Etiology In at least 90% of patients with Cushing’s disease, ACTH excess is caused by a corticotrope pituitary microadenoma, often only a few millimeters in diameter. Pituitary macroadenomas (i.e., tumors >1 cm in size) are found in only 5–10% of patients. Pituitary corticotrope adenomas usually occur sporadically but very rarely can be found in the context of multiple endocrine neoplasia type 1 (MEN 1) (Chap. 408).
Ectopic ACTH production is predominantly caused by occult carcinoid tumors, most frequently in the lung, but also in thymus or pancreas. Because of their small size, these tumors are often difficult to locate. Advanced small-cell lung cancer can cause ectopic ACTH production. In rare cases, ectopic CRH and/or ACTH production has been found to originate from medullary thyroid carcinoma or pheochromocytoma, the latter co-secreting catecholamines and ACTH.
The majority of patients with ACTH-independent cortisol excess harbor a cortisol-producing adrenal adenoma; intratumor mutations, i.e., somatic mutations in the PKA catalytic subunit PRKACA, have been identified as cause of disease in 40% of these tumors. Adrenocortical carcinomas may also cause ACTH-independent disease and are often large, with excess production of several corticosteroid classes.
A rare but notable cause of adrenal cortisol excess is macronodular adrenal hyperplasia with low circulating ACTH, but with evidence for autocrine stimulation of cortisol production via intraadrenal ACTH production. These hyperplastic nodules are often also characterized by ectopic expression of G protein–coupled receptors not usually found in the adrenal, including receptors for luteinizing hormone, vasopressin, serotonin, interleukin 1, catecholamines, or gastric inhibitory peptide (GIP), the cause of food-dependent Cushing’s. Activation of these receptors results in upregulation of PKA signaling, as physiologically occurs with ACTH, with a subsequent increase in cortisol production. A combination of germline and somatic mutations in the tumor-suppressor gene ARMC5 have been identified as a prevalent cause of Cushing’s due to macronodular adrenal hyperplasia. Germline mutations in the PKA catalytic subunit PRKACA can represent a rare cause of macronodular adrenal hyperplasia associated with cortisol excess.
Mutations in one of the regulatory subunits of PKA, PRKAR1A, are found in patients with primary pigmented nodular adrenal disease (PPNAD) as part of Carney’s complex, an autosomal dominant multiple neoplasia condition associated with cardiac myxomas, hyperlentiginosis, Sertoli cell tumors, and PPNAD. PPNAD can present as micronodular or macronodular hyperplasia, or both. Phosphodiesterases can influence intracellular cAMP and can thereby impact PKA activation. Mutations in PDE11A and PDE8B have been identified in patients with bilateral adrenal hyperplasia and Cushing’s, with and without evidence of PPNAD.
Another rare cause of ACTH-independent Cushing’s is McCune-Albright syndrome, also associated with polyostotic fibrous dysplasia, unilateral café-au-lait spots, and precocious puberty. McCune-Albright syndrome is caused by activating mutations in the stimulatory G protein alpha subunit 1, GNAS-1 (guanine nucleotide binding protein alpha stimulating activity polypeptide 1), and such mutations have also been found in bilateral macronodular hyperplasia without other McCune-Albright features and, in rare instances, also in isolated cortisol-producing adrenal adenomas (Table 406-1; Chap. 426e).
Clinical Manifestations Glucocorticoids affect almost all cells of the body, and thus signs of cortisol excess impact multiple physiologic systems (Table 406-2), with upregulation of gluconeogenesis, lipolysis, and protein catabolism causing the most prominent features. In addition, excess glucocorticoid secretion overcomes the ability of 11β-HSD2 to rapidly inactivate cortisol to cortisone in the kidney, thereby exerting mineralocorticoid actions, manifest as diastolic hypertension, hypokalemia, and edema. Excess glucocorticoids also interfere with central regulatory systems, leading to suppression of gonadotropins with subsequent hypogonadism and amenorrhea, and suppression of the hypothalamic-pituitary-thyroid axis, resulting in decreased thyroid-stimulating hormone (TSH) secretion.
|
SIGNS AND SYMPTOMS OF CUSHING’S SYNDROME |
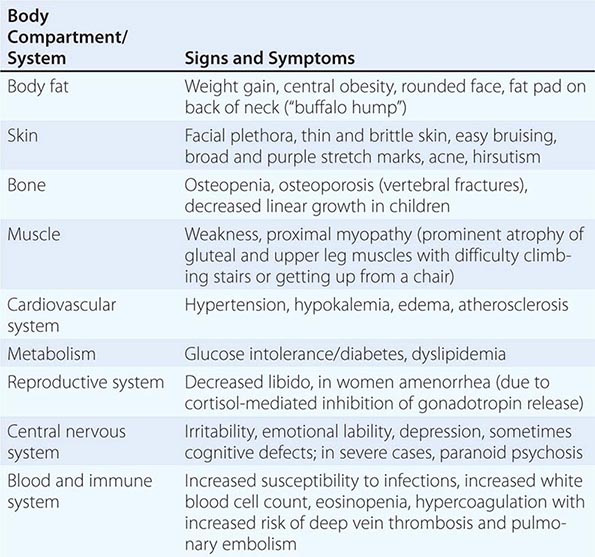
The majority of clinical signs and symptoms observed in Cushing’s syndrome are relatively nonspecific and include features such as obesity, diabetes, diastolic hypertension, hirsutism, and depression that are commonly found in patients who do not have Cushing’s. Therefore, careful clinical assessment is an important aspect of evaluating suspected cases. A diagnosis of Cushing’s should be considered when several clinical features are found in the same patient, in particular when more specific features are found. These include fragility of the skin, with easy bruising and broad (>1 cm), purplish striae (Fig. 406-9), and signs of proximal myopathy, which becomes most obvious when trying to stand up from a chair without the use of hands or when climbing stairs. Clinical manifestations of Cushing’s do not differ substantially among the different causes of Cushing’s. In ectopic ACTH syndrome, hyperpigmentation of the knuckles, scars, or skin areas exposed to increased friction can be observed (Fig. 406-9) and is caused by stimulatory effects of excess ACTH and other POMC cleavage products on melanocyte pigment production. Furthermore, patients with ectopic ACTH syndrome, and some with adrenocortical carcinoma as the cause of Cushing’s, may have a more brisk onset and rapid progression of clinical signs and symptoms.
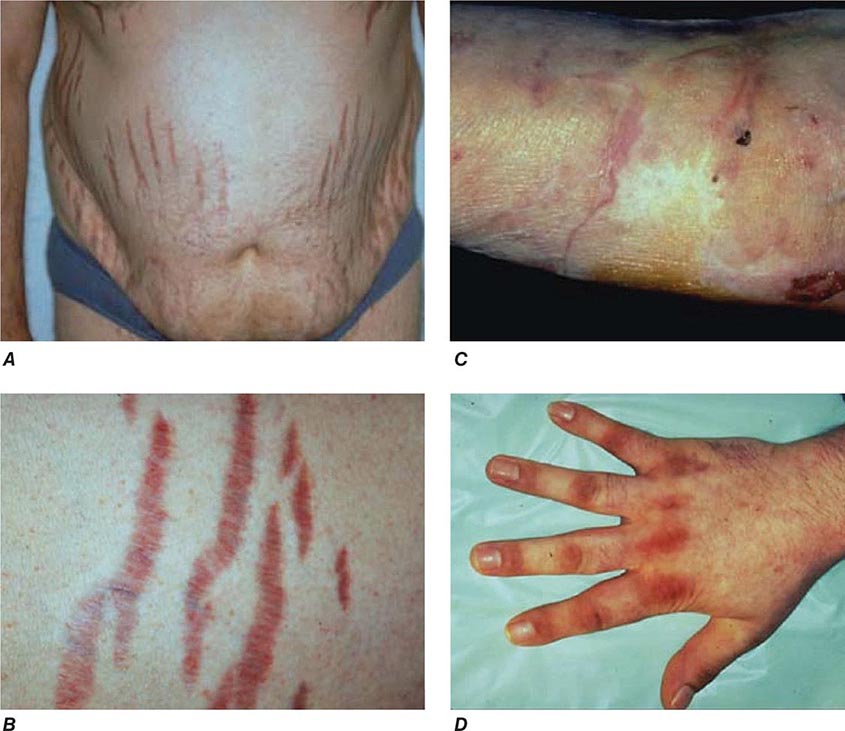
FIGURE 406-9 Clinical features of Cushing’s syndrome. A. Note central obesity and broad, purple stretch marks (B. close-up). C. Note thin and brittle skin in an elderly patient with Cushing’s syndrome. D. Hyperpigmentation of the knuckles in a patient with ectopic adrenocorticotropic hormone (ACTH) excess.
Patients with Cushing’s syndrome can be acutely endangered by deep vein thrombosis, with subsequent pulmonary embolism due to a hypercoagulable state associated with Cushing’s. The majority of patients also experience psychiatric symptoms, mostly in the form of anxiety or depression, but acute paranoid or depressive psychosis may also occur. Even after cure, long-term health may be affected by persistently impaired health-related quality of life and increased risk of cardiovascular disease and osteoporosis with vertebral fractures, depending on the duration and degree of exposure to significant cortisol excess.
Diagnosis The most important first step in the management of patients with suspected Cushing’s syndrome is to establish the correct diagnosis. Most mistakes in clinical management, leading to unnecessary imaging or surgery, are made because the diagnostic protocol is not followed (Fig. 406-10). This protocol requires establishing the diagnosis of Cushing’s beyond doubt prior to employing any tests used for the differential diagnosis of the condition. In principle, after excluding exogenous glucocorticoid use as the cause of clinical signs and symptoms, suspected cases should be tested if there are multiple and progressive features of Cushing’s, particularly features with a potentially higher discriminatory value. Exclusion of Cushing’s is also indicated in patients with incidentally discovered adrenal masses.
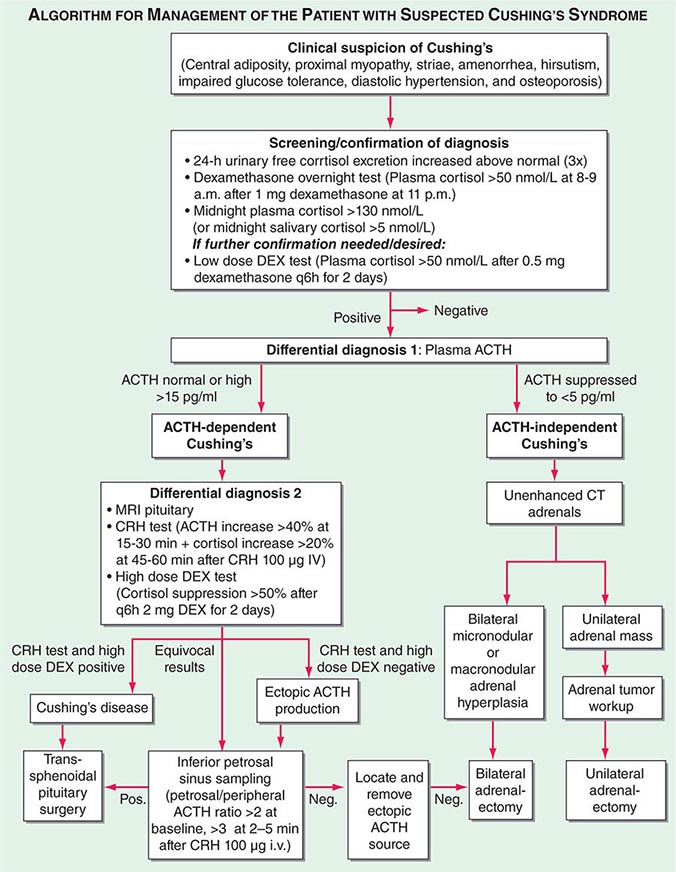
FIGURE 406-10 Management of the patient with suspected Cushing’s syndrome. ACTH, adrenocorticotropic hormone; CRH, corticotropin-releasing hormone; CT, computed tomography; DEX, dexamethasone; MRI, magnetic resonance imaging.
A diagnosis of Cushing’s can be considered as established if the results of several tests are consistently suggestive of Cushing’s. These tests may include increased 24-h urinary free cortisol excretion in three separate collections, failure to appropriately suppress morning cortisol after overnight exposure to dexamethasone, and evidence of loss of diurnal cortisol secretion with high levels at midnight, the time of the physiologically lowest secretion (Fig. 406-10). Factors potentially affecting the outcome of these diagnostic tests have to be excluded such as incomplete 24-h urine collection or rapid inactivation of dexamethasone due to concurrent intake of CYP3A4-inducing drugs (e.g., antiepileptics, rifampicin). Concurrent intake of oral contraceptives that raise CBG and thus total cortisol can cause failure to suppress after dexamethasone. If in doubt, testing should be repeated after 4–6 weeks off estrogens. Patients with pseudo-Cushing states, i.e., alcohol-related, and those with cyclic Cushing’s may require further testing to safely confirm or exclude the diagnosis of Cushing’s. In addition, the biochemical assays employed can affect the test results, with specificity representing a common problem with antibody-based assays for the measurement of urinary free cortisol. These assays have been greatly improved by the introduction of highly specific tandem mass spectrometry.
Differential Diagnosis The evaluation of patients with confirmed Cushing’s should be carried out by an endocrinologist and begins with the differential diagnosis of ACTH-dependent and ACTH-independent cortisol excess (Fig. 406-10). Generally, plasma ACTH levels are suppressed in cases of autonomous adrenal cortisol excess, as a consequence of enhanced negative feedback to the hypothalamus and pituitary. By contrast, patients with ACTH-dependent Cushing’s have normal or increased plasma ACTH, with very high levels being found in some patients with ectopic ACTH syndrome. Importantly, imaging should only be used after it is established whether the cortisol excess is ACTH-dependent or ACTH-independent, because nodules in the pituitary or the adrenal are a common finding in the general population. In patients with confirmed ACTH-independent excess, adrenal imaging is indicated (Fig. 406-11), preferably using an unenhanced computed tomography (CT) scan. This allows assessment of adrenal morphology and determination of precontrast tumor density in Hounsfield units (HU), which helps to distinguish between benign and malignant adrenal lesions.
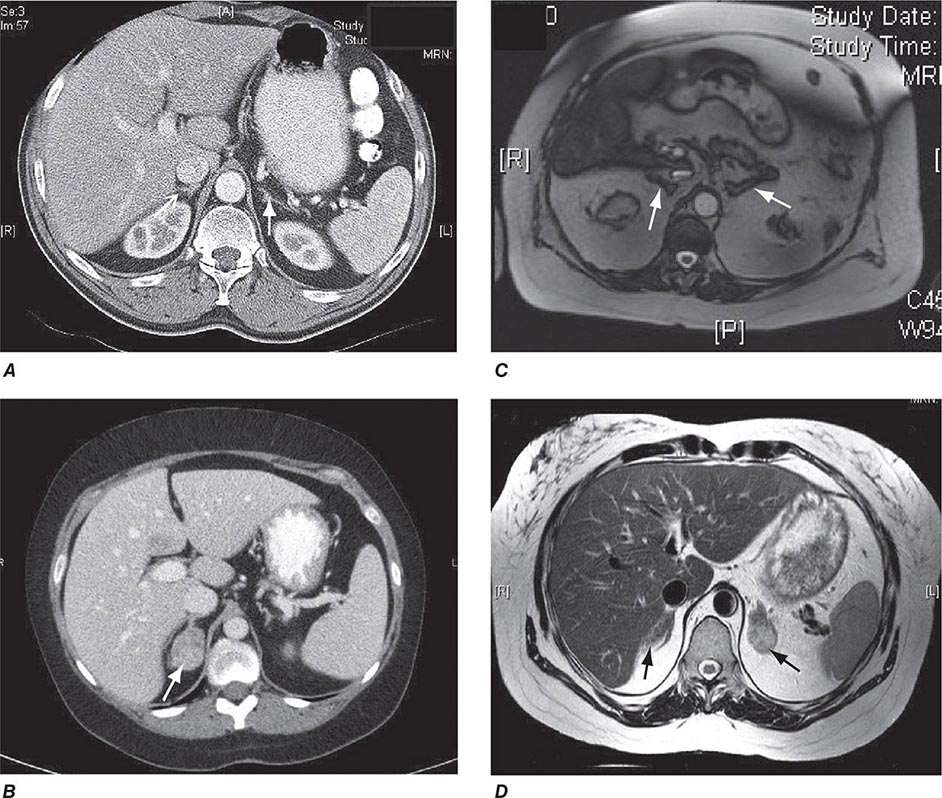
FIGURE 406-11 Adrenal imaging in Cushing’s syndrome. A. Adrenal computed tomography (CT) showing normal bilateral adrenal morphology (arrows). B. CT scan depicting a right adrenocortical adenoma (arrow) causing Cushing’s syndrome. C. Magnetic resonance imaging (MRI) showing bilateral adrenal hyperplasia due to excess adrenocorticotropic hormone stimulation in Cushing’s disease. D. MRI showing bilateral macronodular hyperplasia causing Cushing’s syndrome.
For ACTH-dependent cortisol excess (Chap. 403), a magnetic resonance image (MRI) of the pituitary is the investigation of choice, but it may not show an abnormality in up to 40% of cases because of small tumors below the sensitivity of detection. Characteristically, pituitary corticotrope adenomas fail to enhance following gadolinium administration on T1-weighted MRI images. In all cases of confirmed ACTH-dependent Cushing’s, further tests are required for the differential diagnosis of pituitary Cushing’s disease and ectopic ACTH syndrome. These tests exploit the fact that most pituitary corticotrope adenomas still display regulatory features, including residual ACTH suppression by high-dose glucocorticoids and CRH responsiveness. In contrast, ectopic sources of ACTH are typically resistant to dexamethasone suppression and unresponsive to CRH (Fig. 406-10). However, it should be noted that a small minority of ectopic ACTH-producing tumors exhibit dynamic responses similar to pituitary corticotrope tumors. If the two tests show discordant results, or if there is any other reason for doubt, the differential diagnosis can be further clarified by performing bilateral inferior petrosal sinus sampling (IPSS) with concurrent blood sampling for ACTH in the right and left inferior petrosal sinus and a peripheral vein. An increased central/peripheral plasma ACTH ratio >2 at baseline and >3 at 2–5 min after CRH injection is indicative of Cushing’s disease (Fig. 406-10), with very high sensitivity and specificity. Of note, the results of the IPSS cannot be reliably used for lateralization (i.e., prediction of the location of the tumor within the pituitary), because there is broad interindividual variability in the venous drainage of the pituitary region. Importantly, no cortisol-lowering agents should be used prior to IPSS.
If the differential diagnostic testing indicates ectopic ACTH syndrome, then further imaging should include high-resolution, fine-cut CT scanning of the chest and abdomen for scrutiny of the lung, thymus, and pancreas. If no lesions are identified, an MRI of the chest can be considered because carcinoid tumors usually show high signal intensity on T2-weighted images. Furthermore, octreotide scintigraphy can be helpful in some cases because ectopic ACTH-producing tumors often express somatostatin receptors. Depending on the suspected cause, patients with ectopic ACTH syndrome should also undergo blood sampling for fasting gut hormones, chromogranin A, calcitonin, and biochemical exclusion of pheochromocytoma.
|
CUSHING’S SYNDROME |
Overt Cushing’s is associated with a poor prognosis if left untreated. In ACTH-independent disease, treatment consists of surgical removal of the adrenal tumor. For smaller tumors, a minimally invasive approach can be used, whereas for larger tumors and those suspected of malignancy, an open approach is preferred.
In Cushing’s disease, the treatment of choice is selective removal of the pituitary corticotrope tumor, usually via an endoscopic transsphenoidal approach. This results in an initial cure rate of 70–80% when performed by a highly experienced surgeon. However, even after initial remission following surgery, long-term follow-up is important because late relapse occurs in a significant number of patients. If pituitary disease recurs, there are several options, including second surgery, radiotherapy, stereotactic radiosurgery, and bilateral adrenalectomy. These options need to be applied in a highly individualized fashion.
In some patients with very severe, overt Cushing’s (e.g., difficult to control hypokalemic hypertension or acute psychosis), it may be necessary to introduce medical therapy to rapidly control the cortisol excess during the period leading up to surgery. Similarly, patients with metastasized, glucocorticoid-producing carcinomas may require long-term antiglucocorticoid drug treatment. In case of ectopic ACTH syndrome, in which the tumor cannot be located, one must carefully weigh whether drug treatment or bilateral adrenalectomy is the most appropriate choice, with the latter facilitating immediate cure but requiring life-long corticosteroid replacement. In this instance, it is paramount to ensure regular imaging follow-up for identification of the ectopic ACTH source.
Oral agents with established efficacy in Cushing’s syndrome are metyrapone and ketoconazole. Metyrapone inhibits cortisol synthesis at the level of 11β-hydroxylase (Fig. 406-1), whereas the antimycotic drug ketoconazole inhibits the early steps of steroidogenesis. Typical starting doses are 500 mg tid for metyrapone (maximum dose, 6 g) and 200 mg tid for ketoconazole (maximum dose, 1200 mg). Mitotane, a derivative of the insecticide o,p’DDD, is an adrenolytic agent that is also effective for reducing cortisol. Because of its side effect profile, it is most commonly used in the context of adrenocortical carcinoma, but low-dose treatment (500–1000 mg/d) has also been used in benign Cushing’s. In severe cases of cortisol excess, etomidate can be used to lower cortisol. It is administered by continuous IV infusion in low, nonanesthetic doses.
After the successful removal of an ACTH- or cortisol-producing tumor, the HPA axis will remain suppressed. Thus, hydrocortisone replacement needs to be initiated at the time of surgery and slowly tapered following recovery, to allow physiologic adaptation to normal cortisol levels. Depending on degree and duration of cortisol excess, the HPA axis may require many months or even years to resume normal function.
MINERALOCORTICOID EXCESS
Epidemiology Following the first description of a patient with an aldosterone-producing adrenal adenoma (Conn’s syndrome), mineralocorticoid excess was thought to represent a rare cause of hypertension. However, in studies systematically screening all patients with hypertension, a much higher prevalence is now recognized, ranging from 5 to 12%. The prevalence is higher when patients are preselected for hypokalemic hypertension.
Etiology The most common cause of mineralocorticoid excess is primary aldosteronism, reflecting excess production of aldosterone by the adrenal zona glomerulosa. Bilateral micronodular hyperplasia is somewhat more common than unilateral adrenal adenomas (Table 406-3). Somatic mutations in channels and enzymes responsible for increasing sodium and calcium influx in adrenal zona glomerulosa cells have been identified as prevalent causes of aldosterone-producing adrenal adenomas (Table 406-3) and, in the case of germline mutations, also of primary aldosteronism due to bilateral macronodular adrenal hyperplasia. However, bilateral adrenal hyperplasia as a cause of mineralocorticoid excess is usually micronodular but can also contain larger nodules that might be mistaken for a unilateral adenoma. In rare instances, primary aldosteronism is caused by an adrenocortical carcinoma. Carcinomas should be considered in younger patients and in those with larger tumors, because benign aldosterone-producing adenomas usually measure <2 cm in diameter.
|
CAUSES OF MINERALOCORTICOID EXCESS |
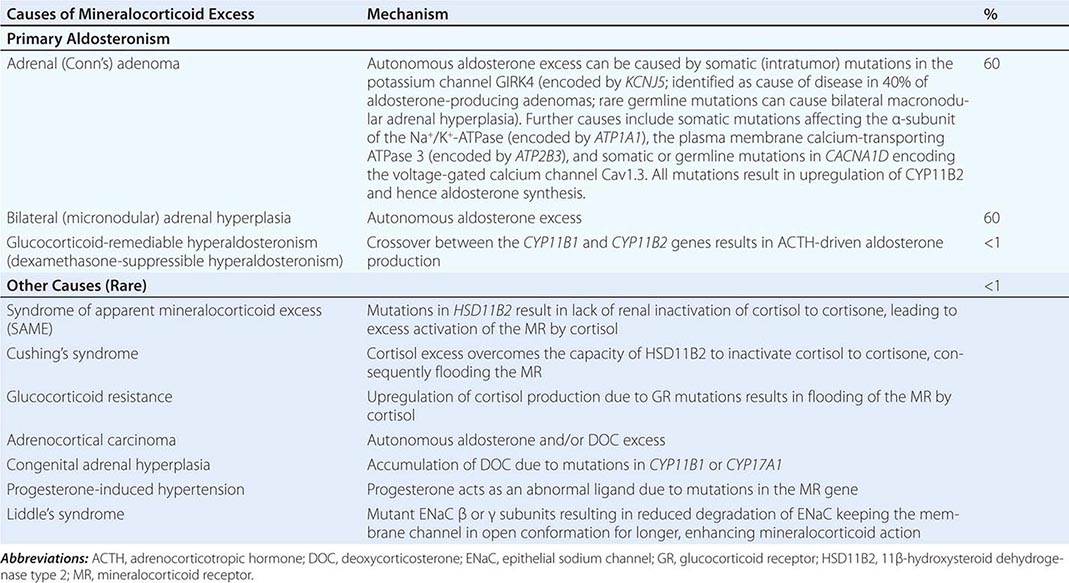
A rare cause of aldosterone excess is glucocorticoid-remediable aldosteronism (GRA), which is caused by a chimeric gene resulting from cross-over of promoter sequences between the CYP11B1 and CYP11B2 genes that are involved in glucocorticoid and mineralocorticoid synthesis, respectively (Fig. 406-1). This rearrangement brings CYP11B2 transcription under the control of ACTH receptor signaling; consequently, aldosterone production is regulated by ACTH rather than by renin. The family history can be helpful because there may be evidence for dominant transmission of hypertension. Recognition of the disorder is important because it can be associated with early-onset hypertension and strokes. In addition, glucocorticoid suppression can reduce aldosterone production.
Other rare causes of mineralocorticoid excess are listed in Table 406-3. An important cause is excess binding and activation of the mineralocorticoid receptor by a steroid other than aldosterone. Cortisol acts as a potent mineralocorticoid if it escapes efficient inactivation to cortisone by 11β-HSD2 in the kidney (Fig. 406-7). This can be caused by inactivating mutations in the HSD11B2 gene resulting in the syndrome of apparent mineralocorticoid excess (SAME) that characteristically manifests with severe hypokalemic hypertension in childhood. However, milder mutations may cause normokalemic hypertension manifesting in adulthood (type II SAME). Inhibition of 11β-HSD2 by excess licorice ingestion also results in hypokalemic hypertension, as does overwhelming of 11β-HSD2 conversion capacity by cortisol excess in Cushing’s syndrome. Deoxycorticosterone (DOC) also binds and activates the mineralocorticoid receptor and can cause hypertension if its circulating concentrations are increased. This can arise through autonomous DOC secretion by an adrenocortical carcinoma, but also when DOC accumulates as a consequence of an adrenal enzymatic block, as seen in congenital adrenal hyperplasia due to CYP11B1 (11β-hydroxylase) or CYP17A1 (17α-hydroxylase) deficiency (Fig. 406-1). Progesterone can cause hypokalemic hypertension in rare individuals who harbor a mineralocorticoid receptor mutation that enhances binding and activation by progesterone; physiologically, progesterone normally exerts antimineralocorticoid activity. Finally, excess mineralocorticoid activity can be caused by mutations in the β or γ subunits of the ENaC, disrupting its interaction with Nedd4 (Fig. 406-7), and thereby decreasing receptor internalization and degradation. The constitutively active ENAC drives hypokalemic hypertension, resulting in an autosomal dominant disorder termed Liddle’s syndrome.
Clinical Manifestations Excess activation of the mineralocorticoid receptor leads to potassium depletion and increased sodium retention, with the latter causing an expansion of extracellular and plasma volume. Increased ENaC activity also results in hydrogen depletion that can cause metabolic alkalosis. Aldosterone also has direct effects on the vascular system, where it increases cardiac remodeling and decreases compliance. Aldosterone excess may cause direct damage to the myocardium and the kidney glomeruli, in addition to secondary damage due to systemic hypertension.
The clinical hallmark of mineralocorticoid excess is hypokalemic hypertension; serum sodium tends to be normal due to the concurrent fluid retention, which in some cases can lead to peripheral edema. Hypokalemia can be exacerbated by thiazide drug treatment, which leads to increased delivery of sodium to the distal renal tubule, thereby driving potassium excretion. Severe hypokalemia can be associated with muscle weakness, overt proximal myopathy, or even hypokalemic paralysis. Severe alkalosis contributes to muscle cramps and, in severe cases, can cause tetany.
Diagnosis Diagnostic screening for mineralocorticoid excess is not currently recommended for all patients with hypertension, but should be restricted to those who exhibit hypertension associated with drug resistance, hypokalemia, an adrenal mass, or onset of disease before the age of 40 years (Fig. 406-12). The accepted screening test is concurrent measurement of plasma renin and aldosterone with subsequent calculation of the aldosterone-renin ratio (ARR) (Fig. 406-12); serum potassium needs to be normalized prior to testing. Stopping antihypertensive medication can be cumbersome, particularly in patients with severe hypertension. Thus, for practical purposes, in the first instance the patient can remain on the usual antihypertensive medications, with the exception that mineralocorticoid receptor antagonists need to be ceased at least 4 weeks prior to ARR measurement. The remaining antihypertensive drugs usually do not affect the outcome of ARR testing, except that beta blocker treatment can cause false-positive results and ACE/AT1R inhibitors can cause false-negative results in milder cases (Table 406-4).
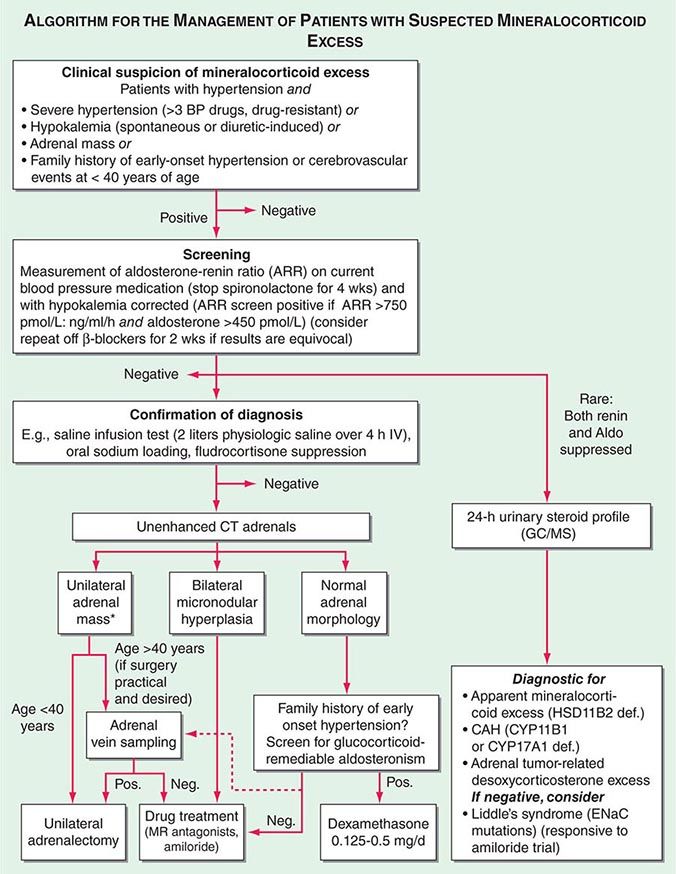
FIGURE 406-12 Management of patients with suspected mineralocorticoid excess. *Perform adrenal tumor workup (see Fig. 406-13). BP, blood pressure; CAH, congenital adrenal hyperplasia; CT, computed tomography; GC/MS, gas chromatography/mass spectrometry; PRA, plasma renin activity.
|
EFFECTS OF ANTIHYPERTENSIVE DRUGS ON THE ALDOSTERONE-RENIN-RATIO (ARR) |
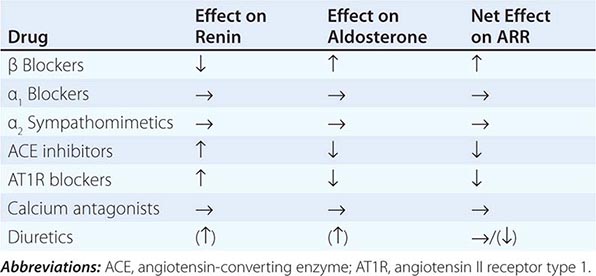
ARR screening is positive if the ratio is >750 pmol/L per ng/mL per hour, with a concurrently high normal or increased aldosterone (Fig. 406-12). If one relies on the ARR only, the likelihood of a false-positive ARR becomes greater when renin levels are very low. The characteristics of the biochemical assays are also important. Some labs measure plasma renin activity, whereas others measure plasma renin concentrations. Antibody-based assays for the measurement of serum aldosterone lack the reliability of tandem mass spectrometry assays, but these are not yet ubiquitously available.
Diagnostic confirmation of mineralocorticoid excess in a patient with positive ARR screening result should be undertaken by an endocrinologist as the tests lack optimized validation. The most straightforward is the saline infusion test, which involves the IV administration of 2 L of physiologic saline over a 4-h period. Failure of aldosterone to suppress below 140 pmol/L (5 ng/dL) is indicative of autonomous mineralocorticoid excess. Alternative tests are the oral sodium loading test (300 mmol NaCl/d for 3 days) or the fludrocortisone suppression test (0.1 mg q6h with 30 mmol NaCl q8h for 4 days); the latter can be difficult because of the risk of profound hypokalemia and increased hypertension. In patients with overt hypokalemic hypertension, strongly positive ARR, and concurrently increased aldosterone levels, confirmatory testing is usually not necessary.
Differential Diagnosis and Treatment After the diagnosis of hyperaldosteronism is established, the next step is to use adrenal imaging to further assess the cause. Fine-cut CT scanning of the adrenal region is the method of choice because it provides excellent visualization of adrenal morphology. CT will readily identify larger tumors suspicious of malignancy but may miss lesions smaller than 5 mm. The differentiation between bilateral micronodular hyperplasia and a unilateral adenoma is only required if a surgical approach is feasible and desired. Consequently, selective adrenal vein sampling (AVS) should only be carried out in surgical candidates with either no obvious lesion on CT or evidence of a unilateral lesion in patients older than 40 years, because the latter patients have a high likelihood of harboring a coincidental, endocrine-inactive adrenal adenoma (Fig. 406-12). AVS is used to compare aldosterone levels in the inferior vena cava and between the right and left adrenal veins. AVS requires concurrent measurement of cortisol to document correct placement of the catheter in the adrenal veins and should demonstrate a cortisol gradient >3 between the vena cava and each adrenal vein. Lateralization is confirmed by an aldosterone/cortisol ratio that is at least twofold higher on one side than the other. AVS is a complex procedure that requires a highly skilled interventional radiologist. Even then, the right adrenal vein can be difficult to cannulate correctly, which, if not achieved, invalidates the procedure. There is also no agreement as to whether the two adrenal veins should be cannulated simultaneously or successively and whether ACTH stimulation enhances the diagnostic value of AVS.
Patients younger than 40 years with confirmed mineralocorticoid excess and a unilateral lesion on CT can go straight to surgery, which is also indicated in patients with confirmed lateralization documented by a valid AVS procedure. Laparoscopic adrenalectomy is the preferred approach. Patients who are not surgical candidates, or with evidence of bilateral hyperplasia based on CT or AVS, should be treated medically (Fig. 406-12). Medical treatment, which can also be considered prior to surgery to avoid postsurgical hypoaldosteronism, consists primarily of the mineralocorticoid receptor antagonist spironolactone. It can be started at 12.5–50 mg bid and titrated up to a maximum of 400 mg/d to control blood pressure and normalize potassium. Side effects include menstrual irregularity, decreased libido, and gynecomastia. The more selective MR antagonist eplerenone can also be used. Doses start at 25 mg bid, and it can be titrated up to 200 mg/d. Another useful drug is the sodium channel blocker amiloride (5–10 mg bid).
In patients with normal adrenal morphology and family history of early-onset, severe hypertension, a diagnosis of GRA should be considered and can be evaluated using genetic testing. Treatment of GRA consists of administering dexamethasone, using the lowest dose possible to control blood pressure. Some patients also require additional MR antagonist treatment.
The diagnosis of nonaldosterone-related mineralocorticoid excess is based on documentation of suppressed renin and suppressed aldosterone in the presence of hypokalemic hypertension. This testing is best carried out by employing urinary steroid metabolite profiling by gas chromatography/mass spectrometry (GC/MS). An increased free cortisol over free cortisone ratio is suggestive of SAME and can be treated with dexamethasone. Steroid profiling by GC/MS also detects the steroids associated with CYP11B1 and CYP17A1 deficiency or the irregular steroid secretion pattern in a DOC-producing adrenocortical carcinoma (Fig. 406-12). If the GC/MS profile is normal, then Liddle’s syndrome should be considered. It is very sensitive to amiloride treatment but will not respond to MR antagonist treatment, because the defect is due to a constitutively active ENaC.
APPROACH TO THE PATIENT: INCIDENTALLY DISCOVERED ADRENAL MASS
Epidemiology Incidentally discovered adrenal masses, commonly termed adrenal “incidentalomas,” are common, with a prevalence of at least 2% in the general population as documented in CT and autopsy series. The prevalence increases with age, with 1% of 40-year-olds and 7% of 70-year-olds harboring an adrenal mass.
Etiology Most solitary adrenal tumors are monoclonal neoplasms. Several genetic syndromes, including MEN 1 (MEN1), MEN 2 (RET), Carney’s complex (PRKAR1A), and McCune-Albright (GNAS1), can have adrenal tumors as one of their features. Somatic mutations in MEN1, GNAS1, and PRKAR1A have been identified in a small proportion of sporadic adrenocortical adenomas. Aberrant expression of membrane receptors (gastric inhibitory peptide, α- and β-adrenergic, luteinizing hormone, vasopressin V1, and interleukin 1 receptors) have been identified in some sporadic cases of macronodular adrenocortical hyperplasia.
The majority of adrenal nodules are endocrine-inactive adrenocortical adenomas. However, larger series suggest that up to 25% of adrenal nodules are hormonally active, due to a cortisol- or aldosterone-producing adrenocortical adenoma or a pheochromocytoma associated with catecholamine excess (Table 406-5). Adrenocortical carcinoma is rare but is the cause of an adrenal mass in 5% of patients. However, the most common cause of a malignant adrenal mass is metastasis originating from another solid tissue tumor (Table 406-5).
|
CLASSIFICATION OF UNILATERAL ADRENAL MASSES |
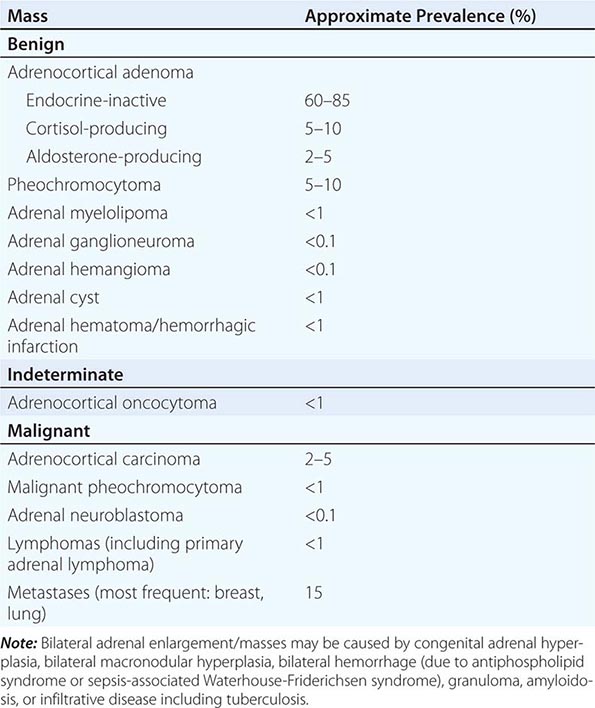
Differential Diagnosis and Treatment Patients with an adrenal mass >1 cm require a diagnostic evaluation. Two key questions need to be addressed: (1) Does the tumor autonomously secrete hormones that could have a detrimental effect on health? (2) Is the adrenal mass benign or malignant?
Hormone secretion by an adrenal mass occurs along a continuum, with a gradual increase in clinical manifestations in parallel with hormone levels. Exclusion of catecholamine excess from a pheochromocytoma arising from the adrenal medulla is a mandatory part of the diagnostic workup (Fig. 406-13). Furthermore, autonomous cortisol and aldosterone secretion resulting in Cushing’s syndrome or primary aldosteronism, respectively, require exclusion. Adrenal incidentalomas can be associated with lower levels of autonomous cortisol secretion, and patients may lack overt clinical features of Cushing’s syndrome. Nonetheless, they may exhibit one or more components of the metabolic syndrome (e.g., obesity, type 2 diabetes, or hypertension). There is ongoing debate about the optimal treatment for these patients with mild or subclinical Cushing’s syndrome. Overproduction of adrenal androgen precursors, DHEA and its sulfate, is rare and most frequently seen in the context of adrenocortical carcinoma, as are increased levels of steroid precursors such as 17-hydroxyprogesterone.
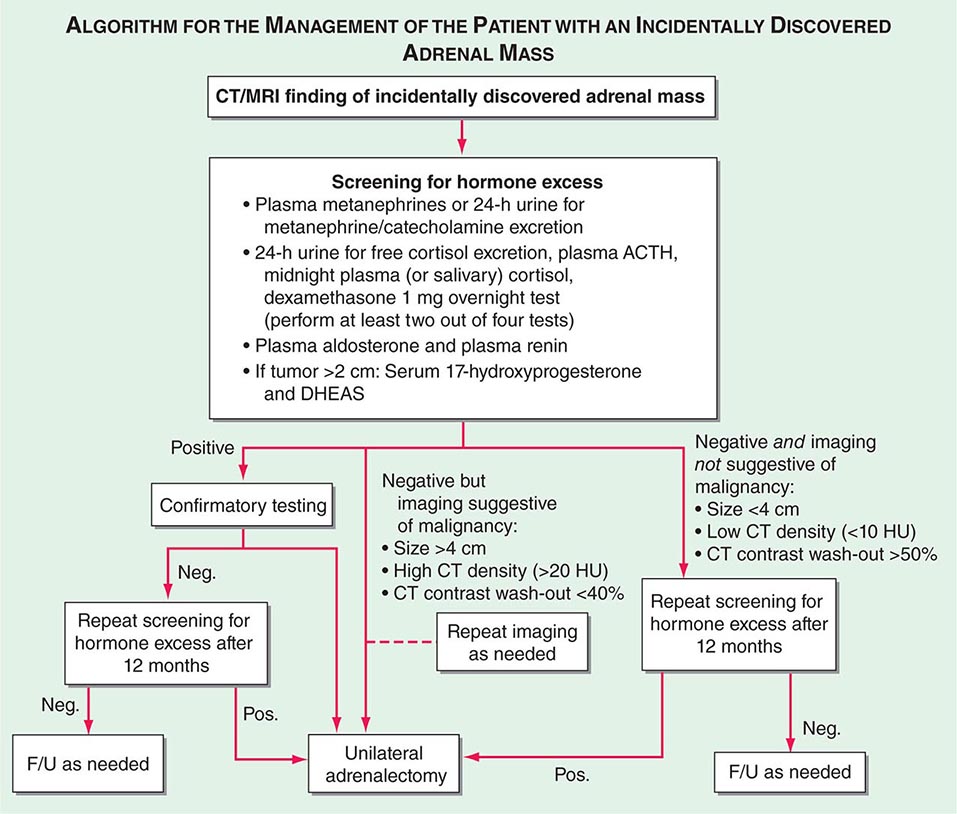
FIGURE 406-13 Management of the patient with an incidentally discovered adrenal mass. CT, computed tomography; F/U, follow-up; MRI, magnetic resonance imaging.
For the differentiation of benign from malignant adrenal masses, imaging is relatively sensitive, although specificity is suboptimal. CT is the procedure of choice for imaging the adrenal glands (Fig. 406-11). The risk of adrenocortical carcinoma, pheochromocytoma, and benign adrenal myelolipoma increases with the diameter of the adrenal mass. However, size alone is of poor predictive value, with only 80% sensitivity and 60% specificity for the differentiation of benign from malignant masses when using a 4-cm cut-off. Metastases are found with similar frequency in adrenal masses of all sizes. Tumor density on unenhanced CT is of additional diagnostic value, with most adrenocortical adenomas being lipid rich and thus presenting with low attenuation values (i.e., densities of <10 HU). By contrast, adrenocortical carcinomas, but also pheochromocytomas, usually have high attenuation values (i.e., densities >20 HU on precontrast scans). Generally, benign lesions are rounded and homogenous, whereas most malignant lesions appear lobulated and inhomogeneous. Pheochromocytoma and adrenomyelolipoma may also exhibit lobulated and inhomogeneous features. Additional information can be obtained from CT by assessment of contrast wash-out after 15 min, which is >50% in benign lesions but <40% in malignant lesions, which usually have a more extensive vascularization. MRI also allows for the visualization of the adrenal glands with somewhat lower resolution than CT. However, because it does not involve exposure to ionizing radiation, it is preferred in children, young adults, and during pregnancy. MRI has a valuable role in the characterization of indeterminate adrenal lesions using chemical shift analysis, with malignant tumors rarely showing loss of signal on opposed-phase MRI.
Fine-needle aspiration (FNA) or CT-guided biopsy of an adrenal mass is almost never indicated. FNA of a pheochromocytoma can cause a life-threatening hypertensive crisis. FNA of an adrenocortical carcinoma violates the tumor capsule and can cause needle track metastasis. FNA should only be considered in a patient with a history of nonadrenal malignancy and a newly detected adrenal mass, after careful exclusion of pheochromocytoma, and if the outcome will influence therapeutic management. It is important to recognize that in 25% of patients with a previous history of nonadrenal malignancy, a newly detected mass on CT is not a metastasis.
Adrenal masses associated with confirmed hormone excess or suspected malignancy are usually treated surgically (Fig. 406-13) or, if adrenalectomy is not feasible or desired, with medication. Preoperative exclusion of glucocorticoid excess is particularly important for the prediction of postoperative suppression of the contralateral adrenal gland, which requires glucocorticoid replacement peri- and postoperatively. If the initial decision is for observation, imaging and biochemical testing should be repeated about a year after the first assessment. However, this may be performed earlier in patients with borderline imaging or hormonal findings. There is no agreement with regard to the required long-term follow-up beyond 1 year in patients with normal biochemistry and no evidence of increased tumor size at follow-up.
ADRENOCORTICAL CARCINOMA
Adrenocortical carcinoma (ACC) is a rare malignancy with an annual incidence of 1–2 per million population. ACC is generally considered a highly malignant tumor; however, it presents with broad interindividual variability with regard to biologic characteristics and clinical behavior. Somatic mutations in the tumor-suppressor gene TP53 are found in 25% of apparently sporadic ACC. Germline TP53 mutations are the cause of the Li-Fraumeni syndrome associated with multiple solid organ cancers including ACC and are found in 25% of pediatric ACC cases; the TP53 mutation R337H is found in almost all pediatric ACC in Brazil. Other genetic changes identified in ACC include alterations in the Wnt/β-catenin pathway and in the insulin-like growth factor 2 (IGF2) cluster; IGF2 overexpression is found in 90% of ACC.
Patients with large adrenal tumors suspicious of malignancy should be managed by a multidisciplinary specialist team, including an endocrinologist, an oncologist, a surgeon, a radiologist, and a histopathologist. FNA is not indicated in suspected ACC: first, cytology and also histopathology of a core biopsy cannot differentiate between benign and malignant primary adrenal masses; second, FNA violates the tumor capsule and may even cause needle canal metastasis. Even when the entire tumor specimen is available, the histopathologic differentiation between benign and malignant lesions is a diagnostic challenge. The most common histopathologic classification is the Weiss score, taking into account high nuclear grade; mitotic rate (>5/HPF); atypical mitosis; <25% clear cells; diffuse architecture; and presence of necrosis, venous invasion, and invasion of sinusoidal structures and tumor capsule. The presence of three or more elements suggests ACC.
Although 60–70% of ACCs show biochemical evidence of steroid overproduction, in many patients, this is not clinically apparent due to the relatively inefficient steroid production by the adrenocortical cancer cells. Excess production of glucocorticoids and adrenal androgen precursors are most common. Mixed excess production of several corticosteroid classes by an adrenal tumor is generally indicative of malignancy.
Tumor staging at diagnosis (Table 406-6) has important prognostic implications and requires scanning of the chest and abdomen for local organ invasion, lymphadenopathy, and metastases. Intravenous contrast medium is necessary for maximum sensitivity for hepatic metastases. An adrenal origin may be difficult to determine on standard axial CT imaging if the tumors are large and invasive, but CT reconstructions and MRI are more informative (Fig. 406-14) using multiple planes and different sequences. Vascular and adjacent organ invasion is diagnostic of malignancy. 18-Fluoro-2-deoxy-D-glucose positron emission tomography (18-FDG PET) is highly sensitive for the detection of malignancy and can be used to detect small metastases or local recurrence that may not be obvious on CT (Fig. 406-14). However, FDG PET is not specific and therefore cannot be used for differentiating benign from malignant adrenal lesions. Metastasis in ACC most frequently occurs to liver and lung.
|
CLASSIFICATION SYSTEM FOR STAGING OF ADRENOCORTICAL CARCINOMA |
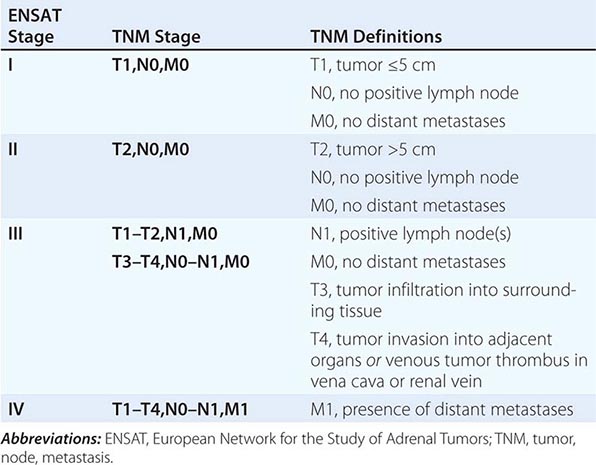
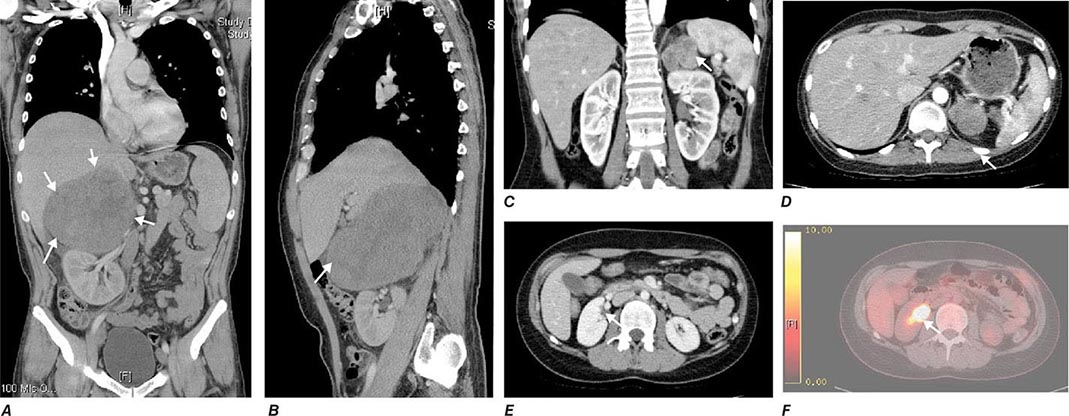
FIGURE 406-14 Imaging in adrenocortical carcinoma. Magnetic resonance imaging scan with (A) frontal and (B) lateral views of a right adrenocortical carcinoma that was detected incidentally. Computed tomography (CT) scan with (C) coronal and (D) transverse views depicting a right-sided adrenocortical carcinoma. Note the irregular border and inhomogeneous structure. CT scan (E) and positron emission tomography/CT (F) visualizing a peritoneal metastasis of an adrenocortical carcinoma in close proximity to the right kidney (arrow).
There is no established grading system for ACC, and the Weiss score carries no prognostic value; the most important prognostic histopathologic parameter is the Ki67 proliferation index, with Ki67 <10% indicative of slow to moderate growth velocity, whereas a Ki67 ≥10% is associated with poor prognosis including high risk of recurrence and rapid progression.
Cure of ACC can only be achieved by early detection and complete surgical removal. Capsule violation during primary surgery, metastasis at diagnosis, and primary treatment in a nonspecialist center are major determinants of poor survival. If the primary tumor invades adjacent organs, en bloc removal of kidney and spleen should be considered to reduce the risk of recurrence. Surgery can also be considered in a patient with metastases if there is severe tumor-related hormone excess. This indication needs to be carefully weighed against surgical risk, including thromboembolic complications, and the resulting delay in the introduction of other therapeutic options. Patients with confirmed ACC and successful removal of the primary tumor should receive adjuvant treatment with mitotane (o,p’DDD), particularly in patients with a high risk of recurrence as determined by tumor size >8 cm, histopathologic signs of vascular invasion, capsule invasion or violation, and a Ki67 proliferation index ≥10%. Adjuvant mitotane should be continued for at least 2 years, if the patient can tolerate side effects. Regular monitoring of plasma mitotane levels is mandatory (therapeutic range 14–20 mg/L; neurotoxic complications more frequent at >20 mg/L). Mitotane is usually started at 500 mg tid, with stepwise increases to a maximum dose of 2000 mg tid in days (high-dose saturation) or weeks (low-dose saturation) as tolerated. Once therapeutic range plasma mitotane levels are achieved, the dose can be tapered to maintenance doses mostly ranging from 1000 to 1500 mg tid. Mitotane treatment results in disruption of cortisol synthesis and thus requires glucocorticoid replacement; glucocorticoid replacement dose should be at least double of that usually used in adrenal insufficiency (i.e., 20 mg tid) because mitotane induces hepatic CYP3A4 activity resulting in rapid inactivation of glucocorticoids. Mitotane also increases circulating CBG, thereby decreasing the available free cortisol fraction. Single metastases can be addressed surgically or with radiofrequency ablation as appropriate. If the tumor recurs or progresses during mitotane treatment, chemotherapy should be considered; the established first-line chemotherapy regimen is the combination of cisplatin, etoposide, and doxorubicin plus continuing mitotane. Painful bone metastasis responds to irradiation. Overall survival in ACC is still poor, with 5-year survival rates of 30–40% and a median survival of 15 months in metastatic ACC.
ADRENAL INSUFFICIENCY
Epidemiology The prevalence of well-documented, permanent adrenal insufficiency is 5 in 10,000 in the general population. Hypothalamic-pituitary origin of disease is most frequent, with a prevalence of 3 in 10,000, whereas primary adrenal insufficiency has a prevalence of 2 in 10,000. Approximately one-half of the latter cases are acquired, mostly caused by autoimmune destruction of the adrenal glands; the other one-half are genetic, most commonly caused by distinct enzymatic blocks in adrenal steroidogenesis affecting glucocorticoid synthesis (i.e., congenital adrenal hyperplasia.)
Adrenal insufficiency arising from suppression of the HPA axis as a consequence of exogenous glucocorticoid treatment is much more common, occurring in 0.5–2% of the population in developed countries.
Etiology Primary adrenal insufficiency is most commonly caused by autoimmune adrenalitis. Isolated autoimmune adrenalitis accounts for 30–40%, whereas 60–70% develop adrenal insufficiency as part of autoimmune polyglandular syndromes (APS) (Chap. 408) (Table 406-7). APS1, also termed APECED (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy), is the underlying cause in 10% of patients affected by APS. APS1 is transmitted in an autosomal recessive manner and is caused by mutations in the autoimmune regulator gene AIRE. Associated autoimmune conditions overlap with those seen in APS2, but may also include total alopecia, primary hypoparathyroidism, and, in rare cases, lymphoma. APS1 patients invariably develop chronic mucocutaneous candidiasis, usually manifest in childhood, and preceding adrenal insufficiency by years or decades. The much more prevalent APS2 is of polygenic inheritance, with confirmed associations with the HLA-DR3 gene region in the major histocompatibility complex and distinct gene regions involved in immune regulation (CTLA-4, PTPN22, CLEC16A). Coincident autoimmune disease most frequently includes thyroid autoimmune disease, vitiligo, and premature ovarian failure. Less commonly, additional features may include type 1 diabetes and pernicious anemia caused by vitamin B12 deficiency.
|
CAUSES OF PRIMARY ADRENAL INSUFFICIENCY |
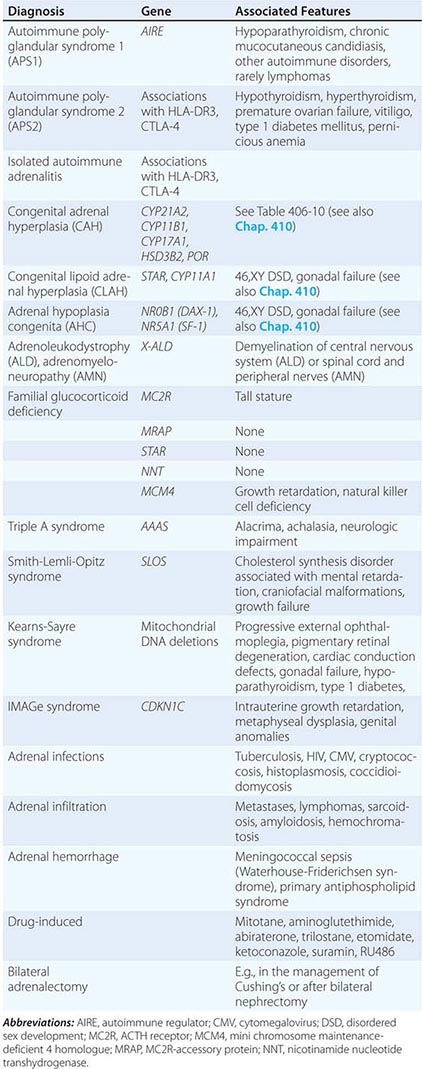
X-linked adrenoleukodystrophy has an incidence of 1:20,000 males and is caused by mutations in the X-ALD gene encoding the peroxisomal membrane transporter protein ABCD1; its disruption results in accumulation of very long chain (>24 carbon atoms) fatty acids. Approximately 50% of cases manifest in early childhood with rapidly progressive white matter disease (cerebral ALD); 35% present during adolescence or in early adulthood with neurologic features indicative of myelin and peripheral nervous system involvement (adrenomyeloneuropathy [AMN]). In the remaining 15%, adrenal insufficiency is the sole manifestation of disease. Of note, distinct mutations manifest with variable penetrance and phenotypes within affected families.
Rarer causes of adrenal insufficiency involve destruction of the adrenal glands as a consequence of infection, hemorrhage, or infiltration (Table 406-7); tuberculous adrenalitis is still a frequent cause of disease in developing countries. Adrenal metastases rarely cause adrenal insufficiency, and this occurs only with bilateral, bulky metastases.
Inborn causes of primary adrenal insufficiency other than congenital adrenal hyperplasia are rare, causing less than 1% of cases. However, their elucidation provides important insights into adrenal gland development and physiology. Mutations causing primary adrenal insufficiency (Table 406-7) include factors regulating adrenal development and steroidogenesis (DAX-1, SF-1), cholesterol synthesis, import and cleavage (DHCR7, StAR, CYP11A1), and elements of the adrenal ACTH response pathway (MC2R, MRAP) (Fig. 406-5), and factors involved in redox regulation (NNT) and DNA repair (MCM4, CDKN1C).
Secondary adrenal insufficiency is the consequence of dysfunction of the hypothalamic-pituitary component of the HPA axis (Table 406-8). Excluding iatrogenic suppression, the overwhelming majority of cases are caused by pituitary or hypothalamic tumors or their treatment by surgery or irradiation (Chap. 403). Rarer causes include pituitary apoplexy, either as a consequence of an infarcted pituitary adenoma or transient reduction in the blood supply of the pituitary during surgery or after rapid blood loss associated with parturition, also termed Sheehan’s syndrome. Isolated ACTH deficiency is rarely caused by autoimmune disease or pituitary infiltration (Table 406-8). Mutations in the ACTH precursor POMC or in factors regulating pituitary development are genetic causes of ACTH deficiency (Table 406-8).
|
CAUSES OF SECONDARY ADRENAL INSUFFICIENCY |
Clinical Manifestations In principle, the clinical features of primary adrenal insufficiency (Addison’s disease) are characterized by the loss of both glucocorticoid and mineralocorticoid secretion (Table 406-9). In secondary adrenal insufficiency, only glucocorticoid deficiency is present, as the adrenal itself is intact and thus still amenable to regulation by the RAA system. Adrenal androgen secretion is disrupted in both primary and secondary adrenal insufficiency (Table 406-9). Hypothalamic-pituitary disease can lead to additional clinical manifestations due to involvement of other endocrine axes (thyroid, gonads, growth hormone, prolactin) or visual impairment with bitemporal hemianopia caused by chiasmal compression. It is important to recognize that iatrogenic adrenal insufficiency caused by exogenous glucocorticoid suppression of the HPA axis may result in all symptoms associated with glucocorticoid deficiency (Table 406-9), if exogenous glucocorticoids are stopped abruptly. However, patients will appear clinically cushingoid as a result of the preceding overexposure to glucocorticoids.
|
SIGNS AND SYMPTOMS OF ADRENAL INSUFFICIENCY |
Abbreviations: AVP, arginine vasopressin; TSH, thyroid-stimulating hormone.
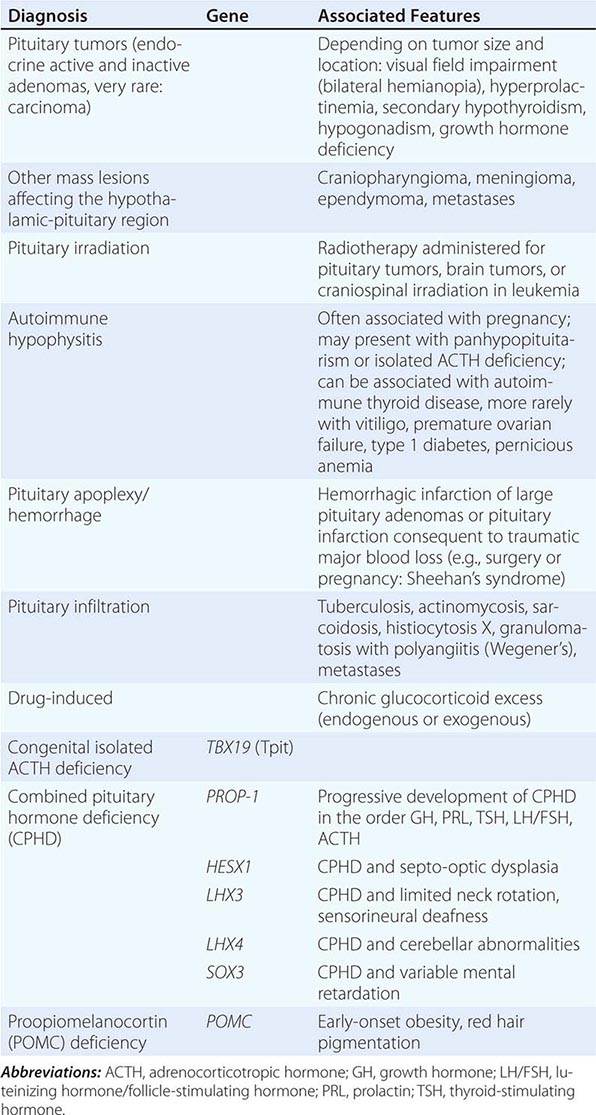
Chronic adrenal insufficiency manifests with relatively nonspecific signs and symptoms such as fatigue and loss of energy, often resulting in delayed or missed diagnoses (e.g., as depression or anorexia). A distinguishing feature of primary adrenal insufficiency is hyperpigmentation, which is caused by excess ACTH stimulation of melanocytes. Hyperpigmentation is most pronounced in skin areas exposed to increased friction or shear stress and is increased by sunlight (Fig. 406-15). Conversely, in secondary adrenal insufficiency, the skin has an alabaster-like paleness due to lack of ACTH secretion.
FIGURE 406-15 Clinical features of Addison’s disease. Note the hyperpigmentation in areas of increased friction including (A) palmar creases, (B) dorsal foot, (C) nipples and axillary region, and (D) patchy hyperpigmentation of the oral mucosa.
Hyponatremia is a characteristic biochemical feature in primary adrenal insufficiency and is found in 80% of patients at presentation. Hyperkalemia is present in 40% of patients at initial diagnosis. Hyponatremia is primarily caused by mineralocorticoid deficiency but can also occur in secondary adrenal insufficiency due to diminished inhibition of antidiuretic hormone (ADH) release by cortisol, resulting in mild syndrome of inappropriate secretion of antidiuretic hormone (SIADH). Glucocorticoid deficiency also results in slightly increased TSH concentrations that normalize within days to weeks after initiation of glucocorticoid replacement.
Acute adrenal insufficiency usually occurs after a prolonged period of nonspecific complaints and is more frequently observed in patients with primary adrenal insufficiency, due to the loss of both glucocorticoid and mineralocorticoid secretion. Postural hypotension may progress to hypovolemic shock. Adrenal insufficiency may mimic features of acute abdomen with abdominal tenderness, nausea, vomiting, and fever. In some cases, the primary presentation may resemble neurologic disease, with decreased responsiveness, progressing to stupor and coma. An adrenal crisis can be triggered by an intercurrent illness, surgical or other stress, or increased glucocorticoid inactivation (e.g., hyperthyroidism).
Diagnosis The diagnosis of adrenal insufficiency is established by the short cosyntropin test, a safe and reliable tool with excellent predictive diagnostic value (Fig. 406-16). The cut-off for failure is usually defined at cortisol levels of <500–550 nmol/L (18–20 μg/dL) sampled 30–60 min after ACTH stimulation; the exact cut-off is dependent on the locally available assay. During the early phase of HPA disruption (e.g., within 4 weeks of pituitary insufficiency), patients may still respond to exogenous ACTH stimulation. In this circumstance, the ITT is an alternative choice but is more invasive and should be carried out only under a specialist’s supervision (see above). Induction of hypoglycemia is contraindicated in individuals with diabetes mellitus, cardiovascular disease, or history of seizures. Random serum cortisol measurements are of limited diagnostic value, because baseline cortisol levels may be coincidentally low due to the physiologic diurnal rhythm of cortisol secretion (Fig. 406-3). Similarly, many patients with secondary adrenal insufficiency have relatively normal baseline cortisol levels but fail to mount an appropriate cortisol response to ACTH, which can only be revealed by stimulation testing. Importantly, tests to establish the diagnosis of adrenal insufficiency should never delay treatment. Thus, in a patient with suspected adrenal crisis, it is reasonable to draw baseline cortisol levels, provide replacement therapy, and defer formal stimulation testing until a later time.
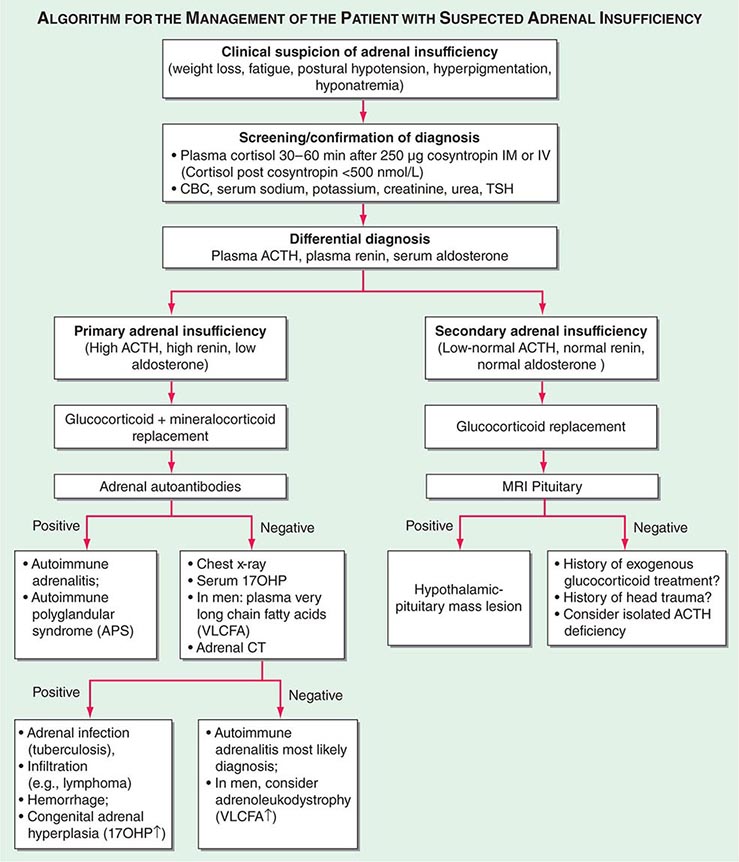
FIGURE 406-16 Management of the patient with suspected adrenal insufficiency. ACTH, adrenocorticotropic hormone; CBC, complete blood count; MRI, magnetic resonance imaging; PRA, plasma renin activity; TSH, thyroid-stimulating hormone.
Once adrenal insufficiency is confirmed, measurement of plasma ACTH is the next step, with increased or inappropriately low levels defining primary and secondary origin of disease, respectively (Fig. 406-16). In primary adrenal insufficiency, increased plasma renin will confirm the presence of mineralocorticoid deficiency. At initial presentation, patients with primary adrenal insufficiency should undergo screening for steroid autoantibodies as a marker of autoimmune adrenalitis. If these tests are negative, adrenal imaging by CT is indicated to investigate possible hemorrhage, infiltration, or masses. In male patients with negative autoantibodies in the plasma, very-long-chain fatty acids should be measured to exclude X-ALD. Patients with inappropriately low ACTH, in the presence of confirmed cortisol deficiency, should undergo hypothalamic-pituitary imaging by MRI. Features suggestive of preceding pituitary apoplexy, such as sudden-onset severe headache or history of previous head trauma, should be carefully explored, particularly in patients with no obvious MRI lesion.
|
TREATMENT |
ACUTE ADRENAL INSUFFICIENCY |
Acute adrenal insufficiency requires immediate initiation of rehydration, usually carried out by saline infusion at initial rates of 1 L/h with continuous cardiac monitoring. Glucocorticoid replacement should be initiated by bolus injection of 100 mg hydrocortisone, followed by the administration of 100–200 mg hydrocortisone over 24 h, either by continuous infusion or by bolus IV or IM injections. Mineralocorticoid replacement can be initiated once the daily hydrocortisone dose has been reduced to <50 mg because at higher doses hydrocortisone provides sufficient stimulation of mineralocorticoid receptors.
Glucocorticoid replacement for the treatment of chronic adrenal insufficiency should be administered at a dose that replaces the physiologic daily cortisol production, which is usually achieved by the oral administration of 15–25 mg hydrocortisone in two to three divided doses. Pregnancy may require an increase in hydrocortisone dose by 50% during the last trimester. In all patients, at least one-half of the daily dose should be administered in the morning. Currently available glucocorticoid preparations fail to mimic the physiologic cortisol secretion rhythm (Fig. 406-3). Long-acting glucocorticoids such as prednisolone or dexamethasone are not preferred because they result in increased glucocorticoid exposure due to extended glucocorticoid receptor activation at times of physiologically low cortisol secretion. There are no well-established dose equivalencies, but as a guide, equipotency can be assumed for 1 mg hydrocortisone, 1.6 mg cortisone acetate, 0.2 mg prednisolone, 0.25 mg prednisone, and 0.025 mg dexamethasone.
Monitoring of glucocorticoid replacement is mainly based on the history and examination for signs and symptoms suggestive of glucocorticoid over- or underreplacement, including assessment of body weight and blood pressure. Plasma ACTH, 24-h urinary free cortisol, or serum cortisol day curves reflect whether hydrocortisone has been taken or not, but do not convey reliable information about replacement quality. In patients with isolated primary adrenal insufficiency, monitoring should include screening for autoimmune thyroid disease, and female patients should be made aware of the possibility of premature ovarian failure. Supraphysiologic glucocorticoid treatment with doses equivalent to 30 mg hydrocortisone or more will affect bone metabolism, and these patients should undergo regular bone mineral density evaluation. All patients with adrenal insufficiency need to be instructed about the requirement for stress-related glucocorticoid dose adjustments. These generally consist of doubling the routine oral glucocorticoid dose in the case of intercurrent illness with fever and bed rest and the need for IV hydrocortisone injection at a daily dose of 100 mg in cases of prolonged vomiting, surgery, or trauma. Patients living or traveling in regions with delayed access to acute health care should carry a hydrocortisone self-injection emergency kit, in addition to their usual steroid emergency cards and bracelets.
Mineralocorticoid replacement in primary adrenal insufficiency should be initiated at a dose of 100–150 μg fludrocortisone. The adequacy of treatment can be evaluated by measuring blood pressure, sitting and standing, to detect a postural drop indicative of hypovolemia. In addition, serum sodium, potassium, and plasma renin should be measured regularly. Renin levels should be kept in the upper normal reference range. Changes in glucocorticoid dose may also impact on mineralocorticoid replacement as cortisol also binds the mineralocorticoid receptor; 40 mg hydrocortisone is equivalent to 100 μg fludrocortisone. In patients living or traveling in areas with hot or tropical weather conditions, the fludrocortisone dose should be increased by 50–100 μg during the summer. Mineralocorticoid dose may also need to be adjusted during pregnancy, due to the antimineralocorticoid activity of progesterone, but this is less often required than hydrocortisone dose adjustment. Plasma renin cannot serve as a monitoring tool during pregnancy, because renin rises physiologically during gestation.
Adrenal androgen replacement is an option in patients with lack of energy, despite optimized glucocorticoid and mineralocorticoid replacement. It may also be indicated in women with features of androgen deficiency, including loss of libido. Adrenal androgen replacement can be achieved by once-daily administration of 25–50 mg DHEA. Treatment is monitored by measurement of DHEAS, androstenedione, testosterone, and sex hormone–binding globulin (SHBG) 24 h after the last DHEA dose.
CONGENITAL ADRENAL HYPERPLASIA
(See also Chap. 410) Congenital adrenal hyperplasia (CAH) is caused by mutations in genes encoding steroidogenic enzymes involved in glucocorticoid synthesis (CYP21A2, CYP17A1, HSD3B2, CYP11B1) or in the cofactor enzyme P450 oxidoreductase that serves as an electron donor to CYP21A2 and CYP17A1 (Fig. 406-1). Invariably, patients affected by CAH exhibit glucocorticoid deficiency. Depending on the exact step of enzymatic block, they may also have excess production of mineralocorticoids or deficient production of sex steroids (Table 406-10). The diagnosis of CAH is readily established by measurement of the steroids accumulating before the distinct enzymatic block, either in serum or in urine, preferably by the use of mass spectrometry–based assays (Table 406-10).
|
VARIANTS OF CONGENITAL ADRENAL HYPERPLASIA |

Mutations in CYP21A2 are the most prevalent cause of CAH, responsible for 90–95% of cases. 21-Hydroxylase deficiency disrupts glucocorticoid and mineralocorticoid synthesis (Fig. 406-1), resulting in diminished negative feedback via the HPA axis. This leads to increased pituitary ACTH release, which drives increased synthesis of adrenal androgen precursors and subsequent androgen excess. The degree of impairment of glucocorticoid and mineralocorticoid secretion depends on the severity of mutations. Major loss-of-function mutations result in combined glucocorticoid and mineralocorticoid deficiency (classic CAH, neonatal presentation), whereas less severe mutations affect glucocorticoid synthesis only (simple virilizing CAH, neonatal or early childhood presentation). The mildest mutations result in the least severe clinical phenotype, nonclassic CAH, usually presenting during adolescence and early adulthood and with preserved glucocorticoid production.
Androgen excess is present in all patients and manifests with broad phenotypic variability, ranging from severe virilization of the external genitalia in neonatal girls (e.g., 46,XX disordered sex development [DSD]) to hirsutism and oligomenorrhea resembling a polycystic ovary syndrome phenotype in young women with nonclassic CAH. In countries without neonatal screening for CAH, boys with classic CAH usually present with life-threatening adrenal crisis in the first few weeks of life (salt-wasting crisis); a simple-virilizing genotype manifests with precocious pseudo-puberty and advanced bone age in early childhood, whereas men with nonclassic CAH are usually detected only through family screening.
Glucocorticoid treatment is more complex than for other causes of primary adrenal insufficiency as it not only needed to replace missing glucocorticoids but also to control the increased ACTH drive and subsequent androgen excess. Current treatment is hampered by the lack of glucocorticoid preparations that mimic the diurnal cortisol secretion profile, resulting in a prolonged period of ACTH stimulation and subsequent androgen production during the early morning hours. In childhood, optimization of growth and pubertal development are important goals of glucocorticoid treatment, in addition to prevention of adrenal crisis and treatment of 46,XX DSD. In adults, the focus shifts to preserving fertility and preventing side effects of glucocorticoid overtreatment, namely, the metabolic syndrome and osteoporosis. Fertility can be compromised in women due to oligomenorrhea/amenorrhea with chronic anovulation as a consequence of androgen excess. Men may develop so-called testicular adrenal rest tumors (Fig. 406-17). These consist of hyperplastic cells with adrenocortical characteristics located in the rete testis and should not be confused with testicular tumors. Testicular adrenal rest tissue can compromise sperm production and induce fibrosis that may be irreversible.

FIGURE 406-17 Imaging in congenital adrenal hyperplasia (CAH). Adrenal computed tomography scans showing homogenous bilateral hyperplasia in a young patient with classic CAH (A) and macronodular bilateral hyperplasia (B) in a middle-aged patient with classic CAH with longstanding poor disease control. Magnetic resonance imaging scan with T1-weighted (C) and T2-weighted (D) images showing bilateral testicular adrenal rest tumors (arrows) in a young patient with salt-wasting congenital adrenal hyperplasia. (Courtesy of N. Reisch.)
|
TREATMENT |
CONGENITAL ADRENAL HYPERPLASIA |
Hydrocortisone is a good treatment option for the prevention of adrenal crisis, but longer acting prednisolone may be needed to control androgen excess. In children, hydrocortisone is given in divided doses at 1–1.5 times the normal cortisol production rate (about 10–13 mg/m2 per day). In adults, if hydrocortisone does not suffice, intermediate-acting glucocorticoids (e.g., prednisone) may be given, using the lowest dose necessary to suppress excess androgen production. For achieving fertility, dexamethasone treatment may be required, but should be only given for the shortest possible time period to limit adverse metabolic side effects. Biochemical monitoring should include androstenedione and testosterone, aiming for the normal sex-specific reference range. 17-Hydroxyprogesterone (17OHP) is a useful marker of overtreatment, indicated by 17OHP levels within the normal range of healthy controls. Glucocorticoid overtreatment may suppress the hypothalamic-pituitary-gonadal axis. Thus, treatment needs to be carefully titrated against clinical features of disease control. Stress dose glucocorticoids should be given at double or triple the daily dose for surgery, acute illness, or severe trauma. Poorly controlled CAH can result in adrenocortical hyperplasia, which gave the disease its name, and may present as macronodular hyperplasia subsequent to long-standing ACTH excess (Fig. 406-17). The nodular areas can develop autonomous adrenal androgen production and may be unresponsive to glucocorticoid treatment.
Mineralocorticoid requirements change during life and are higher in children, explained by relative mineralocorticoid resistance that diminishes with ongoing maturation of the kidney. Children with CAH usually receive mineralocorticoid and salt replacement. However, young adults with CAH should undergo reassessment of their mineralocorticoid reserve. Plasma renin should be regularly monitored and kept within the upper half of the normal reference range.
407 |
Pheochromocytoma |
Pheochromocytomas and paragangliomas are catecholamine-producing tumors derived from the sympathetic or parasympathetic nervous system. These tumors may arise sporadically or be inherited as features of multiple endocrine neoplasia type 2, von Hippel–Lindau disease, or several other pheochromocytoma-associated syndromes. The diagnosis of pheochromocytomas identifies a potentially correctable cause of hypertension, and their removal can prevent hypertensive crises that can be lethal. The clinical presentation is variable, ranging from an adrenal incidentaloma to a hypertensive crisis with associated cerebrovascular or cardiac complications.
EPIDEMIOLOGY
Pheochromocytoma is estimated to occur in 2–8 of 1 million persons per year, and ∼0.1% of hypertensive patients harbor a pheochromocytoma. The mean age at diagnosis is ∼40 years, although the tumors can occur from early childhood until late in life. The classic “rule of tens” for pheochromocytomas states that ∼10% are bilateral, 10% are extra-adrenal, and 10% are malignant.
ETIOLOGY AND PATHOGENESIS
Pheochromocytomas and paragangliomas are well-vascularized tumors that arise from cells derived from the sympathetic (e.g., adrenal medulla) or parasympathetic (e.g., carotid body, glomus vagale) paraganglia (Fig. 407-1). The name pheochromocytoma reflects the black-colored staining caused by chromaffin oxidation of catecholamines; although a variety of terms have been used to describe these tumors, most clinicians use this designation to describe symptomatic catecholamine-producing tumors, including those in extra-adrenal retroperitoneal, pelvic, and thoracic sites. The term paraganglioma is used to describe catecholamine-producing tumors in the skull base and neck; these tumors may secrete little or no catecholamine. In contrast to common clinical parlance, the World Health Organization (WHO) restricts the term pheochromocytoma to adrenal tumors and applies the term paraganglioma to tumors at all other sites.
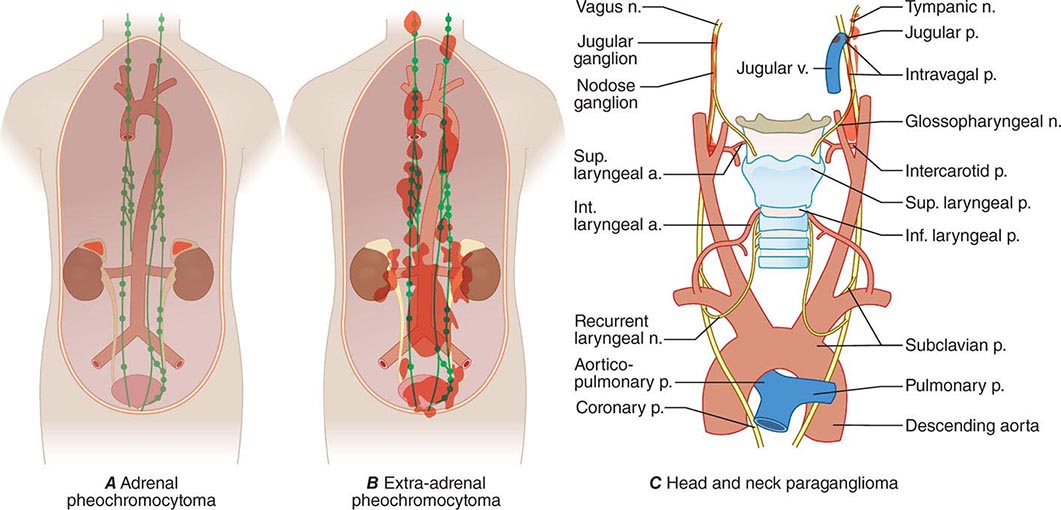
FIGURE 407-1 The paraganglial system and topographic sites (in red) of pheochromocytomas and paragangliomas. (Parts A and B from WM Manger, RW Gifford: Clinical and experimental pheochromocytoma. Cambridge, Blackwell Science, 1996; Part C from GG Glenner, PM Grimley: Tumors of the Extra-adrenal Paraganglion System [Including Chemoreceptors], Atlas of Tumor Pathology, 2nd Series, Fascicle 9. Washington, DC, AFIP, 1974.)
The etiology of sporadic pheochromocytomas and paragangliomas is unknown. However, 25–33% of patients have an inherited condition, including germ-line mutations in the classically recognized RET, VHL, NF1, SDHB, SDHC, and SDHD genes or in the more recently recognized SDHA, SDHAF2, TMEM127, and MAX genes. Biallelic gene inactivation has been demonstrated for the VHL, NF1, and SDH genes, whereas RET mutations activate receptor tyrosine kinase activity. SDH is an enzyme of the Krebs cycle and the mitochondrial respiratory chain. The VHL protein is a component of a ubiquitin E3 ligase. VHL mutations reduce protein degradation, resulting in upregulation of components involved in cell cycle progression, glucose metabolism, and oxygen sensing.
CLINICAL FEATURES
Its clinical presentation is so variable that pheochromocytoma has been termed “the great masquerader” (Table 407-1). Among the presenting manifestations, episodes of palpitation, headache, and profuse sweating are typical, and these manifestations constitute a classic triad. The presence of all three manifestations in association with hypertension makes pheochromocytoma a likely diagnosis. However, a pheochromocytoma can be asymptomatic for years, and some tumors grow to a considerable size before patients note symptoms.
|
CLINICAL FEATURES ASSOCIATED WITH PHEOCHROMOCYTOMA, LISTED BY FR£EQUENCY OF OCCURRENCE |
The dominant sign is hypertension. Classically, patients have episodic hypertension, but sustained hypertension is also common. Catecholamine crises can lead to heart failure, pulmonary edema, arrhythmias, and intracranial hemorrhage. During episodes of hormone release, which can occur at widely divergent intervals, patients are anxious and pale, and they experience tachycardia and palpitations. These paroxysms generally last <1 h and may be precipitated by surgery, positional changes, exercise, pregnancy, urination (particularly with bladder pheochromocytomas), and various medications (e.g., tricyclic antidepressants, opiates, metoclopramide).
DIAGNOSIS
The diagnosis is based on documentation of catecholamine excess by biochemical testing and localization of the tumor by imaging. These two criteria are of equal importance, although measurement of catecholamines or metanephrines (their methylated metabolites) is traditionally the first step in diagnosis.
Biochemical testing Pheochromocytomas and paragangliomas synthesize and store catecholamines, which include norepinephrine (noradrenaline), epinephrine (adrenaline), and dopamine. Elevated plasma and urinary levels of catecholamines and metanephrines form the cornerstone of diagnosis. The characteristic fluctuations in the hormonal activity of tumors results in considerable variation in serial catecholamine measurements. However, most tumors continuously leak O-methylated metabolites, which are detected by measurement of metanephrines.
Catecholamines and metanephrines can be measured by different methods, including high-performance liquid chromatography, enzyme-linked immunosorbent assay, and liquid chromatography/mass spectrometry. When pheochromocytoma is suspected on clinical grounds (i.e., when values are three times the upper limit of normal), this diagnosis is highly likely regardless of the assay used. However, as summarized in Table 407-2, the sensitivity and specificity of available biochemical tests vary greatly, and these differences are important in assessing patients with borderline elevations of different compounds. Urinary tests for metanephrines (total or fractionated) and catecholamines are widely available and are used commonly for initial evaluation. Among these tests, those for the fractionated metanephrines and catecholamines are the most sensitive. Plasma tests are more convenient and include measurements of catecholamines and metanephrines. Measurements of plasma metanephrine are the most sensitive and are less susceptible to false-positive elevations from stress, including venipuncture. Although the incidence of false-positive test results has been reduced by the introduction of newer assays, physiologic stress responses and medications that increase catecholamine levels still can confound testing. Because the tumors are relatively rare, borderline elevations are likely to represent false-positive results. In this circumstance, it is important to exclude dietary or drug-related factors (withdrawal of levodopa or use of sympathomimetics, diuretics, tricyclic antidepressants, alpha and beta blockers) that might cause false-positive results and then to repeat testing or perform a clonidine suppression test (i.e., the measurement of plasma normetanephrine 3 h after oral administration of 300 μg of clonidine). Other pharmacologic tests, such as the phentolamine test and the glucagon provocation test, are of relatively low sensitivity and are not recommended.
|
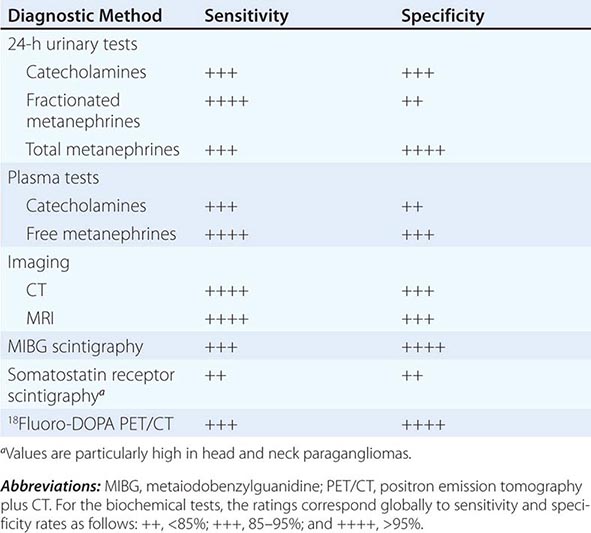
BIOCHEMICAL AND IMAGING METHODS USED FOR DIAGNOSIS OF PHEOCHROMOCYTOMA AND PARAGANGLIOMA |
Diagnostic Imaging A variety of methods have been used to localize pheochromocytomas and paragangliomas (Table 407-2). CT and MRI are similar in sensitivity and should be performed with contrast. T2-weighted MRI with gadolinium contrast is optimal for detecting pheochromocytomas and is somewhat better than CT for imaging extraadrenal pheochromocytomas and paragangliomas. About 5% of adrenal incidentalomas, which usually are detected by CT or MRI, prove to be pheochromocytomas upon endocrinologic evaluation.
Tumors also can be localized by procedures using radioactive tracers, including 131I- or 123I-metaiodobenzylguanidine (MIBG) scintigraphy, 111In-somatostatin analogue scintigraphy, 18F-DOPA positron emission tomography (PET), or 18F-fluorodeoxyglucose (FDG) PET. Because these agents exhibit selective uptake in paragangliomas, nuclear imaging is particularly useful in the hereditary syndromes.
Differential Diagnosis When the possibility of a pheochromocytoma is being entertained, other disorders to consider include essential hypertension, anxiety attacks, use of cocaine or amphetamines, mastocytosis or carcinoid syndrome (usually without hypertension), intracranial lesions, clonidine withdrawal, autonomic epilepsy, and factitious crises (usually from use of sympathomimetic amines). When an asymptomatic adrenal mass is identified, likely diagnoses other than pheochromocytoma include a nonfunctioning adrenal adenoma, an aldosteronoma, and a cortisol-producing adenoma (Cushing’s syndrome).
|
TREATMENT |
PHEOCHROMOCYTOMA |
Complete tumor removal, the ultimate therapeutic goal, can be achieved by partial or total adrenalectomy. It is important to preserve the normal adrenal cortex, particularly in hereditary disorders in which bilateral pheochromocytomas are most likely. Preoperative preparation of the patient is important. Before surgery, blood pressure should be consistently below 160/90 mmHg. Classically, blood pressure has been controlled by α-adrenergic blockers (oral phenoxybenzamine, 0.5–4 mg/kg of body weight). Because patients are volume-constricted, liberal salt intake and hydration are necessary to avoid severe orthostasis. Oral prazosin or intravenous phentolamine can be used to manage paroxysms while adequate alpha blockade is awaited. Beta blockers (e.g., 10 mg of propranolol three or four times per day) can then be added. Other antihypertensives, such as calcium channel blockers or angiotensin-converting enzyme inhibitors, have also been used effectively.
Surgery should be performed by teams of surgeons and anesthesiologists with experience in the management of pheochromocytomas. Blood pressure can be labile during surgery, particularly at the outset of intubation or when the tumor is manipulated. Nitroprusside infusion is useful for intraoperative hypertensive crises, and hypotension usually responds to volume infusion.
Minimally invasive techniques (laparoscopy or retroperitoneoscopy) have become the standard approaches in pheochromocytoma surgery. They are associated with fewer complications, a faster recovery, and optimal cosmetic results. Extra-adrenal abdominal and most thoracic pheochromocytomas also can also be removed endoscopically. Postoperatively, catecholamine normalization should be documented. An adrenocorticotropic hormone test should be used to exclude cortisol deficiency when bilateral adrenal cortex–sparing surgery has been performed.
MALIGNANT PHEOCHROMOCYTOMA
About 5–10% of pheochromocytomas and paragangliomas are malignant. The diagnosis of malignant pheochromocytoma is problematic. The typical histologic criteria of cellular atypia, presence of mitoses, and invasion of vessels or adjacent tissues are insufficient for the diagnosis of malignancy in pheochromocytoma. Thus, the term malignant pheochromocytoma is restricted to tumors with distant metastases, most commonly found by nuclear medicine imaging in lungs, bone, or liver—locations suggesting a vascular pathway of spread. Because hereditary syndromes are associated with multifocal tumor sites, these features should be anticipated in patients with germ-line mutations of RET, VHL, SDHD, or SDHB. However, distant metastases also occur in these syndromes, especially in carriers of SDHB mutations.
Treatment of malignant pheochromocytoma or paraganglioma is challenging. Options include tumor mass reduction, alpha blockers for symptoms, chemotherapy, and nuclear medicine radiotherapy. The first-line choice is nuclear medicine therapy for scintigraphically documented metastases, preferably with 131I-MIBG in 200-mCi doses at monthly intervals over three to six cycles. Averbuch’s chemotherapy protocol includes dacarbazine (600 mg/m2 on days 1 and 2), cyclophosphamide (750 mg/m2 on day 1), and vincristine (1.4 mg/m2 on day 1), all repeated every 21 days for three to six cycles. Palliation (stable disease to shrinkage) is achieved in about one-half of patients. Other chemotherapeutic options are sunitinib and temozolomide/thalidomide. The prognosis of metastatic pheochromocytoma or paraganglioma is variable, with 5-year survival rates of 30–60%.
PHEOCHROMOCYTOMA IN PREGNANCY
Pheochromocytomas occasionally are diagnosed in pregnancy. Endoscopic removal, preferably in the fourth to sixth month of gestation, is possible and can be followed by uneventful childbirth. Regular screening in families with inherited pheochromocytomas provides an opportunity to identify and remove asymptomatic tumors in women of reproductive age.
PHEOCHROMOCYTOMA-ASSOCIATED SYNDROMES
About 25–33% of patients with a pheochromocytoma or paraganglioma have an inherited syndrome. At diagnosis, patients with inherited syndromes are a mean of ∼15 years younger than patients with sporadic tumors.
Neurofibromatosis type 1 (NF1) was the first described pheochromocytoma-associated syndrome (Chap. 118). The NF1 gene functions as a tumor suppressor by regulating the Ras signaling cascade. Classic features of neurofibromatosis include multiple neurofibromas, café au lait spots, axillary freckling of the skin, and Lisch nodules of the iris (Fig. 407-2). Pheochromocytomas occur in only ∼1% of these patients and are located predominantly in the adrenals. Malignant pheochromocytoma is not uncommon.
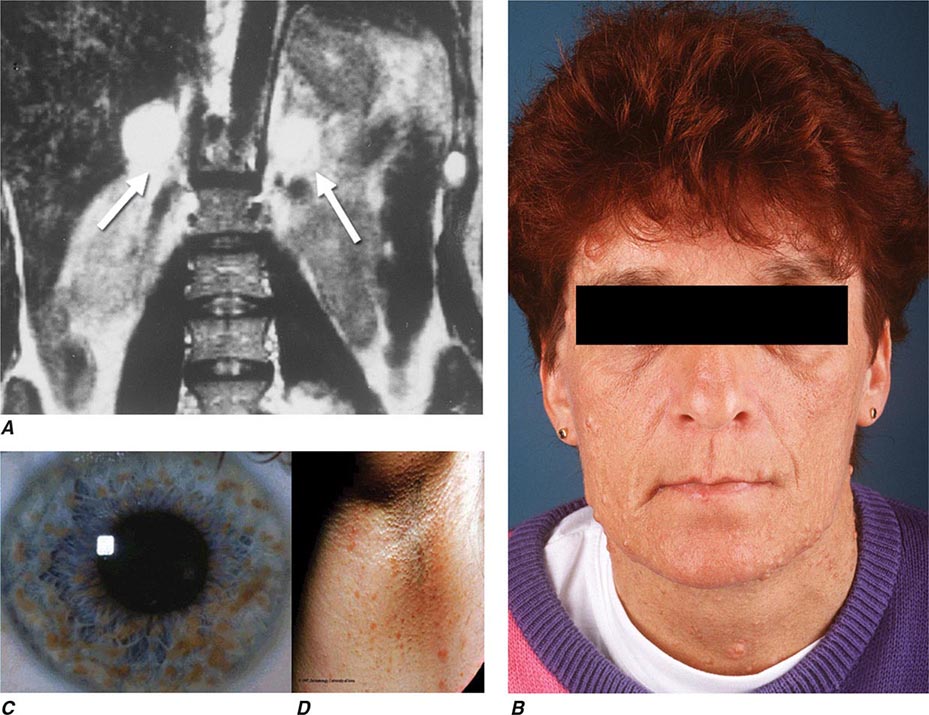
FIGURE 407-2 Neurofibromatosis. A. MRI of bilateral adrenal pheochromocytoma. B. Cutaneous neurofibromas. C. Lisch nodules of the iris. D. Axillary freckling. (Part A from HPH Neumann et al: Keio J Med 54:15, 2005; with permission.)
The best-known pheochromocytoma-associated syndrome is the autosomal dominant disorder multiple endocrine neoplasia type 2 (MEN2) (Chap. 408). Both types of MEN2 (2A and 2B) are caused by mutations in RET (rearranged during transfection), which encodes a tyrosine kinase. The locations of RET mutations correlate with the severity of disease and the type of MEN2 (Chap. 408). MEN2A is characterized by medullary thyroid carcinoma (MTC), pheochromocytoma, and hyperparathyroidism; MEN2B also includes MTC and pheochromocytoma as well as multiple mucosal neuromas, marfanoid habitus, and other developmental disorders, though it typically lacks hyperparathyroidism. MTC is found in virtually all patients with MEN2, but pheochromocytoma occurs in only ∼50% of these patients. Nearly all pheochromocytomas in MEN2 are benign and located in the adrenals, often bilaterally (Fig. 407-3). Pheochromocytoma may be symptomatic before MTC. Prophylactic thyroidectomy is being performed in many carriers of RET mutations; pheochromocytomas should be excluded before any surgery in these patients.
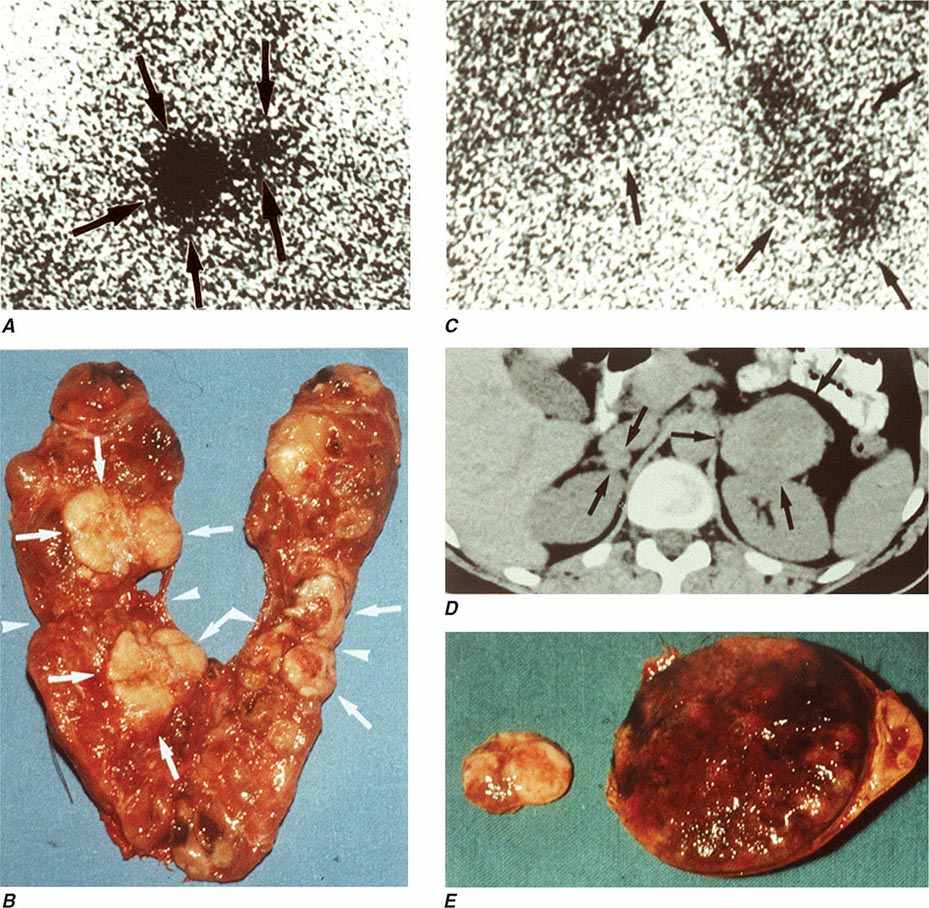
FIGURE 407-3 Multiple endocrine neoplasia type 2. A, B. Multifocal medullary thyroid carcinoma shown by MIBG scintigraphy (A) and operative specimen (B). Arrows demonstrate the tumors; arrowheads show the tissue bridge of the cut specimen. C–E. Bilateral adrenal pheochromocytoma shown by MIBG scintigraphy (C), CT imaging (D), and operative specimens (E). (From HPH Neumann et al: Keio J Med 54:15, 2005; with permission.)
Von Hippel–Lindau syndrome (VHL) is an autosomal dominant disorder that predisposes to retinal and cerebellar hemangioblastomas, which also occur in the brainstem and spinal cord (Fig. 407-4). Other important features of VHL are clear cell renal carcinomas, pancreatic neuroendocrine tumors, endolymphatic sac tumors of the inner ear, cystadenomas of the epididymis and broad ligament, and multiple pancreatic or renal cysts.
FIGURE 407-4 Von Hippel–Lindau disease. A. Retinal angioma. All subsequent panels show findings on MRI: B–D. Hemangioblastomas of the cerebellum (B) in brainstem (C) and spinal cord (D). E. Bilateral pheochromocytomas and bilateral renal clear cell carcinomas F. Multiple pancreatic cysts. (Parts A and D from HPH Neumann et al: Adv Nephrol Necker Hosp 27:361, 1997. © Elsevier. Part B from SH Morgan, J-P Grunfeld [eds]: Inherited Disorders of the Kidney. Oxford, UK, Oxford University Press, 1998. Part F from HPH Neumann et al: Contrib Nephrol 136:193, 2001. © S. Karger AG, Basel.)
The VHL gene (among other genes) encodes an E3 ubiquitin ligase that regulates expression of hypoxia-inducible factor 1. Loss of VHL is associated with increased expression of vascular endothelial growth factor (VEGF), which induces angiogenesis. Although the VHL gene can be inactivated by all types of mutations, patients with pheochromocytoma predominantly have missense mutations. About 20–30% of patients with VHL have pheochromocytomas, but in some families the incidence can reach 90%. The recognition of pheochromocytoma as a VHL-associated feature provides an opportunity to diagnose retinal, central nervous system, renal, and pancreatic tumors at a stage when effective treatment may still be possible.
The paraganglioma syndromes (PGLs) have been classified by genetic analyses of families with head and neck paragangliomas. The susceptibility genes encode subunits of the enzyme succinate dehydrogenase (SDH), a component in the Krebs cycle and the mitochondrial electron transport chain. SDH is formed by four subunits (A–D). Mutations of SDHB (PGL4), SDHC (PGL3), SDHD (PGL1), and SDHAF2 (PGL2) predispose to the PGLs. The transmission of the disease in carriers of SDHB and SDHC germ-line mutations is autosomal dominant. In contrast, in SDHD and SDHAF2 families, only the progeny of affected fathers develop tumors if they inherit the mutation. PGL1 is most common, followed by PGL4; PGL2 and PGL3 are rare. Adrenal, extra-adrenal abdominal, and thoracic pheochromocytomas, which are components of PGL1 and PGL4, are rare in PGL3 and absent in PGL2 (Fig. 407-5). About one-third of patients with PGL4 develop metastases.
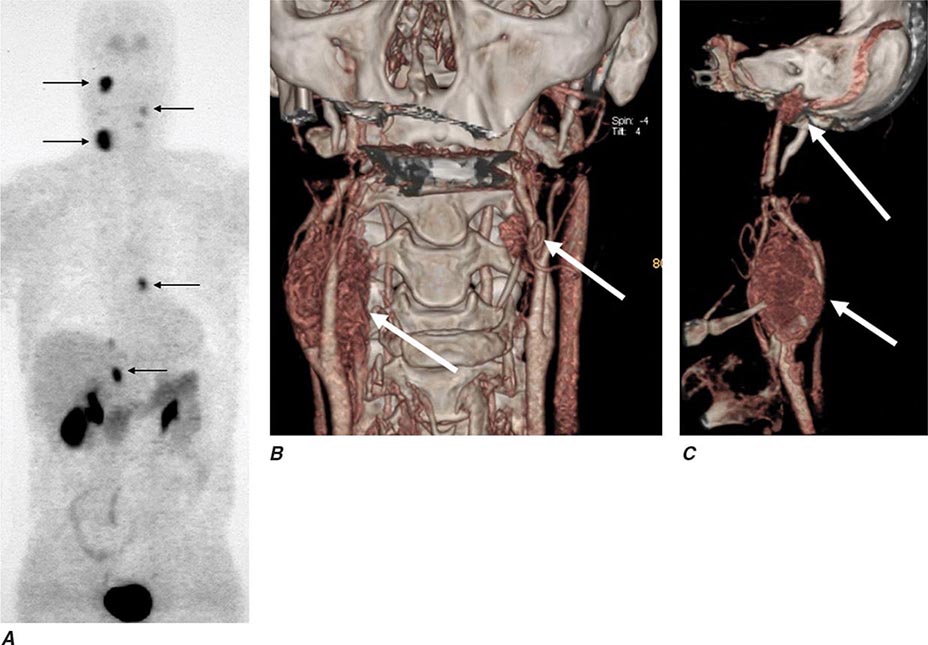
FIGURE 407-5 Paraganglioma syndrome. A patient with the SDHD W5X mutation and PGL1 underwent incomplete resection of a left carotid body tumor. A. 18F-DOPA positron emission tomography demonstrating tumor uptake in the right jugular glomus, the right carotid body, the left carotid body, the left coronary glomus, and the right adrenal gland. Note the physiologic accumulation of the radiopharmaceutical agent in the kidneys, liver, gallbladder, renal pelvis, and urinary bladder. B and C. CT angiography with three-dimensional reconstruction. Arrows point to the paraganglial tumors. (From S Hoegerle et al: Eur J Nucl Med Mol Imaging 30:689, 2003; with permission.)
Familial pheochromocytoma (FP) has been attributed to hereditary, mainly adrenal tumors in patients with germ-line mutations in the genes TMEM127, MAX, and SDHA. Transmission is also autosomal dominant, and mutations of MAX, like those of SDHD, cause tumors only if inherited from the father.
GUIDELINES FOR GENETIC SCREENING OF PATIENTS WITH PHEOCHROMOCYTOMA OR PARAGANGLIOMA
In addition to family history, general features suggesting an inherited syndrome include young age, multifocal tumors, extra-adrenal tumors, and malignant tumors (Fig. 407-6). Because of the relatively high prevalence of familial syndromes among patients who present with pheochromocytoma or paraganglioma, it is useful to identify germ-line mutations even in patients without a known family history. A first step is to search for clinical features of inherited syndromes and to obtain an in-depth, multigenerational family history. Each of these syndromes exhibits autosomal dominant transmission with variable penetrance, but a proband with a mother affected by paraganglial tumors is not predisposed to PLG1 (SDHD mutation carrier). Cutaneous neurofibromas, café au lait spots, and axillary freckling suggest neurofibromatosis. Germ-line mutations in NF1 have not been reported in patients with sporadic pheochromocytomas. Thus, NF1 testing need not be performed in the absence of other clinical features of neurofibromatosis. A personal or family history of MTC or an elevation of serum calcitonin strongly suggests MEN 2 and should prompt testing for RET mutations. A history of visual impairment or tumors of the cerebellum, kidney, brainstem, or spinal cord suggests the possibility of VHL. A personal and/or family history of head and neck paraganglioma suggests PGL1 or PGL4.
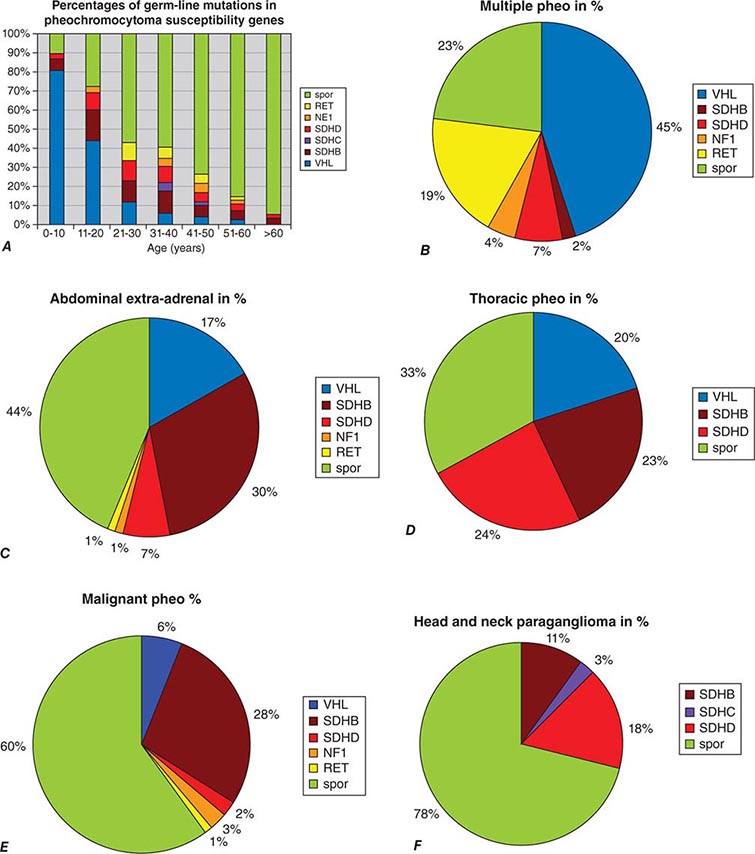
FIGURE 407-6 Mutation distribution in the VHL, RET, SDHB, SDHC, SDHD, and NF1 genes in 2021 patients with pheochromocytomas and paragangliomas from the European-American Pheochromocytoma-Paraganglioma Registry based in Freiburg, Germany, as updated on March 1, 2014. A. Correlation with age. The bars depict the frequency of sporadic (spor) or various inherited forms of pheochromocytoma in different age groups. The inherited disorders are much more common among younger individuals presenting with pheochromocytoma. Patients with mutations in the TMEM127, MAX, and SDHA genes are not included, since they contribute <1% in decades 4–7 only. B–F. Germ-line mutations according to multiple (B), extra-adrenal retroperitoneal (C), thoracic (D), and malignant (E) pheochromocytomas and head and neck paragangliomas (F). (Data from the Freiburg International Pheochromocytoma and Paraganglioma Registry, 2014.)
A single adrenal pheochromocytoma in a patient with an otherwise unremarkable history may still be associated with mutations of VHL, RET, SDHB, or SDHD (in decreasing order of frequency). Two-thirds of extra-adrenal tumors are associated with one of these syndromes, and multifocal tumors occur with decreasing frequency in carriers of RET, SDHD, VHL, and SDHB mutations. About 30% of head and neck paragangliomas are associated with germ-line mutations of one of the SDH subunit genes (most often SDHD) and are rare in carriers of VHL, RET, and TMEM127 mutations (Fig. 407-6F).
Immunohistochemistry is helpful in the preselection of hereditary pheochromocytoma. Negative immunostaining with antibodies to SDHB, TMEM127, and MAX may predict mutations of the SDH, TMEM127, and MAX genes, respectively.
Once the underlying syndrome is diagnosed, the benefit of genetic testing can be extended to relatives. For this purpose, it is necessary to identify the germ-line mutation in the proband and, after genetic counseling, to perform DNA sequence analyses of the responsible gene in relatives to determine whether they are affected (Chap. 84). Other family members may benefit when individuals who carry a germ-line mutation are biochemically screened for paraganglial tumors.
Asymptomatic paraganglial tumors, now often detected in patients with hereditary tumors and their relatives, are challenging to manage. Watchful waiting strategies have been introduced. Head and neck paragangliomas—mainly carotid body, jugular, and vagal tumors—are increasingly treated by radiation, since surgery is frequently associated with permanent palsy of cranial nerves II, VII, IX, X, XI, and XII. Nevertheless, tympanic paragangliomas are symptomatic early, and most of these tumors can easily be resected, with subsequent improvement of hearing and alleviation of tinnitus.
408 |
Multiple Endocrine Neoplasia |
Multiple endocrine neoplasia (MEN) is characterized by a predilection for tumors involving two or more endocrine glands. Four major forms of MEN are recognized and referred to as MEN types 1–4 (MEN 1–4) (Table 408-1). Each type of MEN is inherited as an autosomal dominant syndrome or may occur sporadically; that is, without a family history. However, this distinction between familial and sporadic forms is often difficult because family members with the disease may have died before symptoms developed. In addition to MEN 1–4, at least six other syndromes are associated with multiple endocrine and other organ neoplasias (MEONs) (Table 408-2). These MEONs include the hyperparathyroidism-jaw tumor syndrome, Carney complex, von Hippel-Lindau disease (Chap. 407), neurofibromatosis type 1 (Chap. 118), Cowden’s syndrome, and McCune-Albright syndrome (Chap. 426e); all of these are inherited as autosomal dominant disorders, except for McCune-Albright syndrome, which is caused by mosaic expression of a postzygotic somatic cell mutation (Table 408-2).
|
MULTIPLE ENDOCRINE NEOPLASIA (MEN) SYNDROMES |
|
MULTIPLE ENDOCRINE AND OTHER ORGAN NEOPLASIA SYNDROMES (MEONs) |
A diagnosis of a MEN or MEON syndrome may be established in an individual by one of three criteria: (1) clinical features (two or more of the associated tumors [or lesions] in an individual); (2) familial pattern (one of the associated tumors [or lesions] in a first-degree relative of a patient with a clinical diagnosis of the syndrome); and (3) genetic analysis (a germline mutation in the associated gene in an individual, who may be clinically affected or asymptomatic). Mutational analysis in MEN and MEON syndromes is helpful in clinical practice to: (1) confirm the clinical diagnosis; (2) identify family members who harbor the mutation and require screening for relevant tumor detection and early/appropriate treatment; and (3) identify the ~50% of family members who do not harbor the germline mutation and can, therefore, be alleviated of the anxiety of developing associated tumors. This latter aspect also helps to reduce health care costs by reducing the need for unnecessary biochemical and radiologic investigations.
MULTIPLE ENDOCRINE NEOPLASIA TYPE 1
Clinical Manifestations MEN type 1 (MEN 1), which is also referred to as Wermer’s syndrome, is characterized by the triad of tumors involving the parathyroids, pancreatic islets, and anterior pituitary. In addition, adrenal cortical tumors, carcinoid tumors usually of the foregut, meningiomas, facial angiofibromas, collagenomas, and lipomas may also occur in some patients with MEN 1. Combinations of the affected glands and their pathologic features (e.g., hyperplastic adenomas of the parathyroid glands) may differ in members of the same family and even between identical twins. In addition, a nonfamilial (e.g., sporadic) form occurs in 8–14% of patients with MEN 1, and molecular genetic studies have confirmed the occurrence of de novo mutations of the MEN1 gene in approximately 10% of patients with MEN 1. The prevalence of MEN 1 is approximately 0.25% based on randomly chosen postmortem studies but is 1–18% among patients with primary hyperparathyroidism, 16–38% among patients with pancreatic islet tumors, and <3% among patients with pituitary tumors. The disorder affects all age groups, with a reported age range of 5 to 81 years, with clinical and biochemical manifestations developing in the vast majority by the fifth decade. The clinical manifestations of MEN 1 are related to the sites of tumors and their hormonal products. In the absence of treatment, endocrine tumors are associated with an earlier mortality in patients with MEN 1, with a 50% probability of death by the age of 50 years. The cause of death is usually a malignant tumor, often from a pancreatic neuroendocrine tumor (NET) or foregut carcinoid. In addition, the treatment outcomes of patients with MEN 1–associated tumors are not as successful as those in patients with non–MEN 1 tumors. This is because MEN 1–associated tumors, with the exception of pituitary NETs, are usually multiple, making it difficult to achieve a successful surgical cure. Occult metastatic disease is also more prevalent in MEN 1, and the tumors may be larger, more aggressive, and resistant to treatment.
Parathyroid Tumors (See also Chap. 424) Primary hyperparathyroidism occurs in approximately 90% of patients and is the most common feature of MEN 1. Patients may have asymptomatic hypercalcemia or vague symptoms associated with hypercalcemia (e.g., polyuria, polydipsia, constipation, malaise, or dyspepsia). Nephrolithiasis and osteitis fibrosa cystica (less commonly) may also occur. Biochemical investigations reveal hypercalcemia, usually in association with elevated circulating parathyroid hormone (PTH) (Table 408-3). The hypercalcemia is usually mild, and severe hypercalcemia or parathyroid cancer is a rare occurrence. Additional differences in the primary hyperparathyroidism of patients with MEN 1, as opposed to those without MEN 1, include an earlier age at onset (20–25 years vs 55 years) and an equal male-to-female ratio (1:1 vs 1:3). Preoperative imaging (e.g., neck ultrasound with 99mTc-sestamibi parathyroid scintigraphy) is of limited benefit because all parathyroid glands may be affected, and neck exploration may be required irrespective of preoperative localization studies.
|
BIOCHEMICAL AND RADIOLOGICAL SCREENING IN MULTIPLE ENDOCRINE NEOPLASIA TYPE 1 |

|
TREATMENT |
PARATHYROID TUMORS |
Surgical removal of the abnormally overactive parathyroids in patients with MEN 1 is the definitive treatment. However, it is controversial whether to perform subtotal (e.g., removal of 3.5 glands) or total parathyroidectomy with or without autotransplantation of parathyroid tissue in the forearm, and whether surgery should be performed at an early or late stage. Minimally invasive parathyroidectomy is not recommended because all four parathyroid glands are usually affected with multiple adenomas or hyperplasia. Surgical experience should be taken into account given the variability in pathology in MEN 1. Calcimimetics (e.g., cinacalcet), which act via the calcium-sensing receptor, have been used to treat primary hyperparathyroidism in some patients when surgery is unsuccessful or contraindicated.
Pancreatic Tumors (See also Chap. 113) The incidence of pancreatic islet cell tumors, which are NETs, in patients with MEN 1 ranges from 30 to 80% in different series. Most of these tumors (Table 408-1) produce excessive amounts of hormone (e.g., gastrin, insulin, glucagon, vasoactive intestinal polypeptide [VIP]) and are associated with distinct clinical syndromes, although some are nonfunctioning or nonsecretory. These pancreatic islet cell tumors have an earlier age at onset in patients with MEN 1 than in patients without MEN 1.
Gastrinoma Gastrin-secreting tumors (gastrinomas) are associated with marked gastric acid production and recurrent peptic ulcerations, a combination referred to as the Zollinger-Ellison syndrome. Gastrinomas occur more often in patients with MEN 1 who are older than age 30 years. Recurrent severe multiple peptic ulcers, which may perforate, and cachexia are major contributors to the high mortality. Patients with Zollinger-Ellison syndrome may also suffer from diarrhea and steatorrhea. The diagnosis is established by demonstration of an elevated fasting serum gastrin concentration in association with increased basal gastric acid secretion (Table 408-3). However, the diagnosis of Zollinger-Ellison syndrome may be difficult in hypercalcemic MEN 1 patients, because hypercalcemia can also cause hypergastrinemia. Ultrasonography, endoscopic ultrasonography, computed tomography (CT), nuclear magnetic resonance imaging (MRI), selective abdominal angiography, venous sampling, and somatostatin receptor scintigraphy are helpful in localizing the tumor prior to surgery. Gastrinomas represent more than 50% of all pancreatic NETs in patients with MEN 1, and approximately 20% of patients with gastrinomas will be found to have MEN 1. Gastrinomas, which may also occur in the duodenal mucosa, are the major cause of morbidity and mortality in patients with MEN 1. Most MEN 1 gastrinomas are malignant and metastasize before a diagnosis is established.
|
TREATMENT |
GASTRINOMA |
Medical treatment of patients with MEN 1 and Zollinger-Ellison syndrome is directed toward reducing basal acid output to <10 mmol/L. Parietal cell H+-K+-adenosine triphosphatase (ATPase) inhibitors (e.g., omeprazole or lansoprazole) reduce acid output and are the drugs of choice for gastrinomas. Some patients may also require additional treatment with the histamine H2 receptor antagonists, cimetidine or ranitidine. The role of surgery in the treatment of gastrinomas in patients with MEN 1 is controversial. The goal of surgery is to reduce the risk of distant metastatic disease and improve survival. For a nonmetastatic gastrinoma situated in the pancreas, surgical excision is often effective. However, the risk of hepatic metastases increases with tumor size, such that 25–40% of patients with pancreatic NETs >4 cm develop hepatic metastases, and 50–70% of patients with tumors 2–3 cm in size have lymph node metastases. Survival in MEN 1 patients with gastrinomas <2.5 cm in size is 100% at 15 years, but 52% at 15 years, if metastatic disease is present. The presence of lymph node metastases does not appear to adversely affect survival. Surgery for gastrinomas that are >2–2.5 cm has been recommended, because the disease-related survival in these patients is improved following surgery. In addition, duodenal gastrinomas, which occur more frequently in patients with MEN 1, have been treated successfully with surgery. However, in most patients with MEN 1, gastrinomas are multiple or extrapancreatic, and with the exception of duodenal gastrinomas, surgery is rarely successful. For example, the results of one study revealed that only ~15% of patients with MEN 1 were free of disease immediately after surgery, and at 5 years, this number had decreased to ~5%; the respective outcomes in patients without MEN 1 were better, at 45% and 40%. Given these findings, most specialists recommend a nonsurgical management for gastrinomas in MEN 1, except as noted earlier for smaller, isolated lesions. Treatment of disseminated gastrinomas is difficult. Chemotherapy with streptozotocin and 5-fluorouracil; hormonal therapy with octreotide or lanreotide, which are human somatostatin analogues; hepatic artery embolization; administration of human leukocyte interferon; and removal of all resectable tumor have been successful in some patients.
Insulinoma These β islet cell insulin-secreting tumors represent 10–30% of all pancreatic tumors in patients with MEN 1. Patients with an insulinoma present with hypoglycemic symptoms (e.g., weakness, headaches, sweating, faintness, seizures, altered behavior, weight gain) that typically develop after fasting or exertion and improve after glucose intake. The most reliable test is a supervised 72-h fast. Biochemical investigations reveal increased plasma insulin concentrations in association with hypoglycemia (Table 408-3). Circulating concentrations of C peptide and proinsulin, which are also increased, are useful in establishing the diagnosis. It also is important to demonstrate the absence of sulfonylureas in plasma and urine samples obtained during the investigation of hypoglycemia (Table 408-3). Surgical success is greatly enhanced by preoperative localization by endoscopic ultrasonography, CT scanning, or celiac axis angiography. Additional localization methods may include preoperative and perioperative percutaneous transhepatic portal venous sampling, selective intraarterial stimulation with hepatic venous sampling, and intraoperative direct pancreatic ultrasonography. Insulinomas occur in association with gastrinomas in 10% of patients with MEN 1, and the two tumors may arise at different times. Insulinomas occur more often in patients with MEN 1 who are younger than 40 years, and some arise in individuals younger than 20 years. In contrast, in patients without MEN 1, insulinomas generally occur in those older than 40 years. Insulinomas may be the first manifestation of MEN 1 in 10% of patients, and approximately 4% of patients with insulinomas will have MEN 1.
|
INSULINOMA |
Medical treatment, which consists of frequent carbohydrate meals and diazoxide or octreotide, is not always successful, and surgery is the optimal treatment. Surgical treatment, which ranges from enucleation of a single tumor to a distal pancreatectomy or partial pancreatectomy, has been curative in many patients. Chemotherapy may include streptozotocin, 5-fluorouracil, and doxorubicin. Hepatic artery embolization has been used for metastatic disease.
Glucagonoma These glucagon-secreting pancreatic NETs occur in <3% of patients with MEN 1. The characteristic clinical manifestations of a skin rash (necrolytic migratory erythema), weight loss, anemia, and stomatitis may be absent. The tumor may have been detected in an asymptomatic patient with MEN 1 undergoing pancreatic imaging or by the finding of glucose intolerence and hyperglucagonemia.
|
TREATMENT |
GLUCAGONOMA |
Surgical removal of the glucagonoma is the treatment of choice. However, treatment may be difficult because approximately 50–80% of patients have metastases at the time of diagnosis. Medical treatment with somatostatin analogues (e.g., octreotide or lanreotide) or chemotherapy with streptozotocin and 5-fluorouracil has been successful in some patients, and hepatic artery embolization has been used to treat metastatic disease.
Vasoactive Intestinal Peptide (VIP) Tumors (VIPomas) VIPomas have been reported in only a few patients with MEN 1. This clinical syndrome is characterized by watery diarrhea, hypokalemia, and achlorhydria and is also referred to as the Verner-Morrison syndrome, the WDHA (watery diarrhea, hypokalemia, and achlorhydria) syndrome, or the VIPoma syndrome. The diagnosis is established by excluding laxative and diuretic abuse, by confirming a stool volume in excess of 0.5–1.0 L/d during a fast, and by documenting a markedly increased plasma VIP concentration.
|
TREATMENT |
VIPOMAS |
Surgical management of VIPomas, which are mostly located in the tail of the pancreas, can be curative. However, in patients with unresectable tumor, somatostatin analogues, such as octreotide and lanreotide, may be effective. Streptozotocin with 5-fluorouracil may be beneficial, along with hepatic artery embolization for the treatment of metastases.
Pancreatic Polypeptide-Secreting Tumors (Ppomas) and Nonfunctioning Pancreatic NETs PPomas are found in a large number of patients with MEN 1. No pathologic sequelae of excessive polypeptide (PP) secretion are apparent, and the clinical significance of PP is unknown. Many PPomas may have been unrecognized or classified as nonfunctioning pancreatic NETs, which likely represent the most common enteropancreatic NET associated with MEN 1 (Fig. 408-1). The absence of both a clinical syndrome and specific biochemical abnormalities may result in a delayed diagnosis of nonfunctioning pancreatic NETs, which are associated with a worse prognosis than other functioning tumors, including insulinoma and gastrinoma. The optimum screening method and its timing interval for nonfunctioning pancreatic NETs remain to be established. At present, endoscopic ultrasound likely represents the most sensitive method of detecting small pancreatic tumors, but somatostatin receptor scintography is the most reliable method for detecting metastatic disease (Table 408-3).
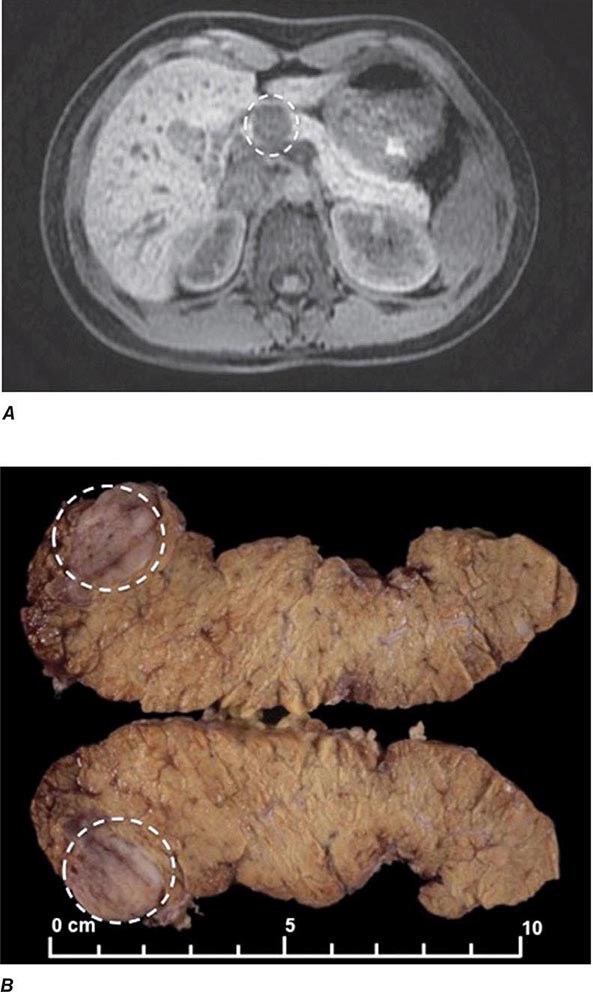
FIGURE 408-1 Pancreatic nonfunctioning neuroendocrine tumor (NET) in a 14-year-old patient with multiple endocrine neoplasia type 1 (MEN 1). A. An abdominal magnetic resonance imaging scan revealed a low-intensity >2.0 cm (anteroposterior maximal diameter) tumor within the neck of pancreas. There was no evidence of invasion of adjacent structures or metastases. The tumor is indicated by white dashed circle. B. The pancreatic NET was removed by surgery, and macroscopic examination confirmed the location of the tumor (white dashed circles) in the neck of the pancreas. Immunohistochemistry showed the tumor to immunostain for chromogranin A, but not gastrointestinal peptides or menin, thereby confirming that it was a nonsecreting NET due to loss of menin expression. (Part A adapted with permission from PJ Newey et al: J Clin Endocrinol Metab 10:3640, 2009.)
|
TREATMENT |
PPOMAS AND NONFUNCTIONING PANCREATIC NETS |
The management of nonfunctioning pancreatic NETs in the asymptomatic patient is controversial. One recommendation is to undertake surgery irrespective of tumor size after biochemical assessment is complete. Alternatively, other experts recommend surgery based on tumor size, using either >1 cm or >3 cm at different centers. Pancreatoduodenal surgery is successful in removing the tumors in 80% of patients, but more than 40% of patients develop complications, including diabetes mellitus, frequent steatorrhea, early and late dumping syndromes, and other gastrointestinal symptoms. However, ~50–60% of patients treated surgically survive >5 years. When considering these recommendations, it is important to consider that occult metastatic disease (e.g., tumors not detected by imaging investigations) is likely to be present in a substantial proportion of these patients at the time of presentation. Inhibitors of tyrosine kinase receptors (TKRs) and of the mammalian target of rapamycin (mTOR) signaling pathway have been reported to be effective in treating pancreatic NETs and in doubling the progression-free survival time.
Other Pancreatic NETs NETs secreting growth hormone–releasing hormone (GHRH), GHRHomas, have been reported rarely in patients with MEN 1. It is estimated that ~33% of patients with GHRHomas have other MEN 1–related tumors. GHRHomas may be diagnosed by demonstrating elevated serum concentrations of growth hormone and GHRH. More than 50% of GHRHomas occur in the lung, 30% occur in the pancreas, and 10% are found in the small intestine. Somatostatinomas secrete somatostatin, a peptide that inhibits the secretion of a variety of hormones, resulting in hyperglycemia, cholelithiasis, low acid output, steatorrhea, diarrhea, abdominal pain, anemia, and weight loss. Although 7% of pancreatic NETs secrete somatostatin, the clinical features of somatostatinoma syndrome are unusual in patients with MEN 1.
Pituitary Tumors (See also Chap. 403) Pituitary tumors occur in 15–50% of patients with MEN 1 (Table 408-1). These occur as early as 5 years of age or as late as the ninth decade. MEN 1 pituitary adenomas are more frequent in women than men and significantly are macroadenomas (i.e., diameter >1 cm). Moreover, about one-third of these pituitary tumors show invasive features such as infiltration of tumor cells into surrounding normal juxtatumoral pituitary tissue. However, no specific histologic parameters differentiate between MEN 1 and non–MEN 1 pituitary tumors. Approximately 60% of MEN 1–associated pituitary tumors secrete prolactin, <25% secrete growth hormone, 5% secrete adrenocorticotropic hormone (ACTH), and the remainder appear to be nonfunctioning, with some secreting glycoprotein subunits (Table 408-1). However, pituitary tumors derived from MEN 1 patients may exhibit immunoreactivity to several hormones. In particular, there is a greater frequency of somatolactotrope tumors. Prolactinomas are the first manifestation of MEN 1 in ~15% of patients, whereas somatotrope tumors occur more often in patients older than 40 years of age. Fewer than 3% of patients with anterior pituitary tumors will have MEN 1. Clinical manifestations are similar to those in patients with sporadic pituitary tumors without MEN 1 and depend on the hormone secreted and the size of the pituitary tumor. Thus, patients may have symptoms of hyperprolactinemia (e.g., amenorrhea, infertility, and galactorrhea in women, or impotence and infertility in men) or have features of acromegaly or Cushing’s disease. In addition, enlarging pituitary tumors may compress adjacent structures such as the optic chiasm or normal pituitary tissue, causing visual disturbances and/or hypopituitarism. In asymptomatic patients with MEN 1, periodic biochemical monitoring of serum prolactin and insulin-like growth factor I (IGF-I) levels, as well as MRI of the pituitary, can lead to early identification of pituitary tumors (Table 408-3). In patients with abnormal results, hypothalamic-pituitary testing should characterize the nature of the pituitary lesion and its effects on the secretion of other pituitary hormones.
|
TREATMENT |
PITUITARY TUMORS |
Treatment of pituitary tumors in patients with MEN 1 consists of therapies similar to those used in patients without MEN 1 and includes appropriate medical therapy (e.g., bromocriptine or cabergoline for prolactinoma; or octreotide or lanreotide for somatotrope tumors) or selective transsphenoidal adenomectomy, if feasible, with radiotherapy reserved for residual unresectable tumor tissue. Pituitary tumors in MEN 1 patients may be more aggressive and less responsive to medical or surgical treatments.
Associated Tumors Patients with MEN 1 may also develop carcinoid tumors, adrenal cortical tumors, facial angiofibromas, collagenomas, thyroid tumors, and lipomatous tumors.
Carcinoid Tumors (See also Chap. 113) Carcinoid tumors occur in more than 3% of patients with MEN 1 (Table 408-1). The carcinoid tumor may be located in the bronchi, gastrointestinal tract, pancreas, or thymus. At the time of diagnosis, most patients are asymptomatic and do not have clinical features of the carcinoid syndrome. Importantly, no hormonal or biochemical abnormality (e.g., plasma chromogranin A) is consistently observed in individuals with thymic or bronchial carcinoid tumors. Thus, screening for these tumors is dependent on radiologic imaging. The optimum method for screening has not been established. CT and MRI are sensitive for detecting thymic and bronchial tumors (Table 408-3), although repeated CT scanning raises concern about exposure to repeated doses of ionizing radiation. Octreotide scintigraphy may also reveal some thymic and bronchial carcinoids, although there is insufficient evidence to recommend its routine use. Gastric carcinoids, of which the type II gastric enterochromaffin-like (ECL) cell carcinoids (ECLomas) are associated with MEN 1 and Zollinger-Ellison syndrome, may be detected incidentally at the time of gastric endoscopy for dyspeptic symptoms in MEN 1 patients. These tumors, which may be found in >10% of MEN 1 patients, are usually multiple and smaller than 1.5 cm. Bronchial carcinoids in patients with MEN 1 occur predominantly in women (male-to-female ratio, 1:4). In contrast, thymic carcinoids in European patients with MEN 1 occur predominantly in men (male-to-female ratio, 20:1), with cigarette smokers having a higher risk for these tumors; thymic carcinoids in Japanese patients with MEN 1 have a less marked sex difference (male-to-female ratio 2:1). The course of thymic carcinoids in MEN 1 appears to be particularly aggressive. The presence of thymic tumors in patients with MEN 1 is associated with a median survival after diagnosis of approximately 9.5 years, with 70% of patients dying as a direct result of the tumor.
|
TREATMENT |
CARCINOID TUMORS |
If resectable, surgical removal of carcinoid tumors is the treatment of choice. For unresectable tumors and those with metastatic disease, treatment with radiotherapy or chemotherapeutic agents (e.g., cisplatin, etoposide) may be used. In addition, somatostatin analogues, such as octreotide or lanreotide, have resulted in symptom improvement and regression of some tumors. Little is known about the malignant potential of gastric type II ECLomas, but treatment with somatostatin analogues, such as octreotide or lanreotide, has resulted in regression of these ECLomas.
Adrenocortical Tumors (See also Chap. 406) Asymptomatic adrenocortical tumors occur in 20–70% of patients with MEN 1 depending on the radiologic screening methods used (Table 408-1). Most of these tumors, which include cortical adenomas, hyperplasia, multiple adenomas, nodular hyperplasia, cysts, and carcinomas, are nonfunctioning. Indeed, <10% of patients with enlarged adrenal glands have hormonal hypersecretion, with primary hyperaldosteronism and ACTH-independent Cushing’s syndrome being encountered most commonly. Occasionally, hyperandrogenemia may occur in association with adrenocortical carcinoma. Pheochromocytoma in association with MEN 1 is rare. Biochemical investigation (e.g., plasma renin and aldosterone concentrations, low-dose dexamethasone suppression test, urinary catecholamines, and/or metanephrines) should be undertaken in those with symptoms or signs suggestive of functioning adrenal tumors or in those with tumors >1 cm. Adrenocortical carcinoma occurs in approximately 1% of MEN 1 patients but increases to >10% for adrenal tumors larger than 1 cm.
|
TREATMENT |
ADRENOCORTICAL TUMORS |
Consensus has not been reached about the management of MEN 1–associated nonfunctioning adrenal tumors, because the majority are benign. However, the risk of malignancy increases with size, particularly for tumors with a diameter >4 cm. Indications for surgery for adrenal tumors include: size >4 cm in diameter; atypical or suspicious radiologic features (e.g., increased Hounsfield unit on unenhanced CT scan) and size of 1–4 cm in diameter; or significant measurable growth over a 6-month period. The treatment of functioning (e.g., hormone-secreting) adrenal tumors is similar to that for tumors occurring in non–MEN 1 patients.
Meningioma Central nervous system (CNS) tumors, including ependymomas, schwannomas, and meningiomas, have been reported in MEN 1 patients (Table 408-1). Meningiomas are found in <10% of patients with other clinical manifestations of MEN 1 (e.g., primary hyperparathyroidism) for >15 years. The majority of meningiomas are not associated with symptoms, and 60% do not enlarge. The treatment of MEN 1–associated meningiomas is similar to that in non–MEN 1 patients.
Lipomas Subcutaneous lipomas occur in >33% of patients with MEN 1 (Table 408-1) and are frequently multiple. In addition, visceral, pleural, or retroperitoneal lipomas may occur in patients with MEN 1. Management is conservative. However, when surgically removed for cosmetic reasons, they typically do not recur.
Facial Angiofibromas and Collagenomas The occurrence of multiple facial angiofibromas in patients with MEN 1 may range from >20 to >90%, and occurrence of collagenomas may range from 0 to >70% (Table 408-1). These cutaneous findings may allow presymptomatic diagnosis of MEN 1 in the relatives of a patient with MEN 1. Treatment for these cutaneous lesions is usually not required.
Thyroid Tumors Thyroid tumors, including adenomas, colloid goiters, and carcinomas, have been reported to occur in >25% of patients with MEN 1. However, the prevalence of thyroid disorders in the general population is high, and it has been suggested that the association of thyroid abnormalities in patients with MEN 1 may be incidental. The treatment of thyroid tumors in MEN 1 patients is similar to that for non–MEN 1 patients.
![]() Genetics and Screening The MEN1 gene is located on chromosome 11q13 and consists of 10 exons, which encode a 610–amino acid protein, menin, that regulates transcription, genome stability, cell division, and proliferation. The pathophysiology of MEN 1 follows the Knudson two-hit hypothesis with a tumor-suppressor role for menin. Inheritance of a germline MEN1 mutation predisposes an individual to developing a tumor that arises following a somatic mutation, which may be a point mutation or more commonly a deletion, leading to loss of heterozygosity (LOH) in the tumor DNA. The germline mutations of the MEN1 gene are scattered throughout the entire 1830-bp coding region and splice sites, and there is no apparent correlation between the location of MEN1 mutations and clinical manifestations of the disorder, in contrast with the situation in patients with MEN 2 (Table 408-1). More than 10% of MEN1 germline mutations arise de novo and may be transmitted to subsequent generations. Some families with MEN 1 mutations develop parathyroid tumors as the sole endocrinopathy, and this condition is referred to as familial isolated hyperparathyroidism (FIHP). However, between 5 and 25% of patients with MEN 1 do not harbor germline mutations or deletions of the MEN1 gene. Such patients with MEN 1–associated tumors but without MEN1 mutations may represent phenocopies or have mutations involving other genes. Other genes associated with MEN 1–like features include: CDC73, which encodes parafibromin, whose mutations result in the hyperparathyroid-jaw tumor syndrome; the calcium-sensing receptor gene (CaSR), whose mutations result in familial benign hypocalciuric hypercalcemia (FBHH); and the aryl hydrocarbon receptor interacting protein gene (AIP), a tumor suppressor located on chromosome 11q13 whose mutations are associated with familial isolated pituitary adenomas (FIPA). Genetic testing to determine the MEN1 mutation status in symptomatic family members within a MEN 1 kindred, as well as to all index cases (e.g., patients) with two or more endocrine tumors, is advisable. If an MEN1 mutation is not identified in the index case with two or more endocrine tumors, then clinical and genetic tests for other disorders such as hyperparathyroid-jaw tumor syndrome, FBHH, FIPA, MEN 2, or MEN 4 should be considered, because these patients may represent phenocopies for MEN 1.
Genetics and Screening The MEN1 gene is located on chromosome 11q13 and consists of 10 exons, which encode a 610–amino acid protein, menin, that regulates transcription, genome stability, cell division, and proliferation. The pathophysiology of MEN 1 follows the Knudson two-hit hypothesis with a tumor-suppressor role for menin. Inheritance of a germline MEN1 mutation predisposes an individual to developing a tumor that arises following a somatic mutation, which may be a point mutation or more commonly a deletion, leading to loss of heterozygosity (LOH) in the tumor DNA. The germline mutations of the MEN1 gene are scattered throughout the entire 1830-bp coding region and splice sites, and there is no apparent correlation between the location of MEN1 mutations and clinical manifestations of the disorder, in contrast with the situation in patients with MEN 2 (Table 408-1). More than 10% of MEN1 germline mutations arise de novo and may be transmitted to subsequent generations. Some families with MEN 1 mutations develop parathyroid tumors as the sole endocrinopathy, and this condition is referred to as familial isolated hyperparathyroidism (FIHP). However, between 5 and 25% of patients with MEN 1 do not harbor germline mutations or deletions of the MEN1 gene. Such patients with MEN 1–associated tumors but without MEN1 mutations may represent phenocopies or have mutations involving other genes. Other genes associated with MEN 1–like features include: CDC73, which encodes parafibromin, whose mutations result in the hyperparathyroid-jaw tumor syndrome; the calcium-sensing receptor gene (CaSR), whose mutations result in familial benign hypocalciuric hypercalcemia (FBHH); and the aryl hydrocarbon receptor interacting protein gene (AIP), a tumor suppressor located on chromosome 11q13 whose mutations are associated with familial isolated pituitary adenomas (FIPA). Genetic testing to determine the MEN1 mutation status in symptomatic family members within a MEN 1 kindred, as well as to all index cases (e.g., patients) with two or more endocrine tumors, is advisable. If an MEN1 mutation is not identified in the index case with two or more endocrine tumors, then clinical and genetic tests for other disorders such as hyperparathyroid-jaw tumor syndrome, FBHH, FIPA, MEN 2, or MEN 4 should be considered, because these patients may represent phenocopies for MEN 1.
The current guidelines recommend that MEN1 mutational analysis should be undertaken in: (1) an index case with two or more MEN 1–associated endocrine tumors (e.g., parathyroid, pancreatic, or pituitary tumors); (2) asymptomatic first-degree relatives of a known MEN1 mutation carrier; and (3) first-degree relatives of a MEN1 mutation carrier with symptoms, signs, or biochemical or radiologic evidence for one or more MEN 1–associated tumors. In addition, MEN1 mutational analysis should be considered in patients with suspicious or atypical MEN 1. This would include individuals with parathyroid adenomas before the age of 30 years or multigland parathyroid disease; individuals with gastrinoma or multiple pancreatic NETs at any age; or individuals who have two or more MEN 1–associated tumors that are not part of the classical triad of parathyroid, pancreatic islet, and anterior pituitary tumors (e.g., parathyroid tumor plus adrenal tumor). Family members, including asymptomatic individuals who have been identified to harbor a MEN1 mutation, will require biochemical and radiologic screening (Table 408-3). In contrast, relatives who do not harbor the MEN1 mutation have a risk of developing MEN 1–associated endocrine tumors that is similar to that of the general population; thus, relatives without the MEN1 mutation do not require repeated screening.
Mutational analysis in asymptomatic individuals should be undertaken at the earliest opportunity and, if possible, in the first decade of life because tumors have developed in some children by the age of 5 years. Appropriate biochemical and radiologic investigations (Table 408-3) aimed at detecting the development of tumors should then be undertaken in affected individuals. Mutant gene carriers should undergo biochemical screening at least once per annum and also have baseline pituitary and abdominal imaging (e.g., MRI or CT), which should then be repeated at 1- to 3-year intervals (Table 408-3). Screening should commence after 5 years of age and should continue for life because the disease may develop as late as the eighth decade. The screening history and physical examination elicit the symptoms and signs of hypercalcemia, nephrolithiasis, peptic ulcer disease, neuroglycopenia, hypopituitarism, galactorrhea and amenorrhea in women, acromegaly, Cushing’s disease, and visual field loss and the presence of subcutaneous lipomas, angiofibromas, and collagenomas. Biochemical screening should include measurements of serum calcium, PTH, gastrointestinal hormones (e.g., gastrin, insulin with a fasting glucose, glucagon, VIP, PP), chromogranin A, prolactin, and IGF-I in all individuals. More specific endocrine function tests should be undertaken in individuals who have symptoms or signs suggestive of a specific clinical syndrome. Biochemical screening for the development of MEN 1 tumors in asymptomatic members of families with MEN 1 is of great importance to reduce morbidity and mortality from the associated tumors.
MULTIPLE ENDOCRINE NEOPLASIA TYPE 2 AND TYPE 3
Clinical Manifestations MEN type 2 (MEN 2), which is also called Sipple’s syndrome, is characterized by the association of medullary thyroid carcinoma (MTC), pheochromocytomas, and parathyroid tumors (Table 408-1). Three clinical variants of MEN 2 are recognized: MEN 2A, MEN 2B, and MTC only. MEN 2A, which is often referred to as MEN 2, is the most common variant. In MEN 2A, MTC is associated with pheochromocytomas in 50% of patients (may be bilateral) and with parathyroid tumors in 20% of patients. MEN 2A may rarely occur in association with Hirschsprung’s disease, caused by the absence of autonomic ganglion cells in the terminal hindgut, resulting in colonic dilatation, severe constipation, and obstruction. MEN 2A may also be associated with cutaneous lichen amyloidosis, which is a pruritic lichenoid lesion that is usually located on the upper back. MEN 2B, which is also referred to as MEN 3, represents 5% of all cases of MEN 2 and is characterized by the occurrence of MTC and pheochromocytoma in association with a Marfanoid habitus; mucosal neuromas of the lips, tongue, and eyelids; medullated corneal fibers; and intestinal autonomic ganglion dysfunction leading to multiple diverticulae and megacolon. Parathyroid tumors do not usually occur in MEN 2B. MTC only (FMTC) is a variant in which MTC is the sole manifestation of the syndrome. However, the distinction between FMTC and MEN 2A is difficult and should only be considered if there are at least four family members above the age of 50 years who are affected by MTC but not pheochromocytomas or primary hyperparathyroidism. All of the MEN 2 variants are due to mutations of the rearranged during transfection (RET) protooncogene, which encodes a TKR. Moreover, there is a correlation between the locations of RET mutations and MEN 2 variants. Thus, ~95% of MEN 2A patients have mutations involving the cysteine-rich extracellular domain, with mutations of codon 634 accounting for ~85% of MEN 2A mutations; FMTC patients also have mutations of the cysteine-rich extracellular domain, with most mutations occurring in codon 618. In contrast, ~95% of MEN 2B/MEN 3 patients have mutations of codon 918 of the intracellular tyrosine kinase domain (Table 408-1 and Table 408-4).
|
RECOMMENDATIONS FOR TESTS AND SURGERY IN MEN 2 AND MEN 3a |
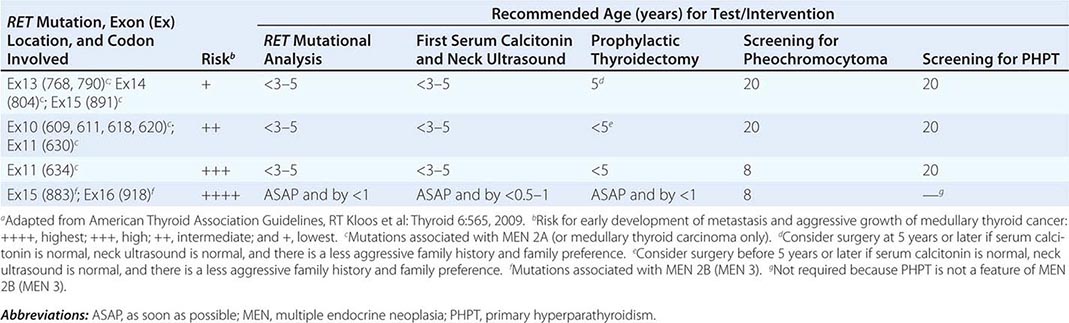
Medullary Thyroid Carcinoma MTC is the most common feature of MEN 2A and MEN 2B and occurs in almost all affected individuals. MTC represents 5–10% of all thyroid gland carcinomas, and 20% of MTC patients have a family history of the disorder. The use of RET mutational analysis to identify family members at risk for hereditary forms of MTC has altered the presentation of MTC from that of symptomatic tumors to a preclinical disease for which prophylactic thyroidectomy (Table 408-4) is undertaken to improve the prognosis and ideally result in cure. However, in patients who do not have a known family history of MEN 2A, FMTC, or MEN 2B, and therefore have not had RET mutational analysis, MTC may present as a palpable mass in the neck, which may be asymptomatic or associated with symptoms of pressure or dysphagia in >15% of patients. Diarrhea occurs in 30% of patients and is associated either with elevated circulating concentrations of calcitonin or tumor-related secretion of serotonin and prostaglandins. Some patients may also experience flushing. In addition, ectopic ACTH production by MTC may cause Cushing’s syndrome. The diagnosis of MTC relies on the demonstration of hypercalcitoninemia (>90 pg/mL in the basal state); stimulation tests using IV pentagastrin (0.5 mg/kg) and or calcium infusion (2 mg/kg) are rarely used now, reflecting improvements in the assay for calcitonin. Neck ultrasonography with fine-needle aspiration of the nodules can confirm the diagnosis. Radionucleotide thyroid scans may reveal MTC tumors as “cold” nodules. Radiography may reveal dense irregular calcification within the involved portions of the thyroid gland and in lymph nodes involved with metastases. Positron emission tomography (PET) may help to identify the MTC and metastases (Fig. 408-2). Metastases of MTC usually occur to the cervical lymph nodes in the early stages and to the mediastinal nodes, lung, liver, trachea, adrenal, esophagus, and bone in later stages. Elevations in serum calcitonin concentrations are often the first sign of recurrence or persistent disease, and the serum calcitonin doubling time is useful for determining prognosis. MTC can have an aggressive clinical course, with early metastases and death in approximately 10% of patients. A family history of aggressive MTC or MEN 2B may be elicited.
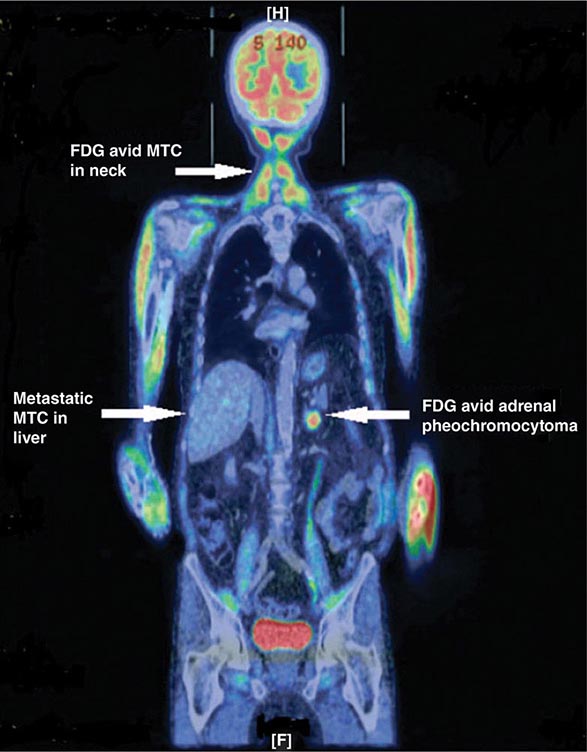
FIGURE 408-2 Fluorodeoxyglucose (FDG) positron emission tomography scan in a patient with multiple endocrine neoplasia type 2A, showing medullary thyroid cancer (MTC) with hepatic and skeletal (left arm) metastasis and a left adrenal pheochromocytoma. Note the presence of excreted FDG compound in the bladder. (Reproduced with permission from A Naziat et al: Clin Endocrinol [Oxf] 78:966, 2013.)
|
TREATMENT |
MEDULLARY THYROID CARCINOMA |
Individuals with RET mutations who do not have clinical manifestations of MTC should be offered prophylactic surgery between the ages of <1 and 5 years. The timing of surgery will depend on the type of RET mutation and its associated risk for early development, metastasis, and aggressive growth of MTC (Table 408-4). Such patients should have a total thyroidectomy with a systematic central neck dissection to remove occult nodal metastasis, although the value of undertaking a central neck dissection has been subject to debate. Prophylactic thyroidectomy, with life-long thyroxine replacement, has dramatically improved outcomes in patients with MEN 2 and MEN 3, such that ~90% of young patients with RET mutations who had a prophylactic thyroidectomy have no evidence of persistent or recurrent MTC at 7 years after surgery. In patients with clinically evident MTC, a total thyroidectomy with bilateral central resection is recommended, and an ipsilateral lateral neck dissection should be undertaken if the primary tumor is >1 cm in size or there is evidence of nodal metastasis in the central neck. Surgery is the only curative therapy for MTC. The 10-year survival in patients with metastatic MTC is ~20%. For inoperable MTC or metastatic disease, the tyrosine kinase inhibitors, vandetanib and cabozantinib, have improved the progression-free survival times. Other types of chemotherapy are of limited efficacy, but radiotherapy may help to palliate local disease.
Pheochromocytoma (See also Chap. 407) These noradrenaline- and adrenaline-secreting tumors occur in >50% of patients with MEN 2A and MEN 2B and are a major cause of morbidity and mortality. Patients may have symptoms and signs of catecholamine secretion (e.g., headaches, palpitations, sweating, poorly controlled hypertension), or they may be asymptomatic with detection through biochemical screening based on a history of familial MEN 2A, MEN 2B, or MTC. Pheochromocytomas in patients with MEN 2A and MEN 2B differ significantly in distribution when compared with patients without MEN 2A and MEN 2B. Extra-adrenal pheochromocytomas, which occur in 10% of patients without MEN 2A and MEN 2B, are observed rarely in patients with MEN 2A and MEN 2B. Malignant pheochromocytomas are much less common in patients with MEN 2A and MEN 2B. The biochemical and radiologic investigation of pheochromocytoma in patients with MEN 2A and MEN 2B is similar to that in non–MEN 2 patients and includes the measurement of plasma (obtained from supine patients) and urinary free fractionated metanephrines (e.g., normetanephrine and metanephrines measured separately), CT or MRI scanning, radionuclide scanning with meta-iodo-(123I or 131I)-benzyl guanidine (MIBG), and PET using (18F)-fluorodopamine or (18F)-fluoro-2-dexoxy-D-glucose (Fig. 408-2).
|
TREATMENT |
PHEOCHROMOCYTOMA |
Surgical removal of pheochromocytoma, using α and β adrenoreceptor blockade before and during the operation, is the recommended treatment. Endoscopic adrenal-sparing surgery, which decreases postoperative morbidity, hospital stay, and expense, as opposed to open surgery, has become the method of choice.
Parathyroid Tumors (See also Chap. 424) Parathyroid tumors occur in 10–25% of patients with MEN 2A. However, >50% of these patients do not have hypercalcemia. The presence of abnormally enlarged parathyroids, which are unusually hyperplastic, is often seen in the normocalcemic patient undergoing thyroidectomy for MTC. The biochemical investigation and treatment of hypercalcemic patients with MEN 2A is similar to that of patients with MEN 1.
![]() Genetics and Screening To date, approximately 50 different RET mutations have been reported, and these are located in exons 5, 8, 10, 11, 13, 14, 15, and 16. RET germline mutations are detected in >95% of MEN 2A, FMTC, and MEN 2B families, with Cys634Arg being most common in MEN 2A, Cys618Arg being most common in FMTC, and Met918Thr being most common in MEN 2B (Tables 408-1 and 408-4). Between 5 and 10% of patients with MTC or MEN 2A–associated tumors have de novo RET germline mutations, and ~50% of patients with MEN 2B have de novo RET germline mutations. These de novo RET germline mutations always occur on the paternal allele. Approximately 5% of patients with sporadic pheochromocytoma have a germline RET mutation, but such germline RET mutations do not appear to be associated with sporadic primary hyperparathyroidism. Thus, RET mutational analysis should be performed in: (1) all patients with MTC who have a family history of tumors associated with MEN 2, FMTC, or MEN 3, such that the diagnosis can be confirmed and genetic testing offered to asymptomatic relatives; (2) all patients with MTC and pheochromocytoma without a known family history of MEN 2 or MEN 3; (3) all patients with MTC, but without a family history of MEN 2, FMTC, or MEN 3, because these patients may have a de novo germline RET mutations; (4) all patients with bilateral pheochromocytoma; and (5) patients with unilateral pheochromocytoma, particularly if this occurs with increased calcitonin levels.
Genetics and Screening To date, approximately 50 different RET mutations have been reported, and these are located in exons 5, 8, 10, 11, 13, 14, 15, and 16. RET germline mutations are detected in >95% of MEN 2A, FMTC, and MEN 2B families, with Cys634Arg being most common in MEN 2A, Cys618Arg being most common in FMTC, and Met918Thr being most common in MEN 2B (Tables 408-1 and 408-4). Between 5 and 10% of patients with MTC or MEN 2A–associated tumors have de novo RET germline mutations, and ~50% of patients with MEN 2B have de novo RET germline mutations. These de novo RET germline mutations always occur on the paternal allele. Approximately 5% of patients with sporadic pheochromocytoma have a germline RET mutation, but such germline RET mutations do not appear to be associated with sporadic primary hyperparathyroidism. Thus, RET mutational analysis should be performed in: (1) all patients with MTC who have a family history of tumors associated with MEN 2, FMTC, or MEN 3, such that the diagnosis can be confirmed and genetic testing offered to asymptomatic relatives; (2) all patients with MTC and pheochromocytoma without a known family history of MEN 2 or MEN 3; (3) all patients with MTC, but without a family history of MEN 2, FMTC, or MEN 3, because these patients may have a de novo germline RET mutations; (4) all patients with bilateral pheochromocytoma; and (5) patients with unilateral pheochromocytoma, particularly if this occurs with increased calcitonin levels.
Screening for MEN 2/MEN 3–associated tumors in patients with RET germline mutations should be undertaken annually and include serum calcitonin measurements, a neck ultrasound for MTC, plasma and 24-h urinary fractionated metanephrines for pheochromocytoma, and albumin-corrected serum calcium or ionized calcium with PTH for primary hyperparathyroidism. In patients with MEN 2–associated RET mutations, screening for MTC should begin by 3 to 5 years; for pheochromocytoma by 20 years; and for primary hyperparathyroidism by 20 years of age (Table 408-4).
MULTIPLE ENDOCRINE NEOPLASIA TYPE 4
Clinical Manifestations Patients with MEN 1–associated tumors, such as parathyroid adenomas, pituitary adenomas, and pancreatic NETs, occurring in association with gonadal, adrenal, renal, and thyroid tumors have been reported to have mutations of the gene encoding the 196–amino acid cyclin-dependent kinase inhibitor (CK1) p27 kip1 (CDNKIB). Such families with MEN 1–associated tumors and CDNKIB mutations are designated to have MEN 4 (Table 408-1). The investigations and treatments for the MEN 4–associated tumors are similar to those for MEN 1 and non–MEN 1 tumors.
![]() Genetics and Screening To date, eight different MEN 4–associated mutations of CDNKIB, which is located on chromosome 12p13, have been reported, and all of these are associated with a loss of function. These MEN 4 patients may represent ~3% of the 5–10% of patients with MEN 1 who do not have mutations of the MEN1 gene. Germline CDNKIB mutations may rarely be found in patients with sporadic (i.e., nonfamilial) forms of primary hyperparathyroidism.
Genetics and Screening To date, eight different MEN 4–associated mutations of CDNKIB, which is located on chromosome 12p13, have been reported, and all of these are associated with a loss of function. These MEN 4 patients may represent ~3% of the 5–10% of patients with MEN 1 who do not have mutations of the MEN1 gene. Germline CDNKIB mutations may rarely be found in patients with sporadic (i.e., nonfamilial) forms of primary hyperparathyroidism.
HYPERPARATHYROIDISM-JAW TUMOR SYNDROME (SEE ALSO CHAP. 424)
Clinical Manifestations Hyperparathyroidism-jaw tumor (HPT-JT) syndrome is an autosomal dominant disorder characterized by the development of parathyroid tumors (15% are carcinomas) and fibro-osseous jaw tumors. In addition, some patients may also develop Wilms’ tumors, renal cysts, renal hematomas, renal cortical adenomas, papillary renal cell carcinomas, pancreatic adenocarcinomas, uterine tumors, testicular mixed germ cell tumors with a major seminoma component, and Hürthle cell thyroid adenomas. The parathyroid tumors may occur in isolation and without any evidence of jaw tumors, and this may cause confusion with other hereditary hypercalcemic disorders, such as MEN 1. However, genetic testing to identify the causative mutation will help to establish the correct diagnosis. The investigation and treatment for HPT-JT-associated tumors are similar to those in non-HPT-JT patients, except that early parathyroidectomy is advisable because of the increased frequency of parathyroid carcinoma.
![]() Genetics and Screening The gene that causes HPT-JT is located on chromosome 1q31.2 and encodes a 531–amino acid protein, parafibromin (Table 408-2). Parafibromin is also referred to as cell division cycle protein 73 (CDC73) and has a role in transcription. Genetic testing in families helps to identify mutation carriers who should be periodically screened for the development of tumors (Table 408-5).
Genetics and Screening The gene that causes HPT-JT is located on chromosome 1q31.2 and encodes a 531–amino acid protein, parafibromin (Table 408-2). Parafibromin is also referred to as cell division cycle protein 73 (CDC73) and has a role in transcription. Genetic testing in families helps to identify mutation carriers who should be periodically screened for the development of tumors (Table 408-5).
|
HPT-JT SCREENING GUIDELINES |
VON HIPPEL-LINDAU DISEASE (SEE ALSO CHAP. 407)
Clinical Manifestations von Hippel-Lindau (VHL) disease is an autosomal dominant disorder characterized by hemangioblastomas of the retina and CNS; cysts involving the kidneys, pancreas, and epididymis; renal cell carcinomas; pheochromocytomas; and pancreatic islet cell tumors. The retinal and CNS hemangioblastomas are benign vascular tumors that may be multiple; those in the CNS may cause symptoms by compressing adjacent structures and/or increasing intracranial pressure. In the CNS, the cerebellum and spinal cord are the most frequently involved sites. The renal abnormalities consist of cysts and carcinomas, and the lifetime risk of a renal cell carcinoma (RCC) in VHL is 70%. The endocrine tumors in VHL consist of pheochromocytomas and pancreatic islet cell tumors. The clinical presentation of pheochromocytoma in VHL disease is similar to that in sporadic cases, except there is a higher frequency of bilateral or multiple tumors, which may involve extra-adrenal sites in VHL disease. The most frequent pancreatic lesions in VHL are multiple cyst-adenomas, which rarely cause clinical disease. However, nonsecreting pancreatic islet cell tumors occur in <10% of VHL patients, who are usually asymptomatic. The pancreatic tumors in these patients are often detected by regular screening using abdominal imaging. Pheochromocytomas should be investigated and treated as described earlier for MEN 2. The pancreatic islet cell tumors frequently become malignant, and early surgery is recommended.
![]() Genetics and Screening The VHL gene, which is located on chromosome 3p26-p25, is widely expressed in human tissues and encodes a 213–amino acid protein (pVHL) (Table 408-2). A wide variety of germline VHL mutations have been identified. VHL acts as a tumor-suppressor gene. A correlation appears to exist between the type of mutation and the clinical phenotype; large deletions and protein-truncating mutations are associated with a low incidence of pheochromocytomas, whereas some missense mutations in VHL patients are associated with pheochromocytoma (referred to as VHL type 2C). Other missense mutations may be associated with hemangioblastomas and RCC but not pheochromocytoma (referred to as VHL type 1), whereas distinct missense mutations are associated with hemangioblastomas, RCC, and pheochromocytoma (VHL type 2B). VHL type 2A, which refers to the occurrence of hemangioblastomas and pheochromocytoma without RCC, is associated with rare missense mutations. The basis for these complex genotype-phenotype relationships remains to be elucidated. One major function of pVHL, which is also referred to as elongin, is to downregulate the expression of vascular endothelial growth factor (VEGF) and other hypoxia-inducible mRNAs. Thus, pVHL, in complex with other proteins, regulates the expression of hypoxia-inducible factors (HIF-1 and HIF-2) such that loss of functional pVHL leads to a stabilization of the HIF protein complexes, resulting in VEGF overexpression and tumor angiogenesis. Screening for the development of pheochromocytomas and pancreatic islet cell tumors is as described earlier for MEN 2 and MEN 1, respectively (Tables 408-3 and 408-4).
Genetics and Screening The VHL gene, which is located on chromosome 3p26-p25, is widely expressed in human tissues and encodes a 213–amino acid protein (pVHL) (Table 408-2). A wide variety of germline VHL mutations have been identified. VHL acts as a tumor-suppressor gene. A correlation appears to exist between the type of mutation and the clinical phenotype; large deletions and protein-truncating mutations are associated with a low incidence of pheochromocytomas, whereas some missense mutations in VHL patients are associated with pheochromocytoma (referred to as VHL type 2C). Other missense mutations may be associated with hemangioblastomas and RCC but not pheochromocytoma (referred to as VHL type 1), whereas distinct missense mutations are associated with hemangioblastomas, RCC, and pheochromocytoma (VHL type 2B). VHL type 2A, which refers to the occurrence of hemangioblastomas and pheochromocytoma without RCC, is associated with rare missense mutations. The basis for these complex genotype-phenotype relationships remains to be elucidated. One major function of pVHL, which is also referred to as elongin, is to downregulate the expression of vascular endothelial growth factor (VEGF) and other hypoxia-inducible mRNAs. Thus, pVHL, in complex with other proteins, regulates the expression of hypoxia-inducible factors (HIF-1 and HIF-2) such that loss of functional pVHL leads to a stabilization of the HIF protein complexes, resulting in VEGF overexpression and tumor angiogenesis. Screening for the development of pheochromocytomas and pancreatic islet cell tumors is as described earlier for MEN 2 and MEN 1, respectively (Tables 408-3 and 408-4).
NEUROFIBROMATOSIS
Clinical Manifestations Neurofibromatosis type 1 (NF1), which is also referred to as von Recklinghausen’s disease, is an autosomal dominant disorder characterized by the following manifestations: neurologic (e.g., peripheral and spinal neurofibromas); ophthalmologic (e.g., optic gliomas and iris hamartomas such as Lisch nodules); dermatologic (e.g., café au lait macules); skeletal (e.g., scoliosis, macrocephaly, short stature, and pseudoarthrosis); vascular (e.g., stenoses of renal and intracranial arteries); and endocrine (e.g., pheochromocytoma, carcinoid tumors, and precocious puberty). Neurofibromatosis type 2 (NF2) is also an autosomal dominant disorder but is characterized by the development of bilateral vestibular schwannomas (acoustic neuromas) that lead to deafness, tinnitus, or vertigo. Some patients with NF2 also develop meningiomas, spinal schwannomas, peripheral nerve neurofibromas, and café au lait macules. Endocrine abnormalities are not found in NF2 and are associated solely with NF1. Pheochromocytomas, carcinoid tumors, and precocious puberty occur in about 1% of patients with NF1, and growth hormone deficiency has been also reported. The features of pheochromocytomas in NF1 are similar to those in non-NF1 patients, with 90% of tumors being located within the adrenal medulla and the remaining 10% at an extra-adrenal location, which often involves the para-aortic region. Primary carcinoid tumors are often periampullary and may also occur in the ileum but rarely in the pancreas, thyroid, or lungs. Hepatic metastases are associated with symptoms of the carcinoid syndrome, which include flushing, diarrhea, bronchoconstriction, and tricuspid valve disease. Precocious puberty is usually associated with the extension of an optic glioma into the hypothalamus with resultant early activation of gonadotropin-releasing hormone secretion. Growth hormone deficiency has also been observed in some NF1 patients, who may or may not have optic chiasmal gliomas, but it is important to note that short stature is frequent in the absence of growth hormone deficiency in patients with NF1. The investigation and treatment for tumors are similar to those undertaken for each respective tumor type in non-NF1 patients.
![]() Genetics and Screening The NF1 gene, which is located on chromosome 17q11.2 and acts as a tumor suppressor, consists of 60 exons that span more than 350 kb of genomic DNA (Table 408-2). Mutations in NF1 are of diverse types and are scattered throughout the exons. The NF1 gene product is the protein neurofibromin, which has homologies to the p120GAP (GTPase activating protein) and acts on p21ras by converting the active GTP bound form to its inactive GDP form. Mutations of NF1 impair this downregulation of the p21ras signaling pathways, which in turn results in abnormal cell proliferation. Screening for the development of pheochromocytomas and carcinoid tumors is as described earlier for MEN 2 and MEN 1, respectively (Tables 408-3 and 408-4).
Genetics and Screening The NF1 gene, which is located on chromosome 17q11.2 and acts as a tumor suppressor, consists of 60 exons that span more than 350 kb of genomic DNA (Table 408-2). Mutations in NF1 are of diverse types and are scattered throughout the exons. The NF1 gene product is the protein neurofibromin, which has homologies to the p120GAP (GTPase activating protein) and acts on p21ras by converting the active GTP bound form to its inactive GDP form. Mutations of NF1 impair this downregulation of the p21ras signaling pathways, which in turn results in abnormal cell proliferation. Screening for the development of pheochromocytomas and carcinoid tumors is as described earlier for MEN 2 and MEN 1, respectively (Tables 408-3 and 408-4).
CARNEY COMPLEX
Clinical Manifestations Carney complex (CNC) is an autosomal dominant disorder characterized by spotty skin pigmentation (usually of the face, labia, and conjunctiva), myxomas (usually of the eyelids and heart, but also the tongue, palate, breast, and skin), psammomatous melanotic schwannomas (usually of the sympathetic nerve chain and upper gastrointestinal tract), and endocrine tumors that involve the adrenals, Sertoli cells, somatotropes, thyroid, and ovary. Cushing’s syndrome, the result of primary pigmented nodular adrenal disease (PPNAD), is the most common endocrine manifestation of CNC and may occur in one-third of patients. Patients with CNC and Cushing’s syndrome often have an atypical appearance by being thin (as opposed to having truncal obesity). In addition, they may have short stature, muscle and skin wasting, and osteoporosis. These patients often have levels of urinary free cortisol that are normal or increased only marginally. Cortisol production may fluctuate periodically with days or weeks of hypercortisolism; this pattern is referred to as “periodic Cushing’s syndrome.” Patients with Cushing’s syndrome usually have loss of the circadian rhythm of cortisol production. Acromegaly, the result of a somatotrope tumor, affects ~10% of patients with CNC. Testicular tumors may also occur in one-third of patients with CNC. These may either be large-cell calcifying Sertoli cell tumors, adrenocortical rests, or Leydig cell tumors. The Sertoli cell tumors occasionally may be estrogen-secreting and lead to precocious puberty or gynecomastia. Some patients with CNC have been reported to develop thyroid follicular tumors, ovarian cysts, or breast duct adenomas.
![]() Genetics and Screening CNC type 1 (CNC1) is due to mutations of the protein kinase A (PKA) regulatory subunit 1 α (R1α) (PPKAR1A), a tumor suppressor, whose gene is located on chromosome 17q.24.2 (Table 408-2). The gene causing CNC type 2 (CNC2) is located on chromosome 2p16 and has not yet been identified. It is interesting to note, however, that some tumors do not show LOH of 2p16 but instead show genomic instability, suggesting that this CNC gene may not be a tumor suppressor. Screening and treatment of these endocrine tumors are similar to those described earlier for patients with MEN 1 and MEN 2 (Tables 408-3 and 408-4).
Genetics and Screening CNC type 1 (CNC1) is due to mutations of the protein kinase A (PKA) regulatory subunit 1 α (R1α) (PPKAR1A), a tumor suppressor, whose gene is located on chromosome 17q.24.2 (Table 408-2). The gene causing CNC type 2 (CNC2) is located on chromosome 2p16 and has not yet been identified. It is interesting to note, however, that some tumors do not show LOH of 2p16 but instead show genomic instability, suggesting that this CNC gene may not be a tumor suppressor. Screening and treatment of these endocrine tumors are similar to those described earlier for patients with MEN 1 and MEN 2 (Tables 408-3 and 408-4).
COWDEN’S SYNDROME
Clinical Manifestations Multiple hamartomatous lesions, especially of the skin, mucous membranes (e.g., buccal, intestinal, and colonic), breast, and thyroid are characteristic of Cowden’s (CWD) syndrome, which is an autosomal dominant disorder. Thyroid abnormalities occur in two-thirds of patients with CWD syndrome, and these usually consist of multinodular goiters or benign adenomas, although <10% of patients may have a follicular thyroid carcinoma. Breast abnormalities occur in >75% of patients and consist of either fibrocystic disease or adenocarcinomas. The investigation and treatment for CWD tumors are similar to those undertaken for non-CWD patients.
![]() Genetics and Screening CWD syndrome is genetically heterogenous, and six types (CWD1–6) are recognized (Table 408-2). CWD is due to mutations of the phosphate and tensin homologue deleted on chromosome 10 (PTEN) gene, located on chromosome 10q23.31. CWD2 is caused by mutations of the succinate dehydrogenase subunit B (SDHB) gene, located on chromosome 1p36.13; and CWD3 is caused by mutations of the SDHD gene, located on chromosome 11q13.1. SDHB and SDHD mutations are also associated with pheochromocytoma. CWD4 is caused by hypermethylation of the Killin (KLLN) gene, the promoter of which shares the same transcription site as PTEN on chromosome 10q23.31. CWD5 is caused by mutations of the phosphatidylinositol 3-kinase catalytic alpha (PIK3CA) gene on chromosome 3q26.32, and CWD6 is caused by mutations of the V-Akt murine thymoma viral oncogene homolog 1 (AKT1) gene on chromosome 14q32.33. Screening for thyroid abnormalities entails neck ultrasonography and fine-needle aspiration with analysis of cell cytology.
Genetics and Screening CWD syndrome is genetically heterogenous, and six types (CWD1–6) are recognized (Table 408-2). CWD is due to mutations of the phosphate and tensin homologue deleted on chromosome 10 (PTEN) gene, located on chromosome 10q23.31. CWD2 is caused by mutations of the succinate dehydrogenase subunit B (SDHB) gene, located on chromosome 1p36.13; and CWD3 is caused by mutations of the SDHD gene, located on chromosome 11q13.1. SDHB and SDHD mutations are also associated with pheochromocytoma. CWD4 is caused by hypermethylation of the Killin (KLLN) gene, the promoter of which shares the same transcription site as PTEN on chromosome 10q23.31. CWD5 is caused by mutations of the phosphatidylinositol 3-kinase catalytic alpha (PIK3CA) gene on chromosome 3q26.32, and CWD6 is caused by mutations of the V-Akt murine thymoma viral oncogene homolog 1 (AKT1) gene on chromosome 14q32.33. Screening for thyroid abnormalities entails neck ultrasonography and fine-needle aspiration with analysis of cell cytology.
MCCUNE-ALBRIGHT SYNDROME (SEE ALSO CHAP. 426e)
Clinical Manifestations McCune-Albright syndrome (MAS) is characterized by the triad of polyostotic fibrous dysplasia, which may be associated with hypophosphatemic rickets; café au lait skin pigmentation; and peripheral precocious puberty; other endocrine abnormalities include thyrotoxicosis, which may be associated with a multinodular goiter, somatotrope tumors, and Cushing’s syndrome (due to adrenal tumors). Investigation and treatment for each endocrinopathy are similar to those used in patients without MAS.
![]() Genetics and Screening MAS is a disorder of mosaicism that results from postzygotic somatic cell mutations of the G protein α stimulating subunit (Gsα), encoded by the GNAS1 gene, located on chromosome 20q13.32 (Table 408-2). The Gsα mutations, which include Arg201Cys, Arg201His, Glu227Arg, or Glu227His, are activating and are found only in cells of the abnormal tissues. Screening for hyperfunction of relevant endocrine glands and development of hypophosphatemia, which may be associated with elevated serum fibroblast growth factor 23 (FGF23) concentrations, is undertaken in MAS patients.
Genetics and Screening MAS is a disorder of mosaicism that results from postzygotic somatic cell mutations of the G protein α stimulating subunit (Gsα), encoded by the GNAS1 gene, located on chromosome 20q13.32 (Table 408-2). The Gsα mutations, which include Arg201Cys, Arg201His, Glu227Arg, or Glu227His, are activating and are found only in cells of the abnormal tissues. Screening for hyperfunction of relevant endocrine glands and development of hypophosphatemia, which may be associated with elevated serum fibroblast growth factor 23 (FGF23) concentrations, is undertaken in MAS patients.
ACKNOWLEDGMENTS
The author is grateful to the Medical Research Council (UK) for support and to Mrs. Tracey Walker for typing the manuscript.
409 |
Autoimmune Polyendocrine Syndromes |
Polyglandular deficiency syndromes have been given many different names, reflecting the wide spectrum of disorders that have been associated with these syndromes and the heterogeneity of their clinical presentations. The name used in this chapter for this group of disorders is autoimmune polyendocrine syndrome (APS). In general, these disorders are divided into two major categories, APS type 1 (APS-1) and APS type 2 (APS-2). Some groups have further subdivided APS-2 into APS type 3 (APS-3) and APS type 4 (APS-4) depending on the type of autoimmunity involved. For the most part, this additional classification does not clarify our understanding of disease pathogenesis or prevention of complications in individual patients. Importantly, there are many nonendocrine disease associations included in these syndromes, suggesting that although the underlying autoimmune disorder predominantly involves endocrine targets, it does not exclude other tissues. The disease associations found in APS-1 and APS-2 are summarized in Table 409-1. Understanding these syndromes and their disease manifestations can lead to early diagnosis and treatment of additional disorders in patients and their family members.
|
DISEASE ASSOCIATIONS WITH AUTOIMMUNE POLYENDOCRINE SYNDROMES |
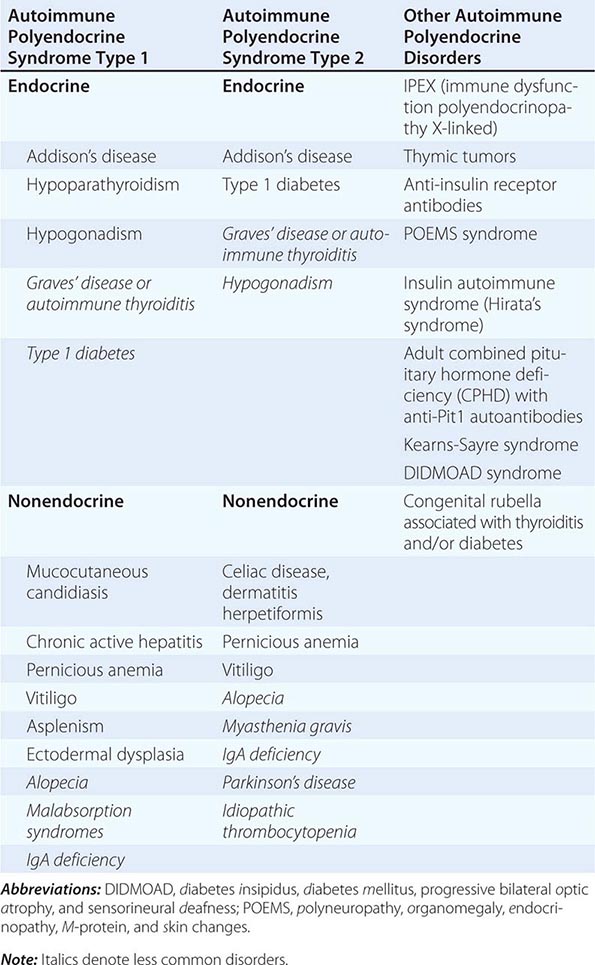
APS-1
APS-1 (Online Mendelian Inheritance in Man [OMIM] 240300) has also been called autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED). Mucocutaneous candidiasis, hypoparathyroidism, and Addison’s disease form the three major components of this disorder. However, as summarized in Table 409-1, many other organ systems can be involved over time. APS-1 is rare, with fewer than 500 cases reported in the literature. It is an autosomal recessive disorder caused by mutations in the AIRE gene (autoimmune regulator gene) found on chromosome 21. This gene is most highly expressed in thymic medullary epithelial cells (mTECs) where it appears to control the expression of tissue-specific self-antigens (e.g., insulin). Deletion of this regulator leads to decreased expression of tissue-specific self-antigens and is hypothesized to allow autoreactive T cells to avoid clonal deletion, which normally occurs during T cell maturation in the thymus. The AIRE gene is also expressed in epithelial cells found in peripheral lymphoid organs, but its role in these extrathymic cells remains controversial. A number of mutations have been described in this gene, and there is a higher frequency within certain ethnic groups including Iranian Jews, Sardinians, Finns, Norwegians, and Irish.
Clinical Manifestations APS-1 develops very early in life, often in infancy (Table 409-2). Chronic mucocutaneous candidiasis without signs of systemic disease is often the first manifestation. It affects the mouth and nails more frequently than the skin and esophagus. Chronic oral candidiasis can result in atrophic disease with areas suggestive of leukoplakia, which can pose a risk for future carcinoma. The etiology is associated with anticytokine autoantibodies (anti-IL-17A, -IL-17F, and -IL-22) related to T helper (TH) 17 T cells and depressed production of these cytokines by peripheral blood mononuclear cells. Hypoparathyroidism usually develops next, followed by adrenal insufficiency. The time from development of one component of the disorder to the next can be many years, and the order of disease appearance is variable.
|
COMPARISON OF APS-1 AND APS-2 |
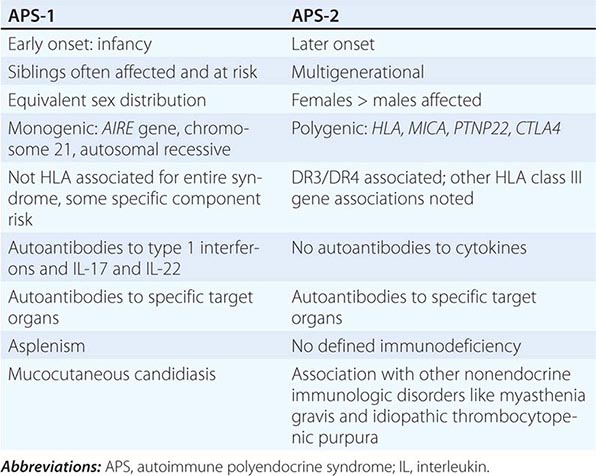
Chronic candidiasis is nearly always present and is not very responsive to treatment. Hypoparathyroidism is found in >85% of cases, and Addison’s disease is found in nearly 80%. Gonadal failure appears to affect women more than men (70% vs 25%, respectively), and hypoplasia of the dental enamel also occurs frequently (77% of patients). Other endocrine disorders that occur less frequently include type 1 diabetes (23%) and autoimmune thyroid disease (18%). Nonendocrine manifestations that present less frequently include alopecia (40%), vitiligo (26%), intestinal malabsorption (18%), pernicious anemia (31%), chronic active hepatitis (17%), and nail dystrophy. An unusual and debilitating manifestation of the disorder is the development of refractory diarrhea/obstipation that may be related to autoantibody-mediated destruction of enterochromaffin or enterochromaffin-like cells. The incidence rates for many of these disorders peak in the first or second decade of life, but the individual disease components continue to emerge over time. Therefore, prevalence rates may be higher than originally reported.
Diagnosis The diagnosis of APS-1 is usually made clinically when two of the three major component disorders are found in an individual patient. Siblings of individuals with APS-1 should be considered affected even if only one component disorder has been detected due to the known inheritance of the syndrome. Genetic analysis of the AIRE gene should be undertaken to identify mutations. Initial sequencing may detect the common mutations, but rare mutations are continually being noted, and an initial negative genetic analysis should not dissuade one from the clinical diagnosis until more extensive DNA sequencing can be performed. Detection of anti–interferon α and anti–interferon ο antibodies can identify nearly 100% of cases with APS-1. The autoantibody arises independent of the type of AIRE gene mutation and is not found in other autoimmune disorders.
Diagnosis of each underlying disorder should be done based on their typical clinical presentations (Table 409-3). Mucocutaneous candidiasis may present throughout the gastrointestinal tract, and it may be detected in the oral mucosa or from stool samples. Evaluation by a gastroenterologist to examine the esophagus for candidiasis or secondary stricture may be merited based on symptoms. Other gastrointestinal manifestations of APS-1, including malabsorption and obstipation, may also bring these young patients to the attention of gastroenterologists for first evaluation. Specific physical examination findings of hyperpigmentation, vitiligo, alopecia, tetany, and signs of hyper- or hypothyroidism should be considered as signs of development of component disorders.
|
CLINICAL FEATURES AND RECOMMENDED FOLLOW-UP FOR APS-1 AND APS-2 |
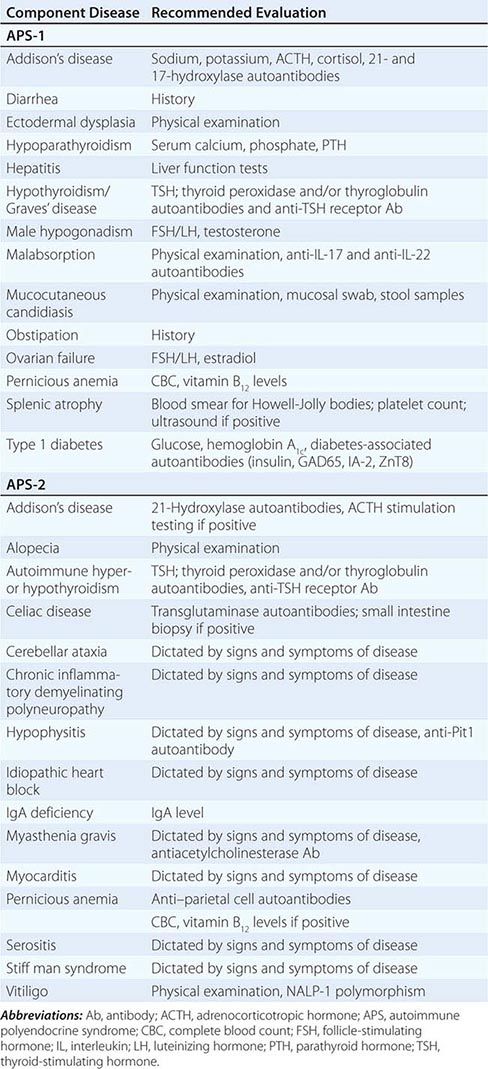
The development of disease-specific autoantibody assays can help confirm disease and also detect risk for future disease. For example, where possible, detection of anticytokine antibodies to interleukin (IL) 17 and IL-22 would confirm the diagnosis of mucocutaneous candidiasis due to APS-1. The presence of anti-21-hydroxylase antibody or anti-17-hydroxylase antibody (which may be found more commonly in adrenal insufficiency associated with APS-1) would confirm the presence or risk for Addison’s disease. Other autoantibodies found in type 1 diabetes (e.g., anti-GAD65), pernicious anemia, and other component conditions should be screened for on a regular basis (6- to 12-month intervals depending on the age of the subject).
Laboratory tests, including a complete metabolic panel, phosphorous and magnesium, thyroid-stimulating hormone (TSH), adrenocorticotropic hormone (ACTH; morning), hemoglobin A1c, plasma vitamin B12 level, and complete blood count with peripheral smear looking for Howell-Jolly bodies (asplenism), should also be performed at these time points. Detection of abnormal physical findings or test results should prompt subsequent examinations of the relevant organ system (e.g., presence of Howell-Jolly bodies indicates need for ultrasound of spleen).
|
TREATMENT |
APS-1 |
Therapy of individual disease components is carried out as outlined in other relevant chapters. Replacement of deficient hormones (e.g., adrenal, pancreas, ovaries/testes) will treat most of the endocrinopathies noted. Several unique issues merit special emphasis. Adrenal insufficiency can be masked by primary hypothyroidism by prolonging the half-life of cortisol. The caveat therefore is that replacement therapy with thyroid hormone can precipitate an adrenal crisis in an undiagnosed individual. Hence, all patients with hypothyroidism and the possibility of APS should be screened for adrenal insufficiency to allow treatment with glucocorticoids prior to the initiation of thyroid hormone replacement. Treatment of mucocutaneous candidiasis with ketoconazole in an individual with subclinical adrenal insufficiency may also precipitate adrenal crisis. Furthermore, mucocutaneous candidiasis may be difficult to eradicate entirely. Severe cases of disease involvement may require systemic immunomodulatory therapy, but this is not commonly needed.
APS-2
APS-2 (OMIM 269200) is more common than APS-1 with a prevalence of 1 in 100,000. It has a gender bias and occurs more often in female patients with a ratio of at least 3:1 compared to male patients. In contrast to APS-1, APS-2 often has its onset in adulthood with a peak incidence between 20 and 60 years of age. It shows a familial, multigenerational heritage (Table 409-2). The presence of two or more of the following endocrine deficiencies in the same patient defines the presence of APS-2: primary adrenal insufficiency (Addison’s disease; 50–70%), Graves’ disease or autoimmune thyroiditis (15–69%), type 1 diabetes mellitus (T1D; 40–50%), and primary hypogonadism. Frequently associated autoimmune conditions include celiac disease (3–15%), myasthenia gravis, vitiligo, alopecia, serositis, and pernicious anemia. These conditions occur with increased frequency in affected patients but are also are found in their family members (Table 409-3).
![]() Genetic Considerations The overwhelming risk factor for APS-2 has been localized to the genes in the human lymphocyte antigen complex on chromosome 6. Primary adrenal insufficiency in APS-2, but not APS-1, is strongly associated with both HLA-DR3 and HLA-DR4. Other class I and class II genes and alleles, such as HLA-B8, HLA-DQ2 and HLA-DQ8, and HLA-DR subtype such as DRB1*0404, appear to contribute to organ-specific disease susceptibility (Table 409-4). HLA-B8- and HLA-DR3-associated illnesses include selective IgA deficiency, juvenile dermatomyositis, dermatitis herpetiformis, alopecia, scleroderma, autoimmune thrombocytopenia purpura, hypophysitis, metaphyseal osteopenia, and serositis.
Genetic Considerations The overwhelming risk factor for APS-2 has been localized to the genes in the human lymphocyte antigen complex on chromosome 6. Primary adrenal insufficiency in APS-2, but not APS-1, is strongly associated with both HLA-DR3 and HLA-DR4. Other class I and class II genes and alleles, such as HLA-B8, HLA-DQ2 and HLA-DQ8, and HLA-DR subtype such as DRB1*0404, appear to contribute to organ-specific disease susceptibility (Table 409-4). HLA-B8- and HLA-DR3-associated illnesses include selective IgA deficiency, juvenile dermatomyositis, dermatitis herpetiformis, alopecia, scleroderma, autoimmune thrombocytopenia purpura, hypophysitis, metaphyseal osteopenia, and serositis.
|
APS-2 AND OTHER POLYENDOCRINE DISORDER ASSOCIATIONS |
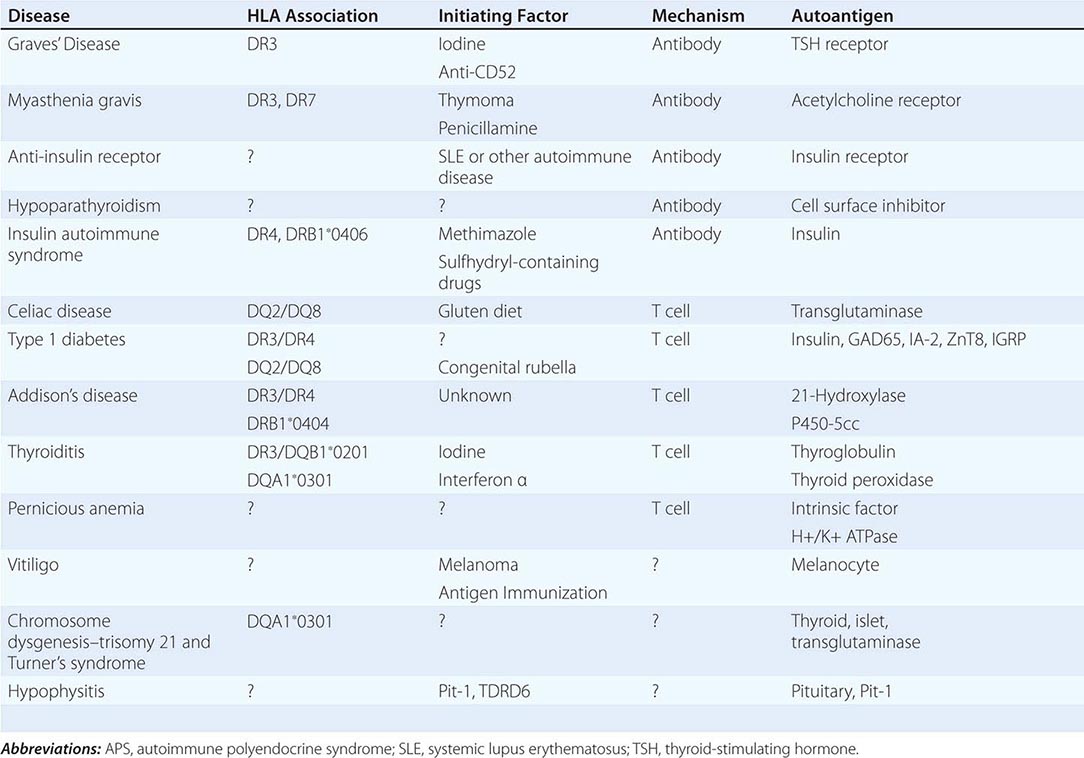
Several other immune genes have been proposed to be associated with Addison’s disease and therefore with APS-2 (Table 409-3). The “5.1” allele of a major histocompatibility complex (MHC) gene is an atypical class I HLA molecule MIC-A. The MIC-A5.1 allele has a very strong association with Addison’s disease that is not accounted for by linkage disequilibrium with DR3 or DR4. Its role is complicated because certain HLA class I genes can offset this effect. PTPN22 codes for a polymorphism in a protein tyrosine phosphatase, which acts on intracellular signaling pathways in both T and B lymphocytes. It has been implicated in T1D, Addison’s disease, and other autoimmune conditions. CTLA4 is a receptor on the T cell surface that modulates the activation state of the cell as part of the signal 2 pathway. Polymorphisms of this gene appear to cause downregulation of the cell surface expression of the receptor, leading to decreased T cell activation and proliferation. This appears to contribute to disease in Addison’s disease and potentially other components of APS-2. Allelic variants of the IL-2Rα are linked to development of T1D and autoimmune thyroid disease and could contribute to the phenotype of APS-2 in certain individuals.
Diagnosis When one of the component disorders is present, a second associated disorder occurs more commonly than in the general population (Table 409-3). There is controversy as to which tests to use and how often to screen individuals for disease. A strong family history of autoimmunity should raise suspicion in an individual with an initial component diagnosis. The development of a rarer form of autoimmunity, such as Addison’s disease, should prompt more extensive screening for other linked disorders compared to the diagnosis of autoimmune thyroid disease, which is relatively common.
Circulating autoantibodies, as previously discussed, can precede the development of disease by many years but would allow the clinician to follow the patient and identify the disease onset at its earliest time point (Tables 409-3 and 409-4). For each of the endocrine components of the disorder, appropriate autoantibody assays are listed and, if positive, should prompt physiologic testing to diagnose clinical or subclinical disease. For Addison’s disease, antibodies to 21-hydroxylase antibodies are highly diagnostic for risk of adrenal insufficiency. However, individuals may take many years to develop overt hypoadrenalism. Screening of 21-hydroxylase antibody–positive patients can be performed measuring morning ACTH and cortisol on a yearly basis. Rising ACTH values over time or low morning cortisol in association with signs or symptoms of adrenal insufficiency should prompt testing via the cosyntropin stimulation test (Chap. 406). T1D can be screened for by measuring autoantibodies including anti-insulin, anti-GAD65, anti-IA-2, and anti-ZnT8. Risk for progression to disease can be based on the number of antibodies, and in some cases the titer (insulin autoantibody), as well as other metabolic factors (impaired oral glucose tolerance test). National Institutes of Health–sponsored trial groups such as Type 1 Diabetes TrialNet are screening first- and second-degree family members for these autoantibodies and identifying prediabetic individuals who may qualify for intervention trials to change the course of the disease prior to onset.
Screening tests for thyroid disease can include anti–thyroid peroxidase (TPO) or anti-thyroglobulin autoantibodies or anti-TSH receptor antibodies for Graves’ disease. Yearly measurements of TSH can then be used to follow these individuals. Celiac disease can be screened for using the anti–tissue transglutaminase (tTg) antibody test. For those <20 years of age, testing every 1–2 years should be performed, whereas less frequent testing is indicated after the age of 20 because the majority of individuals who develop celiac disease have the antibody earlier in life. Positive tTg antibody test results should be confirmed on repeat testing, followed by small-bowel biopsy to document pathologic changes of celiac disease. Many patients have asymptomatic celiac disease that is nevertheless associated with osteopenia and impaired growth. If left untreated, symptomatic celiac disease has been reported to be associated with an increased risk of gastrointestinal malignancy, especially lymphoma.
The knowledge of the particular disease associations should guide other autoantibody or laboratory testing. A complete history and physical examination should be performed every 1–3 years including CBC, metabolic panel, TSH, and vitamin B12 levels to screen for most of the possible abnormalities. More specific tests should be based on specific findings from the history and physical.
|
TREATMENT |
APS-2 |
With the exception of Graves’ disease, the management of each of the endocrine components of APS-2 involves hormone replacement and is covered in detail in the chapters on adrenal (Chap. 406), thyroid (Chap. 405), gonadal (Chaps. 411 and 412), and parathyroid disease (Chap. 424). As noted for APS-1, adrenal insufficiency can be masked by primary hypothyroidism and should be considered and treated as discussed above. In patients with T1D, decreasing insulin requirements or hypoglycemia, without obvious secondary causes, may indicate the emergence of adrenal insufficiency. Hypocalcemia in APS-2 patients is more likely due to malabsorption than hypoparathyroidism.
Immunotherapy for autoimmune endocrine disease has been reserved for T1D, for the most part, reflecting the lifetime burden of the disease for the individual patient and society. Although several immunotherapies (e.g., modified anti-CD3, rituximab, abatacept) can prolong the honeymoon phase of T1D, none has achieved long-term success. Active research using new approaches and combination therapy may change the treatment of this disease or other autoimmune conditions that share similar pathways. Furthermore, treatment of subclinical disease diagnosed by the presence of autoantibodies may provide a mechanism to preempt the development of overt disease and is the subject of active basic and clinical research.
IPEX
Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked disease (IPEX; OMIM 304790) is a rare X-linked recessive disorder. The disease onset is in infancy and is characterized by severe enteropathy, T1D, and skin disease, as well as variable association with several other autoimmune disorders. Many infants die within the first days of life, but the course is variable, with some children surviving for 12–15 years. Early onset of T1D, often at birth, is highly suggestive of the diagnosis because nearly 80% of IPEX patients develop T1D. Although treatment of the individual disorders can temporarily improve the situation, treatment of the underlying immune deficiency is required and includes immunosuppressive therapy generally followed by hematopoietic stem cell transplantation. Transplantation is the only life-saving form of therapy and can be fully curative by normalizing the imbalanced immune system found in this disorder.
IPEX is caused by mutations in the FOXP3 gene, which is also mutated in the Scurfy mouse, an animal model that shares much of the phenotype of IPEX patients. The FOXP3 transcription factor is expressed in regulatory T cells designated CD4+CD25+FOXP3+ (Treg). Lack of this factor causes a profound deficiency of this Treg population and results in rampant autoimmunity due to the lack of peripheral tolerance normally provided by these cells. Certain mutations may lead to varying forms of expression of the full syndrome, and there are rare cases where the FOXP3 gene is intact but other genes involved in this pathway (e.g., CD25, IL-2Rα) may be causative.
THYMIC TUMORS
Thymomas and thymic hyperplasia are associated with several autoimmune diseases, with the most common being myasthenia gravis (44%) and red cell aplasia (20%). Graves’ disease, T1D, and Addison’s disease may also be associated with thymic tumors. Patients with myasthenia gravis and thymoma may have unique anti–acetylcholine receptor autoantibodies. Many thymomas lack AIRE expression within the thymoma, and this could be a potential factor in the development of autoimmunity. In support of this concept, thymoma is the one other disease with “frequent” development of anticytokine antibodies and mucocutaneous candidiasis in adults. The majority of tumors are malignant, and temporary remissions of the autoimmune condition can occur with resection of the tumor.
ANTI-INSULIN RECEPTOR ANTIBODIES
This is a very rare disorder where severe insulin resistance (type B) is caused by the presence of anti-insulin receptor antibodies. It is associated with acanthosis nigricans, which can also be associated with other forms of less severe insulin resistance. About one-third of patients have an associated autoimmune illness such as systemic lupus erythematosus or Sjögren’s syndrome. Therefore, the presence of antinuclear antibodies, elevated erythrocyte sedimentation rate, hyperglobulinemia, leukopenia, and hypocomplementemia may accompany the presentation. The presence of anti-insulin receptor autoantibodies leads to marked insulin resistance, requiring more than 100,000 units of insulin to be given daily with only partial control of hyperglycemia. Patients can also have severe hypoglycemia due to partial activation of the insulin receptor by the antibody. The course of the disease is variable, and several patients have had spontaneous remissions. Therapy targeting B lymphocytes including rituximab, cyclophosphamide, and pulse steroids can induce remission of the disease.
INSULIN AUTOIMMUNE SYNDROME (HIRATA’S SYNDROME)
The insulin autoimmune syndrome, associated with Graves’ disease and methimazole therapy (or other sulfhydryl-containing medications), is of particular interest due to a remarkably strong association with a specific HLA haplotype. Such patients with elevated titers of anti-insulin autoantibodies frequently present with hypoglycemia. In Japan, the disease is restricted to HLA-DR4-positive individuals with DRB1*0406. Curiously, a recent report demonstrated that five out of six Caucasian patients taking lipoic acid (sulfhydryl group) who developed insulin autoimmune syndrome were primarily DRB1*0403 (which is related to DRB1*0406); the sixth was DRB1*0406. In Hirata’s syndrome the anti-insulin autoantibodies are often polyclonal. Discontinuation of the medication generally leads to resolution of the syndrome over time.
POEMS SYNDROME
POEMS (polyneuropathy, organomegaly, endocrinopathy, M-protein, and skin changes; also known as Crow-Fukase syndrome; OMIM 192240) patients usually present with a progressive sensorimotor polyneuropathy, diabetes mellitus (50%), primary gonadal failure (70%), and a plasma cell dyscrasia with sclerotic bony lesions. Associated findings can be hepatosplenomegaly, lymphadenopathy, and hyperpigmentation. Patients often present in the fifth to sixth decade of life and have a median survival after diagnosis of less than 3 years. The syndrome is assumed to be secondary to circulating immunoglobulins, but patients have excess vascular endothelial growth factor as well as elevated levels of other inflammatory cytokines such as IL1-β, IL-6, and tumor necrosis factor α. A small series of patients have been treated with thalidomide, leading to a decrease in vascular endothelial growth factor. Hyperglycemia responds to small, subcutaneous doses of insulin. The hypogonadism is due to primary gonadal disease with elevated plasma levels of follicle-stimulating hormone and luteinizing hormone. Temporary resolution of the features of POEMS, including normalization of blood glucose, may occur after radiotherapy for localized plasma cell lesions of bone or after chemotherapy, thalidomide, plasmapheresis, autologous stem cell transplantation, or treatment with all-trans-retinoic acid.
OTHER DISORDERS
Other diseases can exhibit polyendocrine deficiencies, including Kearns-Sayre syndrome, DIDMOAD syndrome (diabetes insipidus, diabetes mellitus, progressive bilateral optic atrophy, and sensorineural deafness; also termed Wolfram’s syndrome), Down’s syndrome or trisomy 21 (OMIM 190685), Turner’s syndrome (monosomy X, 45,X), and congenital rubella.
Kearns-Sayre syndrome (OMIM 530000) is a rare mitochondrial DNA disorder characterized by myopathic abnormalities leading to ophthalmoplegia and progressive weakness in association with several endocrine abnormalities, including hypoparathyroidism, primary gonadal failure, diabetes mellitus, and hypopituitarism. Crystalline mitochondrial inclusions are found in muscle biopsy specimens, and such inclusions have also been observed in the cerebellum. Antiparathyroid antibodies have not been described; however, antibodies to the anterior pituitary gland and striated muscle have been identified, and the disease may have autoimmune components. These mitochondrial DNA mutations occur sporadically and do not appear to be associated with a familial syndrome.
Wolfram’s syndrome (OMIM 222300, chromosome 4; OMIM 598500, mitochondrial) is a rare autosomal recessive disease that is also called DIDMOAD. Neurologic and psychiatric disturbances are prominent in most patients and can cause severe disability. The disease is caused by defects in wolframin, a 100-kDa transmembrane protein that has been localized to the endoplasmic reticulum and is found in neuronal and neuroendocrine tissue. Its expression induces ion channel activity with a resultant increase in intracellular calcium and may play an important role in intracellular calcium homeostasis. Wolfram’s syndrome appears to be a slowly progressive neurodegenerative process, and there is nonautoimmune selective destruction of the pancreatic beta cells. Diabetes mellitus with an onset in childhood is usually the first manifestation. Diabetes mellitus and optic atrophy are present in all reported cases, but expression of the other features is variable.
Down’s syndrome, or trisomy 21 (OMIM 190685), is associated with the development of T1D, thyroiditis, and celiac disease. Patients with Turner’s syndrome also appear to be at increased risk for the development of thyroid disease and celiac disease. It is recommended to screen patients with trisomy 21 and Turner’s syndrome for associated autoimmune diseases on a regular basis.
SECTION 2 |
REPRODUCTIVE ENDOCRINOLOGY |
410 |
Disorders of Sex Development |
Sex development begins in utero but continues into young adulthood with the achievement of sexual maturity and reproductive capability. The major determinants of sex development can be divided into three components: chromosomal sex, gonadal sex (sex determination), and phenotypic sex (sex differentiation) (Fig. 410-1). Variations at each of these stages can result in disorders (or differences) of sex development (DSDs) (Table 410-1). In the newborn period, approximately 1 in 4000 babies require investigation because of ambiguous (atypical) genitalia. Urgent assessment is required, because some causes such as congenital adrenal hyperplasia (CAH) can be associated with life-threatening adrenal crises. Support for the parents and clear communication about the diagnosis and management options are essential. The involvement of an experienced multidisciplinary team is important for counseling, planning appropriate investigations, and discussing long-term well-being. DSDs can also present at other ages and to a range of health professionals. Subtler forms of gonadal dysfunction (e.g., Klinefelter’s syndrome [KS], Turner’s syndrome [TS]) often are diagnosed later in life by internists. Because these conditions are associated with a variety of psychological, reproductive, and potential medical consequences, an open dialogue must be established between the patient and health care providers to ensure continuity and attention to these issues.
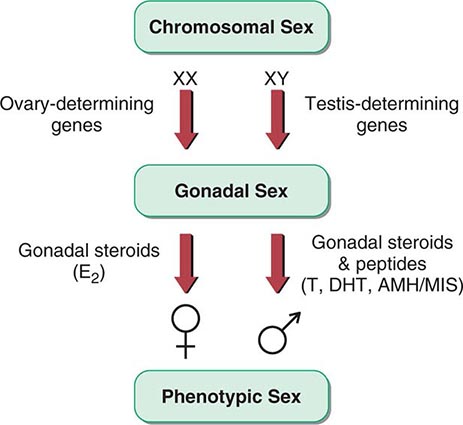
FIGURE 410-1 Sex development can be divided into three major components: chromosomal sex, gonadal sex, and phenotypic sex. DHT, dihydrotestosterone; MIS, müllerian-inhibiting substance also known as anti-müllerian hormone, AMH; T, testosterone.
|
CLASSIFICATION OF DISORDERS OF SEX DEVELOPMENT (DSDS) |
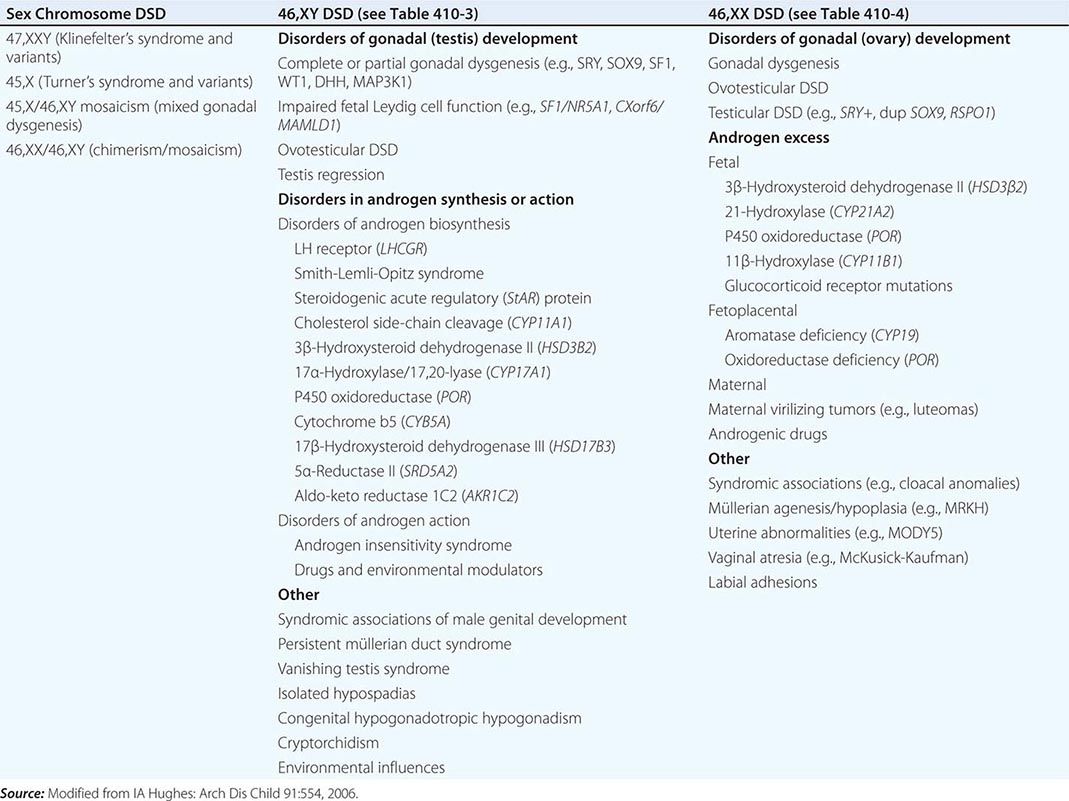
SEX DEVELOPMENT
Chromosomal sex, defined by a karyotype, describes the × and/or Y chromosome complement (46,XY; 46,XX) that is established at the time of fertilization. The presence of a normal Y chromosome determines that testis development will occur even in the presence of multiple × chromosomes (e.g., 47,XXY or 48,XXXY). The loss of an × chromosome impairs gonad development (45,X or 45,X/46,XY mosaicism). Fetuses with no × chromosome (45,Y) are not viable.
Gonadal sex refers to the histologic and functional characteristics of gonadal tissue as testis or ovary. The embryonic gonad is bipotential and can develop (from ~42 days after conception) into either a testis or an ovary, depending on which genes are expressed (Fig. 410-2). Testis development is initiated by expression of the Y chromosome gene SRY (sex-determining region on the Y chromosome) that encodes an HMG box transcription factor. SRY is expressed transiently in cells destined to become Sertoli cells and serves as a pivotal switch to establish the testis lineage. Mutation of SRY prevents testis development in 46,XY individuals, whereas translocation of SRY in 46,XX individuals is sufficient to induce testis development and a male phenotype. Other genes are necessary to continue testis development. SOX9 (SRY-related HMG-box gene 9) is upregulated by SRY in the developing testis but is suppressed in the ovary. WT1 (Wilms’ tumor–related gene 1) acts early in the genetic pathway and regulates the transcription of several genes, including SFI (NR5A1), DAX1 (NR0B1), and AMH (encoding müllerian-inhibiting substance [MIS]). SF1 encodes steroidogenic factor 1, a nuclear receptor that functions in cooperation with other transcription factors to regulate a large array of adrenal and gonadal genes, including SOX9 and many genes involved in steroidogenesis. SF1 mutations causing loss of function are found in ~10% of XY patients with gonadal dysgenesis and impaired androgenization. In contrast, duplication of a related gene DAX1 also impairs testis development, revealing the exquisite sensitivity of the testis-determining pathway to gene dosage effects. DAX1 loss-of-function mutations cause adrenal hypoplasia, hypogonadotropic hypogonadism, and testicular dysgenesis. In addition to the genes mentioned above, studies of humans and mice indicate that at least 30 other genes are also involved in gonad development (Fig. 410-2). These genes encode an array of signaling molecules and paracrine growth factors in addition to transcription factors.
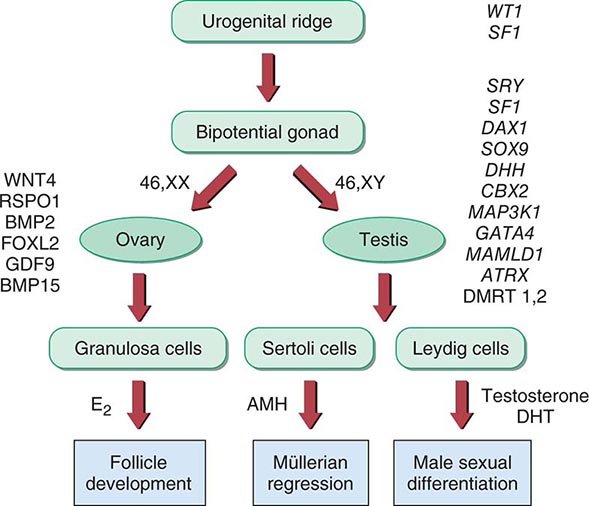
FIGURE 410-2 The genetic regulation of gonadal development. AMH, anti-müllerian hormone (müllerian-inhibiting substance); ATRX, α-thalassemia, mental retardation on the X; BMP2 and 15, bone morphogenic factors 2 and 15; CBX2, chromobox homologue 2; DAX1, dosage sensitive sex-reversal, adrenal hypoplasia congenita on the × chromosome, gene 1; DHH, desert hedgehog; DHT, dihydrotestosterone; DMRT 1,2, doublesex MAB3-related transcription factor 1,2; FOXL2, forkhead transcription factor L2; GATA4, GATA binding protein 4; GDF9, growth differentiation factor 9; MAMLD1, mastermind-like domain containing 1; MAP3K1, mitogen-activated protein kinase kinase kinase 1; RSPO1, R-spondin 1; SF1, steroidogenic factor 1 (also known as NR5A1); SOX9, SRY-related HMG-box gene 9; SRY, sex-determining region on the Y chromosome; WNT4, wingless-type MMTV integration site 4; WT1, Wilms’ tumor–related gene 1.
Although ovarian development once was considered a “default” process, it is now clear that specific genes are expressed during the earliest stages of ovary development. Some of these factors may repress testis development (e.g., WNT4, R-spondin-1) (Fig. 410-2). Once the ovary has formed, additional factors are required for normal follicular development (e.g., follicle-stimulating hormone [FSH] receptor, GDF9). Steroidogenesis in the ovary requires the development of follicles that contain granulosa cells and theca cells surrounding the oocytes (Chap. 412). Thus, there is relatively limited ovarian steroidogenesis until puberty.
Germ cells also develop in a sex dimorphic manner. In the developing ovary, primordial germ cells (PGCs) proliferate and enter meiosis, whereas they proliferate and then undergo mitotic arrest in the developing testis. PGC entry into meiosis is initiated by retinoic acid that activates STRA8 (stimulated by retinoic acid 8) and other genes involved in meiosis. The developing testis produces high levels of CYP26B1, an enzyme that degrades retinoic acid, preventing PGC entry into meiosis. Approximately 7 million germ cells are present in the fetal ovary in the second trimester, and 1 million remain at birth. Only 400 are ovulated during a woman’s reproductive life span (Chap. 412).
Phenotypic sex refers to the structures of the external and internal genitalia and secondary sex characteristics. The developing testis releases anti-müllerian hormone (AMH; also known as müllerian-inhibiting substance [MIS]) from Sertoli cells and testosterone from Leydig cells. AMH is a member of the transforming growth factor (TGF) β family and acts through specific receptors to cause regression of the müllerian structures from 60–80 days after conception. At ~60–140 days after conception, testosterone supports the development of wolffian structures, including the epididymides, vasa deferentia, and seminal vesicles. Testosterone is the precursor for dihydrotestosterone (DHT), a potent androgen that promotes development of the external genitalia, including the penis and scrotum (65–100 days, and thereafter) (Fig. 410-3). The urogenital sinus develops into the prostate and prostatic urethra in the male and into the urethra and lower portion of the vagina in the female. The genital tubercle becomes the glans penis in the male and the clitoris in the female. The urogenital swellings form the scrotum or the labia majora, and the urethral folds fuse to form the shaft of the penis and the male urethra or the labia minora. In the female, wolffian ducts regress and the müllerian ducts form the fallopian tubes, uterus, and upper segment of the vagina. A female phenotype will develop in the absence of the gonad, but estrogen is needed for maturation of the uterus and breast at puberty.
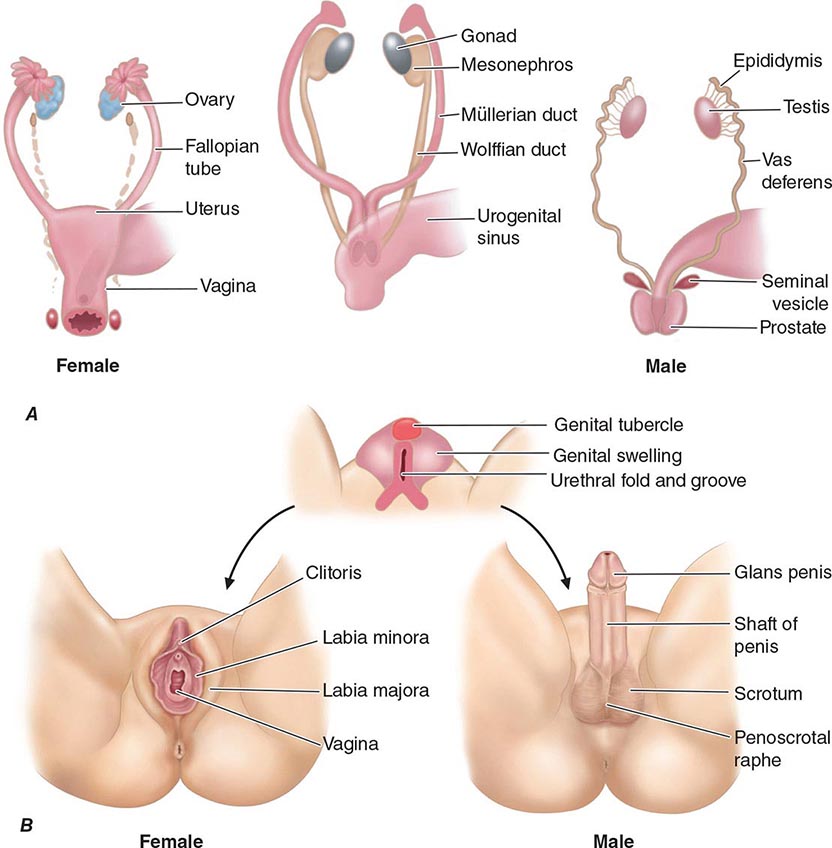
FIGURE 410-3 Sex development. A. Internal urogenital tract. B. External genitalia. (After E Braunwald et al [eds]: Harrison’s Principles of Internal Medicine, 15th ed. New York, McGraw-Hill, 2001.)
DISORDERS OF CHROMOSOMAL SEX
Variations in sex chromosome number and structure can present as DSDs (e.g., 45,X/46,XY). KS (47,XXY) and TS (45,X) do not usually present with genital ambiguity but are associated with gonadal dysfunction (Table 410-2).
|
CLINICAL FEATURES OF CHROMOSOMAL DISORDERS OF SEX DEVELOPMENT (DSD) |
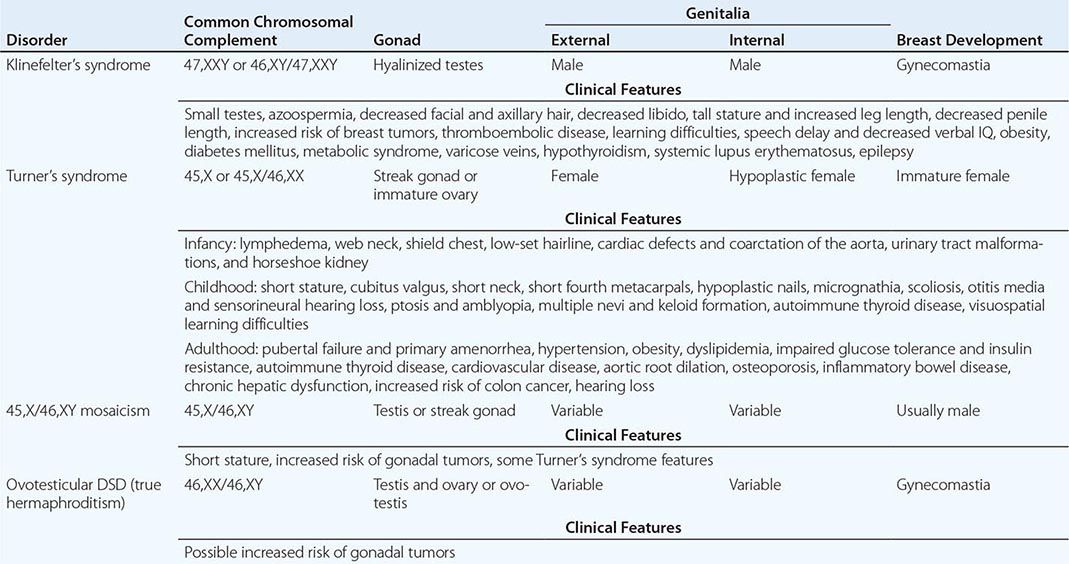
KLINEFELTER’S SYNDROME (47,XXY)
Pathophysiology The classic form of KS (47,XXY) occurs after meiotic nondisjunction of the sex chromosomes during gametogenesis (40% during spermatogenesis, 60% during oogenesis) (Chap. 83e). Mosaic forms of KS (46,XY/47,XXY) are thought to result from chromosomal mitotic nondisjunction within the zygote and occur in at least 10% of individuals with this condition. Other chromosomal variants of KS (e.g., 48,XXYY, 48,XXXY) have been reported but are less common.
Clinical Features KS is characterized by small testes, infertility, gynecomastia, tall stature/increased leg length, and hypogonadism in phenotypic males. It has an incidence of at least 1 in 1000 men, but approximately 75% of cases are not diagnosed. Of those who are diagnosed, only 10% are identified prepubertally, usually because of small genitalia or cryptorchidism. Others are diagnosed after puberty, usually based on impaired androgenization and/or gynecomastia. Developmental delay, speech difficulties, and poor motor skills may be features but are variable, especially in adolescence. Later in life, body habitus or infertility leads to the diagnosis. Testes are small and firm (median length 2.5 cm [4 mL volume]; almost always <3.5 cm [12 mL]) and typically seem inappropriately small for the degree of androgenization. Biopsies are not usually necessary but typically reveal seminiferous tubule hyalinization and azoospermia. Other clinical features of KS are listed in Table 410-2. Plasma concentrations of FSH and luteinizing hormone (LH) are increased in most adults with 47,XXY, and plasma testosterone is decreased (50–75%), reflecting primary gonadal failure. Estradiol is often increased, likely because of chronic Leydig cell stimulation by LH and aromatization of androstenedione by adipose tissue; the increased ratio of estradiol-to-testosterone results in gynecomastia (Chap. 411). Patients with mosaic forms of KS have less severe clinical features, have larger testes, and sometimes achieve spontaneous fertility.
|
TREATMENT |
KLINEFELTER’S SYNDROME |
Growth, endocrine function, and bone mineralization should be monitored, especially from adolescence. Educational and psychological support is important for many individuals with KS. Androgen supplementation improves virilization, libido, energy, hypofibrinolysis, and bone mineralization in men with low testosterone levels but may occasionally worsen gynecomastia (Chap. 411). Gynecomastia can be treated by surgical reduction if it causes concern (Chap. 411). Fertility has been achieved by using in vitro fertilization in men with oligospermia or with intracytoplasmic sperm injection (ICSI) after retrieval of spermatozoa by testicular sperm extraction techniques. In specialized centers, successful spermatozoa retrieval using this technique is possible in >50% of men with nonmosaic KS. Results may be better in younger men. After ICSI and embryo transfer, successful pregnancies can be achieved in ~50% of these cases. The risk of transmission of this chromosomal abnormality needs to be considered, and preimplantation screening may be desired, although this outcome is much less common than originally predicted. Long-term monitoring of men with KS is important given the increased risk of breast cancer, cardiovascular disease, metabolic syndrome, and autoimmune disorders. Because most men with KS are never diagnosed, it is important that all internists consider this diagnosis in men with these features who might be seeking medical advice for other conditions.
TURNER’S SYNDROME (GONADAL DYSGENESIS; 45,X)
Pathophysiology Approximately one-half of women with TS have a 45,X karyotype, about 20% have 45,X/46,XX mosaicism, and the remainder have structural abnormalities of the × chromosome such as × fragments, isochromosomes, or rings. The clinical features of TS result from haploinsufficiency of multiple × chromosomal genes (e.g., short stature homeobox, SHOX). However, imprinted genes also may be affected when the inherited × has different parental origins.
Clinical Features TS is characterized by bilateral streak gonads, primary amenorrhea, short stature, and multiple congenital anomalies in phenotypic females. It affects ~1 in 2500 women and is diagnosed at different ages depending on the dominant clinical features (Table 410-2). Prenatally, a diagnosis of TS usually is made incidentally after chorionic villus sampling or amniocentesis for unrelated reasons such as advanced maternal age. Prenatal ultrasound findings include increased nuchal translucency. The postnatal diagnosis of TS should be considered in female neonates or infants with lymphedema, nuchal folds, low hairline, or left-sided cardiac defects and in girls with unexplained growth failure or pubertal delay. Although limited spontaneous pubertal development occurs in up to 30% of girls with TS (10%, 45,X; 30–40%, 45,X/46,XX) and ~2% reach menarche, the vast majority of women with TS develop complete ovarian insufficiency. Therefore, this diagnosis should be considered in all women who present with primary or secondary amenorrhea and elevated gonadotropin levels.
|
TREATMENT |
TURNER’S SYNDROME |
The management of girls and women with TS requires a multidisciplinary approach because of the number of potentially involved organ systems. Detailed cardiac and renal evaluation should be performed at the time of diagnosis. Individuals with congenital heart defects (CHDs) (30%) (bicuspid aortic valve, 30–50%; coarctation of the aorta, 30%; aortic root dilation, 5%) require long-term follow-up by an experienced cardiologist, antibiotic prophylaxis for dental or surgical procedures, and serial magnetic resonance imaging (MRI) of aortic root dimensions, because progressive aortic root dilation is associated with increased risk of aortic dissection. Individuals found to have congenital renal and urinary tract malformations (30%) are at risk for urinary tract infections, hypertension, and nephrocalcinosis. Hypertension can occur independently of cardiac and renal malformations and should be monitored and treated as in other patients with essential hypertension. Clitoral enlargement or other evidence of virilization suggests the presence of covert, translocated Y chromosomal material and is associated with increased risk of gonadoblastoma. Regular assessment of thyroid function, weight, dentition, hearing, speech, vision, and educational issues should be performed during childhood. Otitis media and middle-ear disease are prevalent in childhood (50–85%), and sensorineural hearing loss becomes progressively common with age (70–90%). Autoimmune hypothyroidism (15–30%) can occur in childhood but has a mean age of onset in the third decade. Counseling about long-term growth and fertility issues should be provided. Patient support groups are active throughout the world and can play an invaluable role.
Short stature can be an issue for some girls because untreated final height rarely exceeds 150 cm in nonmosaic 45,X TS. High-dose recombinant growth hormone stimulates growth rate in children with TS and is occasionally combined with low doses of the nonaromatizable anabolic steroid oxandrolone (up to 0.05 mg/kg per day) in an older child (>9 years). However, final height increments are often about 5–10 cm, and individualization of treatment response to regimens may be beneficial. Girls with evidence of ovarian insufficiency require estrogen replacement to induce breast and uterine development, support growth, and maintain bone mineralization. Most physicians now initiate low-dose estrogen therapy (one-tenth to one-eighth of the adult replacement dose) to induce puberty at an age-appropriate time (~12 years). Doses of estrogen are increased gradually to allow development over a 2- to 4-year period. Progestins are added later to regulate withdrawal bleeds. Some women with TS have achieved successful pregnancy after ovum donation and in vitro fertilization but are high risk, and cardiac assessment is required. Long-term follow-up of women with TS involves careful surveillance of sex hormone replacement and reproductive function, bone mineralization, cardiac function and aortic root dimensions, blood pressure, weight and glucose tolerance, hepatic and lipid profiles, thyroid function, and hearing. This service is provided by a dedicated TS clinic in some centers.
45,X/46,XY MOSAICISM (MIXED GONADAL DYSGENESIS)
The phenotype of individuals with 45,X/46,XY mosaicism (sometimes called mixed gonadal dysgenesis) can vary considerably. Some have a predominantly female phenotype with somatic features of TS, streak gonads, and müllerian structures, and are managed as TS with a Y chromosome. Most 45,X/46,XY individuals have a male phenotype and testes, and the diagnosis is made incidentally after amniocentesis or during investigation of infertility. In practice, most newborns referred for assessment have atypical genitalia and variable somatic features. Management is complex and needs to be individualized. A female sex-of-rearing is often assigned if uterine structures are present, gonads are intraabdominal, and phallic development is incomplete. In such situations, gonadectomy usually is considered to prevent further androgen secretion at puberty and prevent risk of gonadoblastoma (up to 25%). Individuals raised as males usually require reconstructive surgery for hypospadias and removal of dysgenetic or streak gonads if the gonads cannot be brought down into the scrotum. Scrotal testes can be preserved but require regular examination for tumor development and sonography at the time of puberty. Biopsy for carcinoma in situ is recommended in adolescence, and testosterone supplementation may be required to support androgenization in puberty or if low testosterone is detected in adulthood. Height potential is usually attenuated; some children receive recombinant growth hormone using TS protocols. Screening for cardiac, renal, and other TS features should be considered, and psychological support offered for the family and young person.
OVOTESTICULAR DSD
Ovotesticular DSD (formerly called true hermaphroditism) occurs when both an ovary and a testis—or when an ovotestis—are found in one individual. Most individuals with this diagnosis have a 46,XX karyotype, especially in sub-Saharan Africa, and present with ambiguous genitalia at birth or with breast development and phallic development at puberty. A 46,XX/46,XY chimeric karyotype is less common and has a variable phenotype.
DISORDERS OF GONADAL AND PHENOTYPIC SEX
Disorders of gonadal and phenotypic sex can result in underandrogenization of individuals with a 46,XY karyotype (46,XY DSD) and the excess androgenization of individuals with a 46,XX karyotype (46,XX DSD) (Table 410-1). These disorders cover a spectrum of phenotypes ranging from “46,XY phenotypic females” or “46,XX phenotypic males” to individuals with atypical genitalia.
46,XY DSD
Underandrogenization of the 46,XY fetus (formerly called male pseudohermaphroditism) reflects defects in androgen production or action. It can result from disorders of testis development, defects of androgen synthesis, or resistance to testosterone and DHT (Table 410-1).
Disorders of Testis Development • TESTICULAR DYSGENESIS Pure (or complete) gonadal dysgenesis (Swyer’s syndrome) is associated with streak gonads, müllerian structures (due to insufficient AMH/MIS secretion), and a complete absence of androgenization. Phenotypic females with this condition often present because of absent pubertal development and are found to have a 46,XY karyotype. Serum sex steroids, AMH/MIS, and inhibin B are low, and LH and FSH are elevated. Patients with partial gonadal dysgenesis (dysgenetic testes) may produce enough MIS to regress the uterus and sufficient testosterone for partial androgenization, and therefore usually present in the newborn period with atypical genitalia. Gonadal dysgenesis can result from mutations or deletions of testis-promoting genes (WT1, CBX2, SF1, SRY, SOX9, MAP3K1, DHH, GATA4, ATRX, ARX, DMRT) or duplication of chromosomal loci containing “antitestis” genes (e.g., WNT4/RSPO1, DAX1) (Table 410-3). Among these, deletions or mutations of SRY and heterozygous mutations of SF1 (NR5A1) appear to be most common but still account collectively for <25% of cases. Associated clinical features may be present, reflecting additional functional roles for these genes. For example, renal dysfunction occurs in patients with specific WT1 mutations (Denys-Drash and Frasier’s syndromes), primary adrenal failure occurs in some patients with SF1 mutations, and severe cartilage abnormalities (campomelic dysplasia) are the predominant clinical feature of SOX9 mutations. A family history of DSD, infertility, or early menopause is important because mutations in SF1/NR5A1 can be inherited from a mother in a sex-limited dominant manner (which can mimic X-linked inheritance). In some cases, a woman may later develop primary ovarian insufficiency because of the effect of SF1 on the ovary. Intraabdominal dysgenetic testes should be removed to prevent malignancy, and estrogens can be used to induce secondary sex characteristics and uterine development in 46,XY individuals raised as females, if it is felt that a female gender identity is established. Absent (vanishing) testis syndrome (bilateral anorchia) reflects regression of the testis during development. The etiology is unknown, but the absence of müllerian structures indicates adequate secretion of AMH early in utero. In most cases, androgenization of the external genitalia is either normal or slightly impaired (e.g., small penis, hypospadias). These individuals can be offered testicular prostheses and should receive androgen replacement in adolescence.
|
SELECTED GENETIC CAUSES OF 46,XY DISORDERS OF SEX DEVELOPMENT (DSDs) |
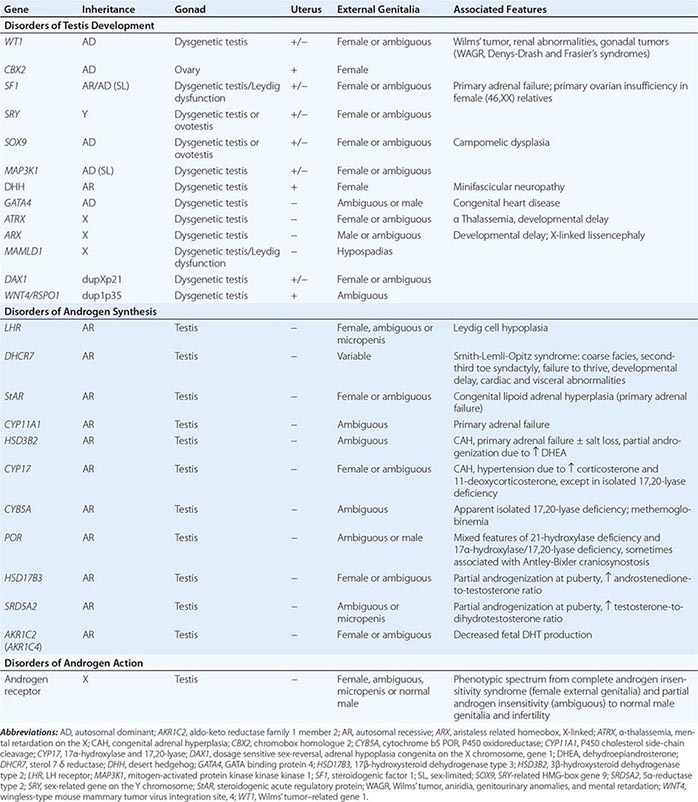
Disorders of Androgen Synthesis Defects in the pathway that regulates androgen synthesis (Fig. 410-4) cause underandrogenization of the 46,XY fetus (Table 410-1). Müllerian regression is unaffected because Sertoli cell function is preserved. Most of these conditions can present with a spectrum of genital phenotypes, ranging from female-typical external genitalia or clitoromegaly in the more severe situations to penoscrotal hypospadias or a small phallus in others.
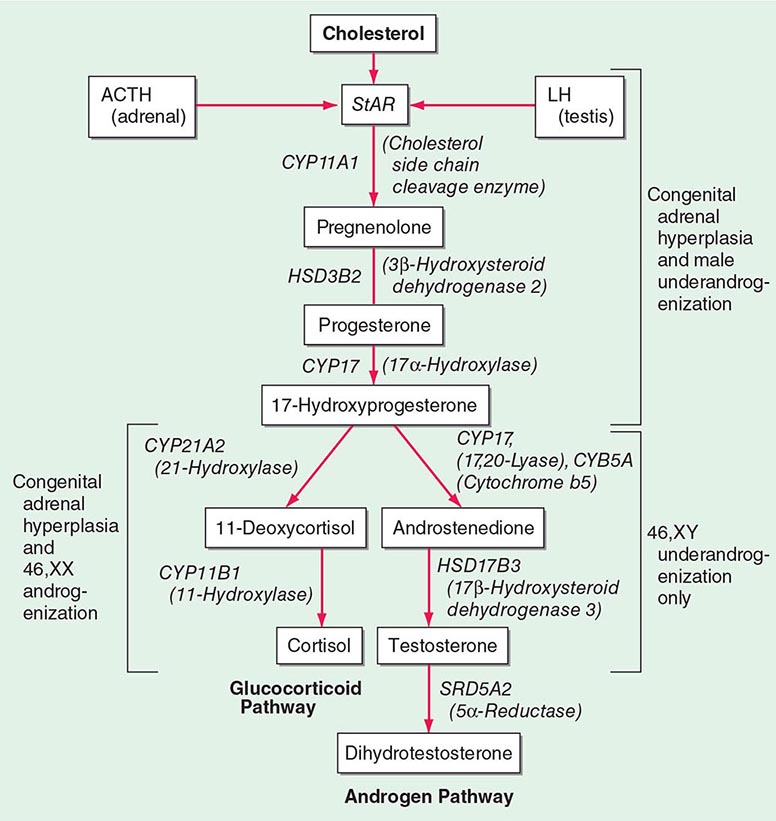
FIGURE 410-4 Simplified overview of glucocorticoid and androgen synthesis pathways. Defects in CYP21A2 and CYP11B1 shunt steroid precursors into the androgen pathway and cause androgenization of the 46,XX fetus. Testosterone is synthesized in the testicular Leydig cells and converted to dihydrotestosterone peripherally. Defects in enzymes involved in androgen synthesis result in underandrogenization of the 46,XY fetus. StAR, steroidogenic acute regulatory protein. (After E Braunwald et al [eds]: Harrison’s Principles of Internal Medicine, 15th ed. New York, McGraw-Hill, 2001.)
LH RECEPTOR Mutations in the LH receptor (LHCGR) cause Leydig cell hypoplasia and androgen deficiency, due to impaired actions of human chorionic gonadotropin in utero and LH late in gestation and during the neonatal period. As a result, testosterone and DHT synthesis are insufficient for complete androgenization.
STEROIDOGENIC ENZYME PATHWAYS Mutations in steroidogenic acute regulatory protein (StAR) and CYP11A1 affect both adrenal and gonadal steroidogenesis (Fig. 410-4) (Chap. 406). Affected individuals (46,XY) usually have severe early-onset salt-losing adrenal failure and a female phenotype, although later-onset milder variants have been reported. Defects in 3β-hydroxysteroid dehydrogenase type 2 (HSD3β2) also cause adrenal insufficiency in severe cases, but the accumulation of dehydroepiandrosterone (DHEA) has a mild androgenizing effect, resulting in ambiguous genitalia or hypospadias. Salt loss occurs in many but not all cases. Patients with CAH due to 17α-hydroxylase (CYP17) deficiency have variable underandrogenization and develop hypertension and hypokalemia due to the potent salt-retaining effects of corticosterone and 11-deoxycorticosterone. Patients with complete loss of 17α-hydroxylase function often present as phenotypic females who fail to enter puberty and are found to have inguinal testes and hypertension in adolescence. Some mutations in CYP17 selectively impair 17,20-lyase activity without altering 17α-hydroxylase activity, leading to underandrogenization without mineralocorticoid excess and hypertension. Disruption of the coenzyme, cytochrome b5 (CYB5A), can present similarly, and methemoglobinemia is usually present. Mutations in P450 oxidoreductase (POR) affect multiple steroidogenic enzymes, leading to impaired androgenization and a biochemical pattern of apparent combined 21-hydroxylase and 17α-hydroxylase deficiency, sometimes with skeletal abnormalities (Antley-Bixler craniosynostosis). Defects in 17β-hydroxysteroid dehydrogenase type 3 (HSD17β3) and 5α-reductase type 2 (SRD5A2) interfere with the synthesis of testosterone and DHT, respectively. These conditions are characterized by minimal or absent androgenization in utero, but some phallic development can occur during adolescence due to the action of other enzyme isoforms. Individuals with 5α-reductase type 2 deficiency have normal wolffian structures and usually do not develop breast tissue. At puberty, the increase in testosterone induces muscle mass and other virilizing features despite DHT deficiency. Some individuals change gender from female to male at puberty. Thus, the management of this disorder is challenging. DHT cream can improve prepubertal phallic growth in patients raised as male. Gonadectomy before adolescence and estrogen replacement at puberty can be considered in individuals raised as females who have a female gender identity. Disruption of alternative pathways to fetal DHT production might also present with 46,XY DSD (AKR1C2/AKR1C4).
Disorders of Androgen Action • ANDROGEN INSENSITIVITY SYNDROME Mutations in the androgen receptor cause resistance to androgen (testosterone, DHT) action or the androgen insensitivity syndrome (AIS). AIS is a spectrum of disorders that affects at least 1 in 100,000 46,XY individuals. Because the androgen receptor is X-linked, only 46,XY offspring are affected if the mother is a carrier of a mutation. XY individuals with complete AIS (formerly called testicular feminization syndrome) have a female phenotype, normal breast development (due to aromatization of testosterone), a short vagina but no uterus (because MIS production is normal), scanty pubic and axillary hair, and a female gender identity and sex role behavior. Gonadotropins and testosterone levels can be low, normal, or elevated, depending on the degree of androgen resistance and the contribution of estradiol to feedback inhibition of the hypothalamic-pituitary-gonadal axis. AMH/MIS levels in childhood are normal or high. Most patients present with inguinal hernias (containing testes) in childhood or with primary amenorrhea in late adolescence. Gonadectomy sometimes is offered for girls diagnosed in childhood, because there is a low risk of malignancy, and estrogen replacement is prescribed. Alternatively, the gonads can be left in situ until breast development is complete and removed because of tumor risk. Some adults with complete AIS decline gonadectomy, but should be counseled about the risk of malignancy, especially because early detection of premalignant changes by imaging or biomarkers is currently not possible. The use of graded dilators in adolescence is usually sufficient to dilate the vagina for sexual intercourse.
Partial AIS (Reifenstein’s syndrome) results from androgen receptor mutations that maintain residual function. Patients often present in infancy with penoscrotal hypospadias and small undescended testes and with gynecomastia at the time of puberty. Those individuals raised as males usually require hypospadias repair in childhood and may need breast reduction in adolescence. Some boys enter puberty spontaneously. High-dose testosterone has been given to support development if puberty does not progress, but long-term data are limited. More severely underandrogenized patients present with clitoral enlargement and labial fusion and may be raised as females. The surgical and psychosexual management of these patients is complex and requires active involvement of the parents and the patient during the appropriate stages of development. Azoospermia and male-factor infertility also have been described in association with mild loss-of-function mutations in the androgen receptor.
OTHER DISORDERS AFFECTING 46,XY MALES
Persistent müllerian duct syndrome is the presence of a uterus in an otherwise phenotypic male. This condition can result from mutations in AMH or its receptor (AMHR2). The uterus may be removed, but only if damage to the vasa deferentia and blood supply can be avoided. Isolated hypospadias occurs in ~1 in 250 males and is usually repaired surgically. Most cases are idiopathic, although evidence of penoscrotal hypospadias, poor phallic development, and/or bilateral cryptorchidism requires investigation for an underlying DSD (e.g., partial gonadal dysgenesis, mild defect in testosterone action, or even severe forms of 46,XX CAH). Unilateral undescended testes (cryptorchidism) affect more than 3% of boys at birth. Orchidopexy should be considered if the testis has not descended by 6–9 months of age. Bilateral cryptorchidism occurs less frequently and should raise suspicion of gonadotropin deficiency or DSD. A small subset of patients with cryptorchidism may have mutations in the insulin-like 3 (INSL3) gene or its receptor LGR8 (also known as GREAT), which mediates normal testicular descent. Syndromic associations and intrauterine growth retardation also occur relatively frequently in association with impaired testicular function or target tissue responsiveness, but the underlying etiology of many of these conditions is unknown.
46,XX DSD
Inappropriate androgenization of the 46,XX fetus (formerly called female pseudohermaphroditism) occurs when the gonad (ovary) contains androgen-secreting testicular material or after increased androgen exposure, which is usually adrenal in origin (Table 410-1).
46,XX Testicular/Ovotesticular DSD Testicular tissue can develop in 46,XX testicular DSD (46,XX males) after translocation of SRY, duplication of SOX9, or defects in RSPO1 (Table 410-4).
|
SELECTED GENETIC CAUSES OF 46,XX DISORDERS OF SEX DEVELOPMENT (DSDs) |
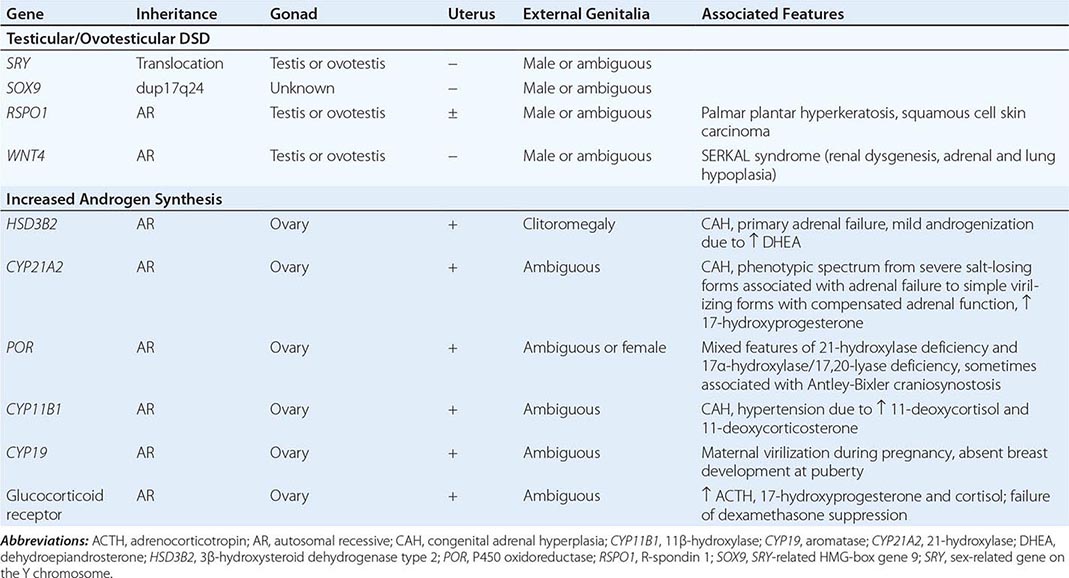
Increased Androgen Exposure • 21-HYDROXYLASE DEFICIENCY (CONGENITAL ADRENAL HYPERPLASIA) The classic form of 21-hydroxylase deficiency (21-OHD) is the most common cause of CAH (Chap. 406). It has an incidence between 1 in 10,000 and 1 in 15,000 and is the most common cause of androgenization in chromosomal 46,XX females (Table 410-4). Affected individuals are homozygous or compound heterozygous for severe mutations in the enzyme 21-hydroxylase (CYP21A2). This mutation causes a block in adrenal glucocorticoid and mineralocorticoid synthesis, increasing 17-hydroxyprogesterone and shunting steroid precursors into the androgen synthesis pathway (Fig. 410-4). Glucocorticoid insufficiency causes a compensatory elevation of adrenocorticotropin (ACTH), resulting in adrenal hyperplasia and additional synthesis of steroid precursors proximal to the enzymatic block. Increased androgen synthesis in utero causes androgenization of the 46,XX fetus in the first trimester. Ambiguous genitalia are seen at birth, with varying degrees of clitoral enlargement and labial fusion. Excess androgen production causes gonadotropin-independent precocious puberty in males with 21-OHD.
The salt-wasting form of 21-OHD results from severe combined glucocorticoid and mineralocorticoid deficiency. A salt-wasting crisis usually manifests between 5 and 21 days of life and is a potentially life-threatening event that requires urgent fluid resuscitation and steroid treatment. Thus, a diagnosis of 21-OHD should be considered in any baby with atypical genitalia with bilateral nonpalpable gonads. Males (46,XY) with 21-OHD have no genital abnormalities at birth but are equally susceptible to adrenal insufficiency and salt-losing crises.
Females with the classic simple virilizing form of 21-OHD also present with genital ambiguity. They have impaired cortisol biosynthesis but do not develop salt loss. Patients with nonclassic 21-OHD produce normal amounts of cortisol and aldosterone but at the expense of producing excess androgens. Hirsutism (60%), oligomenorrhea (50%), and acne (30%) are the most common presenting features. This is one of the most common recessive disorders in humans, with an incidence as high as 1 in 100 to 500 in many populations and 1 in 27 in Ashkenazi Jews of Eastern European origin.
Biochemical features of acute salt-wasting 21-OHD are hyponatremia, hyperkalemia, hypoglycemia, inappropriately low cortisol and aldosterone, and elevated 17-hydroxyprogesterone, ACTH, and plasma renin activity. Presymptomatic diagnosis of classic 21-OHD is now made by neonatal screening tests for increased 17-hydroxyprogesterone in many centers. In most cases, 17-hydroxyprogesterone is markedly increased. In adults, ACTH stimulation (0.25 mg of cosyntropin IV) with assays for 17-hydroxyprogesterone at 0 and 30 min can be useful for detecting nonclassic 21-OHD and heterozygotes (Chap. 406).
|
TREATMENT |
CONGENITAL ADRENAL HYPERPLASIA |
Acute salt-wasting crises require fluid resuscitation, IV hydrocortisone, and correction of hypoglycemia. Once the patient is stabilized, glucocorticoids must be given to correct the cortisol insufficiency and suppress ACTH stimulation, thereby preventing further virilization, rapid skeletal maturation, and the development of polycystic ovaries. Typically, hydrocortisone (10–15 mg/m2 per day in three divided doses) is used in childhood with a goal of partially suppressing 17-hydroxyprogesterone (100 to <1000 ng/dL). The aim of treatment is to use the lowest glucocorticoid dose that adequately suppresses adrenal androgen production without causing signs of glucocorticoid excess such as impaired growth and obesity. Salt-wasting conditions are treated with mineralocorticoid replacement. Infants usually need salt supplements up to the first year of life. Plasma renin activity and electrolytes are used to monitor mineralocorticoid replacement. Some patients with simple virilizing 21-OHD also benefit from mineralocorticoid supplements. Parents and patients should be educated about the need for increased doses of steroids during sickness, and patients should carry medic alert systems.
Steroid treatment for older adolescents and adults varies depending on lifestyle, age, and factors such as a desire to optimize fertility. Hydrocortisone remains a useful approach, but treatment with prednisolone at night may provide more complete ACTH suppression. Steroid doses should be adjusted to individual requirements because overtreatment can result in iatrogenic Cushing’s-like features, including weight gain, insulin resistance, hypertension, and osteopenia. Because it is long acting, dexamethasone given at night is useful for ACTH suppression but is often associated with more side effects, making hydrocortisone or prednisolone preferable for most patients. Androstenedione and testosterone may be useful measurements of long-term control, with less fluctuation than 17-hydroxyprogesterone. Mineralocorticoid requirements often decrease in adulthood, and doses should be reassessed and reduced to avoid hypertension in adults. In very severe cases, adrenalectomy has been advocated but incurs the risks of surgery and total adrenal insufficiency.
Girls with significant genital androgenization due to classic 21-OHD usually undergo vaginal reconstruction and sometimes clitoral reduction (maintaining the glans and nerve supply), but the optimal timing of these procedures is debated, as is the need for the individual to be able to consent. There is a higher threshold for undertaking clitoral surgery in some centers because long-term sensation and ability to achieve orgasm can be affected, but the long-term results of newer techniques are not yet known. Full information about all options should be provided. If surgery is performed in infancy, surgical revision or regular vaginal dilatation may be needed in adolescence or adulthood, and long-term psychological support and psychosexual counseling may be appropriate. Women with 21-OHD frequently develop polycystic ovaries and have reduced fertility, especially when control is poor. Fecundity is achieved in 60–90% of women with good metabolic control, but ovulation induction (or even adrenalectomy) may be required. Dexamethasone should be avoided in pregnancy. Men with poorly controlled 21-OHD may develop testicular adrenal rests and are at risk for reduced fertility. Prenatal treatment of 21-OHD by the administration of dexamethasone to mothers is still under evaluation. However, pending methods to diagnose the disorder early in pregnancy, both affected and nonaffected fetuses will be exposed because treatment is started ideally before 6 to 7 weeks. The long-term effects of prenatal dexamethasone exposure on fetal development are still under evaluation, and current guidelines recommend full informed consent before treatment, ideally in a protocol that allows long-term follow-up of all children treated. Newer techniques such as cell-free fetal DNA testing may potentially reduce treatment of nonaffected fetuses.
The treatment of other forms of CAH includes mineralocorticoid and glucocorticoid replacement for salt-losing conditions (e.g., StAR, CYP11A1, HSD3β2), suppression of ACTH drive with glucocorticoids in disorders associated with hypertension (e.g., CYP17, CYP11B1), and appropriate sex hormone replacement in adolescence and adulthood, when necessary.
OTHER CAUSES Increased androgen synthesis can also occur in CAH due to defects in POR, 11β-hydroxylase (CYP11B1), and 3β-hydroxysteroid dehydrogenase type 2 (HSD3B2) and with mutations in the genes encoding aromatase (CYP19) and the glucocorticoid receptor. Increased androgen exposure in utero can occur with maternal virilizing tumors and with ingestion of androgenic compounds.
OTHER DISORDERS AFFECTING 46,XX FEMALES
Congenital absence of the vagina occurs in association with müllerian agenesis or hypoplasia as part of the Mayer-Rokitansky-Kuster-Hauser (MRKH) syndrome (rarely caused by WNT4 mutations). This diagnosis should be considered in otherwise phenotypically normal females with primary amenorrhea. Associated features include renal (agenesis) and cervical spinal abnormalities.
GLOBAL CONSIDERATIONS
 The approach to a child or adolescent with ambiguous genitalia or another DSD requires cultural sensitivity, as the concepts of sex and gender vary widely. Rare genetic DSDs can occur more frequently in specific populations (e.g., 5α-reductase type 2 in the Dominican Republic). Different forms of CAH also show ethnic and geographic variability. In many countries, appropriate biochemical tests may not be readily available, and access to appropriate forms of treatment and support may be limited.
The approach to a child or adolescent with ambiguous genitalia or another DSD requires cultural sensitivity, as the concepts of sex and gender vary widely. Rare genetic DSDs can occur more frequently in specific populations (e.g., 5α-reductase type 2 in the Dominican Republic). Different forms of CAH also show ethnic and geographic variability. In many countries, appropriate biochemical tests may not be readily available, and access to appropriate forms of treatment and support may be limited.
411 |
Disorders of the Testes and Male Reproductive System |
The male reproductive system regulates sex differentiation, virilization, and the hormonal changes that accompany puberty, ultimately leading to spermatogenesis and fertility. Under the control of the pituitary hormones—luteinizing hormone (LH) and follicle-stimulating hormone (FSH)—the Leydig cells of the testes produce testosterone and germ cells are nurtured by Sertoli cells to divide, differentiate, and mature into sperm. During embryonic development, testosterone and dihydrotestosterone (DHT) induce the wolffian duct and virilization of the external genitalia. During puberty, testosterone promotes somatic growth and the development of secondary sex characteristics. In the adult, testosterone is necessary for spermatogenesis, stimulation of libido and normal sexual function, and maintenance of muscle and bone mass. This chapter focuses on the physiology of the testes and disorders associated with decreased androgen production, which may be caused by gonadotropin deficiency or by primary testis dysfunction. A variety of testosterone formulations now allow more physiologic androgen replacement. Infertility occurs in ~5% of men and is increasingly amenable to treatment by hormone replacement or by using sperm transfer techniques. For further discussion of sexual dysfunction, disorders of the prostate, and testicular cancer, see Chaps. 67, 115, and 116, respectively.
DEVELOPMENT AND STRUCTURE OF THE TESTIS
The fetal testis develops from the undifferentiated gonad after expression of a genetic cascade that is initiated by the SRY (sex-related gene on the Y chromosome) (Chap. 410). SRY induces differentiation of Sertoli cells, which surround germ cells and, together with peritubular myoid cells, form testis cords that will later develop into seminiferous tubules. Fetal Leydig cells and endothelial cells migrate into the gonad from the adjacent mesonephros but may also arise from interstitial cells that reside between testis cords. Leydig cells produce testosterone, which supports the growth and differentiation of wolffian duct structures that develop into the epididymis, vas deferens, and seminal vesicles. Testosterone is also converted to DHT (see below), which induces formation of the prostate and the external male genitalia, including the penis, urethra, and scrotum. Testicular descent through the inguinal canal is controlled in part by Leydig cell production of insulin-like factor 3 (INSL3), which acts via a receptor termed Great (G protein–coupled receptor affecting testis descent). Sertoli cells produce müllerian-inhibiting substance (MIS), which causes regression of the müllerian structures, including the fallopian tube, uterus, and upper segment of the vagina.
NORMAL MALE PUBERTAL DEVELOPMENT
Although puberty commonly refers to the maturation of the reproductive axis and the development of secondary sex characteristics, it involves a coordinated response of multiple hormonal systems including the adrenal gland and the growth hormone (GH) axis (Fig. 411-1). The development of secondary sex characteristics is initiated by adrenarche, which usually occurs between 6 and 8 years of age when the adrenal gland begins to produce greater amounts of androgens from the zona reticularis, the principal site of dehydroepiandrosterone (DHEA) production. The sex maturation process is greatly accelerated by the activation of the hypothalamic-pituitary axis and the production of gonadotropin-releasing hormone (GnRH). The GnRH pulse generator in the hypothalamus is active during fetal life and early infancy but is restrained until the early stages of puberty by a neuroendocrine brake imposed by the inhibitory actions of glutamate, γ-aminobutyric acid (GABA), and neuropeptide Y. Although the pathways that initiate reactivation of the GnRH pulse generator at the onset of puberty have been elusive, mounting evidence supports involvement of GPR54, a G protein–coupled receptor that binds an endogenous ligand, kisspeptin. Individuals with mutations of GPR54 fail to enter puberty, and experiments in primates demonstrate that infusion of the ligand is sufficient to induce premature puberty. Kisspeptin signaling plays an important role in mediating the feedback action of sex steroids on gonadotropin secretion and in regulating the tempo of sexual maturation at puberty. Leptin, a hormone produced by adipose cells, plays a permissive role in the resurgence of GnRH secretion at the onset of puberty, as leptin-deficient individuals also fail to enter puberty (Chap. 415e). The adipocyte hormone leptin, gut hormone ghrelin, neuropeptide Y, and kisspeptin integrate the signals originating in energy stores and metabolic tissues with mechanisms that control onset of puberty through regulation of GnRH secretion. Energy deficit and excess and metabolic stress are associated with disturbed reproductive maturation and timing of pubertal onset.




