Chapter 46 Motor Neuron Diseases
On July 4, 1939, approximately 100 years after Sir Charles Bell first identified the motor function of the corticospinal tract and published pathologic findings from a middle-aged woman with limb paralysis, intact sensation, and anterior spinal cord degeneration, Lou Gehrig tearfully announced his retirement from baseball to a packed crowd in Yankee Stadium, proclaiming himself “the luckiest man in the world.”9,113 The United States watched as their beloved baseball hero nicknamed “the iron horse” suffered the ravaging effects of amyotrophic lateral sclerosis (ALS), the motor neuron disease (MND) with which his name remains synonymous. In 1941, at the age of 36, he succumbed to the disease, leaving his wife Eleanor to establish the ALS division of the Muscular Dystrophy Association in her pursuit of an illusive cure.
Classification of Motor Neuron Diseases
No universally accepted classification system exists for MNDs. Often the diseases are stratified into categories based on whether dysfunction is localized to UMNs, LMNs, or both. This strategy is complicated by the atypical MNDs such as progressive bulbar palsy (PBP), primary muscular atrophy (PMA), and progressive lateral sclerosis (PLS). These blur boundaries because their course often progresses from exclusively UMN or LMN involvement to frank ALS. Other strategies sort MND into categories based on pathologic process or group them under the umbrella terms typical and atypical motor neuron disease. Being familiar with the various organizational strategies is helpful when navigating the literature because it enables the reader to identify an author’s vocabulary bias and avoid unnecessary confusion (Box 46-1).
BOX 46-1 Motor Neuron Disorders
Upper and Lower Motor Neuron Disorders
Amyotrophic Lateral Sclerosis
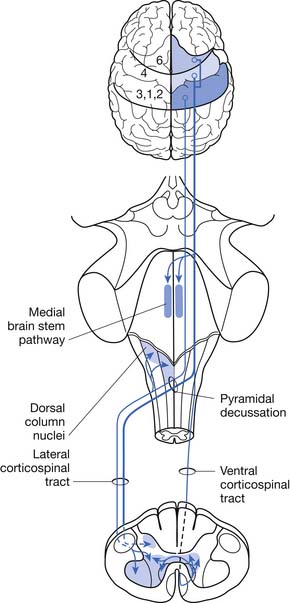
ALS can be defined as a rapidly progressive neurodegenerative disease characterized by weakness, spasticity, and muscular atrophy with subsequent respiratory compromise leading to premature death. It is caused by the destruction of motor neurons in the primary motor cortex, brain stem, and spinal cord (Figure 46-1).8,113,139 ALS was identified as a clinical entity by Charcot in 1874 based on the gross histologic and clinical findings from 5 autopsies and 15 patient cases.113 “Amyotrophy” refers to muscular atrophy occurring from the degeneration of anterior horn cells in the spinal cord with muscle fiber denervation. “Lateral sclerosis” describes the resultant hardening of the anterior and lateral corticospinal tracts caused by replacement of dying motor neurons with subsequent gliosis.139
Onset of ALS is insidious and most commonly presents with painless asymmetric limb weakness. ALS most often afflicts people between 40 and 60 years of age with a mean age of onset of 58 years.100,106,109 Five percent of cases have onset before age 30.139 Men are affected more commonly than women with a ratio of 1.5:1.0. The worldwide prevalence is 5 to 7/100,000, making ALS one of the most common neuromuscular diseases in the world.40
Familial Amyotrophic Lateral Sclerosis
The vast majority of ALS cases are presumably acquired and occur sporadically. Approximately 5% to 10% of all ALS cases, however, are familial (FALS) and most commonly have an autosomal dominant inheritance pattern, although autosomal recessive, X-linked, and mitochondrial inheritance patterns have been reported.104,117,121 The age of onset of FALS occurs a decade earlier than sporadic cases, and progression of the disease is more rapid.80,93,134 Males and females are equally affected. About 20% of FALS cases result from a copper–zinc superoxide dismutase (SOD1) gene defect.9,110,134 These mutations are thought to cause disease by leading to a toxic gain of function from the abnormal tertiary structure, misfolding, of the protein rather than direct impairment in the antioxidant function of the SOD1 enzyme.8,119 More than 100 unique SOD1 mutations have been described.8,69,119,139 Other disease-causing genetic mutations have more recently been identified. These mutations have been found in genes encoding for angiogenin, chromatin-modifying protein, dynactin, vesicle-associated membrane protein, and TAR DNA-binding protein.139
Juvenile Amyotrophic Lateral Sclerosis
Juvenile-onset ALS by definition presents before age 25.139 It is a rarely occurring form of FALS. The progression of the disease is typically much slower than adult-onset ALS and can present initially with either UMN or LMN signs. Inevitably the disease progresses to encompass both UMNs and LMNs. Patients often maintain their ability to ambulate into midlife and can have a normal life span.
ALS2 is also inherited as an autosomal recessive trait. The disease-causing mutation has been linked to chromosome 2q33.41,139 Thus far, nine distinct mutations have been identified, all in the gene encoding for the protein alsin. Each mutation results in a premature stop codon causing production of a truncated, poorly functional protein product. Current research suggests that alsin is involved in vesicle transport and membrane trafficking.41 Disease onset typically begins before age 10. Prominent symptoms include limb and facial spasticity accompanied by pseudobulbar affect.
The autosomal dominant form of juvenile ALS, ALS4, presents with severe distal muscle weakness and pyramidal signs in the absence of bulbar and sensory abnormalities. It is caused by a mutation in the senataxin gene found on chromosome 9q34.41,139 The senataxin protein is known to have a role in RNA processing.41
Sporadic Amyotrophic Lateral Sclerosis
The etiology of sporadic ALS is unknown and likely multifactorial with a complex interplay of pathogenic cellular mechanisms.44,118 In a recent review by Wijesekera and Leigh139 published in 2009, they summarized nine cellular mechanisms for which there is mounting evidence to implicate their role in the pathogenesis of motor neuron degeneration. Included were genetic factors (already discussed), excitotoxicity resulting from excessive glutamate activity in the brain and spinal cord causing calcium influx into neurons with subsequent neuronal death, oxidative stress with accumulation of reactive oxygen species, mitochondrial dysfunction, impaired axonal transport, neurofilament aggregation, protein aggregation causing cytoplasmic inclusions, inflammatory dysfunction with abnormal microglial and dendritic cell activation, and deficits in neurotrophic factors with dysfunction of signaling pathways causing early cell apoptosis.
Population studies from the second half of the twentieth century suggested that the incidence of ALS was increasing. This was probably due, in large part, to increased life span and better recognition of the diagnosis.94,95 More recent population studies from multiple European countries have all demonstrated stable incidence.1,5,42,55,81 Many epidemiologic studies have been undertaken to find causal associations with minimal success. Consistent association between cigarette smoking and ALS, however, has been demonstrated.63 In a population-based case–control study conducted in three counties of western Washington state from 1990 to 1994, a twofold increase in risk was associated with a history of cigarette smoking, and a greater than threefold increased risk was observed for current smokers. Further, the authors found that alcohol consumption was not associated with increased risk of ALS; dietary fat intake was associated with an increased risk; and dietary fiber intake was associated with a decreased risk.96,97 Interestingly, consumption of antioxidant vitamins from diet or supplement sources did not alter the risk, but glutamate intake was associated with an increased risk of ALS.83,200,201 The finding that cigarette smoking and glutamate consumption increase risk for ALS appears consistent with current etiologic theories that implicate glutamate excitotoxicity and oxidative stress in the pathogenesis of ALS.
The hypothesis for an environmental cause of sporadic ALS has partially been spurred by evidence of considerable disease clustering demonstrated most profoundly in the Western Pacific region of the world.40,94,95 The prevalence in this region is 50 to 100 times higher than elsewhere. These populations include the Chamorro people of Guam and Marianas Island, the Kii Peninsula of Honshu Island, and the Auyu and Jakai people of southwest New Guinea, in whom ALS is associated with parkinsonism and dementia.139 Other sporadic cluster cases have been reported but without obvious environmental or causal factors.40
Clinical Features
Other signs and symptoms frequently associated with ALS are cachexia, fatigue, and musculoskeletal complaints. The term ALS cachexia refers to a phenomenon experienced by some patients in which weight loss occurs in excess of that caused by muscle atrophy and reduced caloric intake. Both subcutaneous fat and peritoneal fat are lost, presumably because of acceleration of the basal metabolic rate.116 In patients with ALS cachexia, more than 20% of body weight is typically lost over a 6-month period. Many patients with ALS feel an overwhelming sense of muscle fatigue, which is probably due to a combination of blocking of neuromuscular transmission in reinnervated nerve terminal sprouts and impairment of excitation contraction coupling.116 Some patients seek initial medical attention because of fractures or sprains that do not heal. In reality, these patients probably sustained their initial injury because of a fall or other injury (e.g., sprained ankle) that occurred because of underlying muscle weakness; they were then unable to recover to their premorbid level of function because of that weakness.
Poor prognostic factors include older age at time of onset, bulbar and/or pulmonary dysfunction early in the clinical course of the disease, short period from symptom onset to diagnosis, and predominance of LMN findings at diagnosis.40,100,107,109 More women than men present with bulbar symptoms, and the progression of bulbar palsy appears to be more rapid in women.96,97 Overall median survival from onset of symptoms in bulbar dominant cases is 2 to 3 years.39,73,100 In limb-onset ALS, it is 3 to 5 years.39,73,100 Young males with ALS can have a longer life expectancy, but overall the median 50% survival rate is 2.5 years after diagnosis. Survival rates will vary to a degree depending on the patient’s decision to use or not use mechanical ventilation and a feeding tube. Nonetheless, by 5 years postdiagnosis the overall survival rate is between 4% and 30%.39,40,100,109 Only 4% of patients will survive longer than 10 years.139
Atypical, “ALS-like” MNDs have been reported infrequently as a remote complication of several malignancies, including lymphoma, breast cancer, and small cell carcinoma of the lung.35,127 These likely represent paraneoplastic syndromes and not a true manifestation of ALS.111 Irrespective, patients with atypical MND should be screened for malignancy.
Selected Upper Motor Neuron Disorders
Progressive Lateral Sclerosis
PLS is a rare sporadic disorder of progressive spasticity with mostly spinal and occasionally bulbar region onset.64 Etiology of the disease is unknown. PLS can be clinically difficult to distinguish from hereditary spastic paraplegia (HSP) and is most reliably identified by the lack of familial inheritance.30 Onset of symptoms usually begins in the fifth decade; however, a juvenile variant also exists.30
Clinical Features
Spasticity is the most common presenting symptom. It is progressive and more often begins in the legs versus the arms or bulbar muscles. Asymmetric limb onset and spasticity involving the upper limbs or bulbar region are more common manifestations of PLS and can be diagnostically helpful in distinguishing PLS from the symmetric lower limb involvement observed in HSP.120 Limb wasting is rare in PLS and occurs in only 2% of patients.130 If disease onset is after the age of 45, prominent pseudobulbar symptoms with labile emotional affect can be problematic. Disease progression is usually slower than ALS, occurring over years to decades.120,130 Approximately 45% percent of patients diagnosed with PLS will eventually develop LMN symptoms and progress to ALS.64 For this reason, PLS is often considered a variant of ALS with early dominant UMN dysfunction as opposed to being a separate clinical entity. If the patient remains free of muscle wasting or other LMN symptoms for 4 years after diagnosis, he or she is less likely to progress to ALS, heralding better prognosis with greatly increased life expectancy.64,130
Hereditary Spastic Paraplegia
Like PLS, HSP is a disorder of UMNs and presents most commonly with progressive spasticity, weakness of the lower limbs, hypertonic urinary bladder, and impaired vibration sense. It is a genetically heterogenous group of neurodegenerative disorders in which the most severely affected neurons are those of the spinal cord.114 Caudal to rostral degeneration of the corticospinal tract and mild involvement of the dorsal columns occurs with progression of the disease.114 Population studies done in Ireland showed a prevalence of 1.27/100,000. Inheritance patterns include autosomal dominant, recessive, and X-linked forms. To date, 32 HSP loci and 11 HSP-related genes have been identified. They are classified as spastic gait locus 1 (SPG) through SPG33.57,114
Diagnosis is based on the clinical characteristics of the disease, neurologic examination demonstrating involvement of the corticospinal tract in both lower extremities, family history, and identification of a pathogenic mutation in a disease-causing gene. Sporadic cases occur providing the clinician with a diagnostic challenge. In a recent study by Brugman et al.,30 they determined that differentiation of sporadic presentations of HSP from PLS based solely on clinical characteristics was unreliable and therefore depends predominantly on genetic test results. Information on commercially available genetic tests and diagnostic laboratory locations can be found at http://www.genetests.org.30
Clinical Features
HSP is also classified by clinical characteristics and can be divided into “pure” or “complex” presentations. “Pure” HSP is considered the more classic pattern with spasticity, urinary disturbance, and vibration sense impairment.57,114 Onset occurs at any age, from early childhood through late adulthood, and progresses slowly over many years without exacerbations, remissions, or periods of abrupt worsening. Disability from spasticity and weakness is common, but life expectancy remains normal.57,114
“Complex” HSP is characterized by a combination of the above symptoms with concomitant neurologic disorders, such as seizures, impaired cognition, dementia, extrapyramidal disturbance, or peripheral neuropathy in the absence of other coexisting disorders such as diabetes mellitus, etc.57,114
Selected Lower Motor Neuron Disorders
Chronic Progressive
Progressive Muscular Atrophy
Progressive muscular atrophy by definition is a sporadic degenerative disease selectively affecting the anterior horn cells without signs of UMN involvement. Significant similarities between the natural history of progressive muscular atrophy and ALS have been observed, and debate continues as to whether progressive muscular atrophy should be considered its own clinical entity or a variant in the ALS disease spectrum.71,89 In a postmortem study of 12 patients thought to have progressive muscular atrophy, 50% of the autopsies demonstrated characteristic findings of ALS with degeneration of the corticospinal tract and ubiquitinated inclusions.71 Progressive muscular atrophy is a rare disease with unknown etiology.
Clinical Features
In a prospective study of 37 patients diagnosed with progressive muscular atrophy, published in 2007 by Visser et al.,136 the median age of disease onset was 57 years. The most common presenting symptom was distal limb weakness with muscle atrophy. The arms were affected slightly more often than the legs with either asymmetric or symmetric presentation. Bulbar symptoms were not typically evident at diagnosis but developed over the course of the disease in 43% of the patients. The onset of bulbar symptoms portended a more rapid decline with early death. During the 18 months of study, 35% of patients developed UMN signs progressing to an ALS phenotype. The 5-year survival rate was 45% with median survival of 56 months. Poor prognostic indicators included a forced vital capacity (FVC) less than 90% predicted at the time of diagnosis and decline in the FVC within the first 6 months after diagnosis.
Spinal Muscular Atrophy
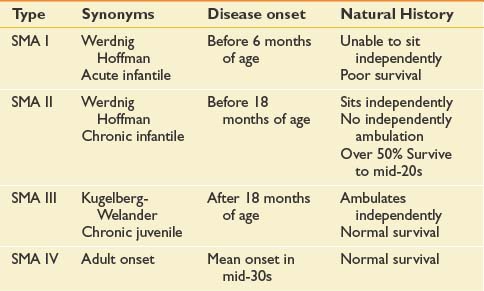
Many forms of spinal muscular atrophy (SMA) exist, all of which involve selective destruction of anterior horn cells. The various forms of SMA are clinically heterogenous, with some rare forms affecting distal or bulbar muscles only. The spinal muscular atrophies are mostly disorders with childhood onset and are usually inherited as autosomal recessive traits (Table 46-1). SMA is relatively common with an incidence of about 10 in 100,000 live births and a carrier frequency of 1 in 50.102
In 1990 the gene responsible for childhood-onset SMA was mapped to chromosome 5q11.2-13.3.31 Both the causative gene, named survival motor neuron 1 (SMN1, telomeric copy), along with a disease-modifying gene, survival motor neuron 2 (SMN2, centromeric copy) (32 to 35) were identified. The survival motor neuron (SMN) protein is ubiquitously expressed in all cells and tissues, with high levels in the nervous system, especially in the spinal cord.15 In about 95% of SMA patients, both copies of SMN1 exon 7 are absent or mutated. Recent data indicate that SMN1 deficiency alters stoichiometry of small nuclear ribonucleoproteins and leads to splicing defects for numerous genes in all cells, including motor neurons.141
Normal structural differences in the SMN2 gene cause frequent but not absolute exon 7 skipping during the splicing of SMN2 transcripts.24,140 The full-length transcripts of SMN1 and SMN2 encode proteins with an identical sequence. Without normal transcription of exon 7, the SMN protein product is less stable and more rapidly degraded, causing the neurodegenerative process. At least one copy of the SMN2 gene must be present in the setting of homozygous SMN1 mutations; otherwise, embryonic lethality occurs. The copy number of SMN2 varies in the population, and this variation appears to have important modifying effects on SMA disease severity.129 It appears an inverse trend exists between the number of SMN2 copies and phenotypic severity. Substantial variations in SMA phenotype and disease severity, however, can exist between patients with similar SMN2 copy numbers, suggesting other factors are likely involved. In the remaining 5% of SMA-affected patients, other small or subtle mutations have been identified.129,140
Clinical Features
The most common forms of SMA are often referred to as types I, II, and III.32 SMA I, also known as Werdnig-Hoffman disease or acute, infantile-onset SMA, is a severe disorder often resulting in death before age 2 years, although recent studies have reported an increase in longevity likely resulting from better medical management of disease sequelae.83 Children with SMA I never attain the ability to sit independently (Figure 46-2). SMA II, also referred to as early-onset, intermediate SMA or chronic Werdnig-Hoffman disease, is less severe, with signs and symptoms becoming apparent in the first 6 to 18 months of life. These children will eventually attain the ability to sit independently but do not ambulate without assistance. SMA III, also known as Kugelberg-Welander disease, is a chronic, later-onset disorder, associated with significantly less morbidity. In SMA III, all early developmental milestones including the ability to ambulate independently are acquired (Figure 46-3). Signs and symptoms of SMA III usually become apparent between ages 5 to 15 years. Patients can be identified initially because of frequent falling and difficulty climbing stairs. In previous studies looking at SMA II and SMA III over a 10-year period, SMA II subjects showed marked weakness and progressive decline of strength, whereas SMA III subjects had a relatively static or very slowly progressive course and were far stronger. In both SMA II and SMA III, proximal weakness was greater than distal. Joint contractures, severe progressive scoliosis, and restrictive lung disease were present in most of the SMA II individuals, but these complications were less frequently identified in SMA III.32 Hand tremor, tongue fasciculations, and areflexia are common. Limb fasciculations occur more predominantly in SMA III. Intellectual function is preserved, and care should be taken to provide the child with SMA access to educationally stimulating environments.
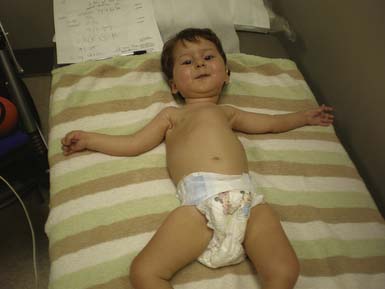
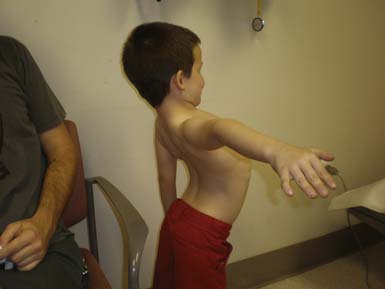
SMA IV, also known as adult-onset SMA, has a mean age of onset in the mid-30s. It can be inherited as either an autosomal recessive or dominant trait.59,171 The disease clinically appears much like SMA III with slowly progressive proximal limb weakness and fasciculations. Deep tendon reflexes are either absent or depressed. Adult-onset SMA and SMA III patients can live normal life spans with a relatively benign disease course. Many of the rehabilitative modalities discussed in this chapter are applicable to this population. Further, with the rapid advancement of rehabilitation technology, many SMA II patients are now living well in to adulthood, and successful pregnancies have been reported in this population.33
Spinal and Bulbar Muscular Atrophy (Kennedy Disease)
Spinal bulbar muscular atrophy (SBMA) is often classified within the SMAs. It is, however, not associated with abnormalities in the SMN gene. It is a hereditary adult-onset disease that causes preferential degeneration of LMNs leading to weakness and atrophy of bulbar, facial, and limb muscles.12,58,128 SBMA is caused by a novel mutation, the expansion of a trinucleotide, cytosine–adenine–guanine (CAG) repeat, in the first exon of the androgen receptor gene and has X-linked recessive inheritance.12,24,58,128 Because CAG is transcribed to a glutamine, it is known as one of a few polyglutamine diseases. Unaffected individuals have a CAG repeat size that ranges between 5 and 35, whereas symptomatic individuals always have a repeat size of 40 or greater.24 Testosterone-driven accumulation of the mutated androgen receptor protein collects in nuclei and cytoplasm of motor neurons, resulting in their degeneration and loss.58
SBMA has some clinical variability; however, phenotypic expression does not appear to correlate with the length of CAG repeats. This is in contrast to myotonic muscular dystrophy and fragile X syndrome, where increased numbers of tandem triplet repeats correlate directly with disease severity.6
Clinical Features
Disease onset usually occurs after the age of 30 and is variable. Cases with onset in late adulthood have been reported. Patients present with amyotrophic, proximal, or distal weakness and wasting of the facial, bulbar, and limb muscles, sensory impairment, and endocrinologic disturbances, such as androgen resistance, gynecomastia, elevated testosterone or progesterone, and reduced fertility. Reflexes are typically decreased. There might be mild increases in creatine kinase. The course is slowly progressive with the ability to ambulate lost only late in life. Few patients require ventilatory support, and life expectancy is only slightly reduced.12,24,58,128
SBMA is clinically similar to ALS and often initially misdiagnosed. Degeneration of sensory neurons of the dorsal root ganglia is a common sign associated with SBMA, often preceding the onset of motor dysfunction in clear contrast to ALS. Electrodiagnostic studies demonstrating abnormal sensory nerve action potentials with spontaneous activity on electromyography (EMG) in patients with clinical signs similar to ALS should prompt further evaluation for SBMA. Another interesting difference to note is that Onuf’s nucleus, an androgen-sensitive spinal cord motor neuron nucleus that innervates the external sphincter muscles of the anus and urethra, is spared in ALS but degenerates in SBMA.6,45 Commercially available molecular genetic tests are now available for both SMA and SBMA.
Infectious
Poliomyelitis and Postpolio Syndrome
Acute poliomyelitis is a disease of the anterior horn neurons of the spinal cord and brain stem caused by poliovirus. The infection leads to the development of acute flaccid paralysis, which can be bulbar or spinal in distribution. The poliovirus is a small RNA virus belonging to the enterovirus group of the picornavirus family. Three antigenically distinct strains each have the potential to cause disease. Type I accounts for approximately 85% of cases progressing to paralytic illness. Only about 1% of patients who contract the disease will develop acute flaccid paralysis, also known as paralytic polio.74 Transmission occurs via fecal–oral route. The virus multiplies in the gastrointestinal tract during an incubation period lasting from days to weeks depending on the mode of infection (e.g., vaccine-induced vs. oral). The virus is shed in both saliva and feces during the incubation period. Further invasion of intestinal lymphoid tissues leads to hematologic spread and potential central nervous system involvement. The severity of the disease typically falls within a spectrum including no obvious manifestation of the disease, viremia without nervous system involvement, viremia with meningeal irritation but without paralysis, or paralytic disease with or without respiratory failure.74
Before the development of the polio vaccine, poliomyelitis was the most common cause of acute flaccid paralysis and disability in the United States. In the Northern Hemisphere epidemics were most common during the summer months. Peak incidence occurred during epidemics in the first half of the twentieth century, culminating in 1952 with approximately 58,000 cases reported in the United States.74 Children under age 15 were most commonly affected. Public fear and desperation led to the inception of the March of Dimes in the 1930s with a mission to eradicate the disease.84 The March of Dimes’ aggressive campaign raised public funds to support research and development of a vaccine. On April 12, 1955, Dr. Jonas Salk sparked celebrations around the world by announcing the results from the largest field trial in medical history.74 Salk had injected 325,000 second-graders with a trivalent inactivated polio vaccine, gave the same number of children a placebo, and monitored an even larger control group for disease. The vaccine nearly eliminated subsequent infection in the treated cohort and forged the path to conquer polio with initiation of the then-largest vaccination campaign in U.S. history. Since 1979 there have been no cases of wild-type poliovirus infection reported within the United States.
An oral vaccine developed by Albert Sabin from live attenuated poliovirus was introduced in 1963.74 Although the oral vaccine was less expensive to manufacture and simple to dispense, in January 2000, the Centers for Disease Control and Prevention (CDC) and the American Academy of Pediatrics recommended vaccination with the inactivated polio vaccine in an attempt to avoid further cases of paralytic polio in nonimmunized contacts of children receiving the live oral virus.7,38
In 1988, the World Health Organization, UNICEF, Rotary International, and the U.S. CDC launched the Global Polio Eradication Effort to rid the rest of the world of polio. Since the launch of this initiative, the worldwide incidence has declined. There remain four countries, however, where wild poliovirus is still endemic. Political instability in regions near the Pakistan/Afghanistan border and resistance of the population to polio immunization in northern Nigeria have resulted in insufficient vaccine coverage and remaining infections. In northern India low oral polio vaccine efficacy has thwarted efforts to eradicate the disease.43 In 2008, cases of poliomyelitis were reported in countries where none occurred in 2007. These countries included Sudan, Benin, Ethiopia, Burkina Faso, and Nepal, illustrating the importance of continued widespread vaccination prevention programs.50
Clinical Features
In patients with overt manifestations of infection, symptoms consist of fever, malaise, myalgia, sore throat, and gastrointestinal upset. Aseptic meningitis with headache, back pain, and stiff neck develop with increasing severity of the disease. In the approximately 1% of patients who progress to paralytic disease, localized fasciculations with intensely painful myalgias occur after 2 to 5 days of illness.74 Asymmetric weakness and atrophy affecting the legs more often than either the arms or bulbar muscles progress to flaccid paralysis. Dysautonomia including labile blood pressure, cardiac arrhythmia, and gastrointestinal and urinary dysfunction can require emergent medical intervention and is associated with higher rates of mortality.74 Respiratory failure often develops rapidly if infection involves the medullary respiratory center or causes weakness of respiratory muscles. Paralysis remains static for several days to weeks followed by slow recovery over months to years. Improvement of strength after acute paralytic polio occurs both by recovery of some neurons and sprouting from remaining axons innervating locallydenervated muscle fibers.84 The enlarged motor units can be up to 8 times the normal size. Recovery is often incomplete with residual weakness and disability.84
Late effects of poliomyelitis occur more commonly in patients with history of paralytic polio. Farbu et al.56 prospectively examined 85 patients with late effects of polio and found that the most common complaints were pain, muscular weakness, and fatigue. They identified loss of function resulting from degenerative joint disease with and without nerve entrapment in 53% of patients. Compensatory mobility patterns secondary to limb weakness and side-to-side growth disparity, as well as overuse of unaffected limbs, have been blamed for increased incidence of symptomatic osteoarthritis.
Of the 85 patients studied, only 26% met criteria for the diagnosis of postpolio syndrome (PPS; Box 46-2). Gradual or sudden onset of new progressive weakness and decreased muscular endurance after a period of at least 15 years of functional stability suggest PPS. The etiology of PPS is unknown. Proposed mechanisms for the development of PPS include distal degeneration of surviving enlarged neurons resulting from increased metabolic demand, degeneration of terminal axonal sprouts resulting in muscle fiber denervation, neuromuscular junction dysfunction as demonstrated by increased jitter on single fiber EMG, and loss of neurons through the normal aging process.56,74,84 Careful medical evaluation should be undertaken to exclude other possible and potentially treatable causes of the patient’s complaints.
BOX 46-2 Criteria for Diagnosis of Postpolio Syndrome
Modified from the March of Dimes: International conference on post-polio syndrome identifying best practices in diagnosis and care, May 19-20, 2000. Available at: http://www.marchofdimes.com/files/PPSreport.pdf.
Clinical Evaluation in Motor Neuron Diseases
History
The evaluation of a patient suspected of having MND begins with a detailed history and general physical and neurologic examination. The history should establish the age at the time of onset and the initial presenting symptoms. The clinician should identify the pattern of weakness and/or spasticity. Are symptoms symmetric versus asymmetric, distal versus proximal, limb or bulbar predominant? Asking the patient what activities have become more difficult can give clues to the pattern of weakness. For example, a patient with proximal weakness might complain of difficulty brushinghis or her hair or climbing stairs. It is also important to determine the rate and pattern of progression of symptoms because these can give diagnostic and prognostic clues. A detailed past medical, social, and family history should be obtained, exploring the potential of immune-mediated, toxic, infectious, or familial etiology.
Physical Examination
On neurologic examination, one is looking for evidence of UMN and LMN dysfunction (Box 46-3). The mental status, nonmotor cranial nerve function, sensory examination, and cerebellar examinations should be normal in the patient with ALS but can be abnormal in atypical MND. Findings of upper motor involvement on examination include spasticity and hyperreflexia, indicated by abnormal spread and amplitude of reflexes, clonus, or by the presence of reflexes despite muscle atrophy as a result of LMN loss. The gold standard used to diagnose UMN pathology is the presence of pathologic reflexes, such as the Babinski sign, Hoffman sign, jaw jerk, and palmomental and snout reflexes.28 If the toe extensors are paralyzed, visualization of contraction of the tensor fascia lata when an attempt is made to elicit a Babinski response has the same significance as great toe extension. Recently, it has been suggested that the corneomandibular reflex might be a more sensitive and specific indicator than the jaw jerk of UMN pathology in the bulbar region.10
LMN findings on examination include weakness, atrophy, hypotonia, hyporeflexia, and fasciculations. Head drop is a manifestation of muscle weakness often seen in ALS, although it can be seen in other neuromuscular disorders. ALS and myasthenia gravis are the two most common causes of head drop (Figure 46-4). Fasciculations are common in lower MND and can occur in the tongue and the extremities.

Electrodiagnostic Testing
No biomarkers are currently available for the diagnosis of ALS. Diagnosis is based on clinical findings requiring the presence of signs of UMN and LMN degeneration and the progression of the symptoms from a body region to another. Electrodiagnostic testing is considered an extension of the physical examination and should be performed on patients suspected of having an MND. Purely UMN disorders will have normal electrodiagnostic studies. The various forms of MND with involvement of the LMN share several electrodiagnostic features. General electrodiagnostic testing characteristics of MND include normal sensory nerve conduction studies with the exception of Kennedy disease, normal or low motor amplitudes depending on disease stage, and normal distal motor latencies and conduction velocities. With profound loss of motor amplitude, conduction velocities can decrease as low as 25% below the lower limit of normal because of loss of the fastest conducting fibers. Motor nerve conduction studies, including proximal stimulation sites to assess for conduction block resulting from peripheral neuropathy (e.g., multifocal motor neuropathy with conduction block) should be included in the electrodiagnostic plan.48
The needle electrode examination shows a decreased recruitment pattern, either normal size or large motor unit action potentials with or without evidence of remodeling depending on the specific disease process, and abnormal spontaneous activity including positive sharp waves, fibrillation potentials, fasciculations, and complex repetitive discharges.48 The electrodiagnostic study should include needle examination of the thoracic paraspinal muscles.49 Choosing to sample muscles below the T6 root level helps to avoid confounding results caused by multilevel root innervation from the lower cervical segments. Other muscles that can be sampled that are perhaps unique to MND include bulbar, facial, masticatory, and rectus abdominus muscles. These can be useful to find evidence of involvement in additional body regions needed to meet the El Escorial criteria for the diagnosis of ALS.48
Other electrodiagnostic tests to consider in the setting of an MND evaluation include single-fiber EMG, which might demonstrate increased jitter resulting from immature sprouting of nerve terminals during reinnervation in ALS. Motor unit number estimation assesses the size of a motor unit and can be followed over time for changes from motor neuron loss and subsequent sprouting of remaining neurons leading to enlarged motor units. Motor unit number estimation is being evaluated as a potential outcome measure in ALS clinical trials.48 Transcranial magnetic stimulation holds promise in evaluation of UMN dysfunction; however, it is not widely available, and this limits its clinical use.48
El Escorial Criteria
The El Escorial Criteria for diagnosing ALS were developed by a task force of the World Federation of Neurology in 1990 to ensure inclusion of more homogeneous patient populations in ALS clinical trials.27 These criteria have been used to enroll patients in most of the recent clinical trials. The criteria were revised in 1998 and again at the international symposium held in Awaji-shima, Japan, in December 2006 with the intent to improve the speed and certainty of diagnosis.27,48 The current criteria classify the certainty level of the diagnosis of ALS as falling into one of three categories: definite, probable, and possible. The category of “Laboratory Supported Probable ALS” is no longer included based on the consensus panel’s decision that clinical features of neurogenic change and neurogenic EMG findings should have the same diagnostic significance in an individual muscle and can be considered together in a single limb to meet the required abnormalities for diagnosis of ALS.48
The El Escorial criteria divide the motor system into four regions: bulbar, cervical, thoracic, and lumbosacral. Clinical evidence of UMN and LMN pathology is sought within each region. The certainty level of diagnosis depends on how many regions reveal UMN and/or LMN pathology. Box 46-4 summarizes the schema for placing patients in the three diagnostic categories.
Laboratory Evaluation and Other Diagnostic Tests
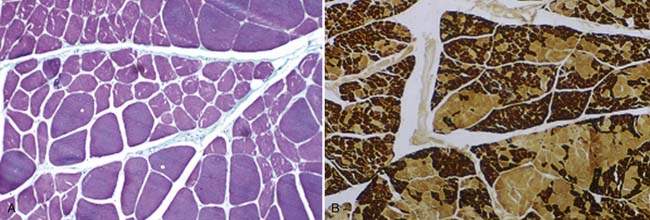
In most neuromuscular clinics, a routine panel of laboratory tests is performed for all patients suspected of having ALS (Box 46-5). The rationale behind performing this battery of tests is to assess the general health of the patient and exclude treatable conditions. The differential diagnosis, developed after the history and physical examination, can suggest that more specialized testing be performed. When there is a family history of MND, genetic testing is often warranted. Muscle biopsy might be considered to exclude myopathies. Findings on muscle biopsy consistent with neurogenic atrophy include fiber-type grouping with increased fiber size variability, and clusters of muscle fiber nuclei caused by denervation and subsequent fiber loss called pyknotic clumps (Figure 46-5). These findings are consistent with but not diagnostic for ALS.
BOX 46-5 Laboratory Evaluation of the Patient With Suspected Motor Neuron Disease
Neuroimaging
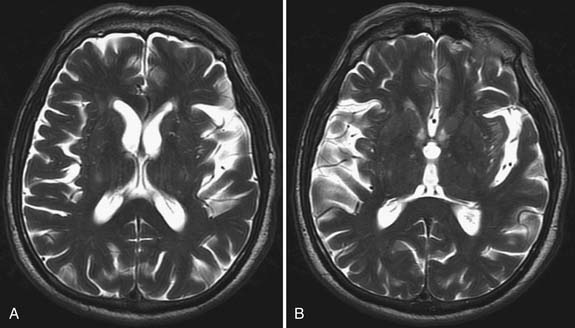
Although MRI is generally not performed to confirm a diagnosis of ALS, a few associated abnormalities have been reported. Rarely, spinal cord and motor cortex atrophy is apparent. Corticospinal tract hyperintensity with T2 imaging has been observed in a few younger patients with a predominance of UMN signs and rapidly progressive disease (Figure 46-6).92 Diffusion tensor imaging is being evaluated for its usefulness in diagnosing and following the progression of MND.3,125
General Principles of Rehabilitation and Disease Management
Although currently incurable, adult MNDs are not untreatable. The goals of rehabilitation and palliative care are to maximize functional capacities, prolong or maintain independent function and mobility, inhibit or prevent physical deformity, and provide full access to community, ensuring good quality of life. In ALS, this includes addressing end-of-life issues.
Initial confirmation of the diagnosis is critical and is a primary responsibility of the consulting neuromuscular disease specialist. Because of the ominous prognosis of ALS, a confirmatory second opinion should always be sought. A physiatrist is well suited to direct the rehabilitation team and oversee a comprehensive, goal-oriented treatment plan.61,62 At initial evaluation the patient should be thoroughly educated about the expected outcome and what problems might be encountered. The physician should then assess the patient’s goals and orchestrate a rehabilitative and ultimately a palliative program that matches those goals. In ALS, palliative care should be aimed at maximizing the patient’s comfort and quality of life but not necessarily extending life.
Disease-Modifying Pharmacologic Treatment
Riluzole
Despite clinical use for nearly 20 years, riluzole, a 2-amino-6-(trifluoromethoxy) benzothiazole, remains the only Food and Drug Administration–approved medication proven to slow the progression of ALS. Pharmacologic mechanisms of riluzole include interference with N-methyl-d-aspartic acid (NMDA) receptor–mediated responses, stabilization of the inactivated state of voltage-dependent sodium channels, inhibition of glutamate release from synaptic terminals, and activation of extracellular glutamate uptake.20,21 A Cochrane Database Review concluded 100 mg of riluzole daily prolongs median survival by about 2 to 3 months based on analysis of four randomized controlled trials.90 Recent studies using large registries suggest a greater benefit, ranging 4 to 20 months. Although the American Academy of Neurology practice guideline recommends the use of riluzole for nonventilated ALS patients, analysis of the ALS C.A.R.E. database found that 41% of the cohort was not prescribed this medication, largely because of the expense.26,105 The drug is generally well tolerated, with the most common side effects being asthenia, nausea, and an increase in serum alanine aminotransferase.19,21 Liver function should be monitored during therapy.
Experimental Therapies and Clinical Trials
The National Institutes of Health sponsored a high-throughput drug screening program looking for drugs with the potential for slowing progression of ALS. Ceftriaxone was identified to increase both brain expression of astroglial glutamate transporter GLT1 and its biochemical and functional activity, and delay loss of neurons and muscle strength. Its central nervous system penetration and long half-life are well known, obviating need for extensive safety trials.112 Clinical trials are currently ongoing.
Memantine is a noncompetitive NMDA receptor antagonist. It has been shown to protect neurons against NMDA- or glutamate-induced toxicity in vitro. Treatment of SOD1G93A mice significantly delayed disease progression and increased life span.137 At the time of this writing the safety studies are completed, and efficacy studies are enrolling patients.
Lithium prevents neurodegeneration by promoting autophagy, through inhibition of inositol-monophosphatase, rescues spinal cord mitochondria, and facilitates the clearance of α-synuclein, ubiquitin, and SOD1. It delayed disease onset and progression in G93A transgenic mice and increased survival and slowed progression in humans over 15 months compared with controls in a small study with somewhat atypical slowly progressing patients.60 Larger trials are currently underway.
Other drugs currently in trial are ONO-2506, an enantiomeric homologue of valproate that restores disturbed astrocyte functions, and talampanel, a noncompetitive modulator of α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid glutamate receptors.99,132 Cannabinoids afford protection against oxidative damage induced by free radicals produced by glutamate.2,23,65–67 Administration of Δ-9-THC before and after the onset of ALS symptoms slowed disease progression and prolonged survival in animals compared with untreated controls.138
Leuprorelin acetate, a luteinizing hormone-releasing hormone analogue that reduces testosterone release from the testes and induces androgen deprivation, has recently completed Phase 2 clinical trials in patients with SBMA.12,13,128 The drug was deemed safe and successfully decreased toxic accumulation of mutant androgen receptor (AR). Patients treated with the drug for 144 weeks exhibited significantly greater functional scores than those treated with placebo.12,13 Large-scale Phase 3 trials have yet to begin enrolling.
Sodium phenylbutyrate, hydroxyurea, and valproic acid combined with carnitine are currently in trials for treatment of SMA. For up-to-date information on clinical trials, explore the website http://www.clinicaltrials.gov.
Role of Exercise
Skeletal muscle weakness is the sine qua non of all adult MND, including ALS, and is the ultimate cause of the majority of clinical problems associated with these diseases (see also Chapter 18). Only a precious few studies of exercise in ALS have been done.47 In other more slowly progressive neuromuscular diseases, however, a 12-week moderate resistance (30% of maximum isometric force) exercise program resulted in strength gains ranging from 4% to 20% without any notable deleterious effects.4 In the same population, however, a 12-week high resistance (training at the maximum weight a subject could lift 12 times) exercise program showed no further added beneficial effect compared with the moderate resistance program, and there was evidence of overwork weakness in some of the subjects.4 Because of the active, on-going muscle degeneration in most cases of ALS, and to a lesser extent in SMA, SBMA, and PPS, the risk for overwork weakness is great, and exercise should be prescribed cautiously and with a common sense approach. Patients should be advised not to exercise to exhaustion, which can produce more muscle damage and dysfunction.75 Patients participating in an exercise program should be cautioned of the warning signs of overwork weakness, which include feeling weaker rather than stronger within 30 minutes after exercise and/or having excessive muscle soreness 24 to 48 hours after exercise. Other warning signs include severe muscle cramping, heaviness in the extremities, prolonged shortness of breath, and an increase in fasciculations.75
Given the lack of any apparent contraindication, aerobic exercise training is recommended for patients with ALS as long as it can be performed safely without a risk of falling or injury. In addition to the physical benefits, this form of exercise often has a beneficial effect on mood, psychologic well-being, appetite, and sleep. Pool therapy is often an ideal place for patients with MND to do aerobic exercise, which can be as simple as walking in the water, with the water at midchest height. A recent study demonstrated improvement in heart rate and in pain levels without any adverse events in a group of postpolio patients.108 Aquatic therapy is best done in a pool with a flat, uniform depth floor that is heated to 92° to 95° F. The warmth of the water helps reduce spasticity and facilitates movement. Other interventions can include low-impact aerobic exercise like walking or stationary bicycling to improve cardiovascular performance, increase muscle efficiency, and help fight fatigue.75 Fatigue in ALS is multifactorial and is due, in part, to impaired muscular activation.37,115 Other contributing factors include generalized deconditioning from immobility and clinical depression. Aerobic exercise not only improves physical functioning but also is beneficial in fighting depression and improving pain tolerance. Although there have been few well-controlled studies looking at exercise-induced strength gains in the ALS population, a recent Cochrane Review focused on this. Studies in randomized or quasirandomized controlled trials of people with a diagnosis of definite, probable, probable with laboratory support, or possible ALS (as defined by the El Escorial criteria) were recently published.46 This included progressive resistance or strengthening exercise and endurance or aerobic exercise. The control condition was no exercise or standard rehabilitation management. The primary outcome measure was improvement in functional ability, decrease in disability, or reduction in rate of decline as measured by a validated outcome tool at 3 months. Secondary outcome measures were improvement in psychologic status or quality of life, decrease in fatigue, increase in or reduction in rate of decline of muscle strength (strengthening or resistance studies), increase in or reduction in rate of decline of aerobic endurance (aerobic or endurance studies) at 3 months, and frequency of adverse effects. Two randomized controlled trials met inclusion criteria. The first examined the effects of a twice-daily exercise program of moderate load, endurance exercise versus “usual activities” in 25 people with ALS.53 The second examined the effects of thrice-weekly moderate load and moderate intensity resistance exercises compared with usual care (stretching exercises) in 27 people with ALS.46 After 3 months, when the results of the two trials were combined, there was a significant weighted mean improvement in the Amyotrophic Lateral Sclerosis Functional Rating Scale measure of function in the exercise compared with the control groups (3.21; 95% confidence interval, 0.46 to 5.96) in favor of the exercise group. No statistically significant differences in quality of life, fatigue, or muscle strength were found.
Symptomatic Treatment
Spasticity
Spasticity in ALS is likely induced both at the motor cortex and the spinal cord level (see also Chapter 30). Treatment of spasticity is only necessary if it is intolerably painful or impairs function. The γ-aminobutyric acid (GABA) analogue baclofen acts to facilitate motor neuron inhibition at spinal levels and is a first-line treatment. Initial dosing starts at 5 to 10 mg two to three times a day and can be titrated to doses of 20 mg four times a day. Side effects include weakness, fatigue, and sedation, which might be intolerable. Patients should be cautioned to avoid abrupt discontinuation of the medication because withdrawal can induce seizures. In patients with severe spasticity an intrathecal baclofen pump can be considered. Patients with HSP are good candidates and might have dramatic improvement to near-normalization of their gait with intrathecal baclofen.
Pain
Although not frequently characterized as a major component of ALS, patients experience significant pain (see also Chapters 42 and 43).101 The pain is due largely to immobility. The physician should be aggressive in identifying the source of the patient’s pain and develop a comprehensive treatment strategy. Nonpharmacologic therapeutics include modalities such as ice, heat, soft tissue mobilization, and range of motion exercise. A course of physical therapy to design a home program and provide caregiver education and training in these techniques is frequently appropriate if the patient has adequate caregiver support.
Opioid medications should be reserved for refractory pain, although concern for addiction is relatively pointless in a terminal disease. The medications should be given on a regular dosing schedule and titrated to the point of comfort.87 Oral or sublingual morphine, 10 to 30 mg every 4 hours, is also effective and might help relieve “air hunger” in the terminal stages of the disease. Another option is taking the total dose of immediate-release morphine required to alleviate pain and giving half of that every 12 hours in a controlled-release preparation such as MS Contin. The intramuscular delivery route should be avoided because of muscle wasting. Fentanyl or morphine patches can deliver inconsistent dosing, particularly if there is excessive perspiration. Complications with opioid medication in ALS are respiratory depression and constipation. These side effects might be acceptable in the final phases of life when respiratory insufficiency or severe pain require increased doses of opioids and/or benzodiazepines.
Dysarthria
In the early stages of the disease, or with slowly progressive deterioration of the bulbar muscles, speech therapy can be helpful (see also Chapter 3). The goal is to correct poor compensatory strategies that can worsen speech intelligibility.77 Patients at this stage often are aware of their deficits and overwork with forceful speech in an attempt to be understood. Adaptive strategies using breathing techniques and maintaining a slow speaking rate with an emphasis on increasing the precision of speech production might be helpful and can be taught by a speech-language pathologist.77 Rapidly progressive bulbar dysfunction in MND does not respond well to conventional articulation training and should be approached by prescribing communicative aids rather than traditional ongoing speech therapy.77 An alphabet supplementation or word board works well early on when patients still have reasonable upper limb function. Once patients have lost hand and arm function, developing yes/no or other binary commands with eye-gaze systems can be used, particularly if the patient is using mechanical ventilation. Major recent advancements have been made in devices such as speech synthesizers or multipurpose, multiaccess, computer-based augmentative communication systems. Although expensive, these devices greatly enhance the patient’s ability to communicate when no longer able to phonate. If patients with dysarthria are identified early, their own speech patterns can be recorded and programmed into a communication device.
Laryngospasm
Laryngospasm is a brief paroxysmal contraction of the vocal cords that temporarily interrupts speech and breathing. It often occurs during the night and can be a significant source of anxiety and panic. It occurs more commonly in the later stages of ALS and affects approximately 20% of patients.77 It is more common in SBMA.123 Recurrent laryngospasm was observed in 47% of 49 patients with SBMA but in only 2% of a control group of patients with early-stage ALS.123 It is associated with smoking and gastroesophageal reflux. Treatment includes rapid upright positioning, which can effectively shorten the duration of the spasm. Long-term treatment strategies are directed at reducing laryngeal irritation from reflux of gastric contents and the irritation of cigarette smoke.77 Avoiding large late-night meals combined with the use of a proton pump inhibitor can reduce the frequency of attacks.
Dysphagia and Nutrition
Patients with ALS frequently develop bulbar muscle dysfunction as a result of motor neuron involvement in the brain stem (see also Chapter 27). Dysfunction of the lips, tongue, and pharyngeal and laryngeal muscles can result in an increased risk of aspiration and difficulty with generating adequate glottic closure for effective cough function. Swallowing can become impaired, and ingesting adequate nutrition can be trying for the patient and family alike.245 Choking episodes are common and might even be triggered by saliva. Secretion management is a particularly difficult issue because secretions can become viscous as a result of inadequate hydration and mouth breathing. Malnutrition caused by inadequate protein–calorie intake can occur, and rapid weight loss should signal the clinician to carefully assess the swallowing mechanism.91 Referral should be made to a speech therapist who can perform dynamic video-assisted swallow evaluations, as well as instruct patients and their families in techniques to reduce the risk of aspiration.77
Most patients with ALS are deficient in energy intake.52 Both muscle and fat mass tend to decrease with disease progression. In addition, some patients become hypermetabolic. The etiology of this change in metabolism is not entirely clear, but it might be related to increased respiratory effort. Inadequate nutrition is problematic because it can cause greater muscle catabolism and increase fatigue. Causes of malnutrition include arm weakness and inability to feed oneself, increased time required to eat, loss of appetite because of depression, bulbar muscle dysfunction, and dysphagia. In a prospective study of French patients with ALS, those who were malnourished had a 7.7-fold increased risk of death.131
Dysphagia can initially be managed by instruction in compensatory strategies. Patients should be instructed to avoid distractions during mealtime. Swallowing techniques (see Chapter 27) such as a double swallow, chin tuck, head turning, and supraglottic swallowing can be used.77 ALS patients have greater difficulty with thin liquids and dry crumbling foods. Dietary modification such as thickening of liquids, moistening solids, and modifying temperature and texture can be helpful. Patients can often maintain nutrition orally by adding high-calorie liquid supplements to their diet. A registered dietician can offer recommendations for appropriate nutritional supplementation.
When nutrition cannot be maintained orally, either partial or complete nutritional support can be provided via a feeding tube. In the past, most centers used percutaneous endoscopic gastrostomy (PEG) tubes, but many centers now use radiologically inserted gastrostomy (RIG) tubes. RIG tubes seem to be easier and safer to insert in patients with low vital capacities.86 Indications for PEG or RIG include aspiration pneumonia, loss of more than 10% of body weight, and the impairment of quality of life because of the time required to maintain nutrition orally. Feeding tube placement typically stabilizes weight and prolongs survival.88,91 It is important to follow respiratory function and plan for gastrostomy tube placement before the patient’s FVC falls below 30% predicted.
Sialorrhea (drooling) is caused by inadequate handling of secretions rather than the amount of them. In fact, salivation in ALS appears to be less than in unaffected individuals.91
Bracing and Equipment
The patient’s ability to ambulate changes during the course of the disease. Initial therapy might require the use of ankle–foot orthoses to improve foot drop. Ankle–foot orthoses molded in a neutral position can prolong ambulation and decrease the risk of injury and falls (see Chapter 15). Wheeled walkers or quad canes can help prevent falls. Once the patient is no longer able to ambulate community distances, a mobility device should be provided. Wheelchairs should provide adequate lumbar support and have pressure relief cushioning (gel foam). The chair should be properly fitted to prevent pressure ulcers and provide adequate support for the spine. The wheelchair should have both the ability to “tilt-n-space” and recline to aid with pressure relief and positioning. Simply giving the patient a prescription for a wheelchair often results in the patient receiving a standard manual chair that does not fit properly. Using a team approach with physical and/or occupational therapists and the durable medical equipment provider is the best way to ensure that the proper wheelchair is prescribed and obtained. A power wheelchair, although expensive, is justified because it will prolong independent mobility and markedly improve quality of life.
Other useful equipment includes hand-held showers, bath tub benches, grab bars, raised toilet seat, commode chair, ADL aids (e.g., sock aid, grabbers), sliding board, pneumatic patient lift, and wheelchair ramps (see Chapters 23 and 26). An occupational therapist can help determine which, if any, of these devices will be useful to the patient. Other simple suggestions, such as moving the patient’s bedroom to the first floor and removing loose rugs or covering slippery floors, are helpful and can be made during an in-home evaluation by the occupational therapist.
Frequently, severe weakness in the neck flexors and extensors will cause a “floppy head” associated with severe neck pain and tightness. This might be helped by a soft cervical collar or a Freeman or Headmaster-type collar, which is a wire frame with padding over the pressure points (see Chapter 16).
Respiratory Management
Cough is an essential airway protection reflex. The individual with ALS might experience cough impairment in any of the stages of cough, including reduction in the inspired volume because of diaphragm weakness, inability to close the glottis completely because of bulbar muscle dysfunction, and inability to compress and expel intrathoracic gas because of expiratory muscle weakness. Maximal expiratory pressure, a commonly used clinical measure of expiratory muscle strength, does not correlate well with the presence or absence of cough generation.18 Inspiratory muscle strength also does not correlate well with cough generation.18 Peak cough flow is considered the best noninvasive assessment of cough function.133 A measured peak cough flow of less than 160 L/min during acute respiratory illness or 270 L/min in the absence of respiratory illness is associated with poor cough and a high risk of respiratory infection. Interventions to improve cough include teaching the caregivers manually assisted cough or use of manual insufflation—a one-way valve mouthpiece circuit combined with a self-inflating resuscitator bag used to insufflate the lungs by applying a series of breath-stacking maneuvers. When bulbar function is good but the patient has significant expiratory muscle weakness, mechanical insufflation–exsufflation (Cough Assist Device, Respironics, Inc., Murrysville, PA) can be used to augment cough function (see Chapter 34).
ALS often affects the inspiratory muscles, including the diaphragm and external intercostal muscles. This leads to a reduction in respiratory muscle strength, restrictive lung disease, and ultimately carbon dioxide retention and frank respiratory failure. In rare cases, respiratory muscle dysfunction leading to respiratory failure can be the presenting clinical picture for the ALS patient. The symptoms of respiratory muscle insufficiency usually occur gradually over time and can defy diagnosis.
Pulmonary function testing is invaluable in assessing the level of respiratory impairment and should be used to follow disease progression and assess prognosis in ALS. Respiratory muscle strength measures appear to correlate with dyspnea in ALS patients even when there is near-normal vital capacity.72,79,133 In patients in whom oral bulbar weakness limits the ability to accurately measure maximum inspiratory pressure by mouth, sniff nasal inspiratory pressure has been found to be a reliable alternative.133 Both maximal inspiratory pressure (MIP) and sniff nasal inspiratory pressure can be inaccurate measures of inspiratory muscle strength when significant bulbar weakness affects the test maneuver because of inadequate oral seal or upper airway collapse.
Nocturnal hypoventilation and sleep-disordered breathing are common problems for patients with ALS. These can occur even when respiratory muscle function is only mildly affected and daytime gas exchange remains normal.14,25 Neural output to the respiratory muscles decreases during sleep. Even mild muscle weakness coupled with the normal decreases in ventilatory drive can result in nocturnal hypoventilation and disturbed sleep architecture. Nocturnal pulse oximetry or formal sleep studies can be very helpful in elucidating sleep-disturbed breathing in these patients.
Predicting when respiratory failure will occur in the patient with ALS is important to plan appropriate clinical interventions and to help patients and their families address crucial decisions concerning long-term mechanical ventilation and end-of-life issues. Unfortunately, accurately predicting impending respiratory failure is a difficult task. Assessing symptoms of respiratory insufficiency such as dyspnea and orthopnea on each visit is important. Objective measurements of pulmonary function can be helpful but are not entirely predictive of either impending respiratory failure or death. Arterial blood gases are usually not helpful because PaCO2 can be maintained until immediately before respiratory failure. Most authors agree that severe restrictive lung disease with an FVC of less than 50% should prompt careful discussions with the patient concerning the type of medical interventions wanted in the event of respiratory failure.18 FVC should be measured in both the seated and the supine positions because it is often lower in the supine position and contributes to nocturnal hypoventilation. This can easily be performed in the clinic with a portable spirometer. Patients with oral muscle weakness might need to perform spirometry maneuvers through an air cushion facemask to obtain reliable measurements.
Mechanical ventilator support with noninvasive positive pressure ventilation (NPPV) has been shown to be effective in improving quality and duration of life.11,25,82,88 Improvements in cognitive function have been shown in ALS patients receiving nocturnal NPPV.98 Recently a randomized controlled trial of noninvasive ventilation was done in a cohort of ALS patients measuring survival and quality of life.82 Ninety-two patients were assessed every 2 months and randomly assigned to noninvasive ventilation or standard care when either orthopnea developed with a maximum inspiratory pressure of less than 60% of predicted, or when symptomatic hypercarbia occurred. NPPV improved quality of life and survival in all patients without bulbar symptoms, as well as in a subset of patients with mild bulbar symptoms. In patients with more severe bulbar symptoms, NPPV produced some improvement in quality of life but did not improve survival.
The Practice Parameters of the American Academy of Neurology suggest that all patients with ALS and respiratory symptoms or an FVC less than 50% predicted should be offered the use of NPPV.91 NPPV is usually initiated at night because of the high frequency of sleep-disordered breathing. Patients will often progress to using NPPV during the day as their disease progresses. Portable daytime NPPV can be most easily provided by using a bilevel pressure ventilator in conjunction with a less obtrusive interface, either a nasal cannula or nasal pillow interface. A small subset of patients with slow-progressing limb-onset disease and no bulbar symptoms can benefit from portable daytime mouthpiece ventilation (MPV). Portable MPV can be supported using a bilevel pressure ventilator but is most effectively administered using a pressure-triggered volume-cycled home ventilator. The benefits of MPV include progressive ventilatory support and sigh and cough augmentation by the use of breath-stacking maneuvers. Unfortunately, NPPV is only a temporizing measure. Most patients with ALS will at some point develop bulbar symptoms that are severe enough that they will be unable to continue use of NPPV because of aspiration pneumonia or failure of NPPV to ventilate the patient effectively despite its constant use. At this point, consideration for invasive ventilation might be the only option for continued survival.
Invasive ventilation involves placement of a tracheostomy or laryngeal diversion for direct airway access, and use of a small, usually volume-cycled home ventilator. A laryngeal diversion (laryngotracheal separation) is an alternative procedure, in which the proximal trachea is either oversewn or hooked side on end into the esophagus and the distal trachea is brought out through a stoma in the neck. This procedure has the advantage of completely preventing aspiration, although phonation is no longer possible.36 Tracheostomy is clearly a life-prolonging intervention, and patient survival can be extended indefinitely. Invasive ventilation has no effect on the progression of the disease, however, and patients can develop complete paresis of all muscles, including the extraocular muscles that produces a “locked-in” syndrome in which communication is no longer possible. The costs of invasive ventilation to patients and families in both financial and emotional terms are significant.17 Family members provide much of the care for these patients at home and might have to relinquish employment outside the home to do so. Despite this, many patients with ALS report a good quality of life while receiving mechanical ventilation.18
Most patients who undergo invasive mechanical ventilation do so in the setting of emergent hospitalization without having planned in advance for this eventuality. Oppenheimer103 reviewed experience with 50 ALS patients on invasive mechanical ventilation and found that only 4 (8%) of the patients had chosen tracheostomy in advance, before acute respiratory failure and emergent intubation.
Depression and Anxiety
Reactive clinical depression is expected in patients diagnosed with MND.70 Once the diagnosis is confirmed, patients should be counseled with respect to the prognosis to allow time for grieving, anger, and ultimately “acceptance” of their disease, which is important for the mental well-being of patients and their families.85,101 The practitioner should keep in mind that the time around diagnosis is often associated with high levels of anxiety that can undermine quality of life.135 Antidepressant medicine should be offered to patients because it can provide assistance with mood elevation, appetite stimulation, and sleep.78
Good family, social, and religious support systems are all helpful and should be encouraged. The patient should be referred to a support group. The most prominent consumer-driven organizations facilitating support groups for people with ALS are the MDA and ALSA. In addition, referral to a psychiatrist or clinical psychologist with experience in treating depression associated with terminal and/or chronic disease might be necessary.78 Depression in the spouse, significant other, family, or friends should not be overlooked and should be treated as needed.
End-of-Life Issues
The physician must consider that the patient with advanced ALS might not want life-sustaining therapies. Life-sustaining therapy, defined as any artificial device or intervention that compensates for the failure of an organ system which would normally result in death, is the patient’s choice and not the physician’s. The most obvious example of this would be mechanical ventilation, but this also includes artificial hydration and nutrition. In the United States, a mentally competent patient has the right to refuse any prescribed treatment. The physician should always respect and foster the patient’s autonomy and self-direction with respect to these types of interventions.22,103 An advanced health care directive is absolutely necessary.
Early in the disease a social worker should be consulted to help arrange a durable power of attorney to a responsible family member. In most states this can be done by a paralegal for a nominal fee. A living will should be drafted that clearly outlines the patient’s wishes regarding the extent of medical intervention desired.17 This is particularly important with respect to entering hospice-level care. By the time hospice care is being considered, patients typically have spent time with grief and anger, and have begun ”acceptance” of the outcome. Many ALS patients are still hesitant about enrolling in hospice, however, because it implies that the disease has reached the end stage.34,76
Quality of life studies have shown a frequent lack of adequate communication between the physician and patient, as well as a poor perception (both positive and negative) on the part of physicians of the level of quality of life of their patients.101 It takes a great deal of time to explain end-of-life issues, including the available treatment options and choices. Without this investment of time from the clinician, the patient is frequently unaware of the available services that can improve quality of life.
The most appropriate level of care for the ALS patient can change frequently, and patients should be followed closely. An advanced ALS patient is unfortunately often told: “There is nothing that can be done,” when in fact optimizing in-home care and hospice can maximize quality of life for patients and provide a more comfortable and less painful end of life. Krivickas et al.153 documented that most ALS patients appear to receive insufficient in-home care. Of 98 advanced ALS patients studied, only 9 received hospice home care, 24 received nonhospice home care, and 7 received both hospice and nonhospice home care. The remaining 58 patients received no in-home care. Among ALS primary caregivers of the patients with tracheostomy, 42% felt physically unwell, and 48% felt psychologically unwell. The authors concluded that home and hospice care received by ALS patients is typically inadequate because it starts too late to relieve the burden placed on family caregivers. Hospice provides an interdisciplinary team of professionals whose mission is to support the patient and the family through their remaining days together.
Hospice organizations have guidelines for entry of ALS patients and often allow for early entry during the advanced stages of the disease.124 The guidelines often require physicians to make some estimate of life expectancy, which is very difficult to do in ALS and is something for which most physicians are probably ill prepared. Inexperienced clinicians perceive hospice as only for “near-terminal” patients. ALS patients, however, might be in that state for a prolonged period, and hospice care can ease suffering considerably.
Lack of physician knowledge of the services provided by hospice is widespread.29,54 Physicians not familiar with the care of terminal patients might not be comfortable with the aggressive use of opiates and benzodiazepines advocated by hospice clinicians for the control of symptoms in ALS. The physician can find it difficult to give carte blanche orders for effective titration of these types of medications, which ease air hunger and anxiety in the end-stage ALS patient.

1. Abhinav K., Stanton B., Johnston C., et al. Amyotrophic lateral sclerosis in South-East England: a population–based study. The South-East England register for amyotrophic lateral sclerosis (SEALS Registry). Neuroepidemiology. 2007;29(1-2):44-48.
2. Abood M.E., Rizvi G., Sallapudi N., et al. Activation of the CB1 cannabinoid receptor protects cultured mouse spinal neurons against excitotoxicity. Neurosci Lett. 2001;309:197-201.
3. Agosta F., Rocca M.A., Valsasina P., et al. A longitudinal diffusion tensor MRI study of the cervical cord and brain in amyotrophic lateral sclerosis patients. J Neurol Neurosurg Psychiatry. 2009;80:53-55.
4. Aitkens S.G., McCrory M.A., Kilmer D.D., et al. Moderate resistance exercise program: its effects in slowly progressive neuromuscular disease. Arch Phys Med Rehabil. 1993;74(7):711-715.
5. Alonso A., Logroscino G., Jick S.S., et al. Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. Eur J Neurol. 2009;16(6):745-751.
6. Amato A.A., Prior T.W., Barohn R.J., et al. Kennedy disease: a clinicopathologic correlation with mutations in the androgen receptor gene. Neurology. 1993;43(4):791-794.
7. American Academy of Pediatrics (AAP). Poliovirus Infections. In: Pickering L.K., editor. 2006 Red book: report of the Committee on Infectious Diseases, ed 27, Elk Grove Village, IL, American Academy of Pediatrics, 2006.
8. Andersen P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep. 2006;6(1):37-46.
9. Andersen P.M., Nilsson P., Keranen M.-L., et al. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia. Brain. 1997;120:1723-1737.
10. Aramideh M., Ongerboer de Visser B.W. Brainstem reflexes: electrodiagnostic techniques, physiology, normative data, and clinical applications. Muscle Nerve. 2002;26(1):14-30.
11. Bach J.R. Amyotrophic lateral sclerosis: predictors for prolongation of life by noninvasive respiratory aids. Arch Phys Med Rehabil. 1995;76(9):828-832.
12. Banno H., Katsuno M., Suzuki K., et al. Neuropathology and therapeutic intervention in spinal and bulbar muscular atrophy. Int J Mol Sci. 2009;10(3):1000-1012.
13. Banno H., Katsuno M., Suzuki K., et al. Phase 2 trial of leuprorelin in patients with spinal and bulbar muscular atrophy. Ann Neurol. 2009;65(2):140-150.
14. Barthlen G.M., Lange D.J. Unexpectedly severe sleep and respiratory pathology in patients with amyotrophic lateral sclerosis. Eur J Neurol. 2000;7(3):299-302.
15. Battaglia G., Princivalle A., Forti F., et al. Expression of the SMN gene, the spinal muscular atrophy determining gene, in the mammalian central nervous system. Hum Mol Genet. 1997;6:1961-1971.
16. Belsh J.M., Schiffman P.L., editors. Amyotrophic lateral sclerosis: diagnosis and management for the clinician. Armonk, NY: Futura Publishing Co., 1996. 192
17. Benditt J., Smith T., Boitano L.T. Empowering the individual with ALS at the end-of-life: disease-specific advanced care planning. Muscle Nerve. 2001;24:1706-1709.
18. Benditt J.O. Management of pulmonary complications in neuromuscular disease. Phys Med Rehabil Clin N Am. 1998;9(1):167-185.
19. Bensimon G., Doble A. The tolerability of riluzole in the treatment of patients with amyotrophic lateral sclerosis. Expert Opin Drug Saf. 2004;3(6):525-534.
20. Bensimon G., Lacomblez L., Delumeau J.C., et al. A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. J Neurol. 2002;249(5):609-615.
21. Bensimon G., Lacomblez L., Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585-591.
22. Bernat J.L. Ethical and legal issues in the management of amyotrophic lateral sclerosis; in home: a way of maintaining control for the person with ALS/MND. Palliat Med. 1993;7(4 suppl):65-68.
23. Bilsland L.G., Dick J.R., Pryce G., et al. Increasing cannabinoid levels by pharmacological and genetic manipulation delay disease progression in SOD1 mice. FASEB J. 2006;20:1003-1005.
24. Boda B., Mas C., Giudicelli C., et al. Survival motor neuron SMN1 and SMN2 gene promoters: identical sequences and differential expression in neurons and non-neuronal cells. Eur J Hum Genet. 2004;12(9):729-737.
25. Bourke S.C., Tomlinson M., Williams T.L., et al. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomized controlled trial. Lancet Neurol. 2006;5:140-147.
26. Bradley W.G., Anderson F., Gowda N., et al. Changes in the management of ALS since the publication of the AAN ALS practice parameter 1999. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(4):240-244.
27. Brooks B. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limit of amyotrophic lateral sclerosis” workshop contributors. J Neurol Sci. 1994;124:96-107.
28. Brooks B., Sanjak M., Roelke K., et al. Phase 2B randomized dose ranging clinical trial of tamoxifen, a selective estrogen receptor modulator [SERM], in ALS: sensitivity analyses of discordance between survival and functional outcomes with long-term follow-up. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6(suppl 1):118.
29. Brooks B. R: Defining optimal management in ALS: from first symptoms to announcement. Neurology. 1999;53(suppl 5):S1-S3.
30. Brugman F., Veldink J.H., Franssen H., et al. Differentiation of hereditary spastic paraparesis from primary lateral sclerosis in sporadic adult-onset upper motor neuron syndromes. Arch Neurol. 2009;66(4):509-514.
31. Brzustowicz L.M., Lehner T., Castilla L.H., et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5Q11.2-13.3. Nature. 1990;344:540-541.
32. Carter G.T., Abresch R.T., Fowler W.M., et al. Profiles of neuromuscular disease: spinal muscular atrophy. Am J Phys Med Rehabil. 1995;74(5):S150-S159.
33. Carter G.T., Bonekat H.W., Milio L. Successful pregnancies in the presence of spinal muscular atrophy: two case reports. Arch Phys Med Rehabil. 1994;75(2):229-231.
34. Carter G.T., Butler L.M., Abresch R.T., et al. Expanding the role of hospice in the care of amyotrophic lateral sclerosis. Am J Hosp and Pall Care. 1999;16(6):707-710.
35. Carter G.T., Fritz R.C. Pancreatic adenocarcinoma presenting as a monomelic motor neuronopathy. Muscle Nerve. 1997;20:103-105.
36. Carter G.T., Johnson E.R., Bonekat H.W., et al. Laryngeal diversion in the treatment of intractable aspiration in motor neuron disease. Arch Phys Med Rehabil. 1992;73(7):680-682.
37. Carter G.T., Miller R.G. Comprehensive management of amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 1998;9(1):271-284.
38. Centers for Disease Control and Prevention (CDC): The pink book: epidemiology and prevention of vaccine-preventable diseases, ed 10, Washington, DC, 2007, Public Health Foundation.
39. Chancellor A.M., Slattery J.M., Fraser H., et al. The prognosis of adult-onset motor neuron disease: a prospective study based on the Scottish Motor Neuron Disease Register. J Neurol. 1993;240:339-346.
40. Chancellor A.M., Warlow C.P. Adult onset motor neuron disease: worldwide mortality, incidence, and distribution since 1950. J Neurol Neurosurg Psychiatry. 1992;55(12):1106-1115.
41. Chen Y.Z., Bennett C.L., Huynh H.M., et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet. 2004;74:1128-1135.
42. Chio A., Mora G., Calvo A., et al. Epidemiology of ALS in Italy: a 10-year prospective population-based study. Neurology. 2009;72(8):725-731.
43. Chumakov K., Ehrenfeld E. New generation of inactivated poliovirus vaccines for universal immunization after eradication of poliomyelitis. Clin Infect Dis. 2008;47(12):1587-1592.
44. Cozzolino M., Ferri A., Carri M.T. Amyotrophic lateral sclerosis: from current developments in the laboratory to clinical implications. Antioxid Redox Signal. 2008;10:405-443.
45. Czaplinski A., Yen A.A., Simpson E.P., et al. Slower disease progression and prolonged survival in contemporary patients with amyotrophic lateral sclerosis: is the natural history of amyotrophic lateral sclerosis changing? Arch Neurol. 2007;64(3):458-459.
46. Dalbello-Haas V.P., Florence J.M., Kloos A.D., et al. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology. 2007;68:2003-2007.
47. Dalbello-Haas V.P., Florence J., Krivickas L. Therapeutic exercise for people with amyotrophic lateral sclerosis or motor neuron disease. Cochrane Database Syst Rev. 2008;16(2):CD005229.
48. de Carvalho M., Dengler R., Eisen A., et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119:497-503.
49. de Carvalho M., Pinto S., Swash M. Motor unit changes in thoracic paraspinal muscles in amyotrophic lateral sclerosis. Muscle Nerve. 2009;39(1):83-86.
50. Desai D., Pelletier L., Garner M., et al. Increase in poliomyelitis cases in Nigeria. CMAJ. 2008;179(9):930.
51. Desnuelle C., Dib M., Garrel C., et al. A double-blind, placebo-controlled randomized clinical trial of alpha-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. ALS riluzole-tocopherol Study Group, Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(1):9-18.
52. Desport J.C., Preux P.M., Truong C.T., et al. Nutritional assessment and survival in ALS patients. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(2):91-96.
53. Drory V.E., Goltsman E., Reznik J.G., et al. The value of muscle exercise in patients with amyotrophic lateral sclerosis. J Neurol Sci. 2001;191:133-137.
54. Enck R.E. Hospice: the next step. Am J Hosp Palliat Care. 1999;16(2):436-437.
55. Fang F., Valdimarsdottir U., Bellocco R., et al. Amyotrophic lateral sclerosis in Sweden, 1991-2005. Arch Neurol. 2009;66(4):515-519.
56. Farbu E., Rekand T., Gilhus N.E. Post-polio syndrome and total health status in a prospective hospital study. Eur J Neurol. 2003;10(4):407-413.
57. Fink JK: Hereditary spastic paraplegia overview. http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=hsp. Accessed May 15, 2009.
58. Finsterer J. Bulbar and spinal muscular atrophy (Kennedy’s disease): a review. Eur J Neurol. 2009;16(5):556-561.
59. Fishbeck K.H., Ionasecu V., Ritter A.W. Localization of the gene for X-linked spinal muscular atrophy. Neurology. 1986;36:1595.
60. Fornai F., Longone P., Cafaro L., et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2008;105(6):2052-2057.
61. Fowler W.M., Carter G.T. Kraft GH: Role of physiatry in the management of neuromuscular disease. Phys Med Rehabil Clin N Am.. 1998;9(1):1-8., v
62. Francis K., Bach J.R., DeLisa J.A. Evaluation and rehabilitation of patients with adult motor neuron disease. Arch Phys Med Rehabil. 1999;80:951-963.
63. Gallo V., Bueno-De-Mesquita H.B., Vermeulen R., et al. Smoking and risk for amyotrophic lateral sclerosis: analysis of the EPIC cohort. Ann Neurol. 2009;65(4):378-385.
64. Gordon P.H., Cheng B., Katz I.B., et al. The natural history of primary lateral sclerosis. Neurology. 2006;66(5):647-653. 14
65. Guzman M., Sanchez C., Galve-Roperh I. Control of the cell survival/death decision by cannabinoids. J Mol Med. 2001;78(11):613-625.
66. Hampson A.J., Grimaldi M., Axelrod J., et al. Cannabidiol and (−)Δ9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci U S A. 1998;95(14):8268-8273.
67. Hampson A.J., Grimaldi M., Lolic M., et al. Neuroprotective antioxidants from marijuana. Ann N Y Acad Sci. 2000;899:274-282.
68. Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1(8334):1151-1155.
69. Hosler B.A., Brown R.H. Copper/zinc superoxide dismutase mutations and free radical damage in amyotrophic lateral sclerosis. Adv Neurol. 1995;680:41-46.
70. Hunter M.D., Robinson I.C., Neilson S. The functional and psychological status of patients with amyotrophic lateral sclerosis: some implications for rehabilitation. Disabil Rehabil. 1993;15(3):119-126.
71. Ince P.G., Evans J., Knopp M., et al. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology. 2003;60(8):1252-1258.
72. Kang S.W., Bach J.R. Maximum insufflation capacity: vital capacity and cough flows in neuromuscular disease. Am J Phys Med Rehabil. 2000;79(3):222-227.
73. Katz J.S., Wolfe G.I., Andersson P.B., et al. Brachial amyotrophic diplegia: a slowly progressive motor neuron disorder. Neurology. 1999;53:1071-1076.
74. Kidd D., Williams A.J., Howard R.S. Poliomyelitis. Postgrad Med J. 1996;72:641-647.
75. Kilmer D.D., McCrory M.A., Wright N.C., et al. The effect of a high resistance exercise program in slowly progressive neuromuscular disease. Arch Phys Med Rehabil. 1994;75(5):560-563.
76. Krivickas L.S., Shockley L., Mitsumoto H. Homecare of patients with amyotrophic lateral sclerosis (ALS). J Neurol Sci. 1997;152(suppl 1):S82-S89.
77. Kuhnlein P., Gdynia H.J., Sperfeld A.D., et al. Diagnosis and treatment of bulbar symptoms in amyotrophic lateral sclerosis. Nat Clin Pract Neurol. 2008;4(7):366-374.
78. Kurt A., Nijboer F., Matuz T., et al. Depression and anxiety in individuals with amyotrophic lateral sclerosis: epidemiology and management. CNS Drugs. 2007;21(4):279-291.
79. Lechtzin N., Shade D., Clawson L., et al. Supramaximal inflation improves lung compliance in subjects with amyotrophic lateral sclerosis. Chest. 2006;129(5):1322-1329.
80. Li T.M., Alberman E., Swash M. Comparison of sporadic and familial disease amongst 580 cases of motor neuron disease. J Neurol Neurosurg Psychiatry. 1988;51:778-784.
81. Logroscino G., Traynor B.J., Hardiman O., et al. Descriptive epidemiology of amyotrophic lateral sclerosis: new evidence and unsolved issues. J Neurol Neurosurg Psychiatry. 2008;79(1):6-11.
82. Lyall R.A., Donaldson N., Fleming T., et al. A prospective study of quality of life in ALS patients treated with noninvasive ventilation. Neurology. 2001;57(1):153-156.
83. Manna M.M., Kalra M., Wong B., et al. Survival probabilities of patients with childhood spinal muscle atrophy. J Clin Neuromuscul Dis. 2009;10(3):85-89.
84. March of Dimes International Conference on Post-Polio Syndrome: identifying best practices in diagnosis and care. http// www.marchofdimes.com/files/PPSreport.pdf. Accessed June 9, 2009.
85. Meininger V. Breaking bad news in amyotrophic lateral sclerosis. Palliat Med. 1993;7(Suppl 4):37-40.
86. Meininger V., Bensimon G., Bradley W.R., et al. Efficacy and safety of xaliproden in amyotrophic lateral sclerosis: results of two phase III trials, Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(2):107-117.
87. Mennini T., De Paola M., Bigini P., et al. Nonhematopoietic erythropoietin derivatives prevent motoneuron degeneration in vitro and in vivo. Mol Med. 2006;12(7-8):153-160.
88. Meriggioli M.N., Rowin J. Distinguishing clinical and electrodiagnostic features of X-linked bulbospinal neuronopathy. Muscle Nerve. 1999;22:1693-1697.
89. Meyer T., Munch C., van Landeghem F.K., et al. Progressive muscle atrophy. A rarely diagnosed variant of amyotrophic lateral sclerosis. Nervenarzt. 2007;78(12):1383-1388.
90. Miller R.G., Mitchell J.D., Lyon M., et al. Riluzole for amyotrophic lateral sclerosis [ALS/motor neuron disease (MND). Cochrane Database Syst Rev. 2007;24(1):CD001447.
91. Miller R.G., Rosenberg J.A., Gelinas D.F., et al. Practice parameter: the care of the patient with amyotrophic lateral sclerosis (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology: ALS Practice Parameters Task Force. Neurology. 1999;52(7):1311-1323.
92. Mitsumoto H., Davidson M., Moore D., et al. Percutaneous endoscopic gastrostomy (PEG) in patients with ALS and bulbar dysfunction. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:177-181.
93. Mulder D.W., Kurland L.T., Offord K.P., et al. Familial adult motor neuron disease: amyotrophic lateral sclerosis. Neurology. 1986;36:511-517.
94. Neilson S., Robinson I., Alperovitch A. Rising amyotrophic lateral sclerosis mortality in France 1968-1990: increased life expectancy and inter-disease competition as an explanation. J Neurol. 1994;241(7):448-455.
95. Neilson S., Robinson I., Nymoen E.H. Longitudinal analysis of amyotrophic lateral sclerosis mortality in Norway, 1966-1989: evidence for a susceptible subpopulation. J Neurol Sci. 1994;122(2):148-154.
96. Nelson L.M., Matkin C., Longstreth W.T.Jr., et al. Population-based case-control study of amyotrophic lateral sclerosis in western Washington State. II. Diet. Am J Epidemiol. 2000;151(2):164-173. 15
97. Nelson L.M., McGuire V., Longstreth W.T.Jr., et al. Population-based case-control study of amyotrophic lateral sclerosis in western Washington State. I. Cigarette smoking and alcohol consumption. Am J Epidemiol. 2000;151(2):156-163. 15
98. Newsom-Davis I.C., Lyall R.A., Leigh P.N., et al. The effect of non-invasive positive pressure ventilation (NIPPV) on cognitive function in amyotrophic lateral sclerosis (ALS): a prospective study. J Neurol Neurosurg Psychiatry. 2001;71(4):482-487.
99. Nilsson M., Hansson E., Ronnback L. Interactions between valproate, glutamate, aspartate, and GABA with respect to uptake in astroglial primary cultures. Neurochem Res. 1992;17:327-332.
100. Norris F., Sheperd R., Denys E., et al. Onset, natural history and outcome in idiopathic adult motor neuron disease. J Neurol Sci. 1993;118(1):48-55.
101. Nygren I., Askmark H. Self-reported quality of life in amyotrophic lateral sclerosis. J Palliat Med. 2006;9(2):304-308.
102. Ogino S., Leonard D.G., Rennert H., et al. Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet. 2002;110(4):301-307.
103. Oppenheimer E.A. Decision-making in the respiratory care of amyotrophic lateral sclerosis: should home mechanical ventilation be used? Palliat Med. 1993;7(4):49-64.
104. Pasinelli P., Brown R.H. Molecular biology of amyotrophic lateral sclerosis: Insights from genetics. Nat Genet. 2006;7:710-723.
105. Practice advisory on the treatment of amyotrophic lateral sclerosis with riluzole. report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 1997;49(3):657-659.
106. Pradas J., Finison L., Andres P.L., et al. The natural history of amyotrophic lateral sclerosis and the use of natural history controls in therapeutic trials. Neurology. 1993;43(4):751-755.
107. Qureshi M., Shui A., Dibernado A.B., et al. Medications and laboratory parameters as prognostic factors in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2008;9(6):369-374.
108. Rekend T., Albrektsen G., Langeland N., et al. Risk of symptoms related to late effects of poliomyelitis. Acta Neurol Scand. 2000;101(3):153-158.
109. Ringel S.P., Murphy J.R., Alderson M.K., et al. The natural history of amyotrophic lateral sclerosis. Neurology. 1993;43(7):1316-1322.
110. Rosen D.R., Siddique T., Patterson D., et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59-62.
111. Rosenfeld M.R., Posner J.B. Paraneoplastic motor neuron disease. Adv Neurol. 1991;56:445-459.
112. Rothstein J.D., Patel S., Regan M.R., et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433(7021):73-77.
113. Rowland L.P. How amyotrophic lateral sclerosis got its name. The clinical-pathologic genius of Jean-Martin Charcot. Arch Neurol. 2001;58:512-515.
114. Salinas S., Proukakis C., Crosby A., et al. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7(12):1127-1138.
115. Sharma K.R., Kent-Braun J.A., Majumdar S., et al. Physiology of fatigue in amyotrophic lateral sclerosis. Neurology. 1995;45(4):733-740.
116. Sharma K.R., Miller R.G. Electrical and mechanical properties of skeletal muscle underlying increased fatigue in patients with amyotrophic lateral sclerosis. Muscle Nerve. 1996;19:1391-1400.
117. Shaw C.E., Al-Chalabi A. Susceptibility genes in sporadic ALS: Separating the wheat from the chaff by international collaboration. Neurology. 2006;67:738-739.
118. Shaw P.J. Molecular and cellular pathways of neurodegeneration in motor neuron disease. J Neurol Neurosurg Psychiatry. 2005;76:1046-1057.
119. Siddique T., Deng H. Genetics of ALS. Hum Mol Genet. 1996:1465-1470.
120. Singer M.A., Statland J.M., Wolfe G.I., et al. Primary lateral sclerosis. Muscle Nerve. 2007;35(3):291-302.
121. Sobue I., Saito N., Iida M., et al. Juvenile type of distal and segmental muscular atrophy of the upper extremities. Ann Neurol. 1978;3:429-432.
122. Sperfeld A.D., Hanemann C.O., Ludolph A.C., et al. Laryngospasm: an underdiagnosed symptom of X-linked spinobulbar muscular atrophy. Neurology. 2005;64(4):753-754.
123. Standards and Accreditation Committee Medical Guidelines Task Force. Medical guidelines for determining prognosis in selected non-cancer diseases. J Nat Hosp Org. 1996;11:47-63.
124. Stanton B.R., Shinhmar D., Turner M.R., et al. Diffusion tensor imaging in sporadic and familial (D90A SOD1) forms of amyotrophic lateral sclerosis. Arch Neurol. 2009;66(1):109-115.
125. Strand E.A., Miller R.M., Yorkston K.M., et al. Management of oral-pharyngeal dysphagia symptoms in amyotrophic lateral sclerosis. Dysphagia. 1996;11:129-139.
126. Stubgen J- P. Neuromuscular disorders in systemic malignancy and its treatment. Muscle Nerve. 1995;18:636-648.
127. Suzuki K., Katsuno M., Banno H., et al. Pathogenesis-targeting therapeutics for spinal and bulbar muscular atrophy (SBMA). Symposium: Clinicopathological aspects of neuromuscular disorders-a new horizon. Neuropathology. 2009;29(4):509-516.
128. Swoboda K.J., Prior T.W., Scott C.B., et al. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann Neurol. 2005;57(5):704-712.
129. Tartaglia M.C., Rowe A., Findlater K., et al. Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis: examination of symptoms and signs at disease onset and during follow-up. Arch Neurol. 2007;64(2):232-236.
130. Thornton F.J., Fotheringham T., Alexander M., et al. Amyotrophic lateral sclerosis: enteral nutrition provision–endoscopic or radiologic gastrostomy? Radiology. 2002;224(3):713-717.
131. Traynor B.J., Bruijn L., Conwit R., et al. Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology. 2006;67(1):20-27.
132. Trebbia G., Lacombe M., Fermanian C., et al. Cough determinants in patients with neuromuscular disease. Respir Physiol Neurobiol. 2005;146(2-3):291-300.
133. Veltema A.N., Roos R.A., Bruyn G.W. Autosomal dominant adult amyotrophic lateral sclerosis: a six generation Dutch family. J Neurol Sci. 1990;97:93-115.
134. Vignola A., Guzzo A., Calvo A., et al. Anxiety undermines quality of life in ALS patients and caregivers. Eur J Neurol. 2008;15(11):1231-1236.
135. Visser J., van den Berg-Vos R., Franssen H., et al. Disease course and prognostic factors of progressive muscular atrophy. Arch Neurol. 2007;64:522-528.
136. Wang R., Zhang D. Memantine prolongs survival in an amyotrophic lateral sclerosis mouse model. Eur J Neurosci. 2005;22(9):2376-2380.
137. Weydt P., Hong S., Witting A., et al. Cannabinol delays symptom onset in SOD1 transgenic mice without affecting survival. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:182-184.
138. Wijesekera L.C., Leigh P.N. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3.
139. Wirth B., Brichta L., Schrank B., et al. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet. 2006;119(4):422-428.
140. Zhang Z., Lotti F., Dittmar K., et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585-600.
Abel E.L. Football increases the risk for Lou Gehrig’s disease, amyotrophic lateral sclerosis. Percept Mot Skills. 2007;104(3 Pt 2):1251-1254.
Amtmann D., Weydt P., Johnson K.L., et al. Survey of cannabis use in patients with amyotrophic lateral sclerosis. Am J Hosp Palliat Care. 2004;21(2):95-104.
Andreadou E., Christodoulou K., Manta P., et al. Familial asymmetric distal upper limb amyotrophy (Hirayama disease): report of a Greek family. Neurologist. 2009;15(3):151-160.
Angelov D.N., Waibel S., Guntinas-Lichius O., et al. Therapeutic vaccine for acute and chronic motor neuron diseases: implications for amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2003;100(8):4790-4795. 15
Attarian S., Vedel J.P., Pouget J., et al. Progression of cortical and spinal dysfunctions over time in amyotrophic lateral sclerosis. Muscle Nerve. 2008;37(3):364-367.
Bach J.R. Amyotrophic lateral sclerosis: communication status and survival with ventilatory support. Arch Phys Med Rehabil. 1993;72(6):343-349.
Baker D., Pryce G., Croxford J.L., et al. Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature. 2000;404(6773):84-87.
Bdnf Study. GROUP: A controlled trial of recombinant methionyl human BDNF in ALS: The BDNF Study Group [Phase III]. Neurology. 1999;52(7):1427-1433.
Beal M. Mitochondria and the pathogenesis of ALS. Brain. 2000;123:1291-1292.
Beck M., Flachenecker P., Magnus T., et al. Autonomic dysfunction in ALS: a preliminary study on the effects of intrathecal BDNF. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6(2):100-103.
Beghi E., Millul A., Logroscino G., et al. SLALOM GROUP: Outcome measures and prognostic indicators in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2008;9(3):163-167.
Bello-Haas V.D., Florence J.M., Kloos A.D., et al. A randomized controlled trial of resistance exercise in individuals with ALS. Neurology. 2007;68(23):2003-2007.
Boireau A., Meunier M., Doble A. 3-Nitropropionic acid exacerbates [3H]GABA release evoked by glucose deprivation in rat striatal slices. J Pharm Pharmacol. 1996;48(1):85-89.
Boireau A., Meunier M., Imperato A. Ouabain-induced increase in dopamine release from mouse striatal slices is antagonized by riluzole. J Pharm Pharmacol. 1998;50(11):1293-1297.
Boitano L.J., Jordan T., Benditt J.O. Noninvasive ventilation allows gastrostomy tube placement in patients with advanced ALS. Neurology. 2001;56(3):413-414.
Borasio G.D., Gelinas D.F., Yanagisawa N. Mechanical ventilation in amyotrophic lateral sclerosis: a cross-cultural perspective. J Neurol. 1998;245(Suppl 2):S7-12.
Borasio G.D., Robberecht W., Leigh P.N., et al. A placebo-controlled trial of insulin-like growth factor-I in amyotrophic lateral sclerosis. European ALS/IGF-I Study Group, Neurology. 1998;51(2):583-586.
Bosboom W.M., Vrancken A.F., van den Berg L.H., et al. Drug treatment for spinal muscular atrophy types II and III. Cochrane Database Syst Rev. 2009;(1):CD006282.
Brown I.R. Heat shock proteins and protection of the nervous system. Ann N Y Acad Sci. 2007;1113:147-158.
Canu W., Billiard M., Baldy-Mouliner M. Fasting plasma and CSF amino acid levels in ALS. Acta Neurol Scand. 1993;88(1):51-55.
Carter G.T., Rosen B.S. Marijuana in the management of amyotrophic lateral sclerosis. Am J Hosp Palliat Care. 2001;18(4):264-270.
Chari A., Paknia E., Fischer U. The role of RNP biogenesis in spinal muscular atrophy. Curr Opin Cell Biol. 2009;21(3):387-393.
Chio A., Bottacchi E., Buffa C., et al. Positive effects of tertiary centres for amyotrophic lateral sclerosis on outcome and use of hospital facilities. J Neurol Neurosurg Psychiatry. 2006;77:948-950.
Conda M.T., Oliveira A.S., Quadros A.A., et al. Post-polio syndrome: epidemiologic and prognostic aspects in Brazil. J Proteomics. 2009;71(6):670-681.
Corcia P., Camu W., Praline J., et al. The importance of the SMN genes in the genetics of sporadic ALS. Amyotroph Lateral Scler. 2009;6:1-5.
Cornblath D.R., Kuncl R.W. Nerve conduction studies in amyotrophic lateral sclerosis. Muscle Nerve. 1992;15:1111-1115.
Cronin S., Hardiman O., Traynor B.J. Ethnic variations in the incidence of ALS: a systematic review. Neurology. 2007;68(13):1002-1007.
Cudkowicz M.E., Shefner J.M., Schoenfeld D.A., et al. A randomized, placebo-controlled trial of topiramate in amyotrophic lateral sclerosis. Neurology. 2003;61:456-459.
Cudkowicz M.E., Shefner J.M., Schoenfeld D.A., et al. Trial of celecoxib in amyotrophic lateral sclerosis. Ann Neurol. 2006;60:22-24.
Deda H., Inci M.C., Kurekci A.E., et al. Treatment of amyotrophic lateral sclerosis patients by autologous bone marrow-derived hematopoietic stem cell transplantation: a 1-year follow-up. Cryotherapy. 2009;11(1):18-25.
del Aguila M.A., Longstreth W.T.Jr., McGuire V., et al. Prognosis in amyotrophic lateral sclerosis: a population-based study. Neurology. 2003;60(5):813-819.
Dettmers C.D., Fatepour C. Sympathetic skin response abnormalities in amyotrophic lateral sclerosis. Muscle Nerve. 1993;16:930-934.
Di Marzo, Bisogno T., De Petrocellis L. Endocannabinoids: new targets for drug development. Curr Pharm Des. 2000;6(13):1361-1380.
Ding Y., Wang X.B., Le C.J. A clinical research of Hirayama disease. Zhonghua Nei Ke Za Zhi. 2008;47(12):991-994.
Dumitru D. Central Nervous System Disorders. In: Dumitru D., editor. Electrodiagnostic Medicine. Philadelphia: Hanley and Belfus, 1995.
Ebens A., Brose K., Leonardo E.D., et al. Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron. 1996;17(6):1157-1172. 98
Eisen A. Amyotrophic lateral sclerosis is a multifactorial disease. Muscle Nerve. 1995;18(7):741-752.
Eisen A., Schulzer M., MacNeil M., et al. Duration of amyotrophic lateral sclerosis is age dependent. Muscle Nerve. 1993;16:27-32.
Estevez A.G., Stutzmann J.M., Berbeito L. Protective effect of riluzole on excitatory amino acid-mediated neurotoxicity in motoneuron-enriched cultures. Eur J Pharmacol. 1995;280(1):47-53.
Ferrante K.L., Shefner J., Zhang H., et al. Tolerance of high-dose (3,000 mg/day) coenzyme Q10 in ALS. Neurology. 2005;65:1834-1836.
Ferrante M.A., Wilbourn A.J. The characteristic electrodiagnostic features of Kennedy disease. Muscle Nerve. 1997;20:323-329.
Fields H.L. Relief of unnecessary suffering. In: Fields H.L., Liebeskind J.C., editors. Pharmacologic approaches to the treatment of chronic pain: new concepts and critical issues, vol. 1. Seattle: International Association for the Study of Pain Press, 1994.
Fumagalli E., Funicello M., Rauen T., et al. Riluzole enhances the activity of glutamate transporters GLAST. GLT1 and EAAC1, Eur J Pharmacol. 2008;578(2-3):171-176.
Ganzini L., Johnston W.S., McFarland B.H., et al. Attitudes of patients with amyotrophic lateral sclerosis and their caregivers toward assisted suicide. N Engl J Med. 1998;339(14):967-973.
Garrity-Moses M.E., Teng Q., Liu J., et al. Neuroprotective adeno-associated virus Bcl-xL gene transfer in models of motor neuron disease. Muscle Nerve. 2005;32(6):734-744.
Ghadge G.D., Slusher B.S., Bodner A., et al. Glutamate carboxypeptidase II inhibition protects motor neurons from death in familial amyotrophic lateral sclerosis models. Proc Natl Acad Sci U S A. 2003;100(16):9554-9559.
Gordon P.H., Moore D.H., Gelinas D.F., et al. Placebo-controlled phase I/II studies of minocycline in amyotrophic lateral sclerosis. Neurology. 2004;62:1845.
Gordon P.H., Moore D.H., Miller R.G., et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6:1045-1047.
Gordon P.H., Moore D.H., Miller R.G., et al. (Western ALS Study Group). Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6(12):1045-1053.
Graf M., Ecker D., Horowski R., et al. German vitamin E/ALS Study Group. High dose vitamin E therapy in amyotrophic lateral sclerosis as add-on therapy to riluzole: results of a placebo-controlled double-blind study. J Neural Transm. 2005;112(5):649-660.
Gurney M.E., Fleck T.J., Himes C.S., et al. Riluzole preserves motor function in a transgenic model of familial amyotrophic lateral sclerosis. Neurology. 1998;50(1):62-66.
Hampson A. Cannabinoids as Neuroprotectants Against Ischemia. In: Grotenhermen F., Russo E., editors. Cannabis and cannabinoids: pharmacology, toxicology, and therapeutic potential. New York: Haworth Integrative Healing Press, 2002.
Hatcher L.B. Postpolio syndrome: unusual disease in rural family practice. Can Fam Physician. 1995;41:637-645.
Hausmanowa-Petrusewicz I.A., Friedman I.S., Kowalski J., et al. Spontaneous motor unit firing in spinal muscular atrophy of childhood. Electromyogr Clin Neurophysiol. 1987;27:259-264.
Hausmanowa-Petrusewicz I.A., Karwanska A. Electromyographic findings in different forms of infantile and juvenile proximal spinal muscular atrophy. Muscle Nerve. 1986;9:37-46.
Hausmanowa-Petrusewicz I., Vrbová G. Spinal muscular atrophy: a delayed development hypothesis. Neuroreport. 2005;16(7):657-661. 12
Heemskerk J. High throughput drug screening. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(Suppl 1):19.
http://www.als-mda.org/publications/als/als4_3.html#amgen 2008. Accessed February 3, 2009.
Hu J.H., Zhang H., Wagey R., et al. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J Neurochem. 2003;85:432-442.
Hubert J.P., Delumeau J.C., Glowinski J., et al. Antagonism by riluzole of entry of calcium evoked by NMDA and veratridine in rat cultured granule cells: evidence for a dual mechanism of action. Br J Pharmacol. 1994;113(1):261-267.
Ishigaki A., Aoki M., Nagai M., et al. Intrathecal delivery of hepatocyte growth factor from amyotrophic lateral sclerosis onset suppresses disease progression in rat amyotrophic lateral sclerosis model. J Neuropathol Exp Neurol. 2007;66(11):1037-1044.
Jackson C.E., Gronseth G., Rosenfeld J., et al. Randomized double-blind study of botulinum toxin type B for sialorrhea in ALS patients. Muscle Nerve. 2009;39(2):137-143.
Kadoyama K., Funakoshi H., Ohya W., et al. Hepatocyte growth factor [HGF] attenuates gliosis and motoneuronal degeneration in the brainstem motor nuclei of a transgenic mouse model of ALS. Neurosci Res. 2007;59(4):446-456.
Kasarkis E.J., Dominic K., Oddone E.Z. The National Registry of Veterans with Amyotrophic Lateral Sclerosis: Department of Veterans Affairs Cooperative Studies Program (CSP) #500a. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(uppl 1):129-132.
Kaspar B.K., Lladó J., Sherkat N., et al. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301(5634):839-842.
Keir S.D., Xiao X., Li J., et al. Adeno-associated virus-mediated delivery of glial cell line-derived neurotrophic factor protects motor neuron-like cells from apoptosis. J Neurovirol. 2001;7(5):437-446.
Kennedy W.R., Alter M., Sung J.H. Progressive proximal spinal and bulbar muscular atrophy of late onset: and X-linked, recessive trait. Neurology. 1968;18:671.
Kiaei M., Petri S., Kipiani K., et al. Thalidomide and its analogue lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2006;26:2467-2473.
Kieran D., Kalmar B., Dick J.R., et al. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10(4):402-405.
Kim K., Moore D.H., Makriyannis A., et al. AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur J Pharmacol. 2006;542(1-3):100-105. 7
Klivenyi P., Ferrante R.J., Matthews R.T., et al. Neuroprotective effects of creatine in a transgenic animal model of amyotrophic lateral sclerosis. Nat Med. 1999;5(3):347-350.
Koh S.H., Kim Y., Kim H.Y., et al. Recombinant human erythropoietin suppresses symptom onset and progression of G93A-SOD1 mouse model of ALS by preventing motor neuron death and inflammation. Eur J Neurosci. 2007;25(7):1923-1930.
Kothari M.J., Rutkove S.B. Coexistent entrapment neuropathies in patients with amyotrophic lateral sclerosis. Arch Phys Med Rehabil. 1996;77:1186-1188.
Krivickas L.S. Pulmonary function and respiratory failure. In: Mitsumoto H., Chad D.A., Pioro E.P., editors. Amyotrophic lateral sclerosis. Philadelphia: F.A. Davis; 1998:382-404.
Lacomblez L., Bensimon G., Leigh P.N., et al. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347(9013):1425-1431.
Lambert E. Electromyography in amyotrophic lateral sclerosis. In: Norris F., Kurland L., editors. Motor neuron disease. New York: Grune and Stratton, 1969.
Lambert E., Mulder D. Electromyographic studies in amyotrophic lateral sclerosis. Mayo Clin Proc. 1957;332:441-446.
Lange D.J., Felice K.J., Festoff B.W., et al. Recombinant human insulin-like growth factor-I in ALS: description of a double-blind, placebo-controlled study. North American ALS/IGF-I Study Group. Neurology. 1996;47(4 Suppl 2):S93-S95.
Langmore S.E., Schatz M.A., Olsen N. Fiberoptic endoscopic examination of swallowing safety: a new procedure. Dysphagia. 1988;2:216-219.
Larsen K.E., Benn S.C., Ay I., et al. A glial cell line-derived neurotrophic factor [GDNF]:tetanus toxin fragment C protein conjugate improves delivery of GDNF to spinal cord motor neurons in mice. Brain Res. 2006;1120(1):1-12.
Lawler J.M., Barnes W.S., Wu G., et al. Direct antioxidant properties of creatine. Biochem Biophys Res Commun. 2002;290(1):47-52.
Lechtzin N., Shade D., Clawson L., et al. Supramaximal inflation improves lung compliance in subjects with amyotrophic lateral sclerosis. Chest. 2006;129(5):1322-1329.
Lehmann K., Sunnerhagen K.S., Willen C. Postural control in persons with late effects of polio. Acta Neurol Scand. 2006;113(1):55-61.
Li B., Xu W., Luo C., et al. VEGF-induced activation of the PI3-K/Akt pathway reduces mutant SOD1-mediated motor neuron cell death. Brain Res Mol Brain Res. 2003;111(1-2):155-164.
Lim M., Mace A., Nouraei S.A., et al. Botulinum toxin in the management of sialorrhoea: a systematic review. Clin Otolaryngol. 2006;31(4):267-272.
Lund M.L., Lexell J. Perceived participation in life situations in persons with late effects of polio. J Rehabil Med. 2008;40(8):659-664.
MacKenzie A.E., Jacob P., Surh L., et al. Genetic heterogeneity in spinal muscular atrophy: a link age analysis-based assessment. Neurology. 1994;44:919-924.
Manabe Y., Nagano I., Gazi M.S., et al. Glial cell line-derived neurotrophic factor protein prevents motor neuron loss of transgenic model mice for amyotrophic lateral sclerosis. Neurol Res. 2003;25(2):195-200.
Mandler R.N., Anderson F.A.Jr., Miller R.G., et al. The ALS Patient Care Database: insights into end-of-life care in ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001 Dec;2(4):203-208.
Martin D., Thompson M.A., Nadler J.V. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol. 1993;250(3):473-476.
Mazzini L., Mareschi K., Ferrero I., et al. Autologous mesenchymal stem cells: clinical applications in amyotrophic lateral sclerosis. Neurol Res. 2006;28(5):523-526.
Mazzini L., Mareschi K., Ferrero I., et al. Stem cell treatment in amyotrophic lateral sclerosis. J Neurol Sci. 2008;265(1-2):78-83.
Meijer J.W., van Kujik A.A., Geurts A.C., et al. Acute deterioration of bulbar function after botulinum toxin treatment for sialorrhoea in amyotrophic lateral sclerosis. Am J Phys Med Rehabil. 2008;87(4):321-324.
Miller R., Bradley W., Cudkowicz M., et al. TCH346 Study Group. Phase II/III randomized trial of TCH346 in patients with ALS. Neurology. 2007;69(8):776-784.
Miller R.G., Anderson F., Brooks B.R., et al. Outcomes research in amyotrophic lateral sclerosis: lessons learned from the amyotrophic lateral sclerosis clinical assessment, research, and education database. Ann Neurol. 2009;65(uppl 1):S24-28.
Miller R.G., Petajan J.H., Bryan W.W., et al. A placebo-controlled trial of recombinant human ciliary neurotrophic [rhCNTF] factor in amyotrophic lateral sclerosis. rhCNTF ALS Study Group. Ann Neurol. 1996;39(2):256-260.
Miller T.M., Kaspar B.K., Kops G.J., et al. Virus-delivered small RNA silencing sustains strength in amyotrophic lateral sclerosis. Ann Neurol. 2005;57(5):773-776.
Misra U.K., Kalita J., Mishra V.N., et al. A clinical, magnetic resonance imaging, and survival motor neuron gene deletion study of Hirayama disease. Arch Neurol. 2005;62(1):120-123.
Mitchell J.D., Borasio G.D. Amyotrophic lateral sclerosis. Lancet. 2007;369(9578):2031-2041.
Mitchell J.D., Wokke J.H., Borasio G.D. Recombinant human insulin-like growth factor I [rhIGF-I] for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2007;(4):CD002064.
Mitsumoto H., Floyd A., Tang M.X., et al. Transcranial magnetic stimulation for upper motor neuron involvement in amyotrophic lateral sclerosis (ALS). Suppl Clin Neurophysiol. 2006;59:327-332.
Nagano I., Shiote M., Murakami T., et al. Beneficial effects of intrathecal IGF-1 administration in patients with amyotrophic lateral sclerosis. Neurol Res. 2005;27(7):768-772.
Ochs G., Penn R.D., York M., et al. A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(3):201-206.
Pertwee R.G. Cannabinoid receptor ligands: clinical and neuropharmacological considerations, relevant to future drug discovery and development. Expert Opin Investig Drugs. 2000;9(7):1553-1571.
Plaitakis A., Constantakakis E. Altered metabolism of excitatory amino acids, N-acetyl-aspartate-glutamate in amyotrophic lateral sclerosis. Brain Res Bull. 1993;30:381-386.
Preisler N., Andersen G., Thogersen F., et al. Effect of aerobic training in patients with spinal and bulbar muscular atrophy (Kennedy disease). Neurology. 2009;72(4):317-323.
Raman C., McAllister S.D., Rizvi G., et al. Amyotrophic lateral sclerosis: delayed disease progression in mice by treatment with a cannabinoid. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;5:33-39.
Rekand T., Korv J., Farbu E., et al. Lifestyle and late effects after poliomyelitis. A risk factor study of two populations. Acta Neurol Scand. 2004;109(2):120-125.
Ross M.A., Miller R.G., Berchert L., Parry G. Toward earlier diagnosis of amyotrophic lateral sclerosis: revised criteria: rhCNTF ALS Study Group. Neurology. 1998;50(3):768-772.
Rothstein J.D., Martin I.J., Kuncl R.W. Decreased glutamate transport by the brain and spinal cor d in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464-1468.
Rothstein J.D., Van Kammen M., Levey A.L., et al. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73-84.
Rowland L.P. What’s in a name? Amyotrophic lateral sclerosis, motor neuron disease, and allelic heterogeneity. Ann Neurol. 1998;43:691-694.
Sagot Y., Toni N., Perrelet D., et al. An orally active anti-apoptotic molecule (CGP 3466B) preserves mitochondria and enhances survival in an animal model of motoneuron disease. Br J Pharmacol. 2000;131(4):721-728.
Sejvar J.J., Holman R.C., Bresee J.S., et al. Amyotrophic lateral sclerosis mortality in the United States, 1979-2001. Neuroepidemiology. 2005;25(3):144-152.
Shefner J.M., Cudkowicz M.E., Schoenfeld D., et al. A clinical trial of creatine in ALS. Neurology. 2004;63:1656-1658.
Shoesmith C.L., Findlater K., Rowe A., et al. Prognosis of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry. 2007;78:629-631.
Shoji H., Ken I., Kutoku Y., et al. Two brothers with very late onset of muscle weakness in X-linked recessive spinal and bulbar muscular atrophy. Rinsho Shinkeigaku. 2009;49(1):22-26.
Silani V., Cova L., Corbo M., et al. Stem-cell therapy for amyotrophic lateral sclerosis. Lancet. 2004;364:200-202.
Simmons Z. Management strategies for patients with amyotrophic lateral sclerosis from diagnosis through death. Neurologist. 2005;11(5):257-270.
Sun W., Funakoshi H., Nakamura T. Overexpression of HGF retards disease progression and prolongs life span in a transgenic mouse model of ALS. J Neurosci. 2002;22(15):6537-6548.
Swift T. Commentary: electrophysiology of progressive spinal muscular atrophy. In: Gamstorp I., Sarnat H., editors. Progressive spinal muscular atrophies. New York: Raven Press, 1984.
Swoboda K.J., Scott C.B., Reyna S.P., et al. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS One. 2009;4(5):e5268.
Takahashi N., Shimada T., Ishibashi Y., et al. Cardiac involvement in Kugelberg-Welander disease: a case report and review. Am J Med Sci. 2006;332(6):354-356.
Thomas A.G., Wozniak K.M., Tsukamoto T., et al. Glutamate carboxypeptidase II (NAALADase) inhibition as a novel therapeutic strategy. Adv Exp Med Biol. 2006;576:327-337.
Thoren-Jonsson A.L., Willen C., Sunnerhagen K.S., et al. Changes in ability, perceived difficulty and use of assistive devices in everyday life:a 4-year follow-up study in people with late effects of polio. Acta Neurol Scand. 2009;120(5):324-330.
Tikka T., Fiebich B.L., Goldsteins G., et al. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21(8):2580-2588.
Traynor B.J., Alexander M., Corr B., et al. Effects of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: a population based study, 1996-2000. J Neurol Neurosurg Psychiatry. 2003;74(9):1258-1261.
Turgeon V.L., Houenou L.J. Prevention of thrombin-induced motoneuron degeneration with different neurotrophic factors in highly enriched cultures. J Neurobiol. 1999;38(4):571-580.
Tysnes O.B., Vollset S.E., Larsen J.P., et al. Prognostic factors and survival in amyotrophic lateral sclerosis. Neuroepidemiology. 1994;13(5):226-235.
Vender R.L., Mauger D., Walsh S., et al. Respiratory systems abnormalities and clinical milestones for patients with amyotrophic lateral sclerosis with emphasis upon survival. Amyotroph Lateral Scler. 2007;8(1):36-41.
Verschueren A., Monnier A., Attarian S., et al. Enteral and parenteral nutrition in the later stages of ALS: an observational study. Amyotroph Lateral Scler. 2009;10(1):42-46.
Wang L.J., Lu Y.Y., Muramatsu S., et al. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis J Neurosci. 2002;22(16):6920-6928.
Weishaupt J.H., Bartels C., Pölking E., et al. Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J Pineal Res. 2006;41(4):313-323.
Weisskopf M.G., Morozova N., O’Reilly E.J., et al. Prospective study of chemical exposures and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2009;80(5):558-561.
Wicks P., Abrahams S., Masi D., et al. Prevalence of depression in a 12-month consecutive sample of patients with ALS. Eur J Neurol. 2007 Sep;14(9):993-1001.
Willen C., Sunnerhagen K.S., Grimby G. Dynamic water exercise in individuals with late poliomyelitis. Arch Phys Med Rehabil. 2001;82(1):66-72.
Workman E., Saieva L., Carrel T.L., et al. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum Mol Genet. 2009;10(12):2215-2229.
Yanagisawa N., Tashiro K., Tohgi H., et al. Efficacy and safety of riluzole in patients with amyotrophic lateral sclerosis: double-blind placebo-controlled study in Japan. Igakuno Ayumi. 1997;182:851-866.
Yoshino H., Kimura A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph Lateral Scler. 2006;7(4):241-245.
Zhao P., Ignacio S., Beattie E.C., et al. Altered presymptomatic AMPA and cannabinoid receptor trafficking in motor neurons of ALS model mice: implications for excitotoxicity. Eur J Neurosci. 2008;27(3):572-579.
Zheng C., Nennesmo I., Fadeel B., et al. Vascular endothelial growth factor prolongs survival in a transgenic mouse model of ALS. Ann Neurol. 2004;56(4):564-567.
Zhou B., Chen L., Fan D., et al. Clinical features of Hirayama disease in mainland China. Amyotroph Lateral Scler. 2009;1:1-7.
Zhu S., Stavrovskaya I.G., Drozda M., et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74-78.
Ziemann U., Winter M. Impaired motor cortex inhibition in patients with amyotrophic lateral sclerosis. Evidence from paired transcranial magnetic stimulation. Neurology. 1998;49(5):1292-1298.
Zinman L., Sadeghi R., Gawel M. Are statin medications safe in patients with ALS? Amyotroph Lateral Scler. 2008;9(4):223-228.
Zoccolella S., Beghi E., Palagano G., et al. ALS multidisciplinary clinic and survival. Results from a population-based study in Southern Italy. J. Neurol. 2007;254:1107-1109.
Zoccolella S., Beghi E., Palagano G., et al. Predictors of long survival in amyotrophic lateral sclerosis: a population-based study. J Neurol Sci. 2008;268(1-2):28-32. 15
Zurn A.D., Henry H., Schluep M., et al. Evaluation of an intrathecal immune response in amyotrophic lateral sclerosis patients implanted with encapsulated genetically engineered xenogeneic cells. Cell Transplant. 2000;9(4):471-484.