[level-membership-for-hematology-oncology-and-palliative-medicine-category]31
Myeloma
Diagnosis and staging
In asymptomatic (‘smouldering’) myeloma there is generally a serum monoclonal protein >30 g/L and/or bone marrow clonal plasma cells >10% but no related organ or tissue impairment (Figs 31.1 and 31.2). A diagnosis of symptomatic myeloma requires evidence of such impairment; typically increased calcium, renal insufficiency, anaemia, or bone lesions (Table 31.1 and Fig 31.3). Bony disease is increasingly assessed by MRI scanning in addition to traditional X-rays (‘skeletal survey’) (Fig 31.4). Patients who have a paraprotein in the serum but who do not meet the criteria for myeloma are diagnosed as having MGUS. They have a rate of progression to myeloma of 1% per year. Monoclonal gammopathy is associated with other diseases such as lymphoma, non-haematopoietic malignancies and connective tissue disorders but it is also quite common in healthy elderly people (approximately 5% over 70 years of age).
Table 31.1
Diagnostic criteria for symptomatic myeloma
Monoclonal protein in serum and/or urine (Fig 31.3)
Bone marrow clonal plasma cells1 or plasmacytoma (Fig 31.2)
Related organ or tissue impairment (end organ damage including bone lesions)
1If flow cytometry is performed most plasma cells (>90%) will show a ‘neoplastic’ phenotype.
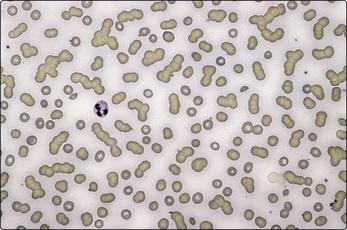
Fig 31.1 The blood film in myeloma.
There is marked rouleaux formation and increased background staining.
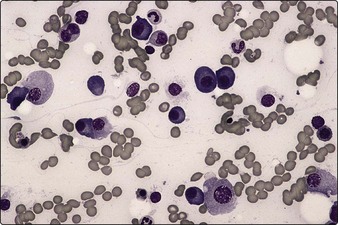
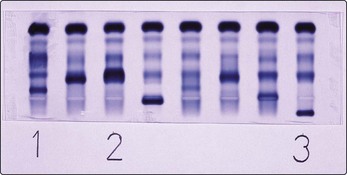
Fig 31.3 Electrophoretic strip showing serum paraprotein bands.
Patient 1 has an IgM paraprotein (Waldenström’s macroglobulinaemia), patient 2 IgA myeloma and patient 3 IgG myeloma.
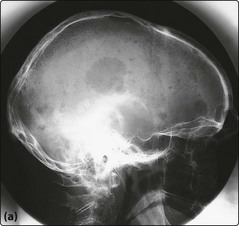
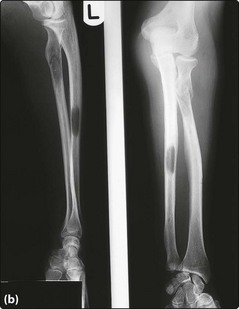
Fig 31.4 X-rays of the skull (a) and left radius and ulna (b) in a patient with myeloma.
Numerous lytic lesions are seen.
The prognosis of myeloma can be predicted from presenting clinical and laboratory features (Table 31.2). The combination of a high β2-microglobulin level and a low albumin level carries a particularly poor prognosis. Cytogenetic and molecular genetic profiles of the malignant cells also predict myeloma behaviour. Hyperdiploid and t(11;14) mutations define standard risk disease while non-hyperdiploid, t(4;14), del(17p) and del(13q) mutations indicate inferior outcome.
Waldenström’s macroglobulinaemia
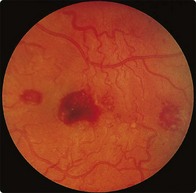
This disease is a form of indolent lymphoma. It is appropriately considered with myeloma as the malignant cells, which show features of lymphocytes and plasma cells, secrete an IgM paraprotein. Patients may complain only of fatigue, but high IgM levels can lead to the ‘hyperviscosity syndrome’, with confusion and neurological symptoms. In these cases retinal examination reveals engorged veins, haemorrhages, exudates (Fig 31.5) and rarely papilloedema. Other possible physical signs include lymphadenopathy and hepatosplenomegaly. Where treatment is required, options include chemotherapy (e.g. chlorambucil or fludarabine) and monoclonal antibodies (e.g. rituximab). Significant hyperviscosity requires plasmapheresis.







[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]31
Myeloma
Diagnosis and staging
In asymptomatic (‘smouldering’) myeloma there is generally a serum monoclonal protein >30 g/L and/or bone marrow clonal plasma cells >10% but no related organ or tissue impairment (Figs 31.1 and 31.2). A diagnosis of symptomatic myeloma requires evidence of such impairment; typically increased calcium, renal insufficiency, anaemia, or bone lesions (Table 31.1 and Fig 31.3). Bony disease is increasingly assessed by MRI scanning in addition to traditional X-rays (‘skeletal survey’) (Fig 31.4). Patients who have a paraprotein in the serum but who do not meet the criteria for myeloma are diagnosed as having MGUS. They have a rate of progression to myeloma of 1% per year. Monoclonal gammopathy is associated with other diseases such as lymphoma, non-haematopoietic malignancies and connective tissue disorders but it is also quite common in healthy elderly people (approximately 5% over 70 years of age).
Table 31.1
Diagnostic criteria for symptomatic myeloma
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
