SECTION V MULTIFOCAL NEUROLOGICAL DISEASE AND ITS MANAGEMENT
BACTERIAL INFECTIONS – MENINGITIS
Pathology
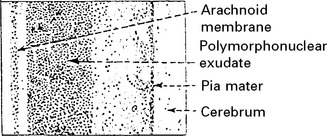
A purulent exudate most evident in the basal cisterns extends throughout the subarachnoid space.
The underlying brain, although not invaded by bacteria, becomes congested, oedematous and ischaemic.
The integrity of the pia mater normally protects against brain abscess formation.
Clinical
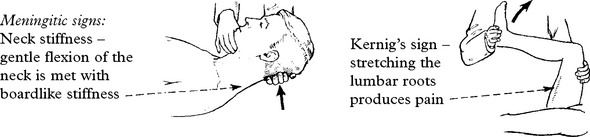
The classical clinical triad is fever, headache and neck stiffness.
| Prodromal features (variable) | Meningitic symptoms |
|---|---|
| A respiratory infection otitis media or pneumonia associated with muscle pain | Severe frontal/occipital headache |
| Stiff neck | |
| Photophobia. |
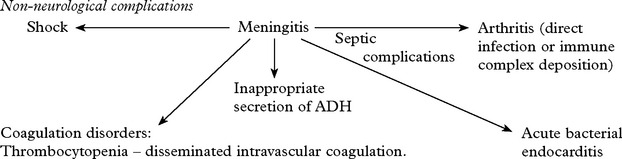
Systemic signs: – High fever. Transient purpuric or petechial skin rash in meningococcal meningitis.
Features specific to causative bacteria
| Haemophilus meningitis | Meningococcal meningitis | Pneumococcal meningitis |
|---|---|---|
Investigations

Treatment
| Neonates (above 1 month) |
Therapy after organism identification
| ORGANISM | ANTIBIOTIC | ALTERNATIVE THERAPY |
|---|---|---|
| Haemophilus | Ampicillin or 3rd generation cephalosporin according to sensitivities |
BACTERIAL INFECTIONS – CNS TUBERCULOSIS
The basal meninges are generally most severely affected.
Clinical features
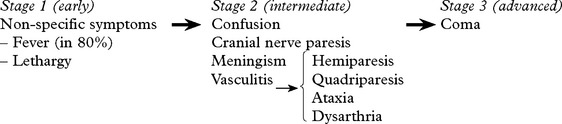
Staging is useful for predicting outcome.
Seizures may occur at the onset. Involuntary movements (chorea, myoclonus) occur in 10%.
Untreated, the illness may progress from phase 1 to death over a 3-week period.
Arachnoiditis inflammatory exudate may result in hydrocephalus/dementia/blindness.
TUBERCULOUS MENINGITIS
Investigations
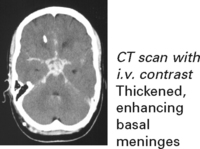
Diagnosis
Diagnosis is based on the clinical presentation with characteristic CSF findings.
DIFFERENTIAL DIAGNOSIS – Viral meningoencephalitis – Subacute/chronic meningitis (see pages 517–8).
Treatment
If suspect, commence antituberculous treatment.
Recommended treatment programme:
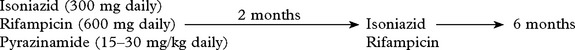
Drug resistance suspected due to previous antituberculous therapy, e.g.
→ Add a fourth drug – streptomycin (1 g daily) or ethambutal (25 mg/kg daily).
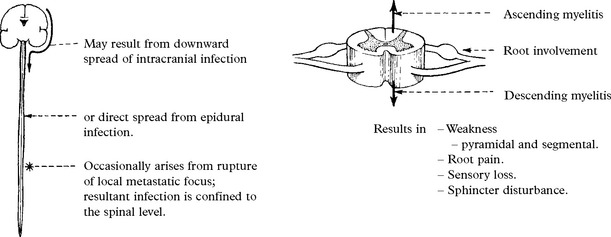
OTHER FORMS OF CNS TUBERCULOUS INFECTION

SPIROCHAETAL INFECTIONS OF THE NERVOUS SYSTEM
SYPHILIS
This infectious disease is caused by the spirochaete Treponema pallidum. Entry is by:
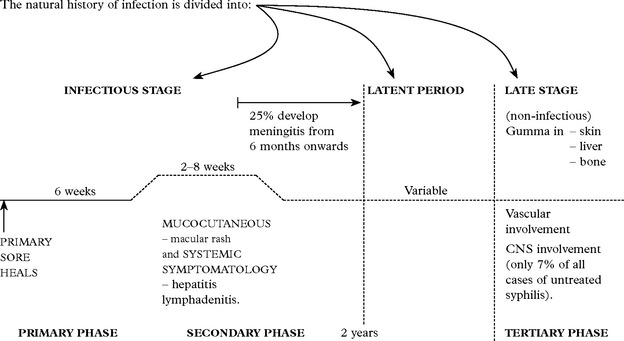
Not all patients untreated in the secondary phase progress to the tertiary phase.
In HIV patients the neurological complications occur earlier and advance more quickly.
Investigations
SPIROCHAETAL INFECTION – NEUROSYPHILIS
ACUTE SYPHILITIC MENINGITIS: Three clinical forms are recognised:
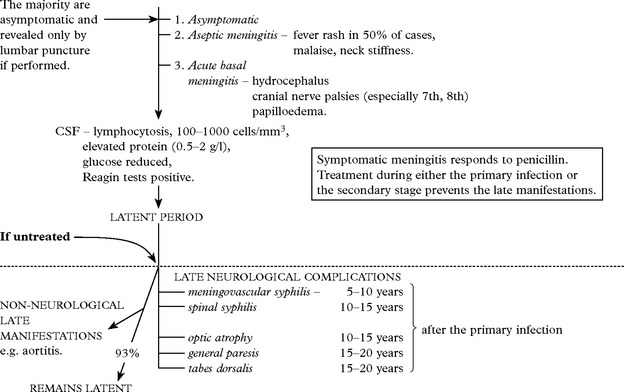
Late neurological complications occur in only 7% of untreated cases.
MENINGOVASCULAR SYPHILIS
‘Early’ late manifestation resulting in an obliterative endarteritis and periarteritis.
OCULAR MANIFESTATIONS
Meningitis around optic nerve with subpial necrosis may be the only manifestation of late syphilis.
Presents as a constriction of the visual fields with a progressive pallor of the optic disc:
Neuroretinitis, uveitis and chorioretinitis occur, especially in HIV patients.
GENERAL PARESIS
Argyll Robertson pupils may be present (see page 146).
CSF – lymphocytes 50/mm3, protein ↑ 0.5–2 g/l, gammaglobulin ↑.
Reagin tests in CSF positive in the majority.
Treatment in the preparalytic phase will halt progression in 40%.
TABES DORSALIS
Posterior spinal root and posterior column dysfunction account for symptoms.
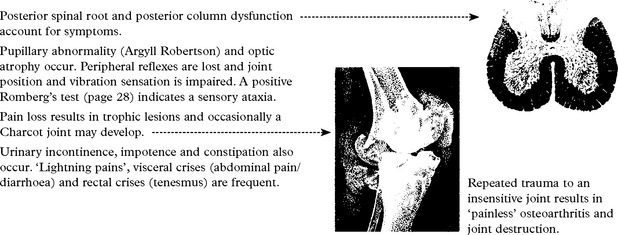
SYPHILITIC GUMMA presenting as an intracranial mass is extremely rare.
TREATMENT OF NEUROSYPHILIS
| Penicillin G. | 2–4 megaunits i.v. | (When patient sensitive to penicillin |
| or | 4-hourly for 10 days. | ↓ |
| Procaine Penicillin | 600 000 units i.m. daily for 15 days. | erythromycin or tetracycline may be given orally over 30 days.) |
| Benzathine Penicillin | 2–4 megaunits i.m. weekly × 3. |
CSF follow up: CSF is checked initially and at 6 monthly intervals until normal.
Cell count and degree of positivity of VDRL are the best indicators of persistent infection.
SPIROCHAETAL INFECTION
LYME DISEASE (NEUROBORRELIOSIS)


Treatment
Stage 1 – Oral antibiotics: penicillin, erythromycin or tetracycline.
Stage 2 – I.V. penicillin G. 20 million units for 10 days (or ceftriaxone).
If symptoms persist – wrong diagnosis with misleading titres, or – immune mediated damage.
Steroids can be used in late stages when symptoms have not responded to antibiotics.

PARASITIC INFECTIONS OF THE NERVOUS SYSTEM – PROTOZOA
TOXOPLASMOSIS
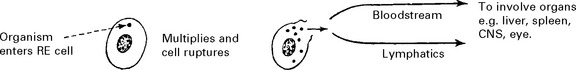
A world-wide parasitic infection affecting many species, including man.
Organism: An anaerobic intracellular protozoan, Toxoplasma gondii.
 Retinal pigment epithelium becomes hyperplastic – densely pigmented areas result.
Retinal pigment epithelium becomes hyperplastic – densely pigmented areas result.Organisms are seldom identified.
VIRAL INFECTIONS
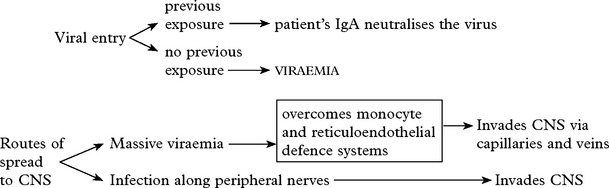
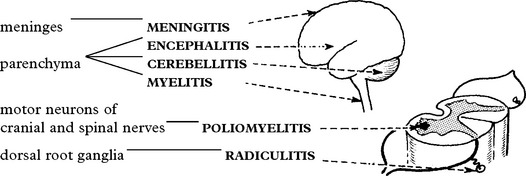
MENINGITIS
| Common causal viruses – | Rare causal viruses – |
| ENTEROVIRUSES | LYMPHOCYTIC CHORIOMENINGITIS |
| MUMPS VIRUS | HUMAN IMMUNODEFICIENCY VIRUS (HIV) |
| HERPES SIMPLEX (subtype 2) | WEST NILE VIRUS |
| EPSTEIN-BARR VIRUS (EBV) |
VIRAL INFECTIONS – MENINGITIS
Clinical features of acute aseptic meningitis
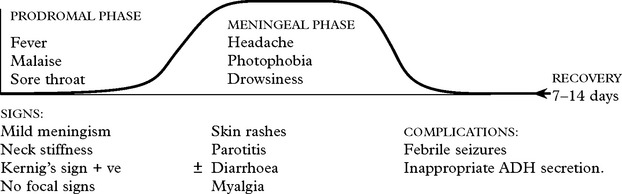
Spread is by the faecal/oral route.
Mumps – affects children/young adults. Winter/spring incidence.
Human Immunodeficiency Virus (HIV) – suspect in high risk groups (page 515). HIV antibodies are often absent and develop 1–3 months later during convalescence.
VIRAL INFECTIONS – PARENCHYMAL
General: pyrexia, myalgia, etc.
Specific to causative virus, e.g. features of infectious mononucleosis (Epstein-Barr).
Meningeal involvement (slight) → neck stiffness, cellular response in CSF.
Signs and symptoms of parenchymal involvement – focal and/or diffuse.

In general, the illness lasts for some weeks.
Prognosis is uncertain and depends on the causal virus as do neurological sequelae.
Herpes Simplex (HSV) and Varicella-Zoster (VZV) commonly cause disease in humans.
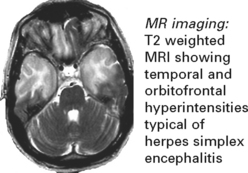
HERPES SIMPLEX ENCEPHALITIS
HSV-1 is the commonest cause of sporadic encephalitis.
One third occur due to primary infection; two thirds have pre-existing antibodies (reactivation).


VIRAL INFECTIONS – REYE’S SYNDROME
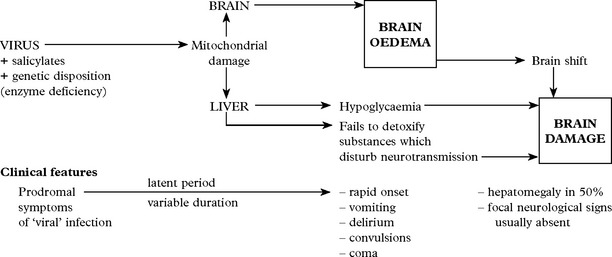
Differential diagnosis
Consider other causes of raised intracranial pressure in childhood, especially
Treatment
Treatment aims at lowering intracranial pressure with the aid of intracranial pressure monitoring (see page 52). In addition, blood glucose must be maintained and any associated coagulopathy treated. Reduction of ammonia may be achieved by peritoneal dialysis or exchange transfusion.
VIRAL INFECTIONS – CHRONIC DISORDERS
In these disorders the infection results in a chronic progressive neurological condition.
The evidence of a viral etiology is:
Direct – finding of inclusion bodies, demonstration of viral particles or isolation of virus.
SUBACUTE SCLEROSING PANENCEPHALITIS (SSPE)
Caused by measles-like paramyxovirus – isolated from brain biopsy.
Less common with the availability of widespread primary measles vaccination.
Stage 1: Behavioural problem, declining school performance, progression → dementia
Stage 2: Chorioretinitis, myoclonic jerks, seizures, ataxia, dystonia
Stage 3: Lapses into rigid comatose state

Accompanying features of infection, i.e. pyrexia, leucocytosis, are absent.
Investigations

EEG – shows periodic high voltage slow wave complexes on a low voltage background trace.
PROGRESSIVE RUBELLA PANENCEPHALITIS
Similar to SSPE with a fatal outcome, caused by rubella virus.
| Presents at a later age (10–15 years) | CSF shows high γ globulin. |
| Progressive dementia. | Antibodies elevated in serum and CSF to rubella. |
| Ataxia. Spasticity. Myoclonus. | Biopsy does not show inclusion bodies. |
| Treatment: No effective treatment |
PRION DISEASES
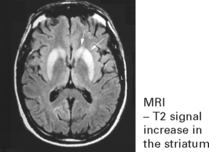
Investigation
EEG – 1–2 Hz triphasic sharp waves with periodic complexes
CSF – increase in protein 14–3–3 (a protein kinase inhibitor)
[The combined EEG and CSF findings, where positive, have diagnostic sensitivity/specificity of 97%]
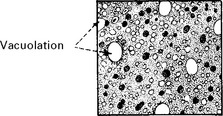
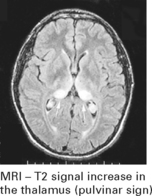
VIRAL INFECTIONS – MYELITIS AND POLIOMYELITIS
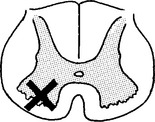

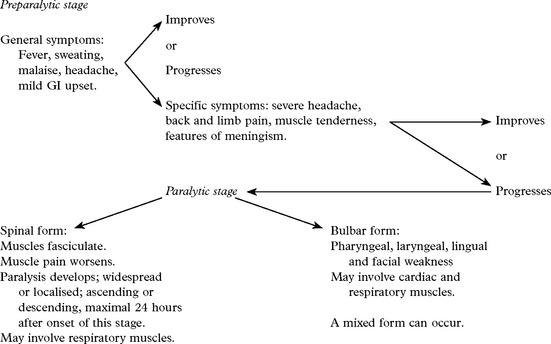
VIRAL INFECTIONS – POLIOMYELITIS

VIRAL INFECTIONS – VARICELLA-ZOSTER INFECTION
| Conditions caused: |
SHINGLES
This occurs after virus reactivation, dormant after the primary infection (chickenpox).
Clinical features

Cranial nerve ganglia involvement:
Diagnosis: Based on clinical features. Virus DNA can be detected in vesicular fluid by PCR.
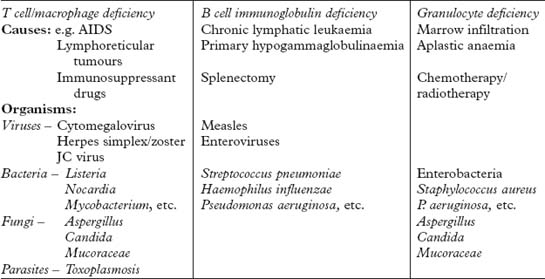
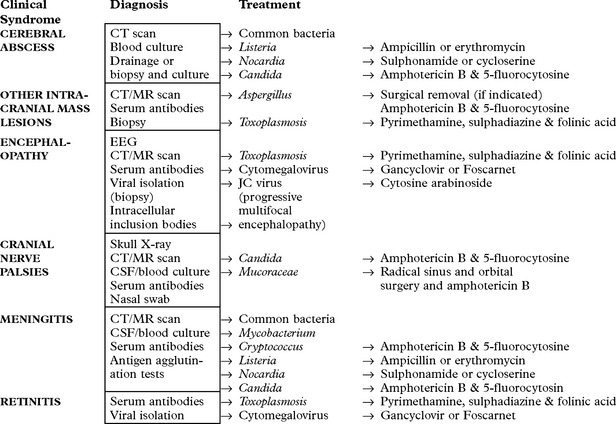
ACQUIRED IMMUNODEFICIENCY SYNDROME (AIDS)

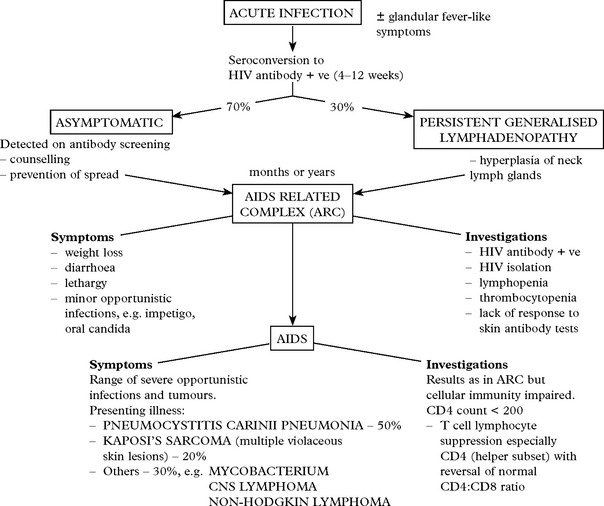
NEUROLOGICAL PRESENTATIONS OF HIV INFECTION
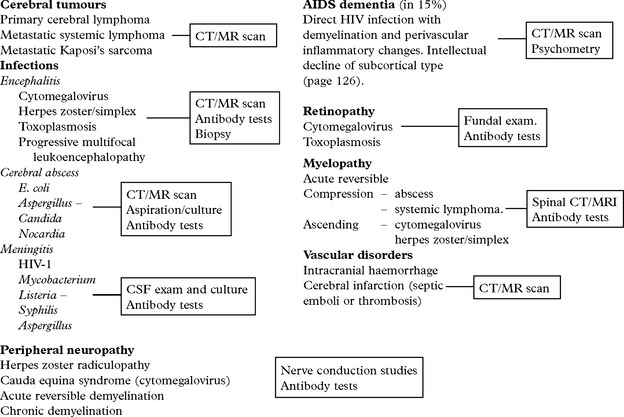
SUBACUTE/CHRONIC MENINGITIS
Chronic meningitis is associated with certain CSF findings.

DEMYELINATING DISEASE
The neuroectodermal cells comprise:
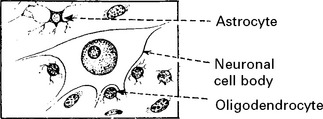
MULTIPLE SCLEROSIS
The disease occurs most commonly in temperate climates and prevalence differs at various latitudes:
| Latitude (°N) | Rate/100 000 (adjusted) | |
| Orkneys and Shetland……… | 60 | 309 |
| England (Cornwall)………… | 51 | 63 |
| Italy (Bari)…………… | 41 | 13 |
Multiple sclerosis is usually characterised by:
Symptoms at onset


Loss of vision
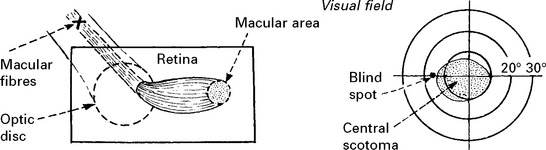
Acute bilateral optic neuritis: less common than unilateral disease and progression to MS not as likely. Occasionally followed by a transverse myelitis (Neuromyelitis optica, page 529). Examination of mitochondrial DNA distinguishes from Leber’s hereditary optic neuropathy (page 551).
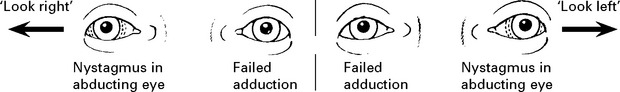
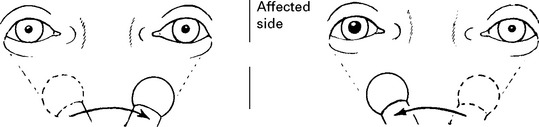
CLINICAL COURSE
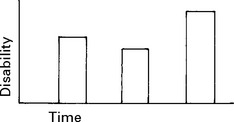
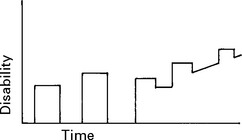
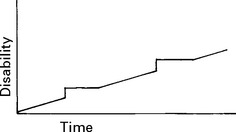
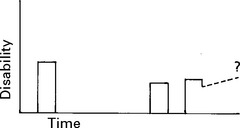
INVESTIGATIONS
The development of imaging and laboratory testing has advanced diagnostic accuracy.
Neurophysiological: may detect a second asymptomatic lesion (see page 54).
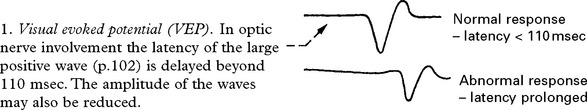
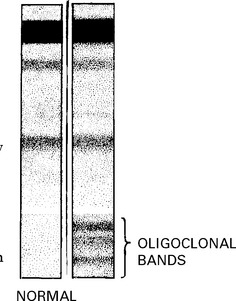
MRI
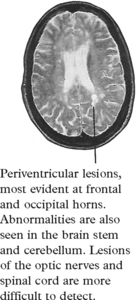
Diagnosis requires two or more episodes of symptoms attributable to demyelination, at least 30 days apart at different sites in the central nervous system, and the exclusion of alternative pathologies. Research criteria have been developed for clinical studies which combine clinical features with investigation findings. The McDonald criteria (2001) allow for MRI scan evidence of the development of new lesions to lead to a diagnosis after a single clinical episode.
Conditions with similar CSF profile to MS (presence of oligoclonal bands)
| Spasticity | Drugs: baclofen (GABA derivative); dantrolene (direct action on muscle); tizanidine (α2 adrenergic agonist); botulinum toxin |
| Physiotherapy and splinting | |
| Intrathecal baclofen | |
| Urinary symptoms | Detrusor instability – anticholinergics (oxybutinin, tolterodine); if severe intravesical botulinum toxin injections |
| Nocturia – desmopressin spray | |
| Incomplete bladder emptying – intermittent self catheterisation | |
| Bowel symptoms | Dietary manipulation; laxatives; suppositories/enemas |
| Pain | Analgesics; anticonvulsants; antidepressants; NSAIDs; transcutaneous electrical nerve stimulation |
| Paroxysmal symptoms | Seizures – anticonvulsants |
| Fatigue | Amantadine; modafinil |
| Depression | Clinical psychology; antidepressants, tricyclic or SSRI |
| Tremor | Betablockers; primidone; if severe deep brain stimulation |
| Ataxia | Walking aids; physiotherapy |
OTHER DEMYELINATING DISEASES
NEUROMYELITIS OPTICA (Devic’s disease)
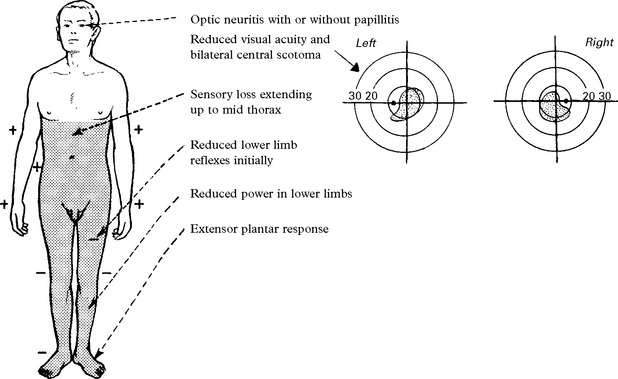
ACUTE DISSEMINATED ENCEPHALOMYELITIS (ADEM) (postinfectious encephalomyelitis)

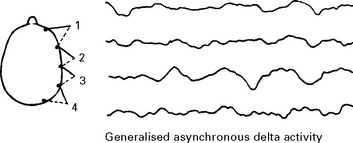
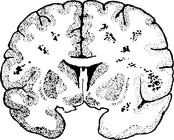
NEUROLOGICAL COMPLICATIONS OF DRUGS AND TOXINS
Introduction
Diagnostic suspicion is especially dependent upon history:
History and examination
When acute intoxication is suspected, the following clinical features are supportive.
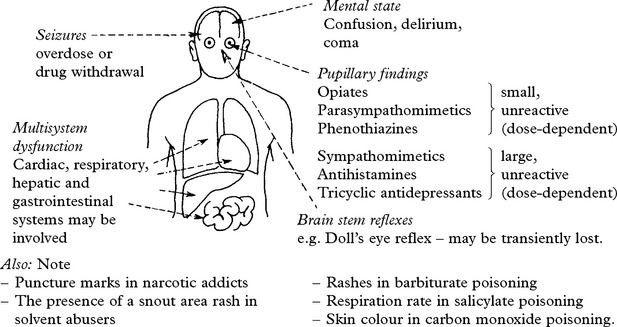
DRUG-INDUCED NEUROLOGICAL SYNDROMES
HEADACHE
PERIPHERAL NEUROPATHY
SPECIFIC SYNDROMES OF DRUGS AND TOXINS
Treatment of acute intoxication is symptomatic; there are no specific antidotes.
Industrial exposure to hydrocarbons produces similar symptoms.

COMPLICATIONS OF RECREATIONAL DRUG ABUSE
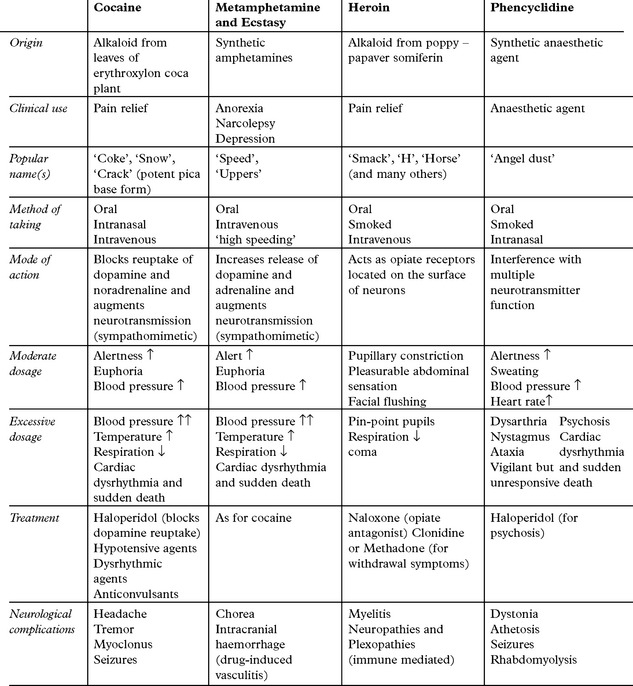
All intravenous drug abusers are at risk of HIV infection and its complications (page 515)
METABOLIC ENCEPHALOPATHIES
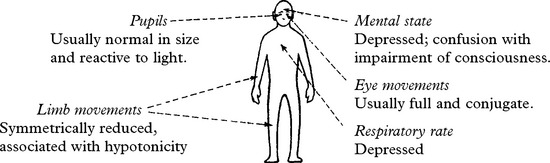
These features are characteristic but exceptions occur in specific encephalopathies –
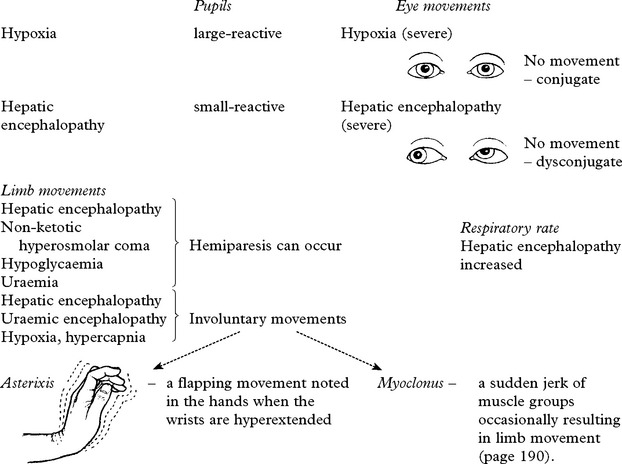
CLASSIFICATION AND BIOCHEMICAL EVALUATION
Many metabolic disturbances cause an acquired encephalopathy in adults.
Drugs and toxins producing encephalopathy are dealt with separately (page 533).
Laboratory assessment of suspected metabolic encephalopathy
All patients should have a basic biochemical screen:
SPECIFIC ENCEPHALOPATHIES
Pathology
As a consequence of high metabolic demand, some areas are more susceptible than others.
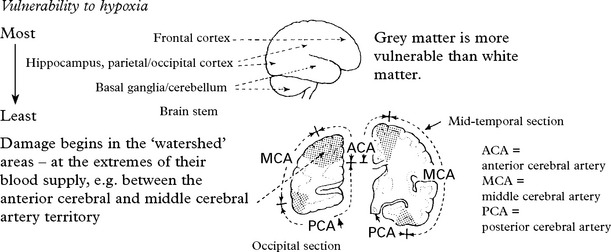
Microscopic changes depend upon the delay between the hypoxic event and death.
| Immediate: | At 48 hours: | At several days/weeks: |
|---|---|---|
| Scattered petechial haemorrhages. | Cerebral oedema associated with petechial haemorrhage. | Necrosis in cortical grey matter and globus pallidus with associated astrocytic proliferation. The cerebellum and brain stem may also be affected. |
Clinical features:
e.g. Severe hypoxia from circulatory arrest
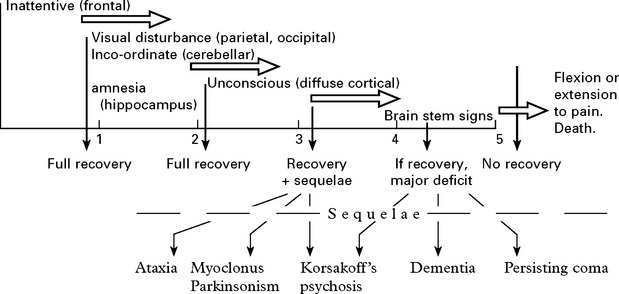
HYPERCAPNIC ENCEPHALOPATHY: the consequence of an elevated arterial carbon dioxide level.
– insulin secreting tumour – insulinoma
– hepatic disease with reduction of liver glycogen.
Clinical features: These, as with hypoxia, depend upon the duration and severity of hypoglycaemia.
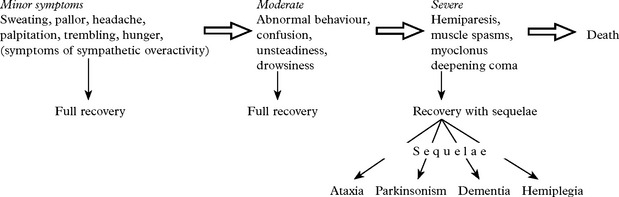
Repeated mild to moderate episodes may result in a chronic cerebellar ataxia.
HYPERGLYCAEMIC ENCEPHALOPATHY
Two types of encephalopathy develop as a consequence of hyperglycaemia:
| Diabetic ketoacidotic coma | Diabetic hyperosmolar non-ketotic coma |
|---|---|
| Accumulation of acetone and ketone bodies in blood results in acidosis. Hyperventilation ensues with a reduction in PCO2 and HCO−3 Osmotic diuresis due to hyperglycaemia results in dehydration. | This results from the hyperosmolar effect of severe hyperglycaemia. Reduction of the intracellular compartment results. Involuntary movements, seizures and hemiparesis may occur. Vascular thrombosis is not uncommon. Ketoacidosis is mild or does not occur. |
| The neurological presentation is that of confusion, progression to coma and, if untreated, death. |
HEPATIC ENCEPHALOPATHY
Neurological signs and symptoms secondary to hepatic dysfunction may arise in:

The encephalopathy is progressive.
Pathology
Hepatocerebral degeneration (Wilson’s disease) (see page 373) is an abnormality of copper metabolism and leads to a deposition of copper in the brain, particularly the basal ganglia, and the liver. This leads to a slowly progressive neurological disorder. This can vary but the predominant features include dementia, dysarthria and ataxia, primitive reflexes, choreoathetosis, myoclonus, tremor and pyramidal signs.

NUTRITIONAL DISORDERS
INTRODUCTION
As a rule, nutritional disorders of the nervous system present clinically in a symmetrical manner.

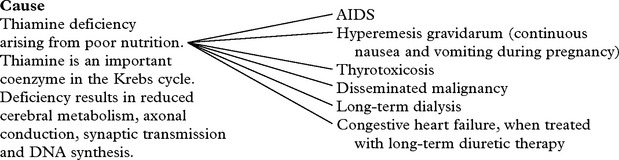
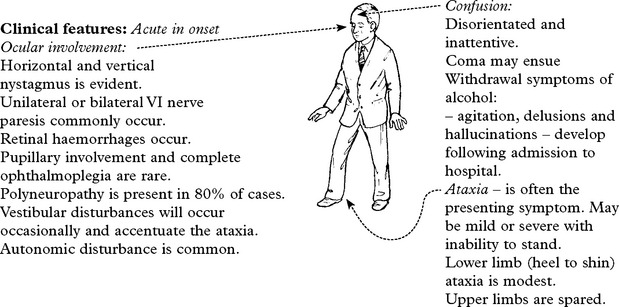
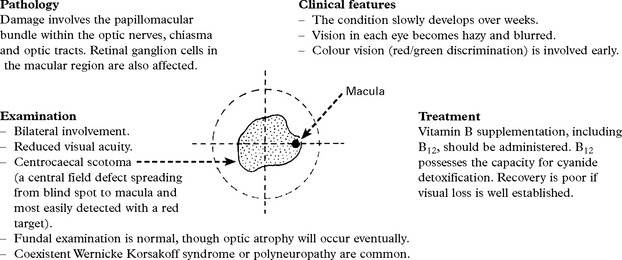
WERNICKE KORSAKOFF SYNDROME
B12 DEFICIENCY – SUBACUTE COMBINED DEGENERATION OF THE SPINAL CORD

Clinical features
Onset is subacute, though can be acutely precipitated by exposure to nitrous oxide anaesthesia.

Examination
Mini mental status examination (MMSE) test may suggest dementia.
NUTRITIONAL POLYNEUROPATHY
Pathology
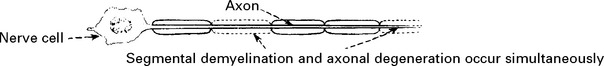
The distal portions of nerves are initially affected.
Anterior horn cells and dorsal root ganglion cells undergo chromatolysis.
Vagus nerve and sympathetic trunk involvement occurs in severe cases.
Diagnosis
Suggested by nutritional/alcohol history.
Nerve conduction studies reveal mildly reduced motor and sensory conduction velocities.
Differential diagnosis
Consider other causes of subacute or chronic sensorimotor neuropathy (see page 436).
ALCOHOL RELATED DISORDERS
ALCOHOL MYOPATHY
There are two forms of alcoholic muscle disease

NON-METASTATIC MANIFESTATIONS OF MALIGNANT DISEASE
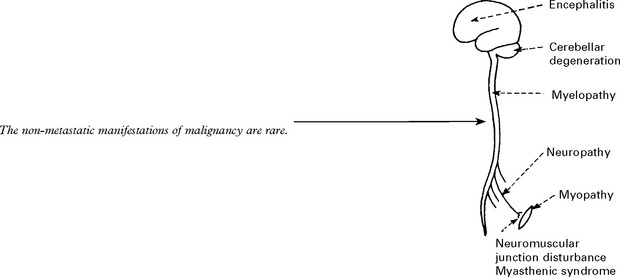
LIMBIC ENCEPHALITIS
Investigations.
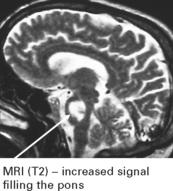
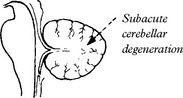
CEREBELLAR DEGENERATION (anti-Yo syndrome) associated with breast or ovarian carcinoma.
Investigations
MRI shows cortical and vermal cerebellar atrophy.
CSF is mildly abnormal and anti-Yo antibodies are present in 50% of suspected cases.
Sensorimotor neuropathy: A mixed neuropathy with weakness and sensory loss. The syndrome may predate the recognition of the underlying neoplasm. Rate of progression is slow and predominantly motor forms may be mistaken for ALS (page 555) associated with Hodgkin’s and other lymphomas.
Rarely an acute neuropathy indistinguishable from postinfectious polyneuropathy occurs.
NECROTISING MYELOPATHY:
Flaccid paraplegia develops subacutely. Spinal MRI may show a swollen cord. Mechanism is uncertain.
MYOPATHY
Muscle weakness in malignancy takes several forms.
Proximal myopathy: A slowly progressive syndrome with weakness of proximal limb muscles.
Inflammatory myopathy (polymyositis/dermatomyositis) (see page 474):
Cachetic myopathy occurs in terminally ill, wasted patients.
THE MYASTHENIC SYNDROME (Lambert-Eaton syndrome)

PROGRESSIVE BLINDNESS
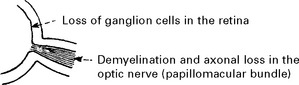
LEBER’S HEREDITARY OPTIC NEUROPATHY (LHON)
Leber’s optic neuropathy is a familial disorder of maternal inheritance with a tendency to affect males significantly more than females. It is classified as a mitochondrial disorder due to DNA mutation (page 481). Most individuals have one of three point mutations of mitochondrial DNA (mtDNA).
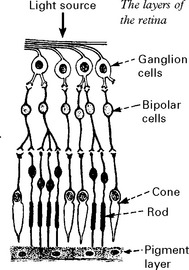
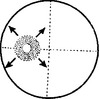
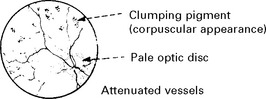
RETINITIS PIGMENTOSA

Clinical features
The electroretinogram – recording the electrical activity of the retina – is eventually lost.
PROGRESSIVE ATAXIA
The degenerative disorders manifested by progressive ataxia are termed spinocerebellar-ataxias.
RECESSIVELY INHERITED ATAXIAS
ATAXIA TELANGIECTASIA (Louis-Barr Syndrome)
Pathologically, widespread cerebellar Purkinje and granular cell loss occurs.
Malignant neoplasms (lymphoreticular tumours) occur in 10%.
Death occurs in second or third decade from infection or malignancy (often lymphoma).
RECESSIVELY INHERITED ATAXIA
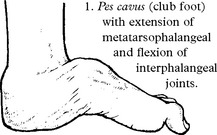
FRIEDREICH’S ATAXIA
Clinical features
Posterior column involvement results in loss of vibration and proprioception in the extremities.
Dorsal root and peripheral nerve involvement results in absent lower limb reflexes.
DOMINANTLY INHERITED AND OTHER ATAXIAS
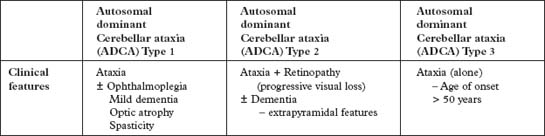
IDIOPATHIC LATE ONSET ATAXIA
| Type 1 – Age of onset 35–55 years – ataxia ± dementia, spasticity |
| Type 2 – Age of onset > 55 years – mid-line ataxia sparing speech/limbs |
| Type 3 – Age of onset 50–60 years – ataxia, titubation and tremor |
MOTOR NEURON DISEASE/AMYOTROPHIC LATERAL SCLEROSIS (ALS)
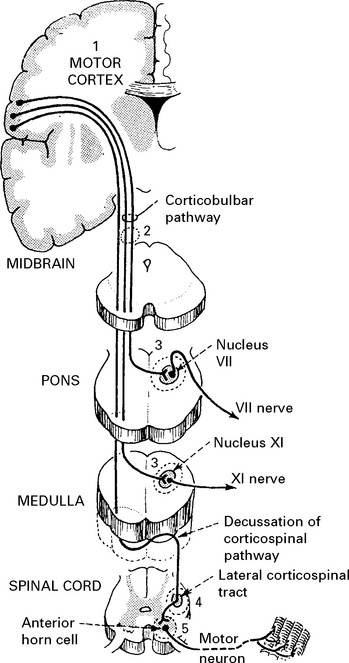
Different levels of the nervous system are involved:
The term AMYOTROPHIC LATERAL SCLEROSIS (ALS) is used synonymously with motor neuron disease.
MOTOR NEURON DISEASE/ALS
AETIOLOGY
The cause of motor neuron disease is unknown. Several possibilities have been suggested:
Limb-onset disease
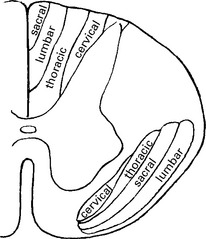
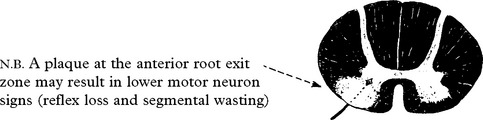
Limb-onset ALS results from involvement of corticospinal tracts and anterior horn cells.
Signs of corticospinal tract degeneration lead to:
Anterior horn cell involvement leads to muscle atrophy, weakness and fasciculations.
The patient may be aware of fasciculation.
Muscle cramps are common. Weakness is not as severe as the degree of wasting suggests.

MOTOR NEURON DISEASE/ALS
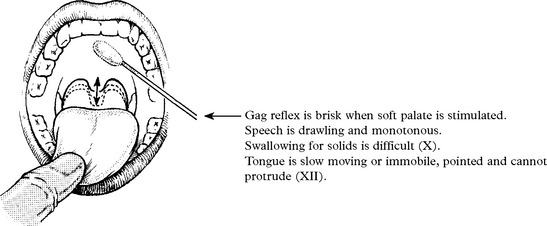
Bulbar-onset disease = Progressive bulbar palsy
Degeneration of corticobulbar pathways to V, VII, X, XI and XII cranial nerve motor nuclei (with sparing of III, IV and VI) leads to an apparent weakness of the muscles of mastication and expression, the patient has difficulty in chewing and the face is expressionless. The jaw jerk (page 15) is exaggerated.
Food and fluid enter nasopharynx when swallowing – palatal weakness (X).

Less common clinical presentations
Occasionally patients can present with:

Hexosaminidase deficiency (autosomal recessive disorder) may mimic ALS.
Hyperthyroidism and hyperparathyroidism produce muscle wasting and hyperreflexia.
Investigations
EMG reveals denervation with fibrillation.
MRI (or myelography) where appropriate excludes foramen magnum or spinal cord compression.
Thyroid and calcium studies exclude endocrine or metabolic disease.
Diagnostic criteria (El Escorial criteria for MND/ALS – World Federation of Neurology)
TREATMENT
Symptomatic treatment:
Anarthria and dysarthria: – Speech assessment and communication aids when indicated.
Dysphagia and aspiration: – Percutaneous endoscopic gastrostomy (PEG).
Nutrition: – Estimate calorific content and supplement diet with vitamins.
Muscle weakness: – Physiotherapy, walking aids. Splints, etc.
INHERITED MOTOR NEURON DISORDERS
SPINAL MUSCULAR ATROPHIES (SMAs)
With an incidence of 1/10000, the offspring of patients have a disease risk of approximately 1%.
Type I – Werdnig Hoffman disease (Acute Infantile SMA)
This is an autosomal recessive disorder.

Reduced fetal movements in late pregnancy with weakness and hypotonia at birth.

All motor milestones are delayed; 95% of all patients are dead by 18 months.
Type II – Kugelberg Welander disease (Late infantile or juvenile SMA)
Pathological features similar to Werdnig Hoffman disease.
Distal and scapuloperoneal forms
Differentiation from CMT types I and II (page 444) and scapuloperoneal dystrophy (page 470) is clinically difficult and separation may only be possible on histological and neurophysiological grounds.
NEUROCUTANEOUS SYNDROMES
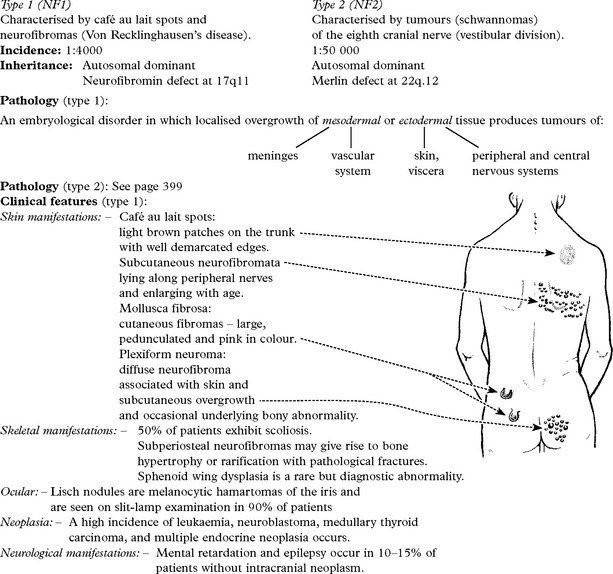
TUBEROUS SCLEROSIS
Characterised by cutaneous, neurologic, renal, skeletal, cardiac and pulmonary abnormalities.