Multiple endocrine neoplasia
MEN 1
A clinical diagnosis of MEN 1 can be made if the patient has at least two of the following:





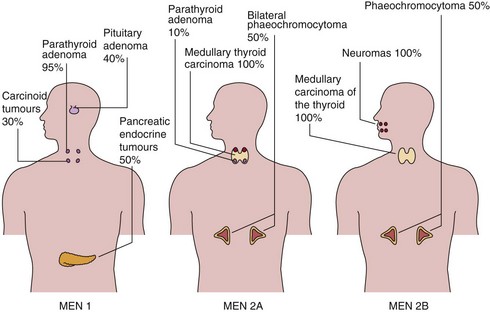
The approximate frequencies of these tumours in MEN 1 are shown in Figure 71.1.
MEN 2
MEN 2A
Medullary carcinoma of the thyroid (MCT) is present, often with phaeochromocytoma or hyperparathyroidism (Fig 71.1). Phaeochromocytoma is bilateral in about 50% of affected cases.
Screening and treatment
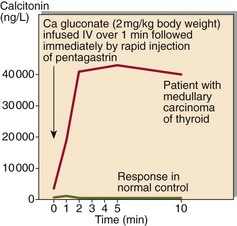
All carriers of MEN mutations will develop one or other of the associated endocrine tumours. Periodic biochemical screening has an important role to play in the follow-up of identified carriers. However, definitive confirmation or exclusion of an individual’s MEN predisposition requires genetic testing. In some cases provocation tests may be necessary (Fig 71.2).
The APUD concept
Carcinoid and pancreatic islet cell tumours are seen in MEN syndromes but also occur sporadically. They arise from specialized neuroendocrine cells that have the capacity for amine precursor uptake and decarboxylation (APUD). Some of the peptides and amines secreted by these cells act like classical hormones, being delivered through the bloodstream, while others are local paracrine regulators or neurotransmitters (Table 71.1). Overproduction of peptides or amines by tumours gives rise to associated tumour syndromes.
Table 71.1
Selected molecules which regulate gastrointestinal function
| Substance | Type of regulator | Major action |
| Gastrin | Hormone | Gastric acid and pepsin secretion |
| Cholecystokinin (CCK) | Hormone | Pancreatic enzyme secretion |
| Secretin | Hormone | Pancreatic bicarbonate secretion |
| Gastric inhibitory polypeptide (GIP) | Hormone | Enhances glucose mediated insulin release, inhibits gastric acid secretion |
| Vasoactive intestinal polypeptide (VIP) | Neurotransmitter | Smooth muscle relaxation. Stimulates pancreatic bicarbonate secretion |
| Motilin | Hormone | Initiates interdigestive intestinal motility |
| Somatostatin | Hormone | Numerous inhibitory effects |
| Neurotransmitter | ||
| Paracrine | ||
| Pancreatic polypeptide (PP) | Hormone | |
| Paracrine | Inhibits pancreatic bicarbonate and protein secretion | |
| Enkephalins | Neurotransmitter | Opiate-like actions |
| Substance P | Neurotransmitter | Contraction of smooth muscle |
| Paracrine |
Carcinoid tumours
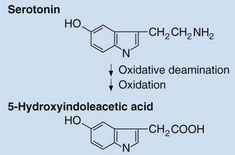
These most commonly arise in the appendix and ileocaecal region where the fore- and mid-gut meet. Carcinoid tumours may convert as much as half of the dietary intake of tryptophan into serotonin, secretion of which causes distinctive clinical effects, known as the carcinoid syndrome. This is characterized by flushing, diarrhoea and sometimes valvular heart disease. Intestinal carcinoid tumours only produce carcinoid syndrome if they have metastasized to the liver, whereas extra-intestinal carcinoids, e.g. bronchial, which do not drain into the portal circulation, may cause it even in the absence of metastasis. Serotonin may be measured directly in plasma or platelets, but the diagnosis is more often made by measurement in urine of its metabolite, 5-hydroxyindoleacetic acid (Fig 71.3).
Others
Gastrinomas, VIPomas, glucagonomas and somatostatinomas are all either rare or very rare.
 Clinical note
Clinical noteMultiple endocrine neoplasia





