Mycobacteria
1. Describe the general characteristics of the Mycobacterium spp., including oxygen requirements, staining patterns and cell morphology, artificial media required for cultivation and growth, and pigmentation.
2. Explain the chemical composition of the bacterial cell wall.
3. Explain the microscopic staining characteristics of Mycobacterium spp. using the Gram stain and acid-fast staining methods.
4. List the most common pathogenic species in the Mycobacterium genus and state the natural habitat, mode of transmission, and reservoir for each.
5. Differentiate M. tuberculosis clinical infections based on the signs and symptoms of the following: primary infection, latent infection, disseminated infection, and reactivation.
6. Compare the current safety and containment methods recommended for handling mycobacterial infectious materials and routine bacteriology in a diagnostic laboratory.
7. Describe the purified protein derivative (PPD; also referred to as the tuberculin skin test). What is the significance of a positive result?
8. List the clinical specimens acceptable for recovery of mycobacteria and describe the limitations of recovery from each type of specimen.
9. Justify the use of DNA probes and molecular sequencing or amplification methods to identify Mycobacterium spp.
10. Evaluate the effectiveness of the staining procedures—Kinyoun, Ziehl-Neelsen, and fluorescent staining (auramine-rhodamine or acridine orange)—for identifying mycobacteria.
11. Describe the requirements for using digestion and decontamination procedures to improve the recovery of Mycobacterium spp.
12. Explain the limitations of digestion and decontamination procedures.
13. Explain the methods commonly used for biochemical identification of Mycobacterium spp. (i.e., niacin, nitrate, urease, modified catalase, Tween 80, tellurite, arylsulfatase, thiophene-2-carboxylic acid hydrazide [TCH], and 5% NaCl tests), including the purpose, principle, and control organisms used for each.
14. Describe the role of the human immunodeficiency virus (HIV) and acquired immunodeficiency syndrome (AIDS) in the dissemination and/or pathogenesis of infections with Mycobacterium spp.
15. Explain the recommended susceptibility testing methods and state when susceptibility testing is required or recommended for Mycobacterium spp.
For the most part, mycobacteria can be divided into two major groups, based on fundamental differences in epidemiology and association with disease: those belonging to the Mycobacterium tuberculosis complex and those referred to as nontuberculous mycobacteria (NTM) (Box 43-1).
Mycobacterium Tuberculosis Complex
Epidemiology and Pathogenesis
Epidemiology
M. tuberculosis is the cause of most cases of human tuberculosis, particularly in developed countries. An estimated 1.7 billion people, or one third of the world’s population, are infected with M. tuberculosis. This reservoir of infected individuals results in 8 million new cases of tuberculosis and 2.9 million deaths annually. Tuberculosis continues to be a public health problem in the United States. An additional complicating factor in the management of tuberculosis is the increasing incidence of co-infection with the human immunodeficiency virus (HIV). HIV-associated tuberculosis remains a significant challenge to world health, with an estimated 1.1 million individuals living with HIV-associated tuberculosis. In the United States, tuberculosis typically is found among the poor, homeless, intravenous (IV) drug users, alcoholics, the elderly, or medically underserved populations. Although the organisms belonging to the M. tuberculosis complex have numerous characteristics in common, including extreme genetic homogeneity, they differ in certain epidemiologic aspects (Table 43-1).
TABLE 43-1
Epidemiology of Organisms Belonging to M. tuberculosis Complex That Cause Human Infections
| Organism | Habitat | Primary Route of Transmission | Distribution |
| M. tuberculosis | Patients with cavitary disease are primary reservoir | Person to person by inhalation of droplet nuclei: droplet nuclei containing the organism (infectious aerosols, 1 to 5 µm) are produced when people with pulmonary tuberculosis cough, sneeze, speak, or sing; infectious aerosols may also be produced by manipulation of lesions or processing of clinical specimens in the laboratory. Droplets are so small that air currents keep them airborne for long periods; once inhaled, they are small enough to reach the lungs’ alveoli* | Worldwide |
| M. bovis | Humans and a wide range of host animals, such as cattle, nonhuman primates, goats, cats, buffalo, badgers, possums, dogs, pigs, and deer | Ingestion of contaminated milk from infected cows†; airborne transmission‡ | Worldwide |
| M. africanum | Humans§ | Inhalation of droplet nuclei | East and West tropical Africa; some cases have been identified in the United States |
| M. caprae | Humans rarely; predominately infects a wide range of animals | Inhalation of droplet nuclei | Europe |
| M. microti | Humans rarely; small animals (e.g., voles and other wild rodents) | Inhalation of droplet nuclei | Europe; Great Britain, Netherlands |
| M. canettii | Natural reservoir has not been clearly defined. Rarely infects humans. | Unclear | Africa |
| M. pinnipedii | Humans rarely; predominantly infects a wide range of animals | Unclear | Europe |
*Infection occasionally can occur through the gastrointestinal tract or skin.
†The incidence has decreased significantly in developed countries since the introduction of universal pasteurization of milk and milk products and the institution of effective control programs for cattle.
‡Can be transmitted human to human, animal to human, and human to animal.
Pathogenesis
The pathogenesis of tuberculosis caused by organisms of the M. tuberculosis complex is discussed in Chapter 69. Inhalation of a single viable organism has been shown to lead to infection, although close contact is usually necessary. Of those who become infected with M. tuberculosis, 15% to 20% develop disease. The disease usually occurs some years after the initial infection, when the patient’s immune system breaks down for some reason other than the presence of tuberculosis bacilli in the lung. In a small percentage of infected hosts, the disease becomes systemic, affecting a variety of organs.
Nontuberculous Mycobacteria
The NTM include all mycobacterial species that do not belong to M. tuberculosis complex. Currently, approximately 130 species of nontuberculous mycobacteria have been recognized. The members of this large group of mycobacteria have been known by several names (Box 43-2). Significant geographic variability is seen both in the prevalence of and the species responsible for NTM disease. As previously mentioned, NTM are present everywhere in the environment and sometimes colonize the skin and respiratory and gastrointestinal tracts of healthy individuals. Little is known about how infection is acquired, but some mechanisms appear to be trauma, inhalation of infectious aerosols, and ingestion; a few diseases are nosocomial or are acquired as an iatrogenic infection. In contrast to M. tuberculosis complex, NTM are not usually transmitted from person to person, nor does isolation of these organisms necessarily mean they are associated with a disease process. Interpretation of a positive NTM culture is complicated, because these organisms are widely distributed in nature, their pathogenic potential varies greatly from one species to another, and humans can be colonized by these mycobacteria without necessarily developing infection or disease. With few exceptions, little is known about the pathogenesis of infections caused by these bacterial agents.
In 1959 Runyon1 classified NTM into four groups (Runyon groups I to IV) based on the phenotypic characteristics of the various species, most notably the growth rate and colonial pigmentation (Table 43-2). Runyon’s system first categorizes the slow-growing NTM (Runyon groups I to III) and then the rapid-growers (Runyon group IV). One other NTM, M. leprae, which cannot be cultivated on artificial media, is also reviewed. (As with many classification schemes, the Runyon classification does not always hold true. For example, some NTM can be either a photochromogen or a nonphotochromogen.)
TABLE 43-2
Runyon Classification of Nontuberculous Mycobacteria (NTM)
| Runyon Group Number | Group Name | Description |
| I | Photochromogens | NTM colonies that develop pigment on exposure to light after being grown in the dark and take longer than 7 days to appear on solid media |
| II | Scotochromogens | NTM colonies that develop pigment in the dark or light and take longer than 7 days to appear on solid media |
| III | Nonphotochromogens | NTM colonies that are nonpigmented regardless of whether they are grown in the dark or light and take longer than 7 days to appear on solid media |
| IV | Rapid growers | NTM colonies that grow on solid media and take fewer than 7 days to appear |
Slow-Growing Nontuberculous Mycobacteria
Photochromogens
The photochromogens (Table 43-3) are slow-growing NTM that produce colonies that require light to form pigment.
TABLE 43-3
Characteristics of Nontuberculous Mycobacteria—Photochromogens
| Organism | Epidemiology | Pathogenicity | Type of Infection |
| M. kansasii | Infection more common in white males; natural reservoir is tap water; aerosols are involved in transmission | Potentially pathogenic | Chronic pulmonary disease; extrapulmonary diseases, such as cervical lymphadenitis and cutaneous disease |
| M. asiaticum | Not commonly encountered (primarily seen in Australia) | Potentially pathogenic | Pulmonary disease |
| M. marinum | Natural reservoirs are freshwater and saltwater as a result of contamination from infected fish and other marine life. Transmission is by contact with contaminated water and organism entry by means of trauma or small breaks in the skin; associated with aquatic activity usually involving fish | Potentially pathogenic | Cutaneous disease; bacteremia |
| M. intermedium | Unknown | Potentially pathogenic | Pulmonary disease |
| M. novocastrense | Unknown | Potentially pathogenic | Cutaneous disease |

Scotochromogens
The scotochromogens (Table 43-4) are slow-growing NTM that produce pigmented colonies whether grown in the dark or the light. The epidemiology of the potentially pathogenic scotochromogens has not been definitively described. In contrast to potentially pathogenic nonphotochromogens, these agents are rarely recovered in the clinical laboratory.
TABLE 43-4
Characteristics of Nontuberculous Mycobacteria—Scotochromogens
| Organism | Epidemiology/Habitat | Pathogenicity | Type of Infection |
| M. szulgai | Water and soil | Potentially pathogenic | Pulmonary disease, predominantly in middle-aged men; cervical adenitis; bursitis |
| M. scrofulaceum | Raw milk, soil, water, dairy products | Potentially pathogenic | Cervical adenitis in children, bacteremia, pulmonary disease, skin infections |
| M. interjectum | Unknown | Potentially pathogenic | Chronic lymphadenitis, pulmonary disease |
| M. heckeshornense | Unknown | Potentially pathogenic | Pulmonary disease (rare) |
| M. tusciae | Unknown—isolated from tap water | Potentially pathogenic | Cervical lymphadenitis (rare) |
| M. kubicae | Unknown | Potentially pathogenic | Pulmonary disease |
| M. gordonae | Tap water, water, soil | Nonpathogenic* | NA |
| M. cookie | Sphagnum moss, surface waters in New Zealand | Nonpathogenic* | NA |
| M. hiberniae | Sphagnum moss, soil in Ireland | Nonpathogenic* | NA |
NA, Not applicable.
Nonphotochromogens
The nonphotochromogens (Table 43-5) are slow-growing NTM that produce unpigmented colonies whether grown in the dark or the light. Of the organisms in this group, M. terrae complex (M. terrae, M. triviale, and M. nonchromogenicum) and M. gastri are considered nonpathogenic for humans. The other nonphotochromogens are considered potentially pathogenic, and many are frequently recovered in the clinical laboratory. The nonphotochromogens belonging to Mycobacterium avium complex are frequently isolated in the clinical laboratory and are able to cause infection in the human host.
TABLE 43-5
| Organism | Epidemiology | Type of Infection |
| M. avium complex | Environmental sources, including natural waters, and soil | Patients without AIDS: Pulmonary infections in patients with preexisting pulmonary disease; cervical lymphadenitis; and disseminated disease* in immunocompromised patients who are HIV negative Patients with AIDS: Disseminated disease |
| M. xenopi† | Water, especially hot water taps in hospitals; believed to be transmitted in aerosols | Primarily pulmonary infections in adults; less common, extrapulmonary infections (bone, lymph nodes, sinus tract) and disseminated disease |
| M. ulcerans | Stagnant tropical waters; also harbored in an aquatic insect’s salivary glands; infections occur in tropical or temperate climates | Indolent cutaneous and subcutaneous infections (African Buruli ulcer or Australian Bairnsdale ulcer) |
| M. malmoense | Most cases from England, Wales, and Sweden. Rarely isolated from patients infected with HIV. Little is known about epidemiology; to date, isolated only from humans and captured armadillos | Chronic pulmonary infections, primarily in patients with preexisting disease; cervical lymphadenitis in children; less common, infections of the skin or bursae |
| M. genovense | Isolated from pet birds and dogs. Mode of acquisition unknown | Disseminated disease in patients with AIDS (wasting disease characterized by fever, weight loss, hepatosplenomegaly, anemia) |
| M. haemophilum | Unknown | Disseminated disease; cutaneous infections in immunosuppressed adults; mild and limited skin infections in preadolescence or early adolescence; cervical lymphadenitis in children |
| M. heidelbergense | Unknown | Lymphadenitis in children; also isolated from sputum, urine, and gastric aspirate |
| M. shimoidei | To date has not been isolated from environmental sources; few case reports, but widespread geographically | Tuberculosis-like pulmonary infection; disseminated disease |
| M. simiae | Tap water and hospital water tanks; rarely isolated | Tuberculosis-like pulmonary infection |
AIDS, Acquired immunodeficiency syndrome; HIV, human immunodeficiency virus.
*Disseminated disease can involve multiple sites, such as bone marrow, lungs, liver, lymph nodes.
Other Nonphotochromogens.
Several other mycobacterial species that are considered nonphotochromogens are potentially pathogenic in humans. The epidemiology and spectrum of disease for these organisms are summarized in Table 43-5. In addition to the species in this table, other, newer species of mycobacteria that are nonphotochromogens have been described, such as M. celatum and M. conspicuum. These newer agents appear to be potentially pathogenic in humans.
Rapidly Growing Nontuberculous Mycobacteria (RGM)
General Characteristics
The large group of organisms that constitute the RGM is divided into six major groups of potentially pathogenic species, based on pigmentation and molecular studies (see Box 43-1). Unlike the majority of other mycobacteria, most rapid-growers can grow on routine bacteriologic media and on media specific for cultivation of mycobacteria. On Gram staining, these organisms appear as weakly gram-positive rods resembling diphtheroids.
Spectrum of Disease
The spectrum of disease caused by the most commonly encountered rapid-growers is summarized in Table 43-6. The most common infection associated with RGM is posttraumatic wound infection. An increase in wound infections has been associated with planktonic M. abscessus, which can be identified as a rough colonial phenotype on artificial media; these organisms are capable of infecting macrophages. The smooth colonial phenotype typically is identified in biofilms and lacks infectivity.
TABLE 43-6
Common Types of Infections Caused by Rapidly Growing Mycobacteria
| Organism | Common Types of Infection |
| M. abscessus subsp. abscessus | Disseminated disease, primarily in immunocompromised individuals; skin and soft tissue infections; pulmonary infections; postoperative infections |
| M. fortuitum | Postoperative infections in breast augmentation and median sternotomy; skin and soft tissue infections; pulmonary infections, usually single. localized lesions. Central nervous system (CNS) disease is rare but has high morbidity and mortality |
| M. chelonae | Skin and soft tissue infections, postoperative wound infections, keratitis |
| Less Common Types of Infection (More Than 10 Cases) | |
| M. peregrinum | Skin and soft tissue infections; bacteremia |
| M. mucogenicum | Posttraumatic wound infections, catheter-related sepsis, health care associated |
| M. smegmatis | Skin or soft tissue infections; less frequently, pulmonary infections |
| M. abscessus subsp. bolletii | Health care–associated infections, skin and soft tissue infections, pulmonary infections |
| M. boenickei | Bone and joint infections |
| M. canariasense | Bacteremia |
| M. cosmeticum | Pulmonary and urosepsis |
| M. goodii | Bone and joint infections, osteomyelitis |
| M. houstonense | Bone and joint infections |
| M. immunogenum | Hypersensitivity pneumonitis |
| M. neoaurum (closely related to M. lacticola) | Catheter-related sepsis |
| M. porcinum | Surgical site infection |
| M. senegalense | Catheter-related sepsis |
| Rare Infections (Fewer Than 10 Cases) | |
| M. aubagnense | Various opportunistic health care–associated infections |
| M. brisbanense | Various opportunistic health care–associated infections |
| M. brumae | Various opportunistic health care–associated infections |
| M. elephantis | Various opportunistic health care–associated infections |
| M. mageritense | Skin and soft tissue infections |
| M. monacense | Various opportunistic health care–associated infections |
| M. moriokaense | Various opportunistic health care–associated infections |
| M. neworleansense | Various opportunistic health care–associated infections |
| M. novocastrense | Various types of opportunistic health care–associated infections |
| M. phocaicum | Catheter-related sepsis |
| M. septicum | Various opportunistic health care–associated infections |
| M. setense | Bone and joint infections |
| M. wolinskyi | Skin and soft tissue infections, bone infection, osteomyelitis |
Laboratory Diagnosis of Mycobacterial Infections
Specimen Collection and Transport
Urine Specimens
Early morning voided urine specimens (40 mL minimum) in sterile containers should be submitted daily for at least 3 days. The collection procedure is the same as for collecting a clean-catch midstream urine specimen (see Chapter 73). The 24-hour urine specimen is undesirable because of excessive dilution, higher contamination, and difficulty in concentrating. Catheterization should be used only if a midstream voided specimen cannot be collected.
Blood Specimens
Immunocompromised patients, particularly those infected with HIV, can have disseminated mycobacterial infection; most of these infections are caused by M. avium complex. A blood culture positive for MAC is always associated with clinical evidence of disease. Recovery of mycobacteria is improved with blood collection in either a broth or the Isolator lysis-centrifugation system (see Chapter 68). Some studies have indicated that the lysis-centrifugation system is advantageous, because quantitative data can be obtained with each blood culture; in patients with AIDS, quantitation of such organisms can be used to monitor therapy and determine the prognosis. However, the necessity of quantitative blood cultures remains unclear.
Specimen Processing
Processing to recover acid-fast bacilli from clinical specimens involves several complex steps, each of which must be carried out with precision. Specimens from sterile sites can be inoculated directly to media (small volume) or concentrated to reduce volume. Other specimens require decontamination and concentration. A processing scheme is shown in Figure 43-1, and the procedures are explored in detail in the following discussions.
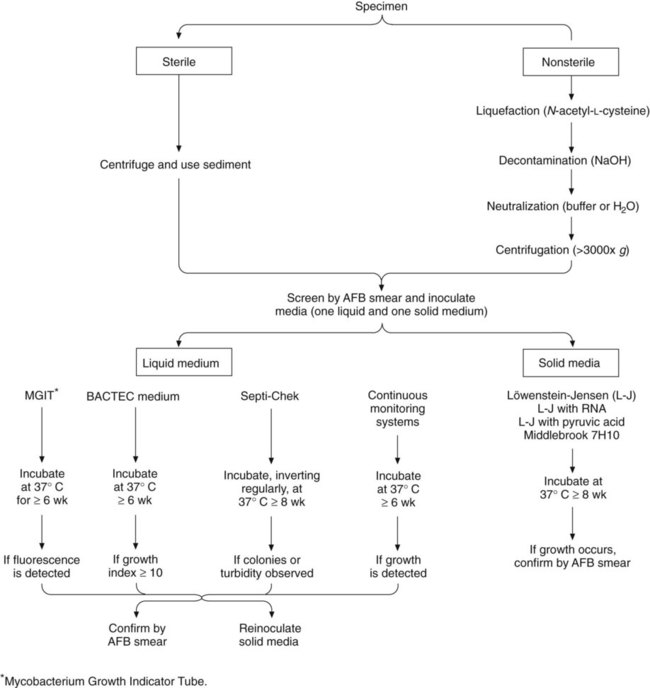
Inadequate Specimens and Rejection Criteria
Overview.
Commonly used digestion-decontamination methods are the NaOH method, the Zephiran-trisodium phosphate method, and the N-acetyl-L-cysteine (NALC)–2% NaOH method. The NALC-NaOH method is presented in detail in Procedure 43-1, which can be found on the Evolve site. Another decontaminating procedure that uses oxalic acid is very useful for treating specimens known to harbor gram-negative rods, particularly Pseudomonas and Proteus spp., which are extremely troublesome contaminants. It is important to note that oxalic acid, NaOH, and mild hydrogen chloride (HCl) may reduce the recovery of M. ulcerans.
Procedure 43-1 N-Acetyl-L-Cysteine–Sodium Hydroxide Method for Liquefaction and Decontamination of Specimens




Add 50 mL of solution B to 50 mL of solution A and adjust the pH to 6.8.
2. Work in a biologic safety cabinet (BSC) and wear protective clothing, gloves, and a mask. Transfer a maximum of 10 mL of sputum, urine, or other fluid to be processed to a sterile, disposable, plastic, 50-mL conical centrifuge tube with a leak-proof and aerosol-free plastic screw cap. Tubes with easily visible volume indicator marks are best.
3. Add an equal volume of freshly prepared digestant to the tube; take great care when pouring the digestant not to touch the lip of the specimen container, because this might inadvertently transfer positive material to a negative specimen. Tighten the cap completely.
4. Vortex the specimen for approximately 15 seconds or for a maximum of 30 seconds; make sure to create a vortex in the liquid and not merely agitate the material. Check for homogeneity by inverting the tube. If clumps remain, vortex the specimen intermittently while the rest of the specimens are being digested. An extra pinch of NALC crystals may be necessary to liquefy mucoid sputa.
5. Start a 15-minute timer when the first specimen is finished being vortexed. Continue digesting the other specimens, noting the time required for the entire run. The digestant should remain on the specimens for a maximum exposure of 20 minutes.
6. After 15 minutes of digestion, add enough phosphate buffer to reach within 1 cm of the top, screw the cap tightly closed, and invert the tube to mix the solutions and stop the digestion process. Addition of this solution also reduces the specific gravity of the specimen, aiding sedimentation of the bacilli during centrifugation.
7. Centrifuge all tubes at 3600× g for 15 minutes, using aerosol-free, sealed centrifuge cups.
8. Carefully pour off the supernatant into a splash-proof container. To ensure that the specimen does not run down the outside of the tube after pouring, wipe the lip of the tube with Amphyl- or phenol-soaked gauze to absorb drips. Be careful not to touch the lip of any tube to another container. It is helpful to watch the sediment carefully as the supernatant is being decanted, because a very mucoid sediment may be loose and may pour out with the supernatant. If the sediment begins to slip, stop decanting and use a sterile capillary pipette to remove the supernatant without losing the sediment.
9. Suspend the sediment in 1 to 2 mL of phosphate buffer, pH 6.8 buffer (with bovine serum albumin [BSA]).
10. Inoculate the sediment to culture media and prepare slides.
Direct Detection Methods
Microscopy
Acid-Fast Stains
When Gram stained, mycobacteria usually appear as slender, poorly stained, beaded, gram-positive bacilli (Figure 43-2); sometimes they appear as “gram neutral,” or “gram-ghosts,” by failing to take up either crystal violet or safranin. Acid-fastness is affected by the age of colonies, the medium on which growth occurs, and exposure to ultraviolet light. Rapidly growing species appear to be acid-fast variable.
Three types of staining procedures are used in the laboratory for rapid detection and confirmation of acid-fast bacilli: fluorochrome, Ziehl-Neelsen, and Kinyoun. Smears for all methods are prepared in the same way (see Procedure 43-2 on the Evolve site).
Methods
Fluorochrome Stain.
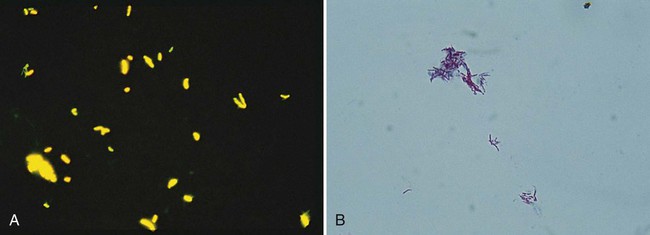
Fluorochrome staining is the screening procedure recommended for laboratories that have a fluorescent (ultraviolet) microscope (see Procedure 43-3 on the Evolve site). Fluorochrome stain is more sensitive than the conventional carbolfuchsin stains, because the fluorescent bacilli stand out brightly against the background (Figure 43-3). Because the smear can be examined initially at lower magnifications (×250 to ×400), more fields can be visualized in a short period. In addition, a positive fluorescent smear may be restained using the conventional Ziehl-Neelsen or Kinyoun procedure, thereby saving the time needed to make a fresh smear. Screening of specimens with rhodamine or rhodamine-auramine results in a higher yield of positive smears and substantially reduces the time needed to examine smears.
Fuchsin Acid-Fast Stains.
The classic carbolfuchsin stain (Ziehl-Neelsen) requires heating of the slide for better penetration of the stain into the mycobacterial cell wall; hence, it is also known as the hot stain procedure (see Procedure 6-3 on the Evolve site). With Ziehl-Neelsen staining, Mycobacterium spp. appear red or have a red-blue, beaded appearance, whereas nonmycobacteria appear blue.
Procedure 6-4, which can be found on the Evolve site, describes the Kinyoun acid-fast stain. The method is similar to Ziehl-Neelsen staining, but no heat is used (see Figure 43-3); this technique is known as the cold stain procedure. If present, typical acid-fast bacilli appear as purple to red, slightly curved, short or long rods (2 to 8 µm); they also may appear beaded or banded (M. kansasii). For some nontuberculous species, such as M. avium complex, they appear pleomorphic, usually coccoid.
Examination, Interpretation, and Reporting of Smears.
When acid-fast organisms are observed on a smear, the report should include information about the type of staining method used and the quantity of organisms. The recommended interpretations and ways to report smear results are shown in Table 43-7.
TABLE 43-7
| Number of AFB Seen Fuchsin Stain (1000× Magnification) |
Number of AFB Seen Fluorochrome Stain (450× Magnification) |
Number of AFB Seen Fluorochrome Stain (250× Magnification) |
Report |
| 0 | 0 | 0 | No AFB seen |
| 1-2/300 fields | 1-2/70 fields | 1-2/30 fields | Doubtful; request another specimen |
| 1-9/100 fields | 2-18/50 fields | 1-9/10 fields | 1+ |
| 1-9/10 fields | 4-36/10 fields | 1-9/field | 2+ |
| 1-9/field | 4-36/field | 10-90/field | 3+ |
| >9/field | >36/field | >90/field | 4+ |

AFB, Acid-fast bacilli.
Modified from Kent PT, Kubica GP: Public health mycobacteriology: a guide for the level III laboratory, US Department of Health and Human Service, Public Health Service, Washington, DC, 1985, Centers for Disease Control and Prevention; and Versalovic J: Manual of clinical microbiology, ed 10, 2011, Washington, DC, ASM Press.
Genetic Sequencing and Nucleic Acid Amplification
Currently no molecular assays are available for direct detection of nontuberculous mycobacteria. In 2004, the Centers for Disease Control established a national tuberculosis genotyping system. Details and updates are available at http://www.cdc.gov/tb/programs/default.htm.
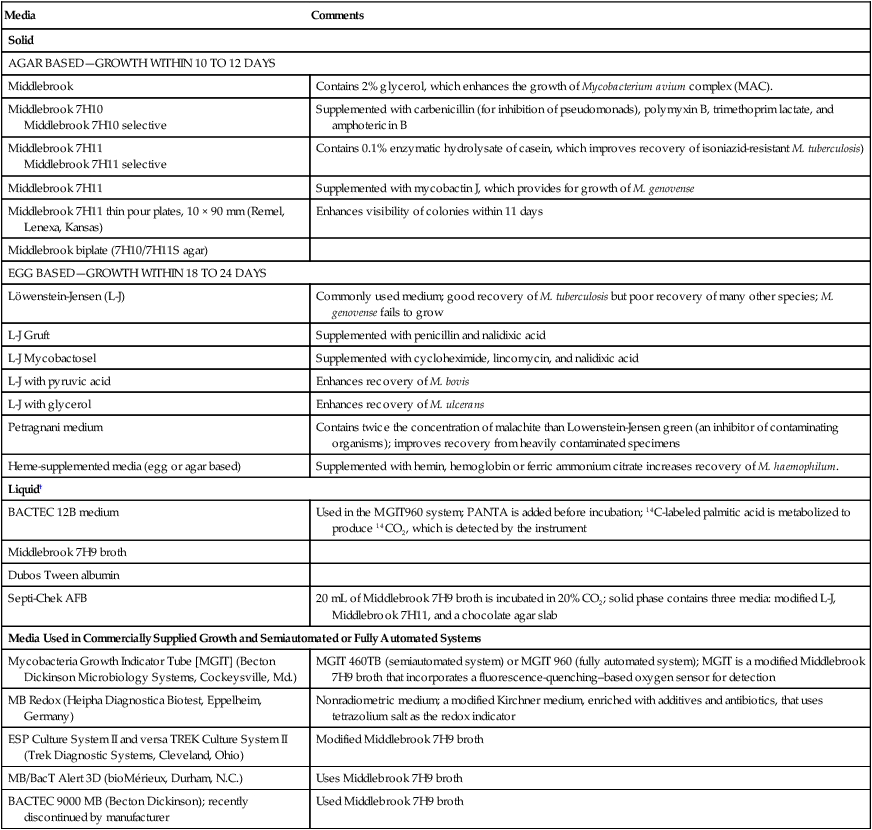
Solid Media
Solid media, such as those listed in Box 43-3, are recommended because of the development of characteristic, reproducible colonial morphology, good growth from small inocula, and a low rate of contamination. Optimally, at least two solid media (a serum [albumin] agar base medium, [e.g., Middlebrook 7H10] and an egg-potato base medium [e.g., Löwenstein-Jensen, or L-J]) should be used for each specimen (these media are available from commercial sources). All specimens must be processed appropriately before inoculation. It is imperative to inoculate test organisms to commercially available products for quality control (see Procedure 43-4 on the Evolve site).
Box 43-3 Suggested Media for Cultivation of Mycobacteria from Clinical Specimens*
| Media | Comments |
| Solid | |
| AGAR BASED—GROWTH WITHIN 10 TO 12 DAYS | |
| Middlebrook | Contains 2% glycerol, which enhances the growth of Mycobacterium avium complex (MAC). |
| Middlebrook 7H10 Middlebrook 7H10 selective |
Supplemented with carbenicillin (for inhibition of pseudomonads), polymyxin B, trimethoprim lactate, and amphotericin B |
| Middlebrook 7H11 Middlebrook 7H11 selective |
Contains 0.1% enzymatic hydrolysate of casein, which improves recovery of isoniazid-resistant M. tuberculosis) |
| Middlebrook 7H11 | Supplemented with mycobactin J, which provides for growth of M. genovense |
| Middlebrook 7H11 thin pour plates, 10 × 90 mm (Remel, Lenexa, Kansas) | Enhances visibility of colonies within 11 days |
| Middlebrook biplate (7H10/7H11S agar) | |
| EGG BASED—GROWTH WITHIN 18 TO 24 DAYS | |
| Löwenstein-Jensen (L-J) | Commonly used medium; good recovery of M. tuberculosis but poor recovery of many other species; M. genovense fails to grow |
| L-J Gruft | Supplemented with penicillin and nalidixic acid |
| L-J Mycobactosel | Supplemented with cycloheximide, lincomycin, and nalidixic acid |
| L-J with pyruvic acid | Enhances recovery of M. bovis |
| L-J with glycerol | Enhances recovery of M. ulcerans |
| Petragnani medium | Contains twice the concentration of malachite than Lowenstein-Jensen green (an inhibitor of contaminating organisms); improves recovery from heavily contaminated specimens |
| Heme-supplemented media (egg or agar based) | Supplemented with hemin, hemoglobin or ferric ammonium citrate increases recovery of M. haemophilum. |
| Liquid† | |
| BACTEC 12B medium | Used in the MGIT960 system; PANTA is added before incubation; 14C-labeled palmitic acid is metabolized to produce 14CO2, which is detected by the instrument |
| Middlebrook 7H9 broth | |
| Dubos Tween albumin | |
| Septi-Chek AFB | 20 mL of Middlebrook 7H9 broth is incubated in 20% CO2; solid phase contains three media: modified L-J, Middlebrook 7H11, and a chocolate agar slab |
| Media Used in Commercially Supplied Growth and Semiautomated or Fully Automated Systems | |
| Mycobacteria Growth Indicator Tube [MGIT] (Becton Dickinson Microbiology Systems, Cockeysville, Md.) | MGIT 460TB (semiautomated system) or MGIT 960 (fully automated system); MGIT is a modified Middlebrook 7H9 broth that incorporates a fluorescence-quenching–based oxygen sensor for detection |
| MB Redox (Heipha Diagnostica Biotest, Eppelheim, Germany) | Nonradiometric medium; a modified Kirchner medium, enriched with additives and antibiotics, that uses tetrazolium salt as the redox indicator |
| ESP Culture System II and versa TREK Culture System II (Trek Diagnostic Systems, Cleveland, Ohio) | Modified Middlebrook 7H9 broth |
| MB/BacT Alert 3D (bioMérieux, Durham, N.C.) | Uses Middlebrook 7H9 broth |
| BACTEC 9000 MB (Becton Dickinson); recently discontinued by manufacturer | Used Middlebrook 7H9 broth |
*For optimal recovery of mycobacteria, a minimum combination of liquid medium and solid media is recommended.
†Tween 80 added to liquid media acts as a surfactant, breaking up clumps of organisms and increasing recovery rates.
Example of Interpreting Quality Control Test Results of Decontamination and Concentration Procedure
| SPUTUM specimen | |||||||
| Unprocessed-Quantification of Growth | Processed Quantification of Growth | ||||||
| Sputum Sample | 104 | 103 | 102 | 104 | 103 | 102 | Interpretation |
| 1 | 3+ | 2+ | 50-100 colonies | 2+ | 1+ or 2+ | Approximately 10 colonies | Media and decontamination procedures acceptable |
| 2 | 3+ | 2+ | 50-100 colonies | 1+ | 0 | 0 | Media acceptable; procedures too toxic |
| 3 | 2+ or 1+ | 2+ or 1+ | 0 | 1+ or 0 | 1+ or 0 | 0 | One or more of the media are not supporting growth of acid-fast bacilli (AFB) adequately |
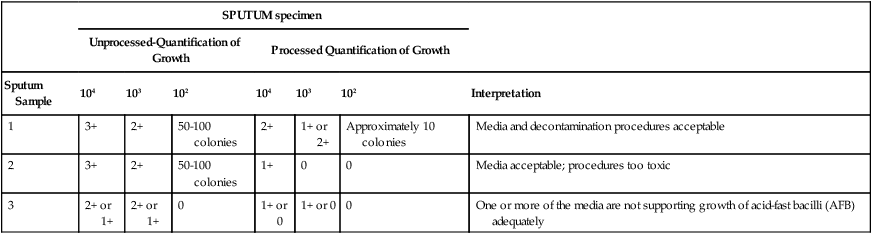
0, No growth; 1+, scanty, barely discernible countable colonies; 2+, dense, discrete growth, not countable; 3+, confluent, abundant growth.
Cultures are examined weekly for growth. Contaminated cultures are discarded and reported as “contaminated, unable to detect presence of mycobacteria”; additional specimens are also requested. If available, sediment may be recultured after enhanced decontamination or by inoculating the sediment to a more selective medium. Most isolates appear between 3 and 6 weeks; a few isolates appear after 7 or 8 weeks of incubation. When growth appears, the rate of growth, pigmentation, and colonial morphology are recorded. The typical colonial appearance of M. tuberculosis and other mycobacteria is shown in Figure 43-4. After 8 weeks of incubation, negative cultures (those showing no growth) are reported, and the cultures are discarded.

Liquid Media
In general, use of a liquid media system reduces the turnaround time for isolation of acid-fast bacilli to approximately 10 days, compared with 17 days or longer for conventional solid media. Several different systems are available for culturing and detecting the growth of mycobacteria in liquid media. The most commonly used systems are summarized in Table 43-8. Growth of mycobacteria in liquid media, regardless of the type, requires 5% to 10% CO2; CO2 is either already provided in the culture vials or is added according to the manufacturer’s instructions. When growth is detected in a liquid medium, acid-fast staining of a culture aliquot is performed to confirm the presence of acid-fast bacilli, and the material is subcultured to solid agar. Gram staining can also be performed if contamination is suspected.
TABLE 43-8
Liquid Media Systems Commonly Used to Culture and Detect the Growth of Mycobacteria
| System | Basic Principles of Detection |
| BACTEC 460 TB (Becton Dickinson Diagnostic Systems, Cockeysville, Md.) | Culture medium contains 14C-labeled palmitic acid. If present in the broth, mycobacteria metabolize the 14C-labeled substrates and release radioactively labeled 14CO2 in the atmosphere, which collects above the broth in the bottle. The instrument withdraws this carbon dioxide (CO2)-containing atmosphere and measures the amount of radioactivity present. Bottles that yield a radioactive index, called a growth index, greater than or equal to 10 are considered positive. |
| Septi-Chek AFB System (Becton Dickinson) | Biphasic culture system made up of a modified Middlebrook 7H9 broth with a three-sided paddle containing chocolate, egg-based, and modified 7H11 solid agars. After inoculation, a supplement is added to the liquid that includes glucose, glycerol, oleic acid, pyridoxal HCl, catalase, albumin, and antibiotics (PANTA). The bottle is inverted regularly to inoculate the solid media. Growth is detected by observing the three-sided paddle. |
| Mycobacteria Growth Indicator Tube (MGIT) (Becton Dickinson) | A culture tube contains Middlebrook 7H9 broth and a fluorescent compound embedded in a silicone sensor. Growth is detected visually using an ultraviolet light. Oxygen (O2) diminishes the fluorescent output of the sensor; therefore, O2 consumption by organisms present in the medium is detected as an increase in fluorescence under ultraviolet (UV) light at 365 nm. The MGIT medium is supplemented with oleic acid-albumin-dextrose and PANTA before incubation. |
| Continuous Growth Monitoring Systems | |
| Versa TREK (TREK Diagnostic Systems, Cleveland, Ohio) | Organisms are cultured in a modified Middlebrook 7H9 broth with enrichment and a cellulose sponge to increase the culture’s surface area. The instrument detects growth by monitoring pressure changes that occur as a result of O2 consumption or gas production by the organisms as they grow. |
| BacT/Alert System (bioMérieux, Durham, N.C.) | Organisms are cultured in modified Middlebrook 7H9 broth. The instrument detects growth by monitoring CO2 production by means of a colorimetric CO2 sensor in each bottle. |
| BACTEC 9000 MB (Becton Dickinson- recently discontinued) | Organisms are cultured in a modified Middlebrook 7H9 broth. The instrument detects growth by monitoring O2 consumption by means of a fluorescent sensor. |
| BACTEC MGIT 960 (Becton Dickinson) | See above for basic principle (MGIT). The instrument detects growth by monitoring O2 consumption by means of a fluorescent sensor. |
| MB Redox (Heipha Diagnostica Biotest, Eppelheim, Germany) | This is a nonradiometric medium. It is a modified Kirchner medium enriched with additives and antibiotics, and tetrazolium salt is the redox indicator. AFB are identified as pink to purple, pinhead-sized particles. |
Approach to Identification
Conventional Phenotypic Tests
Growth Characteristics.
Preliminary identification of mycobacterial isolates depends on the organisms’ rate of growth, colonial morphology (see Figure 43-4), colonial texture, pigmentation and, in some instances, the permissive incubation temperatures of mycobacteria. Despite the limitations of phenotypic tests, the mycobacterial growth characteristics are helpful for determining a preliminary identification (e.g., an isolate appears as rapidly growing mycobacteria). To perform identification procedures, quality control organisms should be tested along with unknowns (Table 43-9). The commonly used quality control organisms can be maintained in broth at room temperature and transferred monthly. In this way they are always be available for inoculation to test media along with suspensions of the unknown mycobacteria being tested.
TABLE 43-9
Controls and Media Used for Biochemical Identification of Mycobacteria
| CONTROL ORGANISMS | RESULT | ||||||
| Biochemical Test | Positive | Negative | Positive | Negative | Medium Used | Duration | Incubation Conditions |
| Niacin | M. tuberculosis | M. intracellulare | Yellow | No color change | 0.5 mL DH2O | 15-30 min | Room temperature |
| Nitrate | M. tuberculosis | M. intracellulare | Pink or red | No color change | 0.3 mL DH2O | 2 hours | 37°C bath |
| Urease | M. fortuitum | M. avium | Pink or red | No color change | Urea broth for AFB | 1, 3, and 5 days | 37°C incubator (without CO2) |
| 68°C Catalase | M. fortuitum or M. gordonae | M. tuberculosis | Bubbles | No bubbles | 0.5 mL phosphate buffer (pH, 7.0) | 20 min | 68°C bath |
| SQ Catalase | M. kansasii or M. gordonae | M. avium | >45 mm | <45 mm | Commercial medium | 14 days | 37°C incubator (with CO2) |
| Tween 80 | M. kansasii | M. intracellulare | Pink or red | No color change | 1 mL DH2O | 5 or 10 days | 37°C incubator (in the dark, without CO2) |
| Tellurite | M. avium | M. tuberculosis | Smooth, fine, black precipitate (smokelike action) | Gray clumps (no smokelike action) | Middlebrook 7H9 broth | 7, then 3 additional days | 37°C incubator (with CO2) |
| Arylsulfatase | M. fortuitum | M. intracellulare | Pink or red | No color change | Wayne’s arylsulfatase medium | 3 days | 37°C incubator (without CO2) |
| 5% NaCl | M. fortuitum | M. gordonae | Substantial growth | Little or no growth | Commercial slant with and without 5% NaCl | 28 days | 37°C incubator (with CO2) |
| TCH | M. bovis | M. tuberculosis | No growth (i.e., susceptible) | Growth (i.e., resistant or ≥1% of colonies are resistant) | TCH slant | 3 weeks | 37°C incubator (with CO2) |
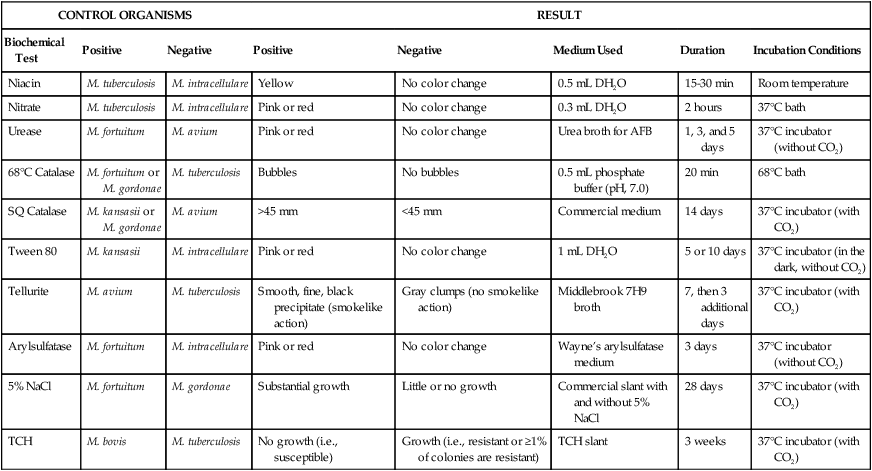
AFB, Acid-fast bacilli; CO2, carbon dioxide; DH2O, distilled water; NaCl, sodium chloride; SQ, semiquantitative; TCH, thiophene-2-carboxylic acid hydrazide.
Growth Rate.
The rate of growth is an important criterion for determining the initial category of an isolate. Rapid-growers usually produce colonies within 3 to 4 days after subculture. However, even a rapid-grower may take longer than 7 days to initially produce colonies because of inhibition by a harsh decontaminating procedure. Therefore, the growth rate (and pigment production) must be determined by subculture (see Procedure 43-5 on the Evolve site). The dilution of the organism used to assess the growth rate is critical. Even slow-growing mycobacteria appear to produce colonies in less than 7 days if the inoculum is too heavy. One organism particularly likely to exhibit false-positive rapid growth is M. flavescens. This species therefore serves as an excellent quality control organism for this procedure.
Pigment Production.
As previously discussed, mycobacteria may be categorized into three groups based on pigment production. Procedure 43-5, which can be found on the Evolve site, describes how to determine pigment production. To achieve optimum photochromogenicity, colonies should be young, actively metabolizing, isolated, and well aerated. Although some species (e.g., M. kansasii) turn yellow after a few hours of light exposure, others (e.g., M. simiae) may take prolonged exposure to light. Scotochromogens produce pigmented colonies even in the absence of light, and colonies often become darker with prolonged exposure to light (Figure 43-5). One member of this group, M. szulgai, is peculiar in that it is a scotochromogen at 35° C and nonpigmented when grown at 25° to 30° C. For this reason, all pigmented colonies should be subcultured to test for photoactivated pigment at both 35° C and 25° to 30° C. Nonchromogens are not affected by light.
Biochemical Testing.
Once categorized into a preliminary subgroup based on its growth characteristics, an organism must be definitively identified to species or complex level. Although conventional biochemical tests can be used for this purpose, new methods (discussed later in this section) have replaced biochemical tests for identifying mycobacterial species because of the previously discussed limitations of phenotypic testing. Although key biochemical tests are still discussed in this edition, the reader must be aware that this approach to identification ultimately will be replaced by molecular methods. Table 43-10 summarizes distinctive properties of the more commonly cultivable mycobacteria isolated from clinical specimens; key biochemical tests for each of the major mycobacterial groupings, including M. tuberculosis complex, are listed in Table 43-11. The following sections address key biochemical tests.
TABLE 43-10
Distinctive Properties of Commonly Cultivable Mycobacteria Encountered in Clinical Specimens
| Group/Complex | Species | Optimal Temp (°C) | Usual Colonial Morphologya | Niacin | Growth on TCH (10 mg/mL)b | Nitrate Reduction | Semi quantitative Catalase (>45 mm) | 68° C Catalase | Tween Hydrolysis, 5 Days | Tellurite Reduction | Tolerance to 5% NaCl | Arylsulfatase, 3 Days | Iron Uptake | Growth on MacConkey Agar | Urease | Pyrazinamidase, 4 Days |
| M. tuberculosis complex | M. tuberculosis | 37 | R | + | + | + | − | − | −c | 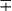 |
− | − | − | − | ± | + |
| M. bovis | 37 | Rt | − | − | − | − | − | − | 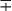 |
− | − | − | − | ± | − | |
| M. africanum | 37 | R | V | V | V | − | − | − | − | − | − | − | − | + | − | |
| Photochromogens | M. marinum | 30 | S/SR | 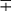 |
+ | − | − | − | + | 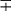 |
− | − |  d d |
− | −/+ | + |
| M. kansasii | 35 | SR/S | − | + | + | + | + | + |  |
− | − | − | − | + | − | |
| M. simiae | 37 | S | ± | + | − | + | + | − | + | − | − | − | ± | + | ||
| M. asiaticum | 37 | S | − | + | − | + | + | + | − | − | − | − | − | − | ||
| Scotochromogens | M. scrofulaceum | 37 | S | − | + | − | + | + | − |  |
− | − | V | − | V | ± |
| M. szulgai | 37 | S or R | − | + | + | + | + |  c c |
± | − | − | V | − | + | + | |
| M. gordonae | 37 | S | − | + | − | + | + | + | − | − | − | V | − | V | ± | |
| Nonphotochromogens | M. avium complex | 35-37 | St/R | − | + | − | + | ± | − | + | − | − | − | 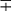 |
− | + |
| M. genavensee | 37 | St | − | + | − | − | + | + | − | − | + | + | ||||
| M. gastri | 35 | S/SR/R | − | + | − | + | − | + | 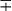 |
− | − | − | − | 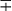 |
− | |
| M. malmoense | 30 | S | − | + | − | − | ± | + | + | − | − | − | − | + | ||
| M. haemophilumf | 30 | R | − | + | − | − | − | − | − | − | − | − | − | − | + | |
| M. shimoidei | 37 | R | − | + | − | − | − | + | − | − | − | − | − | + | ||
| M. ulcerans | 30 | R | − | + | − | − | + | − | − | − | − | − | ||||
| M. flavescensg | 37 | S | − | + | + | + | + | + | 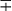 |
+ | − | − | − | + | + | |
| M. xenopih | 42 | Sf | − | + | − | − | + | − | 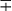 |
− | − | ± | − | − | V | |
| M. terrae complex M. terrae M. trivialei M. nonchromogenicum |
35 | SR | − | − | + | + | + | + | − | − | − | − | V | − | V | |
| Rapidly growing | M. fortuitum group | 28-30 | Sf/Rf | − | + | + | + | + | V | + | + | + | + | + | + | + |
| M. chelonae | 28-30 | S/R | −/+ | + | − | + | V | V | + | − | − | + | + | + | + | |
| M. abscessus | 28-30 | S/R | − | − | + | V | V | + | + | − | + | + | + | |||
| M. smegmatis | 28-30 | R/S | − | + | + | + | + | + | + | + | + | − | − |
Plus and minus signs indicate the presence or absence, respectively, of the feature; blank spaces indicate either that the information is not currently available or that the property is unimportant.
V, Variable; ±, usually present;  , usually absent.
, usually absent.
See Versalovic J: Manual of clinical microbiology, ed 10, Washington, DC, 2011, ASM Press, for biochemical reactions of other mycobacterial species and for additional biochemical reactions on the mycobacteria included in this table.
aR, Rough; S, smooth; SR, intermediate in roughness; t, thin or transparent; f, filamentous extensions.
bTCH, Thiophene-2-carboxylic acid hydrazide.
cTween hydrolysis may be positive at 10 days.
dArylsulfatase, 14 days, is positive.
eRequires mycobactin for growth on solid media.
fRequires hemin as a growth factor.
gYoung cultures may be nonchromogenic or have only pale pigment that may intensify with age.
hStrains of M. xenopi can be nonphotochromogenic or scotochromogenic.
iM. triviale is tolerant to 5% NaCl, and a rare isolate may grow on MacConkey agar.
TABLE 43-11
Key Biochemical Reactions to Help Differentiate Organisms Belonging to the Same Mycobacterial Group
| Mycobacterial Group | Key Biochemical Tests |
| M. tuberculosis complex | Niacin, nitrate reduction; susceptibility to thiophene-2-carboxylic acid hydrazide (TCH) if M. bovis is suspected |
| Photochromogens | Tween 80 hydrolysis, nitrate reduction, pyrazinamidase, 14-day arylsulfatase, urease, niacin |
| Scotochromogens | Permissive growth temperature, Tween 80 hydrolysis, nitrate reduction, semiquantitative catalase, urease, 14-day arylsulfatase |
| Nonphotochromogens | Heat-resistant and semiquantitative catalase activity, nitrate reduction, Tween 80 hydrolysis, urease, 14-day arylsulfatase, tellurite reduction, acid phosphatase activity |
| Rapidly growing | Growth on MacConkey agar, nitrate reduction, Tween 80 hydrolysis, 3-day arylsulfatase, iron uptake |
Niacin.
Niacin (nicotinic acid) plays an important role in the oxidation-reduction reactions that occur during mycobacterial metabolism. Although all species produce nicotinic acid, M. tuberculosis accumulates the largest amount. (M. simiae and some strains of M. chelonae also produce niacin.) Niacin therefore accumulates in the medium in which these organisms are growing. A positive niacin test (see Procedure 43-6 on the Evolve site) is preliminary evidence that an organism that exhibits a buff-colored, slow-growing, rough colony may be M. tuberculosis (Figure 43-6). However, this test is not sufficient to confirm identification. If sufficient growth is present on an initial L-J slant (the egg-base medium enhances accumulation of free niacin), a niacin test can be performed immediately. If growth on the initial culture is scant, the subculture used for growth rate determination can be used. If this culture yields only rare colonies, the colonies should be spread around with a sterile cotton swab (after the growth rate has been determined) to distribute the inoculum over the entire slant. The slant then is incubated until light growth over the surface of the medium is visible. For reliable results, the niacin test should be performed only from cultures on L-J medium that are at least 3 weeks old and show at least 50 colonies; otherwise, enough detectable niacin might not have been produced.

Nitrate Reduction.
This test is valuable for identifying M. tuberculosis, M. kansasii, M. szulgai, and M. fortuitum. The ability of acid-fast bacilli to reduce nitrate is influenced by the age of the colonies, temperature, pH, and enzyme inhibitors. Although rapid-growers can be tested within 2 weeks, slow-growers should be tested after 3 to 4 weeks of luxuriant growth. Commercially available nitrate strips yield acceptable results only with strongly nitrate-positive organisms, such as M. tuberculosis. This test may be tried first because of its ease of performance. The M. tuberculosis–positive control must be strongly positive in the strip test, or the test results are unreliable. If the paper strip test is negative or if the control test result is not strongly positive, the chemical procedure (see Procedure 43-7 on the Evolve site) must be carried out using strong and weakly positive controls.
Catalase.
• The semiquantitative catalase test is based on the relative activity of the enzyme, as determined by the height of a column of bubbles of oxygen (Figure 43-7) formed by the action of untreated enzyme produced by the organism. Based on the semiquantitative catalase test, mycobacteria are divided into two groups: those that produce less than 45 mm of bubbles and those that produce more than 45 mm of bubbles.

• The heat-stable catalase test is based on the ability of the catalase enzyme to remain active after heating (i.e., it is a measure of the enzyme’s heat stability). When heated to 68° C for 20 minutes, the catalase of M. tuberculosis, M. bovis, M. gastri, and M. haemophilum becomes inactivated.
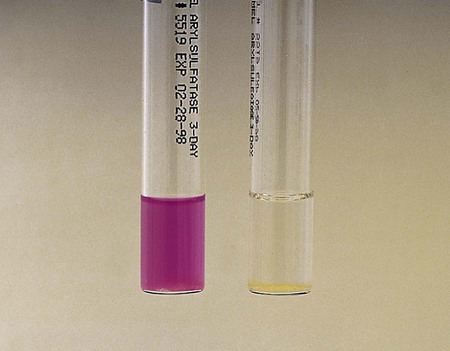
Arylsulfatase.
The enzyme arylsulfatase is present in most mycobacteria. Test conditions can be varied to differentiate different forms of the enzyme. The rate at which this enzyme breaks down phenolphthalein disulfate into phenolphthalein (which forms a red color in the presence of sodium bicarbonate) and other salts helps to differentiate certain strains of mycobacteria. The 3-day test is particularly useful for identifying the potentially pathogenic rapid-growers M. fortuitum and M. chelonae. Slow-growing M. marinum and M. szulgai are positive in the 14-day test (Figure 43-8).
Other Tests.
Other tests are often performed to make more subtle distinctions between species (see Table 43-11). However, performing all the procedures necessary for definitive identification of mycobacteria is not cost-effective for routine clinical microbiology laboratories; therefore, specimens that require further testing can be forwarded to regional laboratories.
Antimicrobial Susceptibility Testing and Therapy
M. Tuberculosis Complex
Conventional Methods
Four general methods are used throughout the world to determine the susceptibility of M. tuberculosis isolates to various antituberculous agents (Table 43-12). Initial isolates of M. tuberculosis are tested against five antimicrobials, which are referred to as primary drugs (Box 43-4.) If resistance to any of the primary drugs is detected, a second battery of agents is tested (Box 43-4).
TABLE 43-12
| Method | Principle |
| Absolute concentration | For each drug tested, a standardized inoculum is inoculated to control (drug free) media and media containing several appropriately graded drug concentrations. Resistance is expressed as the lowest concentration of drug that inhibits all or almost all of the growth; that is, the minimum inhibitory concentration (MIC) |
| Resistance ratio | The resistance of the test organism is compared with that of a standard laboratory strain. The two strains are tested in parallel by inoculating a standard inoculum to media containing twofold serial dilutions of the drug. Resistance is expressed as the ratio of the MIC of the test strain divided by the MIC for the standard strain for each drug |
| Proportion | For each drug tested, several dilutions of standardized inoculum are inoculated onto control and drug-containing agar media. The extent of growth in the absence or presence of drug is compared and expressed as a percentage. If growth at the critical concentration of a drug is >1%, the isolate is considered clinically resistant. This is the standard method for all drugs except pyrazinamide. |
| Commercial systems approved for use by the FDA: BACTEC 460TB (Becton Dickinson, Sparks, Md.); BACTEC MGIT 960 (Becton Dickinson); VersaTREK (Trek Diagnostic Systems, Cleveland, Ohio); MB/BacT Alert 3D (bioMérieux, Durham, N.C.) | Using the principles of the agar proportion method, these methods use liquid media. Growth is indicated by the amount of 14C-labeled carbon dioxide (CO2) released (as measured by the BACTEC 460 instrument) or the amount of fluorescence or gas produced (as measured by the MB/BacT Alert 3D and VersaTREK systems, respectively). For each drug tested, a standardized inoculum is inoculated into a drug-free and a drug-containing vial. The rate and amount of CO2 produced in the absence or (with the BACTEC MGIT 960) presence of drug are then compared. The BACTEC 460TB is being replaced in many laboratories by the MGIT 960 to avoid hazardous waste disposal of radioactive materials, in addition to the cross-contamination that may occur with the 460TB. |
| Alternate methods and molecular methods | Several molecular methods have been developed that identify the mutations in the rifampin-resistant gene (rpoB). Greater than 96% of rifampin resistance correlates to mutations in an 81 bp segment. Additional molecular methods have been developed for the identification of resistance to isoniazid, ethambutol, and pyrazinamide. Molecular methods should be followed up with culture, especially for confirmation of second-line drug resistance and in XDR TB. |
FDA, U.S. Food and Drug Administration. XDR TB, extensively drug-resistant tuberculosis.
Nontuberculous Mycobacteria
Susceptibility testing should be performed on clinically significant, rapidly growing mycobacteria (Table 43-13). Skin and soft tissue infections, if susceptible, are treated with clarithromycin and at least one additional drug based on susceptibility testing. Pulmonary infections with M. abscessus should also be treated with a multidrug regimen that includes clarithromycin, if susceptible, and then additional drugs based on susceptibility testing.
TABLE 43-13
CLSI Recommendations for Susceptibility Testing of Nontuberculous Mycobacteria
microdilution or BACTEC
Second line: Moxifloxacin or linezolid
Isolates typically are intrinsically resistant to isoniazid and pyrazinamide
Broth based
If rifampin resistant, test rifabutin, ethambutol, isoniazid, linezolid, moxifloxacin, streptomycin, clarithromycin, amikacin, ciprofloxacin, and trimethoprim-sulfamethoxazole
Agar disk elution
Broth microdilution
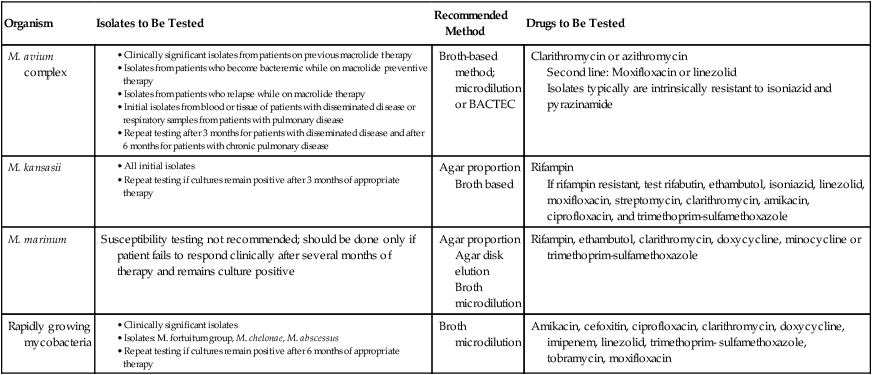
CLSI, Clinical and Laboratory Standards Institute.






