal disease as opposed to normal marrow or heterogeneous involvement.32–34 With successful treatment, the marrow pattern should become more normal.24
Positron-Emission Tomography
The purpose of PET/CT varies depending on the stage of disease. In newly diagnosed patients, it helps find sites of disease, both osseous and in the soft tissues, that may be occult by conventional radiography. In this way, it can help distinguish a solitary plasmacytoma from multiple myeloma.35 Kim and colleagues36 found PET to be positive in 13 of 14 known plasmacytomas, and in 1 of 21 patients with suspected SBP, PET imaging resulted in upstaging of the disease to multiple myeloma by disclosing previously unsuspected additional disease. Depending on the equipment available, PET/CT will usually cover at least the head, trunk, and proximal humeri and femurs, and it may be able to include literally the entire body, so like large–field of view MRI, it is a way to include most or all of the body in a more sensitive screening than is available with a bone survey. In patients who have been treated, PET/CT is used to identify sites of disease progression and to distinguish the osseous lesions that are still apparent anatomically but are no longer metabolically active from those that are resistant to treatment and remain active37,38 (Figure 28-8). Complete suppression of abnormal PET activity after treatment may be linked to longer progression-free survival (PFS) than for patients without complete suppression.39 As compared with MRI, PET/CT tends to be less sensitive to diffuse marrow infiltration and perhaps more sensitive to the presence of focal lesions.40
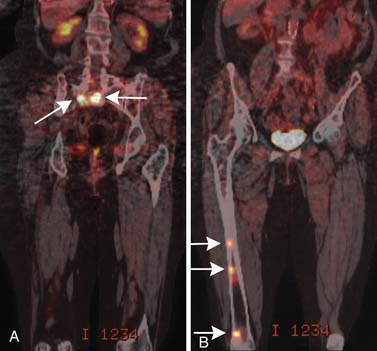
Both MRI and PET/CT are more sensitive than conventional radiography, yet each has limitations.38,41 Short and associates42 compared whole body MRI and PET/CT with bone marrow aspiration. Bone marrow aspiration was taken as the gold standard, although it can also suffer from sampling error if the myelomatous cells are patchy in distribution rather than fairly widespread through the marrow. For the 34 sets of paired MRI and PET/CT studies they obtained in 24 patients, MRI had an overall accuracy rate of 65% and PET/CT had an overall accuracy rate of 74%. When both MRI and PET/CT were either positive or negative, the combined accuracy rate rose to 81%. The routine use of both examinations is probably not justified owing to their financial impact.
Both the presence and the number of focal lytic bone lesions, particularly those that are FDG-avid and that remain so after induction therapy, may be of prognostic significance. Bartel and coworkers39 found that the presence of more than three lytic lesions correlated with inferior survival while complete suppression of FDG avidity during induction therapy correlated with improved outcome.
Treatment*
Approximately 20% of patients are diagnosed with multiple myeloma by chance while in an asymptomatic phase. For these patients without CRAB criteria defining symptomatic disease, the standard of care is frequent follow-up without treatment. Currently, no trial has determined a survival advantage for early initiation of therapy in patients with asymptomatic myeloma; thus, treatment is indicated at the time of progression to symptomatic disease by CRAB criteria.
For decades, melphalan and prednisone (MP) induction therapy and vincristine-doxorubicin-dexamethasone (VAD) for relapsing multiple myeloma remained the standard of care for patients with symptomatic disease.43,44 In the 1990s, myeloablative therapy with autologous stem cell support (AuSCT) provided the first major improvement in survival for myeloma patients and subsequently became the standard of care for eligible patients.45
The introduction of thalidomide in 1999 to therapy for myeloma provided not only a new therapeutic avenue for patients with myeloma but also enthusiasm for investigation of novel pathways and mechanisms of action of the disorder.46 Subsequently, the proteosome inhibitor bortezomib and the immunomodulatory derivative (ImiD) lenalidomide in combination with or without dexamethasone have demonstrated improved response rates, PFS and overall survival (OS) for patients with multiple myeloma.47–49 Thus, it is not unexpected that these combinations now provide the backbone of induction therapy for this disorder. Treatment with either bortezomib or lenalidomide in combination with dexamethasone for induction results in overall response rates of approximately 80% with complete response (CR) rates of 10% to 20%.50–52 Recently, these backbone combinations have been combined with each other, or with drugs like cyclophosphamide, doxorubicin/liposomal doxorubicin, melphalan, and thalidomide. These three drug regimens have improved overall response rates for induction therapy to 85% to 100% and CR rates of approximately 25% to 45%.53–56 Similarly, four drug regimens such as the “Evolution” regimen (bortezomib, lenalidomide, cyclophosphamide, dexamethasone) may also result in overall response rates of 90% to 100% and improved CR rates of 30% to 40%.57 There are no results from large randomized trials that address whether two-, three-, or four-drug induction regimens produce the longest OS for patients with newly diagnosed, symptomatic myeloma. A preliminary report of a small randomized trial from the Mayo Clinic suggests that, while the four-drug Evolution regimen produces an increased depth of response compared with cyclophosphamide, bortezomib, and dexamethasone (CyBorD), toxicity, including mortality, may be worse with the four-drug regimen, suggesting randomized trials on a large scale are necessary to address the question of whether it is better to treat with combinations utilizing more drugs for induction or to preserve the naïveté of some agents for use at relapse.58 For patients eligible for consolidation with myeloablative therapy with AuSCT, randomized trials have demonstrated an improvement in OS compared with chemotherapy alone; similar results have been noted at first relapse and for primary refractory disease.45,59,60 Whether the same results will be obtained with randomized trials in the era of novel agents is unknown, but regimens utilizing novel agent induction regimens followed by AuSCT have resulted in some of the highest CR rates noted to date. A trial of MP, MP-thalidomide (MPT), and MPT followed by AuSCT in patients 65 years old or older was the first trial to indicate that novel agent induction therapy was superior to therapy including AuSCT for elderly patients.61 Although this trial demonstrated superiority in OS for the MPT arm, the intensity of the myeloablative therapy was approximately half that compared with standard AuSCT, leaving the question of AuSCT in patients 65 years old or older unanswered. For patients ineligible for myeloablative therapy, the VISTA trial [bortezomib-melphalan-prednisone [VMP]) demonstrated excellent response rates and PFS/OS benefits even among patients with high-risk chromosomal abnormalities.20
Prolonged therapy with melphalan may impair stem cell collection and should be avoided in transplant candidates; radiation to large areas of marrow should also be avoided for similar reasons.62 Stem cell harvest may also be impaired after prolonged therapy with lenalidomide; thus, stem cell harvest should proceed early in the course of therapy with this agent (or if performed later, cyclophosphamide mobilization may be given) for transplant-eligible patients.62
Key Points Treatment
MGUS: No specific treatment. Monitor for progression to myeloma.
SBP: Radiation with curative intent.
Asymptomatic myeloma: No treatment needed until progression to symptomatic myeloma.
• Chemotherapy [bortezomib or lenolidomide, usually with dexamethasone with or without another agent (i.e., thalidomide, cyclophosphamide, doxorubicin and melphalan in various combinations) to induce remission followed, when possible, by myeloablative therapy and AuSCT].
• Radiation therapy for palliation of selective symptomatic focal lesions. (Avoid if possible in stem cell candidate.)
• Bisphosphonate therapy for 2 years for patients with lytic bone disease, to be resumed if there is progression of bone disease.
Surveillance
Response criteria for myeloma have often varied between institutions and investigators, prompting the European Bone Marrow Transplant (EBMT) group63 to propose uniform response criteria that were later revised by the IMWG.14 In the revised criteria, partial response (PR) is determined by at least a 50% reduction in serum myeloma protein and at least a 90% reduction in Bence-Jones proteinuria, along with additional criteria for special circumstances.14 CR is determined by complete absence of serum and urine monoclonal protein by immunofixation and less than 5% marrow plasma cells, among other criteria for less common circumstances. Other categories include stringent complete response (sCR) including additional normalization of free light chains and absence of clonal marrow plasma cells, and very good partial response (VGPR). After achieving a PR or better, the question of maintenance therapy arises. The best studies have been performed in the post-AuSCT setting and include improved survival for maintenance therapy with thalidomide compared with no maintenance therapy for patients achieving less than VGPR.45 Other trials with either thalidomide or lenalidomide have shown benefits in depth of response and PFS, without benefit in OS, although as data mature, results may change. These trials have usually not reported results based on subgroups to see whether benefits are limited to certain patients (e.g., those with < VGPR).64 Some investigators have, however, noted worse survival after thalidomide maintenance therapy in patients with deletion of 17p.65 The question of maintenance therapy remains unresolved, although for patients achieving less than VGPR, maintenance therapy appears warranted based on survival benefits and may be considered in all patients based on benefits in depth of response and PFS. The benefits of maintenance therapy for patients not receiving AuSCT are less clear, although recent reports demonstrate similar benefits.66
Patients should be monitored every 2 to 3 months (although, if continued on maintenance therapy, this may be more frequent) until relapse with continued monitoring of serum and urine protein electrophoretic studies, serum free-light chain analyses, routine chemistries and blood counts, particularly focusing on hemoglobin and creatinine, and bone x-rays annually or for persistent pain. Criteria for relapse have also been proposed by the IMWG and include recurrence of paraprotein, marrow plasmacytosis of 5% or greater, or progression and/or new lytic lesions for relapse from CR and for VGPR or less, a 25% increase in monoclonal protein, marrow plasmacytosis 10% or greater with similar bone parameters; an absolute rise in monoclonal serum protein of 0.5 g/dL or greater, and of at least 200 mg/day of Bence-Jones protein is required.14 Parameters for clinical relapse have also been proposed by this group.
Patients with bone disease should receive therapy with bisphosphonates after performing a careful dental examination to evaluate for predisposing factors for osteonecrosis of the jaw (ONJ).67 Careful monitoring for ONJ and renal function (with dose reduction as appropriate) is also necessary and bisphosphonates should be held if complications occur. The ideal duration of bisphosphonate therapy is not clear, but a consensus panel has recommended 2 years of therapy, followed by resumption of bisphosphonates for progression of bony disease.67 Vertebroplasty or kyphoplasty may be performed for eligible patients with vertebral compression factors; it is important to note that compression fractures may occur after response, without progression of disease.
Patients with neuropathy secondary to thalidomide or bortezomib, and much less frequently, lenalidomide, may find symptoms alleviated with dose reduction or, if severe, cessation of the offending agent and initiation of drugs for symptom control, such as gabapentin, pregabalin, duloxetine, or acetyl-l-carnitine. Prophylactic valacyclovir is recommended for patients receiving treatment with bortezomib owing to an increased incidence of herpes zoster,68 and prophylactic anticoagulation with aspirin, warfarin, or low-molecular-weight heparin per IMWG guidelines is recommended for patients on thalidomide or lenalidomide, particularly in combination with steroids and/or anthracyclines.69
Key Points Imaging algorithm for multiple myeloma
• Start with the skeletal survey. (Whole body radiography or low-dose CT are possible alternatives.)
• If positive, perform focal imaging of any areas that may be at risk of pathologic fracture. Consider either PET/CT or MRI of the spine or whole body as a baseline to allow an anatomic method of monitoring for response to therapy.
• If negative, obtain MRI of the spine (consider whole body MRI or PET/CT if available) to look for radiographically occult marrow infiltration or focal lesions.
Promising Therapies
The success of the reversible proteasome inhibitor bortezomib has led to further investigation of more specific and/or other targets within the ubiquitin-proteasome system (UPS). Carfilzomib is a second-generation, irreversible proteasome inhibitor that has a higher degree of chymotrypsin-like activity and a lower affinity for caspase- and trypsin-like binding sites within the proteasome.70 Because of these differences in affinity for proteasome-binding sites, differences in the toxicity and efficacy profile compared with bortezomib appeared feasible. Initial data from a phase II trial have demonstrated significant activity, with only rare neuropathy, with carfilzomib alone.71 Because proteasome inhibition has provided such a successful new avenue of treatment for patients with myeloma, it is natural to consider a more convenient, oral route of administration for these agents, and phase I testing on at least one of these drugs has begun.
After the introduction of thalidomide as an active agent in myeloma, it became clear that side effects such as neuropathy and thromboembolic events were frequent. The ImiDs were developed in an attempt to improve upon the activity of thalidomide while reducing the complications. The ImiD lenalidomide has demonstrated significant activity and a different side effect profile than that of thalidomide, leading to its U.S. Food and Drug Administration (FDA) approval for previously treated disease in combination with dexamethasone. Recently, another ImiD, pomalidomide, has demonstrated significant activity in combination with dexamethasone, even among patients who previously received thalidomide or lenalidomide.72 Toxicity included myelosuppression, one thrombotic event (on aspirin), and mild neuropathy in 30%, but only one patient developed grade 3 or higher neuropathy.
Preclinical studies have also indicated that inhibitors of histone deacetylase, KSP, Nedd-8 activating enzymes, and monoclonal antibodies such as elotuzamab, among other agents, also show promise and have currently entered clinical investigation.73–77
Leukemia
Epidemiology and Risk Factors
Acute Lymphoblastic Leukemia
ALL is the most common form of leukemia among children. It accounts for 80% of all childhood leukemia and 20% of leukemia in adults.78 The incidence decreases with age, even among children. ALL accounts for 30% of all cancers in younger children but only 6% of all cancers among adolescents.79 The etiology is unknown, although many different factors including genetics, infectious causes, and toxins are under investigation.80,81 Clinical presentation is usually acute, and the most common symptoms include fever, fatigue, bone pain, arthralgia, and abnormal bleeding.81
Acute Myeloid Leukemia
When AML occurs in children, it is usually in infants.80 People with Down syndrome are at a distinctly increased risk, with approximately 1% of children with Down syndrome developing leukemia. This is usually a form of AML that is sufficiently distinct from other variants of AML that it may be referred to as myeloid leukemia of Down syndrome.82 Children with Down syndrome are also at increased risk for ALL.80
Among adults, AML is the most common type of acute leukemia, with an age-adjusted incidence of 3.6 per 100,000 persons. Peak age at diagnosis is approximately 67, so this is primarily a disease of older adults.83
Chronic Lymphocytic Leukemia
In the Western hemisphere, CLL is the most common form of leukemia occurring in adults.84,85 To diagnose CLL, the patient must have an absolute B-lymphocyte count of 5000/mL or greater.84 This leukocytosis may be found on routine laboratory tests run on asymptomatic individuals. Patients who present with symptoms may complain of painless lymphadenopathy or occasionally of typical B symptoms as seen in lymphoma—weakness, night sweats, weight loss, or fever. Some patients will present with symptoms of immune dysfunction, particularly infection or an autoimmune syndrome.84
Chronic Myeloid (or Myelogenous) Leukemia
Peak age for development of CML is 50 to 55; disease incidence varies from one population to another and may be estimated at 0.6 to 2 per 100,000 persons per year. There is a slight male predominance.86 CML is brought on by a single oncogene, bcr-abl1, formed owing to stem cell acquisition of the Philadelphia chromosome that, in turn, is the product of a characteristic translocation between the long arms of chromosomes 9 and 22 causing fusion between the breakpoint cluster regions gene (bcr) and the Abelson tyrosine kinase proto-oncogene (abl1). The resulting oncogene bcr-abl1 produces a protein that acts as a tyrosine kinase. Diagnosis requires evidence via cytogenetics, polymerase chain reactions, or Western blot tests of this characteristic translocation.87–89
Staging
Acute Lymphoblastic Leukemia
ALL is divided into three types: precursor B-cell ALL, mature B-cell ALL (Burkitt’s ALL), and T-cell ALL.78 Beyond the categorization based on cell type, patients may also be divided into prognostic categories based on the presence or absence of risk factors. Older age is the single most important risk factor. Long-term cure rates are over 80% for children but only 40% to 50% for adults. Not only is prognosis much better among children than among adults, but younger adults do better than older ones; the prognosis is especially dismal for patients older than 60 years. The unfavorable prognosis of older patients may relate not directly to their age but rather to the greater tendency of older individuals to manifest unfavorable chromosomal states. For example, the Philadelphia chromosome, found in 20% to 30% of adults with ALL, is associated with a worse prognosis than in patients without this mutation.78,79,81,90 Other factors that worsen prognosis include high leukocyte count, CD20 expression in the tumor cells, and longer time to achieve initial CR.78 The International Childhood Acute Lymphoblastic Leukaemia Workshop has recommended division of pediatric patients into four prognostic groups.91 Adult patients are usually divided into just two categories: low or standard risk and high risk.92 These categories pertain to newly diagnosed patients and patients who have completed front-line therapy and have hopefully gone into remission. Once a patient relapses, the prognosis depends on the length of prior remission(s) and the availability of a suitable donor for allogeneic stem cell transplantation.90
Chronic Lymphocytic Leukemia
Two staging systems are used in CLL—the Rai and colleagues system93 and the Binet and associates system.94 The Rai system divides the disease into five stages—0 through IV—whereas the Binet system uses only three stages: low, intermediate, and high risk. There are, however, factors that affect prognosis but that are not included in either of these staging systems. About half of CLL will have undergone mutation in the immunoglobulin heavy chain variable region gene. Patients with this mutation have a favorable prognosis compared with patients lacking this mutation.95,96 Two proteins, ZAP-70 and CD38, may be associated with more aggressive disease and a worse prognosis.84
Acute Myeloid Leukemia
Genetic abnormalities associated with the population of leukemic cells constitute the single most important prognostic factor in AML, and the cytogenetic risk groups can be stratified into favorable, intermediate, and unfavorable strata. Division into these risk groups not only helps predict outcome but also allows tailoring of therapy to individual patients.97
Chronic Myeloid (or Myelogenous) Leukemia
Patients with CML may be divided into prognosis-related categories using the Sokal formulation. This method uses risk factors including age, splenic size, platelet count, and the percentage of blasts in peripheral blood to obtain a Sokal score. On the basis of this score, the patients are divided into low-, intermediate-, and high-risk groups that correlate with a stratified tendency to develop longer-term PFS remission.98
Imaging of the Disease and Its Relapse after Treatment
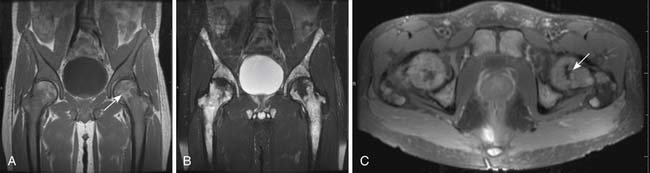
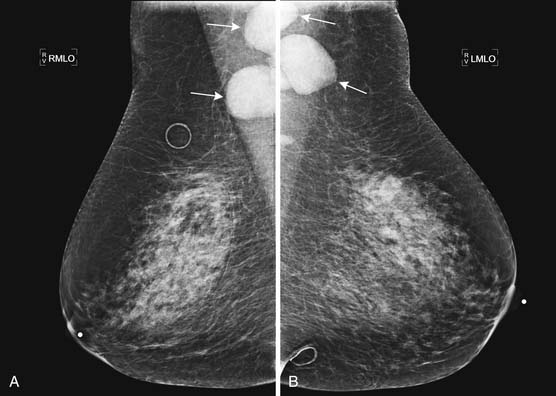
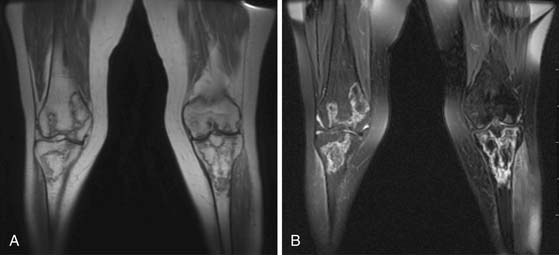
When imaging plays a role in the diagnosis of leukemia, it is often because the leukemia is an unexpected finding discovered when evaluating a symptom. For example, leukemia may present with joint pain. If the joint pain is confined to one or a few joints, an MRI may be ordered to evaluate for causes of joint pain such as mechanical derangement. An acute leukemia may replace normal fatty marrow with a monotonous sea of disease that will appear abnormally dark on T1-weighted images and bright on T2-weighted images and enhance with contrast administration (Figure 28-9). The appearance is not specific, however, because other marrow-replacing disorders, including diffuse myeloma, may have the same appearance. Another common incidental imaging manifestation of leukemia is axillary lymphadenopathy noted in patients with leukemia who undergo screening mammography (Figure 28-10).
Marrow involvement with leukemia is usually assessed clinically rather than with imaging. Zha and coworkers,99 however, have suggested that dynamic contrast–enhanced MRI of spinal bone marrow may prove useful for assessing leukemic marrow involvement. In a mixed group of 26 patients having a variety of hematologic malignancies, 18 of whom had leukemia, these authors found that even at low levels of marrow infiltration (5-25%), time to peak enhancement was significantly shorter and the slope of the time-enhancement curve was significantly greater in patients than in normal controls. There were also statistically significant differences among different grades of infiltration, but this may not be of practical use in individual patients owing to large amounts of overlap between categories.
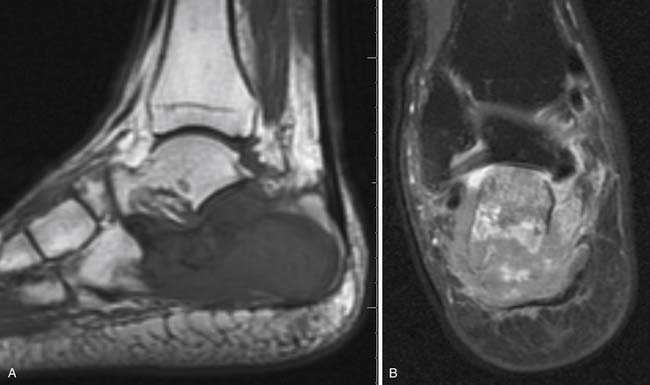
Chloromas, extramedullary masses of leukemia cells, are uncommon on initial evaluation but may occur in up to 21% of patients relapsing after allogeneic stem cell transplantation, especially for myelogenous leukemias. Such a local recurrence may potentially be cured with radiation. The morphologic imaging appearance of these masses is nonspecific, and biopsy is often needed to distinguish them from secondary primary cancers or focal areas of opportunistic infection.100 Extramedullary relapse of AML has been identified using PET/CT. Following successful treatment, the abnormal FDG-avidity resolved.101 In the same way, focal areas of disease within bone marrow have a nonspecific appearance and also commonly need biopsy for diagnosis (Figure 28-11).
The central nervous system is a sanctuary area for leukemia. Relapse in this region may manifest either as focal masses that may be visible on CT or MRI or as leptomeningeal disease (leukemic meningitis). Leptomeningeal involvement is due to sheets of tumor cells spreading over the surface of the brain and/or spinal cord. Intracranially, the tumor cells infiltrate into the sulci and can mimic leptomeningeal hemorrhage or infectious meningitis.102 In the spinal canal, they create an enhancing lumpy coating on the surface of the spinal cord. The main task is to distinguish them from vessels that normally run on the surface of the cord. Ulu and colleagues103 reported leptomeningeal disease in 5 of 15 patients with childhood leukemia. All 5 of these had ALL.
CLL is the most common form of leukemia in adults and, at times, may present with extensive adenopathy (Figure 28-12). Although in CLL imaging may have a greater role than in other leukemias in terms of staging and disease surveillance, it rarely changes management in this disease. Imaging is mainly reserved in CLL for the complications of the disease, either identifying infections these patients are prone to develop owing to their abnormal immune system due to the disease itself and/or therapy or the dreaded complication of Richter’s transformation, in which CLL progresses to other, more aggressive neoplasms, most often large B-cell lymphoma. Differentiating the adenopathy associated with CLL from Richter’s transformation can be difficult with morphologic imaging. However, because the FDG activity in CLL-involved lymph nodes is low grade and with large B-cell lymphoma it is high, imaging with FDG-PET would seem ideal. Indeed, PET/CT was found to have a sensitivity of 91% and specificity of 80% for Richter’s transformation in a study of 37 patients with CLL. If not only Richter’s transformation but also transformation into an accelerated phase of CLL or development of a new malignancy of any sort were considered positive results, then sensitivity and specificity improved to 94% and 90%, respectively.104
Imaging of Complications
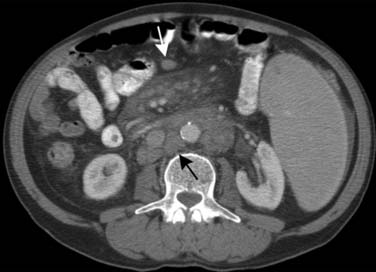
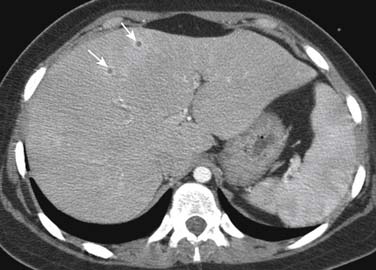
Leukemia patients are immune suppressed both from the disease itself, which affects the function of the immune system, and from the chemotherapy used to treat the disease. The lungs are a common source of infection in these patients, and thus, when a leukemic patient presents with fever, imaging usually starts with a chest radiograph to assess for pneumonia as well as imaging studies that may be directed toward specific symptomatic sites. In addition to ordinary community-acquired bacterial infections, leukemic patients are susceptible to invasive fungal organisms that may involve any area of the body, commonly the lungs and sinuses. Of the fungal organisms, invasive pulmonary aspergillosis is by far the most common. Invasive pulmonary fungal infections, whether due to aspergillosis, mucormycosis, candidiasis, or fusariosis, produce local infarcts in the lungs. Imaging wise, these differ from other bacterial and viral infection in that they produce large (>1 cm) pulmonary nodules.105 Thus, in a leukemic febrile patient, the finding of pulmonary nodules or masses is consistent with invasive fungal pneumonia until proved otherwise. These nodular lesions can be solid or consolidative or have a rim of ground glass surrounding them that is due to surrounding hemorrhage, the so-called halo sign. Mortality is higher than 50% from these invasive fungal pneumonias, and early identification of fungal pneumonia, by the use of CT with early initiation of antifungal agents, improves survival.106 Management of invasive pulmonary aspergillosis in neutropenic patients improved using early thoracic CT scan and surgery (Figure 28-13). In the liver, fungal infection, especially due to Candida, often appears as multiple, small, uniform, low-attenuation regions that may be surrounded by a rim of enhancement (Figure 28-14).
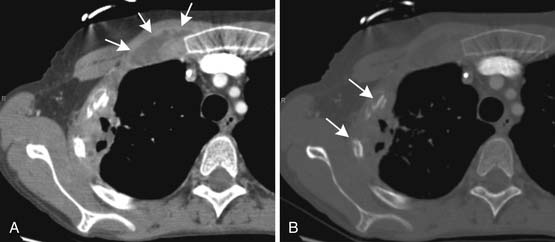
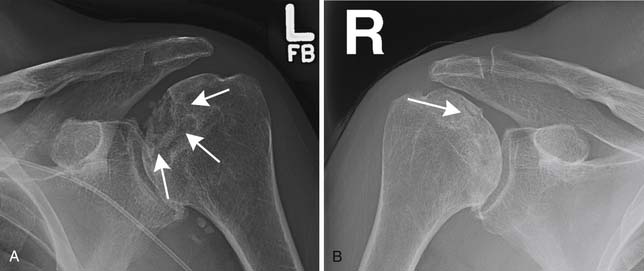
Another common complication in leukemia patients is osteonecrosis. Steroids, which are a part of some chemotherapy regimens for leukemia and which may also play a role in suppressing graft-versus-host disease after stem cell transplantation, predispose to osteonecrosis, as does leukemia itself. Osteonecrosis most often occurs in the femoral heads followed by the humeral heads but can be seen in numerous bones. On radiographs, it may be occult. Later, it may be visible, usually as a vague area of sclerosis in the femoral or humeral head. In long bones, where it is often referred to as an area of bone infarction, it may appear as a jagged area of sclerosis that may be difficult to distinguish from an enchondroma. Bone infarcts, however, are inclined to calcify around their periphery more than centrally, whereas an enchondroma tends to do the opposite. Eventually, osteonecrosis may weaken the bone sufficiently to cause fracturing of the adjacent cortex. This typically occurs at the femoral head and is manifested first by a thin radiolucent rim running just under the articular surface and then by collapse of the articular surface (Figure 28-15). On MRI, osteonecrosis usually appears centrally the same as nearby fatty bone marrow. At the periphery, areas of osteonecrosis are surrounded by a narrow, sharply defined band of contrasting signal, generally dark on T1-weighting and bright on T2-weighting, resembling the guiding outlines in a child’s coloring book (Figure 28-16).
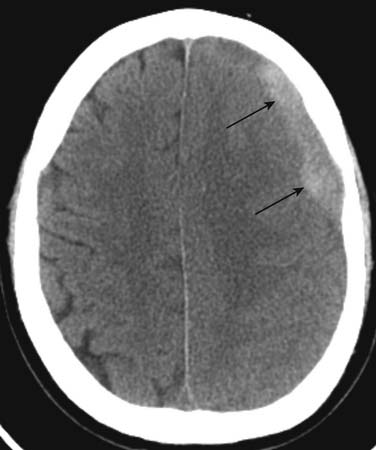
Patients with leukemia can also experience hemorrhage secondary to abnormal clotting mechanisms. If intracranial hemorrhage is suspected, CT is the appropriate means to detect it (Figure 28-17).
Treatment
Leukemia is treated chemically, although the particular type of medication differs from one type of leukemia to another. Radiation therapy can play a role; for example, central nervous system radiation has been used to decrease the likelihood of relapse in that location in childhood ALL, but side effects of this radiation have caused it to be minimized in favor of other treatments such as intrathecal chemotherapy.107,108 Stem cell transplantation is usually reserved for specific situations and is not standard therapy for most patients.
Acute Lymphoblastic Leukemia
Treatment of ALL can be divided into three stages: induction, consolidation, and maintenance. The goal of initial chemotherapy is to induce a CR as quickly as possible. A combination of vincristine, corticosteroid (dexamethasone or prednisone), and an anthracycline will achieve remission in 72% to 92% of patients. Hyper CVAD (hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone) alternating with methotrexate and high-dose cytarabine has also been used recently. In children, L-asparaginase also plays an important role in initial therapy. Because the central nervous system can be the site of relapse in 10% to 16% of patients, intrathecal therapy is also indicated.78
Consolidation regimens vary. For patients with the Philadelphia chromosome, allogeneic stem cell transplantation is recommended for consolidation. For others, consolidation treatment is often a modified continuation of the same drugs used for induction.78
Maintenance therapy is usually given over 3 years and includes daily 6-mercaptopurine, methotrexate weekly, and vincristine and prednisone monthly. No maintenance therapy is used for mature B-cell ALL because these patients have an excellent long-term cure rate after successful induction and relapses are rare after a year.78 Stem cell transplantation may be used for refractory or relapsed ALL.109
Chronic Lymphocytic Leukemia
Patients with asymptomatic, early-stage CLL are monitored for disease progression but are not treated. Early treatment has not been associated with improved outcomes, and because some patients will never require treatment, it is good to avoid the side effects of treatment in these patients.84
Acute Myeloid Leukemia
Patients with AML are treated with standard chemotherapy to achieve remission. This includes cytarabine combined with anthracyclines or anthracenediones to achieve remission followed by additional cytarabine, either alone or in combination, for maintenance. In children and younger adults, intensive cytarabine therapy may be pursued with the goal of cure.82,83 Allogeneic stem cell transplantation is commonly used in remission, primarily for patients with intermediate or unfavorable cytogenetics, to lengthen time to relapse and possibly effect a cure. Indeed, AML is the most common indication for stem cell transplantation.97
In the older adults who make up most of the AML patient population, the relapse rate is high and outcome is poor, with survival often measured in weeks.83 Treatment decisions in these individuals involve both an estimation of the added life span that may result from aggressive therapy and consideration of the patient’s quality of life, which may be better with less aggressive therapy.110
Cytogenetic testing and stratification allow some tailoring of chemotherapeutic regimens. For example, one subtype of AML with favorable cytogenetics is acute promyelocytic leukemia. This subtype has an excellent prognosis if all-trans-retinoic acid and arsenic are added to induction chemotherapy.97 Chen and associates111 have also suggested that dynamic contrast–enhanced MRI of the spinal bone marrow may also help stratify treatment by identifying patients at greater and lesser risk of relapse after achievement of remission.
Chronic Myeloid (or Myelogenous) Leukemia
Because CML is triggered by a single oncogene that, with its resulting protein, has been well-studied, it has lent itself very well to specific, targeted therapies that are distinctly more efficacious than ordinary chemotherapy. The abnormal protein is a tyrosine kinase, and therefore, tyrosine kinase inhibitors are active against CML. Front-line therapy is with imatinib, and almost all patients will go into remission that, with continued therapy, may result in a long period of PFS. In one trial, 61% of patients were still progression-free at 6 years.87,89,98
If relapse occurs, options include continued treatment with higher doses of imatinib or a trial of other tyrosine kinase inhibitors dasatinib or nilotinib.87,89,98 If these are ineffective, or if control is lost, then allogeneic stem cell transplantation may be helpful.87
When CML progresses, it may go into blast crisis, and at that stage, the tyrosine kinase inhibitors are of little use. Allogeneic stem cell transplantation should be attempted.87
Promising Therapies
Most types of leukemia initially respond to therapy, and the patients will achieve a PR or CR. This is followed by a period of minimal residual disease, then eventually refractory relapse or relapses will occur, and the patient may eventually die owing to these relapses. Refractory relapses are likely due to the persistence of a small colony of leukemic stem cells.112 These cells are resistant to therapy because they have a relatively low level of metabolic activity and an impressive group of protective mechanisms. They also tend to migrate to and immobilize themselves in sanctuary areas that protect them from therapy. Although first found in AML, leukemic stem cells have now been found in all major forms of leukemia.113–116
Ultimate cure of leukemia will probably require elimination of leukemic stem cells, and therefore, research is ongoing directed at devising methods of attacking the leukemic stem cells, preferably while sparing the patient’s normal stem cells. One approach has been to target cell surface antigens. No antigen has been identified that is unique to leukemic stem cells, but some antigens have been found that are more active in leukemic stem cells than in normal cells. One of these is CD33, which is mainly expressed in cells of the myeloid lineage. Several experimental therapies are being tested that target CD33. One, for example, is gentuzamab ozogamicin, a combination of a CD33 antibody bound chemically to a toxin. The antibody adheres to the cell membrane, enters the cell, and releases the toxin. The toxin subsequently enters the nucleus, where it causes double-stranded DNA breaks that result in apoptosis. This therapy has been FDA approved in the United States for patients older than 60 years with AML who are not considered candidates for routine chemotherapy, and in a trial produced a 26% remission rate.112,117 In CML also, there is intense interest in investigating methods of attacking the leukemic stem cells.88
Because drugs that are effective with one hematologic malignancy often are effective with others as well, research is also ongoing in application in new settings of drugs already known to be useful in other diseases. In CLL, for example, thalidomide and lenolidomide, both known to be useful against myeloma, have been tested as potential salvage therapy for relapsed or refractory disease. Because one of their effects is to support the immune system, it is hoped that they can stimulate the immune system to battle the disease while also avoiding the infection-promoting effects of immune compromise encountered with many chemotherapies.85
1. Altekruse S.F., Huang L., Cucinelli J.E., et al. Spatial patterns of localized-stage prostate cancer incidence among white and black men in the southeastern United States, 1999-2001. Cancer Epidemiol Biomarkers Prev. 2010;19:1460-1467.
2. Riedel D.A., Pottern L.M. The epidemiology of multiple myeloma. Hematol Oncol Clin North Am.. 1992;6:225-247.
3. Preston D.L., Kusumi S., Tomonaga M., et al. Cancer incidence in atomic bomb survivors. Part III. Leukemia, lymphoma and multiple myeloma, 1950-1987. Radiat Res.. 1994;137:S68-S97.
4. Berenson J.R., Bergsagel P.L., Munshi N. Initiation and maintenance of multiple myeloma. Semin Hematol. 1999;36:9-13.
5. Kristinsson S.Y., Bjorkholm M., Goldin L.R., et al. Patterns of hematologic malignancies and solid tumors among 37,838 first-degree relatives of 13,896 patients with multiple myeloma in Sweden. Int J Cancer. 2009;125:2147-2150.
6. Fonseca R., Bergsagel P.L., Drach J., et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23:2210-2221.
7. International Myeloma Working Group I. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749-757.
8. Sezer O., Heider U., Zavrski I., et al. RANK ligand and osteoprotegerin in myeloma bone disease. Blood. 2003;101:2094-2098.
9. Roodman G.D., Dougall W.C. RANK ligand as a therapeutic target for bone metastases and multiple myeloma. Cancer Treat Rev.. 2008;34:92-101.
10. Sharma S., Nemeth E., Chen Y.H., et al. Involvement of hepcidin in the anemia of multiple myeloma. Clin Cancer Res.. 2008;14:3262-3267.
11. Dimopoulos M.A., Kastritis E., Rosinol L., et al. Pathogenesis and treatment of renal failure in multiple myeloma. Leukemia. 2008;22:1485-1493.
12. Fahey J.L., Scoggins R., Utz J.P., Szwed C.F. Infection, antibody response and gamma globulin components in multiple myeloma and macroglobulinemia. Am J Med. 1963;35:698-707.
13. Cheson B.D., Plass R.R., Rothstein G. Defective opsonization in multiple myeloma. Blood. 1980;55:602-606.
14. Durie B.G., Harousseau J.L., Miguel J.S., et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20:1467-1473.
15. Weber D.M. Solitary bone and extramedullary plasmacytoma. Hematology Am Soc Hematol Educ Program. 2005:373-376.
16. Durie B.G., Salmon S.E. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer. 1975;36:842-854.
17. Greipp P.R., San Miguel J., Durie B.G., et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23:3412-3420.
18. Kastritis E., Zervas K., Symeonidis A., et al. Improved survival of patients with multiple myeloma after the introduction of novel agents and the applicability of the International Staging System (ISS): an analysis of the Greek Myeloma Study Group (GMSG). Leukemia. 2009;23:1152-1157.
19. Jagannath S., Richardson P.G., Sonneveld P., et al. Bortezomib appears to overcome the poor prognosis conferred by chromosome 13 deletion in phase 2 and 3 trials. Leukemia. 2007;21:151-157.
20. San Miguel J.F., Schlag R., Khuageva N.K., et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359:906-917.
21. Dimopoulos M., Terpos E., Comenzo R.L., et al. International myeloma working group consensus statement and guidelines regarding the current role of imaging techniques in the diagnosis and monitoring of multiple myeloma. Leukemia. 2009;23:1545-1556.
22. Mulligan M., Smith S., Talmi D. Whole body radiography for bone survey screening of cancer and myeloma patients. Cancer Invest. 2008;26:916-922.
23. Schmidt G.P., Reiser M.F., Baur-Melnyk A. Whole-body imaging of bone marrow. Semin Musculoskelet Radiol. 2009;13:120-133.
24. Hanrahan C.J., Christensen C.R., Crim J.R. Current concepts in the evaluation of multiple myeloma with MR imaging and FDG PET/CT. Radiographics. 2010;30:127-142.
25. Mulligan M.E. Imaging techniques used in the diagnosis, staging, and follow-up of patients with myeloma. Acta Radiol. 2005;46:716-724.
26. Horger M., Claussen C.D., Bross-Bach U., et al. Whole-body low-dose multidetector row-CT in the diagnosis of multiple myeloma: an alternative to conventional radiography. Eur J Radiol. 2005;54:289-297.
27. Ghanem N., Lohrmann C., Engelhardt M., et al. Whole-body MRI in the detection of bone marrow infiltration in patients with plasma cell neoplasms in comparison to the radiological skeletal survey. Eur Radiol. 2006;16:1005-1014.
28. Walker R., Barlogie B., Haessler J., et al. Magnetic resonance imaging in multiple myeloma: diagnostic and clinical implications. J Clin Oncol. 2007;25:1121-1128.
29. Bäuerle T., Hillengass J., Fechtner K., et al. Multiple myeloma and monoclonal gammopathy of undetermined significance: importance of whole-body versus spinal MR imaging. Radiology. 2009;252:477-485.
30. Baur-Melnyk A., Reiser M.F. Multiple myeloma. Semin Musculoskelet Radiol. 2009;13:111-119.
31. Moulopoulos L.A., Gika D., Anagnostopoulos A., et al. Prognostic significance of magnetic resonance imaging of bone marrow in previously untreated patients with multiple myeloma. Ann Oncol. 2005;16:1824-1828.
32. Moulopoulos L.A., Dimopoulos M.A., Smith T.L., et al. Prognostic significance of magnetic resonance imaging in patients with asymptomatic multiple myeloma. J Clin Oncol. 1995;13:251-256.
33. Dimopoulos M.A., Moulopoulos L.A., Datseris I., et al. Imaging of myeloma bone disease—implications for staging, prognosis and follow-up. Acta Oncol. 2000;39:823-827.
34. Lecouvet F.E., Vande Berg B.C., Michaux L., et al. Stage III multiple myeloma: clinical and prognostic value of spinal bone marrow MR imaging. Radiology. 1998;209:653-660.
35. Nanni C., Rubello D., Zamagni E., et al. 18F-FDG PET/CT in myeloma with presumed solitary plasmocytoma of bone. In Vivo. 2008;22:513-517.
36. Kim P.J., Hicks R.J., Wirth A., et al. Impact of 18F-fluorodeoxyglucose positron emission tomography before and after definitive radiation therapy in patients with apparently solitary plasmacytoma. Int J Radiat Oncol Biol Phys.. 2009;74:740-746.
37. Cholewinski W., Castellon I., Raphael B., Heiba S.I. Value of precise localization of recurrent multiple myeloma with F-18 FDG PET/CT. Clin Nucl Med. 2009;34:1-3.
38. Durie B.G., Waxman A.D., D’Agnolo A., Williams C.M. Whole-body (18)F-FDG PET identifies high-risk myeloma. J Nucl Med. 2002;43:1457-1463.
39. Bartel T.B., Haessler J., Brown T.L., et al. F18-fluorodeoxyglucose positron emission tomography in the context of other imaging techniques and prognostic factors in multiple myeloma. Blood. 2009;114:2068-2076.
40. Delorme S., Baur-Melnyk A. Imaging in multiple myeloma. Eur J Radiol. 2009;70:401-408.
41. Fonti R., Salvatore B., Quarantelli M., et al. 18F-FDG PET/CT, 99mTc-MIBI, and MRI in evaluation of patients with multiple myeloma. J Nucl Med. 2008;49:195-200.
42. Shortt C.P., Gleeson T.G., Breen K.A., et al. Whole-body MRI versus PET in assessment of multiple myeloma disease activity. AJR Am J Roentgenol. 2009;192:980-986.
43. Alexanian R., Haut A., Khan A.U., et al. Treatment for multiple myeloma. Combination chemotherapy with different melphalan dose regimens. JAMA. 1969;208:1680-1685.
44. Barlogie B., Smith L., Alexanian R. Effective treatment of advanced multiple myeloma refractory to alkylating agents. N Engl J Med. 1984;310:1353-1356.
45. Attal M., Harousseau J.L., Stoppa A.M., et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Francais du Myelome. N Engl J Med. 1996;335:91-97.
46. Singhal S., Mehta J., Desikan R., et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341:1565-1571.
47. Richardson P.G., Barlogie B., Berenson J., et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348:2609-2617.
48. Dimopoulos M., Spencer A., Attal M., et al. Lenalidomide plus dexamethasone for relapsed or refractory multiple myeloma. N Engl J Med. 2007;357:2123-2132.
49. Weber D.M., Chen C., Niesvizky R., et al. Lenalidomide plus dexamethasone for relapsed multiple myeloma in North America. N Engl J Med. 2007;357:2133-2142.
50. Jagannath S., Durie B.G., Wolf J.L., et al. Extended follow-up of a phase 2 trial of bortezomib alone and in combination with dexamethasone for the frontline treatment of multiple myeloma. Br J Haematol. 2009;146:619-626.
51. Kropff M.H., Bisping G., Wenning D., et al. Bortezomib in combination with dexamethasone for relapsed multiple myeloma. Leuk Res.. 2005;29:587-590.
52. Rajkumar S.V., Jacobus S., Callander N.S., et al. Lenalidomide plus high-dose dexamethasone versus lenalidomide plus low-dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: an open-label randomised controlled trial. Lancet Oncol. 2009;11:29-37.
53. Anderson K.C., Weller E., Lonial S., et al. Lenalidomide, bortezomib, and dexamethasone in patients with newly diagnosed multiple myeloma (MM): final results of a multicenter phase I/II study. J Clin Oncol (Meeting Abstracts). 2010;28:8016.
54. Reeder C.B., Reece D.E., Kukreti V., et al. Once- versus twice-weekly bortezomib induction therapy with CyBorD in newly diagnosed multiple myeloma. Blood. 2010;115:3416-3417.
55. Oakervee H.E., Popat R., Curry N., et al. PAD combination therapy (PS-341/bortezomib, doxorubicin and dexamethasone) for previously untreated patients with multiple myeloma. Br J Haematol. 2005;129:755-762.
56. Wang M., Giralt S., Delasalle K., et al. Bortezomib in combination with thalidomide-dexamethasone for previously untreated multiple myeloma. Hematology. 2007;12:235-239.
57. Kumar S.K., Flinn I., Noga S.J., et al. Bortezomib, dexamethasone, cyclophosphamide and lenalidomide combination for newly diagnosed multiple myeloma: phase 1 results from the multicenter EVOLUTION study. Leukemia. 2010;24:1350-1356.
58. Kumar S., Flinn I.W., Hari P.N., et al. Novel three- and four-drug combinations of bortezomib, dexamethasone, cyclophosphamide, and lenalidomide, for newly diagnosed multiple myeloma: encouraging results from the multi-center, randomized, phase 2 EVOLUTION study. ASH Annual Meeting Abstracts. 2009;114:127.
59. Fermand J.P., Ravaud P., Chevret S., et al. High-dose therapy and autologous peripheral blood stem cell transplantation in multiple myeloma: up-front or rescue treatment? Results of a multicenter sequential randomized clinical trial. Blood. 1998;92:3131-3136.
60. Alexanian R., Dimopoulos M.A., Delasalle K.B., et al. Myeloablative therapy for primary resistant multiple myeloma. Stem Cells. 1995;13(Suppl 2):118-121.
61. Facon T., Mary J.Y., Hulin C., et al. Melphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99-06): a randomised trial. Lancet. 2007;370:1209-1218.
62. Giralt S., Stadtmauer E.A., Harousseau J.L., et al. International myeloma working group (IMWG) consensus statement and guidelines regarding the current status of stem cell collection and high-dose therapy for multiple myeloma and the role of plerixafor (AMD 3100). Leukemia. 2009;23:1904-1912.
63. Blade J., Samson D., Reece D., et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998;102:1115-1123.
64. McCarthy P.L., Owzar K., Anderson K.C., et al. Phase III intergroup study of lenalidomide versus placebo maintenance therapy following single autologous stem cell transplant (ASCT) for multiple myeloma (MM): CALGB 100104. J Clin Oncol (Meeting Abstracts). 2010;28:8017.
65. Morgan G.J., Jackson G.H., Davies F.E., et al. Maintenance thalidomide may improve progression free but not overall survival: results from the Myeloma IX Maintenance Randomisation. Blood (ASH Annual Meeting Abstracts). 2008;112:656.
66. Mateos M.-V., Oriol A., Martinez J., et al. A prospective, multicenter, randomized, trial of bortezomib/melphalan/prednisone (VMP) versus bortezomib/thalidomide/prednisone (VTP) as induction therapy followed by maintenance treatment with bortezomib/thalidomide (VT) versus bortezomib/prednisone (VP) in elderly untreated patients with multiple myeloma older than 65 years. Blood (ASH Annual Meeting Abstracts). 2009;114:3.
67. Kyle R.A., Yee G.C., Somerfield M.R., et al. American Society of Clinical Oncology 2007 clinical practice guideline update on the role of bisphosphonates in multiple myeloma. J Clin Oncol. 2007;25:2464-2472.
68. Chanan-Khan A., Sonneveld P., Schuster M.W., et al. Analysis of herpes zoster events among bortezomib-treated patients in the phase III APEX study. J Clin Oncol. 2008;26:4784-4790.
69. Palumbo A., Rajkumar S.V., Dimopoulos M.A., et al. Prevention of thalidomide- and lenalidomide-associated thrombosis in myeloma. Leukemia. 2008;22:414-423.
70. Jakubowiak A.J., Benson D.M.Jr., Bensinger W., et al. Elotuzumab in combination with bortezomib in patients with relapsed/refractory multiple myeloma: a phase I study. J Clin Oncol (Meeting Abstracts). 2010;28:8003.
71. Vij R., Siegel D.S., Kaufman J.L., et al. Results of an ongoing open-label, phase II study of carfilzomib in patients with relapsed and/or refractory multiple myeloma (R/R MM). J Clin Oncol (Meeting Abstracts). 2010;28:8000.
72. Lacy M.Q., Hayman S.R., Gertz M.A., et al. Pomalidomide (CC4047) plus low-dose dexamethasone as therapy for relapsed multiple myeloma. J Clin Oncol. 2009;27:5008-5014.
73. Badros A., Burger A.M., Philip S., et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res.. 2009;15:5250-5257.
74. Weber D., Badros A.Z., Jagannath S., et al. Vorinostat plus bortezomib for the treatment of relapsed/refractory multiple myeloma: early clinical experience. Blood (ASH Annual Meeting Abstracts). 2008;112:871.
75. San-Miguel J.F., Sezer O., Siegel D.S., et al. Phase Ib study of oral panobinostat (LBH589) plus intravenous bortezomib in patients (Pts) with relapsed (Rel) or Rel and refractory (Ref) multiple myeloma (MM). J Clin Oncol (Meeting Abstracts). 2010;28:8001.
76. Shah J.J., Cohen A.D., Zonder J.A., et al. Phase I trial of ARRY-520 in relapsed/refractory multiple myeloma (RR MM). J Clin Oncol (Meeting Abstracts). 2010;28:8132.
77. Lonial S., Vij R., Harousseau J., et al. Elotuzumab in combination with lenalidomide and low-dose dexamethasone in relapsed or refractory multiple myeloma: a phase I/II study. J Clin Oncol (Meeting Abstracts). 2010;28:8020.
78. Fullmer A., O’Brien S., Kantarjian H., Jabbour E. Novel therapies for relapsed acute lymphoblastic leukemia. Curr Hematol Malig Rep. 2009;4:148-156.
79. Ribera J.M., Oriol A. Acute lymphoblastic leukemia in adolescents and young adults. Hematol Oncol Clin North Am. 2009;23:1033-1042. vi
80. Eden T. Aetiology of childhood leukaemia. Cancer Treat Rev.. 2010;36:286-297.
81. Onciu M. Acute lymphoblastic leukemia. Hematol Oncol Clin North Am.. 2009;23:655-674.
82. Kaspers G.J., Zwaan C.M. Pediatric acute myeloid leukemia: towards high-quality cure of all patients. Haematologica. 2007;92:1519-1532.
83. Duong H.K., Sekeres M.A. Targeted treatment of acute myeloid leukemia in older adults: role of gemtuzumab ozogamicin. Clin Interv Aging. 2009;4:197-205.
84. Kaufman M., Rubin J., Rai K. Diagnosing and treating chronic lymphocytic leukemia in 2009. Oncology (Huntingt). 2009;23:1030-1037.
85. Awan F.T., Johnson A.J., Lapalombella R., et al. Thalidomide and lenalidomide as new therapeutics for the treatment of chronic lymphocytic leukemia. Leuk Lymphoma. 2010;51:27-38.
86. Rohrbacher M., Hasford J. Epidemiology of chronic myeloid leukaemia (CML). Best Pract Res Clin Haematol. 2009;22:295-302.
87. von Bubnoff N., Duyster J. Chronic myelogenous leukemia: treatment and monitoring. Dtsch Arztebl Int. 2010;107:114-121.
88. Copland M. Chronic myelogenous leukemia stem cells: what’s new? Curr Hematol Malig Rep. 2009;4:66-73.
89. Sullivan C., Peng C., Chen Y., et al. Targeted therapy of chronic myeloid leukemia. Biochem Pharmacol. 2010;80:584-591.
90. Rowe J.M. Prognostic factors in adult acute lymphoblastic leukaemia. Br J Haematol. 2010;150:389-405.
91. Pui C.H., Sallan S., Relling M.V., et al. International Childhood Acute Lymphoblastic Leukemia Workshop. Sausalito, CA, 30 November-1 December 2000. Leukemia. 2001;15:707-715.
92. Hoelzer D., Thiel E., Loffler H., et al. Prognostic factors in a multicenter study for treatment of acute lymphoblastic leukemia in adults. Blood. 1988;71:123-131.
93. Rai K.R., Sawitsky A., Cronkite E.P., et al. Clinical staging of chronic lymphocytic leukemia. Blood. 1975;46:219-234.
94. Binet J.L., Auquier A., Dighiero G., et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer. 1981;48:198-206.
95. Damle R.N., Wasil T., Fais F., et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840-1847.
96. Hamblin T.J., Davis Z., Gardiner A., et al. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848-1854.
97. Hill B.T., Copelan E.A. Acute myeloid leukemia: when to transplant in first complete remission. Curr Hematol Malig Rep. 2010;5:101-108.
98. Saglio G., Baccarani M. First-line therapy for chronic myeloid leukemia: new horizons and an update. Clin Lymphoma Myeloma Leuk. 2010;10:169-176.
99. Zha Y., Li M., Yang J. Dynamic contrast enhanced magnetic resonance imaging of diffuse spinal bone marrow infiltration in patients with hematological malignancies. Korean J Radiol. 2010;11:187-194.
100. Fritz J., Vogel W., Bares R., Horger M. Radiologic spectrum of extramedullary relapse of myelogenous leukemia in adults. AJR Am J Roentgenol. 2007;189:209-218.
101. Rao S., Langston A., Galt J.R., Halkar R.K. Extramedullary acute myeloid leukemia and the use of FDG-PET/CT. Clin Nucl Med. 2009;34:365-366.
102. Konuma T., Ooi J., Takahashi S., et al. Central nervous system leukemia on magnetic resonance imaging. Intern Med. 2008;47:477.
103. Ulu E.M., Tore H.G., Bayrak A., et al. MRI of central nervous system abnormalities in childhood leukemia. Diagn Interv Radiol. 2009;15:86-92.
104. Bruzzi J.F., Macapinlac H., Tsimberidou A.M., et al. Detection of Richter’s transformation of chronic lymphocytic leukemia by PET/CT. J Nucl Med. 2006;47:1267-1273.
105. Escuissato D.L., Gasparetto E.L., Marchiori E., et al. Pulmonary infections after bone marrow transplantation: high-resolution CT findings in 111 patients. AJR Am J Roentgenol. 2005;185:608-615.
106. Caillot D., Casasnovas O., Bernard A., et al. Improved management of invasive pulmonary aspergillosis in neutropenic patients using early thoracic computed tomographic scan and surgery. J Clin Oncol. 1997;15:139-147.
107. Hochberg J., Cairo M.S. Childhood and adolescent lymphoblastic lymphoma: end of the beginning and future directions. Pediatr Blood Cancer. 2009;53:917-919.
108. Nathan P.C., Wasilewski-Masker K., Janzen L.A. Long-term outcomes in survivors of childhood acute lymphoblastic leukemia. Hematol Oncol Clin North Am.. 2009;23:1065-1082. vi-vii
109. Jude V., Chan K.W. Recent advances in hematopoietic stem cell transplantation for childhood acute lymphoblastic leukemia. Curr Hematol Malig Rep. 2010;5:129-134.
110. Deschler B., de Witte T., Mertelsmann R., Lubbert M. Treatment decision-making for older patients with high-risk myelodysplastic syndrome or acute myeloid leukemia: problems and approaches. Haematologica. 2006;91:1513-1522.
111. Chen B.B., Hsu C.Y., Yu C.W., et al. Dynamic contrast-enhanced MR imaging measurement of vertebral bone marrow perfusion may be indicator of outcome of acute myeloid leukemia patients in remission. Radiology. 2011;258:821-831.
112. ten Cate B., de Bruyn M., Wei Y., et al. Targeted elimination of leukemia stem cells: a new therapeutic approach in hemato-oncology. Curr Drug Targets. 2009;11:95-110.
113. Bonnet D., Dick J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-737.
114. Lapidot T., Sirard C., Vormoor J., et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645-648.
115. Castor A., Nilsson L., Astrand-Grundstrom I., et al. Distinct patterns of hematopoietic stem cell involvement in acute lymphoblastic leukemia. Nat Med. 2005;11:630-637.
116. Misaghian N., Ligresti G., Steelman L.S., et al. Targeting the leukemic stem cell: the Holy Grail of leukemia therapy. Leukemia. 2009;23:25-42.
117. Larson R.A., Sievers E.L., Stadtmauer E.A., et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer. 2005;104:1442-1452.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 28 Myeloma and Leukemia
Myeloma
Epidemiology and Risk Factors
Multiple myeloma is a malignant disorder that results from a clonal proliferation of marrow plasma cells that usually results in the production of a monoclonal immunoglobulin in the serum or urine. Approximately 20,180 patients are diagnosed, and 10,650 die, annually in the U.S. with this disorder, making it the second most common hematologic malignancy in the United States.1 The disease most commonly occurs during the seventh and eighth decades of life, is more frequent in men (1.6:1), and has an incidence in African Americans over twice as high as that noted in whites.1
There is no undisputable risk factor for myeloma, but ionizing radiation or work in the paper, pulp, and leather tanning industries and other chemical exposures have been implicated.2 Numerous reports in people with chronic exposure to low-dose ionizing radiation exist, including radiology technicians/physicians prior to routine shielding and radium watch dial painters; perhaps the best evidence was among atomic bomb survivors at Hiroshima in an initial report, but the follow-up of this report failed to confirm those initial findings.3 Similarly, benzene has been implicated, but a thorough retrospective analysis of available reports eliminated this factor as well.4 Hereditary factors have not previously played a prominent role as risk factors for multiple myeloma, but recent reports suggest that at least among certain populations the incidence of monoclonal gammopathy of unknown significance (MGUS)/myeloma may have a familial/genetic association.5
Pathology
Myeloma begins with the clonal expansion of a malignant plasma cell that is usually positive for CD38, CD138, and monoclonal cytoplasmic immunoglobulin with a clonal light chain of either kappa or lambda type. These cells typically have an eccentric nucleus within a large cytoplasm and often have an area of central clearing that corresponds to intracytoplasmic immunoglobulin. Cells of plasmacytic origin are usually negative for other markers of the B-cell lineage such as CD19 and CD20. Recent studies suggest different categories of myeloma, which may separate disease into different levels of outcome.6 Although a heterogeneous array of cytogenetic abnormalities have been described in patients, deletions of chromosomes 13 or 17 p, certain translocations involving chromosome 14, on which the immunoglobulin H (IgH) locus resides [t(4;14), t(14;16)], and chromosome 1 abnormalities have predicted shortened survival; [t(11;14)] has usually been associated with either an average or an improved prognosis and rarely noted to have a subgroup with poor prognosis.6
The abnormal clonal proliferation usually results in the production of monoclonal immunoglobulin in the serum and/or urine; IgG is secreted in approximately 60%, IgA in 20%, IgD in 2%, and IgE in less than 1%; biclonal secretion is rare. Secretion of light chain as the sole monoclonal protein is noted in 18%.7 Previously, 3% of patients were noted to have nonsecretory disease; this number has declined with the advent of serum free light chain (kappa, lambda free kappa:lambda) detection.
Clinical Presentation
Bone disease occurs in nearly 70% of newly diagnosed patients with multiple myeloma and results, in part, from RANKL overexpression and OPG inhibition resulting in unbridled activation of osteoclasts. Other cytokines such as interleukin-1beta (IL-1β), IL-6, and tumor necrosis factor-alpha (TNF-α) also have a role in lytic bone disease.8,9 Progressive bone destruction results in hypercalcemia in approximately 20% of patients with newly diagnosed myeloma.
Marrow infiltration with plasma cells results in a normocytic, normochromic anemia in the majority of patients with previously untreated myeloma. Recently, up-regulation of hepcidin mRNA in myeloma patients has been noted and may play a causative role in this complication.10 In patients with high levels of circulating monoclonal serum protein, stacking of red blood cells on the peripheral blood smear, known as rouleaux formation, may occur.
Nearly one half of patients will develop renal failure at some time during the course of their disease. Cast nephropathy of the distal tubule is the most common cause of this complication, but hypercalcemia, dehydration, hyperuricemia, and concomitant conditions such as amyloidosis and light or heavy chain deposition diseases may also be contributing factors.11
Frequent bacterial infections may also be noted in patients with myeloma due to suppression of uninvolved immunoglobulins, decreased antibody response, and impaired opsonization among other abnormalities reflective of the impaired immune response in patients with multiple myeloma.12,13
Although the diagnosis of myeloma by fixed criteria may be difficult, the International Myeloma Working Group (IMWG) proposed a simple system based on the presence of 10% or greater clonal marrow plasma cells and/or the presence of a serum monoclonal protein 3.0 g/dL or greater.14 The same group developed the “CRAB” (HyperCalcemia, Renal Failure, Anemia, Bone lesions) criteria to determine whether there has been end-organ damage that would classify a patient as having symptomatic disease that would require initiation of treatment.14 These criteria include elevated serum Calcium (≥11.5 mg/dL), Renal insufficiency (creatinine > 2 mg/dL), Anemia (>2 g/dL below the lower limit of normal or < 10 g/dL), Bone lesions (lytic disease or osteoporosis with compression fractures), or other complications (hyperviscosity, amyloidosis, and light chain deposition diseases). Patients without any of these complications are considered asymptomatic and may be observed, without treatment, with frequent follow-up until progression to symptomatic disease.
It is important to distinguish multiple myeloma from related plasma cell dyscrasias such as MGUS and solitary bone plasmacytoma (SBP) or extramedullary plasmacytoma (EMP). The diagnosis of MGUS relies on the presence of fewer than 10% marrow clonal plasma cells and a monoclonal serum protein of less than 3 g/dL in a patient who has no evidence of end-organ/tissue damage, including anemia, hypercalcemia, renal failure, or lytic bone lesions attributable to myeloma and no evidence of a B-cell proliferative disorder.14 SBP or EBP is diagnosed when a single lesion of biopsy-proven clonal plasma cells is detected without evidence of other disease including negative magnetic resonance imaging (MRI) of the spine and bone marrow examination; it is particularly important to identify these patients because 35% to 65% may be curable when given radiation therapy with curative intent.15
Key Points Monoclonal gammopathy of unknown significance
<10% clonal marrow plasma cells
AND serum monoclonal protein less than 3.0 g/dL
• Single lesion of biopsy-proven clonal plasma cells without other evidence of myeloma including negative MRI of the spine and negative bone marrow aspiration.
Staging
Numerous factors have been suggested to determine the prognosis of patients with myeloma. In the 1970s, Durie and Salmon16 proposed a staging system based on the degree of anemia, level of paraprotein, presence or absence of hypercalcemia, lytic lesions, and renal insufficiency that subsequently became the standard staging system for myeloma for several decades (Table 28-1). Although this system was predictive of the degree of myeloma tumor mass, several components were imprecise, such as extent of bone lesions and the inclusion of factors such as anemia, hypercalcemia, and renal function, which may be affected by factors other than myeloma infiltration. Subsequently, multiple new prognostic factors emerged including plasma cell hypodiploidy, C-reactive protein, lactate dehydrogenase, labeling index, cytogenetic abnormalities, and beta-2 microglobulin (β2M).
In 2005, Greipp and coworkers17 proposed the International Staging System (ISS) that separated patients into three stages based on the level of serum albumin and β2M. The data from 10,750 patients were analyzed by univariate and multivariate analysis; the system was designed using half the data set and validated in the other half of the population (Table 28-2). This system effectively identified a group of patients with a relatively short median survival of 29 months (β2M ≥ 5.5 mg/L, stage III), another with longer median survival of 62 months (β2M < 3.5 mg/L and albumin > 3.5 g/dL, stage III), as well as a group with intermediate survival (stage II). Although some patients in the data set may have received thalidomide, the impact on survival of bortezomib, lenalidomide, and multiple combinations utilizing both of these drugs will likely lead to improved median survivals for one or more stages defined by the ISS; the Greek Myeloma Study Group also confirmed the validity of the ISS in the era of novel agents.18 The ISS has subsequently become a standard staging system for patients with myeloma.
Table 28-2 International Staging System for Multiple Myeloma
| STAGE | CRITERIA |
|---|---|
| 1 | β2M < 3.5 mg/L Albumin ≥ 3.5 g/dL |
| 2 | β2M 3.5-5.5 mg/L or β2M < 3.5 mg/L and Albumin < 3.5 g/dL |
| 3 | β2M > 5.5 mg/L |
β2M, beta-2 microglobulin.
From Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23:3412-3420.
Although the Durie-Salmon system and ISS effectively separate patients into different survival categories, the impact of chromosomal abnormalities on prognosis is significant, and some have even suggested a molecular classification of myeloma.6 Certain translocations involving chromosome 14 [t(4;14), t(14;16)] and deletion of chromosome 13 have previously predicted a poor prognosis, but treatment with bortezomib appears to overcome the prognostic impact of these abnormalities; the same data with lenalidomide are premature and need to be validated over time.6,19,20 Deletion of chromosome 17p remains prognostic even in the era of novel agents; other considerations include the poor prognosis of chromosome 1 abnormalities and the possibility of an improved prognosis with t(11;14).6 Although no staging system based on chromosomal abnormalities has been specifically agreed upon, the aforementioned considerations are essential for the determination of prognosis and appropriate treatment decisions.
Imaging
Goals
When a patient is suspected of having an isolated plasmacytoma, imaging initially serves two roles. The first is similar to its role in any focal solid tumor—to define the anatomy of the tumor and its relationship to other structures in order to assist with local treatment planning, which will usually be radiation therapy with curative intent. The second is to determine whether the seemingly isolated plasmacytoma is the only site of disease or whether it is merely the tip of the iceberg in a patient who should properly be classified as having multiple myeloma. To that end, one may weigh and choose between the various forms of systemic imaging, to be discussed later. If no other tumor is found elsewhere, and the patient remains classified as having solitary plasmacytoma, then follow-up imaging also serves two purposes. The first is again similar to its role in other solid tumors—to evaluate the site of the primary disease for evidence of healing or, alternatively, recurrence or progression (Figure 28-1). The second is surveillance for development of multiple myeloma, again by use of systemic imaging along with other criteria.
For patients with multiple myeloma, imaging helps at initial evaluation to distinguish those with asymptomatic or smoldering myeloma, who have no visible bone disease, from those with symptomatic myeloma. Once the patient has been categorized into the symptomatic or asymptomatic group, imaging is used to assess stability versus progression of disease.
Key Points Radiology report
• Identify and characterize focal bone lesions: Number, location, and size when practical. (For multiple lesions, it is often impractical to discuss each separately.)
• Clearly indicate when a bone lesion is thought due to disease other than myeloma, such as a geode due to degenerative joint disease.
• Discuss specific lesions with significantly elevated risk of pathologic fracture.
• Assess subjective bone mineralization—normal, decreased absolutely, decreased allowing for age and gender.
• Look for and discuss compression fractures in the spine.
• Follow-up examinations: Indicate whether the disease is stable or progressive or shows signs of healing to include resolution of fluoro-2-deoxy-D-glucose (FDG) avidity at positron-emission tomography/computed tomography (PET/CT) or decrease in contrast enhancement at MRI.
Techniques
Conventional Radiography
Skeletal surveys have for decades been a mainstay of imaging in multiple myeloma and related diseases and are included in the original Durie and Salmon staging system.16 They consist of a series of images intended to include all the bones that have a reasonable likelihood of developing visible signs of multiple myeloma. There is no generally agreed-upon grouping of images to be included in a skeletal survey, and so there will be variation from one institution to another as to exactly what is included. Based on observation not only of our own practice pattern but also of the assortment of images obtained at outside institutions and then reviewed within our institution, the minimum number and type of views might reasonably be lateral skull, lateral cervical spine, frontal and lateral thoracic and lumbar spine, frontal chest or ribs, frontal pelvis, and frontal views of each humerus and femur. That would be 12 views. The assortment of images suggested by the IMWG21 is listed in a key point text box later. Whole body radiography has been suggested as an alternative to the individual views obtained with skeletal surveys.22
Skeletal surveys have the advantage of being relatively cheap and easy to perform using widely available equipment. They are adequate for finding lytic bone lesions that have reached a sufficient size. Generally that means loss of 30% to 50% of the bone mineralization.23,24 For an individual lesion, the size needed for it to be visible will depend greatly on which bone is involved and where the lesion is in the bone. For example, if a femoral lesion is located along the lateral or medial margin of the bone, it will erode the endosteal surface of the cortex and create a scalloped border that will be noticeable earlier than a similarly sized lesion located either anteriorly or posteriorly, assuming that the bone is being imaged in the frontal projection. Lesions in the pelvis and sacrum often have to attain a size of several centimeters before they are noticed because overlying bowel gas may mask them. In the skull, the presence of normal lucencies due to venous lakes can make it difficult to decide whether a particular lucent area is normal or abnormal.
Skeletal surveys are obtained as part of the initial diagnostic workup of patients with MGUS or suspected SBP because the presence of unexpected lytic bone lesions would necessitate recategorization, probably to multiple myeloma. Skeletal surveys are also obtained in patients with myeloma to help determine the stage of disease when diagnosed. If the skeletal survey shows lytic lesions, more advanced imaging may be unnecessary. The skeletal survey is also useful for searching for progression after treatment.25
Skeletal surveys and other conventional radiographs can reveal disease progression but are of limited utility in assessing response to therapy because lytic bone lesions heal very slowly, if at all24 (Figures 28-2 and 28-3).
Another use of conventional radiography is in assessing the risk of pathologic fracture once a lytic lesion has been identified by any means. For this one usually wants both frontal and lateral views of the area in question (Figure 28-4).
Computed Tomography
CT provides cross-sectional radiographic images of the body and can serve many purposes. Myeloma’s most common anatomic manifestation is in the bones. CT can demonstrate smaller lytic lesions than would be apparent by conventional radiography and can, like conventional radiography, help estimate the risk of pathologic fracture21–23 (Figure 28-5). With attention to the difference in attenuation between tumor (water density) and marrow (fat density), CT can also demonstrate marrow involvement. Despite these capabilities, CT is not commonly used for evaluation of osseous myeloma, although low-dose whole body CT has been suggested as an alternative to the skeletal survey.26 CT is also quite useful for detection and characterization of the uncommon soft tissue manifestations of myeloma or complications of its treatment.
Magnetic Resonance Imaging
MRI is also a cross-sectional imaging technique that differs from CT in having inherently lower spatial resolution but greater contrast resolution, an advantage in imaging the bone marrow. Because of this, its most typical use in myeloma is in staging assessment of bone marrow involvement. Myeloma can involve any bone, but for assessing a large amount of bone marrow in one MRI examination, spine MRI has been traditionally used for myeloma assessment. With the advancement of new techniques using coils adapted to image large areas, imaging from the skull base to the proximal thighs can be done, including assessment of the entire body in one scan. This whole body MRI examination gives a very broad view of the marrow at the cost of spatial resolution. Whole body MRI has recently been tested as a staging method for plasma cell neoplasms. Ghanem and colleagues27 compared whole body MRI and skeletal surveys in patients with either myeloma or MGUS. Among 54 patients, whole body MRI found bone marrow involvement in 10 who had negative skeletal surveys. Walker and associates28 also found MRI to be more sensitive to focal bone lesions than skeletal survey. Evaluating 100 patients with MGUS or myeloma, Bäuerle and coworkers29 compared spinal MRI with whole body MRI and found 9 patients with isolated extra-axial disease that was identified only with whole body MRI.
Patterns of myeloma involvement seen on MRI include normal marrow (which does not exclude low-level infiltration of myelomatous cells), focal involvement (Figure 28-6), diffuse homogeneous involvement (Figure 28-7), and heterogeneous involvement, with combinations of fatty marrow and tiny areas of myeloma creating a salt-and-pepper appearance.30–32 Shorter time to progression and worse response to therapy have been found with diffuse and focal disease as opposed to normal marrow or heterogeneous involvement.32–34 With successful treatment, the marrow pattern should become more normal.24
Positron-Emission Tomography
The purpose of PET/CT varies depending on the stage of disease. In newly diagnosed patients, it helps find sites of disease, both osseous and in the soft tissues, that may be occult by conventional radiography. In this way, it can help distinguish a solitary plasmacytoma from multiple myeloma.35 Kim and colleagues36 found PET to be positive in 13 of 14 known plasmacytomas, and in 1 of 21 patients with suspected SBP, PET imaging resulted in upstaging of the disease to multiple myeloma by disclosing previously unsuspected additional disease. Depending on the equipment available, PET/CT will usually cover at least the head, trunk, and proximal humeri and femurs, and it may be able to include literally the entire body, so like large–field of view MRI, it is a way to include most or all of the body in a more sensitive screening than is available with a bone survey. In patients who have been treated, PET/CT is used to identify sites of disease progression and to distinguish the osseous lesions that are still apparent anatomically but are no longer metabolically active from those that are resistant to treatment and remain active37,38 (Figure 28-8). Complete suppression of abnormal PET activity after treatment may be linked to longer progression-free survival (PFS) than for patients without complete suppression.39 As compared with MRI, PET/CT tends to be less sensitive to diffuse marrow infiltration and perhaps more sensitive to the presence of focal lesions.40
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
