[level-membership-for-anesthesiology-category]
Muscle Function and Neuromuscular Blockade
At first, muscle relaxants were used only occasionally, in small doses, as an adjuvant to aid in the management of a difficult case; they were not used routinely. A tracheal tube was not always used, the lungs were not ventilated artificially and residual block was not routinely reversed; all of these caused significant morbidity and mortality, as demonstrated in the retrospective study by Beecher & Todd (1954). By 1946, however, it was appreciated that using drugs such as curare in larger doses allowed the depth of anaesthesia to be lightened, and it was suggested that incremental doses should also be used during prolonged surgery, rather than deepening anaesthesia – an entirely new concept at that time. The use of routine tracheal intubation and artificial ventilation then evolved.
PHYSIOLOGY OF NEUROMUSCULAR TRANSMISSION
Acetylcholine, the neurotransmitter at the neuromuscular junction, is released from presynaptic nerve endings on passage of a nerve impulse (an action potential) down the axon to the nerve terminal. The neurotransmitter is synthesized from choline and acetylcoenzyme A by the enzyme choline acetyltransferase and stored in vesicles in the nerve terminal. The action potential depolarizes the nerve terminal to release the neurotransmitter; entry of Ca2 + ions into the nerve terminal is a necessary part of this process, promoting further acetylcholine release. On the arrival of an action potential, the storage vesicles are transferred to the active zones on the edge of the axonal membrane, where they fuse with the terminal wall to release the acetylcholine (Fig. 6.1). Three proteins, synaptobrevin, syntaxin and synaptosome-associated protein SNAP-25, are involved in this process. These proteins along with vesicle membrane-associated synaptotagmins cause the docking, fusion and release (exocytosis) of acetylcholine from the vesicles. There are about 1000 active sites at each nerve ending and any one nerve action potential leads to the release of 200–300 vesicles. In addition, small quanta of acetylcholine, equivalent to the contents of one vesicle, are released at the neuromuscular junction spontaneously, causing miniature end-plate potentials (MEPPs) on the postsynaptic membrane, but these are insufficient to generate a muscle action potential.
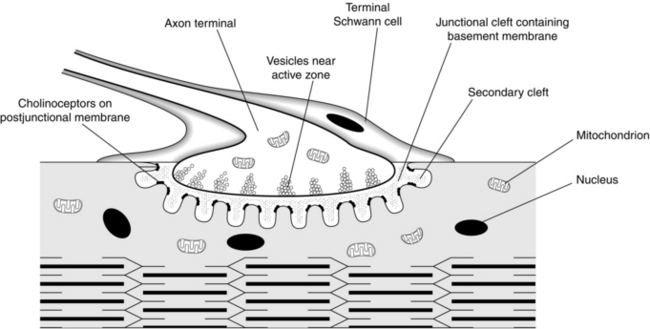
FIGURE 6.1 The neuromuscular junction with an axon terminal, containing vesicles of acetylcholine. The neurotransmitter is released on arrival of an action potential and crosses the junctional cleft to stimulate the postjunctional receptors on the shoulders of the secondary clefts. (Reproduced with kind permission of Professor WC Bowman.)
The active sites of release are aligned directly opposite the acetylcholine receptors on the junctional folds of the postsynaptic membrane, lying on the muscle surface. The junctional cleft, the gap between the nerve terminal and the muscle membrane, has a width of only 60 nm. It contains the enzyme acetylcholinesterase, which is responsible for the ultimate breakdown of acetylcholine. This enzyme is also present, in higher concentrations, in the junctional folds in the postsynaptic membrane (Fig. 6.1). The choline produced by the breakdown of acetylcholine is taken up across the nerve membrane to be reused in the synthesis of the transmitter.
The nicotinic acetylcholine receptors on the postsynaptic membrane are organized in discrete clusters on the shoulders of the junctional folds (Fig. 6.1). Each cluster is about 0.1 μm in diameter and contains a few hundred receptors. Each receptor consists of five subunits, two of which, the alpha (α; MW = 40 000 Da), are identical. The other three, slightly larger subunits, are the beta (β), delta (δ) and epsilon (ε). In fetal muscle, the epsilon is replaced by a gamma (γ) subunit. Each subunit of the receptor is a glycosated protein – a chain of amino acids – coded by a different gene. The receptors are arranged as a cylinder which spans the membrane, with a central, normally closed, channel – the ionophore (Fig. 6.2). Each of the α subunits carries a single acetylcholine binding region on its extracellular surface. They also bind neuromuscular blocking drugs.
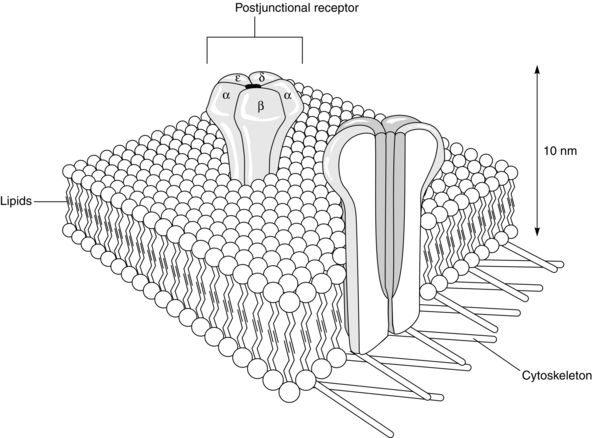
FIGURE 6.2 Two postjunctional receptors, embedded in the lipid layer of the postsynaptic muscle membrane. The α, β, ε and δ subunits are demonstrated on the surface of one receptor and the ionophore is seen in cross-section on the other receptor. On stimulation of the two α subunits by two molecules of acetylcholine, the ionophore opens to allow the passage of the end-plate current. (Reproduced with kind permission of Professor WC Bowman.)
Activation of the receptor requires both α sites to be occupied, producing a structural change in the receptor complex that opens the central channel running between the receptors for a very short period, about 1 ms (Fig. 6.2). This allows movement of cations such as Na+, K+, Ca2 + and Mg2 + along their concentration gradients. The main change is influx of Na+ ions, the end-plate current, followed by efflux of K+ ions. The summation of this current through a large number of receptor channels lowers the transmembrane potential of the end-plate region sufficiently to depolarize it and generate a muscle action potential sufficient to allow muscle contraction.
Acetylcholine receptors are also present on the presynaptic area of the nerve terminal. These are of a slightly different structure to the postsynaptic nicotinic receptors (α3β2). It is thought that a positive feedback mechanism exists for the further release of acetylcholine, such that some of the released molecules of acetylcholine stimulate these presynaptic receptors, producing further mobilization of the neurotransmitter to the readily releasable sites, ready for the arrival of the next nerve stimulus (Fig. 6.3). Acetylcholine activates sodium channels on the prejunctional nerve membrane, which in turn activate voltage-dependent calcium channels (P-type fast channels) on the motor neurone causing an influx of calcium into the nerve cytoplasm to promote further acetylcholine release.
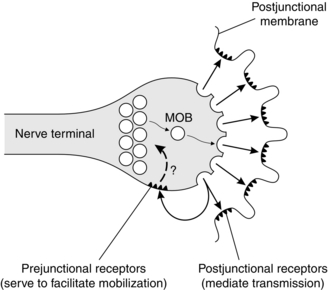
FIGURE 6.3 Acetylcholine receptors are present on the shoulders of the axon terminal, as well as on the postjunctional membrane. Stimulation of the prejunctional receptors mobilizes (MOB) the vesicles of acetylcholine to move into the active zone, ready for release on arrival of another nerve impulse. The mechanism requires Ca2 + ions. (Reproduced with kind permission of Professor WC Bowman.)
PHARMACOLOGY OF NEUROMUSCULAR TRANSMISSION
Depolarizing Neuromuscular Blocking Agents
Succinylcholine Chloride (Suxamethonium)
This quaternary ammonium compound is comparable to two molecules of acetylcholine linked together (Fig. 6.4). The two quaternary ammonium radicals, N+(CH3)3 have the capacity to cling to each of the α units of the postsynaptic acetylcholine receptor, altering its structural conformation and opening the ion channel, but for a longer period than does a molecule of acetylcholine. Administration of succinylcholine therefore results in an initial depolarization and muscle contraction, termed fasciculation. As this effect persists, however, further action potentials cannot pass down the ion channels and the muscle becomes flaccid; repolarization does not occur.
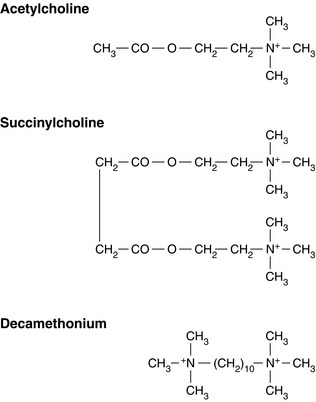
FIGURE 6.4 The chemical structures of acetylcholine and succinylcholine. The similarity between the structure of succinylcholine and two molecules of acetylcholine can be seen. The structure of decamethonium is also shown. The quaternary ammonium radicals N+(CH3)3 cling to the α subunits of the postsynaptic receptor.
Inherited Factors: The exact structure of plasma cholinesterase is determined genetically, by autosomal genes, and this has been completely defined. Several abnormalities in the amino acid sequence of the normal enzyme, usually designated 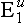 , are recognized. The most common is produced by the atypical gene,
, are recognized. The most common is produced by the atypical gene, 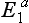 , which occurs in about 4% of the Caucasian population. Thus a patient who is a heterozygote for the atypical gene
, which occurs in about 4% of the Caucasian population. Thus a patient who is a heterozygote for the atypical gene 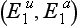 demonstrates a longer effect from a standard dose of succinylcholine (about 30 min). If the individual is a homozygote for the atypical gene
demonstrates a longer effect from a standard dose of succinylcholine (about 30 min). If the individual is a homozygote for the atypical gene 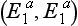 , the duration of action of succinylcholine may exceed 2 h. Other, rarer, abnormalities in the structure of plasma cholinesterase are also recognized, e.g. the fluoride
, the duration of action of succinylcholine may exceed 2 h. Other, rarer, abnormalities in the structure of plasma cholinesterase are also recognized, e.g. the fluoride 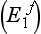 and silent
and silent 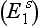 genes. The latter has very little capacity to metabolize succinylcholine and thus neuromuscular block in the homozygous state
genes. The latter has very little capacity to metabolize succinylcholine and thus neuromuscular block in the homozygous state 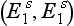 lasts for at least 3 h. In such patients, non-specific esterases gradually clear the drug from plasma.
lasts for at least 3 h. In such patients, non-specific esterases gradually clear the drug from plasma.


Acquired Factors: In these instances, the structure of plasma cholinesterase is normal but its activity is reduced. Thus, neuromuscular block is prolonged by only minutes rather than hours. Causes of reduced plasma cholinesterase activity include:








Side-Effects of Succinylcholine
Muscle Pains: These occur especially in the patient who is ambulant soon after surgery, such as the day-case patient. The pains, thought possibly to be caused by the initial fasciculations, are more common in young, healthy patients with a large muscle mass. They occur in unusual sites, such as the diaphragm and between the scapulae, and are not relieved easily by conventional analgesics. The incidence and severity may be reduced by the use of a small dose of a non-depolarizing muscle relaxant given immediately before administration of succinylcholine, e.g. gallamine 10 mg (which is thought to be most efficacious in this respect) or atracurium 2.5 mg. However, this technique, termed pre-curarization or pretreatment, reduces the potency of succinylcholine, necessitating administration of a larger dose to produce the same effect. Many other drugs have been used in an attempt to reduce the muscle pains, including lidocaine, calcium, magnesium and repeated doses of thiopental, but none is completely reliable.
Increased Intraocular Pressure: This is thought to be caused partly by the initial contraction of the external ocular muscles and contracture of the internal ocular muscles after administration of succinylcholine. It is not reduced by pre-curarization. The effect lasts for as long as the neuromuscular block and concern has been expressed that it may be sufficient to cause expulsion of the vitreal contents in the patient with an open eye injury. This is unlikely. Protection of the airway from gastric contents must take priority in the patient with a full stomach in addition to an eye injury, as inhalation of gastric contents may threaten life.
Increased Intragastric Pressure: In the presence of a normal lower oesophageal sphincter, the increase in intragastric pressure produced by succinylcholine should be insufficient to produce regurgitation of gastric contents. However, in the patient with incompetence of this sphincter from, for example, hiatus hernia, regurgitation may occur.
Hyperkalaemia: It has long been recognized that administration of succinylcholine during halothane anaesthesia increases the serum potassium concentration by 0.5 mmol L–1. This effect is thought to be caused by muscle fasciculation. It is probable that the effect is less marked with the newer potent inhalational agents, e.g. isoflurane, sevoflurane. A similar increase occurs in patients with renal failure, but as these patients may already have an elevated serum potassium concentration, such an increase may precipitate cardiac irregularities and even cardiac arrest.
Cardiovascular Effects: Succinylcholine has muscarinic in addition to nicotinic effects, as does acetylcholine. The direct vagal effect (muscarinic) produces sinus bradycardia, especially in patients with high vagal tone, such as children and the physically fit. It is also more common in the patient who has not received an anticholinergic agent (such as glycopyrrolate) or who is given repeated increments of succinylcholine. It is advisable to use an anticholinergic routinely if it is planned to administer more than one dose of succinylcholine. Nodal or ventricular escape beats may develop in extreme circumstances.
Characteristics of Depolarizing Neuromuscular Block
 a decreased response to a single, low-voltage (1 Hz) twitch stimulus applied to a peripheral nerve is detected. Tetanic stimulation (e.g. at 50 Hz) produces a small, but sustained, response.
a decreased response to a single, low-voltage (1 Hz) twitch stimulus applied to a peripheral nerve is detected. Tetanic stimulation (e.g. at 50 Hz) produces a small, but sustained, response.
 if four twitch stimuli are applied at 2 Hz over 2 s (train-of-four stimulus), followed by a 10-s interval before the next train-of-four, no decrease in the height of successive stimuli is noted (Fig. 6.5).
if four twitch stimuli are applied at 2 Hz over 2 s (train-of-four stimulus), followed by a 10-s interval before the next train-of-four, no decrease in the height of successive stimuli is noted (Fig. 6.5).
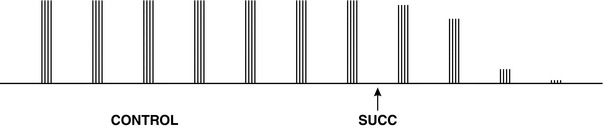
FIGURE 6.5 The train-of-four twitch response recorded before (CONTROL) and after a dose of succinylcholine. Before administration of succinylcholine 1 mg kg–1, four twitches of equal height are visible. After giving the drug (SUCC), the height of all four twitches decreases equally; no ‘fade’ of the train-of-four is seen. Within 1 min, the trace has been ablated.




Non-Depolarizing Neuromuscular Blocking Agents
Benzylisoquinolinium Compounds
Tubocurarine Chloride: This is the only naturally occurring muscle relaxant. It is derived from the bark of the South American plant Chondrodendron tomentosum which has been used for centuries by South American Indians as an arrow poison. It was the first non-depolarizing neuromuscular blocking agent to be used in humans, by Griffith and Johnson in Montreal, in 1942. The intubating dose is 0.5–0.6 mg kg–1. It has a slow onset of action and a prolonged duration of effect (Table 6.1), and its effects are potentiated by inhalational agents and prior administration of succinylcholine. It has a marked propensity to produce histamine release and thus hypotension, with possibly a compensatory tachycardia. In large doses, it may also produce ganglion blockade, which potentiates these cardiovascular effects. It is excreted unchanged through the kidneys, with some biliary excretion. It is no longer available in the UK.
TABLE 6.1
Time to 95% Depression of the Twitch Response, After a Dose of 2 × ED95 of a Neuromuscular Blocking Drug (When Tracheal Intubation Should be Possible), and Time to 20–25% Recovery, When an Anti-Cholinesterase May be Used Reliably to Reverse Residual Block Produced by a Non-Depolarizing Drug
| 95% Twitch Depression (s) | 20–25% Recovery (min) | |
| Succinylcholine | 60 | 10 |
| Tubocurarine | 220 | 80 + |
| Alcuronium | 420 | 70 |
| Gallamine | 300 | 80 |
| Atracurium | 110 | 43 |
| Cisatracurium | 150 | 45 |
| Doxacurium | 250 | 83 |
| Mivacurium | 170 | 16 |
| Pancuronium | 220 | 75 |
| Vecuronium | 180 | 33 |
| Pipecuronium | 300 | 95 |
| Rocuronium | 75 | 33 |
| Rapacuronium | < 75 | 15 |
Alcuronium Chloride: This drug is a semi-synthetic derivative of toxiferin, an alkaloid of calabash curare. It has less histamine-releasing properties, and therefore cardiovascular effect, than tubocurarine, although it may have some vagolytic effect, producing a mild tachycardia. It also has a slow onset time and nearly as long a duration of effect as tubocurarine (Table 6.1). It is almost entirely excreted unchanged through the kidneys. The intubating dose is 0.2–0.25 mg kg–1. Before the advent of atracurium and vecuronium, this inexpensive agent was used widely, but now its popularity has declined and it is no longer available commercially in the UK.
Gallamine Triethiodide: This synthetic substance is a trisquaternary amine. It was first used in France in 1948. The intubating dose in adults is of the order of 160 mg. It has a similar onset to, but slightly shorter duration of action than, tubocurarine, and is excreted almost entirely by the kidneys. Consequently, it should not be used in patients with renal impairment. Being more lipid-soluble than bisquaternary amines, it crosses the placenta to a significant degree and should not be used in obstetric practice. Gallamine has potent vagolytic properties and produces some direct sympathomimetic stimulation. Thus, it increases pulse rate and arterial pressure.
Atracurium Besylate: This drug, introduced into clinical practice in 1982, was developed by Stenlake at Strathclyde University. Quaternary ammonium compounds break down spontaneously at varying temperature and pH, a phenomenon recognized for over 100 years and known as Hofmann degradation. Many such substances also have neuromuscular blocking properties, and atracurium was developed in the search for such an agent which broke down at body temperature and pH. Hofmann degradation may be considered as a ‘safety net’ in the sick patient with impaired liver or renal function, because atracurium is still cleared from the body. Some renal excretion occurs in the healthy patient (10%), as does ester hydrolysis in the plasma; probably only about 45% of the drug is eliminated by Hofmann degradation in the normal patient.
Atracurium (and vecuronium) was developed in an attempt to obtain a non-depolarizing agent which had a more rapid onset, was shorter-acting and had fewer cardiovascular effects than the older agents. Atracurium 0.5 mg kg–1 does not produce neuromuscular block as rapidly as succinylcholine; the onset time is 2.0–2.5 min, depending on the dose used (Table 6.1). However, recovery occurs more rapidly from it than after use of the older non-depolarizing agents and atracurium may be reversed easily 20–25 min after administration of a dose of 2 × ED95 (0.45 mg kg–1). The drug does not have any direct cardiovascular effect, but may release histamine (about a third of that released by tubocurarine) and may therefore produce a local wheal and flare around the injection site, especially if a small vein is used. This may be accompanied by a slight reduction in arterial pressure. It can produce anaphylaxis, but to a lesser degree than succinylcholine.
Cisatracurium: This is the most recently introduced benzylisoquinolinium neuromuscular blocker. It is of particular interest because it is an example of the development of a specific isomer of a drug to produce a ‘clean’ substance with the desired clinical actions but with reduced side-effects. Cisatracurium is the 1R-cis 1’R-cis isomer of atracurium, and one of 10 isomers of the parent compound. It is three to four times more potent than atracurium (ED95 = 0.05 mg kg–1) and has a slightly slower onset and longer duration of action. Its main advantage is that it does not release histamine and therefore is associated with greater cardiovascular stability. It undergoes even more Hofmann degradation than atracurium. Because a lower dose of this more potent drug is given, it produces less laudanosine than an equipotent dose of atracurium. It is therefore particularly useful in the critically ill patient requiring prolonged infusion of a neuromuscular blocking drug.
Doxacurium Chloride: This bisquaternary ammonium compound is only available in the USA. It undergoes a small amount of metabolism in the plasma by cholinesterase (6%), but is excreted mainly through the kidneys. It is the most potent non-depolarizing neuromuscular blocking agent available; an intubating dose is only 0.05 mg kg–1. It has a very slow onset of action (Table 6.1) and a prolonged and unpredictable duration of effect. However, it has no cardiovascular effects and may therefore be of use during long surgical procedures in which cardiovascular stability is required, e.g. cardiac surgery.
Mivacurium Chloride: This drug is metabolized by plasma cholinesterase at 88% of the rate of succinylcholine. An intubating dose (2 × ED95 = 0.15 mg kg–1) has a similar onset of action to an equipotent dose of atracurium, but in the presence of normal plasma cholinesterase, recovery after mivacurium is much faster (Table 6.1) and administration of an anticholinesterase may not be necessary (if neuromuscular function is being monitored and good recovery can be demonstrated). Full recovery in such circumstances takes about 20–25 min, but the drug may be antagonized easily within 15 min. Mivacurium is useful particularly for surgical procedures requiring muscle relaxation in which even atracurium and vecuronium seem too long-acting, and when it is desirable to avoid the side-effects of succinylcholine, e.g. for bronchoscopy, oesophagoscopy, laparoscopy or tonsillectomy. The drug produces a similar amount of histamine release to atracurium.
Aminosteroid Compounds
Pancuronium Bromide: This bisquaternary amine, the first steroid muscle relaxant used clinically, was developed by Savege and Hewitt and marketed in 1964. The intubating dose is 0.1 mg kg–1, which takes 3–4 min to reach its maximum effect (Table 6.1). The clinical duration of action of the drug is long, especially in the presence of potent inhalational agents or renal dysfunction, as 60% of a dose of the drug is excreted unchanged through the kidneys. It is also deacetylated in the liver; some of the metabolites have neuromuscular blocking properties.
Vecuronium Bromide: This steroidal agent was developed in an attempt to reduce the cardiovascular effects of pancuronium. It is similar in structure to the older drug, differing only in the loss of a methyl group from one quaternary ammonium radical. Thus it is a monoquaternary amine. An intubating dose of 0.1 mg kg–1 produces profound neuromuscular block within 3 min, which is slightly longer than the onset time of atracurium, but shorter than those of tubocurarine and pancuronium. This dose produces clinical block for about 30 min. Vecuronium rarely produces histamine release, nor does it have any direct cardiovascular effects, although it allows the cardiac effects of other anaesthetic agents, such as bradycardia produced by the opioids, to go unchallenged. Vecuronium is excreted through the kidneys (30%), although to a lesser extent than pancuronium, and undergoes hepatic deacetylation; the deacetylated metabolites have neuromuscular blocking properties. Repeated doses should be used with care in patients with renal or hepatic disease because they accumulate.
Pipecuronium Bromide: This analogue of pancuronium was developed in Hungary in 1980 and is marketed in Eastern Europe and the USA. The intubating dose is 0.07 mg kg–1. The onset time and time to recovery from block are similar to those of pancuronium (Table 6.1), and excretion of the drug through the kidneys is significant (66%). In contrast to pancuronium, pipecuronium possesses marked cardiovascular stability, having no vagolytic or sympathomimetic effects. It may therefore be useful during major surgery in patients with cardiac disease.
Rocuronium Bromide: This monoquaternary amine has a very rapid onset of action for a non-depolarizing muscle relaxant. It is six to eight times less potent than vecuronium but has approximately the same molecular weight; consequently, a greater number of drug molecules may reach the postjunctional receptors within the first few circulations, enabling faster development of neuromuscular block. In a dose of 0.6 mg kg–1, good or excellent intubating conditions are achieved within 60–90 s; this is only slightly slower than the onset time of succinylcholine. The clinical duration is 30–45 min. At higher doses, such as 0.9 mg kg–1, rocuronium has an onset time similar to succinylcholine, albeit with a greater range of effect. In such doses, however, rocuronium is a very long-acting drug, lasting about 90 min.
Rapacuronium Bromide: This was the last aminosteroid to become available. It is less potent than rocuronium (2 × ED90 = 1.15 mg kg–1) and in equipotent doses has an even more rapid onset of action (< 75 s). It is cleared rapidly from the plasma by hepatic uptake and deacetylation and thus has a shorter duration of effect than rocuronium, 12–15 min (Table 6.1). As with the deacetylation of pancuronium and vecuronium, a metabolite of rapacuronium has neuromuscular blocking properties (Org 9488). This may prolong the effect of incremental doses of the drug.
Characteristics of Non-Depolarizing Neuromuscular Block
 decreased response to a low-voltage twitch stimulus (e.g. 1 Hz) which, if repeated, decreases further in amplitude. This effect, which is in contrast to that produced by a depolarizing drug, also occurs to a greater degree when the train-of-four (TOF) twitch response is applied, and even more so with higher, tetanic rates of stimulation. It is referred to as ‘fade’ or decrement.
decreased response to a low-voltage twitch stimulus (e.g. 1 Hz) which, if repeated, decreases further in amplitude. This effect, which is in contrast to that produced by a depolarizing drug, also occurs to a greater degree when the train-of-four (TOF) twitch response is applied, and even more so with higher, tetanic rates of stimulation. It is referred to as ‘fade’ or decrement.
 post-tetanic potentiation (PTP) or facilitation (PTF) of the twitch response may be demonstrated (Fig. 6.6).
post-tetanic potentiation (PTP) or facilitation (PTF) of the twitch response may be demonstrated (Fig. 6.6).
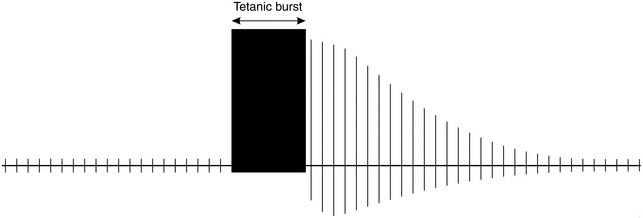
FIGURE 6.6 A 5-s burst of tetanus (50 Hz), applied after a run of single twitch stimuli, causes a transient increase in the height of subsequent twitches, although they gradually decrease to their former height; this is post-tetanic potentiation (PTP) or facilitation (PTF). The number of twitches detectable after the burst of tetanus is referred to as the ‘post-tetanic count’.
 neuromuscular block is reversed by administration of an anticholinesterase.
neuromuscular block is reversed by administration of an anticholinesterase.

REVERSAL AGENTS
Neostigmine: This drug combines reversibly with acetylcholinesterase by formation of an ester linkage. Neostigmine is excreted largely unchanged through the kidneys and has a half-life of about 45 min. It is presented in brown vials because it breaks down on exposure to light. Neostigmine potentiates the action of acetylcholine wherever it is a neurotransmitter, including all cholinergic nerve endings; thus, it produces bradycardia, salivation, sweating, bronchospasm, increased intestinal motility and blurred vision. These cholinergic effects may be reduced by simultaneous administration of an anticholinergic agent such as atropine or glycopyrrolate. The usual dose of neostigmine is of the order of 0.035 mg kg–1, in combination with either atropine 0.015 mg kg–1 or glycopyrrolate 0.01 mg kg–1. Neostigmine takes at least 2 min to have an initial effect, and recovery from neuromuscular block is maximally enhanced by 10 min.
Edrophonium: This anticholinesterase forms an ionic bond with the enzyme but does not undergo a chemical reaction with it. The effect is therefore more short-lived than with neostigmine, of the order of only a few minutes. Edrophonium has a quicker onset of action than neostigmine, producing signs of recovery within 1 min. However, its effects are more evanescent; when edrophonium is given in the presence of profound neuromuscular block, the degree of neuromuscular block may increase after an initial period of recovery. The dose of edrophonium is 0.5–1.0 mg kg–1.
Pyridostigmine: This drug has a slower onset time than neostigmine or edrophonium, and also a longer duration of action. It is used more frequently as oral therapy in patients with myasthenia gravis than in anaesthesia.
Physostigmine: This anticholinesterase, also known as eserine, is a tertiary amine and is more lipid-soluble than the other carbamate esters. It is therefore absorbed more easily from the gastrointestinal tract, and also crosses the blood–brain barrier.
Organophosphorus Compounds: These substances are irreversible inhibitors of acetylcholinesterase; by phosphorylation of the enzyme, they produce a very stable complex which is resistant to reactivation or hydrolysis. Synthesis of new enzyme must occur before recovery. These agents, which include di-isopropylfluorophosphonate (DFP) and tetraethylpyrophosphate (TEPP), are used as insecticides and chemical warfare agents. They are absorbed readily through the lungs and skin. Poisoning is not uncommon among farm workers. Muscarinic effects such as salivation, sweating and bronchospasm are combined with nicotinic effects, such as muscle weakness. Central nervous effects such as tremor and convulsions may occur, as may unconsciousness and respiratory failure. Reactivators of acetylcholinesterase are used to treat this form of poisoning: they include pralidoxime and obidoxime. Atropine, anticonvulsants and artificial ventilation may be necessary. Chronic exposure may produce polyneuritis. Carbamates such as pyridostigmine are used prophylactically in those threatened by chemical warfare with these compounds.
Cyclodextrins
A novel approach to reversal of neuromuscular block was therefore developed. Sugammadex (Org 25969), a γ-cyclodextrin, was designed to chelate or encapsulate rocuronium (and to a lesser extent vecuronium) in the plasma, preventing its access to the nicotinic receptor and encouraging dissociation from it. Sugammadex consists of eight oligosaccharides arranged in a cylindrical structure to encapsulate all four steroidal rings of rocuronium completely (Fig. 6.7). This cylindrical structure is known as a toroid. The hydrophilic external tails on the toroid are negatively charged, attracting the quaternary nitrogen group on the muscle relaxant and drawing it into the lipophilic core of sugammadex. The complex of sugammadex and rocuronium is excreted in the urine and has no muscarinic effect: the use of anticholinergic agents is unnecessary. Sugammadex seems to be devoid of adverse cardiovascular effects, although prolongation of the QT interval has been reported anecdotally. It acts three times as rapidly as neostigmine in reversing neuromuscular block produced by rocuronium.
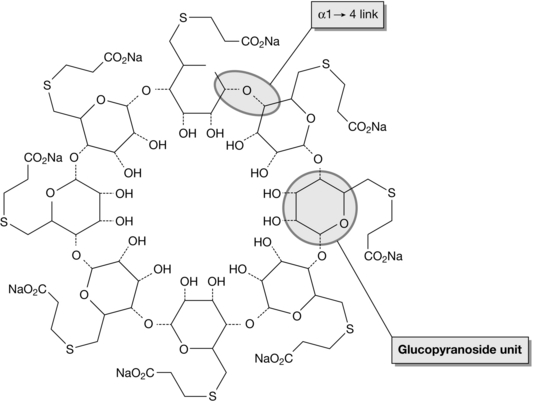
FIGURE 6.7 The cyclical structure of sugammadex consisting of eight glucopyranoside units linked by oxygen radicals (example marked by circle). The negatively charged hydrophilic chains on the outside of the molecule attract rocuronium to the core of the toroid. (Downloaded from Wikipedia, freely accessible.)
Sugammadex became available in the UK in 2008. Its high cost has limited its use.
NEUROMUSCULAR MONITORING
Electromyography
The electromyographic response of a muscle is measured in response to the same electrical stimulus, using recording electrodes similar to ECG pads placed over the motor point of the stimulated muscle. For instance, if the ulnar nerve is stimulated, the recording electrodes are placed over the motor point of adductor pollicis in the thumb (Fig. 6.8). A compound muscle action potential may be recorded. Although primarily a research tool, there are several simple clinical instruments, such as the Datex Relaxograph, which give a less accurate, but similar recording. Maintaining the exact position of the hand is not as essential with electromyography as with mechanomyography.
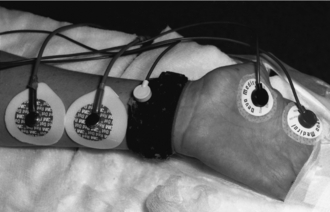
FIGURE 6.8 Positioning of the hand necessary to obtain an electromyographic recording of the response of the adductor pollicis muscle to stimulation of the ulnar nerve is demonstrated. An earth electrode is placed round the wrist. Two recording electrodes are placed over the muscle on the hand; the distal one lies over the motor point.
Modes of Stimulation
Train-of-Four (TOF) Twitch Response
In an attempt to assess the degree of neuromuscular block clinically, Ali et al (1971)described a development of the twitch response which, it was hoped, would be more sensitive than repeated single twitches and did not require a control response. Four stimuli (at 2 Hz) are applied over 2 s, with at least a 10-s gap between each TOF. On administration of a small dose of a non-depolarizing muscle relaxant, fade of the amplitude of the TOF may be visible. The ratio of the amplitude of the fourth to the first twitch is called the train-of-four ratio (TOFR). In the presence of a larger dose of such a drug, the fourth twitch disappears first, then the third, followed by the second and, finally, the first twitch (Fig. 6.9A). On recovery from neuromuscular block, the first twitch appears first, then the second (when the first twitch has recovered to about 20% of control), then the third, and finally the fourth (Fig. 6.9B).
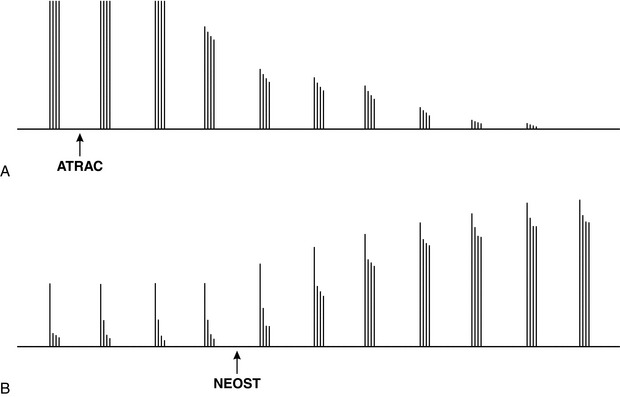
FIGURE 6.9 (A) After administration of a non-depolarizing muscle relaxant (in this instance, atracurium (ATRAC), the decrease in height of the fourth twitch of the TOF response is more marked than the decrease in height of the third twitch, which is more marked than the decrease in the second, which is greater than the decrease in the first. The effect is known as ‘fade’. Within 2 min, the TOF response has been ablated completely. (B) On recovery, the first twitch response appears first, then the second, the third and finally the fourth. Marked fade is present, but after administration of an anticholinesterase (neostigmine (NEOST)) recovery of all four twitches occurs rapidly.
Post-Tetanic Potentiation or Facilitation
This method of monitoring was developed in an attempt to assess more profound degrees of neuromuscular block produced by non-depolarizing neuromuscular blocking agents. If a single twitch stimulus is applied to the nerve with little or no neuromuscular response, but after a 5 s delay a burst of 50-Hz tetanus is given for 5 s, the effect of a further twitch stimulus 3 s later is enhanced (Fig. 6.6). In the presence of profound block, the effect of repeated single twitches applied after the tetanus until the response disappears can be counted; this is termed the post-tetanic count. The augmentation of the twitch is thought to be caused by presynaptic mobilization of acetylcholine as a result of the positive feedback effect of the run of tetanus.
Double-Burst Stimulation (DBS)
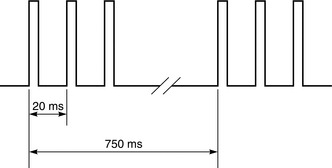
In an attempt to develop a clinical tool which would allow more accurate assessment of residual block by visual or tactile means than fade of the TOF response, Viby-Mogensen suggested the application of two or three short bursts of 50-Hz tetanus, each comprising two or three impulses separated by a 750-ms interval. Each square-wave impulse lasts for 0.2 ms (Fig. 6.10). If records of the fade of the DBS and the TOF response are compared, they are very similar, but there is evidence to suggest that visual assessment of DBS in the later stages of recovery (TOFR < 0.6), is more accurate.
Indications for Neuromuscular Monitoring
 during prolonged anaesthesia, when repeated increments of neuromuscular blocking agents are required
during prolonged anaesthesia, when repeated increments of neuromuscular blocking agents are required
 when infusions of muscle relaxants are given (including in the ITU)
when infusions of muscle relaxants are given (including in the ITU)
 in the presence of renal or hepatic dysfunction
in the presence of renal or hepatic dysfunction
 in patients with neuromuscular disorders
in patients with neuromuscular disorders
 in patients with a history of sensitivity to a muscle relaxant or poor recovery from block
in patients with a history of sensitivity to a muscle relaxant or poor recovery from block
 when poor reversal of neuromuscular block is encountered unexpectedly.
when poor reversal of neuromuscular block is encountered unexpectedly.
Ali, H.H., Utting, J.E., Gray, T.C. Quantitative assessment of residual antidepolarizing block. II. Br. J. Anaesth. 1971;43:478–485.
Appiah-Ankam, J., Hunter, J.M. Pharmacology of neuromuscular blocking drugs. Continuing Education in Anaesthesia Critical Care and Pain. 2004;4:2–7.
Khirwadkar, R., Hunter, J.M. Neuromuscular physiology and pharmacology: an update. Continuing Education in Anaesthesia Critical Care and Pain. 2012;12:237–244.
King, J.M., Hunter, J.M. Physiology of the neuromuscular junction. Continuing Education in Anaesthesia Critical Care and Pain. 2002;2:129–133.
Srivastava, A., Hunter, J.M. Reversal of neuromuscular block. Br. J. Anaesth. 2009;103:115–129.
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Muscle Function and Neuromuscular Blockade
At first, muscle relaxants were used only occasionally, in small doses, as an adjuvant to aid in the management of a difficult case; they were not used routinely. A tracheal tube was not always used, the lungs were not ventilated artificially and residual block was not routinely reversed; all of these caused significant morbidity and mortality, as demonstrated in the retrospective study by Beecher & Todd (1954). By 1946, however, it was appreciated that using drugs such as curare in larger doses allowed the depth of anaesthesia to be lightened, and it was suggested that incremental doses should also be used during prolonged surgery, rather than deepening anaesthesia – an entirely new concept at that time. The use of routine tracheal intubation and artificial ventilation then evolved.
PHYSIOLOGY OF NEUROMUSCULAR TRANSMISSION
Acetylcholine, the neurotransmitter at the neuromuscular junction, is released from presynaptic nerve endings on passage of a nerve impulse (an action potential) down the axon to the nerve terminal. The neurotransmitter is synthesized from choline and acetylcoenzyme A by the enzyme choline acetyltransferase and stored in vesicles in the nerve terminal. The action potential depolarizes the nerve terminal to release the neurotransmitter; entry of Ca2 + ions into the nerve terminal is a necessary part of this process, promoting further acetylcholine release. On the arrival of an action potential, the storage vesicles are transferred to the active zones on the edge of the axonal membrane, where they fuse with the terminal wall to release the acetylcholine (Fig. 6.1). Three proteins, synaptobrevin, syntaxin and synaptosome-associated protein SNAP-25, are involved in this process. These proteins along with vesicle membrane-associated synaptotagmins cause the docking, fusion and release (exocytosis) of acetylcholine from the vesicles. There are about 1000 active sites at each nerve ending and any one nerve action potential leads to the release of 200–300 vesicles. In addition, small quanta of acetylcholine, equivalent to the contents of one vesicle, are released at the neuromuscular junction spontaneously, causing miniature end-plate potentials (MEPPs) on the postsynaptic membrane, but these are insufficient to generate a muscle action potential.
FIGURE 6.1 The neuromuscular junction with an axon terminal, containing vesicles of acetylcholine. The neurotransmitter is released on arrival of an action potential and crosses the junctional cleft to stimulate the postjunctional receptors on the shoulders of the secondary clefts. (Reproduced with kind permission of Professor WC Bowman.)
The active sites of release are aligned directly opposite the acetylcholine receptors on the junctional folds of the postsynaptic membrane, lying on the muscle surface. The junctional cleft, the gap between the nerve terminal and the muscle membrane, has a width of only 60 nm. It contains the enzyme acetylcholinesterase, which is responsible for the ultimate breakdown of acetylcholine. This enzyme is also present, in higher concentrations, in the junctional folds in the postsynaptic membrane (Fig. 6.1). The choline produced by the breakdown of acetylcholine is taken up across the nerve membrane to be reused in the synthesis of the transmitter.
The nicotinic acetylcholine receptors on the postsynaptic membrane are organized in discrete clusters on the shoulders of the junctional folds (Fig. 6.1). Each cluster is about 0.1 μm in diameter and contains a few hundred receptors. Each receptor consists of five subunits, two of which, the alpha (α; MW = 40 000 Da), are identical. The other three, slightly larger subunits, are the beta (β), delta (δ) and epsilon (ε). In fetal muscle, the epsilon is replaced by a gamma (γ) subunit. Each subunit of the receptor is a glycosated protein – a chain of amino acids – coded by a different gene. The receptors are arranged as a cylinder which spans the membrane, with a central, normally closed, channel – the ionophore (Fig. 6.2). Each of the α subunits carries a single acetylcholine binding region on its extracellular surface. They also bind neuromuscular blocking drugs.
FIGURE 6.2 Two postjunctional receptors, embedded in the lipid layer of the postsynaptic muscle membrane. The α, β, ε and δ subunits are demonstrated on the surface of one receptor and the ionophore is seen in cross-section on the other receptor. On stimulation of the two α subunits by two molecules of acetylcholine, the ionophore opens to allow the passage of the end-plate current. (Reproduced with kind permission of Professor WC Bowman.)
Activation of the receptor requires both α sites to be occupied, producing a structural change in the receptor complex that opens the central channel running between the receptors for a very short period, about 1 ms (Fig. 6.2). This allows movement of cations such as Na+, K+, Ca2 + and Mg2 + along their concentration gradients. The main change is influx of Na+ ions, the end-plate current, followed by efflux of K+ ions. The summation of this current through a large number of receptor channels lowers the transmembrane potential of the end-plate region sufficiently to depolarize it and generate a muscle action potential sufficient to allow muscle contraction.
Acetylcholine receptors are also present on the presynaptic area of the nerve terminal. These are of a slightly different structure to the postsynaptic nicotinic receptors (α3β2). It is thought that a positive feedback mechanism exists for the further release of acetylcholine, such that some of the released molecules of acetylcholine stimulate these presynaptic receptors, producing further mobilization of the neurotransmitter to the readily releasable sites, ready for the arrival of the next nerve stimulus (Fig. 6.3). Acetylcholine activates sodium channels on the prejunctional nerve membrane, which in turn activate voltage-dependent calcium channels (P-type fast channels) on the motor neurone causing an influx of calcium into the nerve cytoplasm to promote further acetylcholine release.
FIGURE 6.3 Acetylcholine receptors are present on the shoulders of the axon terminal, as well as on the postjunctional membrane. Stimulation of the prejunctional receptors mobilizes (MOB) the vesicles of acetylcholine to move into the active zone, ready for release on arrival of another nerve impulse. The mechanism requires Ca2 + ions. (Reproduced with kind permission of Professor WC Bowman.)
PHARMACOLOGY OF NEUROMUSCULAR TRANSMISSION
Depolarizing Neuromuscular Blocking Agents
Succinylcholine Chloride (Suxamethonium)
This quaternary ammonium compound is comparable to two molecules of acetylcholine linked together (Fig. 6.4). The two quaternary ammonium radicals, N+(CH3)3 have the capacity to cling to each of the α units of the postsynaptic acetylcholine receptor, altering its structural conformation and opening the ion channel, but for a longer period than does a molecule of acetylcholine. Administration of succinylcholine therefore results in an initial depolarization and muscle contraction, termed fasciculation. As this effect persists, however, further action potentials cannot pass down the ion channels and the muscle becomes flaccid; repolarization does not occur.
FIGURE 6.4 The chemical structures of acetylcholine and succinylcholine. The similarity between the structure of succinylcholine and two molecules of acetylcholine can be seen. The structure of decamethonium is also shown. The quaternary ammonium radicals N+(CH3)3 cling to the α subunits of the postsynaptic receptor.
Inherited Factors: The exact structure of plasma cholinesterase is determined genetically, by autosomal genes, and this has been completely defined. Several abnormalities in the amino acid sequence of the normal enzyme, usually designated , are recognized. The most common is produced by the atypical gene, , which occurs in about 4% of the Caucasian population. Thus a patient who is a heterozygote for the atypical gene demonstrates a longer effect from a standard dose of succinylcholine (about 30 min). If the individual is a homozygote for the atypical gene , the duration of action of succinylcholine may exceed 2 h. Other, rarer, abnormalities in the structure of plasma cholinesterase are also recognized, e.g. the fluoride and silent genes. The latter has very little capacity to metabolize succinylcholine and thus neuromuscular block in the homozygous state lasts for at least 3 h. In such patients, non-specific esterases gradually clear the drug from plasma.
Acquired Factors: In these instances, the structure of plasma cholinesterase is normal but its activity is reduced. Thus, neuromuscular block is prolonged by only minutes rather than hours. Causes of reduced plasma cholinesterase activity include:
Side-Effects of Succinylcholine
Muscle Pains: These occur especially in the patient who is ambulant soon after surgery, such as the day-case patient. The pains, thought possibly to be caused by the initial fasciculations, are more common in young, healthy patients with a large muscle mass. They occur in unusual sites, such as the diaphragm and between the scapulae, and are not relieved easily by conventional analgesics. The incidence and severity may be reduced by the use of a small dose of a non-depolarizing muscle relaxant given immediately before administration of succinylcholine, e.g. gallamine 10 mg (which is thought to be most efficacious in this respect) or atracurium 2.5 mg. However, this technique, termed pre-curarization or pretreatment, reduces the potency of succinylcholine, necessitating administration of a larger dose to produce the same effect. Many other drugs have been used in an attempt to reduce the muscle pains, including lidocaine, calcium, magnesium and repeated doses of thiopental, but none is completely reliable.
Increased Intraocular Pressure: This is thought to be caused partly by the initial contraction of the external ocular muscles and contracture of the internal ocular muscles after administration of succinylcholine. It is not reduced by pre-curarization. The effect lasts for as long as the neuromuscular block and concern has been expressed that it may be sufficient to cause expulsion of the vitreal contents in the patient with an open eye injury. This is unlikely. Protection of the airway from gastric contents must take priority in the patient with a full stomach in addition to an eye injury, as inhalation of gastric contents may threaten life.
Increased Intragastric Pressure: In the presence of a normal lower oesophageal sphincter, the increase in intragastric pressure produced by succinylcholine should be insufficient to produce regurgitation of gastric contents. However, in the patient with incompetence of this sphincter from, for example, hiatus hernia, regurgitation may occur.
Hyperkalaemia: It has long been recognized that administration of succinylcholine during halothane anaesthesia increases the serum potassium concentration by 0.5 mmol L–1. This effect is thought to be caused by muscle fasciculation. It is probable that the effect is less marked with the newer potent inhalational agents, e.g. isoflurane, sevoflurane. A similar increase occurs in patients with renal failure, but as these patients may already have an elevated serum potassium concentration, such an increase may precipitate cardiac irregularities and even cardiac arrest.
