CHAPTER 17 MUSCLE AND NERVE PATHOLOGY
MUSCLE BIOPSY
Overview













Specimen handling



Histochemical and immunohistochemical stains














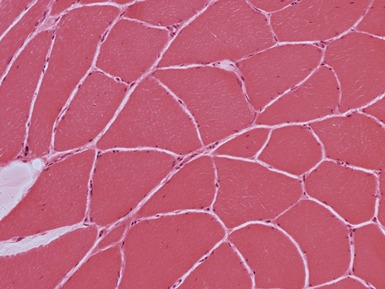
Myopathic changes

 internal nuclei (Fig 17.1)
internal nuclei (Fig 17.1)









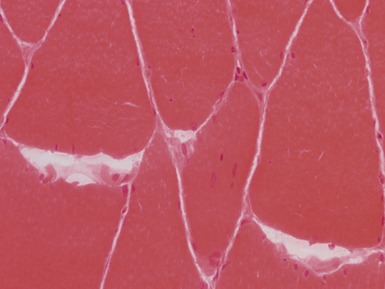
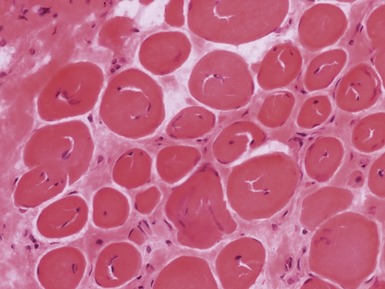
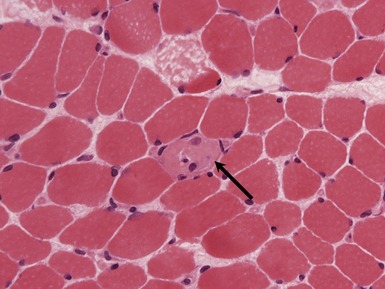
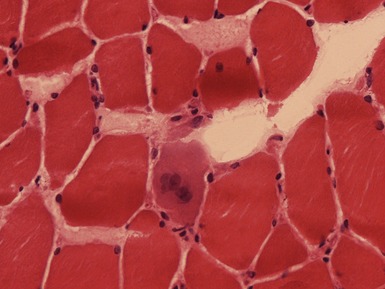
Fig 17.3 Photomicrograph showing a necrotic fiber (arrow). The fiber is pale and has been infiltrated by macrophages.
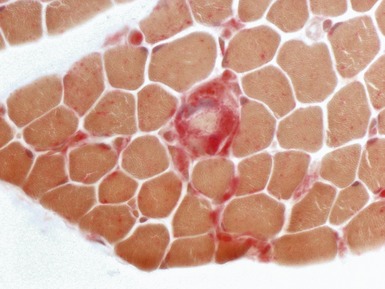





MUSCULAR DYSTROPHY





DYSTROPHINOPATHIES (DUCHENNE AND BECKER MUSCULAR DYSTROPHY)
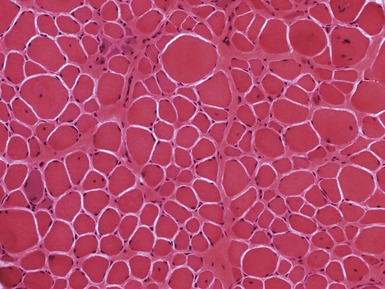
Fig 17.7 Photomicrograph of a muscle biopsy from a patient with Duchenne muscular dystrophy. There is marked variation in fiber size due to a mixture of atrophic and hypertrophic fibers. There are excess internal nuclei, increased endomysial collagen and regenerating fibers (see Fig 17.5 for detail).
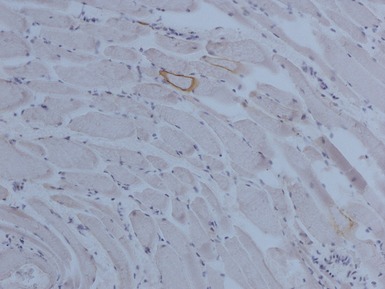
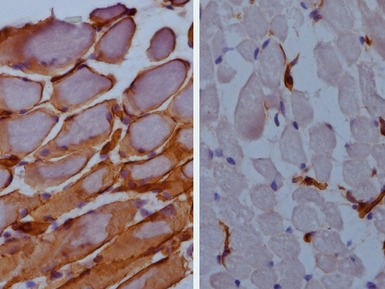






Clinical features





Becker























Immunohistochemical staining












FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY





DEFECTS IN NUCLEAR MEMBRANE PROTEINS (INCLUDING EMERY–DREIFUSS DYSTROPHY)


Clinical features








Limb girdle muscular dystrophy Type 1B (limb girdle muscular dystrophy with arterio-ventricular conduction block)










































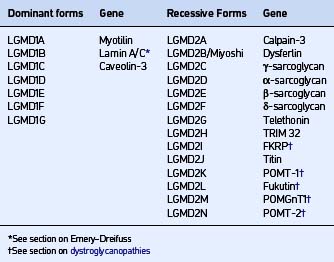
LIMB GIRDLE MUSCULAR DYSTROPHIES (LGMD)



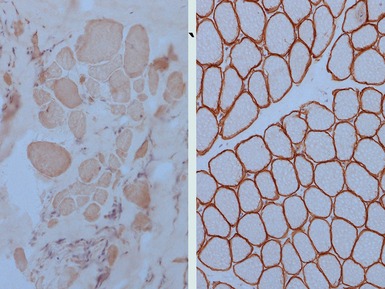
CAVEOLINOPATHIES

















DYSFERLINOPATHIES





Immunohistochemical staining

SARCOGLYCANOPATHIES
Genetics


| Syndrome | Ethnicity | |
|---|---|---|
| α | LGMD2D | No association |
| β | LGMD2E | |
| γ | LGMD2C | |
| Severe childhood autosomal recessive muscular dystrophy (SCARMD) | North Africa | |
| Recessive muscular dystrophy of European Gypsies | European Gypsies | |
| δ | LGMD2F | Brazil |
Clinical features

Immunohistochemical staining








CONGENITAL MUSCULAR DYSTROPHIES

DYSTROGLYCANOPATHIES
Clinical features



















MEROSIN DEFICIENCY
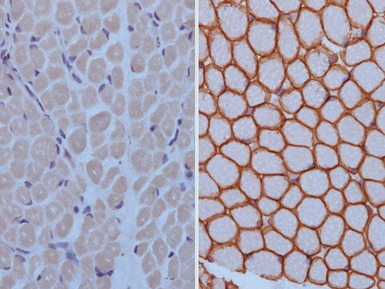


Immunohistochemical staining







COLLAGEN VI DEFICIENCY



































CONGENITAL MYOPATHIES
CENTRAL CORE DISEASE
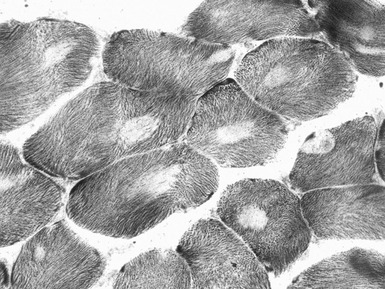

















MULTI-MINI CORE DISEASE































NEMALINE MYOPATHIES

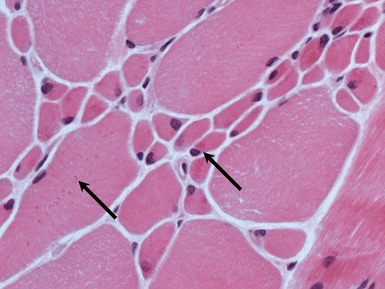
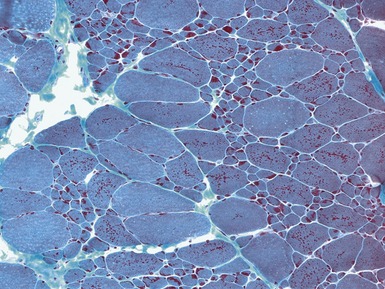
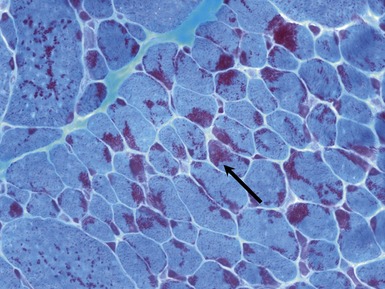
Fig 17.15 Photomicrograph of a muscle biopsy from a patient with a nemaline myopathy. The red inclusions are nemaline rods (see also Fig 17.16) (Gomori trichrome stain).
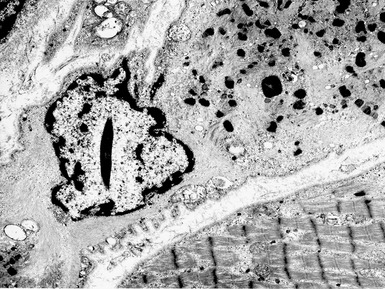
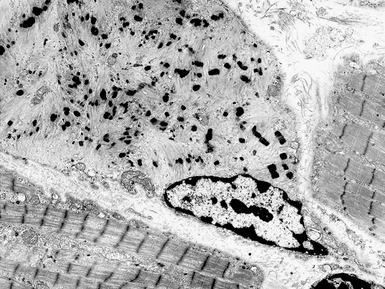
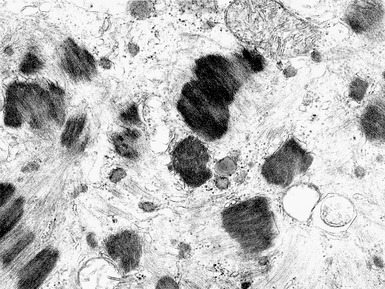





























X-LINKED MYOTUBULAR MYOPATHY

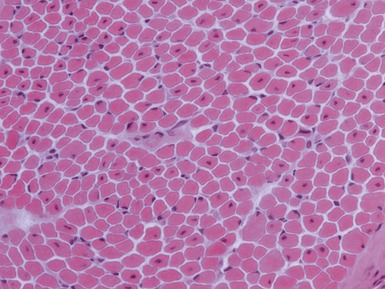
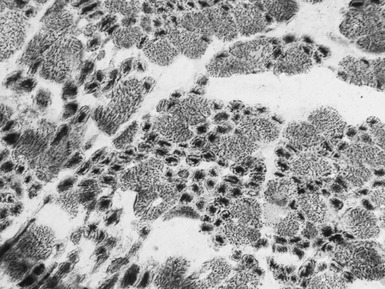
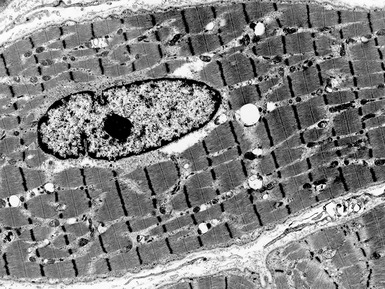
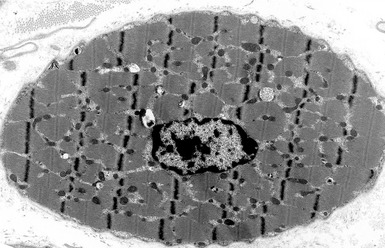







Histopathological features










CENTRONUCLEAR MYOPATHIES


































CONGENITAL FIBER TYPE DISPROPORTION


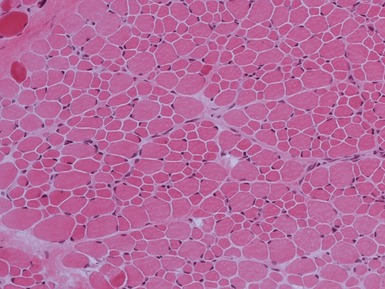
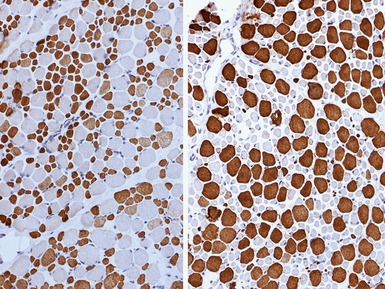






























MYOFIBRILLAR/DESMIN MYOPATHIES AND RELATED DISORDERS
MYOFIBRILLAR (DESMIN) MYOPATHIES







































METABOLIC MYOPATHIES AND RELATED DISEASES
ALPHA-GLUCOSIDASE DEFICIENCY / ACID MALTASE DEFICIENCY / POMPE’S DISEASE / TYPE II GLYCOGEN STORAGE DISEASE
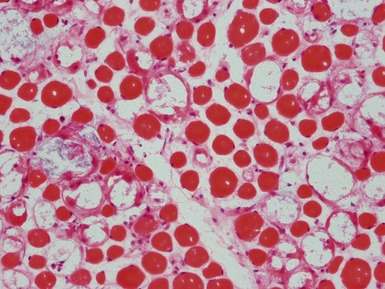
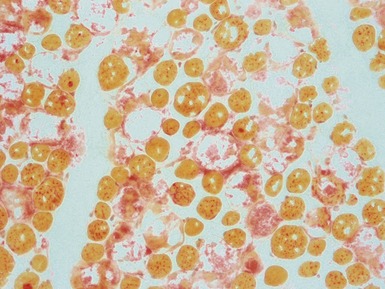
Fig 17.27 Photomicrograph of a muscle biopsy (same case as Fig 17.26) from a patient with Pompe’s disease. The large vacuoles show positive staining for acid phosphatase (red).

Fig 17.28 Electron micrograph of a muscle biopsy from a patient with Pompe’s disease. There are large collections of sarcoplasmic glycogen (see also Fig 17.29).
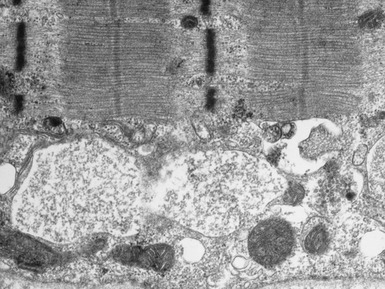
Fig 17.29 Electron micrograph of a muscle biopsy from a patient with Pompe’s disease. There are large collections of membrane-bound glycogen (see also Fig 17.28).
Clinical features




















LAMP-2 DEFICIENCY / DANON’S DISEASE / LYSOSOMAL GLYCOGEN STORAGE DISEASE WITH NORMAL ACID MALTASE











X-LINKED MYOPATHY WITH EXCESSIVE AUTOPHAGY




















GLYCOGEN STORAGE DISEASE IV / POLYGLUCOSAN DISEASE / ANDERSEN’S DISEASE
















DISORDERS ASSOCIATED WITH EXCESS LIPID DEPOSITION ON MUSCLE BIOPSY









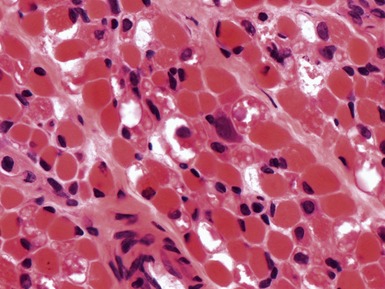
Fig 17.30 Photomicrograph showing large vacuoles due to excess lipid deposition (see Figs 17.31 and 17.32).
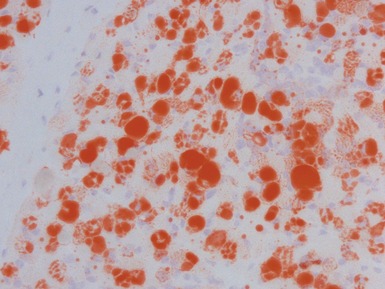

MITOCHONDRIAL DISORDERS (DISORDERS OF OXIDATIVE PHOSPHORYLATION)
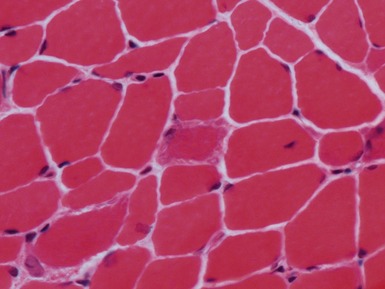
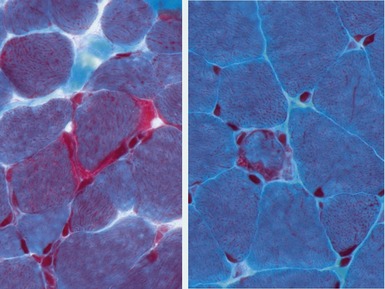
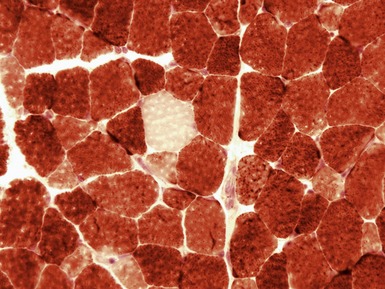
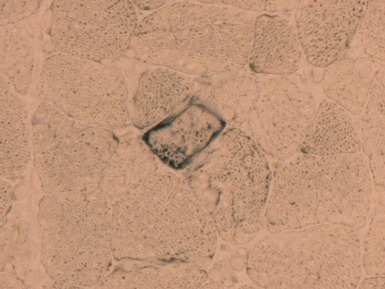
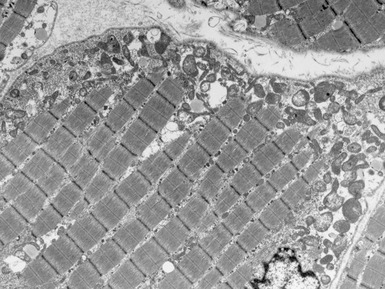
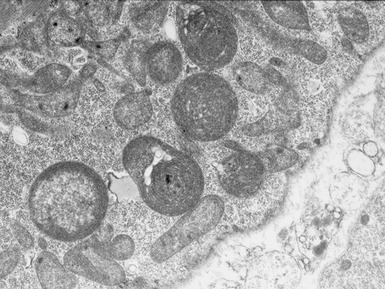


Clinical features

















Histopathological features





Special diagnostic investigations







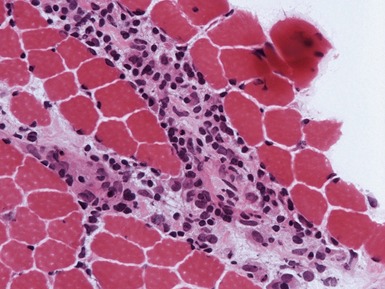
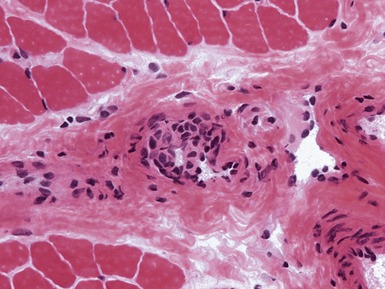
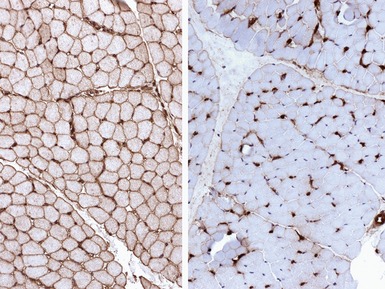
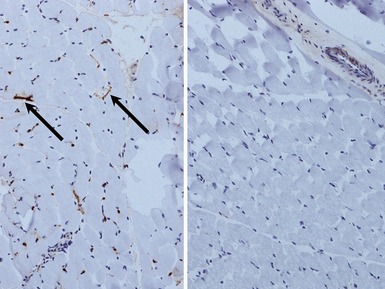
INFLAMMATORY MYOPATHIES
JUVENILE DERMATOMYOSITIS































MISCELLANEOUS MYOPATHIES
TOXIC AND DRUG ASSOCIATED MYOPATHIES







NEUROGENIC DISORDERS

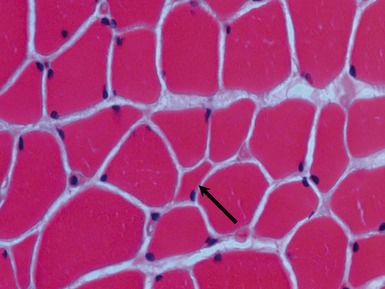
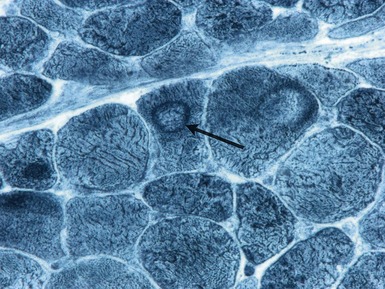
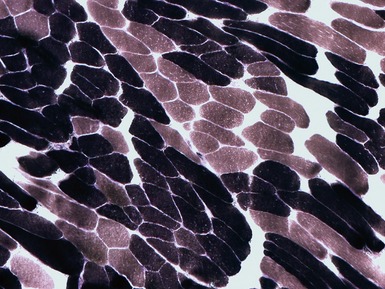
Histopathological features











TYPE 2 FIBER ATROPHY





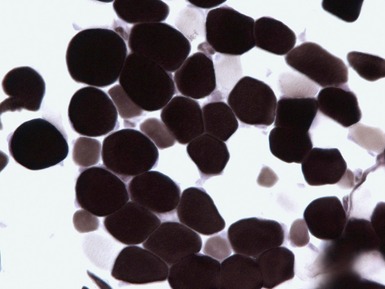
MYOSIN HEAVY CHAIN DEFICIENCY SYNDROME


























ION CHANNEL DISORDERS

MYOTONIA AND PARAMYOTONIA




























NERVE BIOPSY













Argov Z, Mitrani-Rosenbaum S. The hereditary inclusion body myopathy enigma and its future therapy. Neurotherapeutics. 2008;5:633-637.
Brown SC, Piercy RJ, Muntoni F, Sewry CA. Investigating the pathology of Emery–Dreifuss muscular dystrophy. Biochem Soc Trans. 2008;36:1335-1338.
Filosto M, Tomelleri G, Tonin P, et al. Neuropathology of mitochondrial diseases. Bioscience Reports. 2007;27:23-30.
Guglieri M, Straub V, Bushby K, Lochmüller H. Limb-girdle muscular dystrophies. Curr Opin Neurol. 2008;21:576-584.
Jungbluth H, Wallgren-Pettersson C, Laporte J. Centronuclear (myotubular) myopathy. Orphanet J Rare Dis. 2008;3:26.
Klopstock T. Drug-induced myopathies. Curr Opin Neurol. 2008;21:590-595.
Laing NG. Congenital myopathies. Curr Opin Neurol. 2007;20:583-589.
Malicdan MC, Noguchi S, Nonaka I, Saftig P, Nishino I. Lysosomal myopathies: an excessive build-up in autophagosomes is too much to handle. Neuromusc Disord. 2008;18:521-529.
Muntoni F, Torelli S, Brockington M. Muscular dystrophies due to glycosylation defects. Neurotherapeutics. 2008;5:627-632.
Nolte KW, Janecke AR, Vorgerd M, Weis J, Schroder JM. Congenital type IV glycogenosis: the spectrum of pleomorphic polyglucosan bodies in muscle, nerve, and spinal cord with two novel mutations in the GBE1 gene. Acta Neuropathol. 2008;116:491-506.
Oldfors A. Hereditary myosin myopathies. Neuromusc Disord. 2007;17:355-367.
Schessl J, Zou Y, McGrath MJ, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest. 2008;118:904-912.
Selcen D. Myofibrillar myopathies. Curr Opin Neurol. 2008;21:585-589.
Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. 2009;65:83-89.
Sewry CA, Jimenez-Mallebrera C, Muntoni F. Congenital myopathies. Curr Opin Neurol. 2008;21:569-575.
Tawil R. Facioscapulohumeral muscular dystrophy. Neurotherapeutics. 2008;5:601-606.
Treves S, Jungbluth H, Muntoni F, Zorzato F. Congenital muscle disorders with cores: the ryanodine receptor calcium channel paradigm. Curr Opin Pharmacol. 2008;8:319-326.
Van der Ploeg AT, Reuser AJ. Pompe’s disease. Lancet. 2008;372:1342-1353.
Wedderburn L, Varsani H, Li CK, et al. International consensus on a proposed score system for muscle biopsy evaluation in patients with juvenile dermatomyositis: a tool for potential use in clinical trials. Arthritis Rheum. 2007;57:1192-1201.
