[level-membership-for-neurology-category]
Chapter 15 Multiple Sclerosis
Clinical Manifestations
Course
Almost all MS patients follow one of four reasonably distinct courses, disease categories, that consist of multiple attacks, steady deterioration, or several attacks followed by steady deterioration (Fig. 15-1, top). The categories reflect the clinical status as it relates to time. They do not take into account the severity or results of MRIs.
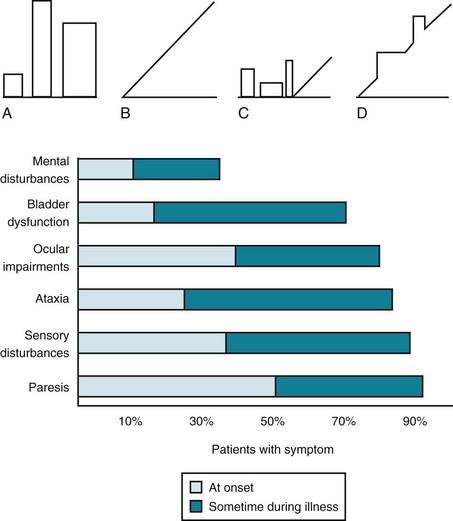
FIGURE 15-1 Top, Graphs of different clinical courses – with severity of multiple sclerosis (MS) attacks (vertical axis) plotted against time (horizontal axis) – reveal four patterns or disease categories: A, Relapsing-remitting; B, primary progressive; C, secondary progressive; D, progressive-relapsing. Bottom, This chart of initial and cumulative manifestations of MS indicates that cognitive impairment develops infrequently at the onset and ultimately less often than physical impairments.
Frequent Symptoms
Lesions in the white-matter tracts of the CNS cause various symptoms during the course of a patient’s illness (Fig. 15-1, bottom). Moreover, simultaneous involvement of two separate CNS areas often produces combinations of disparate symptoms. For example, plaques may simultaneously develop in the cerebellum and thoracic spinal cord, which would cause ataxia and paraparesis.
Cerebellar Signs
As some of their earliest manifestations, MS patients often develop ataxia, intention tremor, and other signs of cerebellar and cerebellar outflow tract injury. When the cerebellum is involved, patients typically develop an ataxic gait (see Fig. 2-13); however, with minimal involvement, patients’ gait impairment may consist only of difficulty walking in a heel-to-toe (tandem gait) pattern. Cerebellar involvement also typically causes scanning speech, a variety of dysarthria analogous to a “speech ataxia,” characterized by irregular cadence and uneven emphasis on words. For example, when asked to repeat a pair of short syllables, such as “ba…ga…ba…ga…,” a patient might place unequal stress on different syllables, blur them together, or pause excessively. Other manifestations of cerebellar involvement include intention tremor (see Fig. 2-11), dysdiadochokinesia, and an irregular, conspicuous, head tremor (titubation).
Decreased Visual Acuity
Neurologists usually attribute visual acuity impairment in MS to inflammation in the retrobulbar portion of the optic nerve, retrobulbar neuritis (optic neuritis) (see Fig. 12-6). Optic neuritis typically causes an irregular area of visual loss in one eye, a scotoma, that often includes the center of vision (Fig. 15-2). It also leads to color desaturation, in which colors, especially red, lose their intensity.
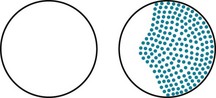
FIGURE 15-2 Optic or retrobulbar neuritis impairs vision in a large, irregular area (scotoma) of the affected eye.
Optic neuritis, as well as other lesions of the optic nerve, causes a readily identifiable, surprising pupillary light reaction (see Fig. 4-2). Swinging a flashlight from the normal eye to the one with optic neuritis will lead to dilation, rather than continued constriction, of both pupils. This paradoxical, unilateral reaction results from less light entering the pupillary reflex arc compared to when the light was shone into the normal eye. Sometimes called the “swinging flashing test,” this abnormality in the afferent limb of the light reflex results in an afferent pupillary defect or “Marcus Gunn” pupil, which neurologists interpret as a sign of optic nerve pathology.
Ocular Motility Abnormalities
MS also causes ocular motility abnormalities, including nystagmus and the characteristic internuclear ophthalmoplegia (INO), which is also known as the medial longitudinal fasciculus (MLF) syndrome. Either brainstem or cerebellar involvement can cause nystagmus. Although it is clinically indistinguishable from nystagmus induced by other conditions (see Chapter 12), MS-induced nystagmus typically occurs in combination with dysarthria and tremor (Charcot’s triad).
In MS-induced INO, MLF demyelination interrupts nerve impulse transmission from the pontine conjugate gaze centers to the oculomotor nuclei (Figs 15-3 and 15-4). The primary symptom of INO is diplopia on lateral gaze because of paresis of the adducting eye. INO is strong evidence of MS; however, systemic lupus erythematosus (SLE or lupus [see later]) and small basilar artery strokes may also cause it. In addition, Wernicke–Korsakoff syndrome, myasthenia gravis, and botulism can produce patterns of ocular muscle weakness that mimic INO. From a physiologic viewpoint, INO is analogous to a disconnection syndrome, such as conduction aphasia, in which communicating links are severed but each neurologic center remains intact (see Chapter 8).
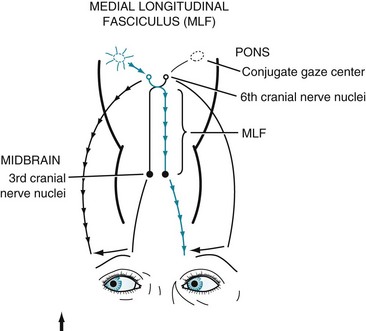
FIGURE 15-3 Under normal conditions, when looking laterally, the pontine conjugate gaze center stimulates the adjacent abducens (sixth) nerve nucleus and, through the medial longitudinal fasciculus (MLF), the contralateral oculomotor (third) nerve nucleus. For example, when looking to the right, as in this illustration, the right pontine gaze center stimulates the right abducens and the left oculomotor nuclei (see Fig. 12-12).
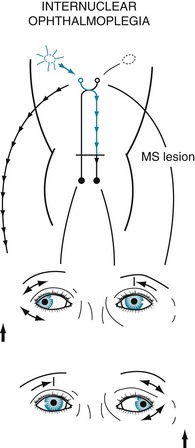
FIGURE 15-4 In internuclear ophthalmoplegia (INO), also known as the MLF syndrome, an interruption of the medial longitudinal fasciculus (MLF) prevents impulses from reaching the oculomotor (third) nuclei. Because those nuclei themselves remain intact, the pupils and eyelids are normal in both eyes. However, when looking to the right, because the left oculomotor nucleus is not stimulated, the left eye fails to adduct. The right eye abducts, but nystagmus develops. With bilateral INO, which is characteristic of multiple sclerosis (MS), neither eye adducts and abducting eyes have nystagmus.
Spinal Cord Symptoms and Signs
Spinal cord involvement also typically leads to urinary incontinence from a combination of spasticity, paresis, and incoordination (dyssynergia) of the bladder sphincter muscles (Fig. 15-5). MS patients initially often have incontinence during sleep and sexual intercourse. As the disease progresses, patients develop intermittent urinary retention and then complete loss of control. They often require intermittent or continuous catheterization, which leads to frequent, chronic, or recurrent urinary tract infections.
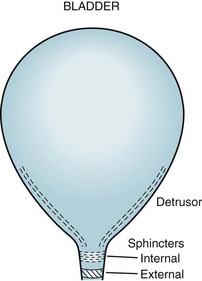
FIGURE 15-5 The urinary outflow of the bladder has two sphincters: an internal sphincter controlled by the autonomic nervous system (ANS), and an external one under voluntary control. Normal urinary bladder emptying (urination) occurs when the detrusor (wall) muscle contracts and both sphincter muscles relax. Purposefully urinating requires voluntary action (to relax the external sphincter) and reflex parasympathetic (ANS) activity (to contract the detrusor and relax the internal sphincter). Urinary retention occurs with either anticholinergic medication or excessive sympathetic activity because both inhibit detrusor contraction and internal sphincter muscle relaxation. Urinary retention also occurs with spinal cord injury because the external sphincter is unable to relax because it is spastic and paretic (dyssynergic).
Erectile dysfunction, decreased desire, and other forms of sexual impairment plague the majority of MS patients (see Chapter 16). About 40% of women with MS do not engage in sexual intercourse. Even before developing erectile dysfunction, men often experience premature or retrograde ejaculation. Sexual dysfunction, with or without urinary incontinence, is attributable to MS involving the spinal cord. With spinal cord damage severe enough to cause paraplegia, men have lowered and abnormal sperm production, but women can conceive and bear children.
Fatigue and Other Important Symptoms
MS-induced fatigue represents a physiologic cause of the chronic fatigue syndrome (see Chapter 6). Although many, but not all, studies found that depression is comorbid, antidepressants do not alleviate MS-induced fatigue.
Various pain syndromes also commonly occur in MS. For example, approximately 2% of MS patients suffer from trigeminal neuralgia (see Chapter 9) and 10% Lhermitte’s sign. Many MS patients have pain in their limbs or trunk. These pains probably arise from MS plaques irritating CNS pain-transmitting fibers in, respectively, the brainstem and cervical spinal cord. As with other forms of neuropathic pain (see Chapter 14), antiepileptic drugs, such as gabapentin and carbamazepine, provide some relief.
Because MS, in general, spares CNS structures that contain little or no myelin, symptoms that originate in gray-matter injury rarely complicate the illness. For example, MS patients seldom develop signs of focal cerebral cortical dysfunction, such as seizures or aphasia. Similarly, because the basal ganglia, like the cerebral cortex, are devoid of myelin, MS patients almost never develop involuntary movement disorders (see Chapter 18).
Psychiatric Comorbidity in MS
Depression
Consultants may also encounter “MS-induced euphoria” – an elevation of mood clearly inappropriate to these patients’ disability. This euphoria is associated with physical deterioration, chronicity of the illness, and at least subtle intellectual impairment, as well as steroid treatment. Some euphoric patients are masking depression or protecting themselves with denial. Others simply sense relief as an MS attack subsides. Whether or not psychologic factors seem to explain the euphoria, extensive cerebral involvement usually underlies it. In particular, pseudobulbar palsy may explain pathological laughter (see Chapter 4).
Cognitive Impairment
Almost all MS patients in the initial phase of their illness have normal cognitive capacity. They satisfactorily complete their day-to-day functions, routine mental status evaluation, and Mini-Mental State Examination (MMSE) (see Fig. 7-1). However, more demanding measures, such as the Wechsler Adult Intelligence Scale (WAIS), Selective Reminding Test, and Halstead Category Test, reveal at least clinically silent deficits in 45–65% of MS patients.
Laboratory Tests
Imaging Studies
MRI – certainly the most valuable test – can readily reveal demyelinated areas indicative of MS plaques. The revised McDonald criteria call for combinations of one gadolinium-enhanced lesion or nine T2-weighted hyperintense MRI lesions located in various regions of the brain, particularly in the periventricular area (Figs 15-6 and 20-25), or spinal cord (Fig. 15-7). Although not pathognomonic, these hyperintensities are detectable in more than 90% of MS patients.
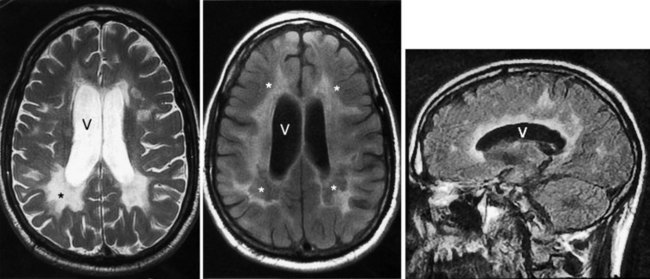
FIGURE 15-6 Left, This axial T2-weighted magnetic resonance imaging (MRI) scan through the cerebrum of a patient with multiple sclerosis (MS) shows multiple plaques (*) concentrated around the ventricles (V), particularly in the posterior regions. The MS plaques are characteristically white (hyperintense), sharply demarcated, and located in the periventricular region. Center, This axial T2-weighted, fluid-attenuated inversion recovery (FLAIR) image of the same study also shows hyperintense lesions (*) surrounding the ventricles (V). FLAIR images, in which cerebrospinal fluid remains black, highlight demyelinated areas. Right, The sagittal T2 FLAIR image of the same study shows the periventricular hyperintensities surrounding the lateral ventricle (V).
Cerebrospinal Fluid
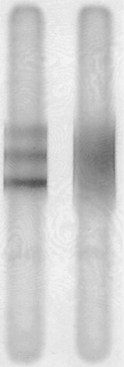
Routine CSF analysis during an MS attack will usually contain protein concentrations that are either normal (40 mg/100 mL) or only slightly elevated, and a mild, nonspecific gamma globulin elevation (9% or greater), but no increase in white blood cells (WBCs). One suggestive feature is that the CSF of 90% of MS patients contains CSF oligoclonal bands that consist of discrete IgG antibodies (Fig. 15-8). However, oligoclonal bands are also present in other inflammatory diseases involving the CNS, such as lupus, chronic meningitis, sarcoidosis, neurosyphilis, Lyme disease, acquired immunodeficiency syndrome (AIDS), and paraneoplastic limbic encephalitis.
Evoked Responses
Visual-evoked responses (VERs) reveal visual pathway lesions. The patient stares at a rapidly flashing pattern on a television screen and a computer averages responses detected over the occipital cortex. Optic neuritis increases the latency or distorts the waveform. Because VERs can indicate the site of an interruption in the visual pathway, they are helpful in distinguishing ocular, cortical, and psychogenic blindness (see Chapter 12).
Therapy
For attacks of MS or optic neuritis, with or without other signs of MS, neurologists generally administer high doses of intravenous steroids, such as methylprednisolone, that shorten attacks and possibly reduce residual disability. Physicians must cautiously administer steroid treatment because it potentially leads to steroid psychosis (see later) or other serious complications (Fig. 15-9).
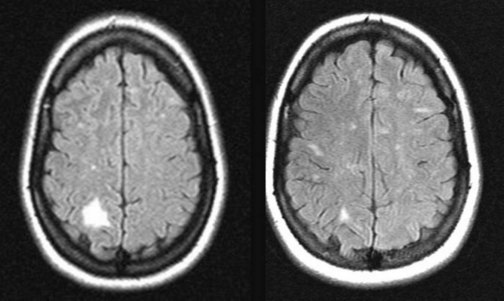
FIGURE 15-9 Left, A 38-year-old woman who had had multiple sclerosis for 9 years developed left-sided paresthesias and left homonymous hemianopsia. Her magnetic resonance imaging (MRI) scan showed a large region of high signal (hyperintensity) in the white matter of her right parietal-occipital cerebrum. Right, Six months after a course of steroids for the exacerbation, her deficits resolved and the MRI lesion almost completely resolved, but new, small lesions appeared.
Immunomodulators are also expensive (see Appendix 2). For example, one population-based study that examined their cost-effectiveness showed that each quality-adjusted year cost $800 000. Immunomodulators are counterindicated in pregnant women. Neoplasms, especially leukemias and lymphomas, on rare occasions complicate long-term immunosuppression in MS patients.
Neurologists treat many individual symptoms. For example, depending on the nature of a patient’s bladder dysfunction, either cholinergic medications, such as bethanechol (Urecholine), or anticholinergic ones, such as oxybutynin (Ditropan), might alleviate incontinence; however, patients with advanced disease may require self-catheterization or sphincter bypass. Although paresis cannot be improved, the accompanying spasticity, which is just as much of an impediment, usually responds to baclofen, diazepam, muscle relaxants, or injections of botulinum toxin (see Chapter 18). Formal exercise programs reduce disability and promote social contacts. Although inhaled cannabis purportedly reduces bladder dysfunction, spasticity, and pain, it impairs stamina and cognition.
Steroid Psychosis
Steroid treatment of MS – as with steroid treatment of lupus, organ transplant rejection, and acute asthma – can induce anxiety, euphoria, mania, depressive symptoms, and psychosis. Likewise, Cushing syndrome and other conditions that generate excessive steroid production can produce these symptoms. Even athletes surreptitiously using steroids for bodybuilding and energy can develop personality and behavioral changes. Steroids often produce insomnia, ravenous appetite, and tremor (see Chapter 18), any of which constitutes the first sign of steroid treatment. The influence of pre-existing psychiatric illness as a risk factor remains controversial.
Conditions That Mimic MS
Demyelinating Diseases That May Mimic MS
Guillain–Barré Syndrome
Most neurologic disorders that mimic MS are peripheral nervous system (PNS) or CNS demyelinating illnesses. Even though it is a demyelinating disease of the PNS rather than of the CNS, Guillain–Barré syndrome may resemble MS because it generally strikes young adults and causes paraparesis or quadriparesis (see Chapter 5). In contrast to MS, Guillain–Barré syndrome is characterized by a single, monophasic attack, lasting several weeks to months, of symmetric, flaccid, areflexic paresis.
Leukodystrophies
Two well-known leukodystrophies are adrenoleukodystrophy (ALD) and metachromatic leukodystrophy (MLD; see Chapter 5). ALD, which is transmitted in a sex-linked pattern, typically first produces neurologic symptoms and adrenal insufficiency in boys between 5 and 15 years old. However, sometimes symptoms emerge only when the men carrying the defective gene reach 20–30 years. When ALD develops at this age, CNS demyelination may cause mania, gait impairment, and eventually dementia. In addition to CNS demyelination, which often has an inflammatory component, MLD causes peripheral neuropathy.
Infections
PML, a DNA JC virus infection, like MS, produces one or more large areas of CNS demyelination (Fig. 15-10) and multiple neurologic deficits. PML is usually a late complication of AIDS and other illnesses characterized by immunologic impairment (see Chapter 7). It also complicates medication-induced immunosuppression for organ transplantation and cancer chemotherapy as well as natalizumab treatment. The presence of CSF JC virus antibodies, which requires polymerase chain reaction testing, confirms the diagnosis of PML and many times obviates the need for a brain biopsy.
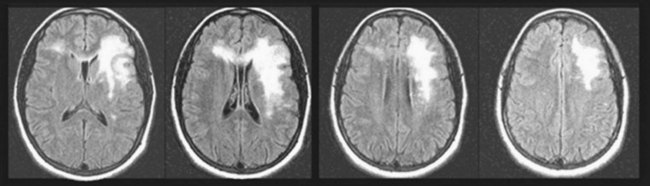
FIGURE 15-10 Surviving 20 years with a kidney transplant and enduring a decade of aggressive immunosuppression, a 40-year-old man insidiously developed right-sided hemiparesis and pseudobulbar speech and affect. His magnetic resonance imaging (MRI) scan showed an extensive area of hyperintensity in his left frontal lobe that extended through his anterior corpus callosum and involved a small area of his frontal lobe. However, unlike the MRI in multiple sclerosis, his MRI showed lesions that were patchy and extensive and not periventricular. The MRI appearance in this setting is indicative of progressive multifocal leukoencephalopathy (PML). He had the right hemiparesis from damage to the corticospinal tract of his left frontal lobe and the pseudobulbar palsy from the bilateral frontal lobe damage. Because the PML spared his cerebral cortex, he did not have aphasia.
Several other CNS infections directly or, through antibodies, indirectly attack CNS myelin and mimic MS. In particular, infection with the human T-lymphotropic virus type 1 (HTLV-1) causes a demyelinating myelitis that particularly affects the corticospinal tracts in the spinal cord (see Fig. 2-15), i.e., HTLV-1 myelitis. Thus, its clinical manifestations resemble MS involving only the spinal cord. HTLV-1, a retrovirus related to the human immunodeficiency virus (HIV), is endemic in the Caribbean islands and some areas of Japan. Sexual intercourse, pregnancy, and contaminated blood transmit HTLV-1, just like HIV; however, fewer than 5% of individuals infected with HTLV-1 develop symptoms. When it occurs, HTLV-1 myelitis typically produces a slowly evolving, painless, MS-like spastic paraparesis in residents of the Caribbean, where MS is uncommon. HTLV-1 infection rarely causes cognitive impairment or personality change. In infected individuals, antibodies to HTLV-1 are detectable in both serum and CSF, which typically shows an atypical lymphocytic pleocytosis.
Toxins
Numerous toxins preferentially attack CNS myelin. For example, a contaminant of certain homemade Italian red wines probably causes degeneration of the heavily myelinated corpus callosum, the Marchiafava–Bignami syndrome. Theoretically at least, Marchiafava–Bignami syndrome can damage the corpus callosum severely enough to produce the split-brain syndrome (see Chapter 8).
Chronic toluene exposure, whether from inadequate industrial ventilation or recreational volatile substance use, damages CNS myelin (see Chapter 5). Although appropriate industrial toluene levels should not produce problems, when individuals regularly inhale high concentrations, they develop cognitive disabilities, personality changes, and MS-like physical findings, such as ataxia, corticospinal tract signs, nystagmus, and even optic nerve impairment. MRI changes from toluene-induced demyelination, which may be more pronounced than ones from MS, correlate with cognitive and physical impairment.
Systemic Lupus Erythematosus
Manifestations of lupus-induced PNS involvement, which occur alone or in conjunction with CNS involvement, include neuropathy and mononeuritis multiplex (see Chapter 5). Studies have attributed both the CNS and PNS neurologic complications to autoantibodies, immune complexes, arteritis or vasculitis, and intrathecal production of cytokines. Many complications clearly reflect cardiac valvular disease, a tendency to develop thromboses, opportunistic infections, hypertension, renal failure, or possibly the elaboration of false neurotransmitters. Sometimes, because steroids remain a mainstay of lupus treatment, steroid psychosis, rather than the underlying disease, explains the eruption of a thought disorder, change in mood, or abnormal behavior.
Spinal Cord Disorders
Insidiously developing, painless paraparesis is a relatively common, important clinical problem (see Chapter 2). Numerous categories of neurologic illnesses in addition to MS that primarily or exclusively affect the spinal cord may cause it:
• Genetic disorders: e.g., spinal cerebellar ataxias and hereditary spastic paraparesis
• Infectious illnesses: e.g., HTLV-1 myelopathy
• Lesions compressing the spinal cord: e.g., cervical spondylosis, spinal meningiomas, and metastatic tumors
• Neurodegenerative illnesses: e.g., amyotrophic lateral sclerosis
• Nutritional deficiencies: e.g., copper deficiency, B12 deficiency (combined system disease).
Amato M, Goretti B, Ghezzi A, et al. Cognitive and psychosocial features of childhood and juvenile MS. Neurology. 2008;70:1891–1897.
Benedict R. Prediction of neuropsychological impairment in multiple sclerosis. Arch Neurol. 2004;61:226–229.
Caine E, Schwid S. Multiple sclerosis, depression and the risk of suicide. Neurology. 2002;59:662–663.
Chanson JB, Zephir H, Collongues N, et al. Evaluation of health-related quality of life, fatigue and depression in neuromyelitis optica. Eur J Neurol. 2011;18:836–841.
Chwastiak L. Depressive symptoms and severity of illness in multiple sclerosis: epidemiologic study of a large community sample. Am J Psychiatry. 2002;159:1862–1868.
Feinstein A. The neuropsychiatry of multiple sclerosis. Can J Psychiatry. 2004;49:157–163.
Figved N, Myhr KM, Larsen JP, et al. Caregiver burden in multiple sclerosis: The impact of neuropsychiatric symptoms. J Neurol Neurosurg Psychiatry. 2007;78:1097–1102.
Fredrickson S, Cheng Q, Jiang GX, et al. Elevated suicide risk among patients with multiple sclerosis in Sweden. Neuroepidemiology. 2003;22:146–152.
Gay MC, Vrignaud P, Garitte C, et al. Predictors of depression in multiple sclerosis patients. Acta Neurol Scand. 2010;121:161–171.
Ghaffar O, Reis M, Pennell N, et al. APOE e4 and the cognitive genetics of multiple sclerosis. Neurology. 2010;74:1611–1618.
Goodin DS, Cohen BA, O’Connor P, et al. Assessment: The use of natalizumab (Tysabri) for the treatment of multiple sclerosis (an evidence-based review): Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2008;71:766–773.
Kale N, Agaoglu J, Tanik O. Neuropsychiatric manifestations in multiple sclerosis: Correlation of fatigue and depression with disease progression. Neurol Res. 2010;32:221–223.
Koch M, Glazenborg A, Uyttenboogaart M, et al. Pharmacologic treatment of depression in multiple sclerosis. Cochrane Database Syst Rev. 2, 2011. CD007295
Koopman WJ, Benbow CL, Vandervoort M. Top 10 needs of people with multiple sclerosis and their significant others. J Neurosci Nurs. 2007;38:369–373.
Krupp LB. Fatigue in Multiple Sclerosis: A Guide to Diagnosis and Management. New York: Demos; 2004.
Langer-Gould A, Huang SM, Gupta R, et al. Exclusive breastfeeding and the risk of postpartum relapses in women with multiple sclerosis. Arch Neurol. 2009;66:958–963.
Lee M, O’Brien P. Pregnancy and multiple sclerosis. J Neurol Neurosurg Psychiatry. 2008;79:1308–1311.
Li J, Johansen C, Bronnum-Hansen H, et al. The risk of multiple sclerosis in bereaved parents: A nationwide cohort study in Denmark. Neurology. 2004;62:726–729.
Major EO. Reemergence of PML in natalizumab-treated patients. N Engl J Med. 2009;361:1041–1043.
Noye K, Bajorska A, Chappel A, et al. Cost-effectiveness of disease-modifying therapy for multiple sclerosis. Neurology. 2011;77:355–363.
Patten SB, Francis G, Metz LM, et al. The relationship between depression and interferon beta-1a therapy in patients with multiple sclerosis. Mult Scler. 2005;11:175–181.
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald Criteria. Ann Neurol. 2011;69:292–302.
Renoux C, Vukusic S, Mikaeloff Y, et al. Natural history of multiple sclerosis with childhood onset. Neurology. 2007;356:2603–2613.
Riise T, Mohr DC, Munger KL, et al. Stress and the risk of multiple sclerosis. Neurology. 2011;76:1866–1871.
Sirois F. Steroid psychosis: a review. Gen Hosp Psychiatry. 2003;25:27–33.
Smith P. Symptomatic treatment of multiple sclerosis using cannabinoids: recent advances. Expert Rev Neurother. 2007;7:1157–1164.
Solomon AJ, Whithan RH. Multiple sclerosis and vitamin D: a review and recommendations. Curr Neurol Neurosci Rep. 2010;10:389–396.
The Optic Neuritis Study Group. Multiple sclerosis risk after optic neuritis. Arch Neurol. 2008;65:727–732.
Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. The relationship of MS to physical trauma and psychologic stress. Neurology. 1999;52:1737–1745.
Zabad RK, Patten SB, Metz LM. The association of depression with disease course in multiple sclerosis. Neurology. 2005;64:359–360.
Abrahams BC, Kaufman DM. Anticipating smallpox and monkeypox outbreaks: Complications of the smallpox vaccine. Neurologist. 2004;10:265–274.
Angell M. Shattuck lecture – Evaluating the health risks of breast implants: The interplay of medical science, the law, and public opinion. N Engl J Med. 1996;334:1513–1518.
Argyriou AA, Makris N. Neuromyelitis optica: A distinct demyelinating disease of the central nervous system. Acta Neurol Scand. 2008;118:209–217.
Burns TM. Neurosarcoidosis. Arch Neurol. 2003;60:1166–1168.
Ferguson JH. Silicone breast implants and neurologic disorders: Report of the Practice Committee of the American Academy of Neurology. Neurology. 1997;48:1504–1507.
Joseph FG, Lammie GA, Scolding NJ. CNS lupus. Neurology. 2007;69:644–654.
Muscal E, Brey RL. Neurologic manifestations of systemic lupus erythematosus in children and adults. Neurol Clin. 2010;28:61–73.
Novak JC, Lovett-Racke AE, Tacke MK. Monoclonal antibody therapies and neurologic disorders. Arch Neurol. 2008;65:1162–1165.
Ramos-Rios R, Berdullas J, Arauxo A, et al. Schizophrenic psychosis at onset of adrenoleukodystrophy. CNS Spectr. 2009;14:711–712.
Sibbitt WL, Brandt JR, Johnson CR, et al. The incidence and prevalence of neuropsychiatric syndromes in pediatric onset systemic lupus erythematosus. J Rheumatol. 2002;29:1536–1542.
Chapter 15 Questions and Answers
c. Because of lesions in his left optic nerve, cerebellum, and spinal cord, even though they are not disseminated in time, the salesman probably has developed MS. Alternative explanations include illnesses that cause widespread central nervous system (CNS) lesions, such as other demyelinating conditions, such as ADEM, lupus, metastatic tumors and abscesses, and exposure to toxins. ADEM usually occurs in young patients and immediately following a viral illness. Although NMO causes paraparesis and visual impairment with an afferent pupillary defect from optic neuritis (see Chapter 12), it would not be as likely to cause his cerebellar signs (dysmetria and dysdiadochokinesia). Spinal cord tumors would create spastic paraparesis but, of course, not visual or cerebellar impairment. Psychogenic disturbances might lead to many of the findings, including feigned Babinski signs; however, patients cannot mimic his afferent pupillary defect. HTLV-1 myelitis typically causes spastic paraparesis but not cerebellar dysfunction or abnormal pupil reactions.
3. After a 2-month course of rehabilitation, the patient became ambulatory and had recovered his vision and coordination. He even returned to work and resumed his social life. Nevertheless, 1 year later, he reported persistent erectile dysfunction and urinary incontinence. Where do those symptoms originate?
d. The loss of proprioception in his legs causes him to have ataxia and a “steppage gait” (see Fig. 2-20). Neurologists might say that he has “sensory ataxia.”
26. A 9-year-old boy has developed social and academic difficulties. He has hyperactive DTRs, clumsiness, and an awkward gait. His older brother had similar symptoms and signs before succumbing to adrenal failure. The patient’s MRI shows extensive demyelination in the brain. Which of the following statements is correct?
34. After two decades of enduring relapsing-remitting MS unresponsive to interferon therapy, a 45-year-old former Broadway actress switched to immunosuppressant treatment with a humanized monoclonal antibody. One year later she developed the rapid development of pseudobulbar speech, cognitive impairment, and left-sided hemiparesis. Her MRI showed new large, demyelinated patches in several subcortical regions. Which is the most likely explanation for her deterioration?
36. A 32-year-old actress, who had been entirely well all of her life, presented to a neurologist with the sudden loss of vision in her left eye and weakness is both legs. Her examination revealed inability to count fingers in her left eye and 20/100 vision in her right eye. She had paraparesis with hyperactive DTRs and bilateral Babinski signs. The MRI of her brain showed signs of demyelination in both optic nerves and in a longitudinal segment of her lower cervical and upper thoracic spinal cord. Which is the most likely diagnosis?
d. Oligodendroglia produce CNS myelin. Schwann cells produce peripheral nervous system (PNS) myelin.
38. In regard to the allegation that silicone breast implants cause MS, which of the following statements is true?
e. There is no credible evidence that silicone breast implants cause neurologic disease.
39. A 22-year-old woman is admitted to a hospital because she has nystagmus, scanning speech, hyperactive DTRs in all limbs, Babinski signs, and a spastic-ataxic gait. She has inappropriate euphoria, impaired recent and remote memory, and impaired judgment. The MRI of her brain shows diffuse loss of myelin in her entire brain and spinal cord. Her HIV test was negative. She smokes two packs of cigarettes each day and admits to daily “huffing” (deliberately inhaling volatile substances for their euphoric effect). Which is the most likely diagnosis?
41. Following exposure to a prairie dog infected with monkeypox, public health officials administered smallpox vaccinations to a 19-year-old waitress and a dozen other young adults. Several days later she developed a high fever, stupor, INO, and spastic paraparesis. From which condition does she probably suffer?
46. A 33-year-old woman, who has just returned from a camping trip, awoke with decreased vision in her right eye. She also had pain in and around that eye when she looked from side to side. An ophthalmologist found that she had a dense central scotoma, visual acuity of 20/400 in her right eye, and one other abnormality. The patient’s pupils were both 4 mm in room light and, when the ophthalmologist aimed a flashlight in her left eye, both pupils constricted to 2 mm; however, when the ophthalmologist swung the light to the right eye, both pupils dilated back to 4 mm. What is the term for this paradoxical pupillary reaction to light?
a. Her pupils both dilated when the flashlight was switched in the “swinging flashlight test” from the normal to the affected eye. This response reflects decreased light input to the entire light reflex arc (see Fig. 4-2). Inflammation of the right optic nerve – optic neuritis – impaired the direct and consensual light reflex when light was shown into the right eye. Neurologists often refer to this afferent pupillary defect as a “Marcus Gunn pupil” and often find that it is a precursor to MS.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 15 Multiple Sclerosis
Clinical Manifestations
Course
Almost all MS patients follow one of four reasonably distinct courses, disease categories, that consist of multiple attacks, steady deterioration, or several attacks followed by steady deterioration (Fig. 15-1, top). The categories reflect the clinical status as it relates to time. They do not take into account the severity or results of MRIs.
FIGURE 15-1 Top, Graphs of different clinical courses – with severity of multiple sclerosis (MS) attacks (vertical axis) plotted against time (horizontal axis) – reveal four patterns or disease categories: A, Relapsing-remitting; B, primary progressive; C, secondary progressive; D, progressive-relapsing. Bottom, This chart of initial and cumulative manifestations of MS indicates that cognitive impairment develops infrequently at the onset and ultimately less often than physical impairments.
Frequent Symptoms
Lesions in the white-matter tracts of the CNS cause various symptoms during the course of a patient’s illness (Fig. 15-1, bottom). Moreover, simultaneous involvement of two separate CNS areas often produces combinations of disparate symptoms. For example, plaques may simultaneously develop in the cerebellum and thoracic spinal cord, which would cause ataxia and paraparesis.
Cerebellar Signs
As some of their earliest manifestations, MS patients often develop ataxia, intention tremor, and other signs of cerebellar and cerebellar outflow tract injury. When the cerebellum is involved, patients typically develop an ataxic gait (see Fig. 2-13); however, with minimal involvement, patients’ gait impairment may consist only of difficulty walking in a heel-to-toe (tandem gait) pattern. Cerebellar involvement also typically causes scanning speech, a variety of dysarthria analogous to a “speech ataxia,” characterized by irregular cadence and uneven emphasis on words. For example, when asked to repeat a pair of short syllables, such as “ba…ga…ba…ga…,” a patient might place unequal stress on different syllables, blur them together, or pause excessively. Other manifestations of cerebellar involvement include intention tremor (see Fig. 2-11), dysdiadochokinesia, and an irregular, conspicuous, head tremor (titubation).
Decreased Visual Acuity
Neurologists usually attribute visual acuity impairment in MS to inflammation in the retrobulbar portion of the optic nerve, retrobulbar neuritis (optic neuritis) (see Fig. 12-6). Optic neuritis typically causes an irregular area of visual loss in one eye, a scotoma, that often includes the center of vision (Fig. 15-2). It also leads to color desaturation, in which colors, especially red, lose their intensity.
FIGURE 15-2 Optic or retrobulbar neuritis impairs vision in a large, irregular area (scotoma) of the affected eye.
Optic neuritis, as well as other lesions of the optic nerve, causes a readily identifiable, surprising pupillary light reaction (see Fig. 4-2). Swinging a flashlight from the normal eye to the one with optic neuritis will lead to dilation, rather than continued constriction, of both pupils. This paradoxical, unilateral reaction results from less light entering the pupillary reflex arc compared to when the light was shone into the normal eye. Sometimes called the “swinging flashing test,” this abnormality in the afferent limb of the light reflex results in an afferent pupillary defect or “Marcus Gunn” pupil, which neurologists interpret as a sign of optic nerve pathology.
Ocular Motility Abnormalities
MS also causes ocular motility abnormalities, including nystagmus and the characteristic internuclear ophthalmoplegia (INO), which is also known as the medial longitudinal fasciculus (MLF) syndrome. Either brainstem or cerebellar involvement can cause nystagmus. Although it is clinically indistinguishable from nystagmus induced by other conditions (see Chapter 12), MS-induced nystagmus typically occurs in combination with dysarthria and tremor (Charcot’s triad).
In MS-induced INO, MLF demyelination interrupts nerve impulse transmission from the pontine conjugate gaze centers to the oculomotor nuclei (Figs 15-3 and 15-4). The primary symptom of INO is diplopia on lateral gaze because of paresis of the adducting eye. INO is strong evidence of MS; however, systemic lupus erythematosus (SLE or lupus [see later]) and small basilar artery strokes may also cause it. In addition, Wernicke–Korsakoff syndrome, myasthenia gravis, and botulism can produce patterns of ocular muscle weakness that mimic INO. From a physiologic viewpoint, INO is analogous to a disconnection syndrome, such as conduction aphasia, in which communicating links are severed but each neurologic center remains intact (see Chapter 8).
FIGURE 15-3 Under normal conditions, when looking laterally, the pontine conjugate gaze center stimulates the adjacent abducens (sixth) nerve nucleus and, through the medial longitudinal fasciculus (MLF), the contralateral oculomotor (third) nerve nucleus. For example, when looking to the right, as in this illustration, the right pontine gaze center stimulates the right abducens and the left oculomotor nuclei (see Fig. 12-12).
FIGURE 15-4 In internuclear ophthalmoplegia (INO), also known as the MLF syndrome, an interruption of the medial longitudinal fasciculus (MLF) prevents impulses from reaching the oculomotor (third) nuclei. Because those nuclei themselves remain intact, the pupils and eyelids are normal in both eyes. However, when looking to the right, because the left oculomotor nucleus is not stimulated, the left eye fails to adduct. The right eye abducts, but nystagmus develops. With bilateral INO, which is characteristic of multiple sclerosis (MS), neither eye adducts and abducting eyes have nystagmus.
Spinal Cord Symptoms and Signs
Spinal cord involvement also typically leads to urinary incontinence from a combination of spasticity, paresis, and incoordination (dyssynergia) of the bladder sphincter muscles (Fig. 15-5). MS patients initially often have incontinence during sleep and sexual intercourse. As the disease progresses, patients develop intermittent urinary retention and then complete loss of control. They often require intermittent or continuous catheterization, which leads to frequent, chronic, or recurrent urinary tract infections.
FIGURE 15-5 The urinary outflow of the bladder has two sphincters: an internal sphincter controlled by the autonomic nervous system (ANS), and an external one under voluntary control. Normal urinary bladder emptying (urination) occurs when the detrusor (wall) muscle contracts and both sphincter muscles relax. Purposefully urinating requires voluntary action (to relax the external sphincter) and reflex parasympathetic (ANS) activity (to contract the detrusor and relax the internal sphincter). Urinary retention occurs with either anticholinergic medication or excessive sympathetic activity because both inhibit detrusor contraction and internal sphincter muscle relaxation. Urinary retention also occurs with spinal cord injury because the external sphincter is unable to relax because it is spastic and paretic (dyssynergic).
Erectile dysfunction, decreased desire, and other forms of sexual impairment plague the majority of MS patients (see Chapter 16). About 40% of women with MS do not engage in sexual intercourse. Even before developing erectile dysfunction, men often experience premature or retrograde ejaculation. Sexual dysfunction, with or without urinary incontinence, is attributable to MS involving the spinal cord. With spinal cord damage severe enough to cause paraplegia, men have lowered and abnormal sperm production, but women can conceive and bear children.
Fatigue and Other Important Symptoms
MS-induced fatigue represents a physiologic cause of the chronic fatigue syndrome (see Chapter 6). Although many, but not all, studies found that depression is comorbid, antidepressants do not alleviate MS-induced fatigue.
Various pain syndromes also commonly occur in MS. For example, approximately 2% of MS patients suffer from trigeminal neuralgia (see Chapter 9) and 10% Lhermitte’s sign. Many MS patients have pain in their limbs or trunk. These pains probably arise from MS plaques irritating CNS pain-transmitting fibers in, respectively, the brainstem and cervical spinal cord. As with other forms of neuropathic pain (see Chapter 14), antiepileptic drugs, such as gabapentin and carbamazepine, provide some relief.
Because MS, in general, spares CNS structures that contain little or no myelin, symptoms that originate in gray-matter injury rarely complicate the illness. For example, MS patients seldom develop signs of focal cerebral cortical dysfunction, such as seizures or aphasia. Similarly, because the basal ganglia, like the cerebral cortex, are devoid of myelin, MS patients almost never develop involuntary movement disorders (see Chapter 18).
Psychiatric Comorbidity in MS
Depression
Consultants may also encounter “MS-induced euphoria” – an elevation of mood clearly inappropriate to these patients’ disability. This euphoria is associated with physical deterioration, chronicity of the illness, and at least subtle intellectual impairment, as well as steroid treatment. Some euphoric patients are masking depression or protecting themselves with denial. Others simply sense relief as an MS attack subsides. Whether or not psychologic factors seem to explain the euphoria, extensive cerebral involvement usually underlies it. In particular, pseudobulbar palsy may explain pathological laughter (see Chapter 4).
Cognitive Impairment
Almost all MS patients in the initial phase of their illness have normal cognitive capacity. They satisfactorily complete their day-to-day functions, routine mental status evaluation, and Mini-Mental State Examination (MMSE) (see Fig. 7-1). However, more demanding measures, such as the Wechsler Adult Intelligence Scale (WAIS), Selective Reminding Test, and Halstead Category Test, reveal at least clinically silent deficits in 45–65% of MS patients.
Laboratory Tests
Imaging Studies
MRI – certainly the most valuable test – can readily reveal demyelinated areas indicative of MS plaques. The revised McDonald criteria call for combinations of one gadolinium-enhanced lesion or nine T2-weighted hyperintense MRI lesions located in various regions of the brain, particularly in the periventricular area (Figs 15-6 and 20-25), or spinal cord (Fig. 15-7). Although not pathognomonic, these hyperintensities are detectable in more than 90% of MS patients.
FIGURE 15-6 Left, This axial T2-weighted magnetic resonance imaging (MRI) scan through the cerebrum of a patient with multiple sclerosis (MS) shows multiple plaques (*) concentrated around the ventricles (V), particularly in the posterior regions. The MS plaques are characteristically white (hyperintense), sharply demarcated, and located in the periventricular region. Center, This axial T2-weighted, fluid-attenuated inversion recovery (FLAIR) image of the same study also shows hyperintense lesions (*) surrounding the ventricles (V). FLAIR images, in which cerebrospinal fluid remains black, highlight demyelinated areas. Right, The sagittal T2 FLAIR image of the same study shows the periventricular hyperintensities surrounding the lateral ventricle (V).
Cerebrospinal Fluid
Routine CSF analysis during an MS attack will usually contain protein concentrations that are either normal (40 mg/100 mL) or only slightly elevated, and a mild, nonspecific gamma globulin elevation (9% or greater), but no increase in white blood cells (WBCs). One suggestive feature is that the CSF of 90% of MS patients contains CSF oligoclonal bands that consist of discrete IgG antibodies (Fig. 15-8). However, oligoclonal bands are also present in other inflammatory diseases involving the CNS, such as lupus, chronic meningitis, sarcoidosis, neurosyphilis, Lyme disease, acquired immunodeficiency syndrome (AIDS), and paraneoplastic limbic encephalitis.
Evoked Responses
Visual-evoked responses (VERs) reveal visual pathway lesions. The patient stares at a rapidly flashing pattern on a television screen and a computer averages responses detected over the occipital cortex. Optic neuritis increases the latency or distorts the waveform. Because VERs can indicate the site of an interruption in the visual pathway, they are helpful in distinguishing ocular, cortical, and psychogenic blindness (see Chapter 12).
Brainstem auditory-evoked responses (BAERs
[/not-level-membership-for-neurology-category]
