Chapter 52 Multiple Sclerosis
Providers of care to persons with multiple sclerosis (MS) have recently begun to appreciate the value of rehabilitative management techniques because rehabilitation is still the only way to improve function in patients with MS.98
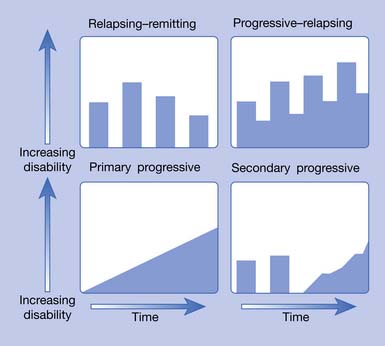
MS is now recognized as a complex disease with at least four pathologic types (Figure 52-1) and four clinical courses (Figure 52-2). As of this printing there are six Food and Drug Administration (FDA)-approved disease-modifying therapies (DMTs) available for the treatment of relapsing–remitting (RR) MS, with more currently in the approval pipeline. All these DMTs are only partially effective in slowing the acquisition of disability. Therefore rehabilitative management continues to play a vital role in the care of MS patients and will continue to do so into the foreseeable future.
Disease Overview
Demographics
MS is the most common cause of nontraumatic disability affecting young adults in the Northern Hemisphere.62 It is believed there are 400,000 persons in the United States with MS, and the prevalence ranges from 40 to 220/100,000, with the highest prevalence in the highest latitudes, although this differential appears to be lessening.113 Similar latitudinal differences are seen throughout other regions in the Northern Hemisphere. In the Southern Hemisphere, the highest prevalence also appears to be in latitudes farthest from the equator, although the much smaller land mass challenges demographic study.115
In certain populations, such as the Chinese, MS is a relatively rare disease, with current estimates at 20/100,000, even in the northern latitudes of the country (Bo Zhao Chong, personal communication, 2001). In other populations, such as native Africans and Native Americans, MS is also rare. African Americans with MS, however, are prone to a more aggressive course than white Americans.31
Approximately 85% of patients have either the RR or secondary progressive (SP) forms. SPMS typically develops after many years of the RR disease stage. When left untreated, 50% of the RRMS patients will have transitioned into SPMS by 10 years.62 More recent data (probably reflecting the widespread use of DMTs) from Tremlett et al.179 found 58% of RRMS patients converted to SPMS at a mean time of 18.9 years.
The RR/SP form of MS is gender dependent. Currently there are 4 times as many females as males with this form of the disease, a situation showing increasing female preponderance.110,136,196 The typical RRMS patient is a white woman in her late 20s who grew up in a temperate latitude and whose ancestors came from northern Europe.115
Ten to 15% of patients with MS have a disease that is progressive from the onset. This type is called primary progressive (PP) MS and has a roughly equal female to male ratio with a much later onset, around age 40.196 The fourth type of MS, progressive–relapsing (PR), is much less common. These four types are graphically represented in Figure 52-2.
Etiology
Epidemiologic studies have given clues about the etiology of MS. The current view is that it is the product of both a genetic predisposition and an early-acquired unknown environmental trigger, referred to by Kurtzke113 as the “MS affective agent” (MSAA). Migration studies indicate that the likelihood of developing the disease depends on where a person spent the first 15 years of life. Such data suggest that either a causative factor was acquired in the more temperate latitudes or a protective factor was acquired in the less temperate latitudes.36,38,196 Current interest has focused on the low levels of vitamin D—which serves as an immune modulator—in northern latitudes (southern latitudes in the Southern Hemisphere) as a geographic factor.30 Another proposed environmental factor is infection with a variety of candidate viruses (Epstein-Barr is the most frequently mentioned).120,172 A single virus might not be responsible for MS in all patients; different viruses (or virus combinations) might represent the particular MSAA in persons with different, but susceptible, human leukocyte antigen (HLA) types.
The genetic predisposition involves the tissue type. Persons with certain specific tissue (HLA) antigens appear to be either vulnerable to the disease or protected from it. Persons of northern European ancestry, especially those with major histocompatibility complex class II allele HLA-DR2 (HLA-DRB1∗1501) genotype, have a greater chance of developing the disease.6 The expression of this haplotype appears to be regulated by vitamin D, which could explain a link between vitamin D deficiency—a condition which is more common away from the equator—and increased risk for MS.150 The apolipoprotein polymorphism APOE-ε4 is also significantly associated with cognitive impairment in patients with MS.164
An attractive hypothesis regarding the etiology of MS is that genetically susceptible individuals could have an aberration in their immune tolerance, allowing environmental antigens (e.g., viruses) to stimulate production of autoreactive T cells. When such antigens are later encountered in adulthood, they might set off an attack against protein fragments of the person’s own myelin (“molecular mimicry”).112,172,187 This molecular mimicry might later develop into a self-perpetuating degenerative loop through the concept of “epitope spreading.”187 This is one among many postulated mechanisms. Reviews of other hypotheses are available.138
Pathophysiology
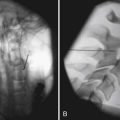
The pathologic hallmark of MS is the presence of multifocal demyelinated plaques scattered throughout the central nervous system, with prominent involvement of the periventricular white matter, optic nerves, brain stem, cerebellum, and cervical spinal cord. Demyelination is accompanied by axonal transection and ovoid body formation.177 Another characteristic feature is that these lesions tend to surround the deep veins of the brain, contributing one of the characteristic magnetic resonance imaging (MRI) features of MS, called Dawson’s fingers. These are linear lesions perpendicular to the long axis of the lateral ventricles (Figure 52-3).91
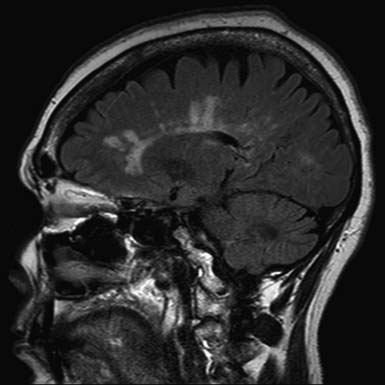
Newer techniques, including magnetic resonance spectroscopy (MRS),111 diffusion-weighted imaging,163 and magnetization transfer ratio (MTR), have shown widespread neuronal deficits in otherwise normal-appearing white matter.188 Progressive development of brain atrophy is a well-known feature of MS. Many studies have shown that atrophy of the brain is present from the earliest stages of the disease and tends to progress as the disease evolves.131 Involvement of gray matter is also evident on dissection but is more difficult to identify with current imaging techniques.34,57,77
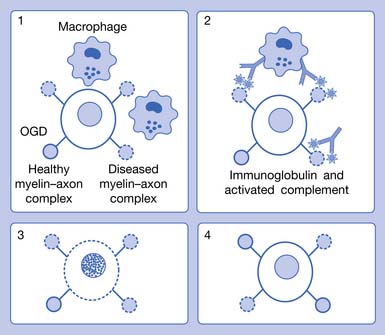
For decades investigators have tried to identify a single pathogenic mechanism that would allow the development of more precise immunotherapy. It now appears that subgroups of patients with MS have different pathogenic mechanisms, which might not correspond to their clinical MS types. The results of an international multicenter collaborative study of MS pathology led by Claudia Lucchinetti of the Mayo Clinic have identified four distinct pathologic patterns of MS (see Figure 52-1). Pattern I appears to be purely cell mediated where T cells produce demyelination; pattern II, the most common type, is similar to I, but also appears to involve additional intralesional immunoglobulins and activated complement (cell- and antibody-mediated); pattern III involves apoptosis of oligodendrocytes with selective loss of myelin-associated glycoprotein; and pattern IV involves primary degeneration of oligodendrocytes.122 Patterns I and II target myelin, and patterns III and IV target oligodendrocytes. Differences in antibody recognition and response to immunotherapy have been identified in these four patterns.119
Clinical Presentation
The classic symptoms of MS are manifold and are listed in decreasing frequency in Box 52-1. No other neurologic disease produces the coexistence of so many problems: weakness, fatigue, spasticity, tremor, ataxia, sensory loss, pain, visual loss, cognitive impairment, depression, and neurogenic organ dysfunction (e.g., neurogenic bladder and bowel). The fact that they progress at an unpredictable rate further complicates the management of MS.
Physicians and patients have recently come to realize that cognitive problems and depression occur earlier and much more frequently than previously appreciated. The point prevalence of depression is now thought to be greater than 40%,20 and lifetime cognitive impairment is greater than 60% to 80%.151 Clinicians now know that for every apparent (motor, sensory, or visual) exacerbation, there are as many as 10 (the exact number becomes greater with increasing MRI magnet strength) “subclinical” new gadolinium-enhancing lesions evident on MRI.69 This disease activity can manifest itself by producing emotional or cognitive symptoms. MS was traditionally viewed as a disease of ambulation (indeed, beyond the lower levels, the Kurtzke Expanded Disability Status Scale [EDSS] is a scale of ambulation114 [see Table 52-3]) but is now being viewed also as a disease producing serious “hidden” disabilities (e.g., fatigue, depression, impaired cognition, and pain).109
Neuroimaging
MRI is the most important diagnostic tool in MS. This is reflected in the new McDonald criteria145 (Table 52-1), which allow the diagnosis of MS to be made without a second clinical attack (e.g., new visual, motor, or sensory symptoms) if there is a new lesion on MRI at any point after the initial MRI. A typical MS lesion is ovoid, more than 3 mm in diameter, and located in the periventricular, corpus callosal, or posterior fossa white matter. These can best be seen on fluid attenuation inversion recovery (FLAIR) imaging or T2-weighted MRI sequences (see Figure 52-3). Pathology indicates that such lesions usually correspond to demyelination, although 40% of these—along with “shadow plaques”—contain some degree of remyelination.7 Because of altered physiologic function in partially remyelinated nerves, however, it is questionable whether remyelinated regions again become functional. The increased current leakage, occurring in partially myelinated nerves, results in increased capacitance. This cannot be readily overcome by physiologically generated action potentials; partially demyelinated nerves do not conduct impulses well.90,123,152
Table 52-1 2005 Revised McDonald Criteria for Multiple Sclerosis
| Clinical (Attacks) | Objective Lesions | Additional Requirements to Make Diagnosis |
|---|---|---|
| 2 or more | 2 or more | None; clinical evidence alone will suffice; additional evidence desirable but must be consistent with MS |
| 2 or more | 1 | Dissemination in space by MRI or 2 or more MRI lesions consistent with MS plus positive CSF or await further clinical attack implicating another site |
| 1 | 2 or more | Dissemination in time by MRI or second clinical attack |
| 1 | 1 |
(progression from onset)
Polman C.H., Reingold S.C., Edan G., et al: Diagnostic criteria for multiple sclerosis: 2005 revisions to the McDonald Criteria, Ann Neurol 58:840-846, 2005.
CSF, Cerebrospinal fluid; MRI, magnetic resonance imaging; MS, multiple sclerosis; VEP, visual evoked potential.
McDonald W.I., Compston A., Edan G., et al: Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis, Ann Neurol 50:121-127, 2001.
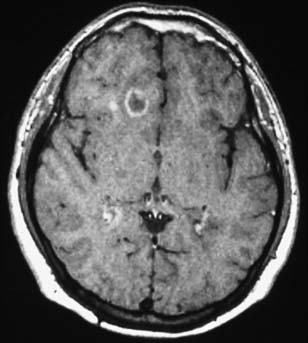
T2 lesions generally persist, whereas 44% of new T1 hypointensities can eventually become isointense and disappear. Enhancing T1 lesions are more reversible than nonenhancing lesions. Ring-enhancing lesions, which can be due to a ring of new inflammation around a previous site of disease, are prone to become persistent T1 “black holes.”157,182 An example of this type of lesion is shown in Figure 52-4. Initial axonal loss and degree of remyelination typically determine the outcome of the lesion.9
Indices of brain atrophy (brain parenchymal fraction and ventricular fraction) show that increased tissue loss is seen in all subtypes of MS. The rate of brain atrophy is higher in younger patients than in older patients.85 Baseline T2 lesion load is a statistically significant predictor of disease-related brain atrophy in patients with established MS.17 A 14-year longitudinal study of a group of patients with “clinically isolated syndrome” (patients who have a symptom that could be MS, but who have not yet developed the second episode in time that is required for the diagnosis) has shown convincingly that baseline T2 lesion load correlates highly with eventual MS diagnosis.11 In this setting, the detection of one or more baseline brain lesions carries a risk of MS of almost 90%. These findings support the use of DMTs in symptomatic patients with MRI abnormalities even before the diagnosis of MS has been established by standard (e.g., McDonald; see Table 52-1) criteria. For those who choose not to take therapy, we recommend having an MRI at least annually to monitor for change in lesion load. Because MS appears to respond best to DMTs in the earliest stages of the disease, there is now an attempt to identify MS as early as possible. Consequently, there is current research on identifying the “radiologically isolated syndrome,” the state before the presentation of the first MS symptoms.143
Other techniques, including those that are now used for research, might soon enter clinical practice. One of these is MRS, which can identify N-acetyl-aspartate (NAA) concentrations. NAA is found in neurons, and a reduction of NAA is a marker for axon damage.111 Another is MTR. These have shown widespread gray matter and neuronal deficits in otherwise normal-appearing white matter.34,77,188
Other than MRI, there are currently no good paraclinical markers to observe patients with established MS. Repeat MRI scans should be obtained when there is uncertainty as to whether a patient is responding well to therapy. These MRIs should include the spinal cord, especially the cervical portion, at least once. Considerable MS activity can occur in the upper spinal cord, but often that area is never imaged. No consensus exists regarding the importance of obtaining routine MRI scans in patients who are clinically doing well on therapy. A case can be made, however, that because MS causes many “hidden disabilities”—such as cognitive impairment—that are less obvious to the observer,109 patients who appear to be “doing well” also might also deserve periodic MRI monitoring, especially if the patient is on submaximal DMT.
Cerebrospinal Fluid Abnormalities
MS remains a clinical diagnosis, but cerebrospinal fluid (CSF) examination can help clarify uncertain cases. White blood cell count and protein levels are normal in two thirds of MS cases; gross elevations of either suggest a different etiology. Immunoglobulin oligoclonal bands are present in 83% to 94% of patients with MS when the test is performed with isoelectric focusing.121 With optic neuritis onset, the presence of oligoclonal bands is also predictive of eventual diagnosis of MS.80
Additional Diagnostic Tests
Evoked potentials (EPs), especially visual evoked potentials (VEP) and somatosensory evoked potentials (SEP), can be used to identify “hidden” lesions, which assist in identifying additional sites of abnormality that are required to make a diagnosis of “multiple” sclerosis. Conduction also demonstrates the state of myelin. Altered or absent myelin causes conduction slowing or conduction block. If conduction slowing through portions of the central neuraxis is found, the diagnosis of a demyelinating disease is strongly supported. Other pathologic conditions that might be mistaken clinically for a demyelinating disease (e.g., vascular lesions or tumor) would likely produce conduction block with attenuation or absence of a response rather than significant conduction slowing.108 Recommendations for the use of SEPs in MS can be found in practice topics published by the American Association of Neuromuscular and Electrodiagnostic Medicine.103
Early studies of VEPs, brain stem auditory evoked responses, and SEPs concluded that VEPs were more sensitive than SEPs in demonstrating central nervous system conduction abnormalities in MS.149,180 Early investigators, however, generally evaluated the short cord SEP pathway available with median nerve testing and did not evaluate SEPs from the lower limbs. Because MS lesions can occur throughout the brain, brain stem, and spinal cord, SEP techniques using the entire central neuraxis are most likely to demonstrate conduction abnormalities. Lower limb SEP testing increases the yield of abnormalities compared with upper limb testing and is comparable with the yield of VEPs.165
VEPs are frequently ordered for the evaluation of a patient with suspected MS to assess the presence of subclinical optic neuritis, in an attempt to confirm the presence of a second site of disease and to confirm that a visual deficit is due to optic neuritis. The technique for these is well established.19 A correlation exists between abnormal VEP latency and MRS-measured NAA levels.71 Optical coherence tomography is an alternative method of assessing the presence of optic neuropathy by measurement of retinal nerve fiber layer thickness.54
Diagnostic Criteria
The revised McDonald criteria (see Table 52-1) represent the current standard of diagnosis.130,145 MRI, CSF, and EP findings can be used to demonstrate dissemination in time and space. In recent years, improvements in MRI techniques have reduced the importance of CSF and EPs in the diagnostic evaluation of MS. The McDonald criteria err on the side of conservancy and might underdiagnose some patients with MS. Examples of this underdiagnosis include those in the early stages of disease or those with primarily spinal cord disease.8,175
For a period after the occurrence of the breakdown in the blood-brain barrier, the affected region is permeable to gadolinium. Consequently, MRI scans taken shortly after an exacerbation (several weeks with a 1.5-tesla magnet; longer with a stronger magnet) after an intravenous injection of gadolinium will show “enhancement” or increased density on T1-weighted images. Such techniques can identify acute lesions, and thereby identify a new lesion in time. It is argued that the observation of both enhancing and nonenhancing lesions confirms the criterion of at least two points in time. Double- and triple-dose gadolinium injections are able to show even more subtle disease activity (seeFigure 52-4).156
Differential Diagnosis
The differential diagnosis for MS is extremely broad and includes metabolic, infectious, vascular, neoplastic, genetic, autoimmune, as well as other central demyelinating disorders (e.g., neuromyelitis optica [NMO] and acute disseminated encephalomyelitis). NMO (Devic disease) is a “demyelinating” disorder consisting of transverse myelitis and optic neuritis that can present similarly to MS. Although similar clinically, it has a different pathogenesis and different treatment, making its differentiation from MS very important. NMO has characteristic features allowing for its differentiation from MS, including longitudinally extensive cord lesions over three or more spinal segments, relatively spared brain MRI, and an associated biomarker, NMO-IgG. NMO-IgG is highly specific (91%) and moderately sensitive (73%) for the diagnosis of NMO.193
Clinically isolated syndrome (CIS) is the term given to the first demyelinating event that might or might not progress to clinically definite MS (CDMS). Typical presentations of CIS include optic neuritis, internuclear ophthalmoplegia, facial sensory loss, sixth nerve palsy, and partial myelopathy. Brain MRI at the time of CIS is predictive of conversion to CDMS with more than 85% of patients with two or more MRI lesions progressing to CDMS, and that risk is greatest in the first 5 years. Brain MRI at presentation of CIS is also predictive of the extent of disability at 10 years. In patients with more than 10 lesions at presentation, 75% show significant functional impairment (EDSS >3 at 10 years), whereas in patients with fewer than 10 lesions at presentation, only 27% demonstrate significant functional impairment (EDSS >3 at 10 years).142
The introduction of the CIS term is clinically relevant as four CIS trials (BENEFIT,86 ETOMS,24 CHAMPS,78 and PRECISE86) have all shown that early treatment with DMT delays progression to CDMS (with a relative risk reduction of about 45% compared with placebo across all studies).
Additional diagnostic tests that can be useful to diagnose MS and the many diseases that can mimic MS can be found in Table 52-2.
| Routine | When Indicated |
|---|---|
| Magnetic resonance imaging: brain and cord | Cerebrospinal fluid studies |
| Antinuclear antibodies | Evoked potentials |
| Lyme titers | HIV |
It should be pointed out that many diseases can mimic MS. Usually (and fortunately) the diagnosis of MS is very clear. But at times, even the most sophisticated MS physician might encounter a disease that appears to be MS but is not. In such cases, Table 52-3 will help sort out the rare imposters from the real disease we are discussing in this chapter.
Table 52-3 Sorting Through the Mimickers of Multiple Sclerosis
| MRI Finding | Disease |
|---|---|
| Brain white matter | |
| Normal | CIS (low risk for MS), NMO (absent or few lesions), ATM |
| Large lesions | AMS (sometimes confluent and perilesional edema), BCS (concentric whorls of alternating rings of enhancement), PACNS (with mass effect) |
| Absent MRI activity at follow-up | ADEM |
| T2-hyperintensity of the temporal pole, U-fibers at the vertex, external capsule and insular regions | CADASIL |
| Diffuse WM involvement | Neuro-Behçet’s disease, HIV encephalitis, small vessel disease, CADASIL |
| Multifocal, asymmetrical lesions starting in a juxtacortical location and progressively enlarging | PML |
| Large lesions with absent or rare mass effect | PML |
| Extensive and bilateral periventricular abnormalities in isolation | Vitamin B12 deficiency, acquired copper deficiency |
| Cortical gray matter | |
| Cortical/subcortical lesions crossing vascular territories | MELAS |
| Prevalent involvement versus white matter | Encephalitis |
| Infiltrating lesions that do not remain in gray or white matter boundaries | Abscesses |
| Deep gray matter | |
| Bilateral lesions | ADEM (at the gray–white matter junction), CADASIL |
| Lacunar infarcts | CADASIL, small-vessel disease |
| T1-hyperintensity of the pulvinar | Fabry’s disease |
| Multiple discrete lesions in the basal ganglia and thalamus | Susac’s syndrome |
| Large and infiltrating basal ganglia lesions | Neuro-Behçet’s disease |
| Infiltrating lesions without respecting gray matter or white matter boundaries | Abscesses |
| T2-hyperintense lesions in the dentate nuclei | AFL (CTX) |
| Spinal cord | |
| Large and swelling lesions | NMO (with corresponding T1 hypointensity), ADEM, ATM, Sjögren’s syndrome |
| Diffuse abnormalities in the posterior columns | Vitamin B12 deficiency, acquired copper deficiency |
| Other | |
| No “occult” changes in the NAWM | NMO, Lyme disease, SID (except in NSLE) |
| Pontine lacunar infarcts | CADASIL, small-vessel disease |
| Dilation of Virchow–Robin spaces | HHC, PACNS |
| Diffuse lactate increase on brain MRS | MELAS |
| Meningeal enhancement | Susac’s syndrome, PACNS, neuro-Behçet’s disease, meningitis, Lyme disease, sarcoidosis |
| Hydrocephalus | Sarcoidosis |
| Absence of optic nerve lesions | PML |
| Regional atrophy | HHC (hippocampus and amygdala), neuro-Behçet’s disease (brain stem) |
ADEM, Acute disseminated encephalomyelitis; AFL (CTX), adult forms of leukoencephalopathy, cerebrotendinous xanthomatosis; AMS, acute Marburg syndrome; ATM, acute transverse myelitis; BCS, Balo’s concentric sclerosis; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CIS, clinically isolated syndrome; HHC, hyperhomocysteinaemia; MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, stroke-like episodes; MRS, magnetic resonance spectroscopy; NAWM, normal appearing white matter; NMO, neuromyelitis optica; NSLE, neuropsychiatric systemic lupus erythematosus; PACNS, primary angiitis of the central nervous system; PML, progressive multifocal leukoencephalopathy; SID, systemic immune-mediated disorders.
Prognosis
A recent natural history study of 2837 MS patients followed prospectively for 22,723 patient years found that progression in MS is slower than previously described. Participants took a median of 27.9 years from onset before requiring a cane. At 15 years after onset, 21% required a cane, increasing to 69% by 40 years. At 30 years after onset, 14% required a wheelchair, increasing to 22% by 40 years. Measured from birth, participants took a median 59 years before requiring a cane. By 50 years of age, 28% required a cane and 6% required a wheelchair. Although men progressed more quickly than women from disease onset, both sexes required a cane at a similar age. This is in contrast to previous studies suggesting that male gender is associated with a worse prognosis. Those with younger age at onset had slower progression but were still significantly younger when requiring a cane compared with those with an older age of onset. This finding also differs from previous reports that younger age of onset is associated with a better prognosis. Male sex and older age at onset were not associated with a worse prognosis in this natural history study. Progression from onset was more rapid in those with a primary progressive course versus those with a relapsing–remitting course. Despite an older age at onset in PPMS, patients were younger when requiring a cane. A primary progressive course was associated with greater progression when measured from both onset and birth. The rate of progression does not differ between PPMS and SPMS once patients reach a level of mild disability. Motor, cerebellar, or brain stem onset symptoms were associated with a more rapid progression from onset compared with sensory or optic neuropathy symptoms; however, patients were of similar age when requiring a cane. Contrary to previous reports, no onset symptom independently predicted a better or worse outcome. Lastly, those progressing rapidly to moderate disability (EDSS 3 in <5 years) required the use of a cane sooner.178
MRI lesion volume at 5 years and the change in volume during the first 5 years correlate with disability scores at 14 years. Early lesion burden might have an important influence on the development of later disability.11
Among the existing measures of clinical status in MS, the most commonly used is Kurtzke’s EDSS, shown in Table 52-4.114 This scale ranges from 0 (no impairment) to 10 (death from MS), with half-point levels along the way. It focuses on mobility more than sensory, bladder, bowel, communicative, or cognitive impairments. In the lower ranges with full ambulation, the so-called functional systems (e.g., sensory, bladder, visual, cognitive) determine the level; in later stages, when ambulation is impaired, gait limitations trump the functional systems. The midrange is heavily weighted on ambulation and vulnerable to interrater and intrarater fluctuations, as well as fluctuations resulting from time of day and other factors. For example, the difference between an EDSS score of 5.5 and 5.0 is the difference in the ability to walk unaided 100 m, but not 200 m. A change in EDSS must be at least two levels (i.e., one full point) to be considered significant because the reliability of half-point changes is poor.119
| Rating | Description∗ |
|---|---|
| 0.0 | Normal neurologic examination |
| 1.0 | No disability; minimal signs on one functional system |
| 1.5 | No disability; minimal signs on two of seven functional systems |
| 2.0 | Minimal disability in one of seven functional systems |
| 2.5 | Minimal disability in two functional systems |
| 3.0 | Moderate disability in one functional system, or mild disability in three or four functional systems, although fully ambulatory |
| 3.5 | Fully ambulatory but with moderate disability in one functional system and mild disability in one or two functional systems; or moderate disability in two functional systems; or mild disability in five functional systems |
| 4.0 | Fully ambulatory without aid, up and about 12 hr/day, despite relatively severe disability; able to walk without aid 500 m |
| 4.5 | Fully ambulatory without aid; up and about much of day; able to work a full day; might otherwise have some limitations of full activity or require minimal assistance; relatively severe disability; able to walk without aid 300 m |
| 5.0 | Ambulatory without aid for about 200 m; disability impairs full daily activities |
| 5.5 | Ambulatory for 100 m; disability precludes full daily activities |
| 6.0 | Intermittent or unilateral constant assistance (cane, crutch, or brace) required to walk 100 m with or without resting |
| 6.5 | Constant bilateral support (cane, crutch, walker, or braces) required to walk 20 m without resting |
| 7.0 | Unable to walk beyond 5 m even with aid; essentially restricted to wheelchair; wheels self, transfers alone; active in wheelchair about 12 hr/day |
| 7.5 | Unable to take more than a few steps; restricted to wheelchair; might need aid to transfer; wheels self but might require motorized chair for full day’s activities |
| 8.0 | Essentially restricted to bed, chair, or wheelchair, but might be out of bed much of day; retains self-care functions; generally effective use of arms |
| 8.5 | Essentially restricted to bed much of day; some effective use of arms; retains some self-care functions |
| 9.0 | Helpless bed patient; can communicate and eat |
| 9.5 | Unable to communicate effectively or to eat or swallow |
| 10.0 | Death |
∗ Functional systems are the visual, brain stem, pyramidal, cerebellar, sensory, bladder and bowel, and mental systems.
Kurtzke J.F.: Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS), Neurology 33:1444-1452, 1983.
Pregnancy and Breastfeeding
Pregnancy raises several complex issues in MS care. First is the question of the hereditary transmission of MS. An increased incidence of MS exists among the offspring of individuals with MS: 3% for girls and 1% for boys. On the other hand, MS does not appear to have a negative effect on the pregnancy itself or fetal outcome.32
Second is the question of what happens to exacerbation rates during and after pregnancy. During pregnancy, relapse rates decrease to about half of what they would be otherwise.27 During the first 3 months postpartum, the relapse rate is higher than normal. The net effect of pregnancy on the course of MS is neutral, and women do not need to make decisions about pregnancy based on fear that it will worsen their disease.
The next question has to do with management of DMT during pregnancy. Given that the interferon-β drugs are all FDA category C for pregnancy (primarily because of increased rates of miscarriage), and given that the risk of relapse is lower during pregnancy, the standard advice is that interferon-β drugs should be stopped before a woman gets pregnant. Glatiramer acetate is FDA category B for pregnancy and might be a safer choice if a woman wishes to continue DMT during pregnancy. Restarting MS therapy after delivery should be delayed until breastfeeding is discontinued. Breastfeeding might decrease the relapse rate by half during the first 6 months postpartum.67 For protection during this vulnerable period, we recommend 1000 mg of intravenous methylprednisolone monthly during the postpartum nontreatment period, extending several months after treatment has again been restarted.
Medical Management
Drug Therapies
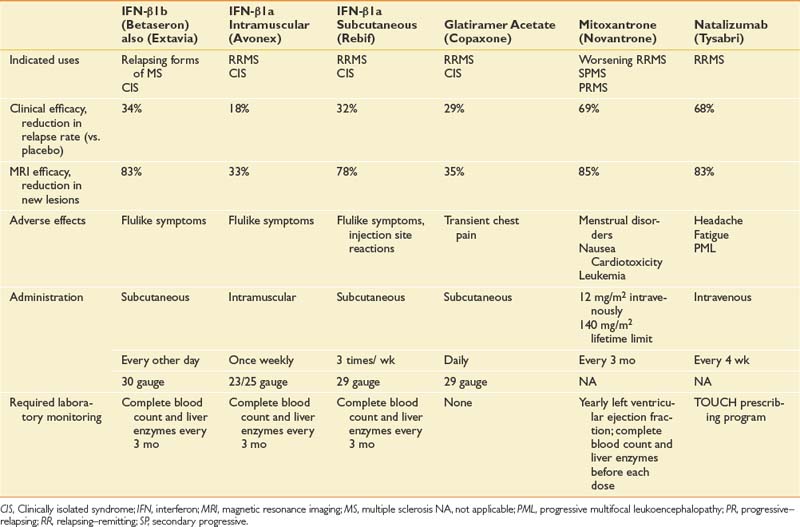
Because MS is an immune-mediated disease, all effective DMTs are immunoactive drugs. These take three forms: DMTs that modify cellular immune responses (immunomodulatory drugs), drugs that interfere with inflammatory cell replication (antiproliferative), and drugs that block extracellular processes. Drugs are also available with multiple actions, including methylprednisolone, which partially protects T-cell migration into the central nervous system and is immunomodulatory at low doses and cytotoxic at high doses. It also reduces edema associated with acute lesions. Table 52-5 outlines the prescribing information for DMT treatments available at the time of the writing of this chapter.
Immunomodulatory Drugs
The initial DMTs were interferon-β. The first was interferon-β1b (Betaseron), which was approved by the FDA for use in RRMS and became available in limited quantities in the United States in 1993.40 This was followed by weekly intramuscular interferon-β1a (Avonex), which gained FDA approval for RRMS in 1996,79 and three times weekly subcutaneous interferon-β1a (Rebif).61,140 Another category of DMT is glatiramer acetate (Copaxone), a polymer of four randomly ordered amino acids (glutamate, lysine, alanine, and tyrosine) in the same molar ratio as in myelin basic protein.81 This is administered subcutaneously daily.
Antiproliferative Drugs
Chemotherapeutic anticancer drugs such as methotrexate, azathioprine, and cyclophosphamide are some of the oldest treatments for MS. These have beneficial results, but the adverse effects limit their use at the most therapeutic dose levels. Data on their effects are limited because it has been difficult to fund large randomized controlled trials with these off-patent medications. More recently, mitoxantrone (Novantrone) has been shown to markedly reduce relapse rate and gadolinium-enhancing MRI lesions in patients with worsening RRMS and SPMS.70 Mitoxantrone has a number of side effects, including menstrual irregularities, nausea, fatigue, and transient leukopenia. The most limiting side effect is that it is cardiotoxic and can cause cardiomyopathy, for which reason the cumulative lifetime dose is limited to 140 mg/m2 (2.5 years with standard dosing).58
Monoclonal Antibodies
The most recently approved DMT, natalizumab (Tysabri), blocks T-cell entry into the central nervous system. It is among the most effective medications (see Table 52-5) but can cause a potentially fatal viral disease—progressive multifocal leukoencephalopathy—in a very small percentage of patients.89,118,197 As a result, in the United States this DMT, which is given by infusion each month, is only administered in approved centers through a strict monitoring program (known as the TOUCH program).
Other monoclonal antibodies (e.g., alemtuzumab, Campath) are being studied with the objective of depleting the host’s autoreactive T cells. They show promise but are also highly toxic.23 Drugs that interfere with lymphocyte purine synthesis (cladribine and mycophenolate mofetil, CellCept) or pyrimidine synthesis (teriflunomide) are being investigated as potential oral agents for MS.53 High-dose immunosuppressive therapy with autologous stem cell rescue is also being studied.23,92,104,105
Corticosteroids
Corticosteroids have been used for many years for MS.147 They can reduce the severity and duration of exacerbations but until recently were not believed to have any long-term effect on the course of the disease. As with chemotherapeutic agents, the toxicity profile of oral steroids was considered too great to use on an ongoing basis. Contrary to previous opinions, a careful recent study suggests that intravenous methylprednisolone might have some long-term benefit. Pulsed intravenous methylprednisolone given several times a year has been shown to slow the development of T1 black holes, delay brain atrophy, and slow progression in RRMS.198 It can also be used in SPMS when other options have been exhausted.63
Rehabilitation
The Rehabilitation Approach
MS has a progressive course that worsens in an uncertain pattern with a variable rate. There can be a week-to-week fluctuation as exacerbations give way to remissions. An exacerbation can occur at any time, and the hallmark of the disease is uncertainty as to what the future might present. Rehabilitation goals for patients with MS can be moving targets. Despite disease-modifying agents, patients tend to get worse over time. In no other neurologic disease are so many functions of the central nervous system affected (see Box 52-1).
MS care is now entering a stage of greater understanding of the disease and realization of the toll it takes on family, friends, job, and community. For example, it is not just that MS can produce cognitive impairment, depression, and motor difficulties. It is also that with progressive neurologic loss diffusely manifested throughout the central nervous system, many central nervous system functions can be performed but require intense focus and require large amounts of patient effort. The summation of central nervous system losses can require considerably increased energy to perform tasks, and multitasking is not easily possible. Brain reorganization that has occurred as a result of plasticity, which allows performance of isolated tasks to continue, does not appear to easily allow multitasking.93
Inpatient Rehabilitation Approach
With rising health care costs and increasing use of outpatient care, MS patients are now only rarely admitted to inpatient rehabilitation services in the United States. This occurs typically only after severe exacerbation or surgery, such as the implantation of a baclofen pump for spasticity management. Recently similar trends are occurring even in European countries, where annual admission of MS patient to inpatient rehabilitation facilities used to be the norm (Claude Varnet, personal communication, 2009).87
Several studies have found that inpatient rehabilitation for MS yields short-term benefits in function, mobility, and several aspects of quality of life.∗ Benefits have generally been most impressive in uncontrolled retrospective trials including patients recovering from exacerbations.1,66,144 Studies that have looked for extended carryover have found that without follow-up therapy, benefits dwindle by 6 to 10 months. This finding was attributed to disease progression in some studies, whereas in others the disease progression was not a possible explanation (because the EDSS remained stable).
Outpatient and Home-Based Exercise
It has been said that every MS patient could benefit from some form of exercise. Exercise has a beneficial effect on MS disability and quality of life. Robust evidence exists that aerobic training improves maximum exercise capacity (VO2max) for ambulatory MS individuals, whereas inactivity makes it worse.55,146,155,190 We know less about exercise effects in semiambulatory and nonambulatory MS individuals, but it appears that they do not receive as much benefit. This might be because they cannot activate enough muscle mass to get a training effect because exercise programs are not designed properly for them, or because their adherence is poor.
For those with a greater degree of disability, multidisciplinary outpatient programs might provide better results than exercise alone, but these are not widely available. Adherence to exercise can yield a partial reduction of MS fatigue. Aerobic exercise is particularly important for the patients with MS who are overweight.73
Every exercise prescription should be tailored to meet individual circumstances. Specific muscle training is recommended for improving focal weaknesses. It has also been advocated when fatigue or heat sensitivity are important issues.42 Focused muscle strengthening with progressive resistive exercises can be effective in motivated individuals with mild, or even severe, impairments.37,101,102 Three sets of 10 repetitions might be an appropriate regimen. With careful selection of muscles (using probably not more than two per limb, and no more than two limbs per session), resistive exercises might still be effective in strengthening, even when weakness is diffuse (see also Chapter 18).
For those whose primary goal is improved gait, exercise must include standing and walking. Aquatic exercise (swimming, water aerobics, and water walking) is an excellent form of integrated exercise, especially where ataxia might create a safety concern. Even those with tetraparesis can use the water buoyancy to facilitate standing and supine swimming with assistance and flotation aids.195 Care should be taken to find a pool that is not too hot (>29° C, 84° F) for those who are sensitive to heat. Combined arm–leg ergometry is useful for people with partial paralysis. Yoga can improve flexibility and reduce spasticity. Outdoor walking, aerobics, and t’ai chi are useful for balance training.76,106
Active exercise should be done at least three times a week. Appropriate, sustained stretching should be done for up to 20 minutes, with emphasis on posterior thigh, calf, and back muscles. Previously sedentary individuals should start aerobic exercises at a comfortable level and increase the duration and intensity of exercise at weekly or monthly intervals.189 Adherence to the exercise program is typically better if a communal or group activity, such as group aerobics, is used.
For markedly disabled MS individuals, the activities of daily living (ADL) might constitute their only regular forms of physical activity (see Chapter 26). In addition to passive stretching and range of motion exercises, it is important to devise simple activities that tap underused strengths to avoid learned disuse and deconditioning. Recreational activities should supplement the amount of exercise derived from ADL.
Management of Spasticity
Spasticity is a velocity-dependent increase in tonic stretch reflexes (muscle tone) resulting from hyperexcitability of the stretch reflex (see also Chapter 30). Spasticity can have a significant functional impact in that it can impair gait and ADL, as well as cause pain. Spasticity is prevalent in the MS population. A cross-sectional study of 20,969 MS patients enrolled in the North American Research Committee on MS patient registry found that 84% of participants reported spasticity and one third of patients modified or eliminated daily activities as a result of it. A linear relationship between spasticity scores and disability was demonstrated with a spasticity level of 2 or greater (defined in the study as “spasticity occasionally forces me to change some of my activities, e.g., once a week or less”) predictive of the need for an assistive device.153
The treatment of spasticity must be directed toward patient and functional goals, not just overall tone reduction. For example, is the goal to improve function (e.g., gait, transfers), improve ease of care (hygiene, dressing), improve positioning (seating), or reduce painful spasms? The patient’s tone must be evaluated in both the resting position and during functional activities. This enables the provider to see the functional impact of the tone and to evaluate how dynamic tone and/or dystonia are contributing. The potential advantages of increased muscle tone must also be considered. For example, extensor tone can facilitate transfers and gait in some individuals. In these cases, treating spasticity could unmask weakness and decrease function. This emphasizes the importance of spasticity management toward achieving functional goals and not just tone reduction.
Management of spasticity starts with a daily stretching program, which reduces muscle tone and limits secondary soft tissue changes that can lead to joint contractures from shortening of the musculotendinous unit. Stretching two joint muscles across both joints is imperative to achieve an adequate stretch (e.g., the gastrocnemius muscle should be stretched with the knee fully extended). Oral medications, most commonly baclofen (Lioresal) and tizanidine (Zanaflex), are first-line treatments to reduce diffuse spasticity (Table 52-6). Their use can be limited by sedating side effects and exacerbation of fatigue and cognitive dysfunction, which are often already present in this MS population. For focal spasticity that is limited to one or two limbs, local treatment with botulinum toxin (Botox) or phenol might be more beneficial. This provides a targeted intervention without the systemic side effects of oral medications. For moderate to severe function-limiting spasticity of the lower limbs, intrathecal baclofen can be considered. Although baclofen pumps are very effective in tone management, their use can be limited by complications (e.g., infection, pump malfunction, overdose, or withdrawal) and high maintenance requirements. Also, they are not an option also in the rare occurrence of primarily upper limb spasticity because the catheter can only safely terminate in the lower spinal canal.
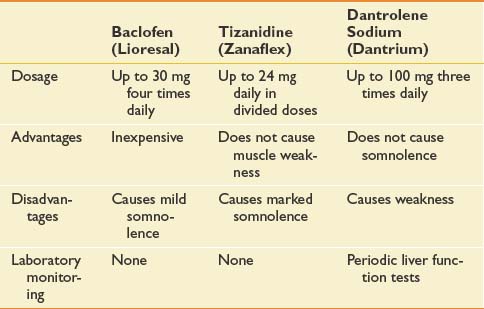
Spasticity treatments have additive effects and are most successful when used in combination. Oral medications work best in conjunction with a stretching program. Treatment of focal spasticity with botulinum toxin is optimized when combined with an appropriate therapy program that is goal specific. For example, botulinum toxin treatment for spastic foot drop should be accompanied by a therapy program consisting of both stretching and gait training.59 After placement of an intrathecal baclofen pump, many patients benefit from inpatient rehabilitation for safe and rapid titration of the pump in conjunction with rehabilitation to optimize functional retraining.
Pain
Pain in MS can be severe and difficult to manage. It is now recognized as a common symptom in patients with MS, with almost 50% of patients experiencing clinically significant pain at some time during the course of the disease, and as the presenting symptom in approximately 20%.135,168 In a recent survey of 442 persons with MS, we found the 3-month point prevalence of bothersome pain to be 44%.41 The average severity of pain was rated at 5.2 on a 10-point scale. Twenty-seven percent claimed severe pain, and 20% believed pain interfered with daily activities (see also Chapter 42).41
Neuropathic Pain
Trigeminal neuralgia (tic douloureux) is a well-known paroxysmal pain syndrome in MS and typically responds well to pharmacologic intervention with anticonvulsants such as carbamazepine (Tegretol) and gabapentin (Neurontin). Milder forms of this pain can be symptomatically ameliorated by using a local anesthetic, such as 20% benzocaine ointment (e.g., over-the-counter Oragel) on the trigger area. Tizanidine and misoprostol are among the other categories of medications for which there is some evidence of efficacy.43,161 Rhizotomy using glycerol, radiofrequency thermal treatment, and surgery are reserved for refractory cases that have failed to respond to other treatments.
Musculoskeletal Pain
MS patients with back pain are typically responsive to standard pain treatments such as spinal orthoses, nonsteroidal antiinflammatory drugs, analgesics, spinal blocks, physical therapy with stretching exercises, and trigger point injections. If the pain is complicated by upper motor neuron symptoms such as spasticity, spasmolytic agents should be also used. If neuropathic radicular pain is suspected, a trial of anticonvulsants is worthwhile (see also Chapter 40).
Pain Related to Upper Motor Neuron Loss
Other techniques for managing the pain associated with muscle spasm include cannabinoids, which appear to have some success in relieving symptoms of muscle pain or spasm and spasticity in some patients with MS.25 For those having both spasticity and severe pain, intrathecal baclofen with morphine or clonidine can be considered, although it is now not recommended very often because the additional analgesic medication can interfere with pump function.106
Fatigue and Sleep
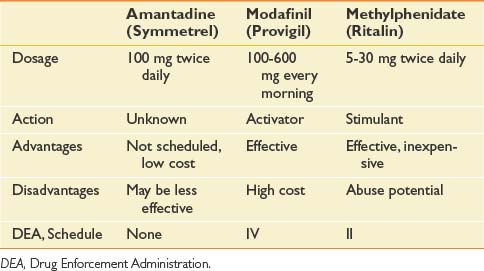
Fatigue was ignored in early descriptions of MS. In the mid-1980s, however, we identified fatigue as the most common symptom of MS and defined its characteristics.50,107 It is most pronounced in the afternoon and is experienced by 77% of patients.107 Treatment of fatigue has recently assumed a much more prominent place in the management of MS. Fatigue has been successfully treated with energy conservation techniques, as well as by a variety of medications, such as amantadine, modafinil, armodafinil, and methylphenidate (see prescribing information in Table 52-7).
Nonmedical treatments of fatigue include techniques to extract heat from the body or by surface cooling. The most effective method appears to be heat extraction using a vest with a circulating coolant flowing through a radiating system. More widely used, and much less expensive, are cooling vests. It has been demonstrated that lowering core body temperature using heat extraction can improve function in repetitive activities that produce fatigue.100,160
The exact mechanism for this reduction of fatigue with cooling is unclear.154 Only an incomplete understanding exists as to the cause of MS fatigue. It does not correlate with MRI lesion burden (although the rate of progression to that level of burden has not been studied).5 Some believe that proinflammatory cytokines contribute to the sense of tiredness.25,46 Recent research from our group has shown association with hormonal and adrenergic changes from cooling, especially an increase in noradrenaline (norepinephrine) (James Bowen, personal communication, 2003). But another factor could be the extensive cortical activity and resultant cognitive energy needed to do life’s tasks as required by the extensive neuroplasticity that develops as a compensatory mechanism as neural loss progresses in the central nervous system.12
Although stimulants such as methylphenidate (Ritalin) and awakeness enhancers such as modafinil (Provigil) and armodafinil (Nuvigil) are effective in reducing the lassitude experienced throughout the day, recent studies of sleep raise questions as to whether their use is in the patient’s long-term interest. If they prevent needed sleep, they might inhibit brain plasticity because slow-wave deep sleep provides the most efficient body milieu for neuroplasticity.132,176
It is striking how well many patients can function with extensive lesion loads and cortical atrophy. These patients typically have a great deal of fatigue, have poor memories (these are the ones who take notes during their medical visits), fight to stay awake much of the day, and cannot multitask. Given the large number of brain neurons destroyed in these patients, it is likely that they have benefitted from brain plasticity, which lays down new memory traces by activating new and undamaged neural pathways. Functional MRI during a simple motor task demonstrates significantly greater activation on the ipsilateral side of the brain of subjects with MS than in control subjects.45 This cortical “spread” indicates plasticity.
Plasticity is a type of new learning, and evidence indicates that such new learning requires sleep.176 During much of sleep, cortical neurons undergo slow oscillations in membrane potential (slow-wave activity on electroencephalogram).75 This appears necessary for the “offline” processing required for new synaptic plasticity.132 Slow-wave activity of sleep is required for efficient consolidation of the fresh memory traces necessary for brain plasticity.124 Studies of sleep deprivation in both experimental animals and human volunteers have demonstrated that new learning requires fairly immediate sleep to “encode” newly learned experiences.65,72,128,159 Both animal and human subjects deprived of adequate sleep after learning a new task do less well on subsequent testing, irrespective of whether they were sleep deprived at the time of testing.13,185
Sleep appears necessary to consolidate new memory traces (plasticity). Given that brain plasticity must be a relatively steady process in a neurodegenerative disease such as MS, this could explain the need for frequent sleep in such patients. Perhaps the MS patient’s desire for sleep is part of this biologic compensatory or repair process.
Patients with MS who need to be alert for vocational or safety reasons might benefit from wakefulness-enhancing drugs. For those without that need, using the traditional rehabilitation energy conservation strategies is recommended. The “4 Ps” of energy conservation are outlined below. Other medical conditions, including depression, might also contribute to fatigue and should be assessed and treated.148
Rehabilitation for Gait, Mobility, and Balance
Gait impairment in MS can be caused by contractures, weakness, spasticity, fatigue, proprioceptive loss, cerebellar or vestibular dysfunction, visual loss, or inability to multitask. Approximately three quarters of individuals with MS have some degree of ambulatory impairment.107,116
Compared with healthy people, patients with MS show decreased stride length; increased steps per minute (cadence); slower free speed walking rates; less rotation at the hips, knees, and ankles (stiffer gait); increased trunk flexion; and reduced vertical lift in center of gravity. Overall, they take short quick steps and lack full joint range of motion.56
The most common activities in which falls occurred were during transfers (e.g., bed, shower, or chair) and ambulation. The most common reasons for falls were trips or slips while walking, and being tired or fatigued. Other notable reasons included not paying attention, rushing or hurrying, not using walking aids, and feeling dizzy or lightheaded.124
In another cross-sectional study of ambulatory patients with MS, 54% reported at least one fall during the previous 2 months, and 32% were recurrent fallers. Impaired balance was the best predictor of falls, followed by use of an assistive device.15 Recommendations for mobility aids should focus on enhancing gait stability. Falls in the home might necessitate home modifications such as grab bars, equipment such as tub benches, and fall alert systems.
Orthoses play an important role in optimizing gait in MS. It is important to prescribe an ankle–foot orthosis (AFO) that will optimally improve a patient’s function with tolerable comfort, which increases the odds of a patient wearing it. Many “exotic” orthoses are available (e.g., functional electrical stimulation orthoses), but unless they improve function, are comfortable, and fit appropriately, patients will not wear them. An AFO for an MS patient must control foot drop and dynamic spasticity. The optimal orthosis for most patients is a custom-molded plastic AFO that can help control the ankle and the knee, reducing genu recurvatum and providing stability. Both articulated and nonarticulated AFOs are available, and the particular type ordered depends on which function is the more important to enhance: some ankle movement or full ankle stability (see also Chapter 15). Hip flexor weakness is a common pattern in MS, and recently an orthosis has been developed to help manage this. It is the hip flexion assist orthosis, which can be of use in improving limb advancement.173 Goal-specific rehabilitation is important for gait retraining in conjunction with the use of an appropriate assistive device and orthosis. Studies suggest that nonspecific exercise programs without some ambulation component are ineffective for enhancing gait.37,56,155
Tremor Management
Some degree of tremor is reported in nearly 30% of patients with MS. It is typically an action tremor and can be one of the most difficult symptoms to manage.2 In one MS population, the median time to tremor was 11 years from disease onset, with arms more often involved than head, trunk, or legs.
One study of inpatient rehabilitation for the treatment of MS-related ataxia and tremor involved eight half-hour sessions of occupational therapy (postural dynamics, adaptive equipment, damping, and weighting) and physical therapy for 8 working days.83 The intervention group significantly improved relative to a wait-listed control group on one ADL scale. Intervention subjects had significantly greater improvement in activity and fatigue.
Weighted wrist cuffs and weighted walkers are the most practical methods for dampening ataxic tremor.1,96 Isoniazid and propranolol can be tried but seldom yield impressive results. THC, a component of marijuana, showed a nonsignificant trend toward subjective benefit in MS tremor in one study.47 Deep brain stimulation, which has an established role in the treatment of parkinsonian tremor, is now being studied as a treatment for MS-related tremor. A review found that deep brain stimulation relieved tremor in more than 80% and improved daily functioning in more than 70% of carefully selected MS cases.194 Long-term hardware complications occur, however, in about one fourth of patients, and not all medical centers are proficient in implanting and managing these stimulators.103
Occupational Therapy for Multiple Sclerosis
People with MS tend to function in daily life below their physical capacities, and cognitive problems do not always account for this discrepancy.167 Occupational therapy can help people with MS meet their potential for independence. The principles of occupational therapy for MS have been described in various review articles.39,110 Occupational therapy has been used extensively in MS research, especially in inpatient trials, where it has been uniformly incorporated into the rehabilitation program (see Chapter 26).∗
A metaanalysis published in 2001 determined that occupational therapy-related treatments have a strong positive effect on MS symptomatology,4 but there have been few investigations of the effects of occupational therapy separate from a multidisciplinary team approach. Occupational therapists are frequently asked to teach the principles of “energy conservation” to MS individuals. These principles have been summarized as the “4 Ps” of planning (organizer, day planner, work planner, activity station in home), prioritizing (is this task necessary?), pacing (budget energy throughout the day and week), and positioning (proper body mechanics, workplace ergonomics).148 Three randomized controlled trials of patient education in energy conservation (weekly or biweekly 1-hour sessions for 6 to 8 weeks) found reductions in fatigue, which in one study was maintained at 6 weeks’ follow-up. Benefits in self-efficacy, quality of life, and social participation were also seen.4,125,126,183 A large randomized controlled trial of individually or group-based counseling in self-care strategies for 268 community-dwelling individuals with MS found some evidence of treatment effect on mental health and vitality, which was used as a proxy for fatigue.141 Although levels of functional independence did not improve, they were maintained in the intervention group but declined in the control group. In a progressive disease such as MS, stabilization and maintenance of function should be considered a positive result of treatment.
Cognition
Cognitive impairment, including memory deficits, represents a significant problem in persons with MS. Comprehensive neuropsychologic testing suggests that more than 40% of a broad spectrum of patients with MS show cognitive impairment of at least some degree of significance.251 In fact, it has recently been shown that measurable cognitive impairment is present in MS as mild as an EDSS of less than 3.5.158 In such patients, cognitive impairment correlates with illness duration.
It is easy for a physician to miss cognitive impairment in an MS patient during an office visit because verbal skills remain relatively intact. A patient with fluent speech and a good vocabulary usually gives the impression of being cognitively normal. An inquiry into the patient’s job status, however, might offer clues that problems exist. Competitive employment, especially in the current economic climate, requires a steady pace of multitasking activities. Cognitive problems in patients with MS might appear earliest in impaired job performance. Neuropsychologic testing of patients with MS, however, usually indicates memory to be the most significant area of impairment. Standard office or bedside memory testing might not elicit this deficit because the patient does not forget information immediately. When asked to recall three items after 5 minutes during a mental status examination, many patients with MS who have serious memory deficits can pass.191 With regard to MRI markers, cognitive impairment correlates with cortical atrophy more than with lesion load.3
Treatment of this problem consists of first identifying the specific deficit, then using pacing, memory books, environmental restructuring, and focusing on substituting for the deficit. Attention must also be paid to treating depression, fatigue, and heat intolerance; all of these can contribute to cognitive impairment. Classic data are conflicting as to whether interferon-β or glatiramer acetate produces cognitive improvement,186,199 but newer data show that only interferon-β1b and natalizumab might support cognitive function. No drugs have been approved by the FDA for improving memory in MS, but a combination of medications used to treat Alzheimer disease (donepezil [Aricept] + memantine [Namenda]) might be helpful.
Mood
Depression is a common problem in patients with MS. We conducted a large survey of 739 patients with MS in King County, Washington, and found the point prevalence of significant depressive symptoms (Center for Epidemiologic Studies Depression Scale 16) to be 41.8%.20 There appear to be two peaks of depression: early in the disease and late in advanced MS. Depression in MS appears to have both situational and organic causes, and MRI studies indicate that organic major depression correlates with T2-weighted lesion volume. Disease in the left medial inferior prefrontal cortex and atrophy of the left anterior temporal region are independent predictors for depression.44 In less-developed countries where treatment is less available, MS depression might be even more common.39
Treatments include antidepressant medications and behavioral intervention such as counseling. A small published study indicated that cognitive behavioral therapy was superior to selective serotonin reuptake inhibitor (SSRI) antidepressants.133 We have conducted a large randomized controlled trial with the SSRI paroxetine (Paxil), which was also negative in an intent-to-treat analysis because of high dropout in the treated group. Paroxetine was of benefit, however, for those completing the study. This suggests that SSRIs can be worth a try in MS patients with depression. Patients experiencing fatigue might be depressed, and patients with this symptom should be screened for depression. When SSRI antidepressants are tried, it is recommended that they be of the energizing type (e.g., paroxetine). They might also be useful in the subset of fatigued patients who also have depression.137 Cognitive impairment can also express itself as depression, and that should be assessed and managed as appropriate.
Speech and Swallowing
General speech performance registers in the normal range in the majority of individuals with MS. The most common speech problem in MS is controlling the volume (either too soft or too loud). Dysarthria is reported in 14% to 19%, and it is most often found in more neurologically impaired patients.33 The typical speech impairment in MS has been characterized as a mixed spastic cerebellar dysarthria, although flaccid dysarthrias are also encountered. Apraxia, anomia, and aphasia are much less common (see also Chapter 3).117
Dysphagia is a potentially life-threatening manifestation of MS (see also Chapter 27). A quantitative water test detected dysphagia in 43% of an MS cohort, almost half of whom had no related complaints.174 Using fiberoptic nasopharyngeal endoscopy, dysphagia was found in 48% of nonambulatory and 11% of ambulatory individuals.14 When videofluoroscopy has been used, most asymptomatic MS individuals have been found to have some degree of abnormality.192 The oral phase of swallowing is more frequently abnormal than the pharyngeal and esophageal phases. Fluids can be more problematic than solids, but for the majority of dysphagic individuals, both are abnormal.35
Heat Sensitivity and Thermoregulation
One of the classic characteristics of patients with MS is heat intolerance.68 It had been noted in the nineteenth century by Uhthoff181 that exercise might precipitate amblyopia in some patients with MS, which would resolve after the patient had stopped exercising. It was thought that the cause for this was increased body temperature induced by exercise. Recent studies, however, do not support this explanation. The temperature of the brain and spinal cord is kept too steady, even during extremes of exercise or cooling, for there to be sufficient change to produce the observed events.68 Many suspect cooling to produce a change in a hormone or circulating agent, producing a possible endocrine or adrenergic effect. Noradrenaline (norepinephrine) and thyroid hormone have been implicated (James Bowen, personal communication, 2003).
Heat intolerance is a very common symptom in MS, and heat extraction is an effective way to treat patients with severe manifestations.100 Although long-term heat extraction might not be a practical solution, avoiding a hot environment is an important component of a rehabilitation strategy. Cooling vests or heat extraction units can be prescribed for patients with heat intolerance. A physician’s “prescription” for environmental air conditioning is another practical and valid management tool for heat-sensitive patients with MS. Sipping a slurry of ice chips or a cool beverage might also be efficacious.
Bladder and Bowel Dysfunction
Bladder symptoms are common in MS and are present from disease onset in 35% of patients; more than 80% of patients with MS eventually have some voiding dysfunction.60 Urinary problems have a major psychologic impact and are among the most socially disabling manifestations of MS.16
Urgency and frequency are slightly more common than obstructive symptoms.10 An initial rehabilitation evaluation of an individual with MS should include a review of urologic symptoms, and clinicians should have a low threshold to check postvoid residual bladder volume. It is controversial whether a urodynamic study is routinely necessary.164 Urodynamic studies reveal a range of findings, with detrusor hyperreflexia in about two thirds of MS patients, the rest having detrusor–sphincter dyssynergia or detrusor hypocontractility.21 If detrusor–sphincter dyssynergia is present initially, it usually does not change with time. Seldom do patients with MS have complete spinal cord interruption, and they usually maintain at least some bladder sensation and residual voluntary micturition. Management involves determining the type of bladder–sphincter dysfunction and helping to maintain continence and avoid upper urinary tract complications. This can be done with such measures as timed voids, Kegel exercises, anticholinergic medication, an intermittent catheterization program, and continent diversions, as indicated (see also Chapter 28).
Bowel dysfunction is another common complaint in MS.74 Constipation is seen most often, with multiple causes that include disease-related reduced parasympathetic input, drug side effects (e.g., anticholinergics and analgesics), immobility, fluid restriction resulting from bladder frequency, and low-residue diet. Fecal incontinence can be caused by a loss of sensation of bowel movement, irregular bowel program, inadequate dietary fiber, or stool that is too soft or liquid. Helpful steps to establish a regular bowel elimination program include advising the patient to do one or more of the following techniques as listed in Box 52-2 (see also Chapter 29).
Improving Social and Vocational Integration
In our view, effective vocational performance in a competitive economy—especially while participating in family and community activities—can represent the greatest challenge for persons with MS. Such vocational performance can challenge the compensatory neuroplasticity and cognitive reserve occurring early in the course of the disease in the brains of persons with MS.12,88,170,171 Observation bears this out; persons with MS are unemployed disproportionate to their physical status and their educational and vocational histories.82 In assessing the neuropsychologic function in regard to employment, it is best to use an instrument specifically designed for this purpose.22 A patient with such impairment will benefit from directed neuropsychologic therapy and job modification, and might deserve serious consideration for disability retirement (see also Chapters 6 and 35).49
Another factor that should be taken into consideration is a patient’s age. As patients get older, a variety of other complications can develop. Many of these are treatable and should not necessarily be attributed to the MS.169 Patients with MS and their physicians sometimes erroneously assume that any symptom that occurs is related to MS, but MS patients are at the same risk as able-bodied individuals of developing common medical problems such as diabetes, heart disease, and arthritis. Perhaps the most likely additional medical problems that are missed in this population are those that produce neurologic symptoms. In our experience, the most common treatable comorbidities that are frequently missed in this population are carpal tunnel syndrome, cervical myelopathy, and lumbosacral spinal stenosis.
Patients with MS live with the disease for the rest of their lives. The better they can preserve and improve their general health, the better they will be.106 Because MS is a lifetime disease, lifetime management and rehabilitation must be used.97
Patients with MS live in a world with an uncertain future of progression, disability, and financial loss. In addition to organic brain dysfunction, the uncertainty of the disease can challenge a patient’s psychologic coping mechanisms.191 Maintaining adequate health insurance is another issue for patients with MS. Many carriers limit coverage for persons with preexisting disease.99 This tendency to limit care often works counter to the patient’s progressive need for more and more health care services as the disease progresses.94,95 All the factors—disease, uncertainty, progression, symptoms, disability, and vocational and social issues—mentioned in this chapter make MS arguably the most challenging disease to rehabilitate, but in many ways one of the rewarding.
1. Aisen M.L., Sevilla D., Fox N. Inpatient rehabilitation for multiple sclerosis. J Neurol Rehabil. 1996;10:43-46.
2. Alusi S.H., Worthington J., Glickman S., et al. A study of tremor in multiple sclerosis. Brain. 2001;124:720-730.
3. Amato M.P., Bartolozzi M.L., Zipoli V., et al. Neocortical volume decrease in relapsing-remitting MS patients with mild cognitive impairment. Neurology. 2004;63:89-93.
4. Baker N.A., Tickle-Degnen L. The effectiveness of physical, psychological, and functional interventions in treating clients with multiple sclerosis: a meta-analysis. Am J Occup Ther. 2001;55:324-331.
5. Bakshi R., Miletich R.S., Henschel K., et al. Fatigue in multiple sclerosis: cross-sectional correlation with brain MRI findings in 71 patients. Neurology. 1999;53:1151-1153.
6. Barcellos L.F., Oksenberg J.R., Green A.J., et al. Genetic basis for clinical expression in multiple sclerosis. Brain. 2002;125:150-158.
7. Barkhof F., Bruck W., De Groot C.J., et al. Remyelinated lesions in multiple sclerosis: magnetic resonance image appearance. Arch Neurol. 2003;60:1073-1081.
8. Barkhof F., Filippi M., Miller D.H., et al. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain. 1997;120(Pt 11):2059-2069.
9. Bitsch A., Kuhlmann T., Stadelmann C., et al. A longitudinal MRI study of histopathologically defined hypointense multiple sclerosis lesions. Ann Neurol. 2001;49:793-796.
10. Bonniaud V., Parratte B., Amarenco G., et al. Measuring quality of life in multiple sclerosis patients with urinary disorders using the Qualiveen questionnaire. Arch Phys Med Rehabil. 2004;85:1317-1323.
11. Brex P.A., Ciccarelli O., O’Riordan J.I., et al. A longitudinal study of abnormalities on MRI and disability from multiple sclerosis. N Engl J Med. 2002;346:158-164.
12. Burke M., Mahurin R., Bowen J.D. Differential brain compensation for cognitive versus motor processes in early relapsing-remitting multiple sclerosis subjects without clinical impairment. Mult Scler. 2006;12:S43-S44.
13. Cajochen C., Knoblauch V., Wirz-Justice A., et al. Circadian modulation of sequence learning under high and low sleep pressure conditions. Behav Brain Res. 2004;151:167-176.
14. Calcagno P., Ruoppolo G., Grasso M.G., et al. Dysphagia in multiple sclerosis: prevalence and prognostic factors. Acta Neurol Scand. 2002;105:40-43.
15. Cattaneo D., De Nuzzo C., Fascia T., et al. Risks of falls in subjects with multiple sclerosis. Arch Phys Med Rehabil. 2002;83:864-867.
16. Chancellor M.B., Blaivas J.G. Urological and sexual problems in multiple sclerosis. Clin Neurosci. 1994;2:189-195.
17. Chard D.T., Brex P.A., Ciccarelli O., et al. The longitudinal relation between brain lesion load and atrophy in multiple sclerosis: a 14 year follow up study. J Neurol Neurosurg Psychiatry. 2003;74:1551-1554.
18. Charil A., Yousry T.A., Rovaris M., et al. MRI and the diagnosis of multiple sclerosis: expanding the concept of “no better explanation.”. Lancet Neurol. 2006;5(10):841-852.
19. Chiappa K.H. Pattern-shift visual evoked potentials: methodology. In: Chiappa K.H., editor. Evoked potentials in clinical medicine. Philadelphia: Lippincott-Raven, 1997.
20. Chwastiak L., Ehde D.M., Gibbons L.E., et al. Depressive symptoms and severity of illness in multiple sclerosis: epidemiologic study of a large community sample. Am J Psychiatry. 2002;159:1862-1868.
21. Ciancio S.J., Mutchnik S.E., Rivera V.M., et al. Urodynamic pattern changes in multiple sclerosis. Urology. 2001;57:239-245.
22. Clemmons D.C., Fraser R.T., Rosenbaum G., et al. An abbreviated neuropsychological battery in multiple sclerosis vocational rehabilitation: findings and implications. Rehabil Psychol. 2004;49:100-105.
23. Coles A., Deans J., Compston A. Campath-1H treatment of multiple sclerosis: lessons from the bedside for the bench. Clin Neurol Neurosurg. 2004;106:270-274.
24. Comi G., Filippi F., Barkhof L., et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised studey. Lancet. 2001;357:1576-1582.
25. Comi G., Leocani L., Rossi P., et al. Physiopathology and treatment of fatigue in multiple sclerosis. J Neurol. 2001;248:174-179.
26. Comi C., Martinelli M., Rodegher L., et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe) study: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374(9700):1503-1511.
27. Confavreux C., Hutchinson M., Hours M.M., et al. Rate of pregnancy-related relapse in multiple sclerosis: Pregnancy in Multiple Sclerosis Group. N Engl J Med. 1998;339:285-291.
28. Consroe P., Musty R., Rein J., et al. The perceived effects of smoked cannabis on patients with multiple sclerosis. Eur Neurol. 1997;38:44-48.
29. Correale J., Ysrraelit M.C., Gaitan M.I. Immunomodulatory effects of Vitamin D in multiple sclerosis. Brain. 2009;132:1146-1160.
30. Craig J., Young C.A., Ennis M., et al. A randomised controlled trial comparing rehabilitation against standard therapy in multiple sclerosis patients receiving intravenous steroid treatment. J Neurol Neurosurg Psychiatry. 2003;74:1225-1230.
31. Cree B.A., Khan O., Bourdette D., et al. Clinical characteristics of African Americans vs Caucasian Americans with multiple sclerosis. Neurology. 2004;63:2039-2045.
32. Damek D.M., Shuster E.A. Pregnancy and multiple sclerosis. Mayo Clin Proc. 1997;72:977-989.
33. Darley F.L., Brown J.R., Goldstein N.P. Dysarthria in multiple sclerosis. J Speech Hear Res. 1972;15:229-245.
34. Davies G.R., Ramio-Torrenta L., Hadjiprocopis A., et al. Evidence for grey matter MTR abnormality in minimally disabled patients with early relapsing-remitting multiple sclerosis. J Neurol Neurosurg Psychiatry. 2004;75:998-1002.
35. De Pauw A., Dejaeger E., D’Hooghe B., et al. Dysphagia in multiple sclerosis. Clin Neurol Neurosurg. 2002;104:345-351.
36. Dean G., Kurtzke J.F. On the risk of multiple sclerosis according to age at immigration to South Africa. BMJ. 1971;3:725-729.
37. DeBolt L.S., McCubbin J.A. The effects of home-based resistance exercise on balance, power, and mobility in adults with multiple sclerosis. Arch Phys Med Rehabil. 2004;85:290-297.
38. Detels R., Visscher B.R., Haile R.W., et al. Multiple sclerosis and age at migration. Am J Epidemiol. 1978;108:386-393.
39. Diaz-Olavarrieta C., Cummings J.L., Velazquez J., et al. Neuropsychiatric manifestations of multiple sclerosis. J Neuropsychiatry Clin Neurosci. 1999;11:51-57.
40. Duquette P., Girard M., Knobler R.L. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. The IFNB Multiple Sclerosis Study Group. Neurology. 1993;43:655-661.
41. Ehde D.M., Gibbons L.E., Chwastiak L., et al. Chronic pain in a large community sample of persons with multiple sclerosis. Mult Scler. 2003;9:605-611.
42. Erickson R.P., Lie M.R., Wineinger M.A. Rehabilitation in multiple sclerosis. Mayo Clin Proc. 1989;64:818-828.
43. Evers S. Misoprostol in the treatment of trigeminal neuralgia associated with multiple sclerosis. Mayo Clin Proc. 2003;64:818-828.
44. Feinstein A., Roy P., Lobaugh N., et al. Structural brain abnormalities in multiple sclerosis patients with major depression. Neurology. 2004;62:586-590.
45. Filippi M., Rocca M.A., Colombo B., et al. Functional magnetic resonance imaging correlates of fatigue in multiple sclerosis. Neuroimage. 2002;15:559-567.
46. Flachenecker P., Bihler I., Weber F., et al. Cytokine mRNA expression in patients with multiple sclerosis and fatigue. Mult Scler. 2004;10:165-169.
47. Fox P., Bain P.G., Glickman S., et al. The effect of cannabis on tremor in patients with multiple sclerosis. Neurology. 2004;62:1105-1109.
48. Francabandera F.L., Holland N.J., Wiesel-Levison P., et al. Multiple sclerosis rehabilitation: inpatient vs. outpatient. Rehabil Nurs. 1988;13:251-253.
49. Fraser R.T., Johnson E.K., Clemmons D.C., et al. Vocational rehabilitation in multiple sclerosis (MS): a profile of clients seeking services. Work. 2003;21:69-76.
50. Freal J.E., Kraft G.H., Coryell J.K. Symptomatic fatigue in multiple sclerosis. Arch Phys Med Rehabil. 1984;65:135-138.
51. Freeman J.A., Langdon D.W., Hobart J.C., et al. Inpatient rehabilitation in multiple sclerosis: do the benefits carry over into the community? Neurology. 1999;52:50-56.
52. Freeman J.A., Langdon D.W., Hobart J.C., et al. The impact of inpatient rehabilitation on progressive multiple sclerosis. Ann Neurol. 1997;42:236-244.
53. Frohman E.M., Brannon K., Racke M.K., et al. Mycophenolate mofetil in multiple sclerosis. Clin Neuropharmacol. 2004;27:80-83.
54. Frohman E.M., Fujimoto J.G., Frohman T.C., et al. Optical coherence tomography: a window into the mechanisms of multiple sclerosis. Nat Clin Pract Neurol. 2008;4:664-675.
55. Gappmaier E., Spencer M.K., White A. Fifteen weeks of aerobic training improves fitness of multiple sclerosis individuals. Med Sci Sports Exerc. 1994;26:S29.
56. Gehlsen G., Beekman K., Assmann N., et al. Gait characteristics in multiple sclerosis: progressive changes and effects of exercise on parameters. Arch Phys Med Rehabil. 1986;67:536-539.
57. Geurts J.J., Barkhof F. Grey matter pathology in multiple sclerosis. Lancet Neurol. 2008;7:841-851.
58. Ghalie R.G., Edan G., Laurent M., et al. Cardiac adverse effects associated with mitoxantrone (Novantrone) therapy in patients with MS. Neurology. 2002;59:909-913.
59. Giovannelli M., Borriello G., Castri P., et al. Early physiotherapy after injection of botulinum toxin increases the beneficial effects on spasticity in patients with multiple sclerosis. Clin Rehabil. 2007;21:331-337.
60. Goldstein I., Siroky M.B., Sax D.S., et al. Neurourologic abnormalities in multiple sclerosis. J Urol. 1982;128:541-545.
61. Goodin D.S., Frohman E.M., Garmany G.P.Jr., et al. Disease modifying therapies in multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Neurology. 2002;58:169-178.
62. Goodkin D. Treatment of progressive forms of multiple sclerosis. In: Burks J., Johnson K.P., editors. Multiple sclerosis: diagnosis, medical management, and rehabilitation. New York: Demos Medical Publishing, 2000.
63. Goodkin D.E., Kinkel R.P., Weinstock-Guttman B., et al. A phase II study of i.v. methylprednisolone in secondary-progressive multiple sclerosis. Neurology. 1998;51:239-245.
64. Goodman A.D., Brown T.R., Krupp L.B., et al. Sustained-release oral fampridine in multiple sclerosis: a randomized double-blind controlled study. Lancet. 2009;373:732-738.
65. Graves L.A., Heller E.A., Pack A.I., et al. Sleep deprivation selectively impairs memory consolidation for contextual fear conditioning. Learn Mem. 2003;10:168-176.
66. Greenspun B., Stineman M., Agri R. Multiple sclerosis and rehabilitation outcome. Arch Phys Med Rehabil. 1987;68:434-437.
67. Gulick E.E. Influence of infant feeding method on postpartum relapse of mothers with MS. Int MS J Care. 2002:183-191.
68. Guthrie T.C., Nelson D.A. Influence of temperature changes on multiple sclerosis: critical review of mechanisms and research potential. J Neurol Sci. 1995;129:1-8.
69. Harris J.O., Frank J.A., Patronas N., et al. Serial gadolinium-enhanced magnetic resonance imaging scans in patients with early, relapsing-remitting multiple sclerosis: implications for clinical trials and natural history. Ann Neurol. 1991;29:548-555.
70. Hartung H.P., Gonsette R., Konig N., et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360:2018-2025.
71. Heide A.C., Kraft G.H., Slimp J.C., et al. Cerebral N-acetylaspartate is low in patients with multiple sclerosis and abnormal visual evoked potentials. AJNR Am J Neuroradiol. 1998;19:1047-1054.
72. Heuer H., Klein W. One night of total sleep deprivation impairs implicit learning in the serial reaction task, but not the behavioral expression of knowledge. Neuropsychology. 2003;17:507-516.
73. Hewson D.C., Phillips M.A., Simpson K.E., et al. Food intake in multiple sclerosis. Hum Nutr Appl Nutr. 1984;38:355-367.
74. Hinds J.P., Eidelman B.H., Wald A. Prevalence of bowel dysfunction in multiple sclerosis: a population survey. Gastroenterology. 1990;98:1538-1542.
75. Huber R., Ghilardi M.F., Massimini M., et al. Local sleep and learning. Nature. 2004;430:78-81.
76. Husted C., Pham L., Hekking A., et al. Improving quality of life for people with chronic conditions: the example of t’ai chi and multiple sclerosis. Altern Ther Health Med. 1999;5:70-74.
77. Inglese M., Ge Y., Filippi M., et al. Indirect evidence for early widespread gray matter involvement in relapsing-remitting multiple sclerosis. Neuroimage. 2004;21:1825-1829.
78. Jacobs L.D., Beck R.W., Simon J.H., et al. Intramuscular interferon beta-la therapy initiated during a first demyelinating event in multiple sclerosis. N Engl J Med. 2000;343:898-904.
79. Jacobs L.D., Cookfair D.L., Rudick R.A., et al. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. The Multiple Sclerosis Collaborative Research Group (MSCRG). Ann Neurol. 1996;39:285-294.
80. Jin Y.P., de Pedro-Cuesta J., Huang Y.H., et al. Predicting multiple sclerosis at optic neuritis onset. Mult Scler. 2003;9:135-141.
81. Johnson K.P., Brooks B.R., Cohen J.A., et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268-1276.
82. Johnson K.P., Klasner E., Amtmann D., et al. Medical, psychological, social, and programmatic barriers to employment for people with multiple sclerosis. J Rehabil. 2004;70:38-50.
83. Jones L., Lewis Y., Harrison J., et al. The effectiveness of occupational therapy and physiotherapy in multiple sclerosis patients with ataxia of the upper limb and trunk. Clin Rehabil. 1996;10:277-282.
84. Jonsson A., Dock J., Ravnborg M.H. Quality of life as a measure of rehabilitation outcome in patients with multiple sclerosis. Acta Neurol Scand. 1996;93:229-235.
85. Kalkers N.F., Ameziane N., Bot J.C., et al. Longitudinal brain volume measurement in multiple sclerosis: rate of brain atrophy is independent of the disease subtype. Arch Neurol. 2002;59:1572-1576.
86. Kappos L., Polman C.H., Freedman M.S., et al. Treatment with interferon beta-Ib delays conversion to clinically definite and McDonald MS in patients with clinically isolated syndromes. Neurology. 2006;67:1242-1249.
87. Kesselring J., Beer S. Rehabilitation in multiple sclerosis. ACNR. 2002;2:1572-1576.
88. Khurana L., Cline M., DiGiacomo A., et al. Cognitive demand affects functional mobility in multiple sclerosis. Mult Scler. 2006;12:S92.
89. Kleinschmidt-DeMasters B.K., Tyler K.L. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353:369-374.
90. Kocsis J.D., Waxman S.G. Demyelination: causes and mechanisms of clinical abnormality and functional recovery. In: Vilnken P.J., Bruyn G.W., Klawans H.L., editors. Demyelinating diseases handbook of clinical neurology. Amsterdam: Elsevier Science, 1985.
91. Koenigsberg R.A., Faro S.H., Hershey B.L., et al. Neuroimaging. In: Goetz C.G., editor. Textbook of clinical neurology. Philadelphia: Saunders, 2003.
92. Korbling M. Peripheral blood stem cells for allogeneic transplantation. In: Thomas E.D., Blume K.G., Forman S.J., editors. Hematopoietic cell transplantation. Oxford: Blackwell Science, 1999.
93. Kraft G.H. Foreword. In: Brown T., Kraft G.H., editors. Physical medicine and rehabilitation clinics of North America: multiple sclerosis: a paradigm shift. Philadelphia: Saunders, 2005.
94. Kraft G.H. Improving health care delivery for persons with multiple sclerosis. In: Kraft G.H., editor. Physical medicine and rehabilitation clinics of North America. Philadelphia: Saunders; 1998:703-715.
95. Kraft G.H. In defense of health care: preserving the capacity for excellence. Ann N Y Acad Sci. 1994;729:39-55. discussion 62–36
96. Kraft G.H. Movement disorders. In: Basmajian J.V., Kirby R.L., editors. Medical rehabilitation. Baltimore: Williams & Wilkins, 1984.
97. Kraft G.H. Multiple sclerosis: future directions in the care and the cure. Neurol Rehabil. 1989;3:61-64.
98. Kraft G.H. Rehabilitation still the only way to improve function in multiple sclerosis. Lancet. 1999;354:2016-2017.
99. Kraft G.H. The 24th Walter J. Zeiter Lecture. Variations on a theme: in defense of health care. Arch Phys Med Rehabil. 1992;73:211-219.
100. Kraft G.H., Alquist A., De Lateur B.J. Effect of microclimate cooling on physical function in multiple sclerosis. Mult Scler Clin Lab Res. 1996;2:114-115.
101. Kraft G.H., Alquist A.D., De Lateur B.J. Effect of resistive exercise on physical function in multiple sclerosis. Rehabil Res Dev Rep. 1995;33:328-329.
102. Kraft G.H., Alquist A., De Lateur B.J. Effect of resistive exercise on strength in multiple sclerosis. Rehabil Res Dev Rep. 1995;33:329-330.
103. Kraft G.H., Aminoff M.J., Baran E.M., et al. Somatosensory evoked potentials: clinical uses: AAEM Somatosensory Evoked Potentials Subcommittee. American Association of Electrodiagnostic Medicine. Muscle Nerve. 1998;21:252-258.
104. Kraft G.H., Bowen J., Cui J., et al. Clinical application of autologous stem cell transplantation in severe multiple sclerosis treated with high-dose immunosuppressive therapy. Neurology. 2002;58:A116.
105. Kraft G.H., Bowen J., Nash R. Functional stabilization of worsening multiple sclerosis patients treated with high-dose immunosuppression and stem cell rescue [abstract]. Arch Phys Med Rehabil. 2002., p 1682
106. Kraft G.H., Catanzaro M.L. Living with MS: a wellness approach. New York: Demos Medical Publishing; 2000.
107. Kraft G.H., Freal J.E., Coryell J.K. Disability, disease duration, and rehabilitation service needs in multiple sclerosis: patient perspectives. Arch Phys Med Rehabil. 1986;67:164-168.
108. Kraft G.H., Janczakowski J., Slimp J.C. Comparison of MRI and SEP in patients with MS and stroke. J Clin Neurophysiol. 1993;10:243.
109. Kraft G.H., Johnson K.L., Yorkston K., et al. Setting the agenda for multiple sclerosis rehabilitation research. Mult Scler. 2008;14:1292-1297.
110. Kraft G.H., McMullen K.A., Bamer A.M. The changing face of multiple sclerosis: differences in gender and symptoms over a 27-year period [abstract]. Int J MS Care. 2009;11:4.
111. Kraft G.H., Richards T.L., Heide A.C. Correlations of evoked potentials with MR imaging and MR spectroscopy in multiple sclerosis. In: Kraft G.H., editor. Physical medicine and rehabilitation clinics of North America. Philadelphia: Saunders, 1998.
112. Kriesel J.D., White A., Hayden F.G., et al. Multiple sclerosis attacks are associated with picornavirus infections. Mult Scler. 2004;10:145-148.
113. Kurtzke J.F. Epidemiology and etiology of multiple sclerosis. In: Brown T., Kraft G.H., editors. Physical medicine and rehabilitation clinics of North America: Multiple Sclerosis a paradigm shift. Philadelphia: Saunders, 2005.
114. Kurtzke J.F. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33:1444-1452.
115. Kurtzke J.F., Wallin M. Epidemiology. In: Burks J., Johnson K.P., editors. Multiple sclerosis: diagnosis, medical management, and rehabilitation. New York: Demos Medical Publishing, 2000.
116. LaBan M.M., Martin T., Pechur J., et al. Physical and occupational therapy in the treatment of patients with multiple sclerosis. In: Kraft G.H., editor. Physical medicine and rehabilitation clinics of North America. Philadelphia: Saunders, 1998.
117. Lacour A., De Seze J., Revenco E., et al. Acute aphasia in multiple sclerosis: a multicenter study of 22 patients. Neurology. 2004;62:974-977.
118. Langer-Gould A., Atlas S.W., Green A.J., et al. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353:375-381.
119. Lassmann H., Bruck W., Lucchinetti C. Heterogeneity of multiple sclerosis pathogenesis: implications for diagnosis and therapy. Trends Mol Med. 2001;7:115-121.
120. Levin L.I., Munger K.L., Rubertone M.V., et al. Temporal relationship between elevation of Epstein-Barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA. 2005;293:2496-2500.
121. Lewitt P.A., Garbern J.Y., Ferrante M.A., et al. Body fluid and tissue analysis. In: Goetz C.G., editor. Textbook of clinical neurology. Philadelphia: Saunders, 2003.
122. Lucchinetti C., Bruck W., Parisi J., et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47:707-717.
123. Lyons K.E., Wilkinson S.B., Overman J., et al. Surgical and hardware complications of subthalamic stimulation: a series of 160 procedures. Neurology. 2004;63:612-616.
124. Maquet P. The role of sleep in learning and memory. Science. 2001;294:1048-1052.
125. Mathiowetz V. Randomized clinical trial of an energy conservation course for persons with MS. Int J MS Care. 2004;6:56.
126. Mathiowetz V., Matuska K.M., Murphy M.E. Efficacy of an energy conservation course for persons with multiple sclerosis. Arch Phys Med Rehabil. 2001;82:449-456.
127. Matsuda P.N., Bamer A., Shumway-Cook A., et al. Falls in multiple sclerosis: prevalence and risk factors. Neurology. 2009;72:A408.
128. McDermott C.M., LaHoste G.J., Chen C., et al. Sleep deprivation causes behavioral, synaptic, and membrane excitability alterations in hippocampal neurons. J Neurosci. 2003;23:9687-9695.
129. McDonald W.I. The pathophysiology of multiple sclerosis. In: McDonald W.I., Silberberg D.H., editors. Multiple sclerosis. London: Butterworths, 1986.
130. McDonald W.I., Compston A., Edan G., et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50:121-127.
131. Miller D.H., Barkhof F., Frank J.A., et al. Measurement of atrophy in multiple sclerosis: pathological basis, methodological aspects and clinical relevance. Brain. 2002;125:1676-1695.
132. Miyamoto H., Hensch T.K. Reciprocal interaction of sleep and synaptic plasticity. Mol Interv. 2003;3:404-417.
133. Mohr D.C., Boudewyn A.C., Goodkin D.E., et al. Comparative outcomes for individual cognitive-behavior therapy, supportive-expressive group psychotherapy, and sertraline for the treatment of depression in multiple sclerosis. J Consult Clin Psychol. 2001;69:942-949.
134. Mostert S., Kesselring J. Effects of a short-term exercise training program on aerobic fitness, fatigue, health perception and activity level of subjects with multiple sclerosis. Mult Scler. 2002;8:161-168.
135. Moulin D.E., Foley K.M., Ebers G.C. Pain syndromes in multiple sclerosis. Neurology. 1988;38:1830-1834.
136. Murray T.J. The history of multiple sclerosis. In: Burks J., Johnson K.P., editors. Multiple sclerosis: diagnosis, medical management, and rehabilitation. New York: Demos Medical Publishing, 2000.
137. Ninan P.T., Hassman H.A., Glass S.J., et al. Adjunctive modafinil at initiation of treatment with a selective serotonin reuptake inhibitor enhances the degree and onset of therapeutic effects in patients with major depressive disorder and fatigue. J Clin Psychiatry. 2004;65:414-420.
138. Noseworthy J.H., Lucchinetti C., Rodriguez M., et al. Multiple sclerosis. N Engl J Med. 2000;343:938-952.
139. Noseworthy J.H., Vandervoort M.K., Wong C.J., et al. Interrater variability with the Expanded Disability Status Scale (EDSS) and Functional Systems (FS) in a multiple sclerosis clinical trial. The Canadian Cooperation MS Study Group. Neurology. 1990;40:971-975.
140. O’Connor P. Key issues in the diagnosis and treatment of multiple sclerosis: an overview. Neurology. 2002;59:S1-S33.
141. O’Hara L., Cadbury H., De S.L., et al. Evaluation of the effectiveness of professionally guided self-care for people with multiple sclerosis living in the community: a randomized controlled trial. Clin Rehabil. 2002;16:119-128.
142. O’Riordan J.I., Thompson A.J., Kingsley D.P., et al. The prognostic value of brain MRI in clinically isolated syndromes of the CNS: a 10-year follow-up. Brain. 1998;121(Pt 3):495-503.
143. Okuda D.T., Mowry E.M., Beheshtian A., et al. Incidental MRI anomalies suggestive of multiple sclerosis: the radiologically isolated syndrome. Neurology. 2009;72:800-805.
144. Olgiati R., Jacquet J., Di Prampero P.E. Energy cost of walking and exertional dyspnea in multiple sclerosis. Am Rev Respir Dis. 1986;134:1005-1010.
145. Polman C.H., Reingold S.C., Edan G., et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria. Ann Neurol. 2005;58:840-846.
146. Ponichtera-Mulcare JA, Mathews T, Barrett PJ: Change in aerobic fitness of individuals with multiple sclerosis during a 6-month training program, Sports Med Train Rehabil 1007; 7:265-272.
147. Poser C.M. Diseases of the myelin sheath. In: Merritt H.H., editor. A textbook of neurology. Philadelphia: Lea & Febiger, 1973.
148. Provance P.G. Symptom Awareness: Mobility Independence and Safety. MSAA Motivator. 2009., pp 56-61
149. Purves S.J., Low M.D., Galloway J., et al. A comparison of visual, brainstem auditory, and somatosensory evoked potentials in multiple sclerosis. Can J Neurol Sci. 1981;8:15-19.
150. Ramagopalan S.V., Maugeri N.J., Handunnetthi L., et al. Expression of the multiple sclerosis-associated MHC class II Allele HLA-DRB1∗1501 is regulated by vitamin D. PLoS Genet. 2009;5:e1000369.
151. Rao S.M., Leo G.J., Bernardin L., et al. Cognitive dysfunction in multiple sclerosis. I. Frequency, patterns, and prediction. Neurology. 1991;41:685-691.
152. Rasminsky M. Pathophysiology of demyelination. Ann N Y Acad Sci. 1984;436:68-85.
153. Rizzo M.A., Hadjimichael O.C., Preiningerova J., et al. Prevalence and treatment of spasticity reported by multiple sclerosis patients. Mult Scler. 2004;10:589-595.
154. Robinson L.R., Kraft G.H., Fitts S.S., et al. Body cooling may not improve somatosensory pathway function in multiple sclerosis. Am J Phys Med Rehabil. 1997;76:191-196.
155. Rodgers M.M., Mulcare J.A., King D.L., et al. Gait characteristics of individuals with multiple sclerosis before and after a 6-month aerobic training program. J Rehabil Res Dev. 1999;36:183-188.
156. Rovaris M., Filippi M. Contrast enhancement and the acute lesion in multiple sclerosis. Neuroimaging Clin N Am. 2000;10:705-716. viii-ix
157. Rovira A., Alonso J., Cucurella G., et al. Evolution of multiple sclerosis lesions on serial contrast-enhanced T1-weighted and magnetization-transfer MR images. AJNR Am J Neuroradiol. 1999;20:1939-1945.
158. Ruggieri R.M., Palermo R., Vitello G., et al. Cognitive impairment in patients suffering from relapsing-remitting multiple sclerosis with EDSS < or = 3.5. Acta Neurol Scand. 2003;108:323-326.
159. Ruskin D.N., Liu C., Dunn K.E., et al. Sleep deprivation impairs hippocampus-mediated contextual learning but not amygdala-mediated cued learning in rats. Eur J Neurosci. 2004;19:3121-3124.
160. Schwid S.R., Petrie M.D., Murray R., et al. A randomized controlled study of the acute and chronic effects of cooling therapy for MS. Neurology. 2003;60:1955-1960.
161. Semenchuk M.R., Sherman S. Effectiveness of tizanidine in neuropathic pain: an open-label study. J Pain. 2000;1:285-292.
162. Shi J., Zhao C.B., Vollmer T.L., et al. APOE epsilon 4 allele is associated with cognitive impairment in patients with multiple sclerosis. Neurology. 2008;70:185-190.
163. Simon J.H. MRI in Multiple Sclerosis. In: Kraft G.H., Brown T.R., editors. Physical medicine and rehabilitation clinics of North America. Philadelphia: Saunders, 2005.
164. Sirls L.T., Zimmern P.E., Leach G.E. Role of limited evaluation and aggressive medical management in multiple sclerosis: a review of 113 patients. J Urol. 1994;151:946-950.
165. Slimp J.C., Janczakowski J., Seed L.J., et al. Comparison of median and posterior tibial nerve somatosensory evoked potentials in ambulatory patients with definite multiple sclerosis. Am J Phys Med Rehabil. 1990;69:293-296.
166. Solari A., Filippini G., Gasco P., et al. Physical rehabilitation has a positive effect on disability in multiple sclerosis patients. Neurology. 1999;52:57-62.
167. Staples D., Lincoln N.B. Intellectual impairment in multiple sclerosis and its relation to functional abilities. Rheumatol Rehabil. 1979;18:153-160.
168. Stenager E., Knudsen L., Jensen K. Acute and chronic pain syndromes in multiple sclerosis. Acta Neurol Scand. 1991;84:197-200.
169. Stern M. Aging with multiple sclerosis. In: Kraft G.H., Christen A., editors. Physical medicine and rehabilitation clinics of North America: aging with a disability. Philadelphia: Saunders, 2004.
170. Sumowski J.F., Chiaravalloti N., Deluca J. Cognitive reserve protects against cognitive dysfunction in multiple sclerosis. J Clin Exp Neuropsychol. 2009:1-14.
171. Sumowski J.F., Chiaravalloti N., Wylie G., et al. Cognitive reserve moderates the negative effect of brain atrophy on cognitive efficiency in multiple sclerosis. J Int Neuropsychol Soc. 2009;15:606-612.
172. Sundstrom P., Juto P., Wadell G., et al. An altered immune response to Epstein-Barr virus in multiple sclerosis: a prospective study. Neurology. 2004;62:2277-2282.
173. Sutliff M.H., Naft J.M., Stough D.K., et al. Efficacy and safety of a hip flexion assist orthosis in ambulatory multiple sclerosis patients. Arch Phys Med Rehabil. 2008;89:1611-1617.
174. Thomas F.J., Wiles C.M. Dysphagia and nutritional status in multiple sclerosis. J Neurol. 1999;246:677-682.
175. Tintore M., Rovira A., Martinez M.J., et al. Isolated demyelinating syndromes: comparison of different MR imaging criteria to predict conversion to clinically definite multiple sclerosis. AJNR Am J Neuroradiol. 2000;21:702-706.
176. Tononi G., Cirelli C. Some considerations on sleep and neural plasticity. Arch Ital Biol. 2001;139:221-241.
177. Trapp B.D., Peterson J., Ransohoff R.M., et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278-285.
178. Tremlett H., Paty D., Devonshire V. Disability progression in multiple sclerosis is slower than previously reported. Neurology. 2006;66:172-177.
179. Tremlett H., Yinshan Z., Devonshire V. Natural history of secondary-progressive multiple sclerosis. Mult Scler. 2008;14:314-324.
180. Trojaborg W., Petersen E. Visual and somatosensory evoked cortical potentials in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1979;42:323-330.
181. Uhthoff W. Untersuchungen über die bei der multiplen Herdsklerose vorkommenden Augenströrungen. Arch Psychiat Nervenkr. 1890;55-116:303-410.
182. van Waesberghe J.H., van Walderveen M.A., Castelijns J.A., et al. Patterns of lesion development in multiple sclerosis: longitudinal observations with T1-weighted spin-echo and magnetization transfer MR. AJNR Am J Neuroradiol. 1998;19:675-683.
183. Vanage S.M., Gilbertson K.K., Mathiowetz V. Effects of an energy conservation course on fatigue impact for persons with progressive multiple sclerosis. Am J Occup Ther. 2003;57:315-323.
184. Vaney C., Blaurock H., Gattlen B., et al. Assessing mobility in multiple sclerosis using the Rivermead Mobility Index and gait speed. Clin Rehabil. 1996;1:216-226.
185. Wang J.H., van den Buuse M., Tian S.W., et al. Effect of paradoxical sleep deprivation and stress on passive avoidance behavior. Physiol Behav. 2003;79:591-596.
186. Weinstein A., Schwid S.R., Schiffer R.B., et al. Neuropsychologic status in multiple sclerosis after treatment with glatiramer. Arch Neurol. 1999;56:319-324.
187. Wekerle H., Hohlfeld R. Molecular mimicry in multiple sclerosis. N Engl J Med. 2003;349:185-186.
188. Werring D.J., Brassat D., Droogan A.G., et al. The pathogenesis of lesions and normal-appearing white matter changes in multiple sclerosis: a serial diffusion MRI study. Brain. 2000;123(Pt 8):1667-1676.
189. White A. Exercise and MS: challenges and opportunities. Mult Scler Q Rep. 2004;23:18-20.
190. White A., Gappmaier E., Mino L. Response to acute exercise before and after 15 weeks of training for multiple sclerosis individuals. Med Sci Sports Exerc. 1994;26:S29.
191. White D., Catanzaro M.L., Kraft G.H. An approach to the psychological aspects of multiple sclerosis: a coping guide for health care providers and families. J Neurol Rehabil. 1993;7:43-52.
192. Wiesner W., Wetzel S.G., Kappos L., et al. Swallowing abnormalities in multiple sclerosis: correlation between videofluoroscopy and subjective symptoms. Eur Radiol. 2002;12:789-792.
193. Wingerchuk D.M., Lennon V.A., Lucchinetti C.F., et al. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805-815.
194. Wishart H.A., Roberts D.W., Roth R.M., et al. Chronic deep brain stimulation for the treatment of tremor in multiple sclerosis: review and case reports. J Neurol Neurosurg Psychiatry. 2003;74:1392-1397.
195. Woods D.A. Aquatic exercise programs for individuals with multiple sclerosis. Clin Kinesiol. 1992. pp 14-20
196. Yorkston K., Johnson K.L., Kuehn C., et al. Age and gender issues related to MS: a survey study. Int J MS Care. 2002;4:94.
197. Yousry T.A., Major E.O., Ryschkewitsch C., et al. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. N Engl J Med. 2006;354:924-933.
198. Zivadinov R., Rudick R.A., De Masi R., et al. Effects of IV methylprednisolone on brain atrophy in relapsing-remitting MS. Neurology. 2001;57:1239-1247.
199. Zivadinov R., Zorzon M., Tommasi M.A., et al. A longitudinal study of quality of life and side effects in patients with multiple sclerosis treated with interferon beta-1a. J Neurol Sci. 2003;216:113-118.