[level-membership-for-critical-care-medicine-category]
21 Multiple Organ Dysfunction Syndrome
After reading this chapter, you should be able to:
• define the common terminology related to multiple organ dysfunction syndrome
• describe the related pathophysiology of multiple organ dysfunction syndrome
• identify the clinical manifestations of multiple organ dysfunction syndrome
• identify patients at risk of developing multiple organ dysfunction, including predictors of mortality
• initiate appropriate monitoring, care planning and evaluation strategies for the patient with multiple organ dysfunction in relation to the current evidence base
• discuss treatment strategies that promote homeostasis in the patient with multiple organ dysfunction syndrome
Introduction
The term multiple organ dysfunction syndrome (MODS) was established by an expert consensus conference in 1992 to describe a continuum of physiologic derangements and subsequent dynamic alterations in organ function that may occur during a critical illness.1,2 Previous terminologies in the literature were confusing. For example, multiple organ failure (MOF) was a term commonly used, but somewhat misleading as normal physiologic function can, in most cases, be restored in survivors of a critical illness who have temporary organ dysfunction.3,4 Although the syndrome affects many organs, it also affects physiological systems such as the haematological, immune and endocrine systems. MODS therefore more accurately describes altered organ function in a critically ill patient who requires medical and nursing interventions to achieve homeostasis.4
MODS is associated with widespread endothelial and parenchymal cell injury because of hypoxic hypoxia, direct cytotoxicity, apoptosis, immunosuppression and coagulopathy.4 Four clinical stages describe a patient with developing MODS:5
1. increasing volume requirements and mild respiratory alkalosis, accompanied by oliguria, hyperglycaemia and increased insulin requirements
2. tachypnoea, hypocapnia and hypoxaemia, with moderate liver dysfunction and possible haematological abnormalities
3. developing shock with azotaemia, acid–base disturbances and significant coagulation abnormalities
4. vasopressor dependence with oliguria or anuria, ischaemic colitis and lactic acidosis.
Cellular damage in various organs in patients who develop MODS begins with the onset of local injury that is then compounded by activation of the innate immune system. This includes a combination of pattern recognition, receptor activation and release of mediators at the microcellular level, leading to episodes of hypotension or hypoxaemia and secondary infections.4,5 The primary therapeutic goal for nursing and medical staff is prompt, definitive control of the source of infection or pro-inflammation6 and early recognition of preexisting factors that may lead to subsequent organ damage away from the initial site of injury. This preemptive therapy is instituted to maintain adequate tissue perfusion and prevent the onset of MODS. Recognition and response to early signs of clinical deterioration are therefore important to minimise further organ dysfunction.
This chapter initially describes the pathophysiology of inflammatory and infective conditions that may lead to multiple organ dysfunction. System responses and specific organ dysfunction are discussed, expanding on dialogue in previous chapters, particularly Chapters 19 and 20. Assessment of the severity of MODS and nursing considerations in the treatment of the MODS patient is presented.
Pathophysiology
The syndrome of multiple organ dysfunction is most closely related to an outcome of sepsis, which was described in Chapter 20. MODS is a state characterised by aberrant cellular responses involving multiple organ systems and sequential processes. The pathogenesis of MODS is complex, simultaneously involving every cell type, neuro-hormonal axis and organ system.7
In brief, hypoxic hypoxia results from altered metabolic regulation of tissue oxygen delivery which contributes to further organ dysfunction. Microcirculatory injury as a result of lytic enzymes, and vasoactive substances (nitric oxide, endothelial growth factor), is compounded by the inability of erythrocytes to navigate the septic microcirculation. Mitochondrial electron transport is affected by endotoxins in sepsis, nitric oxide and TNF-alpha, leading to disordered energy metabolism (see Figure 21.1). This causes cytopathic or histotoxic anoxia (the inability to use oxygen, even when available).8 This context of impaired oxygen utilisation rather than delivery7,8 results from diminished mitochondrial production of cellular energy (ATP), despite normal or even supranormal intra-cellular PO2 levels.9 Cytopathic hypoxia appears resistant to resuscitation measures, and this may ultimately worsen already-existing organ dysfunction. During sepsis or ischaemia, mitochondria respond by facilitating cell death rather than the restoration of homeostasis.7
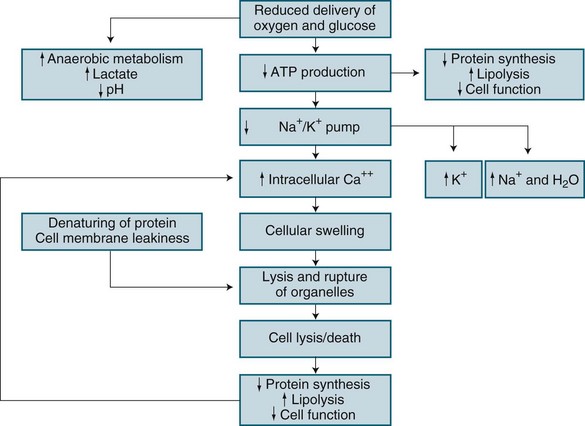
Apoptosis is normal physiological programmed cell death and is the main mechanism to eliminate dysfunctional cells.10 Apoptosis involves chromatin condensation, membrane blebbing, cell shrinkage and subsequent breakdown of cellular components into apoptic bodies. This normally orderly process is deranged in critical illness, leading to tissue or organ bed injury and MODS. Proinflammatory cytokines released in sepsis may delay apoptosis in activated macrophages and neutrophils, but in other tissues, such as gut endothelium, accelerated apoptosis occurs.8
In contrast, necrosis is a form of cell death characterised by cellular swelling and loss of membrane integrity as a result of hypoxia or trauma. Necrosis has been termed ‘cellular energy crisis’,10 and is unregulated resulting in loss of membrane sodium/potassium/ATP-ase pumps. This loss leads to cell swelling, rupture and spillage of intracellular contents into surrounding regions creating collateral damage.10 Necrosis therefore can involve significant amounts of tissue and organ bed damage. Apoptosis differs from necrosis in that it does not seem to involve the recruitment of inflammatory cells or mediators to complete its task. Activation of an enzyme cascade systematically cleaves proteins, including the cell’s nuclear DNA, with the end-result being death of the cell. This requires energy from mitrochondria and if not available necrosis of the cell occurs. Apoptosis and necrosis are processes that if is therefore important to understand in relation to future MODS research.
Increased concentrations of cell-free plasma DNA are present in various clinical conditions such as stroke, myocardial infarction and trauma, a likely result of accelerated cell death. Maximum plasma DNA concentrations correlated significantly with APACHE II scores and maximum SOFA scores (described later in this chapter), with cell-free plasma DNA concentrations higher in hospital non-survivors than in survivors. Using regression analysis, maximum plasma DNA was an independent predictor of hospital mortality.11
Other cellular organelles may also exhibit pathological reactions in MODS. In ischaemia/reperfusion, endoplasmic reticulum loses its ability to process proteins which induces the expression of heat shock proteins,7 affecting transcription of proteins necessary for organ specific functions. For example, liver cell metabolism, renal cell function or cardiac cell contractility may be affected.7 This has led to the controversial concept of a mode of hibernation of cells at the expense of survival of the whole organism.7
Cellular communication is also altered in MODS. Cells normally communicate through highly interactive bidirectional networks. The endothelium acts as a communication interface between cells, organs and systems and is involved in orchestration of systemic responses, including haemodynamic regulation, inflammation and coagulation; oxygen and nutrient delivery; oxidative stress and sensing of psychological stress and neuroendocrine alterations.7 In critical illness, endothelia release molecules that trigger the immune and neuroendocrine systems to produce a generalised inflammatory response.7 The combination of the pathophysiological processes involved with the development of MODS, compensatory mechanisms and the effect on target organs and systems is now discussed.
Systemic Response
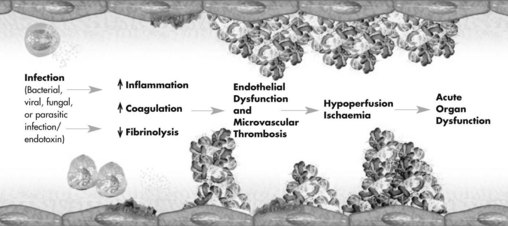
After an overwhelming incident such as trauma, sepsis or non-infectious inflammation, a complex range of interrelated reactions occurs that result in a cascade of responses. The complex host-response generated involves the inflammatory immune systems, hormonal activation and metabolic derangements, resulting in multiple organ system involvement.12,13 These host-responses are initially adaptive to maintain nutrient perfusion to the tissues, however eventually organ systems become dysfunctional and fail, and the body is no longer able to maintain homeostasis16 (see Figure 21.2).
Initially, proinflammatory mediators are released locally to fight foreign antigens and promote wound healing. Antiinflammatory mediators are also released to downregulate the initial response to the insult.14 If the local defence system is overwhelmed, inflammatory mediators appear in the systemic circulation and recruit additional leucocytes to the area of damage. A whole-body stress response ensues, further compounding the situation. If proinflammatory mediators and antiinflammatory response is imbalanced, the patient may develop systemic inflammatory response syndrome (SIRS) and subsequent immunological dissonance15 of organ dysfunction.2,15,16
Intracellular transcription factors, in particular nuclear factor kappa B (NFκB), are important in innate and adaptive immunity,17,18 as they regulate the transcription of genes involved in the inflammatory and acute stress response, leading to expression of TNFα, interleukins and tissue factor.18,19 NFκB therefore plays an important role in response pathways in critical states including hypoxia, ischaemia, haemorrhage, sepsis, shock and MODS.18,20,21
The inflammatory cascade activates a number of prostaglandins and leucotrienes that also have pro- and antiinflammatory effects. Thromboxane A2 plays a role in the acute phase, in part due to stimulation of platelet aggregation, leading to microvascular thrombosis and tissue injury;15 it may also play a role in pulmonary bronchoconstriction and myocardial depression.
The specific pathophysiological concepts of inflammation, oedema and infection are discussed below.
Inflammation
Inflammation is part of innate immunity, a generic response to injury, and is normally an excellent mechanism to localise injury and promote healing.22,23 The basis of this immune response is recognition and an immediate response to an invading pathogen without necessarily having previous exposure to the pathogen.24 Neutrophils, macrophages, natural killer cells, dendrites, coagulation and complement are the principal active components of the innate host response.23
The classic signs of inflammation are:
Nitric oxide and prostaglandins (e.g. prostacyclin), are the primary mediators of vasodilation and inflammation at the injury site.23 Injured endothelium produces molecules that attract leucocytes and facilitate movement to the tissues. White blood cells accumulate by margination (adhesion to endothelium during the early stages of inflammation) and neutrophils accumulate at the injury site, where rolling and adherence to binding molecules on the endothelium occurs with eventual movement across the endothelium into the tissues.23 Different blood components therefore escape the intravascular space and occupy the interstitial space where they play the main role in successive phases of the inflammatory response. The endothelium therefore plays a bidirectional mediating role between blood flow and the interstitial space where inflammation mainly takes place.25 Macrophages, neutrophils and monocytes are responsible for phagocytosis and the production of toxic free radicals to kill invading pathogens.24 The complement system, a collection of 30 proteins circulating in the blood, is also activated, with plasma and membrane proteins acting as adjuncts to inflammatory and immune processes.26 When activated by inflammation and microbial invasion, these processes facilitate lysis (cellular destruction) and phagocytosis (ingestion) of foreign material.23,26
Dysfunction of organ systems often persists after the initial inflammatory response diminishes; this is largely unexplained, although dysoxia (abnormal tissue oxygen metabolism and utilisation) has been implicated.22,27 Hypoxia induces release of IL-6, the main cytokine that initiates the acute phase response. After reperfusion of ischaemic tissues, tissue and neutrophil activation forms reactive oxygen species (e.g. hydrogen peroxide) as a byproduct. These strong oxidants damage other molecules and cell structures that they form,23 resulting in water and sodium infiltrate and cellular oedema.
Oedema
Oedema occurs as a consequence of alterations to tissue endothelium, with increased microvascular permeability (‘capillary leak’). As noted earlier, many mediators, including circulating cytokines, oxygen free-radicals and activated neutrophils, alter the structure of endothelial cells, enabling larger molecules (proteins, water) to cross into the extravascular space.23,28 This response mechanism improves supply of nutrient-rich fluid to the site of injury, but if this becomes systemic, fluid shifts can lead to hypovolaemia, third-spacing (interstitial oedema) or affect other organs (e.g. acute lung injury, ALI).23
Infection and Immune Responses
Infection exists when there is one of the following: positive culture, serology,29 presence of polymorphonuclear leucocytes in a normally sterile body fluid except blood, and clinical focus of infection such as perforated viscus or pneumonia. In sepsis, the most common sites of infection are the lungs (34–54%), intra-abdominal organs (15–28%) and urinary tract (5–10%).30,31 The incidence of bloodstream infections is 30–40%,29 although one-third of cases with septic shock have negative blood cultures; one reason suggested for this is antibiotic administration prior to sample collection.32 The type of infecting organism has also changed over time, with Gram-positive bacteria predominant, accounting for at least one-third of pathogens in septic shock; Gram-negative, fungal, viruses and parasitic organisms are also involved.29 The increasing incidence of resistant organisms, partially as a result of the indiscriminate use of antibiotics, is an ongoing concern.
The immune response to infection has both non-specific and specific actions, with inflammation and coagulation responses intricately linked in sepsis pathophysiology.23,24,33,34 Tissue injury and the production of inflammatory mediators lead to:
• coagulation via the expression of tissue factor and factor VIIa complex (tissue factor pathway; the primary cascade for initiation of coagulation; previously termed the ‘extrinsic’ pathway)28,33–35
• coagulation amplification via factors Xa and Va, leading to massive thrombin formation and fibrin clots (common coagulation pathway).28,33
Note that blood cell injury or platelet contact with endothelial collagen initiates the contact activation pathway (previously termed the ‘intrinsic’ coagulation pathway).33
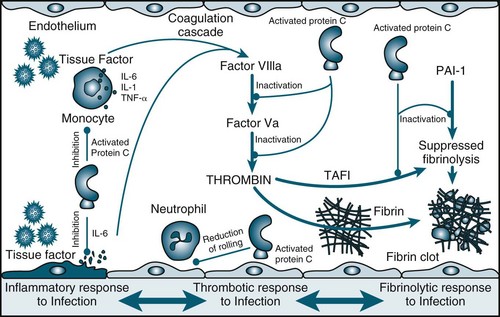
Procoagulation
Tissue factor is a procoagulant glycoprotein-signalling receptor,36 expressed when tissue is damaged or cytokines are released from macrophages or the endothelium (see Figure 21.3). Prothrombin is formed, leading to thrombin and fibrin generation from activated platelets. Resulting clots are stabilised by factor XIII and thrombin-activatable fibrinolysis inhibitor (TAFI).33,36 Fibrinolysis is a homeostatic process that dissolves clots via the plasminogen–tissue plasminogen activator (tPA)–plasmin pathway (involving antithrombin, activated protein C [APC] and tissue factor pathway inhibitor). APC:37
• reduces inflammation by decreasing TNF and NFκB production
• reduces thrombin production when activated via thrombin–thrombomodulin complexes (anticoagulant action)
• inhibits thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 (profibrinolytic action).33,34
APC is consumed in severe sepsis, and thrombomodulin is unable to activate protein C,33,34,37 promoting a proinflammatory, prothrombotic state.34
Endocrine Response
Physiological changes are triggered as a normal response to a stressor. In a critically ill patient, however, chronic activation of the stress response, including the hypothalamic–pituitary–adrenal axis and the sympathetic–adrenal–medullary axis, results in ongoing production of glucocorticoid hormones and catecholamines.17 This response interferes with the regulation of cytokine-producing immune cells, leading to immune dysfunction. Other compensatory mechanisms are instigated in an attempt to maintain supply and perfusion to organs.15
These homeostatic mechanisms are activated through positive or negative feedback systems to counteract stress. When stress is extreme or prolonged, these normal homeostatic mechanisms may be insufficient and a patient may respond through a sequence of physiological changes called the stress response. The stress response occurs in three stages: the alarm reaction, the resistance reaction and exhaustion (see Table 21.1).38
| Response stage | Neurohormonal response | Actions |
|---|---|---|
| Alarm reaction | Hypothalamus |
The alarm reaction (flight-or-fight response)38 is initiated when stress is detected, increasing the amount of glucose and oxygen available to the brain, skeletal muscle and heart. Two-thirds of total blood volume is also redistributed to support central circulation.38 A rise in glucose production and the breakdown of glycogen in skeletal muscle increases circulating glucose levels, providing an immediate energy source. The long-lasting second stage is a resistance reaction, involving hypothalamic, pituitary and adrenal hormone release.38 Response exhaustion occurs when these physiological changes can no longer maintain homeostasis.
Compensatory Mechanisms
Internal equilibrium (homeostasis) is maintained by the nervous and endocrine systems, and these work symbiotically with other compensatory mechanisms, such as endothelial cells, to maintain cellular perfusion. The nervous system responds rapidly to maintain homeostasis by sending impulses to organs to activate neurohormonal responses (see Chapters 16 and 20). Endothelins (ET-1, ET-2, ET-3) are potent vasoconstrictors produced by endothelial cells that regulate arterial pressure.20 The endocrine system works in a slow and sustained manner by secreting hormones, which travel via the blood to end-organs.
An initial acute-adaptive response is activated when an insult or stress occurs. For example, the body senses a disruption of blood flow through baroreceptor and chemoreceptor reflex actions: baroreceptors located in the carotid sinus detect changes in arterial pressure;13 chemoreceptors co-located with the baroreceptors detect O2, CO2 and H+ concentration. When alterations are sensed, the cardiovascular centre in the brain adjusts autonomic outflow accordingly.38 In a patient with decreased tissue perfusion, there is increased peripheral vasoconstriction, contractility and heart rate. Blood flow is shunted to the vital organs (brain, heart, lungs), and away from less vital areas (e.g. gastrointestinal and reproductive organs).39 Important hormonal regulators of blood flow are also activated from decreased blood flow to the kidneys, including adrenocorticotrophic hormone (ACTH), and the renin–angiotensin–aldosterone system (see Chapter 18). Adrenal medullary hormones, adrenaline and noradrenaline, vasopressin (antidiuretic hormone) and atrial natriuretic peptide also regulate blood flow to maintain adequate circulation and tissue oxygenation.13,38,39
Arterial pressure is a major determinant of tissue perfusion as it forces blood through the regional vasculature.20 Hypotension (systolic blood pressure <90 mmHg or mean arterial pressure [MAP] <70 mmHg) results from either low systemic vascular resistance or a low cardiac output.20 Glomerular filtration falls, leading to reduced urine output; low cerebral blood flow results in an altered level of consciousness; and other manifestations reflect low-flow states in other organ systems. To maintain oxygen supply, respirations and heart rate increase to meet organ oxygenation demands.40 Organ dysfunction ensues if balance is not sufficiently restabilised (see Table 21.2).
| Organ system | Clinical parameters |
|---|---|
| Cardiovascular | Patient requires vasopressor support (systolic BP <90 mmHg) or MAP <70 mmHg for 1 hour despite fluid bolus |
| Respiratory | Patient requires mechanical ventilation: P/F ratio <250, PEEP >7.5 cmH2O |
| Renal | Low urine output <0.5 mL/kg/h; raised creatinine >50% from baseline or requiring acute dialysis |
| Haematological | Low platelet count (<1 000 000/mm3) or APTT/PTT > upper limit of normal |
| Metabolic | Low pH with increased lactate (pH <7.3 and plasma lactate > upper limit of normal) |
| Hepatic | Liver enzymes >2 × upper limit of normal |
| CNS | Altered level of consciousness/reduced Glasgow Coma Scale score |
| Gastrointestinal | Translocation of bacteria, possible elevated pancreatic enzymes and cholecystitis |
Organ Dysfunction
Organ dysfunction is a common clinical presentation in ICU. Patients with dysfunction in the respiratory, cardiovascular, hepatic or metabolic systems were 50% more likely to require ICU treatment and had a higher mortality than patients not requiring intensive care.41 Timely identification of organ dysfunction is therefore critical, as early intervention reduces damage and improves recovery in organ systems. As each organ fails, the average risk of death rises by 11–23%, with up to 75% of patients in sepsis clinical trials having at least two failing organs.42 The organ system that most commonly fails is the pulmonary system, followed by the cardiovascular, renal and haematological systems.43 Organ and systems dysfunction are a result of hypoperfusion, inflammation, cellular dysfunction and oedema. Dysfunction of the cardiovascular (Chapters 10 and 12), respiratory (Chapters 14 and 15), renal (Chapter 18), and hepatic and gastrointestinal systems (Chapter 19) have been previously addressed. This next section addresses the haematological, endocrine and metabolic systems. Neurological dysfunction is also common in the patient with MODS and complements previous discussions in Chapter 17.
Haematological Dysfunction
Systemic inflammatory response syndrome (SIRS) and disseminated intravascular coagulation (DIC) have pivotal and synergistic roles in the development of MODS.44 The coagulopathy present in MODS results from deficiencies of coagulation system proteins (e.g. protein C, antithrombin 3 and tissue factor inhibitors).8 Inflammatory mediators initiate direct injury to the vascular endothelium, releasing tissue factor, triggering the extrinsic coagulation cascade and accelerating thrombin production.8 Coagulation factors are activated as a result of endothelial damage with binding of factor XII to the subendothelial surface, activation of factors XI, XII, X, VIII, calcium and phospholipid.8 The final pathway is production of thrombin which converts soluble fibrinogen to fibrin. Fibrin and aggregated platelets form intravascular clots.
Inflammatory cytokines also initiate coagulation though activation of tissue factor (TF), a principal activator of coagulation. Endotoxins increase the activity of inhibitors of clot breakdown (fibrinolysis). Levels of protein C and endogenous activated protein C are decreased in sepsis; this inhibits coagulation cofactors Va and VIIa and acts as an antithrombotic in the microvasculature.8
Microvascular thrombosis that leads to MODS results from two major syndromes: thrombotic microangiopathy (TMA) and disseminated intravascular coagulation (DIC). TMA is characterised by formation of microvascular platelet aggregates and occasionally fibrin formation. Typically there is history of injury to the microvascular endothelium (e.g. thrombotic thrombocytopenic purpura, haemolytic uremic syndrome, haemolytic anaemia, elevated liver enzymes and low platelet syndromes of pregnancy or antiphospholipid antibody syndrome).44 TMA usually presents with normal coagulation profiles such as prothrombin times and partial thromboplastin time.44
Disseminated intravascular coagulation results from widespread activation of tissue factor-dependent coagulation, insufficient control of coagulation and plasminogen-mediated attenuation of fibrinolysis.44 This leads to formation of fibrin clots, consumption of platelets and coagulation proteins, occlusion of the microvasculature, and resultant reductions in cellular tissue oxygen delivery.44 DIC is most commonly a result of trauma or sepsis and is an exaggerated response to normal coagulation aimed at limiting infection, exsanguination and promoting wound healing.44
Thrombocytopenia (a platelet count of <80,000/mm3 or a decrease of ≥50% over the preceding three days) signifies haematological failure,45 with leucocytopenia/cytosis, markers of coagulation and DIC also present.46 Treatment is supportive and aimed at removing the triggering insults. Clinical biomarkers include a simultaneous rise in prothrombin time, APTT and thrombocytopenia.34 A patient may exhibit bleeding from puncture sites (e.g. invasive vascular access), mucous membranes including bowel, or upper gastrointestinal tract. Bruising or other subcutaneous petechiae may be evident. The skin should be protected from trauma.
Primary therapy is directed at the cause of the insult, with SIRS, ischaemia, uraemia, hepatotoxins and sources of infection, injury or necrosis managed concurrently. Aggressive resuscitation includes crystalloid or colloid administration, replacement of blood components and clotting factors using packed cells, platelets, cryoprecipitate and fresh frozen plasma. Endpoints for haemoglobin, platelets and coagulation levels have not been agreed upon and replacement is therefore individualised.47
The role of heparin or fractionated heparin is controversial in the presence of sepsis, particularly in those with overt thromboembolism or extensive fibrin deposition, such as in purpura fulminans or ischaemia in the extremities.47,48 Administration of APC in its role as inhibitor of the coagulation cascade is controversial. A Cochrane review of four studies involving 4911 participants (4434 adults and 477 paediatric patients) identified no reduction in risk of death (28-day mortality) in adult participants with severe sepsis, but was associated with a higher risk of bleeding. Effectiveness was not associated with the degree of severity of sepsis.49 Studies continue into this area of clinical practice.
Endocrine Dysfunctions
Numerous endocrine derangements are noted in critically ill patients, including abnormalities in thyroid, adrenocortical, pancreas, growth and sex hormones. A high thyrotropin (TSH) level is a significant independent predictor of non-survival in critically ill patients,50 while subclinical hypothyroidism has significant negative effects on cardiac function and haemodynamic instability.50,58
Adrenal Insufficiency
Adrenal insufficiency is present in approximately 30% of patients with sepsis or septic shock,32,43,51,52 and is associated with chronic adrenal insufficiency and recent physiological stress, or in new-onset adrenal insufficiency.50 This adrenal insufficiency can be caused by sepsis, surgery, bleeding and head trauma. Adrenal insufficiency as a cause of shock should be considered in any patient with hypotension with no signs of infection, cardiovascular disease or hypovolaemia. Incidence ranges from 0–95%,53 partly because there is no standard definition for adrenal insufficiency.
Eosinophilia (>3% of total white blood count) is reported as a marker of adrenal insufficiency. Methods to diagnose acute adrenal insufficiency include: (1) a single random cortisol level check, or a change in cortisol level after endogenous adrenocorticotrophic hormone (ACTH) is administered; or (2) a short corticotrophin stimulation test with administration of high-dose ACTH. A change in cortisol level (≤9 µg/dL) is considered relative adrenal insufficiency. It is however argued that patients with severe sepsis may have appropriate cortisol levels, but not the reserve function to respond to the stimulation test.31
Steroid Therapy
As septic shock is a major complication of infectious processes, the relationship between the immune, coagulation and neuroendocrine systems has been explored.54 The role of corticosteroids in the treatment of septic shock has led to a number of trials that suggested some survival benefit for low-dose corticosteroid therapy. More research is required, however, because of conflicting findings from individual studies.
Therapy with corticosteroids at a physiological dose, rather than a high dose, followed observations that patients with septic shock who had a reduced response to corticotropin were more likely to have increased mortality, and that pressor response to noradrenaline may be improved by the administration of hydrocortisone.55 A trial exploring steroid use in sepsis demonstrated reduced vasopressor requirements and early lower mortality, but no difference in 1-year survival.52 A multicentre trial demonstrated that hydrocortisone administration did not improve survival in patients with septic shock. Shock reversal was shorter in patients who received hydrocortisone, but there were more episodes of infection including new sepsis and septic shock.56 Although the largest trial of corticosteroids in patients with septic shock, the study was not adequately powered to detect a clinically important treatment and so findings are to be interpreted with caution.55 It is therefore appropriate to reserve corticosteroids for patients with septic shock whose blood pressure is poorly responsive to fluid resuscitation and high dose vasopressor therapy.57 Long-term treatment with corticosteroids may result in an inadequate response of the adrenal axis to subsequent stress such as infection, surgery or trauma, with resulting onset or worsening of shock. Other studies using corticosteroids for adrenal insufficiency in critically ill patients demonstrated lower mortality.e.g. 8
Corticosteroid administration is associated with hyperglycaemia and may affect patient outcomes, necessitating insulin therapy to normalise blood glucose levels. A recent multicentre trial (Corticosteroids and Intensive Insulin Therapy for Septic Shock [COIITS]),54 demonstrated that intensive insulin therapy did not improve in-hospital mortality for patients treated with hydrocortisone and oral fludrocortisones for septic shock.
Glycaemia Control
Hyperglycaemia is common in critically ill patients as a result of stress-induced insulin resistance and accelerated glucose production, and excessive circulating levels of glucagon, growth hormone, sympathomimetics and glucocorticoids (see Chapter 19). An increased caloric intake from parenteral or enteral nutrition will also increase glucose levels. Hyperglycaemia has undesirable effects such as fluid imbalance, immune dysfunction, promoting inflammation, abnormalities in granulocyte adherence, chemotaxis, phagocytosis and intracellular killing.31 Resulting associations between hyperglycaemia and adverse clinical outcomes have been reported in many observational studies. Potential benefits of exogenous insulin administration include normalising immune functional, improving oxygen delivery to ischaemic areas of myocardium, tissue repair and preventing transfusion, dialysis and critical illness polyneuropathy.31 Intensive insulin therapy has also been suggested to improve morbidity, reducing the risk of sepsis, excessive inflammation and multiple organ failure, transfusion requirements and dependence on mechanical ventilation.59
The Normoglycaemia in Intensive Care Evaluation and Survival Using Glucose Algorithm Regulation Study (NICE-SUGAR) examined tight glycaemic control with insulin during critical illness.60 Maintaining blood glucose at less than 10 mmol/L resulted in 10% reduction in 90-day mortality compared to a tighter glycaemic control target (4.5–6.0 mmol/L).60 Lower target blood sugar levels are therefore not recommended for managing glycaemia in critical ill patients.
Hypocalcaemia
Hypocalcaemia is common in patients with SIRS,47 and affects myocardial contractility and neuromuscular functions. The link between neuromuscular changes such as polyneuropathy or polymyopathy and critical illness has not been established beyond early investigations into corticosteroid use, neuromuscular blocking medication administration and prolonged mechanical ventilation.61
Neurological Dysfunction
Recent evidence has highlighted that multiple organ dysfunction can result from severe traumatic brain injury (TBI) or subarachnoid haemorrhage (SAH) (see Chapter 17). Cardiovascular and respiratory dysfunction contribute to mortality in approximately two-thirds of all deaths following severe TBI.62 In non-traumatic SAH the incidence and importance of life-threatening conditions from non-neurological physiology has been identified, including lethal arrhythmias, myocardial ischaemia and dysfunction and neurogenic pulmonary oedema.62 The cause of cardiovascular and respiratory organ dysfunction following these acute severe neurological events is associated with dysfunction of the sympathetic nervous system. Beta blockers may modulate the sympathetic storm resulting from severe neurological injury.62
Critically ill patients may develop a syndrome of neuromuscular dysfunction characterised by generalised muscle weakness and an inability to wean successfully from mechanical ventilation. Critical illness neuromyopathy syndromes (CINM) or ICU-Acquired Weakness (ICU-AW) has been associated with risk factors including hypergylcaemia, SIRS, sepsis, MODS, renal replacement therapy, glucocorticoids, neuromuscular blocking agents and catecholamine administration.63 The risk of CINM/ ICU-AW is nearly 50% in patients with sepsis, MODS or protracted ventilation,63 with short-term survival uncertain. Glycaemic control may be a potential strategy for decreasing CINM/ICU-AW risk.63
Survivors of sepsis-induced multiple organ dysfunction may also suffer long-term cognitive impairment, including alterations in memory, attention, concentration and/or global loss of cognitive function.64 The participation of the brain during sepsis is poorly understood; septic encephalopathy is the more common neurological dysfunction, accounting for up to 70% of brain dysfunctions.64 Chapter 4 described the physical, psychological and cognitive sequelae for survivors of a critical illness during their recovery.
Multiorgan Dysfunction
MODS contributes to significant morbidity and use of intensive care resources worldwide. Patients with MODS have an increased ICU length of stay when compared to high-risk patients without multiple organ involvement.65 The epidemiology of MODS is changing however, with studies in post-injury organ failure indicating a reduction in incidence, disease severity, duration and mortality.65,66 Mortality ten years ago was estimated at 40–60%, rising with subsequent organ dysfunction.42,67,68 More recent data in post-injury MODS indicates a reduction in mortality rates to 14–27%.65,66 This decrease in mortality is occurring despite increasing patient acuity and may reflect improvements in the delivery of critical care.6
Scoring Systems
Organ dysfunction can be a consequence of a primary insult or a secondary insult due to circulating mediators (e.g. the patient with ALI from pneumonia that also has renal dysfunction or failure as a consequence). This is sometimes quantified by scoring systems, traditionally used for predicting mortality but increasingly being explored as clinical management tools.69–71 These systems are continually being tested and modified, to assess organ dysfunction severity and prognosis in an effort to identify patients who will benefit most from timely clinical intervention.71 Scoring systems such as APACHE (acute physiology and chronic health evaluation), SAPS (simplified acute physiology score) and MPM (mortality probability models) account for information relating to a 24-hour cycle of patient data (commonly in the first 24 hours of admission), but do not account for the dynamic nature of many of the factors that affect clinical outcomes.
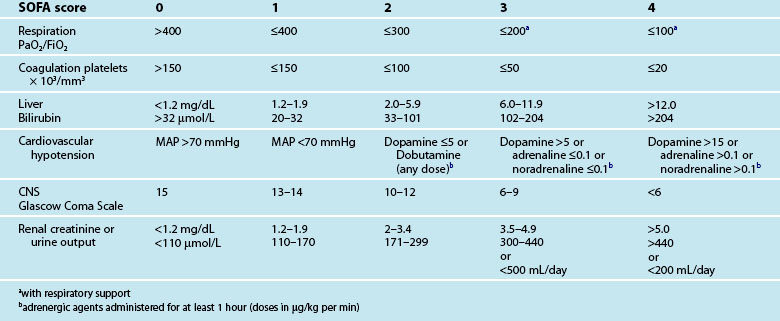
Specific instruments designed to assess organ dysfunction or failure include the sepsis-related/sequential organ failure assessment (SOFA) score, the multiple organ dysfunction score and the logistic organ dysfunction system.70,72 Traditionally SOFA uses the worst values for six commonly measured clinical parameters within a 24-hour period: PaO2/FiO2 (P/F ratio), an index that may be used to characterise acute respiratory distress syndrome;73 platelet count, bilirubin level, blood pressure, Glasgow Coma Scale score, and urine output or creatinine concentration. As the number of dysfunctional organ systems increases, there is a rise in mortality as measured by SOFA scores (see Table 21.3). Many variations of SOFA-based models have emerged in the literature, such as single SOFA scores calculated at admission or at a set time after admission, sequential SOFA scores (mean SOFA score), dynamic SOFA scores and scores of separate SOFA components.69,70 SOFA scores at admission are comparable with severity of illness models such as APACHE or SAPS for predicting mortality.70 SOFA scoring has the advantage of ease of use, as the clinical and laboratory data required are those that are routinely available. As such, the use of dynamic SOFA scoring as a means of monitoring patient response to treatments is being explored.69,71
Other Factors
Biomarkers such as lactate and strong ion gap (SIG) are also being studied as indicators of occult hypoperfusion and severity of organ dysfunction. Blood lactate levels are associated with SOFA scores, particularly in the early stage of ICU admission, supporting early resuscitation as a management strategy to prevent organ dysfunction. Serial lactate scores may therefore be appropriate to guiding optimal oxygen delivery in early resuscitation, with hyperlactataemia a sign of impending organ dysfunction. Prospective, well-controlled studies are however needed to confirm the role of lactate and SIG in MODS management.74–76
Variations in the human DNA sequences can affect the way a person responds to disease. Researchers have studied the gene code for PAI-1 which is a key element in the inhibition of fibrinolysis and is active during acute inflammation77 (the gene most studied is found at the 4G/5G insertion/deletion loci), finding that different aspects bind as either a repressor (5G) or activator (4G) protein. For example, the 4G allele (position on the gene) of the 4G/5G gene sequence variation has been associated with increased susceptibility to community acquired pneumonia and increased mortality in cases of severe pneumonia. It has also been reported to affect the risk of developing severe outcomes and higher mortality in meningococcal sepsis and trauma.77 Among critically ill patients with severe sepsis due to pneumonia, carriers of the PAI-1 4G/5G genotypes have higher risk for MODS and septic shock.77 In future, identification of genetic factors may assist selection of appropriate therapy for the patient at risk.
Nursing Practice
Improvement in patient survival with MODS is thought to be due to improved shock management, awareness of secondary insults, improved critical care management and a better understanding of the risk factors associated with MODS. Current prevention and management strategies therefore focus on efficient shock resuscitation, timely treatment of infection, exclusion of secondary inflammatory insults and organ support.65
Effective Shock Resuscitation
A number of interventions have been recommended to reduce mortality for patients with MODS due to sepsis. The surviving sepsis guidelines (SSG) are based on clinical evidence graded according to the quality of evidence available,76 although there is controversy and dissent regarding some recommendations, particularly Early Goal Directed Therapy (EGDT) (see Table 21.4) (see Chapter 20 for further discussion).
| Item | Target |
|---|---|
| CVP | 8–12 mmHg 12–15 mmHg in mechanically ventilated patient or patient with decreased ventricular compliance |
| MAP | ≥65 mmHg |
| Urine output | ≥0.5 mL/kg/hr |
| ScvO2/ SvO2 | ≥70%/≥65% |
The multicentre, prospective, observational ARISE study (Australasian resuscitation in sepsis evaluation) assessed the resuscitation practices and outcomes in patients presenting to EDs with sepsis with hypoperfusion or septic shock. Overall in-hospital mortality of 23% was comparable to inhospital mortality reported in studies of early EGDT. The study confirmed that protocolised ScvO2-directed EGDT is not routinely practised in Australia or New Zealand, and recommended that EGDT not be adopted in Australia and New Zealand without further multicentre randomised controlled trials.78 While some evidence of the benefits of EGDT from a quality improvement perspective are emerging,79 these benefits may be due to increased awareness of sepsis management rather than EGDT.80 In addition, the complex invasive technologies which underpin EGDT are not practical in resource-limited low- and middle-income countries.81 Early resuscitation in severe sepsis does appear to improve patient outcomes,82 however, the evidence in relation to which components of EGDT are effective is lacking. Trials currently underway to address this issue include the ProCESS (Protocolized Care for Early Septic Shock) and ARISE study.83 See Chapter 20 for further discussion of resuscitation in septic shock.
Early Treatment of Infection
• inappropriate initial antimicrobial therapy was associated with a five-fold decrease in survival to hospital discharge84
• the incidence of early acute kidney injury (AKI) increased with delays in antimicrobial therapy from the onset of hypotension.85
Other single centre studies also supported the SSG recommendation of antimicrobial therapy within the first hour of diagnosing severe sepsis.86 As early antimicrobial administration may be difficult to achieve given competing patient management priorities (e.g. airway management, volume resuscitation, vasopressor administration), systems must be developed to promote early administration.86 Nurses are in a pivotal position to ensure these guidelines or processes are developed, implemented and evaluated.
Practice tip
Tips for promoting early antimicrobial administration in severe sepsis/septic shock:86,89
• Ensure high priority in severe sepsis/septic shock algorithms
• Do not delay antimicrobial administration if difficulty sampling blood cultures
• Ensure adequate supply of antimicrobials in ED and ICU that fit local colonisation patterns
• Utilise appropriate antibiotics that can be given via IV push vs longer infusion
• Emphasise education of staff on the significance of early administration of initial antimicrobial
• Consider other potential barriers to early antimicrobial administration in your facility
Combination antibiotic therapy may offer a survival benefit in septic shock, but may be deleterious to patients with a low mortality risk.87 Certainly antibiotic overuse and misuse is of concern given the emergence of antibiotic resistance.88 Other factors that can lead to antibiotic failure in the critically ill include increased volume of distribution secondary to expanded extracellular volume, transient increased drug clearance due to elevated cardiac output (early sepsis) and increased free-drug levels secondary to reduced serum albumin. Maximum antibiotic dosage levels are therefore recommended in life-threatening infections, as inadequate antibiotic penetration can occur due to impaired vascularity of infected tissue (inhibits delivery of antibiotic), antibiotic antagonism (uncommon but possible with combination therapy) and coexisting unrecognised bacterial infection.89 Nursing assessment of patient response to antibiotic therapy (resolution or exacerbation of signs of sepsis) and surveillance for sites of unrecognised infection is therefore important.
Exclusion of Secondary Insults and Organ Support
Prevention of secondary inflammatory insults and organ support includes a broad range of interventions including use of massive transfusion protocols,90 recognition of abdominal compartment syndrome via urine catheter manometry,65 lung protective ventilation,76 early nutritional support,91,92 glycaemic control,60 haemodynamic support using vasopressors and intropes,76 renal replacement therapy,76 nitric oxide therapy and extracorporeal membrane oxygenation (ECMO). Routine evidence-based measures are also essential, including hygiene, bowel management, pressure area, mouth and eye care and other processes of care (e.g. FAST-HUG; see Chapter 20).
Awareness of the latest evidence that underpins management of these complex patients is important, including emerging therapies such as the use of statins93,94 and ACE-inhibitors95 (see Research vignette). Also note that the third edition of the SSG is due for release in 2012 (see Box 21.1).96 There is a surprising dearth of literature specifically addressing the complex nursing care required by a MODS patient. These patients require highly-skilled nurses who are able to balance competing priorities via ongoing patient assessment, care planning, monitoring and evaluation. The complex care required to nurse the MODS patient is highlighted in the clinical case study.
Box 21.1
Surviving Sepsis campaign
The Surviving Sepsis campaign is an international collaborative formed in 2003 to reduce the mortality of sepsis. Guidelines for the management of severe sepsis and shock were updated in 2008 and offer a comprehensive list of graded recommendations to care for these patients.76 Many of the recommendations for practice have implications for critical care nurses and the multidisciplinary team (see Online resources). The third edition is due for release in 2012.
Summary
Research vignette
Critique
Further, the authors reiterate the association between patients receiving statin therapy and reduced inflammation and subsequent rate of severe sepsis, ICU admission and mortality in patients admitted to hospital with acute bacterial infection. This is further reported as improved outcome in MODS patients receiving statin therapy versus those that did not, attributed to improved endothelial function, reduced inflammation and improved autonomic function. Recent work in the area of statin therapy has been reported demonstrating that chronic statin therapy was associated with decreased mortality in postoperative patients who had major adverse outcomes such as MODS.94 This paper therefore sets the scene to present a strong argument to support the research aims of investigating whether ACEI is associated with reduced mortality in MODS; whether a potential reduction in mortality is seen only in cardiogenic triggered MODS; and whether the time of ACEI application has impact on outcome. The hypothesis was that ACEI therapy could be advantageous for MODS patients despite its blood-pressure-lowering features.
Learning activities
1. Review the coagulation cascade and inflammatory and immune functions of the body.
2. Review the role of the adrenal gland and its relationship to adrenal insufficiency in the patient with MODS.
3. Develop a care plan for Mr Wyland (discussed in the case study) for his ICU stay. Ensure that you include routine cares as well as care specifically targeted at organ support. Discuss your plan with an experienced colleague.
4. List some of the important assessment findings that influenced the care of Mr Wyland during his stay in ICU, e.g. increasing bronchospasm, unstable BGLs, quiet bowel sounds.
5. Think of a patient with MODS who you have recently cared for. Reflect on the important elements of your nursing care that allowed you to effectively manage this patient. Consider what aspects of your care you would like to change when you next care for a complex MODS patient.
6. Review the pharmacology, therapeutic actions and interactions of statins and ACEI. Using the evidence based literature, consider their application in patients with MODS.
The Institute for Healthcare Improvement (IHI) is a non-profit organisation for advancing the quality and value of health care. The sepsis link includes information about improving care and severe sepsis bundles. http://www.ihi.org/ihi/topics/criticalcare/sepsis.
The Surviving Sepsis guidelines webpage provides access to full text documents here and updates. http://www.survivingsepsis.org.
. The US National Institutes of Health clinical trials registry. Search the site for currents trials in MODS http://www.clinicaltrials.gov
Bagshaw S.M, Lapinsky S, Dial S, Arabi Y, Dodek P, et al. Acute kidney injury in septic shock: clinical outcomes and impact of duration of hypotension prior to initiation of antimicrobial therapy. Intensive Care Med. 2009;35(5):871–881.
Kumar A, Roberts D, Wood K, Light B, Parrillo J, et al. Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med. 2006;34(6):1589–1596.
Kumar A, Ellis P, Arabi Y, Roberts D, Light B, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest. 2009;136(5):1237–1248.
Le Manach M, Ibanez Esteves C, Bertrand M, Goarin JP, Riou B, Landals P. Impact of preoperative statin therapy on adverse postoperative outcomes in patients undergoing vascular surgery. Anaesthesiol. 2011;114(1):98–104.
Madách K, Aladzsity I, Szilágyi Á, Fust G, Gál J, et al. 4G/5G polymorphism of PAI-1 gene is associated with multiple organ dysfunction and septic shock in pneumonia induced severe sepsis: prospective, observational, genetic study. Critical Care. 2010;14(2):R79.
Moore FA, Moore EE. The evolving rationale for early enteral nutrition based on paradigms of multiple organ failure: a personal journey. Nutr Clin Practice. 2009;24(3):297–304.
Perel A. Bench-to-bedside review: The initial hemodynamic resuscitation of the septic patient according to Surviving Sepsis Campaign guidelines – does one size fit all? Crit Care. 2008;12(5):223.
1 Jackson W, Gallagher C, Myhand R, Waselenko J. Medical management of patients with multiple organ dysfunction arising from acute radiation syndrome. Brit J Radiol. 2005;27:161–168.
2 Bone R, Balk R, Cerra F, Dellinger R, Fein A, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101(6):1644–1655.
3 Al-Khafaji A, Sharma S. Multisystem organ failure of sepsis. eMedicine Critical Care. 2010. Available from http://emedicine.medscape.com/article/169640-overview
4 Marshall JC. The multiple organ dysfunction syndrome. Surgical treatment: evidence based and problem oriented. http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=surg&part=A5364, 2001. Available from
5 Deitch E. Multiple organ failure: pathophysiology and potential future therapy. Ann Surg. 1992;216(2):117–134.
6 Barie P, Hydo L, Shou J, Eachempati S. Decreasing magnitude of multiple organ dysfunction syndrome despite increasingly severe critical surgical illness: a 17-year longitudinal study. Trauma. 2008;65(6):1227–1235.
7 Papathanassoglou E, Bozas E, Giannakopoloulou M. Multiple organ dysfunction syndrome pathogenesis and care: a complex systems’ theory perspective. British Association of Critical Care Nurses. Nurs Critical Care. 2008;13(5):249–259.
8 Pinsky M, Al Faresi F, Brenner B, Dire D, Filbin M, Flowers F, Gaeta T, et al. Septic shock. eMedicine Critical Care. 2011. Available from http://emedicine.medscape.com/article/168402-overview
9 Fink M. Cytopathic hypoxia in sepsis. Acta Anaesthesiol Scand. 1997;110(Suppl.):87–95.
10 Henke K, Eigisti J. Self-annihilation: a cells story of suicide. Dimens Crit Care Nurs. 2005;24(3):117–119.
11 Saukkonen K, Lakkisto P, Varpula M, Varpula T, Voipio-Pulkki L-M, et al. Association of cell-free plasma DNA with hospital mortality and organ dysfunction in intensive care unit patients. Intens Care Med. 2007;33(9):1624–1627.
12 Mizock BA. Metabolic derangements in sepsis and septic shock. Crit Care Clinics. 2000;16(2):319–336.
13 Singer M, De Santis V, Vitale D, Jeffcoate W. Multiorgan failure is an adaptive, endocrine-mediated, metabolic response to overwhelming systemic inflammation. Lancet. 2004;364(9433):545–548.
14 Jastrow K, Gonzalez E, McGuire M, Suliburk J, Kozar R, et al. Early cytoline production risk stratifies trauma patients for multiple organ failure. J Am Coll Surgeons. 2009;3(209):320–331.
15 Bone RC. Immunologic dissonance: a continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS). Annals Internl Med. 1996;125(8):680–687.
16 Bridges EJ, Dukes S. Cardiovascular aspects of septic shock: pathophysiology, monitoring, and treatment. Crit Care Nurs. 2005;25(2):14–40.
17 Padgett DA, Glaser R. How stress influences the immune response. Trends Immunol. 2003;24(8):444–448.
18 Hubbard WJ, Bland KI, Chaudry IH. The role of the mitochondrion in trauma and shock. Shock. 2004;22(5):395–402.
19 Adrie C, Pinsky MR. The inflammatory balance in human sepsis. Intens Care Med. 2000;26(4):364–375.
20 Magder S, Cernacek P. Role of endothelins in septic, cardiogenic, and hemorrhagic shock. Can J Physiol Pharmacol. 2003;81(6):635–643.
21 Zingarelli B, Sheehan M, Wong HR. Nuclear factor-kappaB as a therapeutic target in critical care medicine. Crit Care Med. 2003;31(Supp):S105–S111.
22 Brealey D, Brand M, Hargreaves I, Heales S, Land J, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360(9328):219–223.
23 Sherwood E, Toliver-Kinsky T. Mechanisms of the inflammatory response. Best Prac Res Clin Anaesthesiol. 2004;18(3):385–405.
24 Weigand M, Horner C. The systemic inflammatory response syndrome. Best Prac Res Clin Anaesthesiol. 2004;18(3):455–475.
25 Arias J-I, Aller M-A, Arias J. Surgical inflammation: a pathophysiological rainbow. J Translation Med. 7(19), 2009. Available from http://www.translational-medicine.com/content/pdf/1479-5876-7-19.pdf
26 Kirschfink M. Controlling the complement system in inflammation. Immunopharmacol. 1997;38(1–2):51–62.
27 Dishart MK, Schlichtig R, Tonnessen TI, Rozenfeld RA, Simplaceanu E, et al. Mitochondrial redox state as a potential detector of liver dysoxia in vivo. J Applied Physiol. 1998;84(3):791–797.
28 Fishel RS, Are C, Barbul A. Vessel injury and capillary leak. Crit Care Med. 2003;31(8):S502–S511.
29 Calandra T, Cohen J. The international sepsis forum consensus conference on definitions of infection in the intensive care unit. Crit Care Med. 2005;33(7):1538–1548.
30 Bernard GR, Macias WL, Joyce DE, Williams MD, Bailey J, Vincent JL. Safety assessment of drotrecogin alfa (activated) in the treatment of adult patients with severe sepsis. Crit Care. 2003;7(2):155–163.
31 Micek ST, Shah RA, Kollef MH. Management of severe sepsis: integration of multiple pharmacologic interventions. Pharmacotherapy. 2003;23(11):1486–1496.
32 Annane D, Bellissant E, Cavaillon JM. Septic shock. Lancet. 2005;365(9453):63–78.
33 Amaral A, Opal SM, Vincent JL. Coagulation in sepsis. Intens Care Med. 2004;30(6):1032–1040.
34 Rice TW, Bernard GR. Drotrecogin alfa (activated) for the treatment of severe sepsis and septic shock. Am J Med Sci. 2004;328(4):205–214.
35 Sharma S, Eschun G. Multisystem organ failure of sepsis. eMedicine Critical Care. 2004. Available from http://www.emedicine.com/med/topic3372.htm
36 Doshi SN, Marmur JD. Evolving role of tissue factor and its pathway inhibitor. Crit Care Med. 2002;30(Suppl):S241–S250.
37 Liaw P. Endogenous protein C activation in patients with severe sepsis. Crit Care Med. 2004;32(5):S214–S218.
38 Tortora G, Grabowski S. Principles of anatomy and physiology, 10th edn. New York: Wiley & Sons; 2003.
39 Hameed SM, Aird WC, Cohn SM. Oxygen delivery. Crit Care Med. 2003;31(12Suppl):S658–S667.
40 Trager T, DeBacker D, Radermacher P. Metabolic alterations in sepsis and vasoactive drug-related metabolic effects. Curr Opin Crit Care. 2003;9(4):271–278.
41 Sundararajan V, Macisaac C, Presneill J, Cade J, Visvanathan K. Epidemiology of sepsis in Victoria, Australia. Crit Care Med. 2005;33(1):71–80.
42 Ferreira FL, Bota DP, Bross A, Melot C, Vincent JL. Serial evaluation of the SOFA score to predict outcome in critically ill patients. JAMA. 2001;286(14):1754–1758.
43 Micek ST, Isakow W, Shannon W, Kollef MH. Predictors of hospital mortality for patients with severe sepsis treated with Drotrecogin alfa (activated). Pharmacotherapy. 2005;25(1):26–34.
44 Gando S. Microvascular thrombosis and multiple organ dysfunction syndrome. Crit Care Med. 2010;38(2Suppl):S35–S42.
45 Department of Health and Aging. Schedule of pharmaceutical benefits [database on the Internet]. Canberra: Department of Health and Aging. Available from http://www.health.gov.au/pbschedule
46 Ely EW, Kleinpell RM, Goyette RE. Advances in the understanding of clinical manifestations and therapy of severe sepsis: an update for critical care nurses. Am J Crit Care. 2003;12(2):120–133.
47 Kinney M, Dunbar S, Brooks-Brunn J, Molter N, Vitello-Cicciu JM. AACN’s clinical reference for critical care nursing. St Louis: Mosby; 1998.
48 Bauer M, editor. Multiple Organ Failure – update on pathophysiology and treatment strategies. Euroanesthesia conference proceedings; Vienna, Austria May 28–31: European Society of Anaesthesiology; 2005.
49 Martí-Carvajal A, Salanti G, Cardona-Zorrilla A. Human recombinant activated protein C for severe sepsis. Cochrane Reviews. 2007. Available from http://www2.cochrane.org/reviews/en/ab004388.html
50 Maldonado L, Murata G, Hershman J, Braunstein G. Do Thyroid Function tests independently predict survival in the critically ill? Thyroid. 1992;2(2):119–123.
51 Annane D, Bellissant E, Bollaert P, Briegel J, Keh D, Kupfer Y. Corticosteroids for treating severe sepsis and septic shock (review). Cochrane Reviews. 2004. Available from http://www2.cochrane.org/reviews/en/ab002243.html
52 Annane D, Sebille V, Charpentier C, Bollaert PE, Francois B, et al. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288(7):862–871.
53 Zaloga G, Marik P. Hypothalamic-pituitary-adrenal insufficiency. Crit Care Clinics. 2001;17(1):25–41.
54 Investigators TCS. Corticosteriod treatment and intensive insulin therapy for septic shock in adults: a randomised controlled trial. JAMA. 2010;303(17):1694–1698.
55 Finfer S. Corticosteriods in septic shock. New Eng J Med. 2008;358(2):188–190.
56 Sprung C, Annane D, Keh D, Moreno R, Singer M, et al. Hydrocortisone therapy for patients with septic shock. New Eng J Med. 2008;358(2):111–124.
57 Mason P, Al-Khafaji A, Milbrandt E, Suffoletto B, Huang D. CORTICUS: the end of unconditional love for steroid use? Critical Care. 2009;13(4):309.
58 Ho H, Chapital AD, Yu M. Hypothyroidism and adrenal insufficiency in sepsis and hemorrhagic shock. Archives of Surgery. 2004;139(11):1199–1203.
59 Vanhorebeek I, De Vos R, Mesotten D, Wouters P, De Wolf-Peeters C, Van den Berghe G. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet. 2005;365(9453):53–59.
60 Finfer S, Chittock D, Yu-Shhuo S, Blair D, Foster D, et al. Intensive versus conventional glucose control in critically ill patients. New Eng J Med. 2009;360(13):1283–1297.
61 Hermans G, De Jonghe B, Bruyninckx F, Van den Berghe G. Clinical Review: critical illness polyneuropathy and myopathy. Critical Care. 2008;12(6):238–247.
62 Kemp CM, Johnson C, Riordan W, Cotton B. How we die: the impact of non neurologic organ dysfunction after severe traumatic brain injury. American Surgeon. 2008;74(9):866–872.
63 Stevens R, Dowdy D, Michaels R, Mendez-Tellez P, Pronovost P, Needham D. Neuromuscular dysfunction acquired in critical illness: a systematic review. Intens Care Med. 2007;33(11):1876.
64 Streck E, Commin C, Barichello T, Quevedo J. The septic brain. Neurochem Res. 2008;33:2171–2177.
65 Dewar D, Mackay P, Balogh Z. Epidemiology of post-injury multiple organ failure in an Australian trauma system. ANZ J Surg. 2009;79:431–436.
66 Ciesla D, Moore E, Johnson J, Burch J, Cothren C, Sauaia A. A 12-year prospective study of postinjury multiple organ failure: has anything changed? Arch Surgery. 2005;140:432–440.
67 Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–1310.
68 Peres Bota D, Melot C, Lopes Ferreira F, Nguyen BV, Vincent J-L. The multiple organ dysfunction score (MODS) versus the sequential organ failure assessment (SOFA) score in outcome prediction. Intens Care Med. 2002;28(11):1619–1624.
69 Anami E, Grion C, Cardoso L, Kauss I, Thomazini M, et al. Serial evaluation of SOFA score in a Brazilian teaching hospital. Intens Crit Care Nurs. 2010;26:75–82.
70 Minne L, Abu-Hanna A, de Jonge E. Evaluation of SOFA-based models for predicting mortality in the ICU: a systematic review. Critical Care. 2008;12(6):R161.
71 Jones A, Trzeciak S, Kline J. The sequential organ failure assessment score for predicting outcome in patients with severe sepsis and evidence of hypoperfusion at the time of emergency department presentation. Crit Care Med. 2009;35(5):1649–1654.
72 Khwannimit B. A comparison of three organ dysfunction scores: MODS, SOFA and LOD for predicting ICU mortality in critically ill patients. J Med Assoc Thai. 2007;90(6):1074–1081.
73 Rice T, Wheeler A, Bernard G, Hayden D, Schoenfeld D, et al. Comparison of the SpO2/FiO2 ratio and the PaO2/FiO2 ratio in patients with Acute Lung Injury or Acute Respiratory Distress Syndrome. Chest. 2007. Available from http://chestjournal.chestpubs.org/content/early/2007/06/15/chest.07-0617.full.pdf+html
74 Honore P, Joannes-Boyau O, Boer W, Collins V. Regional occult hypoperfusion detected by lactate and sequential organ failure assessment subscores: old tools for new tricks? Crit Care Med. 2009;37(8):2477–2478.
75 Jansen T, van Bommel J, Woodward R, Mulder P, Bakker J. Association between blood lactate levels, sequential organ failure assessment subscores, and 28-day mortality during early and late intensive care unit stay: a retrospective observational study. Crit Care Med. 2009;37(8):2369–2374.
76 Dellinger R, Levy M, Carlet J, Bion J, Parker M, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med. 2008;36(1):296–327.
77 Madách K, Aladzsity I, Szilágyi Á, Fust G, Gál J, et al. 4G/5G polymorphism of PAI-1 gene is associated with multiple organ dysfunction and septic shock in pneumonia induced severe sepsis: prospective, observational, genetic study. Crit Care. 2010;14(2):R79.
78 Peake S, Bailey M, Bellomo R, Cameron P, Cross A, et al. Australasian resuscitation of sepsis evaluation (ARISE): a multi-centre, prospective, inception cohort study. Resuscitation. 2009;80:811–818.
79 Levy M, Dellinger R, Townsend S, Linde-Zwirble W, Marshall J, et al. The surviving sepsis campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Crit Care Med. 2010;38(2):367–374.
80 Finfer S. The surviving sepsis campaign: robust evaluation and high-quality primary research is still needed. Crit Care Med. 2010;38(2):683–684.
81 Becker J, et al. Surviving sepsis in low-income and middle-income countries: new directions for care and research. Lancet Infect Dis. 2009;9(9):577–582.
82 Rivers E. Management of sepsis: early resuscitation. Clin Chest Med. 2008;29:689–704.
83 US National Institutes of Health website. ClinicalTrials.gov. [Cited October 2010]. Available from http://clinicaltrials.gov/
84 Kumar A, Ellis P, Arabi Y, Roberts D, Light B, et al. Initiation of inappropriate antimicrobial therapy results in a fivefold reduction of survival in human septic shock. Chest. 2009;136(5):1237–1248.
85 Bagshaw S, Lapinsky S, Dial S, Arabi Y, Dodek P, et al. Acute kidney injury in septic shock: clinical outcomes and impact of duration of hypotension prior to initiation of antimicrobial therapy. Intens Care Med. 2009;35:871–881.
86 Gaieski D, Mikkelsen M, Band R, Pines J, Massone R, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med. 2010;38(4):1045–1053.
87 Kumar A, Safdar N, Kethireddy S, Chateau D. A survival benefit of combination antibiotic therapy for serious infections associated with sepsis and septic shock is contingent only on the risk of death: a meta-analytic/meta-regression study. Crit Care Med. 2010;38(8):1651–1664.
88 Pines J. Timing of antibiotics for acute, severe infections. Emerg Med Clin N Am. 2008;26:245–257.
89 Sharma S, Kumar A. Antimicrobial management of sepsis and septic shock. Clin Chest Med. 2008;29:677–687.
90 Cotton B, Au B, Nunez T, Gunter O, Robertson A, Young P. Predefined massive transfusion protocols are associated with a reduction in organ failure and postinjury complications. Trauma. 2008;66(1):41–47.
91 Vincent J. Metabolic support in sepsis and multiple organ failure: more questions than answers. Crit Care Med. 2007;35(9Suppl):S436–S440.
92 Moore F, Moore E. The evolving rationale for early enteral nutrition based on paradigms of multiple organ failure: a personal journey. Nutrition Clin Prac. 2009;24(3):297–304.
93 Schmidt H, Hennen R, Keller A, Russ M, Müller-Werdan U, et al. Association of statin therapy and increased survival in patients with multiple organ dysfunction syndrome. Intens Care Med. 2006;32:1248–1251.
94 Le Manach M, Ibanez Esteves C, Bertrand M, Goarin JP, Riou B, Landals P. Impact of preoperative statin therapy on adverse postoperative outcomes in patients undergoing vascular surgery. Anaesthesiology. 2011;114(1):98–104.
95 Schmidt H, Hoyer D, Rauchhaus M, Prondzinsky R, Hennen R, et al. ACE-inhibitor therapy and survival among patients with multiorgan dysfunction syndrome (MODS) of cardiac and non-cardiac origin. Int J Cardiol. 2010;140:296–303.
96 Barie PS. Surviving sepsis: something doing by doing something. Crit Care Med. 2010;38(4):1209–1210.
97 Australian College of Critical Care Nurses. National Advanced Life Support Education Package: Pathophysiology of cellular dysfunction. Melbourne: Cambridge Press; 2004.
98 Sepsis.com database. [Cited Oct 2005]. Available from http://www.sepsis.com/index.jsp
99 Vincent J, Moreno R, Takala J, Willats S, De Mendonca A, et al. The SOFA (sepsis related organ failure assessment) score to describe organ dysfunction/failure. Intens Care Med. 1996;22:707–710.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]
21 Multiple Organ Dysfunction Syndrome
After reading this chapter, you should be able to:
• define the common terminology related to multiple organ dysfunction syndrome
• describe the related pathophysiology of multiple organ dysfunction syndrome
• identify the clinical manifestations of multiple organ dysfunction syndrome
• identify patients at risk of developing multiple organ dysfunction, including predictors of mortality
• initiate appropriate monitoring, care planning and evaluation strategies for the patient with multiple organ dysfunction in relation to the current evidence base
• discuss treatment strategies that promote homeostasis in the patient with multiple organ dysfunction syndrome
Introduction
The term multiple organ dysfunction syndrome (MODS) was established by an expert consensus conference in 1992 to describe a continuum of physiologic derangements and subsequent dynamic alterations in organ function that may occur during a critical illness.1,2 Previous terminologies in the literature were confusing. For example, multiple organ failure (MOF) was a term commonly used, but somewhat misleading as normal physiologic function can, in most cases, be restored in survivors of a critical illness who have temporary organ dysfunction.3,4 Although the syndrome affects many organs, it also affects physiological systems such as the haematological, immune and endocrine systems. MODS therefore more accurately describes altered organ function in a critically ill patient who requires medical and nursing interventions to achieve homeostasis.4
MODS is associated with widespread endothelial and parenchymal cell injury because of hypoxic hypoxia, direct cytotoxicity, apoptosis, immunosuppression and coagulopathy.4 Four clinical stages describe a patient with developing MODS:5
1. increasing volume requirements and mild respiratory alkalosis, accompanied by oliguria, hyperglycaemia and increased insulin requirements
2. tachypnoea, hypocapnia and hypoxaemia, with moderate liver dysfunction and possible haematological abnormalities
3. developing shock with azotaemia, acid–base disturbances and significant coagulation abnormalities
4. vasopressor dependence with oliguria or anuria, ischaemic colitis and lactic acidosis.
Cellular damage in various organs in patients who develop MODS begins with the onset of local injury that is then compounded by activation of the innate immune system. This includes a combination of pattern recognition, receptor activation and release of mediators at the microcellular level, leading to episodes of hypotension or hypoxaemia and secondary infections.4,5 The primary therapeutic goal for nursing and medical staff is prompt, definitive control of the source of infection or pro-inflammation6 and early recognition of preexisting factors that may lead to subsequent organ damage away from the initial site of injury. This preemptive therapy is instituted to maintain adequate tissue perfusion and prevent the onset of MODS. Recognition and response to early signs of clinical deterioration are therefore important to minimise further organ dysfunction.
This chapter initially describes the pathophysiology of inflammatory and infective conditions that may lead to multiple organ dysfunction. System responses and specific organ dysfunction are discussed, expanding on dialogue in previous chapters, particularly Chapters 19 and 20. Assessment of the severity of MODS and nursing considerations in the treatment of the MODS patient is presented.
Pathophysiology
The syndrome of multiple organ dysfunction is most closely related to an outcome of sepsis, which was described in Chapter 20. MODS is a state characterised by aberrant cellular responses involving multiple organ systems and sequential processes. The pathogenesis of MODS is complex, simultaneously involving every cell type, neuro-hormonal axis and organ system.7
In brief, hypoxic hypoxia results from altered metabolic regulation of tissue oxygen delivery which contributes to further organ dysfunction. Microcirculatory injury as a result of lytic enzymes, and vasoactive substances (nitric oxide, endothelial growth factor), is compounded by the inability of erythrocytes to navigate the septic microcirculation. Mitochondrial electron transport is affected by endotoxins in sepsis, nitric oxide and TNF-alpha, leading to disordered energy metabolism (see Figure 21.1). This causes cytopathic or histotoxic anoxia (the inability to use oxygen, even when available).8 This context of impaired oxygen utilisation rather than delivery7,8 results from diminished mitochondrial production of cellular energy (ATP), despite normal or even supranormal intra-cellular PO2 levels.9 Cytopathic hypoxia appears resistant to resuscitation measures, and this may ultimately worsen already-existing organ dysfunction. During sepsis or ischaemia, mitochondria respond by facilitating cell death rather than the restoration of homeostasis.7
Apoptosis is normal physiological programmed cell death and is the main mechanism to eliminate dysfunctional cells.10 Apoptosis involves chromatin condensation, membrane blebbing, cell shrinkage and subsequent breakdown of cellular components into apoptic bodies. This normally orderly process is deranged in critical illness, leading to tissue or organ bed injury and MODS. Proinflammatory cytokines released in sepsis may delay apoptosis in activated macrophages and neutrophils, but in other tissues, such as gut endothelium, accelerated apoptosis occurs.8
In contrast, necrosis is a form of cell death characterised by cellular swelling and loss of membrane integrity as a result of hypoxia or trauma. Necrosis has been termed ‘cellular energy crisis’,10 and is unregulated resulting in loss of membrane sodium/potassium/ATP-ase pumps. This loss leads to cell swelling, rupture and spillage of intracellular contents into surrounding regions creating collateral damage.10 Necrosis therefore can involve significant amounts of tissue and organ bed damage. Apoptosis differs from necrosis in that it does not seem to involve the recruitment of inflammatory cells or mediators to complete its task. Activation of an enzyme cascade systematically cleaves proteins, including the cell’s nuclear DNA, with the end-result being death of the cell. This requires energy from mitrochondria and if not available necrosis of the cell occurs. Apoptosis and necrosis are processes that if is therefore important to understand in relation to future MODS research.
Increased concentrations of cell-free plasma DNA are present in various clinical conditions such as stroke, myocardial infarction and trauma, a likely result of accelerated cell death. Maximum plasma DNA concentrations correlated significantly with APACHE II scores and maximum SOFA scores (described later in this chapter), with cell-free plasma DNA concentrations higher in hospital non-survivors than in survivors. Using regression analysis, maximum plasma DNA was an independent predictor of hospital mortality.11
Other cellular organelles may also exhibit pathological reactions in MODS. In ischaemia/reperfusion, endoplasmic reticulum loses its ability to process proteins which induces the expression of heat shock proteins,7 affecting transcription of proteins necessary for organ specific functions. For example, liver cell metabolism, renal cell function or cardiac cell contractility may be affected.7 This has led to the controversial concept of a mode of hibernation of cells at the expense of survival of the whole organism.7
Cellular communication is also altered in MODS. Cells normally communicate through highly interactive bidirectional networks. The endothelium acts as a communication interface between cells, organs and systems and is involved in orchestration of systemic responses, including haemodynamic regulation, inflammation and coagulation; oxygen and nutrient delivery; oxidative stress and sensing of psychological stress and neuroendocrine alterations.7 In critical illness, endothelia release molecules that trigger the immune and neuroendocrine systems to produce a generalised inflammatory response.7 The combination of the pathophysiological processes involved with the development of MODS, compensatory mechanisms and the effect on target organs and systems is now discussed.
Systemic Response
After an overwhelming incident such as trauma, sepsis or non-infectious inflammation, a complex range of interrelated reactions occurs that result in a cascade of responses. The complex host-response generated involves the inflammatory immune systems, hormonal activation and metabolic derangements, resulting in multiple organ system involvement.12,13 These host-responses are initially adaptive to maintain nutrient perfusion to the tissues, however eventually organ systems become dysfunctional and fail, and the body is no longer able to maintain homeostasis16 (see Figure 21.2).
Initially, proinflammatory mediators are released locally to fight foreign antigens and promote wound healing. Antiinflammatory mediators are also released to downregulate the initial response to the insult.14 If the local defence system is overwhelmed, inflammatory mediators appear in the systemic circulation and recruit additional leucocytes to the area of damage. A whole-body stress response ensues, further compounding the situation. If proinflammatory mediators and antiinflammatory response is imbalanced, the patient may develop systemic inflammatory response syndrome (SIRS) and subsequent immunological dissonance15 of organ dysfunction.2,15,16
Intracellular transcription factors, in particular nuclear factor kappa B (NFκB), are important in innate and adaptive immunity,17,18 as they regulate the transcription of genes involved in the inflammatory and acute stress response, leading to expression of TNFα, interleukins and tissue factor.18,19 NFκB therefore plays an important role in response pathways in critical states including hypoxia, ischaemia, haemorrhage, sepsis, shock and MODS.18,20,21
The inflammatory cascade activates a number of prostaglandins and leucotrienes that also have pro- and antiinflammatory effects. Thromboxane A2 plays a role in the acute phase, in part due to stimulation of platelet aggregation, leading to microvascular thrombosis and tissue injury;15 it may also play a role in pulmonary bronchoconstriction and myocardial depression.
The specific pathophysiological concepts of inflammation, oedema and infection are discussed below.
Inflammation
Inflammation is part of innate immunity, a generic response to injury, and is normally an excellent mechanism to localise injury and promote healing.22,23 The basis of this immune response is recognition and an immediate response to an invading pathogen without necessarily having previous exposure to the pathogen.24 Neutrophils, macrophages, natural killer cells, dendrites, coagulation and complement are the principal active components of the innate host response.23
The classic signs of inflammation are:
Nitric oxide and prostaglandins (e.g. prostacyclin), are the primary mediators of vasodilation and inflammation at the injury site.23 Injured endothelium produces molecules that attract leucocytes and facilitate movement to the tissues. White blood cells accumulate by margination (adhesion to endothelium during the early stages of inflammation) and neutrophils accumulate at the injury site, where rolling and adherence to binding molecules on the endothelium occurs with eventual movement across the endothelium into the tissues.23 Different blood components therefore escape the intravascular space and occupy the interstitial space where they play the main role in successive phases of the inflammatory response. The endothelium therefore plays a bidirectional mediating role between blood flow and the interstitial space where inflammation mainly takes place.25 Macrophages, neutrophils and monocytes are responsible for phagocytosis and the production of toxic free radicals to kill invading pathogens.24 The complement system, a collection of 30 proteins circulating in the blood, is also activated, with plasma and membrane proteins acting as adjuncts to inflammatory and immune processes.26 When activated by inflammation and microbial invasion, these processes facilitate lysis (cellular destruction) and phagocytosis (ingestion) of foreign material.23,26
Dysfunction of organ systems often persists after the initial inflammatory response diminishes; this is largely unexplained, although dysoxia (abnormal tissue oxygen metabolism and utilisation) has been implicated.22,27 Hypoxia induces release of IL-6, the main cytokine that initiates the acute phase response. After reperfusion of ischaemic tissues, tissue and neutrophil activation forms reactive oxygen species (e.g. hydrogen peroxide) as a byproduct. These strong oxidants damage other molecules and cell structures that they form,23 resulting in water and sodium infiltrate and cellular oedema.
Oedema
Oedema occurs as a consequence of alterations to tissue endothelium, with increased microvascular permeability (‘capillary leak’). As noted earlier, many mediators, including circulating cytokines, oxygen free-radicals and activated neutrophils, alter the structure of endothelial cells, enabling larger molecules (proteins, water) to cross into the extravascular space.23,28 This response mechanism improves supply of nutrient-rich fluid to the site of injury, but if this becomes systemic, fluid shifts can lead to hypovolaemia, third-spacing (interstitial oedema) or affect other organs (e.g. acute lung injury, ALI).23
Infection and Immune Responses
Infection exists when there is one of the following: positive culture, serology,29 presence of polymorphonuclear leucocytes in a normally sterile body fluid except blood, and clinical focus of infection such as perforated viscus or pneumonia. In sepsis, the most common sites of infection are the lungs (34–54%), intra-abdominal organs (15–28%) and urinary tract (5–10%).30,31 The incidence of bloodstream infections is 30–40%,29 although one-third of cases with septic shock have negative blood cultures; one reason suggested for this is antibiotic administration prior to sample collection.32 The type of infecting organism has also changed over time, with Gram-positive bacteria predominant, accounting for at least one-third of pathogens in septic shock; Gram-negative, fungal, viruses and parasitic organisms are also involved.29 The increasing incidence of resistant organisms, partially as a result of the indiscriminate use of antibiotics, is an ongoing concern.
The immune response to infection has both non-specific and specific actions, with inflammation and coagulation responses intricately linked in sepsis pathophysiology.23,24,33,34 Tissue injury and the production of inflammatory mediators lead to:
• coagulation via the expression of tissue factor and factor VIIa complex (tissue factor pathway; the primary cascade for initiation of coagulation; previously termed the ‘extrinsic’ pathway)28,33–35
• coagulation amplification via factors Xa and Va, leading to massive thrombin formation and fibrin clots (common coagulation pathway).28,33
Note that blood cell injury or platelet contact with endothelial collagen initiates the contact activation pathway (previously termed the ‘intrinsic’ coagulation pathway).33
Procoagulation
Tissue factor is a procoagulant glycoprotein-signalling receptor,36 expressed when tissue is damaged or cytokines are released from macrophages or the endothelium (see Figure 21.3). Prothrombin is formed, leading to thrombin and fibrin generation from activated platelets. Resulting clots are stabilised by factor XIII and thrombin-activatable fibrinolysis inhibitor (TAFI).33,36 Fibrinolysis is a homeostatic process that dissolves clots via the plasminogen–tissue plasminogen activator (tPA)–plasmin pathway (involving antithrombin, activated protein C [APC] and tissue factor pathway inhibitor). APC:37
• reduces inflammation by decreasing TNF and NFκB production
• reduces thrombin production when activated via thrombin–thrombomodulin complexes (anticoagulant action)
• inhibits thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 (profibrinolytic action).33,34
APC is consumed in severe sepsis, and thrombomodulin is unable to activate protein C,33,34,37 promoting a proinflammatory, prothrombotic state.34
Endocrine Response
Physiological changes are triggered as a normal response to a stressor. In a critically ill patient, however, chronic activation of the stress response, including the hypothalamic–pituitary–adrenal axis and the sympathetic–adrenal–medullary axis, results in ongoing production of glucocorticoid hormones and catecholamines.17 This response interferes with the regulation of cytokine-producing immune cells, leading to immune dysfunction. Other compensatory mechanisms are instigated in an attempt to maintain supply and perfusion to organs.15
These homeostatic mechanisms are activated through positive or negative feedback systems to counteract stress. When stress is extreme or prolonged, these normal homeostatic mechanisms may be insufficient and a patient may respond through a sequence of physiological changes called the stress response. The stress response occurs in three stages: the alarm reaction, the resistance reaction and exhaustion (see Table 21.1).38
| Response stage | Neurohormonal response | Actions |
|---|---|---|
| Alarm reaction | Hypothalamus |
The alarm reaction (flight-or-fight response)38 is initiated when stress is detected, increasing the amount of glucose and oxygen available to the brain, skeletal muscle and heart. Two-thirds of total blood volume is also redistributed to support central circulation.38 A rise in glucose production and the breakdown of glycogen in skeletal muscle increases circulating glucose levels, providing an immediate energy source. The long-lasting second stage is a resistance reaction, involving hypothalamic, pituitary and adrenal hormone release.38 Response exhaustion occurs when these physiological changes can no longer maintain homeostasis.
Compensatory Mechanisms
Internal equilibrium (homeostasis) is maintained by the nervous and endocrine systems, and these work symbiotically with other compensatory mechanisms, such as endothelial cells, to maintain cellular perfusion. The nervous system responds rapidly to maintain homeostasis by sending impulses to organs to activate neurohormonal responses (see Chapters 16 and 20). Endothelins (ET-1, ET-2, ET-3) are potent vasoconstrictors produced by endothelial cells that regulate arterial pressure.20 The endocrine system works in a slow and sustained manner by secreting hormones, which travel via the blood to end-organs.
An initial acute-adaptive response is activated when an insult or stress occurs. For example, the body senses a disruption of blood flow through baroreceptor and chemoreceptor reflex actions: baroreceptors located in the carotid sinus detect changes in arterial pressure;13 chemoreceptors co-located with the baroreceptors detect O2, CO2 and H+ concentration. When alterations are sensed, the cardiovascular centre in the brain adjusts autonomic outflow accordingly.38 In a patient with decreased tissue perfusion, there is increased peripheral vasoconstriction, contractility and heart rate. Blood flow is shunted to the vital organs (brain, heart, lungs), and away from less vital areas (e.g. gastrointestinal and reproductive organs).39 Important hormonal regulators of blood flow are also activated from decreased blood flow to the kidneys, including adrenocorticotrophic hormone (ACTH), and the renin–angiotensin–aldosterone system (see Chapter 18). Adrenal medullary hormones, adrenaline and noradrenaline, vasopressin (antidiuretic hormone) and atrial natriuretic peptide also regulate blood flow to maintain adequate circulation and tissue oxygenation.13,38,39
Arterial pressure is a major determinant of tissue perfusion as it forces blood through the regional vasculature.20 Hypotension (systolic blood pressure <90 mmHg or mean arterial pressure [MAP] <70 mmHg) results from either low systemic vascular resistance or a low cardiac output.20 Glomerular filtration falls, leading to reduced urine output; low cerebral blood flow results in an altered level of consciousness; and other manifestations reflect low-flow states in other organ systems. To maintain oxygen supply, respirations and heart rate increase to meet organ oxygenation demands.40 Organ dysfunction ensues if balance is not sufficiently restabilised (see Table 21.2).
| Organ system | Clinical parameters |
|---|---|
| Cardiovascular | Patient requires vasopressor support (systolic BP <90 mmHg) or MAP <70 mmHg for 1 hour despite fluid bolus |
| Respiratory | Patient requires mechanical ventilation: P/F ratio <250, PEEP >7.5 cmH2O |
| Renal | Low urine output <0.5 mL/kg/h; raised creatinine >50% from baseline or requiring acute dialysis |
| Haematological | Low platelet count (<1 000 000/mm3) or APTT/PTT > upper limit of normal |
| Metabolic | Low pH with increased lactate (pH <7.3 and plasma lactate > upper limit of normal) |
| Hepatic | Liver enzymes >2 × upper limit of normal |
| CNS | Altered level of consciousness/reduced Glasgow Coma Scale score |
| Gastrointestinal | Translocation of bacteria, possible elevated pancreatic enzymes and cholecystitis |
Organ Dysfunction
Organ dysfunction is a common clinical presentation in ICU. Patients with dysfunction in the respiratory, cardiovascular, hepatic or metabolic systems were 50% more likely to require ICU treatment and had a higher mortality than patients not requiring intensive care.41 Timely identification of organ dysfunction is therefore critical, as early intervention reduces damage and improves recovery in organ systems. As each organ fails, the average risk of death rises by 11–23%, with up to 75% of patients in sepsis clinical trials having at least two failing organs.42 The organ system that most commonly fails is the pulmonary system, followed by the cardiovascular, renal and haematological systems.43 Organ and systems dysfunction are a result of hypoperfusion, inflammation, cellular dysfunction and oedema. Dysfunction of the cardiovascular (Chapters 10 and 12), respiratory (Chapters 14 and 15), renal (Chapter 18), and hepatic and gastrointestinal systems (Chapter 19) have been previously addressed. This next section addresses the haematological, endocrine and metabolic systems. Neurological dysfunction is also common in the patient with MODS and complements previous discussions in Chapter 17.
[/not-level-membership-for-critical-care-medicine-category]
