Chapter 10 Muscle disorders
Introduction
Muscle diseases include a number of rare, often progressive conditions leading to physical disability and, frequently, reduced life expectancy. Although each condition is rare, the overall prevalence of muscle disease is 1:1000 and individual disorders tend to present at around the same age, although there may be a wide range. Significant improvements in management means that affected children are surviving to middle age where previously they would have died and they are now presenting for care in adulthood (Wagner et al., 2007). Knowledge of these conditions tends to be poor outside the specialist area of paediatric physiotherapy. This chapter gives an overview of the most frequently seen disorders so the physiotherapist is aware of the different diagnoses which can influence overall management. For many conditions, management strategies are transferable and knowledge of the most frequently seen conditions will be relevant even to the most rare disorder. For a comprehensive text on muscle disorders the reader is referred to Dubowitz (1995).
Classification and diagnostic investigations
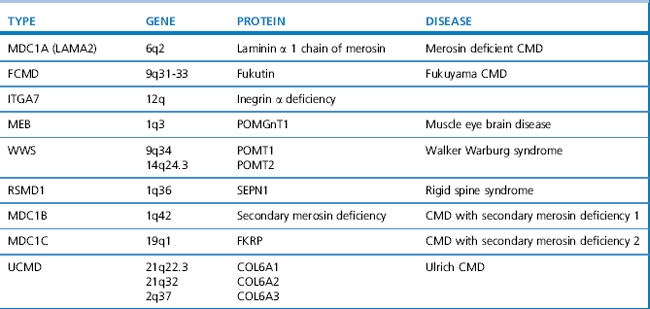
Since the 1980s tremendous advances in genetics and molecular biology have enhanced our understanding of the pathogenesis of muscle disorders. This has led to the availability of genetic testing for many conditions which has broadened our concept of classical phenotypes to include milder, even subclinical presentations. Becker muscular dystrophy (BMD) is a good example, whereby the disorder can result in loss of mobility as early as 16 years of age or as late as the sixth decade (Bradley et al., 1978), yet some patients present only with exertional cramps and myoglobinuria (Gospe et al., 1989). The limb girdle muscular dystrophies (LGMD) are another example. They are a heterogeneous group of disorders classified according to age of onset, inheritance pattern, and molecular and genetic defect, as shown in Table 10.1. The classification of congenital muscular dystrophies (CMD), based on clinical and/or pathological features, is also expanding and several forms are alleic with LGMD as shown in Table 10.2.
Table 10.1 Classification of the limb girdle and sex-linked muscular dystrophies in relation to the genetic defect
| Type | Gene Locus | Gene Product |
|---|---|---|
| Autosomal dominant | ||
| LGMD1A | 5q22-q34 | Myotilin |
| LGMD1B | 1q11-21 | Lamin A/C |
| ADEDMD | 1q11-21 | Lamin A/C |
| LGMD1C | 3p25 | Caveolin 3 |
| Autosomal recessive | ||
| LGMD2A | 15q15.1-q21.1 | Calpain 3 |
| LGMD2B | 2p13 | Dysferlin |
| LGMD2C | 13q12 | γ Sarcoglycan |
| LGMD2D | 17q21 | α Sarcoglycan |
| LGMD2E | 4q12 | β Sarcoglycan |
| LGMD2F | 5q33-q34 | δ Sarcoglycan |
| LGMD2G | 17q11-12 | Telethonin |
| LGMD2H | 9q31-34.1 | Not yet known |
| LGMD2I | 19q13.3 | FKRP |
| LGMD2J | 2q31 | Titin |
| LGMD2K | 9q34.1 | POMT1 |
| LGMD2L | 11p13-p12 | |
| LGMD2M | 9q31 | POMT2 |
| Sex-linked | ||
| Dystrophinopathy | Xp21 | Dystrophin |
| (DMD, BMD) | ||
| EDMD | Xq28 | Emerin |
LGMD, limb girdle muscular dystrophy; ADEDMD, autosomal dominant Emery Dreifuss muscular dystrophy; DMD, Duchenne muscular dystrophy; BMD, Becker muscular dystrophy; EDMD, Emery Dreifuss muscular dystrophy.
The severity of respiratory muscle involvement does not always correlate with the severity of skeletal weakness and it is, therefore, essential that all patients with neuromuscular disorders are monitored regularly for forced vial capacity (FVC) (Griggs et al., 1981; Hough, 1991; also see Ch. 15). Symptoms of incipient respiratory failure include recurrent chest infections, weight loss, early-morning headaches, sweating, disturbed nights and daytime somnolence. A sleep study will confirm nocturnal hypoventilation, and non-invasive respiratory support improves symptoms and can be life saving.
Principles of management
Current therapeutic research is targeted towards finding ways of replacing the abnormal protein. Those individuals with fewer secondary complications of their disorder (e.g. contractures and spinal deformity) will almost certainly derive the greatest benefit if new effective treatments are discovered. Management is based upon the recognition of the patient’s specific needs, together with monitoring for complications of the disorder, e.g. 24-hour cardiac monitoring in EDMD and respiratory monitoring in rigid spine syndrome (RSSI). Corticosteroids have been shown to improve muscle strength in Duchenne muscular dystrophy (DMD), thus prolonging independent walking and are now routinely prescribed from the age of 5 years (Manzur et al., 2008a). But side effects are significant, especially osteoporosis, and must be actively managed.
The muscular dystrophies
The muscular dystrophies are a heterogeneous group of genetically determined disorders associated with progressive degeneration of skeletal muscle. They can be subdivided into a number of different conditions based upon the mode of inheritance, protein, enzyme and/or genetic defect (Tables 10.1). The Xp21 dystrophies or dystrophinopathies are allelic disorders with a wide spectrum of severity with DMD at the most severe end, a group of intermediate patients and BMD at the milder end of the spectrum. X-linked cardiomyopathy (XLDC) is caused by a deletion in the same gene and results in a severe life-limiting cardiomyopathy, but little, if any, muscle weakness. All of these disorders are caused by a defect of the protein dystrophin and are characterized by X-linked inheritance, in which males are affected and females are carriers, although up to 10% of carriers can manifest muscle weakness. Apart from muscle hypertrophy particularly affecting the calf muscles, there is a normal appearance in infancy, although the serum, CK is very high. With advancing age there is progressive muscle weakness and wasting leading to severe physical disability.
Duchenne muscular dystrophy
DMD was first described by Meryon in 1852 and later by Duchenne. It is rapidly progressive and is the most severe of all the muscular dystrophies. The incidence is 1:3500 live male births and there is a prevalence in the population of 1.9–4.8 per 100 000 (Emery, 1991).
Clinical and diagnostic features
By 7–8 years of age, contractures of heel cords and ilio-tibial bands lead to toe walking. Without corticosteroid treatment ambulation is always lost by the 13th birthday and the mean age for loss of ambulation is 9.5 years (Emery & Muntoni, 2003). Prolonged sitting caused by wheelchair dependence leads to the rapid development of flexion contractures of the elbows, hips and knees.
In the early ambulatory phase of DMD, an equinus foot posture is precipitated by relative weakness of the ankle dorsiflexors compared with the better preserved plantar-flexors. Gait analysis has shown that a dynamic equinus is a necessary biomechanical adaptation to maintain knee stability in the presence of gross quadriceps muscle weakness. Forceful action of the ankle plantarflexors provides a torque which opposes knee flexion (Khodadadeh et al., 1986). Thus, contracture of the Achilles tendon, which eventually accompanies disease progression, is secondary to dynamic equinus.
Thus, DMD is a severe life-limiting disease characterized by muscle weakness, contracture, deformity and progressive disability. However, ‘incurable’ is not synonymous with ‘untreatable’. A variety of therapeutic and surgical measures are available that can help to minimize deformity, prolong independent ambulation and maximize functional capabilities. There is evidence that improved management strategies are resulting in increased survival rates (Eagle et al., 2002; Eagle et al., 2007). Corticosteroid treatment results in an increase in muscle strength followed by a slowing down of the dystrophic process (Manzur et al., 2008a). Most boys will die from respiratory or cardiac failure, but the introduction of nocturnal nasal ventilation has improved survival figures, such that the average life expectancy is now 25 years or more with non-invasive ventilation, compared with 19 years without this treatment (Eagle et al., 2002). The principles of successful management are based on an understanding of the natural evolution of patterns of weakness, contracture and deformity, so that intervention can be staged appropriately.
Pathology
Weakness in DMD results from the gradual loss of functional muscle fibres which are replaced by fat and connective tissue due to a lack of dystrophin, a protein encoded by the Xp21 gene, is the primary biochemical defect (Muntoni et al., 2003). Dystrophin is integral within a complex of proteins which stabilizes the integrity of the sarcolemmal membrane, particularly during the stress associated with repeated cycles of contraction and relaxation. Absence of dystrophin in the skeletal and cardiac muscle results in a reduction in permeability of the muscle cell membrane, so allowing excessive quantities of calcium to accumulate within the muscle fibre leading to myofibrillar over-contracture, breakdown of myofibrils and various metabolic disturbances that culminate in muscle fibre degeneration. Dystrophin isoforms are also expressed in Schwann and Purkinge cells found in the brain which is the reason for the high incidence of learning difficulty.
Diagnosis
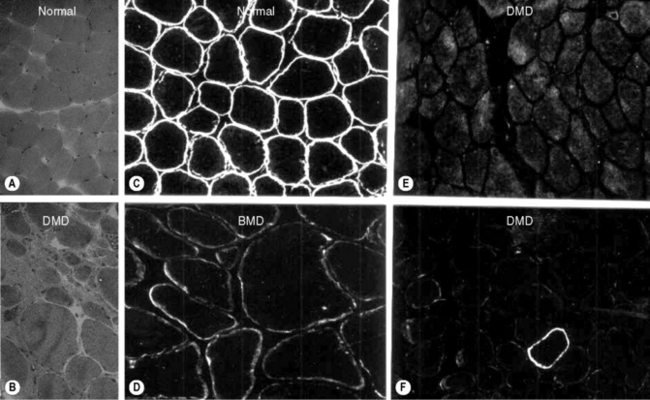
Diagnosis is suspected by finding a raised serum creatine kinase usually in the region of 50–100 times the normal level. A muscle biopsy is necessary to confirm the diagnosis together with DNA analysis. Muscle biopsy can be undertaken as a percutaneous technique using a biopsy needle or as an open procedure, depending on the practice of the investigating unit. Muscle histology demonstrates dystrophic features which include: an increased variation of fibre size, evidence of necrosis with phagocytosis, an increase in central nuclei, hypercontracted eosinophillic hyaline fibres, and an increase in fat and connective tissue (Figure 10.1). Histochemical staining using antibodies to N, C and rod domain epitopes of dystrophin usually show complete absence of the protein, except for occasional revertent fibres (these are fibres which label normally with antibodies to dystrophin; their origin is not understood).
DNA testing from a blood sample will demonstrate a frame-shift deletion within the dystrophin gene in approximately 60% of DMD patients, the rest will result from duplications and nonsense mutations (Mastaglia & Laing, 1996). Confirmation of the diagnosis by DNA testing allows carrier detection and prenatal diagnosis in the affected boy’s mother and female relatives. In some cases DNA analysis is the only test required to confirm the diagnosis of DMD. In BMD the reading frame of the gene is preserved (in-frame). Exceptions to the frame-shift rule occur with some deletions and thus a muscle biopsy is recommended to confirm the diagnosis.
General management
From an early age, tightness and subsequently contractures of the tendo-achilles (TA) develop. Daily passive stretching of the TA and provision of night splints are recommended as soon as any tightening is demonstrated. Later on, when significant pelvic girdle weakness is manifest by a waddling gait, the ilio-tibial bands and hip flexors may start to tighten. Care must be taken to ensure assessment at each outpatient visit with advice to undertake stretching exercises. The priority of physical management is to prolong ambulation for as long as possible, since once ambulation is lost scoliosis and joint contractures develop rapidly. At the point of loss of ambulation, light weight knee-ankle-foot orthoses (KAFOs) can be used to prolong walking (Figure 10.2). This usually involves a minor surgical procedure to percutaneously release any lower limb contractures, usually the TAs, together with an intensive rehabilitation programme. Where there are only moderate contractures of the TAs, serial casting may be an alternative to surgery to allow fitting of the KAFOs (Main et al., 2007). The parents and child must be both strongly motivated and well supported for this form of rehabilitation to be successful.
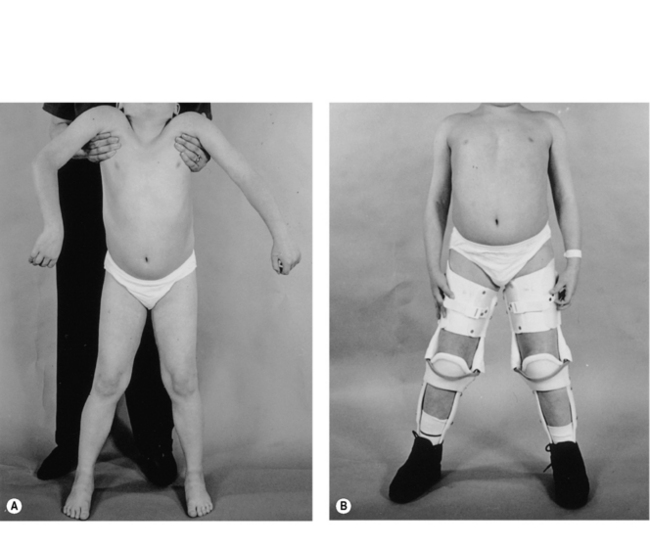
In recent years, there is growing evidence that treatment with corticosteroids (prednisolone or deflazacort) will stabilize muscle function for some time, therefore delaying the loss of ambulation by up to 2 years (Manzur et al., 2008a). Careful monitoring for side-effects is essential, especially weight gain, which may potentially have a negative effect on function. The potential benefit of longer-term steroid treatment has yet to be evaluated, but some open studies suggest preserved respiratory and cardiac function (Manzur et al., 2008a). Premature loss of ambulation can follow lower limb fractures or ligament strains unless active management, such as internal fixation, is instigated to promote early mobility. Immobilizing any joint beyond the initial painful phase should be actively discouraged.
Once independent ambulation is lost, regular use of a standing frame is recommended to maintain good posture and reduce contracture development. Rigid ankle foot orthoses (AFOs) are recommended for daytime use to maintain a good foot position. Likewise it is essential to ensure that the wheelchair provides good back and neck support (Pope, 2002) and, ideally, the controls of an electric wheel chair should be centrally placed. Swimming and hydrotherapy are particularly useful and enjoyed by the boys who find they can move more easily in the water.
A rapidly progressive scoliosis requiring surgical stabilization will develop in up to 95% of boys. There is, however, a reduced incidence and severity of scoliosis in glucocorticosteroid-treated boys (Biggar et al., 2006) which is likely to be due to prolongation of walking and increase in trunk muscle strength. Posterior spinal fusion is highly successful in correcting the deformity, and in improving posture and the quality of life for both patient and carer (Mehdian et al., 1989). The timing of operation is crucial since a decline in vital capacity occurs at the same time as the development of scoliosis. To avoid undue anaesthetic risks the procedure must be undertaken when the vital capacity is greater than 30% of predicted height (Manzur et al., 2008b). Thus surgical correction is usually recommended when the degree of spinal curvature is still relatively mild.
Management of restricted participation
Restricted participation has replaced the term ‘handicap’ in the revised International Classification of Functioning, Disability and Health by the World Health Organization (WHO ICF, 2001; see Ch. 11) and its management must take into account psychosocial issues, mobility and education. Providing an electric wheelchair is essential to providing a degree of independence, but this requires appropriate home adaptations including: widened doors and indoor/outdoor wheelchair access. A hoist is required for lifting in and out of the wheelchair, and the child will require a ground floor bedroom and bathroom with shower and adapted toilet. An electric bed enables the child to alter his position and saves the parents many sleepless nights. An adapted motor vehicle is required for transport to enable the child to drive his own wheelchair in and out of the vehicle.
Respiratory problems
With advancing age, respiratory impairment becomes inevitable and, if not recognized early, is an important cause of unpleasant symptoms or death. Characteristically there is a restrictive defect, with a reduction in total lung capacity caused by a combination of diaphragmatic and intercostal muscle weakness. Chest wall stiffness, recurrent aspiration and an inability to cough effectively compound the respiratory insufficiency leading to an increased frequency of chest infections (Smith et al., 1991). The forced vital capacity is a reliable measure of respiratory function, provided the boy is able to undertake a good technique (in some boys with learning difficulty this may be a problem) (Griggs et al., 1981). The forced vital capacity, when corrected for height, plateaus and then falls progressively on average between 12 and 14 years of age. Once the vital capacity falls below 1 L, in a boy who has reached skeletal maturity, the average life expectancy without treatment is 3 years (Phillips et al., 2001).
Sleep-related respiratory abnormalities play a major role in ventilatory failure, resulting in symptoms of hypercapnia which include: early-morning headache, nausea and sweating, daytime somnolence and a loss of respiratory drive (resulting in rapid deterioration into coma if a high concentration of oxygen is administered). Chronic nocturnal hypoxaemia leads to cor pulmonale (right heart failure), the ECG may show evidence of pulmonary hypertension and right heart strain (Carroll et al., 1991). Once the vital capacity falls below 40% sleep studies should be undertaken at regular intervals to monitor for nocturnal hypoventilation. Treatment by non-invasive nasal ventilation is effective in alleviating symptoms and prolongs survival (Eagle et al., 2002; Jeppesen et al., 2003; Simonds, 2000). There is recent evidence that the cumulative effect of both spinal surgery and nocturnal ventilation further improves survival (Eagle et al., 2007).
Cardiac problems
Post-mortem studies show that all boys with DMD have evidence of cardiomyopathy by 18 years of age. In practice, however, symptomatic cardiomyopathy is less common than might be expected. It has been assumed the sedentary lifestyle of these boys contributes to the lack of symptoms (Hunsaker et al., 1982). Abnormalities of the electrocardiogram are evident from an early age and will be present in all boys by 18 years of age (Nigro et al., 1990); the most common abnormality is a resting tachycardia, which is almost universal. Cardiac arrhythmias occur and may be a cause of early sudden death. When congestive cardiac failure does occur the progression is rapid and relentless. Monitoring with cardiac echo is recommended once ambulation is lost. Early treatment with ACE (angiotensin-converting enzyme) inhibitors has been shown to be beneficial, although the results need to be confirmed (Duboc et al., 2007).
The final illness
Close monitoring and liaison with the general practitioner and palliative care services are paramount. Children’s Respite Hospices play a vital role in supporting families and boys during this difficult time. Patient support groups, such as The Muscular Dystrophy Campaign, frequently fund care workers to support families (see Appendix ‘Associations and Support Groups’).
Becker muscular dystrophy
Becker muscular dystrophy (BMD) is allelic to DMD, but has a milder phenotype (see Table 10.1) and has a prevalence of 1 in 30 000. It is caused by a partial deficiency of the protein dystrophin (Karpati et al., 2001).
Clinical and diagnostic features
BMD has a wide spectrum of severity; at the severest end ambulation may be lost by 16 years of age compared with the mildest form which presents with non-progressive cramps and myoglobinuria (Gospe et al., 1989). Distribution of weakness is similar to DMD, but progression of the disease is much slower and contractures are often less severe than in DMD. As with DMD, muscle hypertrophy, especially of the calves, occurs. Up to 40% of patients will lose ambulation, and prolonging ambulation with long-leg calipers is more difficult than in DMD because of adult height.
In some patients, as with DMD, an unexpected malignant hyperthermia reaction following general anaesthesia may be the first manifestation of the disease. Cardiomyopathy is common and more likely to be symptomatic than in DMD. ECG and echocardiogram abnormalities may be evident in up to 50% of cases (Steare et al., 1992) and many patients have successfully undergone cardiac transplantation (Quinlivan & Dubowitz, 1992).
General management
The management of BMD involves prevention of contractures and prolonging ambulation as with DMD. An active approach to management using low-intensity aerobic exercise has been shown to safely improve fitness and strength in individuals with mild BMD (Sveen et al., 2008). Home adaptations are essential in promoting independence and the patient may require support to continue working in an adapted environment. For those patients who are wheelchair dependent, regular standing, and preventing excessive weight gain and constipation are important. Prevention of respiratory infections by vaccination against influenza and pneumococcus, together with prompt antibiotic treatment of infection are important. Monitoring of respiratory function and overnight oximetry for sleep hypoxaemia are necessary. Symptoms of chronic ventilatory failure should be managed with non-invasive ventilation as for the DMD group. Regular cardiac monitoring with yearly ECGs and cardiac ECHOs every 2 or 3 years is necessary. Early intervention with ACE inhibitors for ventricular dysfunction may be helpful, but as yet has to be fully evaluated. If cardiac symptoms fail to respond to medical treatment, assessment for cardiac transplantation is warranted (Quinlivan & Dubowitz, 1992).
Emery–Dreifuss muscular dystrophy
This is a rare but clinically distinct form of MD. Two modes of inheritance exist. Firstly, a sex-linked form (EDMD) in which mutations in the Emerin gene located at Xq28 lead to a complete absence of the nuclear envelope protein Emerin (Mastaglia & Laing, 1996). Secondly, there is an autosomal dominant form (ADEDMD) in which the defective gene is at 1q11-q23 encoding for another nuclear envelope protein; Lamin A/C (LMNA) (Bonne et al., 1999).
Clinical and diagnostic features
The main feature of the nuclear envelope muscular dystrophies is the predominance of joint contractures and muscle weakness occurring in a scapulo-peroneal distribution. Contractures predominantly affect the neck and spine, elbows and tendo-achilles. Disturbances of cardiac conduction are universal by the second or third decades, and may result in sudden death unless detected and cardiac pacing instituted bb0025(Bialer et al., 1991). Thus, regular ECG and 24-hour ECG monitoring are essential. Muscle weakness is slowly progressive, however many patients retain some ambulation throughout adult life.
General management
The associated weakness is usually mild. However, due to rigidity of the spine throughout its length, the patient is unable to compensate for any hip extensor weakness with a lumbar lordosis, as is commonly seen in many of the other neuromuscular diseases. Instead, the patient maintains the centre of mass with increasing equinus at the ankles, leading to secondary contracture of the Achilles tendon. If these contractures become severe, they may in themselves jeopardize mobility and require percutaneous surgical correction. Management should be aimed at controlling the progression of deformity by appropriate strengthening, passive stretching and splinting techniques (see Ch. 12 & 14).
Limb girdle muscular dystrophy
The limb girdle muscular dystrophies (LGMDs) are a clinically and genetically diverse group of disorders, characterized by the predominance of limb girdle weakness, with or without contractures (Table 10.1). Inheritance can be either dominant or recessive, depending upon the specific disorder. Clinical heterogeneity also occurs within some of these disorders, for example LGMD2B is caused by mutations in the dysferlin gene on chromosome 2p13 and is allelic to Miyoshi distal myopathy (Illarioshkin et al., 1997).
General management
As with other disabling neuromuscular diseases, patients with LGMD need help and support to adapt their environment to suit their needs. A self-raising chair or pneumatic cushion can be invaluable in assisting rising to standing. Adaptations to the patient’s home may be required to ensure appropriate bathroom facilities, hoists or stair lifts. Adaptations to the patient’s vehicle may also be required to maintain independence. Weight gain can be a major problem for some individuals, especially when they are confined to a wheelchair, and may compromise respiration. Encouraging exercise is important for general health and well being; swimming is an excellent example which also helps maintain strength. Low-intensity aerobic training has been show to improve exercise performance in patients with LGMD type 21 (Sveen et al., 2007). Many patients appreciate the benefit of regular passive stretching exercises and the opportunity to stand using either a tilt table or standing frame. Prevention of chest infections with pneumococcal vaccination and yearly flu vaccines is important for any patient with evidence of respiratory muscle weakness.
Congenital muscular dystrophies
There are several recognized forms of congenital muscular dystrophy depending upon the protein/gene defect and/or the association of central abnormalities (see Table 10.2). They are usually caused by autosomal recessive genes and present at birth or in infancy with hypotonia and joint contractures, often involving the spine. The serum CK may be normal in some groups, and elevated in others. Intellect may be normal or impaired depending upon the presence of neuronal migration defects. White matter changes may be seen on magnetic resonance imaging (MRI) in cases with merosin deficiency, although there is normal intellect. Muscle biopsy shows dystrophic features and in some types specific protein abnormalities.
Clinical and diagnostic features
Reduced fetal movements during pregnancy suggest that signs are already present before birth, such infants are frequently born with arthrogryposis. Features in early infancy consist of muscle weakness and generalized hypotonia, ‘floppiness’, poor suck and respiratory difficulty (Kobayashi et al., 1996). In childhood, motor milestones are delayed, with severe and early contractures and often joint deformities. Weakness is greater in the pelvic girdle and upper leg muscles than in the shoulder girdle and upper arm muscles. On the whole, with the exception of Fukuyama congenital muscular dystrophy (FCMD), these conditions are relatively slowly progressive and functional ability can initially improve over time. Contractures at birth are common, and may restrict function to a greater degree than weakness if not controlled. It is particularly important to be vigilant for the insidious development of contractures and to treat them promptly.
Fascioscapulohumeral muscular dystrophy
This condition follows an autosomal dominant pattern of inheritance with a high degree of penetrance but variable expression. The clinical features are always present by 30 years of age. As many as 30% of cases present sporadically and are due to germ line mosaicism. The overall prevalence is estimated to be 1:20 000 (Kissell, 1999). In 95% of affected individuals, a short fragment on the telomeric portion of chromosome 4 (4q35) is identified (Wijmenga et al., 1991), which has been associated with a reduced number of 3.3-kb tandem repeat segments called D4Z4 in a non-protein-encoding region of the gene (Orrell et al., 1999). The size of the D4Z4 segment correlates inversely with clinical severity. The mechanism by which the reduced number of D4Z4 tandem repeats produce disease is unknown.
The age at onset, degree of severity and course of the disease are more variable than in many other neuromuscular diseases. Within a family it may range from someone who has minimal facial weakness with slow progression, to a condition which has a more marked progression of lower limb weakness that can cause severe disability early in life. Examination reveals facial weakness and a characteristic horizontal smile and inability to whistle. Shoulder-girdle weakness is often asymmetrical and more pronounced than lower limb weakness. Scapular winging with characteristic shoulder ‘terracing’ on abduction of the arms reflects weakness of serratus anterior, trapezius and rhomboids (Figure 10.3). The biceps and triceps muscles tend to be affected later.
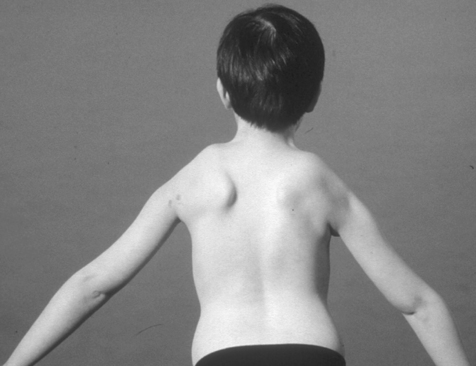
The deltoid muscles are preserved in 50% of cases (Bunch & Siegel, 1993), but even if the deltoids are not involved the muscles lose their mechanical advantage due to lack of shoulder-girdle stability, causing limitation of active abduction and flexion. The patient may compensate surprisingly well, using trick movements to raise the hand above shoulder level, but can be more obviously compromised if the activity involves lifting objects of any weight. Foot drop may occur early in the disease due to peroneal and anterior tibial muscle weakness. Leg muscle weakness may eventually progress to loss of ambulation, which occurs in approximately 20% of cases. Joint contractures are rare and mild.
Cardiac involvement is uncommon and reported to occur in about 5% of cases; atrial arrhythmias are the most usual manifestation (Laforet et al., 1998). Sometimes the severe facial weakness may be mistaken for Moebius syndrome (Miura et al., 1998). The serum CK may be normal or three to five times the normal value. Confirmation of diagnosis can now be made with DNA analysis so that muscle biopsy is rarely necessary. Congenital onset is rare and can be associated with deafness, learning difficulties and severe muscle weakness.
General management
Studies of specific treatment for FSH are limited. Patients often develop a marked lumbar lordosis and low back pain due to pelvic girdle weakness, so that strengthening the pelvic musculature plus passive stretching exercises to minimize hip flexion contractures may be helpful. A recent study of mildy affected patients with FSH showed that low-intensity aerobic training over a 12-week period improved exercise performance (Olsen et al., 2005). Ankle–foot orthoses (AFOs) can be provided to control for foot drop, but may not be tolerated unless the ankle achieves plantigrade and quadriceps strength is at least antigravity (Eagle et al., 2001).
Thoracoscapular fusion can be effective in the long term for patients with scapular winging, provided the deltoid muscle remains functional (Rhee & Ha, 2006). More recently scapulothoracic fixation (scapulopexy) using wires to reposition the scapula over the ribs, without arthrodesis, has been shown to be successful (Gianinni et al., 2007). Both techniques result in improved range of movement and appearance of the shoulder and this is most beneficial for patients whose occupation specifically requires the ability to sustain flexion and abduction.
Myotonic dystrophy
Classical myotonic dystrophy (dystrophia myotonica; DM1) is a dominantly inherited multisystem disease that is relatively common, with a prevalence of 4 per 100 000 (Harper, 1989). DM1 is caused by an expansion of CTG repeats in the DMPK gene on chromosome 19q13 (Friedrich et al., 1987). The size of the expansion determines the severity of the phenotype and contributes to the phenomenon of anticipation, whereby the severity of the condition worsens in successive generations. This is especially obvious with the congenital form of the disease, where a severe and life-threatening phenotype occurs in the newborn infant, in contrast to the often presymptomatic mother.
At the mildest end of the spectrum cataracts or diabetes may be the only feature of the condition. Anaesthetic risks are significant in all cases, depolarizing muscle relaxants can lead to severe laryngospasm, and when given in combination with potent inhalational anaesthetics, a malignant hyperthermia reaction can occur (Moore & Moore, 1987). Myotonia is demonstrable by asking the patient to clench his or her fist and then let go quickly, the patient has to release their grip using the other hand. The EMG shows widespread myotonic discharges, which produce a classical dive-bomber sound caused by gradual fluctuations in frequency and amplitude (Fawcett & Barwick, 1994). The diagnosis is confirmed by DNA analysis.
Spinal muscular atrophies
The spinal muscular atrophies (SMAs) are a group of disorders in which there is degeneration of the anterior horn cells of the spinal cord, resulting in muscle weakness. They are the most common neuromuscular disease in childhood after DMD and affect both sexes. The mode of inheritance is mainly autosomal recessive but can vary, with dominant or X-linked traits (Emery, 1971). The genetic defect for the recessive form is a deletion in exon 7 and or exon 8 of the SMN1 gene on chromosome 5q (Ogino et al., 2004). Weakness is symmetrical, is greater proximally than distally, and the pelvic girdle is weaker than the shoulder girdle. Weakness is generally non-progressive, although as a result of increasing height and weight there may be some loss of functional activities over time. There is no facial weakness and intellectual development is often above normal. Classification of 5q SMA is most usefully based on clinical severity (Dubowitz, 1995):
Mild spinal muscular atrophy (type 3 or Kugelburg–Welander disease)
In Type 3 SMA the ability to walk is achieved at the normal age or slightly late, and the child often presents with a deterioration of motor skills often around the time of the pubertal growth spurt. Proximal weakness is more marked in the lower, rather than the upper limbs, and particularly in the hip abductors. Individuals are therefore dependent on compensatory pelvic and trunk motion for forward propulsion. Strengthening programmes focusing on the pelvic girdle would be functionally beneficial for type 2 and type 3 SMA (Armand et al., 2009). Joint contractures and progressive scoliosis are rare in this group (Carter et al., 1995). Weakness is generally relatively static, but rapid periods of growth may result in loss of ambulation, and rehabilitation can be achieved in some using lightweight KAFOs.
Muscle sodium channel and muscle chloride channel diseases
The muscle channelopathies comprise two groups of disorders – those where the muscle is hyperexcitable, leading to stiffness (myotonia), and those conditions where there is reduced muscle excitability leading to weakness (the periodic paralyses). Both symptoms are caused by a temporary depolarization of the skeletal muscle membrane (Karpati et al., 2001).
Glycogen storage diseases
The glycogen storage diseases (GSDs) are characterized by abnormal glycogen metabolism.
Pompe disease
Pompe disease (acid maltase deficiency, GSD11) is a lysosomal storage disorder affecting muscle glycogen metabolism. The condition can present in infancy with a severe phenotype, resulting in progressive muscle weakness and cardiomyopathy. Death usually occurs by 2 years of age from cardiac and respiratory failure. Early diagnosis is essential for a good outcome with enzyme replacement with myozyme (Nicolino et al., 2009). Less severe forms of GSD11 present during childhood (juvenile-onset) and adulthood. The condition is milder with a slower progression, causing limb girdle weakness, which closely resembles LGMD or mild SMA. Respiratory failure due to both diaphragmatic involvement and cardiomyopathy occurs and can be disproportionate to the skeletal weakness. Regular cardiorespiratory monitoring is therefore recommended. Enzyme replacement therapy is available for all forms of Pompe disease, although the evidence for benefit is currently based upon the treatment of the severe infantile form.
McArdle disease
If exercise is continued despite pain, an electrically silent contracture occurs where the muscle seizes and later becomes swollen. This is followed by myoglobinuria, a dark red/brown or black discoloration of the urine caused by rhabdomyolysis (muscle damage), which, if severe, can cause acute renal failure. The serum CK is invariably raised at rest and there is a lack of rise in venous lactate during a forearm exercise test. Muscle biopsy demonstrates a subsarcolemmal accumulation of glycogen and absent muscle phosphorylase activity. The condition is caused by mutations in the muscle phosphorylase gene on chromosome 11q13 (Beynon et al., 2002). Strenuous anaerobic exercise should be avoided to prevent muscle damage; however, regular gentle aerobic exercise is recommended, to upregulate fatty acid oxidation, thus conditioning the muscles and improving performance.
Inflammatory myopathies
Inflammatory myopathies can be divided into three distinct groups:
DM and PM have an autoimmune aetiology and respond well to immuno-suppressive agents; IBM occurs in older people, it is usually sporadic and does not always respond to immunesuppression. There are two rare subtypes of inherited IBM in younger patients, caused by homozgous mutations in the GNE gene, while in older patients a rare genetic form with mutations in VCP is associated with Pagets disease and dementia (Phadke et al., 2009; Vesa et al., 2009).
DM is an inflammatory muscle condition associated with a characteristic facial rash occurring around the eyes (heliotrope rash). The condition affects children between the ages of 5 and 15 years. A second peak in incidence occurs in middle and old age, and is frequently associated with an underlying malignancy (Callen, 1988). The disease is characterized by muscle pain and proximal weakness, joint contractures can develop rapidly. The serum CK and erythrocyte sedimentation rate may be elevated, and a muscle biopsy confirms the inflammatory process. Treatment includes immune suppression and physiotherapy to minimize contractures.
Physical management of neuromuscular disorders
This section highlights the areas of management which are specific to muscle disorders, both in adulthood and childhood. Details of treatment concepts and techniques are found in Chapters 11 & 12, respectively. A problem-solving approach is preferable and treatment planning should involve the multidisciplinary team (MDT), the patient, parents and carers.
Assessment
Thorough, standardized and regular assessment of neuromuscular patients is essential because of the progressive nature of many conditions and the superimposed effect of growth on children. Assessment will guide clinical management and evaluate therapeutic outcome for research purposes. The key aspects of assessment are measurement of muscle strength and performance and lung function; the principles of assessment are discussed in Chapter 1 (also see Ryerson, 2009; Stokes, 2009).
Measurement of muscle strength
Manual muscle testing
Manual muscle testing is the most widely used means of assessing muscle strength and has been recommended as an outcome measure for therapeutic trials in neuromuscular disease (Brooke et al., 1981). The Medical Research Council scale of grading muscle strength is the most widely known grading system and is based on an ordinal scale of 0 to 5. No special equipment is required and manual muscle testing is a rapid method of determining the distribution and severity of weakness over a large number of muscle groups. However, the major criticism of this method is its subjectivity. There are no standardized joint positions at which testing should be performed and the point at which counterforce is administered is also self-selected. The proportion of maximum strength required to overcome gravity is markedly different between muscle groups (Wiles et al., 1990), and a loss of strength in excess of 50% may develop before weakness can be detected by manual muscle testing (Fisher et al., 1990).
Dynamometry
Force can be measured directly with dynamometers; these quantitative measurements of muscle force are superior to manual muscle testing and provide the most direct method of assessing a particular muscle group. A hand-held dynamometer can measure maximal isometric strength and the results are highly reproducible provided standardized techniques are used and the same observer performs the measurement on each occasion (Bohannon, 1986; Lennon & Ashburn, 1993). Serial measurements of a single patient will be the most useful means of evaluating the distribution and rate of change of muscle weakness, whilst the degree of weakness can be established by comparison with published normal values of muscle strength (Beenacker et al., 2001). Hand-held dynamometers are useful only when muscles are weak, since their use is restricted by the strength of the operator to oppose the patient’s efforts. Strain gauges attached to rigs, and also commercially available isometric and isokinetic machines, are available.
Measurement of joint range
The development of joint contractures should be monitored carefully and joint ROM can be measured using a goniometer. However, caution must be taken with this method as it shows variable inter-rater reliability and measurements should be made by the same assessor where possible (Pandya et al., 1985).
Measurement of functional performance
The quantification of muscle strength has proved to be of value in the assessment and management of many muscle diseases, but it can be seen that this is a measure of impairment and is incomplete without concomitant measures of disability and ultimately the handicap to the patient. Measures of functional performance range from simple tests such as the ability to rise from the floor to more detailed measures of motor ability including gait analysis (D’Angelo et al., 2005; Khodadadeh et al., 1986; Sutherland et al., 1981). These measurements are susceptible to the effects of impairments other than strength, but it is important in terms of patient management to determine whether a change in disability can properly be attributed to a change in the strength of the muscles measured.
Motor ability tests
Classically the most commonly used measures of motor ability have been those developed for DMD by Vignos et al. (1963), Brooke et al. (1981) and the Hammersmith Motor Ability Scale (HMAS) (Scott et al., 1982). The Vignos scale was originally designed as a functional classification and its sensitivity as an objective measure of function is doubtful. The HMAS has been widely used in clinical practice, although there is little published information about its reliability and validity. This scale has recently been modified, is now known as the ‘North Star Ambulatory Assessment’ and been shown to have excellent inter-observer reliability (Mazzone et al., 2009). Its 17 motor activities are biased towards activities involving the lower limbs, making it suitable for assessment of ambulant patients (Table 10.3). The patient sequentially performs a succession of movements that are scored on a 3-point scale which can be completed in 15 minutes. A functional test, the EK scale, for non-ambulant patients with DMD has been shown to be reliable by Steffensen et al. (2002).
| Test Item | Score |
|---|---|
| 1. Stand | |
| 2. Walk (10 m) | |
| 3. Sit to stand from chair | |
| 4. Stand on one leg – R | |
| 5. Stand on one leg – L | |
| 6. Climb step – R | |
| 7. Climb step – L | |
| 8. Descend step – R | |
| 9. Descend step – L | |
| 10. Gets to sitting | |
| 11. Rise from floor | |
| 12. Lifts head | |
| 13. Stand on heels | |
| 14. Jump | |
| 15. Hop – R | |
| 16. Hop – L | |
| 17. Run | |
| TOTAL SCORE |
Scores: 2 – ‘Normal’: achieves goal without assistance; 1 – Modified method but achieves goal independent of physical assistance from another; 0 – Unable to achieve independently. R, right leg; L, left leg
(Reprinted from Mazzone ES, Messina S, Vasco G et al. Reliability of the North Star Ambulatory Assessment in a multicentric setting. Neuromuscular Disorders, 2009; 19:458-461, with permission from Elsevier.)
Timed tests
Timed performance tests are commonly used as supplementary measures of physical performance as a means of reflecting progressive weakness (Brooke et al., 1981). A timed walk over a fixed distance or time, such as the 10 m test or the 6 minute walk test, can reliably detect changes over time (Mazzone et al., 2009). The 10 m walk test can predict loss of ambulation (McDonald et al., 1995). Timing the Gowers’ manoeuvre is a useful measure of changing function and supine lying is a more reproducible starting position than is sitting.
Lung function
Spirometry forms part of the regular assessment to monitor changes in lung function. Inspiratory and expiratory mouth pressures can also be measured to assess the strength of the respiratory muscles. Details of lung function tests can be found elsewhere; see Chapter 15 and also Griggs et al. (1981) and Hough (1991).
Treatment principles
The main principles of treatment are:
Treatment concepts (see Ch. 11) and details of techniques (see Ch. 12) are not discussed here, but the principles relevant to patients with muscle disorders are outlined.
Maintenance of muscle strength
There are very few controlled studies on the effect of exercise in neuromuscular disease and many are not disease specific. The results of mild to moderate resistance exercise programmes in muscular dystrophies have shown limited increases in strength, with no negative effect on muscle function (Ansved, 2003). Greatest effects occur in patients with mild to moderate weakness and in the more slowly progressive conditions, whilst patients with severely weak muscles do not generally benefit from strengthening programmes. It appears in normal subjects that a prerequisite for successful strength training is a high content of type II fibres (Jones et al., 1989). The relative deficiency of type II fibres in DMD (Dubowitz, 1985) may contribute to the poor force-generating capacity of dystrophic muscle and could also be a limiting factor in the eventual benefit of a strengthening programme.
Consistent reductions in maximal or peak oxygen uptake, pulmonary ventilation, work capacity and endurance have been reported in both rapidly and slowly progressive neuromuscular disorders (Kilmer, 2002). Short-term studies in disease-specific patients (Olsen et al., 2005; Sveen et al., 2007; Sveen et al., 2008) indicate low to moderate intensity aerobic exercise is well tolerated with functional improvements in aerobic capacity.
Eccentric muscle training is increasingly being used in the training of athletes to facilitate the development of muscle power, i.e. the rate of force generation. However, eccentric exercise can cause appreciable morphological damage to muscle fibres (Newham et al., 1986) and damage of this nature is commonly seen in the muscles of patients with myopathic diseases. Whilst normal muscle recovers from this damage, eccentric exercise would seem best avoided in muscle disease in favour of more traditional concentric protocols.
Edwards et al. (1987) documented important differences in the rate of progression of various muscle groups and highlighted a particularly rapid loss of force in the hip and knee extensors. Insufficiency of these muscle groups has been shown to be the key deficit in functional decline and gait deterioration in DMD (Sutherland et al., 1981). Whilst maximizing muscle strength to achieve optimal or improved functional ability is a primary objective of treatment, the effect of specific muscle strengthening programmes on function in neuromuscular disorders awaits objective evaluation. To devise a strengthening programme, the required functional gain should be considered and appropriate muscle groups targetted.
It is well accepted that in normal individuals physical exercise increases muscle strength, whilst inactivity causes de-conditioning, and there is also widespread observation amongst clinicians that severe restriction of activity causes rapid weakening of muscle in dystrophic conditions and should be avoided. It is therefore important that the duration of enforced immobilization during any acute illness, and after surgery, should be kept to a minimum so that the patient’s return to mobility is not compromised by muscle atrophy. Exercise and strength training in patients with neuromuscular disorders is discussed in Chapter 18.
Weakness occurs when a muscle is held in a shortened position due to joint deformity, and also when it is contracting over a reduced range (Gossman et al., 1982). The establishment of compensatory postures, long before the development of fixed contracture, means that the muscle is biomechanically disadvantaged earlier than is obvious, since it is continually contracting over a shortened range. This could be a major factor in further progression of the disease as optimal function of the muscle is prevented. Joint positioning during strengthening may therefore be important but research in these patients is still required.
Electrical stimulation of normal muscle can improve strength and fatiguability but evidence that the technique is safe, as well as beneficial, in muscle disorders has yet to be produced (see Chapter 12).
Retarding contracture progression
The management of contractures is one of the major contributions of physiotherapy in neuromuscular disease. The aim is not only to retard the progression of contracture, but also and more importantly to promote or prolong independent ambulation and functional ability. Impairment of mobility caused by contractures compromises the strength of the muscles working across the involved joint or joints. The force-generating ability of a muscle is influenced by the length at which it contracts (Jones & Round, 1990) and thus the strength of a muscle held in a shortened position is reduced. In the presence of profound weakness, the maintenance of full joint range of motion is essential for optimal muscle function.
A sustained programme of night splinting and passive stretching in the early stages of DMD can retard the development of lower limb contractures. Two studies have specifically evaluated the effect of passive stretching and the use of orthoses on the development of contractures. Both concluded that the combination of passive stretching and night splints are more effective than passive stretching alone at delaying contractures and prolonging independent ambulation (Hyde et al., 2000; Scott et al., 1981).
Whilst independently ambulant, the provision of AFOs for control of Achilles tendon contracture should be confined to night use only. Gait analysis has shown that an equinus position of the foot is used as a compensatory manouevre to increase knee stability during walking. Khodadadeh et al. (1986) observed that boys with DMD necessarily adopt a dynamic equinus during gait in order to maintain a knee-extending moment in the presence of gross quadriceps weakness. AFOs intended to correct the foot position by reducing the equinus during walking will have biomechanical effects which will de-stabilize the knee. If there is significant quadriceps weakness the knee will buckle. Thus AFOs used in this way reduce the available compensatory manoeuvres and result in premature loss of ambulation.
Promoting or prolonging ambulation
Following the cessation of independent walking, the duration of useful ambulation in children can be prolonged with an immediate programme of percutaneous Achilles tenotomy and rehabilitation in lightweight KAFOs and intensive physiotherapy (Figure 10.2). The gains in additional walking time have varied in different centres but on average an extra 2 years of walking can be achieved and sometimes up to 4 years (Bakker et al., 2000). This approach is now generally accepted as a means of maintaining mobility after independent walking ceases and has been shown to impede the development of both lower limb contractures (Vignos et al., 1996) and scoliosis (Rodillo et al., 1988).
The accurate timing of intervention and prompt provision of orthoses are crucial to the success of prolonging ambulation (Thompson et al., 2007). The optimal time for the provision of the orthoses is when the child has lost useful walking but is still able to stand or walk a few steps. There is no advantage in providing orthoses earlier than this. Two of the important factors used to predict successful outcome are the absence of severe hip and knee contractures, and the percentage of residual muscle strength (Hyde et al., 1982). Swivel walkers may be appropriate to allow movement over short distances at home or school. Once the child has been wheelchair-bound for even a short time, fixed lower limb deformities and muscle weakness rapidly progress, and therefore any delay in undertaking this programme may compromise a successful outcome.
Management of scoliosis
In DMD, a progressive scoliosis almost always develops once the child loses the ability to walk. The period of most rapid deterioration corresponds most closely with the adolescent growth spurt between the ages of 12 and 15 years (McDonald et al., 1995). Progressive scoliosis is also a threat in the adolescent years of patients with type 3 SMA, but due to the profound weakness that is present from early infancy in patients with type 2 SMA it may become a problem at a much earlier age.
The curve often develops in a paralytic long C pattern in the thoraco-lumbar areas and is associated with increasing pelvic obliquity. It further compromises respiratory capacity, which is already restricted by involvement of the respiratory muscles (Kurz et al., 1983). An increasing scoliosis also leads to difficulty in sitting and maintaining head control, and can cause discomfort and pressure areas. Patients will often need to use their elbows for support in maintaining an upright position, so preventing them from using the arms for other functions such as feeding. Untreated, the scoliosis may cause patients to become bedridden.
One of the major benefits of treatment aimed at maintaining an upright posture is that it will help to delay the progression of scoliosis. Once the patient is dependent on a wheelchair, the main means of managing scoliosis are conservative, using a spinal orthosis, or by spinal fusion. The spine should be monitored carefully where scoliosis is a likely complication of the disease, in conjunction with the respiratory capacity using simple spirometry. Once a curve is clinically apparent, any progression is most accurately measured from radiographs using Cobb’s angle. Prompt provision of a spinal orthosis is advisable, to be worn during the day whilst the patient is upright and should be corrective rather than supportive. It is recognized that spinal bracing is not the definitive treatment in curves that are known to be rapidly progressive, but it is important in slowing the rate of progression of the curvature (Seeger et al., 1990) and can be used effectively for skeletally immature patients in whom spinal fusion is not yet indicated.
Posterior spinal fusion is used widely in the management of progressive scoliosis in neuromuscular disease and provides rigid fixation without the need for post-operative immobilization or orthoses. It is effective in achieving maximum curve correction and minimizing respiratory complications. Early surgery is the treatment of choice while respiratory and cardiac function is adequate to undergo the procedure safely. In DMD vital capacity increases with age and growth in the early years, reaches a plateau, and then declines in the early teens. There is therefore a window of opportunity when surgery can be performed safely and this is usually when the curve is 20–40° (Cobb angle) and forced vital capacity is above 30% (Manzur et al., 2008b). Spinal fusion is associated with slowing of the rate of respiratory decline postoperatively, as well as enhanced comfort and seating (Velasco et al., 2007). It has recently been reported that patients with low vital capacity can safely undergo spinal surgery provided it is undertaken in a specialist centre (Gill et al., 2006; Marsh et al., 2003).
In the immediate postoperative period, respiratory therapy to aid removal of secretions will be necessary in patients with a poor cough (see Ch. 15). It is possible for a lumbar lordosis and dorsal kyphosis to be moulded into the rods (Galasko et al., 1992), which helps prevent loss of head control in sitting when weakness of the neck and trunk musculature is likely to be advanced. However, seating requirements will need to be reassessed postoperatively, for example, due to significant upper limb weakness the child is likely to utilize upper trunk flexion to help get his or her mouth down to his or her hands for feeding purposes. Following surgery the height of wheelchair arm supports or table height will need to be adjusted to compensate for a fused spine.
Management of respiratory complications
Chest infections are a serious complication to vulnerable patients with respiratory muscle weakness and a poor cough. Longstanding weakness may lead to more serious secondary problems including widespread microatelectasis with reduced lung compliance, a ventilation perfusion imbalance, and nocturnal hypoxaemia (Smith et al., 1991).
Chest infections should be treated promptly with physiotherapy, postural drainage, antibiotics and, when appropriate, assisted ventilation (see Ch. 15). Spinal orthoses that control scoliosis may reduce respiratory capacity and should be temporarily removed if causing distress or interfering with treatment. The aim of treatment is to help clear the lungs of secretions effectively in the shortest possible time without causing fatigue. Thoracic expansion exercises will allow increased airflow through small airways and the loosening of secretions, while forced expiration techniques, the use of intermittent positive pressure breathing (IPPB) and assisted coughing will aid removal of secretions (Webber, 1988). Diaphragmatic weakness may limit the use of supine or tipped positions for postural drainage. Parents of children prone to recurrent chest infections can become competent at administering chest physiotherapy but will require support that is readily accessible if children become distressed.
Inspiratory muscle training has been reported by some to improve respiratory force and endurance (Martin et al., 1986; Wanke et al., 1994), an effect which may be dose dependent (Topin et al., 2002), whereas others report no significant effects (Rodillo et al., 1989; Stern et al., 1989). Overall training is best started in the early stages of DMD where there is only moderate impairment of lung function. This approach does not appear to be used regularly in clinical practice and has probably been superceded by the successful use of nocturnal ventilation at a later stage.
Respiratory failure may be precipitated by chest infection or it may occur as a result of increasing nocturnal hypoventilation and hypoxia. The onset is often insidious but symptoms include morning drowsiness, headache or confusion and nighttime restlessness, and can be confirmed by sleep study. Life expectancy is less than 1 year once diurnal hypercapnia develops. Symptoms, quality of life and survival can be improved by non-invasive nasal ventilation (Eagle et al., 2002; Simonds, 2000).
‘Social’ and psychological issues in neuromuscular disorders
A complexity of factors influences management at different stages of the patient’s life. For lifelong disorders, these influences pose similar problems as in other disabling conditions, such as cerebral palsy or spina bifida (see Shepherd, 1995). The need for more training and support for professionals managing these patients was highlighted in a survey by Heap et al. (1996).
Acknowledgements
Dr Quinlivan gratefully acknowledges the support of the Muscular Dystrophy Campaign and the AGSDUK.
Ansved T. Muscular dystrophies: influence of physical conditioning on the disease evolution. Curr. Opin. Clin. Nutr. Metab. Care. 2003;6:435-439.
Armand S., Mercier M., Watelain E., Patte K., Pelissier J., Rivier F. Gait pattern in Duchenne muscular dystrophy. Gait Posture. 2009;29(1):36-41. Epub 2008 Jul 25
Bakker J.P.J., De Groot I.J.M., Beckerman H., de Jong B.A., Lankhorst G.J. The effects of knee-ankle-foot-orthoses in the treatment of Duchenne muscular dystrophy: review of the literature. Clin. Rehabil.. 2000;14:343-359.
Bialer M.G., McDaniel N.L., Kelly T.E. Progression of cardiac disease in Emery–Dreifuss muscular dystrophy. Clin. Cardiol.. 1991;14(5):411-416.
Beenakker E.A., van der Hoeven J.H., Fock J.M., et al. Reference values of maximum isometric force obtained in 270 children aged 4-16 years by hand-held dynamometry. Neuromuscul. Disord.. 2001;11(5):441-446.
Beynon R., Quinlivan R.C.M., Sewry C.A. Selected disorders of carbohydrate metabolism. In: Karpati G., editor. Structural and Molecular Basis of Skeletal Muscle Diseases. Basel: ISN Neuropath Press; 2002:182-188.
Bertorini T.E., et al. Diagnostic Criteria for Duchenne’s Muscular Dystrophy. In: Clinical Evaluation and Diagnostic tests for Neuromuscular Disorders. Butterworth-Heinemann Press; 2002:784.
Biggar W.D., Harris V.A., Eliasoph L., et al. Long term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul. Disord.. 2006;16:249-255.
Bohannon R.W. Test–retest reliability of hand-held dynamometry during a single session of strength assessment. Phys. Ther.. 1986;66:206-209.
Bonne G., Di Barletta M.R., Varnous S., et al. Mutations in the gene encoding lamin A/C cause Autosomal Dominant Emery-Dreifuss muscular dystrophy. Nat. Genet.. 1999;21:285-288.
Bradley W.G., Jones M.Z., Fawcett P.R.W. Becker Type muscular dystrophy. Muscle Nerve. 1978;1:111-132.
Brooke M.H., Griggs M.D., Mendell J.R., et al. Clinical trial in Duchenne dystrophy. 1. The design of the protocol. Muscle Nerve. 1981;4:186-197.
Bunch W.H., Siegel I.M. Scapulothoracic arthrodesis in Fascioscapulohumeral Muscular Dystrophy. J. Bone Joint Surg. Am.. 1993;75A:372-376.
Callen J.P. Malignancy in polymyositis / dermatomyositis. Clinical Dermatology. 1988;2:55-63.
Carroll N., Bain R.J.I., Smith P.E.M., et al. Domiciliary investigation of sleep-related hypoxaemia in Duchenne muscular dystrophy. Eur. Respir. J.. 1991;4:434-440.
Carter G.T., Abresch R.T., Fowler W.M., et al. Profiles of neuromuscular disease: Spinal muscular atrophy. Am. J. Phys. Med. Rehabil.. 1995;74:150-159.
D’Angelo M.G., Berti M., Piccinini L., et al. A comparison of gait in spinal muscular atrophy, type II and Duchenne muscular dystrophy. Gait Posture. 2005;21(4):369-378.
Duboc D., Meune C., Pierre B., Wahbi K., Eymard B., Totain A., et al. Perindopril preventive treatment on mortality in Duchenne muscular dystrophy: 10 years follow-up. Am. Heart J.. 2007;154:596-602.
Dubowitz V. Muscle biopsy: A practical approach, second ed. London: Ballière Tindall, 1985.
Dubowitz V. Muscle disorders in childhood, second ed. London: WB Saunders, 1995.
Eagle M., Baudouin S.V., Chandler C., et al. Survival in Duchenne muscular dystrophy; improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord.. 2002;12:926-930.
Eagle M., Bourke J., Bullock R., et al. Managing Duchenne muscular dystrophy – the additive effect of spinal surgery and home ventilation in improving survival. Neuromuscul. Disord.. 2007;17:470-475.
Eagle M., Peacock C.K., Bushby K., Major R., Clements P. Fascioscapuohumeral muscular dystrophy: gait analysis and effectiveness of ankle foot orthoses (AFOs) (abstract). Neuromuscul. Disord.. 2001;11(6–7):631.
Edwards R.H.T., Chapman S.J., Newham D.J., et al. Practical analysis of variability of muscle function measurements in Duchenne Muscular Dystrophy. Muscle Nerve. 1987;10:6-14.
Emery A.E.H. The nosology of the spinal muscular atrophies. J. Med. Genet.. 1971;8:481-495.
Emery A.E.H. Population frequencies of inherited neuromuscular diseases– a world survey. Neuromuscul. Disord.. 1991;1:19-29.
Emery A.E., Muntoni F. Duchenne Muscular Dystrophy. Oxford: Oxford University Press, 2003.
Fawcett P.R.W., Barwick D.D. The clinical neurophysiology of neuromuscular disease. In: Walton J.N., Karpati G., Hilton-Jones D., editors. Disorders of voluntary muscle. sixth ed. Edinburgh: Churchill Livingstone; 1994:1033-1104.
Fisher N.M., Pendergast D.R., Calkins E.C. Maximal isometric torque of knee extension as a function of muscle length in subjects of advancing age. Arch. Phys. Med. Rehabil.. 1990;71:729-734.
Friedrich U., Brunner H., Smeets D., et al. Three points linkage analysis employing C3 and 19cen markers assign the myotonic dystrophy gene to 19q. Hum. Genet.. 1987;75:291-293.
Galasko C.S.B., Delaney C., Morris P. Spinal stabilisation in Duchenne Muscular Dystrophy. J. Bone Joint Surg.. 1992;74B:210-214.
Gianinni S., Faldini C., Pagkrati S., Grandi G., Digennaro V., Luciani D., et al. Fixation of winged scapula in facioscapulohumeral muscular dystrophy. Clin. Med. Res.. 2007;5(3):155-162.
Gill I., Eagle M., Jwalant S., et al. Correction of neuromuscular scoliosis in patients with pre-existing respiratory failure. Spine. 2006;21:2478-2483.
Gospe S.M., Lazaro R.P., Lava N.S., et al. Familial X linked myalgia and cramps: a non-progressive myopathy associated with a deletion in the dystrophin gene. Neurology. 1989;39:1277-1280.
Gossman M.R., Sahrmann S.A., Rose S.J. Review of length associated changes in muscle. Phys. Ther.. 1982;62:1799-1808.
Griggs R.C., Donohoe K.M., Utell M.J., et al. Evaluation of pulmonary function in neuromuscular disease. Arch. Neurol.. 1981;38:9-12.
Harper P.S. Myotonic dystrophy, second ed. Philadelphia: WB Saunders, 1989.
Heap R.M., Mander M., Bond J., et al. Management of Duchenne muscular dystrophy in the community: views of physiotherapists, GP’s and school teachers. Physiotherapy. 1996;82:258-263.
Hough A. Physiotherapy in respiratory care. London: Chapman & Hall, 1991.
Hunsaker R.H., Fulkerson P.K., Barry F.J., et al. Cardiac function in Duchenne’s muscular dystrophy. Results of 10–year follow up study and noninvasive tests. Am. J. Med.. 1982;73:235-238.
Hyde S.A., Fløytrup I., Glent S., et al. A randomised comparative study of two methods of controlling tendo achilles contracture in Duchenne muscular dystrophy. Neuromuscul. Disord.. 2000;10:257-263.
Hyde S.A., Scott O.M., Goddard C.M., et al. Prolongation of ambulation in Duchenne muscular dystrophy. Physiotherapy. 1982;68:105-108.
Illarioshkin S.N., Ivanova-Smolenskala I.A., Tanaka H. Refined genetic location of the chromosome 2p linked progressive muscular dystrophy gene. Genomics. 1997;42:345-348.
Jeppesen J., Green A., Steffensen B.F., Rahbek J. The Duchenne muscular dystrophy population in Denmark, 1977–2001: prevalence, incidence and survival in relation to the introduction of ventilator use. Neuromuscul. Disord.. 2003;13(10):804-812.
Jones D.A., Round J.M. Skeletal muscle in health and disease. Manchester: Manchester University Press, 1990.
Jones D.A., Rutherford O.M., Parker D.F. Physiological changes in skeletal muscle as a result of strength training. Q. J. Exp. Physiol.. 1989;74:233-256.
Karpati G., Hilton-Jones D., Griggs R.C. Disorders of Voluntary Muscle, seventh ed. Cambridge University Press, 2001.
Khodadadeh S., McClelland M.R., Patrick J.H., et al. Knee moments in Duchenne muscular dystrophy. Lancet. 1986;6(September):544-555.
Kilmer D.D. Response to aerobic exercise training in humans with neuromuscular disease. Am. J. Phys. Med. Rehabil.. 2002;81(Suppl. 11):S187-S195.
Kissell J.T. Fascioscapulohumeral dystrophy. Semin. Neurol.. 1999;19:35-43.
Kobayashi O., Hayashi Y., Arahata K., et al. Congenital muscular dystrophy. Neurology. 1996;46:815-818.
Kurz L.T., Mubarek S.J., Schultz P. Correlation of scoliosis and pulmonary function in Duchenne muscular dystrophy. J. Pediatr. Orthop.. 1983;3:347-353.
Laforet P., De Toma C., Eymard B., et al. Cardiac involvement in genetically confirmed Fascioscapulohumeral muscular dystrophy. Neurology. 1998;51:1454-1456.
Lennon S.M., Ashburn A. Use of myometry in the assessment of neuropathic weakness: testing for reliability in clinical practice. Clin. Rehabil.. 1993;7:125-133.
Main M., Mercuri E., Haliloglu G., et al. Serial casting of the ankles in Duchenne muscular dystrophy: can it be an alternative to surgery? Neuromuscul. Disord.. 2007;17:227-230.
Manzur A.Y., Kuntzer T., Pike M., Swan A. Glucocorticosteroids for Duchenne muscular dystrophy. The Cochrane Library. (1):2008. CD 003725
Manzur A.Y., Kinali M., Muntoni F. Update on the management of Duchenne muscular dystrophy. Arch. Dis. Child.. 2008;93:986-990.
Marsh A., Edge G., Lehovsky J. Spinal fusion in patients with Duchenne’s muscular dystrophy and a low forced vital capacity. Eur. Spine J.. 2003;12(5):507-512.
Martin A.J., Stern L., Yeates J., et al. Respiratory muscle training in Duchenne Muscular Dystrophy. Dev. Med. Child. Neurol.. 1986;28:314-318.
Mastaglia F.L., Laing N.G. Investigation of muscle disease. J. Neurol. Neurosurg. Psychiatry. 1996;60:256-274.
Mazzone E.S., Messina S., Vasco G., et al. Reliability of the North Star Ambulatory Assessment in a multicentric setting. Neuromuscul. Disord.. 2009;19:458-461.
McDonald C.M., Abresch R.T., Carter G.T., et al. Profiles of neuromuscular disease. Duchenne muscular dystrophy. Am. J. Phys. Med. Rehabil.. 1995;74:70-92.
Mehdian H., Shimizu N., Draycott V., et al. Spinal stabilisation for scoliosis in Duchenne muscular dystrophy. Experience with various sublaminar instrumentation systems. Neuro-Orthopedics. 1989;7:74-82.
Miura K., Kumagai T., Matsumoto A., Iriyama E., Watanabe K., Goto K., et al. Two cases of chromosome 4q35-linked early onset fascioscapulohumeral muscular dystrophy with mental retardation and epilepsy. Neuropaediatrics. 1998;29:239-241.
Moore J.K., Moore A.P. Postoperative complications of dystrophia myotonica. Anaesthesia. 1987;42:529-533.
Muntoni F., Torelli S., Ferlini A. Dystrophin and mutations: one gene,several proteins, multiple phenotypes. Lancet Neurol.. 2003;2:731-740.
Newham D.J., Jones D.A., Edwards R.H.T. Plasma creatinine kinase changes after eccentric and concentric contractions. Muscle Nerve. 1986;9:59-63.
Nicolino M., Byrne B., Wraithe J.E., Leslie N., Mendel H., Freyer D.R., et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet. Med.. 2009;11:210-219.
Nigro G., Comi L.I., Politano L., et al. The incidence and evaluation of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol.. 1990;26:271-277.
Ogino S., Wilson R.B., Gold B. New insights on the evolution of the SMN1 and SMN2 region simulation and meta-analysis for allele and haplotype frequency calculations. Eur. J. Hum. Genet.. 2004;12:1015-1023.
Olsen D.B., Orngreen M.C., Vissing J. Aerobic training improves exercise performance in fascioscapulohumeral dystrophy. Neurology. 2005;64:1064-1066.
Orrell R.W., Forrester J.D., Tawil R., et al. Definitive molecular diagnosis of fascioscapulohumeral muscular dystrophy. Neurology. 1999;52:1822-1826.
Pandya S., Florence J.M., King W.M., et al. Reliability of goniometric measurements in patients with Duchenne muscular dystrophy. Phys. Ther.. 1985;65:1339-1342.
Phadke A.P., Jay C., Chen S.J., Haddock C., Wang Z., Yu Y., et al. Safety and in vivo Expression of a GNE-Transgene: A Novel Treatment Approach for Hereditary Inclusion Body Myopathy-2. Gene Regulation and Systems Biology. 2009;3(May):89-101.
Phillips M.F., Quinlivan R.C.M., Edwards R.H.T., et al. Changes in spirometry over time as a prognostic marker in patients with Duchenne Muscular Dystrophy. American Journal of Critical Care Medicine. 2001;164:2191-2194.
Pope P.M. Postural management and special seating. In: Edwards S., editor. Neurological physiotherapy: a problem solving approach. second ed. London: Churchill Livingstone; 2002:189-217.
Quinlivan R.M., Dubowitz V. Cardiac transplantation in Becker muscular dystrophy. Neuromuscul. Disord.. 1992;2(3):165-167.
Rhee Y.G., Ha J.H. Long-term results of scapulothoracic arthrodesis of fascioscapulohumeral muscular dystrophy. J. Shoulder Elbow Surg.. 2006;15(4):445-450.
Rodillo E.B., Fernandez-Bermejo E., Heckmatt J.Z., et al. Prevention of rapidly progressive scoliosis in duchenne muscular dystrophy by prolonging walking with orthoses. J. Child. Neurol.. 1988;3:269-274.
Rodillo E., Noble-Jamieson C.M., Aber V., et al. Respiratory muscle training in Duchenne muscular dystrophy. Arch. Dis. Child.. 1989;64:736-738.
Ryerson S. Neurological assessment: the basis of clinical decision making. In: Lennon S., Stokes M., editors. Pocketbook of Neurological Physiotherapy. Churchill Livingstone; 2009:113-126.
Scott O.M., Hyde S.A., Goddard C., et al. Prevention of deformity in Duchenne muscular dystrophy. Physiotherapy. 1981;67:177-180.
Scott O.M., Hyde S.A., Goddard C., et al. Quantitation of muscle function in children: a prospective study in Duchenne Muscular Dystrophy. Muscle Nerve. 1982;5:291-301.
Seeger B.R., Sutherland A., Clark M.S. Management of scoliosis in Duchenne muscular dystrophy. Arch. Phys. Med. Rehabil.. 1990;65:83-86.
Shepherd R.B. Physiotherapy in Paediatrics, third ed. Oxford: Butterworth Heinnemann, 1995.
Simonds A.K. Nasal ventilation in progressive neuromuscular disease: experience in adults and adolescents. Monaldi Archives of Chest Disease. 2000;55(3):237-241.
Smith P.E.M., Edwards R.H.T., Calverley P.M.A. Mechanisms of sleep-disordered breathing in chronic neuromuscular disease: implications for management. Q. J. Med.. 1991;296:961-973.
Steare S.E., Benatar A., Dubowitz V. Subclinical cardiomyopathy in Becker muscular dystrophy. Br. Heart J.. 1992;68:304-308.
Steffensen B.F., Hyde S.A., Attermann J., et al. Reliability of the EK scale; a functional test for non-ambulatory individuals with Duchenne muscular dystrophy. Adv. Physiother.. 2002;4:47.
Stern L.M., Martin A.J., Jones N., et al. Training inspiratory resistance in Duchenne dystrophy using adapted computer games. Dev. Med. Child. Neurol.. 1989;31:494-500.
Stokes E.K. Outcome measurement. In: Lennon S., Stokes M., editors. Pocketbook of Neurological Physiotherapy. Churchill Livingstone; 2009:192-201.
Sutherland D.H., Olshen R., Cooper L., et al. The pathomechanics of gait in Duchenne muscular dystrophy. Dev. Med. Child. Neurol.. 1981;23:3-22.
Sveen M.L., Jeppesen T.D., Hauersley S., Krag T.O., Vissing J. Edurance training. An effective and safe treatment for patients with LGMD21. Neurology. 2007;68:59-61.
Sveen M.L., Jeppesen T.D., Hauerslev, et al. Endurance training improves fitness and strength in patients with Becker muscular dystrophy. Brain. 2008;131:2824-2831.
Thompson N., Porter S., Morris C. Muscular Dystrophies, Spinal Muscular Atrophies and Peripheral Neuropathies. In: Morris C., Dias L., editors. Paediatric Orthotics. London: Mac Keith Press, 2007.
Topin N., Matecki S., Le Bris S., et al. Dose dependent effect of individualized respiratory muscle training in Duchenne muscular dystrophy. Neuromuscul. Disord.. 2002;12:576-583.
Velasco M.V., Cloin A.A., Zurakowski D., et al. Posterior spinal fusion for scoliosis in Duchenne muscular dystrophy diminishes the rate of respiratory decline. Spine. 2007;4:459-465.
Vesa J., Su H., Watts G.D., Krause S., Walter M.C., Martin B., et al. Valosin containing protein associated inclusion body myopathy: abnormal vacuolization, autophagy and cell fusion in myoblasts. Neuromuscul. Disord.. 2009;19(11):766-772. Epub 2009 Oct 13
Vignos P.J., Spencer G.E., Archibald K.C. Management of progressive muscular dystrophy in childhood. JAMA. 1963;13:89-96.
Vignos P.J., Wagner M.B., Karlinchak B., et al. Evaluation of a program for long-term treatment of Duchenne muscular dystrophy: experience at the University Hospitals of Cleveland. J. Bone Joint Surg. Am.. 1996;78(12):1844-1852.
Wagner K.R., Lechtzin N., Judge D.P. Current treatment of adult Duchenne muscular dystrophy. Biochim. Biophys. Acta. 2007;1772(2):229-237. Epub 2006 Jul 8
Wanke T., Toifl K., Merkle M., et al. Inspiratory muscle training in patients with Duchenne Muscular Dystrophy. Chest. 1994;105:475-482.
Wijmenga C., Padberg G.W., Moerer P., et al. Mapping of fascioscapulohumeral gene to chromosome 4q35-qter by multipoint linkage analysis and in situ hybridization. Genomics. 1991;9:570-575.
Webber B.A. The Brompton Hospital guide to chest physiotherapy. Oxford: Blackwell Scientific Publications, 1988.
Wiles C.M., Karni Y., Nicklin J. Laboratory testing of muscle function in the management of neuromuscular disease. J. Neurol. Neurosurg. Psychol.. 1990;53:384-387.
World Health Organization (WHO). International classification of functioning, disability and health (ICF). Geneva: World Health Organization, 2001. Online. Available: http://www.who.int/classification/icf