Chapter 6 Muscle Disorders
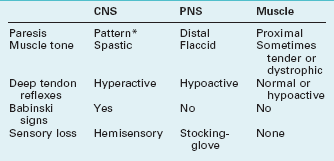
The clinical evaluation can usually distinguish disorders of muscle from those of the central nervous system (CNS) and peripheral nervous system (PNS) (Table 6-1). It can then divide muscle disorders into those of the neuromuscular junction and those of the muscles themselves, myopathies (Box 6-1). Surprisingly, considering their physiologic distance from the brain, several muscle disorders are associated with mental retardation, cognitive decline, personality changes, or use of psychotropic medications.
TABLE 6-1 Signs of Central Nervous System (CNS), Peripheral Nervous System (PNS), and Muscle Disorders
Box 6-1
Common Neuromuscular Junction and Muscle Disorders
Neuromuscular Junction Disorders
Myasthenia Gravis
Neuromuscular Transmission Impairment
Normally, the presynaptic neuron at the neuromuscular junction releases discrete amounts – packets or quanta – of acetylcholine (ACh) across the neuromuscular junction to trigger a muscle contraction (Fig. 6-1). After the muscle contraction, acetylcholinesterase (AChE) (or simply “cholinesterase”) metabolizes ACh.
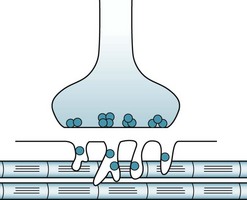
FIGURE 6-1 At the neuromuscular junction, the peripheral nerve endings contain discrete packets or quanta of acetylcholine (ACh) (dark blue). In response to stimulation, presynaptic neurons release about 200 ACh packets. They cross the synaptic cleft of the neuromuscular junction to reach ACh receptor-binding sites, situated deeply in convolutions of the postsynaptic membrane. ACh–receptor interactions open cation channels, thereby inducing an end-plate potential. If this potential reaches a certain magnitude, it triggers an action potential along the muscle fiber. Action potentials open calcium storage sites, which produce muscle contractions.
In myasthenia gravis, the classic neuromuscular junction disorder, ACh receptor antibodies block, impair, or actually destroy ACh receptors (Fig. 6-2). These antibodies predominantly attack ACh receptors located in the extraocular, facial, neck and proximal limb muscles. When binding to antibody-inactivated receptors, ACh produces only weak, unsustained muscle contractions. Another characteristic of the ACh receptor antibodies is that they attack only nicotinic ACh – not muscarinic ACh – receptors. Moreover, they do not penetrate the blood–brain barrier and do not interfere with CNS function. In contrast, they readily pass through the placenta and cause transient myasthenia symptoms in neonates of mothers with myasthenia gravis.
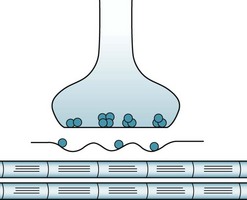
FIGURE 6-2 In myasthenia gravis, acetylcholine (ACh) receptors become abnormally shallow and lose many of their binding sites. The synaptic cleft widens, which further impedes neuromuscular transmission.
In the other therapeutic strategy – restoring the integrity of ACh receptors – neurologists administer steroids, other immunosuppressive medications, plasmapheresis, or intravenous infusions of immunoglobulins (IVIG). (Neurologists also infuse IVIG in Guillain–Barré syndrome [see Chapter 5], a commonly occurring inflammatory PNS illness.)
ACh, unlike dopamine and serotonin, serves as a transmitter at both the neuromuscular junction and the CNS. Also, metabolism instead of reuptake almost entirely terminates its action. Antibodies associated with myasthenia gravis impair neuromuscular junction but not CNS ACh transmission: One reason is that neuromuscular ACh receptors are nicotinic, but cerebral ACh receptors are mostly muscarinic (see Chapter 21).
Physicians caring for myasthenia gravis patients who have almost complete paralysis but normal cognitive status see the stark contrast between impaired neuromuscular junction activity but preserved CNS ACh activity. Similarly, most anticholinesterase medications have no effect on cognitive status or other CNS function because they do not penetrate the blood–brain barrier. One of the few exceptions, physostigmine, penetrates into the CNS where it can preserve ACh concentrations. Thus, researchers proposed physostigmine as a treatment for conditions with low CNS ACh levels, such as Alzheimer disease. However, in various experiments with Alzheimer disease, despite increasing cerebral ACh concentrations, physostigmine produced no clinical benefit (see Chapter 7).
Clinical Features
As their first symptom, almost 90% of patients, who are typically young women or older men, develop diplopia and ptosis. When facial and neck muscle weakness emerges, a nasal tone suffuses patients’ speech and, when attempting to smile, they grimace (Fig. 6-3). These patients have significant trouble whistling and chewing. Neck, shoulder, and swallowing and respiratory muscles weaken as the disease progresses, i.e., myasthenia gravis causes bulbar palsy (see Chapter 4). In severe cases, patients suffer respiratory distress, quadriplegia, and an inability to speak (anarthria). Paralysis can spread and worsen so much that patients reach a “locked-in” state (see Chapter 11).
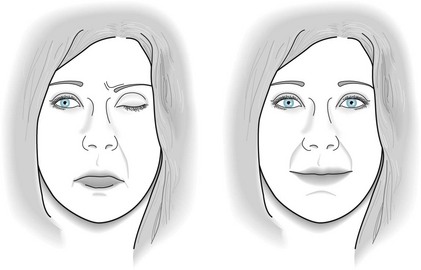
FIGURE 6-3 Left, This young woman described several weeks of intermittent double vision and nasal speech. She had left-sided ptosis and bilateral, asymmetric facial muscle weakness, evident in the loss of the contour of the right nasolabial fold and sagging lower lip. Right, Intravenous administration of the cholinesterase inhibitor edrophonium (Tensilon) 10 mg – the Tensilon test – produces a 60-second restoration of eyelid, ocular, and facial strength. This typically brief but dramatic restoration of her strength resulted from edrophonium transiently inhibiting cholinesterase to increase acetylcholine activity.
Neurologists usually attempt to confirm a clinical diagnosis of myasthenia by performing a Tensilon (edrophonium) test (see Fig. 6-3). Alternatively, they perform the “ice cube test,” which presumably temporarily uncouples toxic antibodies from ACh receptors and, like the Tensilon test, briefly reverses ptosis in myasthenia. They test for serum antibodies to ACh receptors and, in certain circumstances, antibodies to MuSK. They may also perform an electromyogram (EMG). About 5% of patients have underlying hyperthyroidism and 10% have a mediastinal thymoma. If these conditions are present and respond to treatment, myasthenia gravis will usually improve.
Differential Diagnosis
Lesions of the oculomotor nerve (cranial nerve III), which may be a sign of a midbrain infarction (see Fig. 4-9) or nerve compression by a posterior communicating artery aneurysm, also cause extraocular muscle paresis. In addition to their usually having an abrupt and painful onset, these lesions are identifiable by a subtle finding: the pupil will be widely dilated and unreactive to light because of intraocular (pupillary) muscle paresis (see Fig. 4-6). In addition, many other illnesses cause facial and bulbar palsy: amyotrophic lateral sclerosis (ALS), Guillain–Barré syndrome, Lyme disease, Lambert–Eaton syndrome, and botulism.
Lambert–Eaton Syndrome
Lambert–Eaton and botulism also differ in their etiology and, to a certain extent, their clinical manifestations. A toxin causes botulism, but an autoimmune disorder, by directing antibodies against voltage-gated calcium channels, causes Lambert–Eaton. This autoimmune disorder, in turn, is frequently an expression of small cell carcinoma of the lung and occasionally a component of a rheumatologic illness. When associated with any cancer, neurologists consider Lambert–Eaton a paraneoplastic syndrome (see Chapter 19).
Botulism
Ironically, neurologists now routinely turn botulinum-induced paresis to an advantage. They inject pharmaceutically prepared botulinum toxin to alleviate focal dystonias and dyskinesias, such as blepharospasm, spasmodic torticollis, and writer’s cramp (see Chapter 18). Even more ironically, numerous physicians and nonphysicians routinely inject pharmaceutically prepared botulinum toxin into the paper-thin muscles underlying furrows to smooth patients’ skin.
Nerve Gas and Other Wartime Issues
On the other hand, people committing suicide, especially in India, often deliberately drink organophosphate pesticides. Similarly, the nerve gases that threatened soldiers from World War I through the Persian Gulf War bind and inactivate AChE. The common ones – GA, GB, GD, and VX – affect both the CNS and PNS. Some are gaseous, but others, such as sarin (GB), the Tokyo subway poison, are liquid. Several investigators postulated that pyridostigmine caused neurologic symptoms of the “Gulf War syndrome” (see Chapter 5); however, they provided no direct evidence and patients with myasthenia take pyridostigmine for decades with no such untoward effects.
The notion that silicone toxicity from breast implants causes a neuromuscular disorder and other neurologic illness, which is also unfounded, is discussed in the differential diagnosis of multiple sclerosis (see Chapter 15).
Muscle Disease (Myopathy)
Inherited Dystrophies
Duchenne Muscular Dystrophy
Dystrophy typically first affects boys’ thighs and shoulders. The first symptom to emerge is their struggle to stand and walk. Subsequently, even though drastically weak, muscles paradoxically increase in size because fat cells and connective tissue infiltrate them (muscle pseudohypertrophy, Fig. 6-4, top). Instinctively learning Gowers’ maneuver (Fig. 6-4, bottom), boys with the illness arise from sitting only by pulling or pushing themselves upward on their own legs. Usually by age 12 years, when their musculature can no longer support their maturing frame, adolescent boys become wheelchair-bound and eventually develop respiratory insufficiency.
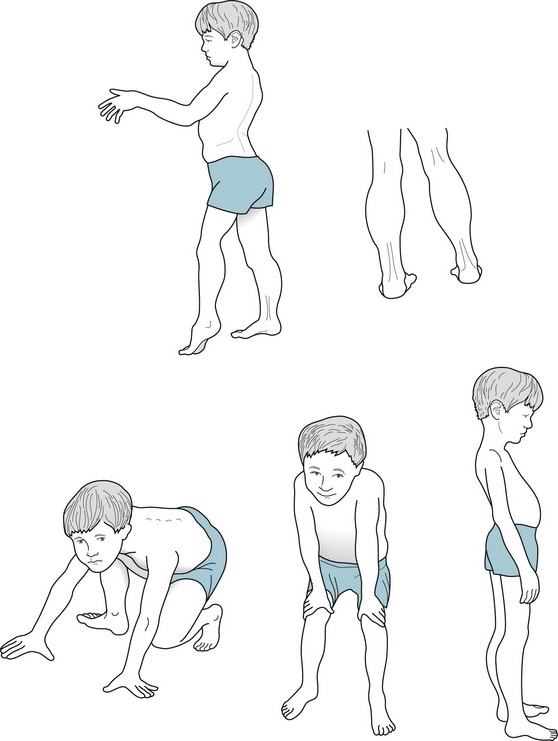
FIGURE 6-4 Top, This 10-year-old boy with typical Duchenne muscular dystrophy has a waddling gait and inability to raise his arms above his head because of weakness of his shoulder and pelvic girdle muscles, i.e., his proximal muscles. His weakened calf muscles show enlargement (pseudohypertrophy) not from exercise but from fat and connective tissue infiltration. He also has exaggeration of the normal inward curve of the lumbar spine, hyperlordosis. Bottom, Gowers’ maneuver, an early sign of Duchenne muscular dystrophy, consists of a young victim pushing his hands against his knees then thighs to reach a standing position. He must use his arms and hands because the disease primarily weakens hip and thigh muscles that normally would be sufficient to allow him to stand.
Myotonic Dystrophy
Myotonic dystrophy is named for its clinical signature, myotonia, which is involuntary prolonged muscle contraction. Myotonia inhibits the release of patients’ grip for several seconds after shaking hands or grasping and turning a doorknob. Neurologists elicit this phenomenon by asking patients to make a fist and then rapidly release it. In addition, if the physician lightly taps a patient’s thenar (thumb base) muscles with a reflex hammer, myotonia causes a prolonged, visible contraction that moves the thumb medially (Fig. 6-5).
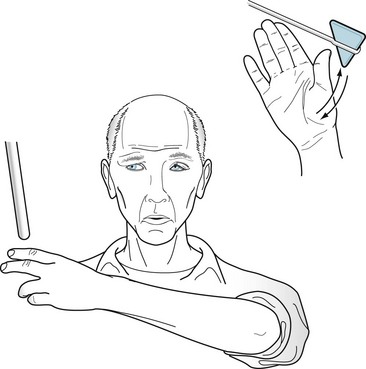
FIGURE 6-5 This 25-year-old man with myotonic dystrophy has the typically elongated, “hatchet” face caused by temporal and facial muscle wasting, frontal baldness, and ptosis. Because of myotonia, a percussion hammer striking his thenar eminence muscles precipitates a forceful, sustained contraction that draws in the thumb for 3–10 seconds. Myotonia also prevents him from rapidly releasing his grasp.
Another feature, caused by facial and temple muscle atrophy, is a sunken and elongated face, ptosis, and a prominent forehead. This distortion forms the distinctive “hatchet face” (see Fig. 6-5). Additional neurologic and nonneurologic manifestations vary. Patients often develop cataracts, cardiac conduction system disturbances, and endocrine organ failure, such as testicular atrophy, diabetes, and infertility. Treatment is limited to replacement of endocrine deficiencies and, by giving phenytoin, quinine, or other medicines, to reducing myotonia.
Genetics
Other disorders that result from different excessive trinucleotide repeats include ones that are inherited in an autosomal recessive pattern (Friedreich ataxia), autosomal dominant pattern (spinocerebellar atrophies and Huntington disease), and sex-linked pattern (fragile X syndrome) (see Chapters 2, 13, and 18, and the Appendix). Whichever the particular trinucleotide base repeat and pattern of inheritance, physicians can easily and reliably diagnose these illnesses in symptomatic and asymptomatic individuals by testing DNA in their white blood cells.
A clinical counterpart of amplification is anticipation: successive generations of individuals who inherit the abnormal gene show signs of the illness at a progressively younger age. For example, a grandfather may not have been diagnosed with myotonic dystrophy until he was 38 years old. At that age, he already had an asymptomatic boy and girl who both carried the gene. The son and daughter typically would not show signs of the illness until they reached 26 years; however, by then, they might each have had several of their own children. Anticipation would be further apparent when affected grandchildren show signs in their teenage years. In the classic example, Huntington disease, dementia appears earlier in life and more severely in successive generations, especially when the father has transmitted the abnormal gene (see Chapter 18).
Metabolic Myopathies
A different disorder involving potassium metabolism is hypokalemic periodic paralysis, in which patients have dramatic attacks, lasting several hours to 2 days, of areflexic quadriparesis. During attacks of hypokalemia, patients remain alert and fully cognizant, breathing normally, and purposefully moving their eyes despite the widespread areflexic paralysis. Contrary to its label, periodic paralysis is irregular and not “periodic.” The attacks tend to occur spontaneously every few weeks, but exercise, sleep, or large carbohydrate meals often precipitate them. Although attacks resemble sleep paralysis and cataplexy (see Chapter 17), they are differentiated by a longer duration and hypokalemia. Hypokalemic periodic paralysis, sleep paralysis, and cataplexy all differ from psychogenic episodes by their areflexia.
Administration of atypical neuroleptics, particularly clozapine, as well as typical dopamine-blocking ones, causes a mostly asymptomatic elevation of CK serum concentrations. In as many as 10% of patients with acute psychosis, the CK concentration increases to fivefold or greater levels. Physicians might find that medication injections, excessive physical activity, or subclinical neuroleptic-induced parkinsonism or dystonia (see Chapter 18) are responsible for this elevation. An asymptomatic, isolated, mild to moderate CK elevation should not automatically trigger a diagnosis of neuroleptic-malignant syndrome (see below); however, physicians should assess the patient for other parameters of muscle breakdown and repeat the CK determination in 48 hours.
Mitochondrial Myopathies
When they generate energy, mitochondria must constantly remove free radicals, which are highly toxic metabolic byproducts. Failure to remove them may lead to Parkinson disease and other illnesses (see Chapter 18).
Another difference between nDNA and mtDNA is that mtDNA is passed to daughter cells’ mitochondria in random, variable mixtures. The daughter cells’ mitochondria inherit variable proportions of normal and abnormal mtDNA. When the proportion of abnormal mtDNA reaches a certain level, the threshold effect, ATP production becomes insufficient for cellular function and symptoms ensue. The variable proportion of normal and abnormal mtDNA in single cells, heteroplasmy (Fig. 6-6), explains why organs typically have variable proportion of abnormal cells and the illnesses’ variable age of onset and clinical features.
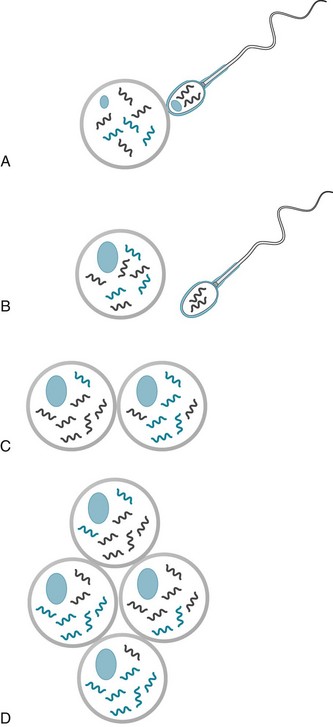
FIGURE 6-6 Heteroplasmy. A, Sperm and egg nuclei each contain an equal complement of chromosomal DNA (nDNA). Sperm mitochondria, containing different DNA (mtDNA), power the flagella. B, After fertilizing an egg and transferring their nDNA to the egg’s nucleus, sperm drop away with their mitochondria and mtDNA intact. Thus, sperm mtDNA, whether mutant or wild (normal), does not enter the fertilized egg. The fertilized egg’s chromosomes undergo mitosis, resulting in equal distribution of nDNA into the two daughter cells. C, Chromosomes divide and distribute nDNA, with or without mutations, equally to daughter cells. In contrast, although mitochondria also duplicate, they do not segregate equally. If mtDNA contains mutations, daughter cells receive unequal distributions of mutant mtDNA. D, The nonrandom, non-Mendelian distribution of mitochondria and their mtDNA in daughter cells ultimately gives different organs variable proportions of mtDNA. Heteroplasmy is defined as the mixture of mitochondria, in a single cell, that contains mutant and normal mtDNA.
A different cause of mitochondria dysfunction is that mutations and other abnormalities in nDNA can impair mtDNA. For example, mutations in nDNA that influence mtDNA probably account for many of the problems underlying Wilson disease (see Chapter 18) and Friedreich ataxia (see Chapter 2). The influence of nDNA on mtDNA can explain why paternally inherited abnormal nDNA can cause malfunction of mtDNA. Moreover, it can explain how a father might transmit an illness characterized by mitochondrial dysfunction to his child.
The primary mitochondrial myopathies, which result from mitochondria having deficiencies in cytochrome oxidase or other enzymes, cause weakness and exercise intolerance, short stature, epilepsy, deafness, and episodes of lactic acidosis. Another group of mitochondrial myopathies, progressive ophthalmoplegia and its related disorders, cause ptosis and other extraocular muscle palsies along with numerous nonneurologic manifestations, such as retinitis pigmentosa, short stature, cardiomyopathy, and endocrine abnormalities. One mitochondrial DNA disorder, Leber optic atrophy, causes hereditary optic atrophy in young men (see Chapter 12).
Neuroleptic Malignant Syndrome (NMS)
1. Delirium and decreased level of consciousness
2. Extrapyramidal signs, especially muscle rigidity, tremors, and dystonic posturing
3. Autonomic hyperactivity with prominent tachycardia and, although not the first sign, high fever.
Recommended treatment, aimed at restoring dopamine activity, has included administering L-dopa, which is a dopamine precursor; dopamine agonists, such as bromocriptine and apomorphine; or amantadine, which enhances dopamine activity (see Chapter 18). A complementary approach has been to administer dantrolene (Dantrium), which restores a normal intracellular calcium distribution. Several articles have proposed administering ECT, but the rationale and results have been unclear. In any case, physicians must provide fluids, antipyretics, and other supportive measures.
Other Causes of Rhabdomyolysis, Hyperthermia, and Altered Mental States
Other Causes
With the appropriate history, physicians probably would not confuse CNS or systemic infections with NMS. The distinction between NMS and meningitis is the most difficult because both conditions cause delirium, fever, and nuchal rigidity. Finally, sometimes neurologists consider catatonia as a manifestation of psychiatric illness in the differential diagnosis of NMS because of the setting and increased muscle tone; however, its course and lack of major autonomic dysfunction set it apart (see Chapter 18).
Laboratory Tests
Nerve Conduction Studies
Nerve conduction studies (NCS) (Fig. 6-7) can determine the site of nerve damage, confirm a clinical diagnosis of polyneuropathy, and distinguish polyneuropathy from myopathy. In addition, they can help separate neuropathies that have resulted from loss of myelin, such as Guillain–Barré syndrome, in which the conduction velocities slow, from those that have resulted from axon damage, such as with chemotherapy, in which the amplitude is reduced.
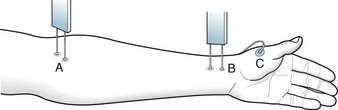
FIGURE 6-7 In determining nerve conduction velocity (NCV), a stimulating electrode placed at two points (A and B) along a nerve excites the appropriate muscle (C). The distance between A and B, divided by the time interval, determines the NCV. In the upper extremities, NCV is approximately 50–60 m/s, and in the lower extremities, 40–50 m/s.
Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352:1112–1120.
Chalk C, Benstead TJ, Keezer M. Medical treatment for botulism. Cochrane Database Syst Rev. 3, 2011. CD008123
Ferro S, Esposito F, Biamonti M, et al. Myasthenia gravis during pregnancy. Expert Rev Neurother. 2008;8:979–988.
Gervais RO, Russell AS, Green P, et al. Effort testing in patients with fibromyalgia and disability incentives. J Rheumatol. 2001;28:1892–1899.
Gilman PK. Triptans, serotonin agonists, and serotonin syndrome (serotonin toxicity): A review. Headache. 2010;50:264–272.
Goetz CG, Bolla KI, Rogers SM. Neurologic health outcomes and Agent Orange: Institute of Medicine report. Neurology. 1994;44:801–809.
Hassani A, Horvath R, Chinnery PF. Mitochondrial myopathies. Developments in treatment. Curr Opin Neurol. 2010;23:459–465.
Hyams KC, Wignall FS, Roswell R. War syndromes and their evaluation: From the U.S. Civil War to the Persian Gulf War. Ann Intern Med. 1996;125:398–405.
Krup LB, Masur DM, Kaufman LD. Neurocognitive dysfunction in the eosinophilia–myalgia syndrome. Neurology. 1993;43:931–936.
Manzur AY, Kuntzer T, Pike M, et al. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. The Cochrane Library. 2009;3:1–71.
Martin CO, Adams HP. Neurologic aspects of biological and chemical terrorism. Arch Neurol. 2003;60:21–25.
Melkersson K. Serum creatine kinase levels in chronic psychosis patients – a comparison between atypical and conventional antipsychotics. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1277–1282.
Meola G, Sansone V, Perani D, et al. Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM-1) and proximal myotonic myopathy. Neuromuscul Dis. 2003;13:813–821.
Morse CG, Kovacs J. Metabolic and skeletal complications of HIV infection. JAMA. 2006;296:844–854.
Shapiro RL, Hatheway C, Swerdlow DL. Botulism in the United States: A clinical and epidemiologic review. Ann Intern Med. 1998;129:221–228.
Turner C, Hilton-Jones D. The myotonic dystrophies: Diagnosis and management. J Neurol Neurosurg Psychiatry. 2010;81:358–367.
Chapter 6 Questions and Answers
c. This is a classic case of myasthenia gravis with ocular, pharyngeal, and neck flexor paresis but no pupil abnormality. She develops diplopia when one or more ocular muscles fatigue. By way of contrast, this pattern of neck flexor paresis, ocular muscle weakness, and ptosis does not occur in MS. Although internuclear ophthalmoplegia frequently occurs in MS, it causes nystagmus in the abducting eye as well as paresis of the adducting eye (see Chapters 12 and 15). As for psychogenic disturbances, people cannot mimic either paresis of one ocular muscle or ptosis. Compression of the third cranial nerve by an expanding aneurysm also produces ptosis and paresis of adduction. However, compression of the third cranial nerve differs from myasthenia because it has a painful onset, and the pupil dilates and loses its reactivity to light. Furthermore, such an aneurysm cannot explain the bulbar palsy.
30. After diagnosing a 75-year-old woman with congestive heart failure, her physician placed her on a low-salt diet and began her on a powerful diuretic. Although the congestive heart failure resolves, she develops somnolence, disorientation, and generalized weakness. Which of the following is the most likely cause of her mental status change?
d. Strychnine competes with GABA and glycine at their postsynaptic receptors.
46. Which one of the following is not a characteristic of the Lambert–Eaton syndrome?
48. Called to a subway station because of a terrorist attack, a physician is confronted with dozens of passengers in a state of panic who all have abdominal cramps, dyspnea, miosis, weakness, and fasciculations. Many passengers are unconscious and several are having seizures. Which medication should she first administer?
49. A friend brings a 52-year-old woman with a history of several episodes of psychosis to the emergency room because she is agitated and confused. She has muscle rigidity and tremulousness, but her neck is supple. Her temperature is 105°F and white blood count 18 000/mm3. Her friend said that her medications had been changed, but could provide no other useful information. A head CT and lumbar puncture revealed no abnormalities. Her urine was dark brown. Of the following tests, which one should be performed next?
57. An 8-year-old girl has episodes of confusion and headaches lasting 1–3 days. Between attacks, she has a normal neurologic examination and unremarkable blood tests, head CT, and head MRI. Also, an EEG during attacks shows no epileptiform discharges and between attacks it shows normal alpha rhythm. Eventually, a physician determines that the serum lactic acid concentration rises markedly during every attack and is normal between them. Which should be the next diagnostic test?
59. A corporation’s chief executive officer develops ALS. His left arm begins to weaken. Then a multinational conglomerate claims the executive is losing his mental capabilities and initiates a hostile takeover bid. Can the stockholders be sure that the ALS is causing his cognitive decline and apparently abnormal behavior?
60. A psychiatrist has been called to evaluate a 30-year-old woman for agitation and bizarre behavior. She had been admitted to an intensive care unit for exacerbation of myasthenia gravis and treated with high-dose anticholinesterase medications (e.g., pyridostigmine [Mestinon] and neostigmine). When no substantial improvement occurred, she was given plasmapheresis. The next day she had regained strength, but was confused and agitated. Which is the most likely cause of her mental status change?
61. The family of a 45-year-old man, who had a history of depressive illness, brings him and his girlfriend to the emergency room. Both are comatose and apneic. Their pupils are mid-sized and reactive. Extraocular movements are normal. The CT, illicit drug screening, blood glucose, and other blood tests are all normal except for an anion gap that proves to be due to a markedly elevated lactic acid concentration. Of the following, which is the most likely intoxicant?
65. A 35-year-old woman, who has had myasthenia for 15 years, has been stable on pyridostigmine (Mestinon) 120 mg QID. After a psychologically stressful situation developed, she began to have cramping abdominal pains, diarrhea, rhinorrhea, and excessive pulmonary secretions. Her face, jaw, and neck muscles weakened. Then her limb muscles weakened. Which of the following is most likely to have developed?
c. Absence of dystrophin in voluntary muscle characterizes Duchenne dystrophy.
81. An 80-year-old shoe salesman has had Parkinson disease for 12 years. For the past several years he has been progressively incapacitated and bedridden. His medication regimen includes levodopa, carbidopa, and antihypertensive medications. About 1 week before the visit, he began to have weakness and lack of appetite. His neurologist, diagnosing progression of his Parkinson disease, added deprenyl to the regimen. Over the next several days, he became confused and febrile. His rigidity increased. Which is the most likely cause of his immediate deterioration?
82. A 25-year-old woman, under the care of a psychotherapist for mild depression, reported that she has recently developed weakness and fatigue but not sleepiness or change in mood. She mentioned that, while she had begun to drink “gallons” of water and various beverages, she felt that she was urinating even more fluid than she was drinking. She denied bulimia and purging. She has lost 10 lb (4.5 kg). With the weakness being her primary symptoms, which would be the next test?
83. A 45-year-old scrap metal worker, who was an illegal immigrant from an underdeveloped country, came to the hospital because of stiffness and spasms of his right leg that had begun 1 week before. While at work 10 days before coming to the hospital, he had sustained a laceration, which remained infected. During the 2 days before he came to the hospital, the stiffness and spasms spread to his lower back and other leg. The spasms came in waves. They followed loud sound, light touch, and his own movement. Despite the spasms, his strength and DTRs were normal. A psychiatry consult was solicited because the spasms seemed voluntary to the housestaff, he had excessive response to stimulation, and the disability potentially offered great secondary gain. Which is the most likely diagnosis?