CHAPTER 14 Motor Disabilities and Multiple Handicapping Conditions
In this chapter, we will discuss two of the more common motor disabilities: cerebral palsy and spina bifida. The term cerebral palsy actually refers to a group of disorders that are characterized by impairments in movement or posture as a result of injury or anomaly of the developing brain. Spina bifida is a generic term for chronic conditions related to defects in the developing neural tube. For each, we discuss definition and classification, prevalence, etiology, strategies for prevention, evaluation, management, and associated health and developmental problems.





CEREBRAL PALSY
Definition and Classification
Classification of cerebral palsy is based on anatomical distribution of the dysfunction, type of neurological involvement, and function. Children may have monoplegia (involvement of a single extremity), hemiplegia (one side involved), diplegia (predominant involvement of legs), or quadriplegia (total body involvement). Motor dysfunction may be asymmetrical or there may be primary involvement of one arm and both legs (triplegia). Neurological type is classified as spastic, dyskinetic (includes dystonia and athetosis), ataxic, and mixed. Some clinicians also include a hypotonic or atonic type of cerebral palsy. The Gross Motor Function Classification System (GMFCS) defines functional status by categorizing children with cerebral palsy into one of five different levels of function primarily on the basis of skills in sitting and walking (Table 14-1).2
| Level I | Walks without restrictions; limitations in advanced skills only |
| Level II | Walks without assistive devices; limited outdoors/community mobility |
| Level III | Walks with assistive mobility devices; limited outdoors/community mobility |
| Level IV | Self-mobility with limitations; transported in wheelchair or use power mobility in outdoors/community |
| Level V | Self-mobility is severely limited, even with the use of assistive technology |
From Palisano R, Rosenbaum P, Walter S, et al: Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol 39:214-223, 1997.
With the current system, it has not been clear when diplegia with significant upper extremity involvement should be classified as quadriplegia or when involvement of one upper extremity and both legs should be classified as asymmetrical quadriplegia, triplegia, or diplegia with superimposed hemiplegia.3 It has also been unclear what various authors have meant by “mixed”; for example, how significant is the spasticity and the dystonia in a child with mixed spastic dystonic quadriplegia? Children with cerebral palsy often have significant oral motor (bulbar) and truncal involvement, and neither is included in the current classification system. Although the GMFCS has become a widely used and validated tool for functional classification,4 it does not address upper extremity function. Finally, studies have varied with regard to which children with known genetic disorders and which young children with acquired brain injury to include or exclude.
In 2004, discussions at the International Workshop on Definition and Classification held in Bethesda, Maryland, led to the publication in 2005 of a proposed new definition and classification system for cerebral palsy (Table 14-2).5 A number of cited factors supported the need for the new definition and classification system, including improved knowledge of brain development, neuroimaging techniques, and measurement tools; the increase in number and quality of outcome studies; and the need to compare children across studies. The authors also noted that the current definition and classification system did not include associated disabilities and chronic health problems, which are common and significantly affect a child’s ability to participate in desired societal roles. The proposed definition is as follows:
TABLE 14-2 Components of a Proposed Classification System for Children with Cerebral Palsy
From Bax M, Goldstein M, Rosenbaum P, et al: Proposed definition and classification of cerebral palsy. Dev Med Child Neurol 47:571-576, 2005.
The proposed classification system addresses most of these issues. The components of the proposed cerebral palsy classification are listed in Table 14-2. Classification requires identification of the predominant tone or movement abnormality and identification of any secondary tone or movement abnormalities. It also requires description of the functional motor abilities in all body areas, although the authors acknowledged the need for research validation of measurement tools for arm and hand function and for speech and oral motor function. This classification eliminates the categorization of diplegia, quadriplegia, and so forth; requires description of the distribution of the dysfunction in all body areas, including the trunk and bulbar regions, and a description of associated health and developmental conditions or deficits; and includes identification of a clearly defined cause if there is one. This addresses a key issue for parents: the cause of the child’s problem. This classification does not resolve the issue of inclusion or exclusion of children with known genetic disorders or the age for inclusion of children with postnatally acquired brain injury. It may exclude some children currently classified as having cerebral palsy who have no or minimal activity limitation. Much work remains to be done to develop a reliable and useful system for both clinical and research practice.
Prevalence
The prevalence of cerebral palsy remains at 1.5 to 2.5 per 1000, and has remained essentially unchanged for a number of decades.6–11 Studies on the prevalence of cerebral palsy have varied in whether they have included children with postnatal causes7,9 and whether they have included children with known causation.11 Improvement in the survival of infants with low birth weight has contributed to an increase in prevalence of cerebral palsy for these children12,13; however, the prevalence of cerebral palsy in children with birth weights of 2500 g or more has remained generally unchanged.14 In one study, investigators reported an increase in the number of children born at term with dyskinetic cerebral palsy.7 In this group, perinatal hypoxic-ischemic encephalopathy (HIE) was present in 71%.
The use of magnetic resonance imaging (MRI) in children with cerebral palsy has expanded the identification of children with developmental defects of the central nervous system. In one study, more than half of the children with cerebral palsy who were born at term had evidence of a prenatal causative factor, and most of them had developmental defects of the brain.15 In addition, 7% to 11% of all children with cerebral palsy who have undergone neuroimaging have been shown to have a central nervous system malformation.16 MRI also has improved the identification of schizencephaly in children with hemiplegia or quadriplegia. Schizencephaly results from probable ischemic injury to the brain at the 10th to 12th weeks of gestation. Only a small number of children with severe bilateral schizencephaly have an apparent genetic disorder.17 Case reports of children with hemiplegia associated with thrombophilic factors18,19 have increased research interest in the more general association of prothrombic factors in children with cerebral palsy. To date, there has been little research support for this association, other than for children who have had neonatal stroke.20–26 Children with identified genetic factors represent only about 1% to 5% of children with cerebral palsy.16,17,27
In a study that excluded postnatal causes, the relative contribution of prenatal factors was 22% and that of perinatal factors was 47%; the remainder of cases were unclassified.9 Of infants with low birth weight in that study, 59% had a perinatal causative factor, primarily periventricular leukomalacia (PVL) and intraventricular hemorrhage. In general, preterm infants with PVL represent about 35% to 40% of children with cerebral palsy.27 These children present with spastic diplegia, and their condition represents a common clinical type of cerebral palsy. Other common clinical types related to perinatal factors are hemiplegia and posthemorrhagic hydrocephalus after premature birth with grade IV intraventricular hemorrhage; congenital spastic hemiplegia and porencephaly visible on MRI scan (prenatal or perinatal factor); dyskinetic quadriplegia with a history of HIE; and athetoid quadriplegia with sensorineural hearing loss and a history of hyperbilirubinemia and kernicterus. Postnatal causative factors are identified in only about 10% of children with cerebral palsy.27
Prevention
Research into the causes of cerebral palsy in preterm infants has focused on two mechanisms of brain damage: insufficient cerebral perfusion and cytokine-mediated damage, potentially triggered by maternal or neonatal infection.12,28 For example, a number of studies have demonstrated an association between chorioamnionitis (infection), inflammatory cytokines, and white matter damage.29–32 Additional studies are needed in order to develop effective prevention strategies—for example, to document that chorioamnionitis actually precedes white matter damage29—and to clarify the role of protective factors such as thyroid hormones or glucocorticoids.12
Prevention strategies for full-term infants have focused on prevention of secondary or reperfusion injury in neonates with HIE. For example, MRI with diffusion-weighted imaging during the first days after birth is contributing to the early identification of full-term infants with significant HIE at high risk for subsequent cerebral palsy, so that neuroprotective strategies can be initiated.33–36 A randomized clinical trial (RCT) of whole-body hypothermia in neonates with moderate and severe HIE has demonstrated reduction in the risk of both death and disability in the experimental group.37
Identification
NEWBORN
In general, cranial ultrasonography and MRI of preterm and full-term infants are more predictive than the clinical examination of the neonate or the identification of individual or combinations of perinatal risk factors. The neurological examination of the newborn is best at demonstrating current status but is poorly predictive of subsequent neurodevelopmental disability. In addition, data from the National Collaborative Perinatal Project demonstrated that perinatal risk factors, whether present alone or in combination, are poor predictors of cerebral palsy.38 Of children with high-risk factors, 97% did not have cerebral palsy, and high-risk factors were present in only 63% of the children with cerebral palsy.39
The finding of persistent ventricular dilatation, cystic PVL, and grades III and IV intraventricular hemorrhage on cranial ultrasonography are highly predictive of subsequent cerebral palsy.40 The timing of cranial ultrasonography in the preterm infant is critical. In one study, cranial ultrasonography detected 29% of abnormalities only after 28 days after birth.41 In the same study, 83% of the children with cerebral palsy at 2 years had major cranial ultrasonographic abnormalities on examinations that were repeated weekly to 40 weeks’ postconceptual age. The practice parameter of the Child Neurology Society recommends cranial ultrasonography at 7 to 14 days and again at 36 to 40 weeks’ postconceptual age.42
The MRI is the imaging study of choice for full-term infants with possible HIE. Diffusion-weighted imaging and magnetic resonance spectroscopy are improving the identification of full-term infants with acute ischemia who are at risk for HIE and subsequent cerebral palsy.33,36 Abnormalities of the thalamus and basal ganglia visible on MRI are highly predictive of subsequent neurodevelopmental problems, including cerebral palsy.38 The role of MRI in preterm infants is evolving. MRI, including diffusion-weighted imaging with attention to the myelination of the posterior limb of the internal capsule at 36 to 40 weeks’ postcenceptual age, may prove to be a valuable addition to cranial ultrasonography in the early detection of PVL and subsequently cerebral palsy. It is superior to cranial ultrasonography in the detection of diffuse PVL in preterm infants40 and may be helpful in evaluation of preterm infants with acute ischemia. The use of MRI diffusion tensor imaging to map white mater pathways and also to identify white matter injury in the neonatal period may be helpful.33 Computed tomography has limited utility in full-term and preterm neonates.40
INFANT AND TODDLER
The accurate identification of infants and toddlers with cerebral palsy depends on repeating examinations at different ages and evaluating the quality of movement patterns, in addition to assessing motor milestones and completing the traditional neurological examination. Important movement patterns include primitive reflexes, such as the asymmetrical tonic neck reflex, the tonic labyrinthine reflex-supine, and the neonatal positive support reflex, which disappear with maturation; and automatic reactions, such as truncal equilibrium responses and parachute responses that appear with increasing age. In addition, it is important to observe the motor patterns used to roll, come to sit, and pull to stand. A number of screening tests incorporate some or most of these items: the Alberta Infant Motor Scale,43,44 the Chandler Movement Assessment of Infants Screening Test,45 the Infant Motor Screen,46 the Milani Comparetti Motor Development Screening Test,47 and the Primitive Reflex Profile.48 Screening tests provide a structure for making accurate observations and can assist with referral decisions.
Other authors have emphasized making careful observations of the generalized movements of very young infants to improve the identification of cerebral palsy.49,50 This approach is based on the work of Heinz Prechtl and involves scoring the video recording of the movement of infants from a few weeks to several months of age. The age for fidgety movements (2 to 4 months) is reported to be the best age for making predictions.50 As with other methods, however, prediction of developmental outcomes are best made on the basis of a longitudinal series of assessments.50
A number of infants continue to present diagnostic challenges. Some preterm infants appear to have spasticity in their legs and a spastic diplegia pattern of cerebral palsy; however, these signs resolve after a year of age. In one study, only 118 of 229 children with a diagnosis of cerebral palsy at 1 year of age still had the diagnosis at 7 years.51 This pattern of development is called transient dystonia. A few infants with mild diplegia, hemiplegia, or extrapyramidal cerebral palsy may be “missed” when examined in the first year. Athetosis and ataxia may not develop until after a year of age. Finally, a few infants present with the signs of cerebral palsy but subsequently are shown to have a progressive disorder. All these issues underline the importance of repeating examinations at different ages.
Evaluation
The diagnostic evaluation of the child with suspected cerebral palsy is best done by an experienced neurodevelopmental team. The goals of the history and physical examination are to confirm the diagnosis, clarify the type and distribution of the neuromotor impairment, review causation and timing, identify associated health issues, and plan for additional evaluations as needed. The practice parameter on the diagnostic assessment of the child with cerebral palsy formulated by the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society recommends neuroimaging in all children with cerebral palsy if the etiology is not established and consideration of coagulation studies in children with hemiplegic cerebral palsy and unexplained hemorrhagic infarction.16 Routine metabolic and genetic studies and routine electroencephalography are not recommended. The MRI is the imaging study of choice. Children who do have central nervous system malformations may benefit from further genetic testing or evaluation. An example is the child with agenesis of the corpus callosum, spastic paraplegia, and mutations in the L1CAM gene.52 An accurate medical diagnosis helps clarify natural history and risk for associated problems such as seizures, helps identify potentially treatable conditions such as dopa-responsive dystonia, and helps identify progressive disorders such as ataxia-telangiectasia.
The practice parameter also recommends screening children for mental retardation, vision impairments, and hearing impairments and monitoring nutrition, growth, and swallowing dysfunction. The revised classification system for children with cerebral palsy5 also includes evaluation of attention and behavior and potential associated impairments: for example, gastrointestinal problems. Table 14-4 lists representative tools for the evaluation of speech and language, gross and fine motor skills, functional motor abilities, overall developmental progress, and attention and behavior. Children with speech and language delay should have formal audiological testing. The results of the diagnostic evaluation form the basis for the development of the initial management plan and may include recommendations for therapy services, as well as adaptive equipment. These are discussed in greater detail later in this chapter.
TABLE 14-4 Representative Instruments for the Developmental Assessment of Children with Cerebral Palsy
DIFFERENTIAL DIAGNOSIS
A number of different disorders may be confused with cerebral palsy, particularly on initial evaluation. These include other static disorders such as habitual toe walking; disorders that manifest neurological progression or deterioration, such as familial spastic paraplegia and ataxia telangiectasia; and potentially treatable disorders, such as dopa-responsive dystonia and glutaric acidemia. The diagnosis of habitual toe walking is one of exclusion.53 Parents of affected children typically report that the children have always walked on their toes. They have no evidence of spasticity, muscle weakness, or other neurological condition. They may or may not present with Achilles tendon contractures and may have a positive family history of toe walking.54 Clinicians may confuse habitual toe walking with mild spastic diplegia; however, electromyography helps differentiate the two in difficult cases.55
A positive family history of cerebral palsy should raise the question of familial or hereditary spastic paraplegia and the need for further genetic evaluation. Dopa-responsive dystonia is often initially misdiagnosed as cerebral palsy. It is an autosomal dominant genetic disorder (arising from mutation in the GCH-1 gene) that is characterized by diurnal variation in motor performance that responds dramatically to low doses of l-dopa.56,57
Management
The impairments of children with cerebral palsy include oral motor dysfunction, joint contractures, hip subluxation and dislocation, and spine changes (scoliosis, kyphosis, and lordosis). Functional problems include feeding dysfunction, delayed and disordered speech, limited independent mobility and written communication, and difficulty performing self-care activities. These impairments, as well as the functional problems of children with cerebral palsy, result from one or more of the following pathophysiological conditions: hypertonicity (spasticity and dystonia) and hypotonia; muscle weakness and easy fatigability; loss of selective motor control; impaired balance; and involuntary movement.
SPECIALIZED EVALUATIONS
Muscle Tone
Hypertonia may result from rigidity, spasticity, dystonia, or a combination of all.58 Hypertonia is defined as abnormally increased resistance to passive movement at a joint. Rigidity is typically not seen in children, and we do not further discuss it. Spasticity is the velocity-dependent increase in resistance to passive movement about a joint, so that resistance increases with increasing speed of the stretch.58 It can be measured with the Ashworth Scale,59 the Modified Ashworth Scale,60 and the Tardieu Scale.61 The Tardieu Scale specifically compares the occurrence of the “catch,” or exaggerated stretch reflex, at low and high speeds. Dystonia refers to involuntary sustained or intermittent muscle contractions that cause twisting or repetitive movement, abnormal postures, or both.58 Dystonia is typically exacerbated by voluntary movement, may vary with posture and type of attempted movement, and is often associated with athetosis. The severity of dystonia can be rated with Barry Albright Dystonia Scale.62 The differentiation of spasticity from dystonia is crucial for treatment planning.
Gait Analysis
The objective measurement of gait parameters in the laboratory has become an essential part of the evaluation for many children with cerebral palsy. Three-dimensional computerized gait analysis can assist with preoperative planning particularly for multilevel orthopedic surgery and can document changes before and after both surgical and nonsurgical treatments.63–67 The components of gait analysis include electromyographic analysis, videotaped assessment of kinematics (joint angles and velocities) and kinetics (joint movements, powers, and ground-reaction forces), force plate analysis, and, at times, oxygen consumption. Standard gait parameters include step and stride length, gait velocity, and cadence. The laboratory gait analysis complements the clinical evaluation of the child.63,68
Quality-of-Life Scales
Quality-of-life measures are particularly important for the families of children with severe cerebral palsy. For example, the treatment goals for a child classified as having level V cerebral palsy on the GMFCS who is receiving intrathecal baclofen therapy may focus on improvement in ease of care and sleep and decrease in pain and discomfort, rather than improvement in functional skills. Pain is a common experience for children and adults with cerebral palsy.69,70 Although there is increasing recognition of the importance of quality-of-life measures and the assessment of pain, there are significant limitations in current measures of quality of life and in health-related quality-of-life scales.71–73 The Child Health Questionnaire is one example of a quality-of-life measure.74–76 Several tools to assess health-related quality of life and pain in children with cerebral palsy are under development.34,72,77,78
Treatment
PHYSICAL AND OCCUPATIONAL THERAPY
The roles of the physical therapist and occupational therapist with children who have cerebral palsy and their families are broad. They provide direct treatment, participate in diagnostic evaluations, recommend braces and assistive devices, and provide training and support to children and caregivers. In general, the indications for physical and occupational therapy treatment services, including regular therapy during the preschool years and subsequent interval physical and occupational therapy services, are to improve strength, endurance, and speed; gait training, particularly with new orthoses or assistive devices; to assess when there is a change in motor skills or emerging skills such as independent walking; postoperative services, when a child is removed from casts after surgery; impending joint contractures; and other situations, such as prescription of a new brace or ambulatory aide.79
The evidence base to support the efficacy of physical and occupational therapy treatment, however, is limited.80,81 A systematic review of 21 studies, including 7 RCTs, by the Treatment Outcomes Committee of the American Academy for Cerebral Palsy and Developmental Medicine revealed no evidence to support the efficacy of neurodevelopmental treatment for young children with cerebral palsy.81 In addition, many treatment studies have provided low levels of evidence of efficacy (i.e., Sackett levels III to V).82 On the other hand, studies have reported the efficacy of physical therapy for muscle strengthening in children with cerebral palsy, including such functional improvements as increase in activity level.83–85 Other studies have reported the benefits of constraint induced therapy, a relatively new therapy for children with hemiplegia.86–89 In this therapy, the child’s unaffected arm is constrained in a cast or by another method, in order to force the child to use the affected hand and arm. The research studies, however, have varied with regard to the method and length of constraint used and outcome measures.
BRACES, ADAPTIVE EQUIPMENT, AND POSITIONING DEVICES
In general, clinicians prescribe lower and upper extremity braces (orthoses) to maintain normal alignment at a joint, prevent deformity, and improve function. There is regional variability in the type of orthoses prescribed for children with cerebral palsy. Unfortunately, the research evidence supporting one brace over another is limited; therefore, clinical experience primarily determines choice. There is considerable evidence documenting the efficacy of ankle-foot orthoses over barefoot walking on gait parameters of children with dynamic equinus.66,80,90–92 In addition, there is limited data on the efficacy of specific types of ankle-foot orthoses (posterior leaf spring, hinged vs. solid) in less impaired children91 and some support for functional improvement with these braces.93
Adaptive seating is crucial in some children with cerebral palsy (GMFCS levels IV and V) for improving function, including feeding and speech; for improving quality of life; for preventing progression of secondary problems such as scoliosis; and for offering an opportunity for safe, independent mobility. Unfortunately, the evidence to support the efficacy of adaptive seating for any of these indications is limited. Studies have documented the benefits of power-drive wheelchairs for children as young as 2 years, who have no other choices for independent mobility.94
TONE MANAGEMENT
Control of hypertonicity or tone management is a significant part of the treatment program for many children with cerebral palsy. Treatment approaches include oral medications, intramuscular injections with botulinum toxin, nerve blocks with phenol or alcohol, intrathecal baclofen, and SDR. Children with marked spasticity and or dystonia are likely to benefit from a combination of these treatments. Decisions are complex and require an experienced multidisciplinary team.95 One goal of early tone management is to prevent orthopedic complications such as flexion contractures in order to avoid the need for subsequent orthopedic surgical procedures. A population-based study from Sweden appears to confirm the appropriateness of this strategy. This study reported a reduction of orthopedic surgery for contracture or skeletal torsion deformity from 40% to 15% in children up to 8 years of age during an aggressive early tone management program.96
Oral Medications
Table 14-5 lists the common oral medications used for the treatment of spasticity and dystonia. These include baclofen, diazepam and other benzodiazepines, dantrolene, and tizanidine and other α2-adrenergic agents for spasticity and levodopa-carbidopa, trihexyphenidyl, and baclofen for dystonia.97 Side effects, including sedation, drowsiness, and weakness, have limited the modest benefits of oral medications. A systematic review of 12 RCTs of oral antispasticity medications concluded that the evidence of efficacy is scarce and weak.98 The authors could make no recommendations to guide clinical practice because of the low methodological quality of the studies, the limited numbers of patients, the short duration of followup, and the failure to include functional outcomes. In a small RCT in India, a single nighttime dose of diazepam significantly reduced tone and improved range of motion in children with cerebral palsy and thus may be of benefit in developing countries with little access to other treatments, such as botulinum toxin and intrathecal baclofen.99 A trial of levodopa-carbidopa is indicated in children with unexplained dystonia, because of the variability in the presentation of children with dopa-responsive dystonia.
TABLE 14-5 Oral Medications for Tone Management in Children with Cerebral Palsy
| Medication | Mechanism of Action |
|---|---|
| Spasticity | |
| Baclofen | GABA agonist |
| Benzodiazepines (diazepam, clonazepam) | GABA agonist |
| Dantrolene | Inhibits calcium release from muscle sarcoplasmic reticulum |
| α2-Adrenergic agonists (tizanidine, clonidine) | Decrease excitatory amino acids, hyperpolarize neurons |
| Gabapentin | Increase brain GABA levels |
| Dystonia | |
| Levodopa-carbidopa | Dopaminergic |
| Trihexyphenidyl | Anticholinergic |
GABA, γ-amino butyric acid.
Adapted from Krach L: Pharmacotherapy of spasticity: Oral medications and intrathecal baclofen. J Child Neurol 16:31-36, 2001.
Botulinum Toxin, Phenol, and Alcohol
Traditionally, phenol and alcohol have been injected into motor points or onto the motor nerves for the reduction of spasticity. These medications cause protein denaturation and axonal degeneration, have an onset of action of hours, and a duration of action of up to 12 months.100,101 Injections may be repeated. Treatment indications include improving the ease of care, improving gait, and treating pain secondary to spasticity. The technical expertise necessary for the procedure and the risk of chronic pain or paresthesia after the procedure limit their use.
Botulinum toxin has become the procedure of choice for neuromuscular blockade because of the ease of administration, low risk of side effects, and rapid onset of action. It interferes with the release of acetylcholine at the neuromuscular junction. The primary limitations to the use of botulinum toxin are the relatively short duration of action (up to 3 months after the initial injection) and the limited number of muscles that can receive injections at one time. Two serotypes (A and B) currently are available for clinical use, and they vary in dosage and duration of action. Consensus dosing guidelines are available.101 In individual studies, investigators have reported significant reduction in spasticity and functional improvement in both the upper and lower extremities. However, systematic reviews of use in the upper extremities and lower extremities have revealed insufficient evidence to support or refute its use.102,103 One group of authors recommends a cautious approach to the use of botulinum toxin injections because the data on long-term outcomes are limited.104
Intrathecal Baclofen
Baclofen is a GABA agonist, and its site of action is the spinal cord. It can be given intrathecally in small doses to maximize benefits with limited side effects. The effects of a single dose of intrathecal baclofen last only a few hours, and so it is given by a continuous-infusion pump. Since initial reports in the early 1990s,105,106 intrathecal baclofen has become widely used for the management of spasticity and dystonia. The Treatment Outcomes Committee of the American Academy for Cerebral Palsy and Developmental Medicine published the results of a systematic review of 14 studies, including one RCT in 2000.107 The review revealed evidence of decreased tone in upper and lower extremities, improved function of upper and lower extremities, improved ease of care and sleep, and decreased pain, as well as decreased truncal tone. Other studies have confirmed the benefits of intrathecal baclofen in children with both spasticity and dystonia.108–111 Despite concerns about the relationship of intrathecal baclofen to progression of hip subluxation and scoliosis and increase in seizures, studies have demonstrated no relationship between intrathecal baclofen and change in seizure frequency112 or hip status.113 Both intrathecal baclofen withdrawal and overdose can be life-threatening emergencies. Table 14-6 reviews the signs and symptoms of baclofen overdose and withdrawal.
TABLE 14-6 Signs and Symptoms of Baclofen Withdrawal and Overdose
| Baclofen Withdrawal | Baclofen Overdose |
|---|---|
Selective Dorsal Rhizotomy
SDR is a neurosurgical procedure for the treatment of spasticity that is not effective for dystonia. It involves severing the dorsal spinal rootlets from the levels of L2 to S1 or S2. However, the number of rootlets cut and other procedural issues has varied significantly from center to center.114 The ideal candidate for SDR appears to be a child who was born prematurely, has spastic diplegia, and is ambulatory with little or no truncal weakness. In the weeks after surgery, most children do manifest significant weakness, and maximum functional improvement does not occur until 6 to 12 months after the procedure. Three RCTs have demonstrated significant reductions in spasticity, and two revealed significant gains in functional skills as measured by the Gross Motor Function Measure, as did a subsequent meta-analysis.115–118
Functional changes after SDR persist over time.119 SDR does not change the need for orthopedic surgery, especially for older children,120 and SDR has no significant effect on the progression of hip subluxation.121 Studies have reported conflicting data on the presence and progression of spinal deformities after SDR.122,123 Of note, the number of children undergoing SDR has significantly fallen concomitantly with an increase in the number of children treated by intrathecal baclofen. There have been few available studies in which researchers compared SDR, intrathecal baclofen, or orthopedic interventions.
ORTHOPEDIC MANAGEMENT
The musculoskeletal problems of children with cerebral palsy include hip subluxation and dislocation, scoliosis and other spinal deformities, flexion contractures, foot and ankle deformities, hand and arm deformities, rotational deformities of the legs, leg length discrepancy, patella alta, osteopenia and fractures, joint pain, and hypertrophic ossification after surgery. Clinical gait abnormalities include the crouched gait and stiff knee gait. Orthopedic surgery is one of the treatment options for most of these issues. Orthopedic surgical procedures are either soft tissue surgical procedures, such as tendon and muscle releases, or bone surgical procedures, such as varus osteotomy of the femur or derotation osteotomy of the tibia. In general, orthopedic surgery is usually delayed until after 5 to 8 years of age, when all aspects of the deformity of the legs may be addressed at one time (multilevel surgery), unless structural issues necessitate earlier surgery to preserve function. For example, lengthening of the Achilles tendon before 8 years of age carries a higher risk for overcorrection,124 and surgery before age 5 years carries the risk of recurrence of the plantar flexion contracture. Traditionally, investigators of surgical outcomes have reported change in the deformity and range of motion but have rarely reported change in function or activity participation.
Hip Subluxation and Dislocation
A common problem for children with spastic diplegia and spastic quadriplegia is hip subluxation and dislocation. As many as 30% of adults with cerebral palsy and untreated hip dislocation have chronic hip pain.125 For this reason, monitoring of the hip status of young children with cerebral palsy is important for detecting progressive hip subluxation early and preventing dislocation and possible pain in the hip. Clinicians monitor children with plain radiographs of the hip, starting at about 18 months of age. The migration percentage, or the percentage of the femoral head that is uncovered by the acetabulum, is the principal measure of hip stability. In hips with a migration percentage of 40% or greater at the time of soft tissue surgery, the migration progresses,126 and in most hips with a preoperative migration percentage of less than 40%, the migration remains reduced. The initial surgical procedure is release of bilateral adductor tendons. Additional procedures may include varus osteotomies of the proximal femur and acetabular augmentation.
Scoliosis
Spinal deformity is a common problem in children with quadriplegia. Hip subluxation and dislocation with an asymmetrical sitting posture may contribute to progression of scoliosis. A spinal curve of 40% is likely to worsen and necessitate surgical stabilization.127 Surgical stabilization is with posterior or combined anterior and posterior instrumentation and fusion. In general, parents and caregivers are pleased with the results of surgery for children with cerebral palsy, particularly with improvement in quality of life and ease of care.128,129 The benefits of surgical stabilization of scoliosis in profoundly involved children, however, remain controversial.
ASSOCIATED PROBLEMS
Table 14-7 lists the associated health problems of children with cerebral palsy.
Adapted from Nickel R: Cerebral palsy. In Nickel RE, Desch LW, eds: The Physician’s Guide to Caring for Children with Disabilities and Chronic Conditions. Baltimore: Paul H. Brookes, 2000, p 146.
Osteopenia
Nonambulatory children with cerebral palsy are at risk for decreased bone mineral density, osteopenia, and recurrent fractures. In one study, 77% of 117 children with moderate and severe cerebral palsy had osteopenia (bone mineral density z-score ≤2 standard deviations), as did all of the participants who were unable to stand and older than 9 years.130 The severity of the cerebral palsy, poor nutritional status, and the use of anticonvulsants increase the risk for osteopenia. The osteopenia in children with cerebral palsy results from the slow rate of growth in bone mineralization and not from loss of bone mineral, as in elderly adults.131 Treatment consists of supplementation with vitamin D and calcium, standing programs,132 and, in rare cases, the use of bisphosphonates. In a small RCT, pamidronate increased bone mineral density by 89% in the experimental group, in comparison with only 9% in the controls.133 The authors noted no major adverse effects.
Oral Motor Dysfunction
Feeding problems are common in children with cerebral palsy and are highly correlated with indicators of poor health and nutrition.134,135 In a study of 3- to 12-year-old children with moderate and severe cerebral palsy, 38% had significantly reduced fat stores; however, standard weight-to-height ratios were poor indices for the reduced fat stores.136 Children with severe oral motor dysfunction may require enteral feeding to maintain adequate nutrition; however, limited information is available on the long-term benefits of gastrostomy feeding. In two systematic reviews, authors reported insufficient evidence for judging the effects of gastrostomy feedings in children with cerebral palsy.137,138 A prospective multicenter cohort study of 57 children with cerebral palsy did demonstrate significant weight gain at 6 and 12 months after gastrostomy placement, and almost all parents reported decreased time in feeding and fewer hospital days.139 The growth of children with cerebral palsy should be monitored with standard anthropomorphic measures, including length, weight, and body mass index; a measure of subcutaneous fat stores, such as the triceps skinfold; and alternative measures of linear growth, such as upper arm length and knee height, as needed.
Gastroesophageal Reflux
Gastroesophageal reflux is common in neurologically impaired children and is also associated with poor nutrition, oral motor dysfunction, and risk for aspiration. The symptoms of gastroesophageal reflux include recurrent vomiting or spitting up, choking and frequent swallowing during feeding, refusal of feedings or apparent early satiety, arching with feeding, torticollis (associated with esophagitis), recurrent respiratory symptoms, episodes of irritability, and frequent nighttime awakenings.140
Gastroesophageal reflux may be ameliorated with small, thickened feedings and careful positioning; however, children with persistent gastroesophageal reflux require medications to decrease gastric acidity, neutralize gastric acid, or increase intestinal motility. Infants with severe gastroesophageal reflux may require a Nissan fundoplication. Because of the risk for symptomatic gastroesophageal reflux, a current controversy is whether children who require placement of a gastrostomy for enteral feeding should undergo fundoplication at the same time. Placement of a percutaneous endoscopic gastrostomy does not appear to increase the risk for gastroesophageal reflux. In one study, 8% of children developed gastroesophageal reflux after percutaneous endoscopic gastrostomy, and it resolved in 38% of the children who had had gastroesophageal reflux preoperatively.141 Percutaneous endoscopic gastrostomy is becoming the gastrostomy of choice because of its low cost, ease of placement, and cosmetic advantage.142
Incontinence, Constipation, and Drooling
The age at successful toilet training is significantly delayed for the majority of children with cerebral palsy,143 and some children continue to have daytime urinary incontinence even though they have established independent bowel control. About one third of children with cerebral palsy have dysfunctional voiding.144 The primary issues are urgency incontinence and, to a lesser extent, hesitancy (i.e., difficulty initiating a urinary stream).144–146 Urodynamic findings include detrusor overactivity, detrusor-sphincter dyssynergia, and low bladder capacity.144 Treatment is individualized and primarily involves use of anticholinergic medications and, in rare cases, intermittent catheterization.
Gastrointestinal problems are common in children with cerebral palsy, and chronic constipation is the most frequent condition identified, with a prevalence of 70% to 90%.147,148 The factors that contribute to constipation include decreased mobility and activity, decreased fluid and fiber intake, difficulty positioning securely on the toilet, side effects of medications, and decreased colonic motility. In one study, all children with constipation and 70% of the children with cerebral palsy without constipation showed an abnormal colonic transit time in at least one segment of the colon.149 Steps in the treatment of chronic constipation and secondary impaction include reviewing positioning/seating for toileting, addressing behavioral issues, making dietary alterations, performing a “clean-out” program for children with impaction (enemas, oral stimulants, or polyethylene glycol), and beginning a daily maintenance program (supplemental fiber and fluid, mineral oil, sorbitol or lactulose, or polyethylene glycol).
Drooling in children with cerebral palsy results from oral motor dysfunction, not from overproduction of saliva.150 Persistent drooling can disrupt school and other day-to-day activities, cause chronic skin irritation, and interfere with social relationships. The treatment of drooling needs to be individualized and includes behavioral approaches, medications, injections of botulinum toxin, and surgical procedures. The goals of treatment are to improve the child’s quality of life and social functioning. Behavioral strategies typically are ineffective when the child is focusing attention on other activities such as schoolwork. The use of medications, botulinum toxin, and possible surgery is usually considered in school-aged children with persistent drooling. In general, anticholinergic medications are the initial treatment, with consideration of botulinum toxin injections and surgery for children who do not respond to medications or who have significant side effects.
Glycopyrrolate (Robinul) is a commonly used medication because it does not appear to have the central nervous system side effects of other anticholinergic medications. A number of studies have reported significant benefit from anticholinergic medications,151–153 including improvement in social functioning.154 However, side effects—primarily constipation, sedation, irritability, and, less frequently, blurred vision and urinary retention—are frequent. The use of intraglandular botulinum toxin injections is a relatively new intervention for drooling. Several studies have documented the effectiveness of these injections.155–157 However, dosage has varied across studies, and the effect persists for only a few months. A single clinical trial has been conducted to compare intraglandular botulinum toxin injections and scopolamine patches.156 The magnitude of response to botulinum toxin was much higher (42% reduction in flow vs. 25%). However, 95% of the children responded to scopolamine, whereas only 49% of the children responded to botulinum toxin. Surgical interventions have included salivary gland excision and salivary duct ligation or rerouting.158–160 There is no consensus on the most appropriate surgical procedure, and postoperative complications are significant. These have included dry mouth with thick saliva, increased caries and other dental problems, and worsening of oral motor skills.161 The data on the use of intraoral appliances to treat drooling are very limited.162
Seizures
In general, the prevalence of epilepsy in children with cerebral palsy varies markedly, depending on the anatomical type of cerebral palsy and whether cerebral palsy is associated with mental retardation. Epilepsy occurs in 20% to 40% of children with mental retardation and cerebral palsy.163 It is more common in children with quadriplegia and more difficult to treat.164 In children with cerebral palsy monitored in a neurology clinic, 60% of children with quadriplegia had intractable epilepsy, in comparison with 27.3% of children with hemiplegia and 16.7% of children with diplegia.165 Newer antiepileptic drugs, procedures such as vagal stimulation, and epilepsy surgery have significantly improved the management of children with cerebral palsy and epilepsy.
Pain
Pain is a significant and understudied problem of children and adults with cerebral palsy. In a study of 100 adults with cerebral palsy, 67 reported one or more chronic pain problems and 19 reported daily pain.166 Similarly, in a study of 43 families, 67% of parents reported that their children had pain within the previous month, and assisted stretching was the daily living activity most often associated with pain.167 In a separate study, 11% of parents with children with cerebral palsy and GMFCS levels III to V reported that their children had daily pain. The pain was correlated with the severity of the motor impairment and with school days missed.168 Assessment of pain in children with cerebral palsy can be difficult, because they may have associated communication or cognitive deficits. McKearnan and colleagues reviewed pain management in detail.69
COMPLEMENTARY AND ALTERNATIVE TREATMENTS
The use of complementary and alternative medicine (CAM) is frequent among children with chronic conditions and disabilities, including cerebral palsy (see Chapter 8E). Of families of children with chronic conditions who received care through a regional center in Arizona, 64% reported that their children used CAM.169 Seventy-six percent of families reported that their children used CAM if their condition was noncorrectable. Similarly, 56% of families attending a cerebral palsy clinic reported that their children used one or more CAM treatments.170 The children with quadriplegia who were nonambulatory used CAM the most often. The study reported massage therapy and aqua therapy as the most frequently used CAM treatments. Table 14-8 lists a number of CAM treatments used by children with cerebral palsy. Unfortunately, there are few rigorous scientific studies of CAM treatments for children with cerebral palsy. Collet and coworkers reported the results of a RCT on the use of hyperbaric oxygen in children with cerebral palsy.171 The researchers reported no significant differences between the experimental and control groups.
TABLE 14-8 Representative Complementary and Alternative Medicine Treatments Used by Families of Children with Cerebral Palsy
The responsibilities of the health care provider are to be familiar with CAM treatments and providers; to provide families with information on the efficacy, safety, and cost of all treatments; and to assist families with evaluation of the effects of a CAM treatment.172–174
DEVELOPMENTAL AND MENTAL HEALTH ISSUES
Table 14-9 lists the common developmental and mental health issues experienced by children with cerebral palsy. Many children have both a physical disability (cerebral palsy) and one or more developmental disabilities. For example, cerebral palsy may be present in association with attention-deficit/hyperactivity disorder and learning disabilities or with mental retardation. Children with cerebral palsy and mental retardation are more likely than those without these conditions to have seizures and other chronic health problems such as gastroesophageal reflux. Adolescents with cerebral palsy are more likely than their peers to report low self-esteem and to be more socially isolated. Although they rate having friends as very important, they have limited contact with friends outside of school and rarely participate in after-school community activities.175,176
TABLE 14-9 Common Developmental and Mental Health Issues in Children with Cerebral Palsy
LIFESPAN ISSUES
A minority of adults with cerebral palsy are fully employed.177,178 Some affected individuals lose the ability to walk, and many report a deterioration in walking ability.177,179 Many do not have access to health insurance and regular health surveillance, although they continue to have problems with neck, back, and joint pain, as well as drooling, dental hygiene issues, constipation, urinary tract infections, and other adult health care issues. The health-related quality of life and the successful participation of individuals with cerebral palsy in all aspects of life depend as much on the treatment of associated health conditions, the development of social skills, and competency in making their own health care decisions as they do on the presence and treatment of motor impairments. It is crucial to take a lifespan approach in working with persons with cerebral palsy to maximize their successful participation and overall quality of life. Preparation for the transition to adult health care, employment, and independent living must begin early in life by encouraging self-care, independence, participation in community activities typical for the child’s age, and the development of self-determination.
SPINA BIFIDA
Definition and Classification
There is limited agreement on how to classify myelomeningocele according to the anatomical or motor functional level of the defect. The relevance of the classification often has to do with the purpose of the study. All authors classify a thoracic defect as high level. Defects at level L1 and L2 are referred to as high lumbar. Most authors refer to defects at L4 and L5 as low lumbar. In some classifications, L3 is included with the high lumbar level; in others, with the low lumbar levels; in yet others it is grouped together with L4 defects to as midlumbar. Most authors agree on using sacral level as another categorical group. According to distribution based on level, approximately 10% to 20% of defects are thoracic, 15% to 20% are high lumbar, 26% to 37% are low lumbar, and 21% to 35% are sacral.180
Prevalence
In the United States, the current prevalence of myelomeningocele is 0.20 per 1000 live births. The prevalence of anencephaly is 0.09 per 1000 live birth.181 The increased awareness of the role of folic acid in reducing the risk of spina bifida has helped to reduce the risk of NTDs. In 1992, the U.S. Public Health Service recommended that women of childbearing age increase consumption of the vitamin folic acid to reduce spina bifida and anencephaly.181a Mandatory fortification of enriched cereal grain products with folic acid by the U.S. Food and Drug Administration began in January 1998. The prevalence of spina bifida decreased by 20% between 1991 and 2001. The prevalence of NTDs decreases from the eastern to western United States.182 The prevalence is lower among African Americans than in white people, and most studies find a higher risk in Hispanic/Latino families.183 A relatively higher prevalence of NTDs in low-income populations may be related to limited access to health care, as well environmental and dietary factors.
Etiology
A combination of multiple risk factors cause NTDs, including dietary, environmental, and genetic factors (Table 14-10).184 Detailed nutritional studies, as well as laboratory research, pointed to folic acid as a likely mediator. Randomized studies conducted in the late 1980s showed that extra intake of folic acid could reduce the risk of NTDs by 50% to 70%.185,186 In countries that enforced enrichment of flour with folic acid, the prevalence of NTDs has declined.183,187 There is also strong evidence for a genetic contribution to the risk of NTD. Prevalence is different among ethnic groups, even when they share a similar environment. In the United States, families of Irish origin have a higher risk of spina bifida, whereas African-American families have a lower risk.188 The recurrence risk is 2% to 4% after a mother has a single child with a NTD. After two affected pregnancies, the risk increases to 11%-15%.184 Studies have identified a variant form of methylenetetrahydrofolate dehydrogenase, 677C-T, as a risk factor for NTDs, but the prevalence of the genotype explains only a small portion of the protective effect of folic acid.189,190 Researchers have found an association of homeobox genes (PAX, PRX, HOX) with NTDs in animals and in some patients.191–194 The understanding of the genetic factors is expected to increase in the near future.
| Risk Factor | Evidence of Risk |
|---|---|
| Nutritional |
Other known risk factors during pregnancy include high fever, valproic acid, carbamazepine, exposure to high doses of vitamin A, maternal diabetes, and obesity.184,195–198 Chromosomal abnormalities, such as trisomies 13 and 18, and a number of other syndromes can manifest with spina bifida.
Evaluation
Currently, screening of pregnant women includes a triple marker screen (α-fetoprotein combined with human chorionic gonadotropin and unconjugated estriol). Routine screening is done ideally between the 15th and 18th weeks of gestation.199 Early diagnosis of open spina bifida or anencephaly can be suspected if the maternal α-fetoprotein level is increased. α-Fetoprotein levels change significantly with gestational age, and the most frequent reason for elevated levels is incorrect dating of the pregnancy. Twin pregnancy can also elevate the α-fetoprotein level. If the level is high, high-resolution ultrasonography should be performed. This study can help to identify other associated abnormalities, such as hydrocephalus, Chiari malformation, and abnormalities of the spine. American College of Obstetricians and Gynecologists guidelines recommend amniocentesis if α-fetoprotein level is high.199a High levels of α-fetoprotein and acetylcholinesterase in amniotic fluid can confirm the diagnosis of NTD. Chromosome analysis should rule out chromosomal abnormalities and aid in prenatal counseling. Routine ultrasonography can detect NTD. Analysis of fetal movements by ultrasonography is not predictive of future function, although the level of the defect is somewhat predictive.200,201 If the spinal defect is located at the thoracic level, it is very likely that the child will have very limited or no movements in the lower extremities. Children with sacral defects have good prognosis for ambulation in most cases. When the defect is in the lumbar area, it is more difficult to determine the prognosis, because one vertebra level higher or one level lower can mean the difference between functional ambulation or no ambulation. MRI may provide a more detailed evaluation of the defect and associated malformations, although it is not recommended as a standard evaluation during pregnancy. In general, surgery for open spina bifida in the fetus has not been effective. Fetal surgery of the spinal defect does appear to reduce the prevalence of hydrocephalus, with no changes in the sensorimotor function.202,203 Investigators are currently evaluating the risk : benefit ratio of a surgery that necessitates two cesarean sections in less than 3 months and the risk of prematurity.204 Fetal surgery to treat hydrocephalus has not yielded satisfactory results. Whenever possible, the delivery of a fetus identified with myelomeningocele should be in a tertiary care center with a readily available experienced team. The benefit of cesarean section over vaginal delivery is controversial.205–207 Once the diagnosis is made, a clinician experienced in myelomeningocele should provide counseling to the family.
Management
INITIAL CARE
After birth and stabilization of cardiopulmonary function, a careful examination should be performed. Sterile gauze should cover the defect, with saline solution to keep it moist. If the child requires placement in the supine position, a donut of sterile gauze may protect the defect. Avoidance of trauma to the sac is important. If the sac is open, it must be closed immediately. When the defect is intact and covered by skin, it can be closed days or weeks later. Initial assessment should include a complete neurological examination. This examination may help predict future motor function, although patients may have temporary loss of movements after the trauma of the spinal cord surgery or may have movements that originate at a spinal level and are not under the control of the cortex. This examination should include the observation of movements in upper and lower extremities and the use of pinpricks to evaluate sensation. The skeletal examination may reveal orthopedic malformations in the spine and lower extremities. These defects are frequent and are the result of the lack of innervations of some groups of muscles. Chromosome analysis and genetic consultation should be performed if the child has other physical abnormalities not related to the defect. Fluorescent in situ hybridization 22q11 analysis is indicated in children with cardiac malformations, cleft palate, or DiGeorge syndrome.208,209 Head ultrasonography or computed tomography should be performed to evaluate for the presence of Arnold-Chiari malformation and hydrocephalus. Daily head circumference measurements should be performed to monitor for the presence of hydrocephalus, the response to shunt insertion, and early detection of shunt malfunction. Symptoms related to Arnold-Chiari malformation, such as feeding or swallowing difficulties or apnea, require special attention. Children may present with early symptoms of neurogenic bladder. The monitoring of urinary output, physical examination to detect bladder distention, and measurement of renal function with creatinine and blood urea nitrogen are important, as is consultation with the urology department. Initial evaluation must include vesicoureterography and renal ultrasonography.210,211 Some urologists advocate the use of urodynamics in the initial evaluation.212 However, spinal cord surgery can temporarily affect bladder dynamics. Monitoring of bowel movements and stool characteristics should include instruction of parents on the risk of constipation. The high prevalence of congenital cardiac defects among children with myelomeningocele suggests the need for echocardiography before discharge.213
INTERDISCIPLINARY CARE
The number and complexity of health issues that require attention underscore the need for a coordinated, multidisciplinary team. Table 14-11 suggests the types of professionals in the team caring for a child with myelomeningocele, as well as main areas of concern. These issues will affect the children at different ages. Neurosurgeons address issues such as closure of the defect and hydrocephalus during the neonatal period. Hydronephrosis necessitates urgent treatment, whereas treatment of urine and bowel incontinence can be deferred until the preschool years. Orthopedic problems rarely necessitate urgent attention, although clubfoot may necessitate early treatment. Control and treatment of joint problems and scoliosis require ongoing followup. Developmental issues may arise at any age. Although severe developmental delay necessitates attention in infants, mild learning problems may not become apparent until adolescence.
TABLE 14-11 Multidisciplinary Participation in Care of Children with Myelomeningocele
| Discipline | Clinical Focus |
|---|---|
| Neurosurgery |
MOTOR FUNCTION
The strength of the movements in the lower extremities allows estimation of the child’s functional level (Table 14-12). Patients with thoracic-level defects have no controlled movements of the lower extremities, and their prognosis for independent ambulation is poor. They may be able to stand with the use of orthoses (parapodium; parawalker) and move with the use of a walker or crutches. Between 5% and 20% of such children may demonstrate household ambulation.180,214 Children with high lumbar motor function of L1, L2, and some L3 have some movements of the hips. The children can ambulate with the use of orthoses: a high-level orthosis, a reciprocating-gait orthosis, or a hip-knee-ankle-foot orthosis. These children need the support of a walker or crutches, and most use a wheelchair for long-distance ambulation. Ambulation is achieved in 52% to 67% of patients with high lumbar or midlumbar defects.180,214,215 The benefit of intensive treatment to achieve ambulation in children with high-level defects is controversial, because older children prefer to use a wheelchair.216,217 Children with L4 motor level defects need low-level braces, either a knee-ankle-foot orthosis or an ankle-foot orthosis, to support their feet; they may later need a wheelchair for long-distance and independent mobility. Children with an L5 motor level defect are functionally independent in most cases, requiring only low-level braces (ankle-foot orthoses). Children with low lumbar defects have a good prognosis; 85% to 95% are able to ambulate.180,214 Children with sacral defects may have weakness of the intrinsic muscles of the feet and have no limitations on ambulation. Motor function can deteriorate with worsening of orthopedic problems, such as scoliosis or contractures, or with neurological injuries caused by shunt complications, such as tethered cord or spasticity.218,219 In a study of 35 adults with sacral defects who had been community ambulators, Brinker and associates found a decline in the ability to walk in 30% of the patients, with 11% of the 35 subjects becoming nonambulators and 13% household ambulators.220
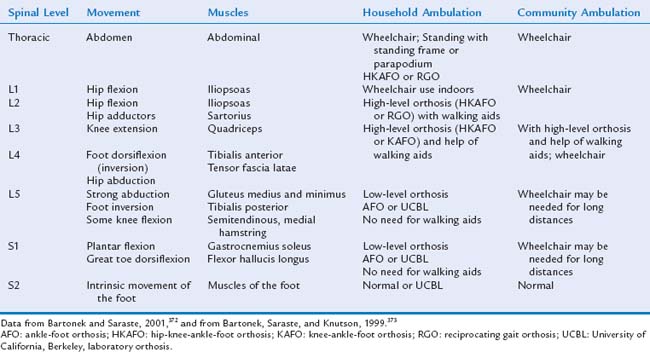
Treatment
PRINCIPLES IN MOTOR MANAGEMENT
The main goal for motor management is to maximize functional abilities. Independent ambulation and self-care are the primary objectives. To achieve these goals, the patient requires good range of motion and an appropriate posture and may need walking aids or a wheelchair. Maintaining range of motion mandates lifelong attention. The appropriate posture depends on the functional level of the myelomeningocele and appropriate orthosis (Table 14-13). Ideally, the treatment plan follows normal developmental stages: upright position, standing, and ambulation. The use of parapodium or standers in children at 12 months with high lumbar and thoracic defects can help achieve a standing posture. Around 2 or 3 years of age, children with high lumbar defects require a high-level orthosis and gait training in order to obtain independent ambulation. The use of a wheelchair provides independent mobility. Periodic physical therapy and occupational therapy assessments should be part of the treatment of all the children with myelomeningocele. Range of motion, muscle strength, and function should be assessed. The provision of regular physical and occupational therapy evaluations of children with myelomeningocele should begin in infancy. Therapy goals include maintenance or improvement of joint range of motion; selection of an appropriate orthosis and assistive devices; monitoring strength and coordination of the upper extremities; training on ambulation and transfers in and out of a wheelchair; selection of an appropriate wheelchair and seating devices; and assisting the family and the patient in solving activities of daily living, such as bathing, toileting, and driving. Periodic assessments must guide treatment and also detect change in function, because multiple neurological and orthopedic complications can negatively affect motor function.
| Condition | Indications | Treatment |
|---|---|---|
| Scoliosis |
ORTHOPEDIC MANAGEMENT
Scoliosis is caused by an imbalance of muscle strength and spine malformations. It is a frequent problem, affecting about 47% to 70% of children with myelomeningocele.221–223 The frequency of scoliosis varies with the level of the spinal defect. It can be as high as 94% among children with thoracic defects and as low as 5% among children with sacral defects.224,225 Once the curvature in scoliosis is greater than 40 degrees, it tends to progress and necessitates surgical treatment. Rapid progression of scoliosis can be a symptom of tethered cord.
Children with thoracic or high lumbar motor function often develop hip dislocation as the strength of the iliopsoas muscle is unopposed. The risk for hip dislocation is 60% to 70% with thoracic defects, 75 to 85% with high lumbar defects, 25% with low lumbar defects, and 3% with sacral defects.214 Children with a poor prognosis for ambulation and no pain do not require surgery.226 Muscle transfer of the iliopsoas or adductors can stabilize the hip and prevent further migration.227,228 Surgical treatment for hip dislocation has yielded mixed results.226 Careful evaluation of the gait pattern, functional abilities, and resources is indicated before surgery. In addition, patients often develop joint contractures as a result of decreased mobility.229 Treatment of contractures is based on the extent that they impair the child’s functioning or interfere with caring for the child.
Some newborns with myelomeningocele have foot malformations related to the level of the defect and muscle innervations.230 Children with sacral defects may have clawed toes and flat feet; those with paralysis below L5 may have calcaneous foot; those with defect below L4 may have equinovarus foot; and those with higher level defects may also have equinovarus.231,232 Nonambulatory children may require surgery to facilitate care and use of shoes. Surgical treatment during infancy carries a high risk of recurrence of the malformation.231
OSTEOPOROSIS MANAGEMENT
Patients with myelomeningocele have decreased bone mineral density, and 22% to 40% develop fractures as a result of osteoporosis.233–237 Although lack of weight bearing can explain osteoporosis in the lower extremities, the etiology of the osteoporosis is not yet understood. For example, investigations of the radial bone revealed significantly lower values of bone density in children with myelomeningocele that are not explained by lack of use.234 Children who are placed in standing position have less osteoporosis and a decreased risk for fractures.238,239 The measurement of bone mineral density can help to identify the patients at greatest risk for multiple fractures. Treatment with oral bisphosphonates can decrease osteoporosis and apparently reduces the incidence of fractures in patients with myelomeningocele.240
NEUROGENIC BLADDER MANAGEMENT
Ureteral Reflux and Hydronephrosis Management
The causes of increased pressure in the bladder include increased activity of the bladder, hypertonic sphincter, and uninhibited contractions of the bladder and sphincter. This increased pressure results in vesicoureteral reflux in 20% of the patients with myelomeningocele. Hydronephrosis occurs in 7% to 30% of such infants. The hyperactivity of the bladder results in poor compliance of the bladder, which can worsen during the first months of life. Between 32% and 45% of children with myelomeningocele and initial normal bladder pressures have abnormal pressures at older ages.241,242 Therefore, normal findings of a urodynamic study after birth do not ensure normal bladder function, and such children require longitudinal monitoring. Most centers conduct periodic evaluations with renal ultrasonography, voiding cystourethrography, and/or urodynamic tests.243 Early treatment of hydronephrosis prevents renal damage. The standard intervention is the use of clean intermittent catheterization (CIC). Some experts advocate starting with CIC in the neonatal period in all children, arguing that they will ultimately require CIC for social continence and an early start will facilitate compliance. The use of anticholinergic medication will increase bladder capacity and decrease hyperactivity of the detrusor. Intravesical instillation of oxybutynin can avoid systemic effects from the medication.244,245 Vesicostomy can be done as a temporary surgery when medical treatment fails.246 Vesicoureteral reflux may resolve after reducing the bladder pressure, although it often requires surgical intervention.
Urinary Infections Management
Incomplete emptying of the bladder and/or ureters results in increased risk for urinary infections. Periodic emptying with CIC helps to reduce the risk of infections. In patients with recurrent urinary tract infections (UTI), the use of prophylactic antibiotics by mouth or by local instillation can help to reduce the number of infections. Detection of UTI can be challenging since abnormal urinalysis with increased white cell or bacteriuria is common in children using CIC. The use of nitrite and leukocyte esterase chemstrip can help as screening tests.247,248 Most centers treat bacteriuria only if the child has other clinical signs or symptoms (fever, dysuria, flank pain, changes in the urinary pattern).243
Social Continence Management
A combination of lack or limited sensation from the bladder, lack of voluntary control of the sphincter and bladder, hypertonic bladder, and/or hypotonic sphincter causes incontinence. Most children with myelomeningocele require an active treatment for social continence. Urinary incontinence can decrease social integration and self perception of subjects with myelomeningocele.249–251 Medical management is usually a combination of CIC and anticholinergic medication. α-Adrenergic agonist medication can help to improve continence when the internal sphincter is hypotonic. About 50% of the children can obtain social continence with medical management.252 Catheterizations should be sufficiently frequent to avoid accidents. Bladder augmentation can increase the bladder capacity when medical treatment fails. This procedure can assist the older child who has a well-established catheterization program, but is unable to obtain continence due to the low volume of the bladder. Augmentation uses a flap obtained from the colon, ileum, or stomach, or by detrusor myotomy to increase bladder volume. Stone formation due to mucous secretion is a frequent complication (18% to 48%) when augmentation is done with colon.253,254 Metabolic acidosis can occur in some children after augmentation.236,255 The use of a ureter for augmentation appears to solve some of the problems from other augmentation techniques.256–258 For patients with weak sphincter, other surgical procedures may be helpful, including the implantation of an artificial sphincter, bladder neck wrap with muscle (sling procedure), or the injection of bulking agents around the neck.259–261 There is no agreement on which procedure has a better outcome. Ileal conduit urinary diversion, a frequent treatment in the past, is now rarely done because of the high number of complications.262,263 If the child is unable to perform self catheterization due to anatomical impairments, a urinary diversion using the appendix (Mitrofanoff procedure) or a tubularization of ileum or sigmoid can be effective in achieving independent continence.264,265 The patient or a caregiver performs catheterizations through a stoma.
Hydronephrosis, chronic pyelonephritis, and associated malformations can affect renal function.266 Approximately 40% of older children and adults with these malformations have abnormal renal function.266,267 Renal transplantation has been successful after renal failure.268,269
NEUROGENIC BOWEL MANAGEMENT
Constipation
Constipation can manifest early in life and necessitates active treatment in most patients with myelomeningocele. When constipation is present, the treatment must be proactive, not delayed until the child has missed a bowel movement or stools for several days. Treatment includes a diet high in fiber and sufficient fluid intake. When the child’s diet has insufficient fiber, it can be added to foods. Clinicians can recommend the use of foods with natural laxative effect, such as prunes, on a routine basis. Some children need laxatives such as polyethylene glycol, bisacodyl, or senna. Bowel training with timed toileting on a daily basis can be effective in achieving continence in cooperative patients who have no constipation and have sufficient abdominal muscles strength. Some patients may require digital stimulation of the rectum to initiate the defecation reflex. Some patients require routine use of suppositories and enemas. The antegrade continence enema procedure may be effective for patients with recalcitrant constipation. The original description by Malone and colleagues consists of a nonrefluxing channel in which the appendix is used to produce a catheterizable colonic stoma.270 If the appendix is not available, options include retubularization of the sigmoid or ileum or a standard gastrostomy button placed in the cecum.265,271,272 A few patients with myelomeningocele have an overactive colon, which results in loose stools and incontinence that is difficult to manage. The antisecretory and antimotility agent loperamide can be helpful for some of these patients. The child and family require an individualized bowel program. Although a systematic approach and a family commitment are keys to a bowel program, they do not guarantee success.
SENSORY FUNCTION AND ITS MANAGEMENT
Children with myelomeningocele lack sensation for touch, pressure, pain, and temperature below the defect. This lack of sensation can be asymmetrical and may not be at the same level as the lack of motor function. Figure 14-1 depicts the dermatomes or areas of the skin supplied by sensory fibers of single posterior spinal roots.
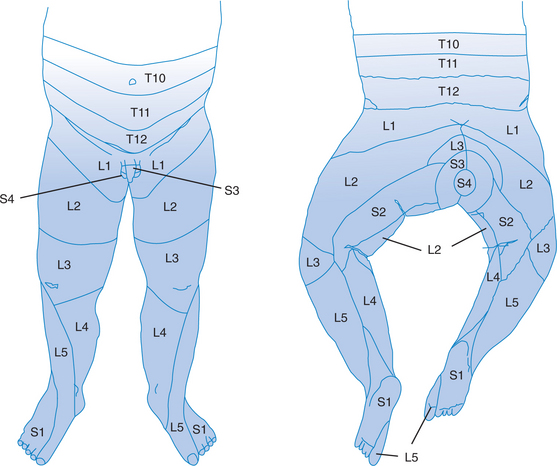
FIGURE 14-1 Dermatomes.
(Data from Foerster A, Haymaker W, Woodhall B: Peripheral Nerve Injury, 2nd ed. Philadelphia: WB Saunders, 1953.)
Pinprick examination can be used periodically to assess the sensory level. Spinal cord complications, such as tethered cord or syringomyelia, can produce loss of sensation, and the confirmation by physical examination can help with diagnosis and treatment decisions. During the sensory examination, the examiner should carefully watch the motor response of the infant. It is important to prevent older children from seeing the pinprick, because they often report positive sensation, even in areas with proven anesthesia. Lack of sensation can result in pressure sores or injuries. In one study, McDonnell found that 35% of adults with myelomeningocele had pressure sores. The location of ulcers and pressure sores varies with the ambulatory status of the child. Children who ambulate in wheelchairs tend to have pressure sores in the gluteal area, whereas those who ambulate upright develop ulcers in the lower extremities.217 In one study, 15% of adults with sacral motor defects lost their ability to ambulate because of complications from skin infections.220 Children do not complain about lack of sensation. From an early age, parents must learn regular care of skin to prevent injuries produced by pressure, cold temperatures, hot temperatures, and friction. Checking the skin daily is important. Older children must learn self-examination. It is critical that patients be instructed to wear new braces and shoes for a very short period, around 20 minutes, and then inspect the skin. Once sores develop, they can take several weeks to heal. In certain situations, patients may require surgical procedures to correct pressure sores.273,274 An estimated $2 million was the cost of the care of patients admitted for treatment of pressure sores in a single institution during a 13-year period.275
NEUROLOGICAL/NEUROSURGICAL MANAGEMENT
Arnold-Chiari Malformation
Arnold-Chiari type II malformation consists of herniation of the tonsils and the contents of the posterior fossa into the foramen magnum. This herniation involves the brainstem, fourth ventricle, and cerebellar vermis. About 5% to 10% of the children with spina bifida present with symptoms related to compression of the brainstem caused by the Arnold-Chiari malformation.276 Anatomopathology studies have demonstrated that compression of the brainstem results in ischemia and hemorrhages, although in some cases, abnormal anatomical findings suggest a developmental anomaly in the brainstem.277 Symptoms include stridor, dysphagia, weakness in the upper extremities, ataxia, and nystagmus.277–279 Mortality is high among patients with abnormal respiratory function that necessitates tracheotomy.277 Shunt evaluation is essential before surgery for posterior fossa decompression is considered.
Some neurosurgeons recommend posterior fossa decompression on an emergency basis as soon the patient shows any symptoms of Arnold-Chiari malformation.280,281 Patients with posterior fossa compression also may require placement of tracheostomy, ventilatory assistance, and gastrostomy. Symptomatic Arnold-Chiari malformation is the most common cause of death in children with spina bifida.
Hydrocephalus
About 85% of children with open spina bifida have hydrocephalus. The treatment is a ventricular shunt, which a neurosurgeon typically places a few days after the child’s birth or simultaneously during the closure of the back during the first surgery. The presence of hydrocephalus is not predictive of future cognitive function except in severe cases, although the accumulation of shunt problems and/or infections increases the risk for cognitive limitations.282 If the shunt is obstructed, the patient often exhibits the classic symptoms of increased intracranial pressure: bulging fontanelle in infants and lethargy, headaches, and vomiting in older children. A shunt can sometimes fail without obvious symptoms, and this failure can manifest with occasional morning headaches or the patient’s awakening with headaches during the night, having occasional morning vomiting, or displaying changes in behavior or learning. Failure occurs in 45% to 60% of placed shunts.283,284 Failure of the shunt can be caused by obstruction (70% to 80%) or infection (20% to 30%).285 Shunt infection rates vary among centers and range from 0% to as high as 15%.283 The shunt has fewer complications when it is placed at the time of the myelomeningocele repair.284 Shunt infection is rare after 6 months of the shunt placement or revision.286 Ventriculoatrial shunts are currently present only in adult patients or patients with intra-abdominal complications that necessitate the removal of a peritoneal shunt. Ventriculoatrial shunts have more complications, including thrombosis of the superior vena cava, pulmonary hypertension, and endocarditis.287,288 Patients with ventriculoatrial shunts require prophylaxis for bacterial endocarditis. Annual monitoring is necessary for patients with hydrocephalus, because shunts can fail after several years of normal function or hydrocephalus can decompensate after many years in patients with stable, nonfunctional (i.e., disconnected) shunts.289,290
Tethered Cord
In patients with myelomeningocele, tethered cord can manifest with progressive scoliosis, back pain, changes in bowel or bladder function, increased spasticity, or loss of motor or sensory function.291 The stretching of the spinal cord produces symptoms resulting from impaired oxidative metabolism on the stretched segments of the spinal cord. Spine MRI demonstrates that almost all patients with myelomeningocele have the end of the spinal cord below L2. For this reason, the low setting of the conus medullaris is not diagnostic of tethered cord.292,293 Tethered cord is a common complication in children with spina bifida (15% to 25%). Periodic evaluation of motor and sensory function and evaluation of the spine can facilitate diagnosis. Children with high-level defects present with tethered cord at younger ages, usually before 6 years of age, whereas diagnosis is often later for children with lower level defects.294 Surgical release is effective in reducing symptoms or stopping the progression in 47% to 80% of cases, and results are better when surgery is performed soon after presentation.291,295,296
Syringomyelia
Asymptomatic hydromelia or syringomyelia is found in 50% of spine MRI scans in children with myelomeningocele. On occasion, the syrinx becomes symptomatic, and patients present with complex upper limb weakness and wasting, sensory disturbance, dysphagia, or ataxia.297 Worsening of symptoms should first prompt a careful evaluation of shunt function. Symptoms and radiological findings often improve after shunt revision. Sometimes, the release of tethered cord or posterior fossa decompression can improve the symptoms and resolve the hydromelia. Some patients require shunting of the syrinx to the subarachnoid space, the pleural space, or to the peritoneum.298,299
Seizures
About 16% to 20% of children with myelomeningocele have seizures at some time.300–302 The number of shunt revisions and additional brain anomalies are associated with increased risk for seizures.300,302 Seizures respond well to medication, and 75% of children with seizures can later discontinue treatment.301 Seizures can be the first symptom of central nervous system complications such as infection, bleeding, or shunt malfunction. Chronic headaches affect 55% to 88% of patients with myelomeningocele and may not be caused by shunt malfunction or complications of Arnold-Chiari malformations.303,304
GROWTH AND NUTRITION MANAGEMENT
Evaluation of linear growth in children with spina bifida requires the use of alternative measurements. Poor growth in the lower extremities, contractures, and scoliosis make the measurement of length or height an inaccurate estimate of linear growth. The measurement of arm span is a good alternative. Clinicians may use an arm span growth chart or plot the arm span on a growth chart of height or length. The latter method is less accurate but readily available.305–307 Periodic measurement of arm span helps identify endocrine disorders such as growth hormone deficiency or hypothyroidism.308 The use of weight, height for weight, or body mass index measures are poor indicators of nutritional status, inasmuch as the body proportions are different for children with different levels of myelomeningocele. The use of other measurements such as arm circumference and skin fold can provide a better estimation of nutrition.309–311 Feeding problems in infants with Arnold-Chiari type II symptoms may necessitate gastrostomy tube placement to maintain nutrition or prevent pulmonary aspiration.312,313,221 These children may have a sensitive gag reflex with intolerance to food with texture.
Obesity
Investigators recognized obesity309,314 as another health problem in myelomeningocele since the early 1970s. The risk for obesity has many contributing factors. Children with myelomeningocele require less energy than normal, particularly if they are nonambulatory.315–318 Social isolation and decreased physical activity increase the risk. Sometimes, medical complications such as pressure sores or surgical procedures cause children to decrease their activity further. In children with myelomeningocele, excessive weight has serious consequences. Obesity can result in the loss of their ability to ambulate. In severe cases, obesity may impair the ability to perform such daily life activities as self-catheterization or toileting. In older teenagers and adults, it jeopardizes their independence, as they may need help with transfers from the wheelchair to the bed or toilet. Pediatricians and parents should monitor the child’s weight from infancy, and they should implement treatment as soon as the child is overweight.
Growth Hormone Deficiency
Children with spina bifida and hydrocephalus have increased risk for endocrinological disorders. Researchers estimate the prevalence of growth hormone deficiency to be between 11% and 18%.319 Clinicians should suspect growth hormone deficiency when the arm span is below the third percentile on the growth chart. Routine evaluation of short children with insulin-like growth factor 1 and insulin-like growth factor binding protein 3 can help with early diagnosis.319 Children treated with growth hormone show a response similar to that of children with idiopathic growth hormone deficiency.308,320,321 With treatment, growth velocity and final height can be close to that expected for age.322 Treatment of growth hormone deficiency can precipitate symptoms of tethered cord; therefore, children require frequent monitoring during treatment.323 Hyperthyroidism has a prevalence of about 3%.324
Precocious Puberty
The prevalence of precocious puberty in children with myelomeningocele is 6% to 18%.324–327 Children with both hydrocephalus and myelomeningocele start puberty 2 years before their peers. Careful genital examination and breast examination during the prepubertal years can assist in early diagnosis and treatment. Untreated precocious puberty can lead to short stature, particularly in women.328
REPRODUCTION AND SEXUAL FUNCTION
Women with myelomeningocele appear to have normal fertility. Information about the risk in individuals with myelomeningocele of having a child with NTD is limited. In one study, investigators examined the outcome of 39 pregnancies from 11 men and 11 women with spina bifida. Four offspring had NTD (two with anencephaly and two with open spina bifida).329 Results should be interpreted cautiously, because most patients in 1975 with open spina bifida had a poor prognosis for survival. Other reported risks among pregnant women with myelomeningocele include urinary tract infections, constipation, decreased mobility, and decubitus. Cesarean section can be more challenging in women with bladder augmentation, ileal conduit, or ventriculoperitoneal shunt.
Sexual Function
Information regarding sexual function and reproduction is scant.329a–329d In general, patients with lower spinal defects have better outcomes. Although the percentage of adults reporting sexual activity is relatively high, study subjects are not representative of all patients with myelomeningocele. In a study by Sandler and associates, who used an objective measurement of penile rigidity, 11 of 15 young men reported erections, whereas objective documentation showed normal erections in only 2 subjects (who had sacral defects), brief and incomplete erections in 7 other subjects, and no response in 6. In this study, patients with lower motor and sensory defects had better outcomes.330 Erection dysfunction can respond to sildenafil.331
CARDIOVASCULAR SYSTEM
The risk for congenital cardiovascular defects is higher in children with myelomeningocele than in a normal population. Ritter and coworkers, in a retrospective study of 105 children who underwent echocardiography before surgery, found that 37% had a cardiac malformation. In their sample, 25% had a secundum atrial septal defect, 9% had ventricular septal defect, and almost 5% had other defects (anomalous pulmonary venous return, tetralogy of Fallot, bicuspid aortic valve, coarctation, and hypoplastic left heart syndrome).213 Kidney damage from repeated pyelonephritis, nephrolithiasis, and hydronephrosis can result in hypertension and/or renal insufficiency.332 In a study of adults with myelomeningocele, 14% had hypertension; 46% had abnormal kidneys as a result of scarring, hydronephrosis, or nephrolithiasis; and 3% had renal failure that necessitated dialysis or kidney transplantation.333 Blood pressure and renal function should be monitored in all patients with myelomeningocele. Patients with ventriculoatrial shunts have an increased risk for pulmonary hypertension. The cause of the pulmonary hypertension may be microembolism from the catheter or an immunological reaction of the pulmonary vessels to proteins from the cerebrospinal fluid.334,335
COGNITIVE AND DEVELOPMENTAL OUTCOME
In general, cognitive function is often poorer for children with high spinal lesions than in those with lower lesions. The prevalence of mental retardation is close to 40% in subjects with thoracic lesions but much lower in the population with sacral lesions. The larger number of brain malformations in children with higher defects mediates the association between level of the lesion and cognitive function.336,337 In the typical cognitive profile of patients with hydrocephalus, verbal skills are better than nonverbal skills.338–340 “Cocktail party syndrome” describes some children with an exaggerated profile of nonverbal learning disability, with verbal expression that significantly exceeds cognitive skills. These children tend to be very friendly, characteristically make inappropriate comments, and appear to understand more than they really do. In clinical practice, it is important to objectively verify verbal comprehension of recommendations or explanations. The interaction between cognitive function and long-term outcome is complex. Low cognitive function is the most significant factor limiting independence.282,341 However, reported quality of life and self-esteem are more closely associated with bowel and bladder functioning.
Attention-deficit/hyperactivity disorder is another frequent diagnosis. The prevalence varies between 34% and 39%.337,342 Response to stimulant medication in children with spina bifida is similar to that in other children with this disorder. Cognitive or behavioral deterioration can occur after central nervous system infections or with chronic shunt malfunction.343 It is therefore important to perform periodic neuropsychological evaluations.
OPHTHALMOLOGICAL ISSUES
Eye motor coordination disorders are very frequent. Strabismus occurs in 42% to 44% of children with spinal lesions; most of these have convergent esotropia.344,345 Optic disc abnormalities, such as papilledema or disc atrophy, occur in 32%. In one study, only 27% of 322 children with myelomeningocele had normal results of eye examinations.344 Not surprisingly, there is a correlation between the degree of mesencephalic abnormalities and problems with eye motor coordination.346,347 The sudden appearance of strabismus, other ocular motility disorders, or papilledema is usually a manifestation of uncontrolled hydrocephalus. Children with myelomeningocele require regular ophthalmological evaluations.
LATEX ALLERGY
Allergy to latex can be a life-threatening condition for some patients with myelomeningocele. It was recognized as a common problem in the early 1990s, after some patients suffered anaphylactic shock during surgery.348,349 The reported prevalence varies, depending on the criteria used to identify patients with allergy. Researchers have reported different prevalence rates of sensitized children with latex allergy, such as 32% to 55% and 15% to 34%.350–352 The clinical manifestation may include redness after contact with objects made from rubber, such as balloons or gloves. Pinprick testing or specific immunoglobulin E or radioallergosorbent testing can help identify asymptomatic children with latex sensitivity. The number of surgical procedures is the major risk factor for latex allergy, along with a personal and family history of atopy.353–355 Because it is important to prevent all contacts with latex products, patients who have had allergic reactions should wear an alert bracelet and carry epinephrine. Patients should be aware of cross-sensitization with fruits from trees, such as kiwi, avocado, and banana.356 Most clinical centers currently avoid the use of latex in the operating rooms and clinics. Avoiding exposure to latex starting with the first surgery may decrease the number of patients with allergy.357
1 National Institute of Child Health and Human Development, National Institutes of Health. Research Plan for the National Center for Medical Rehabilitation Research, NIH Publication No. 93–3509. Washington, DC: U.S. Department of Health and Human Services, Public Health Service, 1993.
2 Palisano R, Rosenbaum P, Walter S, et al. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol. 1997;39:214-223.
3 Shapiro BK. Cerebral palsy: A reconceptualization of the spectrum. J Pediatr. 2004;145(2 Suppl):S3-S7.
4 Morris C, Bartlett D. Gross Motor Function Classification System: Impact and utility. Dev Med Child Neurol. 2004;46:60-65.
5 Bax M, Goldstein M, Rosenbaum P, et al. Proposed definition and classification of cerebral palsy, April 2005. Dev Med Child Neurol. 2005;47:571-576.
6 Prevalence and characteristics of children with cerebral palsy in Europe. Dev Med Child Neurol. 2002;44:633-640.
7 Himmelmann K, Hagberg G, Beckung E, et al. The changing panorama of cerebral palsy in Sweden. IX. Prevalence and origin in the birth-year period 1995–1998. Acta Paediatr. 2005;94:287-294.
8 Kuban KCK, Leviton A. Cerebral palsy. N Engl J Med. 1994;330:188-195.
9 Meberg A, Broch H. Etiology of cerebral palsy. J Perinat Med. 2004;32:434-439.
10 Paneth N. Birth and the origins of cerebral palsy. N Engl J Med. 1986;315:124-126.
11 Williams K, Albermann E. The impact of diagnostic labelling in population-based research into cerebral palsy. Dev Med Child Neurol. 1998;40:182-185.
12 O’Shea TM. Cerebral palsy in very preterm infants: New epidemiological insights. Ment Retard Dev Disabil Res Rev. 2002;8:135-145.
13 Pharoah POD, Cooke T, Rosenbloom L. Acquired cerebral palsy. Arch Dis Child. 1989;64:1013-1016.
14 Munch L. Annotations: Cerebral palsy epidemiology: Where are we now and where are we going? Dev Med Child Neurol. 1992;34:547-555.
15 Truwit CL, Barkovich AJ, Koch TK, et al. Cerebral palsy: MR findings in 40 patients. AJNR Am J Neu-roradiol. 1992;13:67-78.
16 Ashwal S, Russman BS, Blasco PA, et al. Practice parameter: Diagnostic assessment of the child with cerebral palsy: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2004;62:851-863.
17 Granata T, Freri E, Caccia C, et al. Schizencephaly: Clinical spectrum, epilepsy, and pathogenesis. J Child Neurol. 2005;20:313-318.
18 Thorarensen O, Ryan S, Hunter J, et al. Factor V Leiden mutation: An unrecognized cause of hemiplegic cerebral palsy, neonatal stroke, and placental thrombosis. Ann Neurol. 1998;42:372-375.
19 Verdu A, Cazorla MR, Moreno JC, et al. Prenatal stroke in a neonate heterozygous for factor V Leiden mutation. Brain Dev. 2005;27:451-454.
20 Fattal-Valevski A, Kenet G, Kupfermine MJ, et al. Role of thrombophilic risk factors in children with non-stroke cerebral palsy. Thromb Res. 2005;116:133-137.
21 Gibson CS, MacLennan AH, Hague WM, et al. Associations between inherited thrombophilias, gestational age, and cerebral palsy. Am J Obstet Gynecol. 2005;193:1437.
22 Lee J, Croen LA, Backstrand KH, et al. Maternal and infant characteristics associated with perinatal arterial stroke in the infant. JAMA. 2005;293:723-729.
23 Lynch JK, Nelson KB, Curry CJ, et al. Cerebrovascu-lar disorders in children with the factor V Leiden mutation. J Child Neurol. 2001;16:735-744.
24 Mercuri E, Cowan F, Gupte G, et al. Prothrombotic disorders and abnormal neurodevelopmental outcome in infants with neonatal cerebral infarction. Pediatrics. 2001;107:1400-1404.
25 Nelson KB, Lynch JK. Stroke in newborn infants. Lancet Neurol. 2004;3:150-158.
26 Smith RA, Skelton M, Howard M, et al. Is thrombophilia a factor in the development of hemiplegic cerebral palsy? Dev Med Child Neurol. 2001;43:724-730.
27 Paneth N. Etiologic factors in cerebral palsy. Pediatr Ann. 1986;15:193-201.
28 Back SA, Rivkees SA. Emerging concepts in periventricular white matter injury. Semin Perinatol. 2004;28:405-414.
29 Dammann O, Leviton A. Inflammatory brain damage in preterm newborns-Dry numbers, wet lab, and causal inferences. Early Hum Dev. 2004;79:1-15.
30 Hagberg H, Mallard C, Jacobsson B. Role of cytokines in preterm labour and brain injury. BJOG. 2005;112(Suppl 1):16-18.
31 Kent A, Lomas F, Hurrion E, et al. Antenatal steroids may reduce adverse neurological outcome following chorioamnionitis: Neurodevelopmental outcome and chorioamnionitis in premature infants. J Paediatr Child Health. 2005;41:186-190.
32 Willoughby REJr, Nelson KB. Chorioamnionitis and brain injury. Clin Perinatol. 2002;29:603-621.
33 Accardo J, Kammann H, Hoon AHJr. Neuroimaging in cerebral palsy. J Pediatr. 2004;145:S19-S27.
34 Hunt A, Goldman A, Seers K, et al. Clinical validation of the paediatric pain profile. Dev Med Child Neurol. 2004;46:9-18.
35 Khong PL, Tse C, Wong IY, et al. Diffusion-weighted imaging and proton magnetic resonance spectroscopy in perinatal hypoxic-ischemic encephalopathy: Association with neuromotor outcome at 18 months of age. J Child Neurol. 2004;19:872-881.
36 Perlman J. Brain injury in the term infant. Semin Perinatol. 2004;28:415-424.
37 Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med. 2005;353:1574-1584.
38 Palmer F. Strategies for the early diagnosis of cerebral palsy. J Pediatr. 2004;145:S8-S11.
39 Nelson KB, Ellenberg JH. Antecedents of cerebral palsy. Multivariate analysis of risk. N Engl J Med. 1986;315:81-86.
40 Neil JJ, Inder TE. Imaging perinatal brain injury in premature infants. Semin Perinatol. 2004;28:433-443.
41 De Vries LS, Van Haastert IL, Rademaker KJ, et al. Ultrasound abnormalities preceding cerebral palsy in high-risk preterm infants. J Pediatr. 2004;144:815-820.
42 Ment LR, Bada HS, Barnes P, et al. Practice parameter: Neuroimaging of the neonate: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2002;58:1726-1738.
43 Darrah J, Piper M, Watt MJ. Assessment of gross motor skills of at-risk infants: Predictive validity of the Alberta Infant Motor Scale. Dev Med Child Neurol. 1998;40:485-491.
44 Piper MC, Pinnell LE, Darrah J, et al. Construction and validation of the Alberta Infant Motor Scale (AIMS). Can J Public Health. 1994;83(Suppl 2):S46-S50.
45 Chandler L. Screening for movement dysfunction in infancy. Phys Occup Ther Pediatr. 1986;6:171-190.
46 Nickel RE, Renken CA, Gallenstein JA. The Infant Motor Screen. Dev Med Child Neurol. 1989;31:35-42.
47 Ellison PH, Browning CA, Larson B, et al. Development of a scoring system for the Milani-Comparetti and Gidoni method of assessing neurologic abnormality in infancy. Phys Ther. 1983;63:1414-1423.
48 Capute AJ, Palmer FB, Shapiro BK, et al. Primitive Reflex Profile: A quantitation of primitive reflexes in infancy. Dev Med Child Neurol. 1984;26:375-383.
49 Groen SE, de Blecourt AC, Postema K, et al. General movements in early infancy predict neuromotor development at 9 to 12 years of age. Dev Med Child Neurol. 2005;47:731-738.
50 Hadders-Algra M. General movements: A window for early identification of children at high risk for developmental disorders. J Pediatr. 2004;145(2 Suppl):S12-S18.
51 Nelson KB, Ellenberg JH. Children who “outgrew” cerebral palsy. Pediatrics. 1982;69:529-536.
52 Silan F, Ozdemir I, Lissens W. A novel L1CAM mutation with L1 spectrum disorders. Prenat Diagn. 2005;25:57-59.
53 Furrer F, Deonna T. Persistent toe-walking in children. A comprehensive clinical study of 28 cases. Helv Paediatr Acta. 1982;37:301-316.
54 Katz M, Mubarak SJ. Hereditary tendo Achillis contractures. J Pediatr Orthop. 1984;4:711-714.
55 Policy JF, Torburn L, Rinsky LA, et al. Electromyographic test to differentiate mild diplegic cerebral palsy and idiopathic toe-walking. J Pediatr Orthop. 2001;21:784-789.
56 Jan M. Misdiagnoses in children with dopa-responsive dystonia. Pediatr Neurol. 2004;31:298-303.
57 Segawa M, Nomura Y, Nishiyama N. Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann Neurol. 2003;54(Suppl 6):S32-S45.
58 Sanger TD, Delgado MR, Gaebler-Spira D, et al. Classification and definition of disorders causing hypertonia in childhood. Pediatrics. 2003;111(1):e89-e97.
59 Ashworth B. Preliminary trial of carisoprodol in multiple sclerosis. Practitioner. 1964;192:540-542.
60 Bohannon R, Smith MB. Interrater reliability of a modified Ashworth scale of muscle spasticity. Phys Ther. 1987;67:206-207.
61 Boyd RN, Graham HK. Objective measurement of clinical findings in the use of botulinum toxin type A for the management of children with cerebral palsy. Eur J Neurol. 1999;6(Suppl 4):S23-S35.
62 Albright AL, Barry MJ, Painter MJ, et al. Infusion of intrathecal baclofen for generalized dystonia in cerebral palsy. J Neurosurg. 1998;88:73-76.
63 Cook RE, Schneider I, Hazlewood ME, et al. Gait analysis alters decision-making in cerebral palsy. J Pediatr Orthop. 2003;23:292-295.
64 Graham HK, Baker R, Dobson F, et al. Multilevel orthopaedic surgery in group IV spastic hemiplegia. J Bone Joint Surg Br. 2005;87:548-555.
65 Postans NG, Granat MH. Effect of functional electrical stimulation, applied during walking, on gait in spastic cerebral palsy. Dev Med Child Neurol. 2005;47:46-52.
66 Radtka SA, Skinner SR, Johanson ME. A comparison of gait with solid and hinged ankle-foot orthoses in children with spastic diplegic cerebral palsy. Gait Posture. 2005;21:303-310.
67 Saraph V, Zwick EB, Steinwender G, et al. Leg lengthening as part of gait improvement surgery in cerebral palsy: An evaluation using gait analysis. Gait Posture. 2006;23:83-90.
68 Desloovere K, Molenaers G, Feys H, et al. Do dynamic and static clinical measurements correlate with gait analysis parameters in children with cerebral palsy? Gait Posture. 2006;24:302-313.
69 McKearnan KA, Kieckhefer GM, Engel JM, et al. Pain in children with cerebral palsy: A review. J Neurosci Nurs. 2004;36:252-259.
70 Schwartz L, Engel JM, Jensen MP. Pain in persons with cerebral palsy. Arch Phys Med Rehabil. 1999;80:1243-1246.
71 McCarthy ML, Silberstein CB, Atkins EA, et al. Comparing reliability and validity of pediatric instruments for measuring health and well-being of children with spastic cerebral palsy. Dev Med Child Neurol. 2002;44:468-476.
72 Morris C, Kurinczuk JJ, Fitzpatrick R. Child or family assessed measures of activity performance and participation for children with cerebral palsy: A structured review. Child Care Health Dev. 2005;31:397-407.
73 Waters E, Maher E, Salmon L, et al. Development of a condition-specific measure of quality of life for children with cerebral palsy: Empirical thematic data reported by parents and children. Child Care Health Dev. 2005;31:127-135.
74 Landgraf JM, Abetz L, Ware JE. The Child Health Questionnaire Users Manual. Boston: Health Institute, New England Medical Center, 1996.
75 Vargus-Adams J. Health-related quality of life in childhood cerebral palsy. Arch Phys Med Rehabil. 2005;86:940-945.
76 Vitale MG, Roye EA, Choe JC, et al. Assessment of health status in patients with cerebral palsy: What is the role of quality-of-life measures? J Pediatr Orthop. 2005;25:792-797.
77 Hadden KL, von Baeyer CL. Global and specific behavioral measures of pain in children with cerebral palsy. Clin J Pain. 2005;21:140-146.
78 Schneider JW, Gurucharri LM, Gutierrez AL, et al. Health-related quality of life and functional outcome measures for children with cerebral palsy. Dev Med Child Neurol. 2001;43:601-608.
79 Nickel R. Cerebral palsy. In: Nickel RE, Desch LW, editors. The Physician’s Guide to Caring for Children with Disabilities and Chronic Conditions. Baltimore: Paul H. Brookes; 2000:141-184.
80 Boyd RN, Morris ME, Graham HK. Management of upper limb dysfunction in children with cerebral palsy: A systematic review. Eur J Neurol. 2001;8(Suppl 5):150-166.
81 Butler C, Darrah J. Effects of neurodevelopmental treatment (NDT) for cerebral palsy: An AACPDM evidence report. Dev Med Child Neurol. 2001;43:778-790.
82 Harris SR, Roxborough L. Efficacy and effectiveness of physical therapy in enhancing postural control in children with cerebral palsy. Neural Plast. 2005;12:229-243.
83 Dodd KJ, Taylor NF, Damiano DL. A systematic review of the effectiveness of strength-training programs for people with cerebral palsy. Arch Phys Med Rehabil. 2002;83:1157-1164.
84 Dodd KJ, Taylor NF, Graham HK. A randomized clinical trial of strength training in young people with cerebral palsy. Dev Med Child Neurol. 2003;45:652-657.
85 McBurney H, Taylor NF, Dodd KJ, et al. A qualitative analysis of the benefits of strength training for young people with cerebral palsy. Dev Med Child Neurol. 2003;45:658-663.
86 Eliasson AC, Krumlinde-Sundholm L, Shaw K, et al. Effects of constraint-induced movement therapy in young children with hemiplegic cerebral palsy: An adapted model. Dev Med Child Neurol. 2005;47:266-275.
87 Naylor CE, Bower E. Modified constraint-induced movement therapy for young children with hemiplegic cerebral palsy: A pilot study. Dev Med Child Neurol. 2005;47:365-369.
88 Taub E, Ramey SL, DeLuca S, et al. Efficacy of constraint-induced movement therapy for children with cerebral palsy with asymmetric motor impairment. Pediatrics. 2004;113:305-312.
89 Willis JK, Morello A, Davie A, et al. Forced use treatment of childhood hemiparesis. Pediatrics. 2002;110(1 Pt 1):94-96.
90 Buckon CE, Thomas SS, Jakobson-Huston S, et al. Comparison of three ankle-foot orthosis configurations for children with spastic diplegia. Dev Med Child Neurol. 2004;46:590-598.
91 Morris C. A review of the efficacy of lower-limb orthoses used for cerebral palsy. Dev Med Child Neurol. 2002;44:205-211.
92 White H, Jenkins J, Neace WP, et al. Clinically prescribed orthoses demonstrate an increase in velocity of gait in children with cerebral palsy: A retrospective study. Dev Med Child Neurol. 2002;44:227-232.
93 Russell DJ, Gorter JW. Assessing functional differences in gross motor skills in chidlren with cerebral palsy who use an ambulatory aid or orthoses: Can the GMFM-88 help? Dev Med Child Neurol. 2005;47:462-467.
94 Butler C. Powered tots: Augmentative mobility for locomotor disabled youngsters. Tot Line. 1988;4:18-19.
95 Gormley MEJr, Krach LE, Piccini L. Spasticity management in the child with spastic quadriplegia. Eur J Neurol. 2001;8(Suppl 5):127-135.
96 Hagglund G, Andersson S, Duppe H, et al. Prevention of severe contractures might replace multilevel surgery in cerebral palsy: Results of a population-based health care programme and new techniques to reduce spasticity. J Pediatr Orthop B. 2005;14:269-273.
97 Krach L. Pharmacotherapy of spasticity: Oral medications and intrathecal baclofen. J Child Neurol. 2001;16:31-36.
98 Montane E, Vallano A, Laporte JR. Oral antispastic drugs in nonprogressive neurologic diseases: A systematic review. Neurology. 2004;63:1357-1363.
99 Mathew A, Mathew MC, Thomas M, et al. The efficacy of diazepam in enhancing motor function in children with spastic cerebral palsy. J Trop Pediatr. 2005;51:109-113.
100 Gracies JM, Elovic E, McGuire J, et al. Traditional pharmacological treatments for spasticity: Part I. Local treatments. Muscle Nerve Suppl. 1997;6:S61-S91.
101 Tilton A. Injectable neuromuscular blockade in the treatment of spasticity and movement disorders. J Child Neurol. 2003;18(Suppl 1):S50-S66.
102 Ade-Hall RA, Moore AP. Botulinum toxin type A in the treatment of lower limb spasticity in cerebral palsy. Cochrane Database Syst Rev (2). 2000. CD001408
103 Wasiak J, Hoare B, Wallen M. Botulinum toxin A as an adjunct to treatment in the management of the upper limb in children with spastic cerebral palsy. Cochrane Database Syst Rev (4). 2004. CD003469
104 Gough M, Fairhurst C, Shortland AP. Botulinum toxin and cerebral palsy: Time for reflection? Dev Med Child Neurol. 2005;47:709-712.
105 Albright AL, Barron WB, Fasick MP, et al. Continuous intrathecal baclofen infusion for spasticity of cerebral origin. JAMA. 1993;270:2475-2477.
106 Albright AL, Cervi A, Singletary J. Intrathecal bac lofen for spasticity in cerebral palsy. JAMA. 1991;265:1418-1422.
107 Butler C, Campbell S. Evidence of the effects of intrathecal baclofen for spastic and dystonic cerebral palsy. ACPDM Treatment Outcomes Committee Review Panel. Dev Med Child Neurol. 2000;42:634-645.
108 Albright AL, Barry MJ, Shafton DH, et al. Intrathecal baclofen for generalized dystonia. Dev Med Child Neurol. 2001;43:652-657.
109 Bjornson KF, McLaughlin JF, Loeser JD, et al. Oral motor, communication, and nutritional status of children during intrathecal baclofen therapy: A descriptive pilot study. Arch Phys Med Rehabil. 2003;84:500-506.
110 Krach LE, Kriel RL, Gilmartin RC, et al. GMFM 1 year after continuous intrathecal baclofen infusion. Pediatr Rehabil. 2005;8:207-213.
111 Murphy NA, Irwin MC, Hoff C. Intrathecal baclofen therapy in chidlren with cerebral palsy: Efficacy and complications. Arch Phys Med Rehabil. 2002;83:1721-1725.
112 Buonaguro V, Scelsa B, Curci D, et al. Epilepsy and intrathecal baclofen therapy in children with cerebral palsy. Pediatr Neurol. 2005;33:110-113.
113 Krach LE, Kriel RL, Gilmartin RC, et al. Hip status in cerebral palsy after one year of continuous intrathecal baclofen infusion. Pediatr Neurol. 2004;30:163-168.
114 Steinbok P, Kestle JR. Variation between centers in electrophysiologic techniques used in lumbosacral selective dorsal rhizotomy for spastic cerebral palsy. Pediatr Neurosurg. 1996;25:233-239.
115 McLaughlin J, Bjornson K, Temkin N, et al. Selective dorsal rhizotomy: Meta-analysis of three randomized controlled trials. Dev Med Child Neurol. 2002;44:17-25.
116 McLaughlin JF, Bjornson KF, Astley SJ, et al. Selective dorsal rhizotomy: Efficacy and safety in an investigator-masked randomized clinical trial. Dev Med Child Neurol. 1998;40:220-232.
117 Steinbok P, Reiner AM, Beauchamp R, et al. A randomized clinical trial to compare selective posterior rhizotomy plus physiotherapy with physiotherapy alone in children with spastic diplegic cerebral palsy. Dev Med Child Neurol. 1997;39:178-184.
118 Wright FV, Sheil EM, Drake JM, et al. Evaluation of selective dorsal rhizotomy for the reduction of spasticity in cerebral palsy: A randomized controlled trial. Dev Med Child Neurol. 1998;40:239-247.
119 Mittal S, Farmer JP, Al-Atassi B, et al. Long-term functional outcome after selective posterior rhizotomy. J Neurosurg. 2002;97:315-325.
120 O’Brien DF, Park TS, Puglisi JA, et al. Effect of selective dorsal rhizotomy on need for orthopedic surgery for spastic quadriplegic cerebral palsy: Long-term outcome analysis in relation to age. J Neurosurg. 2004;101(1 Suppl):59-63.
121 Hicdonmez T, Steinbok P, Beauchamp R, et al. Hip joint subluxation after selective dorsal rhizotomy for spastic cerebral palsy. J Neurosurg. 2005;103(1 Suppl):10-16.
122 Spiegel DA, Loder RT, Alley KA, et al. Spinal deformity following selective dorsal rhizotomy. J Pediatr Orthop. 2004;24:30-36.
123 Steinbok P, Hicdonmez T, Sawatzky B, et al. Spinal deformities after selective dorsal rhizotomy for spastic cerebral palsy. J Neurosurg. 2005;102(4 Suppl):363-373.
124 Borton DC, Walker K, Pirpiris M, et al. Isolated calf lengthening in cerebral palsy. Outcome analysis of risk factors. J Bone Joint Surg Br. 2001;83:364-370.
125 Knapp DRJr, Cortes H. Untreated hip dislocation in cerebral palsy. J Pediatr Orthop. 2002;22:668-671.
126 Cornell MS, Hatrick NC, Boyd R, et al. The hip in children with cerebral palsy. Predicting the outcome of soft tissue surgery. Clin Orthop Relat Res. 1997;340:165-171.
127 Saito N, Ebara S, Ohotsuka K, et al. Natural history of scoliosis in spastic cerebral palsy. Lancet. 1998;351:1687-1692.
128 Jones KB, Sponseller PD, Shindle MK, et al. Longitudinal parental perceptions of spinal fusion for neuromuscular spine deformity in patients with totally involved cerebral palsy. J Pediatr Orthop. 2003;23:143-149.
129 Tsirikos AI, Chang WN, Dabney KW, et al. Comparison of parents’ and caregivers’ satisfaction after spinal fusion in children with cerebral palsy. J Pediatr Orthop. 2004;24:54-58.
130 Henderson RC, Lark RK, Gurka MJ, et al. Bone density and metabolism in children and adolescents with moderate to severe cerebral palsy. Pediatrics. 2002;110(1 Pt 1):e5.
131 Henderson RC, Kairella JA, Barrington JW, et al. Longitudinal changes in bone density in children and adolescents with moderate to severe cerebral palsy. J Pediatr. 2005;146:769-775.
132 Caulton JM, Ward KA, Alsop CW, et al. A randomized controlled trial of standing programme on bone mineral density in non-ambulant children with cerebral palsy. Arch Dis Child. 2004;89:131-135.
133 Henderson RC, Lark RK, Kecskemethy HH, et al. Bisphosphonates to treat osteopenia in children with quadriplegic cerebral palsy: a randomized, placebo-controlled clinical trial. J Pediatr. 2002;141:644-651.
134 Fung EB, Samson-Fang L, Stallings VA, et al. Feeding dysfunction is associated with poor growth and health status in children w ith cerebral palsy. J A m Diet Assoc. 2002;102:361-373.
135 Samson-Fang L, Fung E, Stallings VA, et al. Relationship of nutritional status to health and societal participation in children with cerebral palsy. J Pediatr. 2002;141:637-643.
136 Samson-Fang LJ, Stevenson RD. Identification of malnutrition in children with cerebral palsy: poor performance of weight-for-height centiles. Dev Med Child Neurol. 2000;42:162-168.
137 Sleigh G, Brocklehurst P. Gastrostomy feeding in cerebral palsy: A systematic review. Arch Dis Child. 2004;89:534-539.
138 Sleigh G, Sullivan PB, Thomas AG. Gastrostomy feeding versus oral feeding alone for children with cerebral palsy. Cochrane Database Syst Rev (2). 2004. CD003943
139 Sullivan PB, Juszczak E, Bachlet AME, et al. Gastrostomy tube feeding in children with cerebral palsy: A prospective, longitudinal study. Dev Med Child Neurol. 2005;47:77-85.
140 Jepsen C, Nickel RE. Nutrition and growth. In: Nickel RE, Desch LW, editors. The Physician’s Guide to Caring for Children with Disabilities and Chronic Conditions. Baltimore: Paul Brookes; 2000:78-99.
141 Saitua F, Acuna R, Herrera P. Percutaneous endoscopic gastrostomy: The technique of choice? J Pediatr Surg. 2003;38:1512-1515.
142 Wadie GM, Lobe TE. Gastroesophageal reflux disease in neurologically impaired children: The role of the gastrostomy tube. Semin Laparosc Surg. 2002;9:180-189.
143 Roijen LE, Postema K, Limbeek VJ, et al. Development of bladder control in children and adolescents with cerebral palsy. Dev Med Child Neurol. 2001;43:103-107.
144 Karaman MI, Kaya C, Caskurlu T, et al. Urodynamic findings in children with cerebral palsy. Int J Urol. 2005;12:717-720.
145 Mayo M. Lower urinary tract dysfunction in cerebral palsy. J Urol. 1992;147:419-420.
146 Reid CJ, Borzyskowski M. Lower urinary tract dysfunction in cerebral palsy. Arch Dis Child. 1993;68:739-742.
147 Agnarsson U, Warde C, McCarthy G, et al. Anorectal function of children with neurological problems. II: Cerebral palsy. Dev Med Child Neurol. 1993;35:903-908.
148 Del Giudice E, Staiano A, Capano G, et al. Gastrointestinal manifestations in children with cerebral palsy. Brain Dev. 1999;21:307-311.
149 Park ES, Park CI, Cho SR, et al. Colonic transit time and constipation in children with spastic cerebral palsy. Arch Phys Med Rehabil. 2004;85:453-456.
150 Senner JE, Logemann J, Zecker S, et al. Drooling, saliva production, and swallowing in cerebral palsy. Dev Med Child Neurol. 2004;46:801-806.
151 Bachrach SJ, Walter RS, Trzcinski K. Use of glycopyr-rodate and other anticholinergic medications for sialorrhea in children with cerebral palsy. Clin Pediatr (Phila). 1998;37:485-490.
152 Blasco PA. Glycopyrrolate treatment of chronic drooling. Arch Pediatr Adolesc Med. 1996;150:932-935.
153 Stern L. Preliminary study of glycopyrrolate in the management of drooling. J Pediatr Child Health. 1997;33:52-54.
154 van der Burg JJ, Jongerius PH, van Limbeek J, et al. Social interaction and self-esteem of children with cerebral palsy after treatment for severe drooling. Eur J Pediatr. 2006;165:37-41.
155 Jongerius PH, Joosten F, Hoogen FJ, et al. The treatment of drooling by ultrasound-guided intraglandular injections of botulinum toxin type A into the salivary glands. Lar yngoscope. 2003;113:107-111.
156 Jongerius PHRJ, van Limbeek J, Gabreels FJ, van Hulst K, van den Hoogen FJ. Botulinum toxin effect on salivary flow rate in children with cerebral palsy. Neurology. 2004;63:1371-1375.
157 Suskind DL, Tilton A. Clinical study of botulinum-A toxin in the treatment of sialorrhea in children with cerebral palsy. Laryngoscope. 2002;112:73-81.
158 Blasco PA. Management of drooling: 10 years after the Consortium on Drooling, 1990. Dev Med Child Neurol. 2002;44:778-781.
159 Hockstein NG, Samadi DS, Gendron K, et al. Sialorrhea: A management challenge. Am Fam Physician. 2004;69:2628-2634.
160 McAloney N, Kerawala CJ, Stassen LF. Mnagement of drooling by transposition of the submandibular ducts and excision of the sublingual glands. J Ir Dent Assoc. 2005;51:126-131.
161 Hallett KB, Lucas JO, Johnston T, et al. Dental health of children with cerebral palsy following sialodocho-plasty. Spec Care Dentist. 1995;15:234-238.
162 Johnson HM, Reid SM, Hazard CJ, et al. Effectiveness of the Innsbruck Sensorimotor Activator and Regulator in improving saliva control in children with cerebral palsy. Dev Med Child Neurol. 2004;46:39-45.
163 Pellock JM, Morton LD. Treatment of epilepsy in the multiply handicapped. Ment Retard Dev Disabil Res Rev. 2000;6:309-323.
164 Kulak W, Sobaniec W, Smigielska-Kuzia J, et al. A comparison of spastic diplegic and tetraplegic cerebral palsy. Pediatr Neurol. 2005;32:311-317.
165 Kulak W, Sobaniec W. Risk factors and prognosis of epilepsy in children with cerebral palsy in northeastern Poland. Brain Dev. 2003;25:499-506.
166 Engel JM, Jensen MP, Hoffman AJ, et al. Pain in persons with cerebral palsy: Extension and cross validation. Arch Phys Med Rehabil. 2003;84:1125-1128.
167 Hadden KL, von Baeyer CL. Pain in children with cerebral palsy: Common triggers and expressive behaviors. Pain. 2002;99:281-288.
168 Houlihan CM, O’Donnell M, Conaway M, et al. Bodily pain and health-related quality of life in children with cerebral palsy. Dev Med Child Neurol. 2004;46:305-310.
169 Sanders H, Davis MF, Duncan B, et al. Use of complementary and alternative medical therapies among children with special health care needs in southern Arizona. Pediatrics. 2003;111:584-587.
170 Hurvitz EA, Leonard C, Ayyangar R, et al. Complementary and alternative medicine use in families of children with cerebral palsy. Dev Med Child Neurol. 2003;45:364-370.
171 Collet JP, Vanasse M, Marois P, et al. Hyperbaric oxygen for children with cerebral palsy: A randomized multicentre trial. HBO-CP Research Group. Lancet. 2001;357:582-586.
172 Liptak G. Complementary and alternative therapies for cerebral palsy. Ment Retard Dev Disabil Res Rev. 2005;11:156-163.
173 Nickel R. The use of complementary and alternative medicine by families of children with disabilities. In: Oken B, editor. Complementary Therapies in Neurology: An Evidence-Based Approach. Boca Raton, FL: Parthenon; 2004:371-389.
174 Rosenbaum P. Controversial treatment of spasticity: Exploring alternative therapies for motor function in children with cerebral palsy. J Child Neurol. 2003;18(Suppl 1):S89-S94.
175 Blum RW, Resnick MD, Nelson R, et al. Family and peer issues among adolescents with spina bifida and cerebral palsy. Pediatrics. 1991;88:280-285.
176 Wadsworth JS, Harper DC. The social needs of adolescents with cerebral palsy. Dev Med Child Neurol. 1993;35:1019-1022.
177 Andersson C, Mattsson E. Adults with cerebral palsy: A survey describing problems, needs, and resources, with special emphasis on locomotion. Dev Med Child Neurol. 2001;43:76-82.
178 Michelsen SI, Uldall P, Kejs AM, et al. Education and employment prospects in cerebral palsy. Dev Med Child Neurol. 2005;47:511-517.
179 Bottos M, Feliciangeli A, Sciuto L, et al. Functional status of adults with cerebral palsy and implications for treatment of children. Dev Med Child Neurol. 2001;43:516-528.
180 Williams EN, Broughton NS, Menelaus MB. Age-related walking in children with spina bifida. Dev Med Child Neurol. 1999;41:446-449.
181 Spina bifida and anencephaly before and after folic acid mandate-United States, 1995–1996 and 1999–2000. MMWR Morb Mortal Wkly Rep. 2004;53:362-365.
181a CDC. Recommendations for the use of folic acid to reduce the number of cases of spina bifidal and other neural tube defects. MMWR. 14(No. RR-14), 1992.
182 Greenberg F, James LM, Oakley GPJr. Estimates of birth prevalence rates of spina bifida in the United States from computer-generated maps. Am J Obstet Gynecol. 1983;145:570-573.
183 Williams LJ, Rasmussen SA, Flores A, et al. Decline in the prevalence of spina bifida and anencephaly by race/ethnicity: 1995–2002. Pediatrics. 2005;116:580-586.
184 Elwood JM, Little J, Elwood J. Epidemiology and control of neural tube defects. Vessey M, editor. Monographs in Epidemiology and Biostatistics, 20. Oxford, UK: Oxford University Press, 1992.
185 Czeizel AE, Dudas I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N Engl J Med. 1992;327:1832-1835.
186 Prevention of neural tube defects: Results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. Lancet. 1991;338:131-137.
187 Lopez-Camelo JS, Orioli IM, da Graca Dutra M, et al. Reduction of birth prevalence rates of neural tube defects after folic acid fortification in Chile. Am J Med Genet A. 2005;135:120-125.
188 Chatkupt S, Skurnick JH, Jaggi M, et al. Study of genetics, epidemiology, and vitamin usage in familial spina bifida in the United States in the 1990s. Neurology. 1994;44:65-70.
189 van der Put NM, Steegers-Theunissen RP, Frosst P, et al. Mutated methylenetetrahydrofolate reductase as a risk factor for spina bifida. Lancet. 1995;346:1070-1071.
190 van der Put NM, van den Heuvel LP, Steegers-Theunissen RP, et al. Decreased methylene tetrahy-drofolate reductase activity due to the 677C←T mutation in families with spina bifida offspring. J Mol Med. 1996;74:691-694.
191 Epstein DJ, Vekemans M, Gros P. Splotch (Sp2H), a mutation affecting development of the mouse neural tube, shows a deletion within the paired homeo-domain of Pax-3.. Cell. 1991;67:767-774.
192 Goulding M, Paquette A. Pax genes and neural tube defects in the mouse. Ciba Found Symp. 1994;181:103-113. Ciba Found Symp. 1994;181:113-117. [discussion]
193 Hol FA, Geurds MP, Chatkupt S, et al. PAX genes and human neural tube defects: An amino acid substitution in PAX1 in a patient with spina bifida. J Med Genet. 1996;33:655-660.
194 Martin JF, Olson EN. Identification of a prx1 limb enhancer. Genesis. 2000;26:225-229.
195 Anderson JL, Waller DK, Canfield MA, et al. Maternal obesity, gestational diabetes, and central nervous system birth defects. Epidemiology. 2005;16:87-92.
196 Layde PM, Edmonds LD, Erickson JD. Maternal fever and neural tube defects. Teratology. 1980;21:105-108.
197 Martinez-Frias ML, Garcia Mazario MJ, Caldas CF, et al. High maternal fever during gestation and severe congenital limb disruptions. Am J Med Genet. 2001;98:201-203.
198 Watkins ML, Rasmussen SA, Honein MA, et al. Maternal obesity and risk for birth defects. Pediatrics. 2003;111(5 Part 2):1152-1158.
199 Muller F. Prenatal biochemical screening for neural tube defects. Childs Nerv Syst. 2003;19:433-435.
199a ACOG practice bulletin neural tube defects. Number 44 July 2003. Int. J. Gynaecol Obstet. 2003;83:122-133.
200 Coniglio SJ, Anderson SM, Ferguson JE2nd. Functional motor outcome in children with myelomeningocele: Correlation with anatomic level on prenatal ultrasound. Dev Med Child Neurol. 1996;38:675-680.
201 Sival DA, Begeer JH, Staal-Schreinemachers AL, et al. Perinatal motor behaviour and neurological outcome in spina bifida aperta. Early Hum Dev. 1997;50:27-37.
202 Bruner JP, Tulipan N, Paschall RL, et al. Fetal surgery for myelomeningocele and the incidence of shunt-dependent hydrocephalus. JAMA. 1999;282:1819-1825.
203 Tulipan N, Bruner JP, Hernanz-Schulman M, et al. Effect of intrauterine myelomeningocele repair on central nervous system structure and function. Pediatr Neurosurg. 1999;31:183-188.
204 Chescheir NC, D’Alton M. Evidence-based medicine and fetal treatment: How to get involved. Obstet Gy necol. 2005;106:610-613.
205 Lewis D, Tolosa JE, Kaufmann M, et al. Elective cesarean delivery and long-term motor function or ambulation status in infants with meningomyelocele. Obstet Gynecol. 2004;103:469-473.
206 Merrill DC, Goodwin P, Burson JM, et al. The optimal route of delivery for fetal meningomyelocele. Am J Obstet Gynecol. 1998;179:235-240.
207 Shurtleff DB, Luthy DA, Nyberg DA, et al. Meningomyelocele: Management in utero and post natum. Ciba Found Symp. 1994;181:270-280. Ciba Found Symp. 1994;181:280-286. [discussion]
208 Nickel RE, Magenis RE. Neural tube defects and deletions of 22q11. Am J Med Genet. 1996;66:25-27.
209 Nickel RE, Pillers DA, Merkens M, et al. Velo-cardio-facial syndrome and DiGeorge sequence with meningomyelocele and deletions of the 22q11 region. Am J Med Genet. 1994;52:445-449.
210 Hopps CV, Kropp KA. Preservation of renal function in children with myelomeningocele managed with basic newborn evaluation and close followup. J Urol. 2003;169:305-308.
211 Wu HY, Baskin LS, Kogan BA. Neurogenic bladder dysfunction due to myelomeningocele: Neonatal versus childhood treatment. J Urol. 1997;157:2295-2297.
212 Snodgrass WT, Adams R. Initial urologic management of myelomeningocele. Urol Clin North Am. 2004;31:427-434. viii
213 Ritter S, Tani LY, Shaddy RE, et al. Are screening echocardiograms warranted for neonates with meningomyelocele? Arch Pediatr Adolesc Med. 1999;153:1264-1266.
214 Iborra J, Pages E, Cuxart A. Neurological abnormalities, major orthopaedic deformities and ambulation analysis in a myelomeningocele population in Catalonia (Spain). Spinal Cord. 1999;37:351-357.
215 Charney EB, Melchionni JB, Smith DR. Community ambulation by children with myelomeningocele and high-level paralysis. J Pediatr Orthop. 1991;11:579-582.
216 Gerritsma-Bleeker CL, Heeg M, Vos-Niel H. Ambulation with the reciprocating-gait orthosis. Experience in 15 children with myelomeningocele or paraplegia. Acta Orthop Scand. 1997;68:470-473.
217 Liptak GS, Shurtleff DB, Bloss JW, et al. Mobility aids for children with high-level myelomeningocele: Parapodium versus wheelchair. Dev Med Child Neurol. 1992;34:787-796.
218 Bartonek A, Saraste H, Samuelsson L, et al. Ambulation in patients with myelomeningocele: A 12-year followup. J Pediatr Orthop. 1999;19:202-206.
219 Schiltenwolf M, Carstens C, Rohwedder J, et al. Results of orthotic treatment in children with myelomeningocele. Eur J Pediatr Surg. 1991;1(Suppl 1):50-52.
220 Brinker MR, Rosenfeld SR, Feiwell E, et al. Myelomeningocele at the sacral level. Long-term outcomes in adults. J Bone Joint Surg Am. 1994;76:1293-1300.
221 Bowman RM, McLone DG, Grant JA, et al. Spina bifida outcome: A 25-year prospective. Pediatr Neurosurg. 2001;34:114-120.
222 Piggott H. The natural history of scoliosis in myelodysplasia. J Bone Joint Surg Br. 1980;62:54-58.
223 Trivedi J, Thomson JD, Slakey JB, et al. Clinical and radiographic predictors of scoliosis in patients with myelomeningocele. J Bone Joint Surg Am. 2002;84:1389-1394.
224 Muller EB, Nordwall A. Prevalence of scoliosis in children with myelomeningocele in western Sweden. Spine. 1992;17:1097-1102.
225 Parsch D, Geiger F, Brocai DR, et al. Surgical management of paralytic scoliosis in myelomeningocele. J Pediatr Orthop B. 2001;10:10-17.
226 Sherk HH, Uppal GS, Lane G, et al. Treatment versus non-treatment of hip dislocations in ambulatory patients with myelomeningocele. Dev Med Child Neurol. 1991;33:491-494.
227 Lorente Molto FJ, Martinez Garrido I. Retrospective review of L3 myelomeningocele in three age groups: Should posterolateral iliopsoas transfer still be indicated to stabilize the hip? J Pediatr Orthop B. 2005;14:177-184.
228 Tosi LL, Buck BD, Nason SS, et al. Dislocation of hip in myelomeningocele. The McKay hip stabilization. J Bone Joint Surg Am. 1996;78:664-673.
229 Wright JG, Menelaus MB, Broughton NS, et al. Natural history of knee contractures in myelomeningocele. J Pediatr Orthop. 1991;11:725-730.
230 Omeroglu S, Peker T, Omeroglu H, et al. Intrauterine structure of foot muscles in talipes equinovarus due to high-level myelomeningocele: A light microscopic study in fetal cadavers. J Pediatr Orthop B. 2004;13:263-267.
231 Frischhut B, Stockl B, Landauer F, et al. Foot deformities in adolescents and young adults with spina bifida. J Pediatr Orthop B. 2000;9:161-169.
232 Hesz N, Wolraich M. Myelodysplasia. In: Wolraich M, editor. The Practical Assessment and Management of Children with Disorders of Development and Learning. Chicago: Year Book Medical; 1987:194-221.
233 Hafez AT, McLorie G, Gilday D, et al. Long-term evaluation of metabolic profile and bone mineral density after ileocystoplasty in children. J Urol. 2003;170(4 Pt 2):1639-1646. 41
234 Quan A, Adams R, Ekmark E, et al. Bone mineral density in children with myelomeningocele. Pediatrics. 1998;102(3):E34.
235 James CC. Fractures of the lower limbs in spina bifida cystica: A survey of 44 fractures in 122 children. Dev Med Child Neurol. 1970;Suppl 22:88.
236 Koch MO, McDougal WS, Hall MC, et al. Long-term metabolic effects of urinary diversion: A comparison of myelomeningocele patients managed by clean intermittent catheterization and urinary diversion. J Urol. 1992;147:1343-1347.
237 Kumar SJ, Cowell HR, Townsend P. Physeal, metaph-yseal, and diaphyseal injuries of the lower extremities in children with myelomeningocele. J Pediatr Orthop. 1984;4:25-27.
238 Anschuetz RH, Freehafer AA, Shaffer JW, et al. Severe fracture complications in myelodysplasia. J Pediatr Orthop. 1984;4:22-24.
239 Rosenstein BD, Greene WB, Herrington RT, et al. Bone density in myelomeningocele: The effects of ambulatory status and other factors. Dev Med Child Neurol. 1987;29:486-494.
240 Sholas MG, Tann B, Gaebler-Spira D. Oral bisphosphonates to treat disuse osteopenia in children with disabilities: A case series. J Pediatr Orthop. 2005;25:326-331.
241 Roach MB, Switters DM, Stone AR. The changing urodynamic pattern in infants with myelomeningocele. J Urol. 1993;150:944-947.
242 Sillen U, Hansson E, Hermansson G, et al. Development of the urodynamic pattern in infants with myelomeningocele. Br J Urol. 1996;78:596-601.
243 Elliott SP, Villar R, Duncan B. Bacteriuria management and urological evaluation of patients with spina bifida and neurogenic bladder: A multicenter survey. J Urol. 2005;173:217-220.
244 Greenfield SP, Fera M. The use of intravesical oxybutynin chloride in children with neurogenic bladder. J Urol. 1991;146(2 Pt 2):532-534.
245 Zerin JM, DiPietro MA, Ritchey ML, et al. Intravesical oxybutinin chloride in children with intermittent catheterization: Sonographic findings. Pediatr Radiol. 1994;24:348-350.
246 Lee MW, Greenfield SP. Intractable high-pressure bladder in female infants with spina bifida: clinical character istics and use of vesicostomy. Urology. 2005;65:568-571.
247 Anderson JD, Chambers GK, Johnson HW. Application of a leukocyte and nitrite urine test strip to the management of children with neurogenic bladder. Diagn Microbiol Infect Dis. 1993;17(1):29-33.
248 Schlager TA, Dilks SA, Lohr JA, et al. Periurethral colonization and urinary leukocytes as markers for bacteriuria in children with neurogenic bladder. Urol Res. 1992;20:361-363.
249 Edwards M, Borzyskowski M, Cox A, et al. Neuropathic bladder and intermittent catheterization: Social and psychological impact on children and adolescents. Dev Med Child Neurol. 2004;46:168-177.
250 Moore C, Kogan BA, Parekh A. Impact of urinary incontinence on self-concept in children with spina bifida. J Urol. 2004;171:1659-1662.
251 Verhoef M, Lurvink M, Barf HA, et al. High prevalence of incontinence among young adults with spina bifida: Description, prediction and problem perception. Spinal Cord. 2005;43:331-340.
252 Knoll M, Madersbacher H. The chances of a spina bifida patient becoming continent/socially dry by conservative therapy. Paraplegia. 1993;31:22-27.
253 Hensle TW, Bingham J, Lam J, et al. Preventing reservoir calculi after augmentation cystoplasty and continent urinary diversion: The influence of an irrigation protocol. BJU Int. 2004;93:585-587.
254 Zhang H, Yamataka A, Koga H, et al. Bladder stone formation after sigmoidocolocystoplasty: Statistical analysis of risk factors. J Pediatr Surg. 2005;40:407-411.
255 Mingin G, Maroni P, Gerharz EW, et al. Linear growth after enterocystoplasty in children and adolescents: A review. World J Urol. 2004;22:196-199.
256 Bellinger MF. Ureterocystoplasty: A unique method for vesical augmentation in children. J Urol. 1993;149:811-813.
257 Bellinger MF. Ureterocystoplasty update. World J Urol. 1998;16:251-254.
258 Wolf JSJr, Turzan CW. Augmentation ureterocystoplasty. J Urol. 1993;149:1095-1098.
259 Godbole P, Bryant R, MacKinnon AE, et al. Endoure-thral injection of bulking agents for urinary incontinence in children. BJU Int. 2003;91:536-539.
260 Godbole P, Mackinnon AE. Expanded PTFE bladder neck slings for incontinence in children: The long-term outcome. BJU Int. 2004;93:139-141.
261 Spiess PE, Capolicchio JP, Kiruluta G, et al. Is an artificial sphincter the best choice for incontinent boys with spina bifida? Review of our long term experience with the AS-800 artificial sphincter. Can J Urol. 2002;9:1486-1491.
262 Crooks KK, Enrile BG. Comparison of the ileal conduit and clean intermittent catheterization for myelomeningocele. Pediatrics. 1983;72:203-206.
263 Heath AL, Eckstein HB. Ileal conduit urinary diversion in children. A long term followup. J Urol (Paris). 1984;90:91-96.
264 Harris CF, Cooper CS, Hutcheson JC, et al. Appendi-covesicostomy: The Mitrofanoff procedure-A 15-year perspective. J Urol. 2000;163:1922-1926.
265 Lemelle JL, Simo AK, Schmitt M. Comparative study of the Yang-Monti channel and appendix for continent diversion in the Mitrofanoff and Malone principles. J Urol. 2004;172(5 Pt 1):1907-1910.
266 Brown S, Marshall D, Patterson D, et al. Chronic pyelonephr itis in association with neuropath ic bladder. Eur J Pediatr Surg. 1999;9(Suppl 1):29-30.
267 Chan YL, Chan KW, Yeung CK, et al. Potential utility of MRI in the evaluation of children at risk of renal scarring. Pediatr Radiol. 1999;29:856-862.
268 Hamdi M, Mohan P, Little DM, et al. Successful renal transplantation in children with spina bifida: Long term single center experience. Pediatr Transplant. 2004;8:167-170.
269 Mendizabal S, Estornell F, Zamora I, et al. Renal transplantation in children with severe bladder dysfunction. J Urol. 2005;173:226-229.
270 Malone PS, Ransley PG, Kiely EM. Preliminary report: The antegrade continence enema. Lancet. 1990;336:1217-1218.
271 Duel BP, Gonzalez R. The button cecostomy for management of fecal incontinence. Pediatr Surg Int. 1999;15:559-561.
272 Herndon CD, Cain MP, Casale AJ, et al. The colon flap/extension Malone antegrade continence enema: An alternative to the Monti-Malone antegrade continence enema. J Urol. 2005;174:299-302.
272a McDonnell GV, McCann JP. Issues of medical management in adults with spina bifida. Childs Nerv Syst. 2000;16:222-227.
273 Krupp S, Kuhn W, Zaech GA. The use of innervated flaps for the closure of ischial pressure sores. Paraplegia. 1983;21:119-126.
274 Thomson HG, Azhar Ali M, Healy H. The recurrent neurotrophic buttock ulcer in the meningomyelocele paraplegic: A sensate flap solution. Plast Reconstr Surg. 2001;108:1192-1196.
275 Harris MB, Banta JV. Cost of skin care in the myelomeningocele population. J Pediatr Orthop. 1990;10:355-361.
276 Verhoef M, Barf HA, Post MW, et al. Secondary impairments in young adults with spina bifida. Dev Med Child Neurol. 2004;46:420-427.
277 McLone DG, Dias MS. The Chiari II malformation: Cause and impact. Childs Nerv Syst. 2003;19:540-550.
278 Dyste GN, Menezes AH, VanGilder JC. Symptomatic Chiari malformations. An analysis of presentation, management, and long-term outcome. J Neurosurg. 1989;71:159-168.
279 Hoffman HJ, Hendrick EB, Humphreys RP. Manifestations and management of Arnold-Chiari malformation in patients with myelomeningocele. Childs Brain. 1975;1:255-259.
280 Pollack IF, Pang D, Albright AL, et al. Outcome following hindbrain decompression of symptomatic Chiari malformations in children previously treated with myelomeningocele closure and shunts. J Neurosurg. 1992;77:881-888.
281 Vandertop WP, Asai A, Hoffman HJ, et al. Surgical decompression for symptomatic Chiari II malformation in neonates with myelomeningocele. J Neurosurg. 1992;77:541-544.
282 Hetherington R, Dennis M, Barnes M, et al. Functional outcome in young adults with spina bifida and hydrocephalus. Childs Nerv Syst. 2006;22:117-124.
283 Albright AL, Pollack IF, Adelson PD, et al. Outcome data and analysis in pediatric neurosurgery. Neurosurgery. 1999;45:101-106.
284 Caldarelli M, Di Rocco C, La Marca F. Shunt complications in the first postoperative year in children with meningomyelocele. Childs Nerv Syst. 1996;12:748-754.
285 Tuli S, Drake J, Lamberti-Pasculli M. Long-term outcome of hydrocephalus management in myelome-ningoceles. Childs Nerv Syst. 2003;19:286-291.
286 Enger PO, Svendsen F, Wester K. CSF shunt infections in children: Experiences from a population-based study. Acta Neurochir (Wien). 2003;145:243-248.
287 Kuffer F. Prophylactic long-term anticoagulant treatment of hydrocephalic patients with ventriculoatrial shunts. Dev Med Child Neurol. 1976;Suppl 37:74-77.
288 Sleigh G, Dawson A, Penny WJ. Cor pulmonale as a complication of ventriculoatrial shunts reviewed. Dev Med Child Neurol. 1993;35:74-78.
289 Lorber J, Pucholt V. When is a shunt no longer necessary? An investigation of 300 patients with hydrocephalus and myelomeningocele: 11–22 year follow up. Z Kinderchir. 1981;34:327-329.
290 Tomlinson P, Sugarman ID. Complications with shunts in adults with spina bifida. BMJ. 1995;311:286-287.
291 Hudgins RJ, Gilreath CL. Tethered spinal cord following repair of myelomeningocele. Neurosurg Focus. 2004;16(2):E7.
292 Naik DR, Emery JL. The position of the spinal cord segments related to the vertebral bodies in children with meningomyelocele and hydrocephalus. Dev Med Child Neurol. 1968;Suppl 16:62-68.
293 Oi S, Yamada H, Matsumoto S. Tethered cord syndrome versus low-placed conus medullaris in an over-distended spinal cord following initial repair for myelodysplasia. Childs Nerv Syst. 1990;6:264-269.
294 Petersen MC. Tethered cord syndrome in myelodysplasia: Correlation between level of lesion and height at time of presentation. Dev Med Child Neurol. 1992;34:604-610.
295 Haberl H, Tallen G, Michael T, et al. Surgical aspects and outcome of delayed tethered cord release. Zentralbl Neurochir. 2004;65:161-167.
296 Sarwark JF, Weber DT, Gabrieli AP, et al. Tethered cord syndrome in low motor level children with myelomeningocele. Pediatr Neurosurg. 1996;25:295-301.
297 Craig JJ, Gray WJ, McCann JP. The Chiari/hydrosy-ringomyelia complex presenting in adults with myelo-meningocoele: An indication for early intervention. Spinal Cord. 1999;37:275-278.
298 Chapman PH, Frim DM. Symptomatic syringomyelia following surgery to treat retethering of lipomyelo-meningoceles. J Neurosurg. 1995;82:752-755.
299 Park TS, Cail WS, Broaddus WC, et al. Lumboperito-neal shunt combined with myelotomy for treatment of syringohydromyelia. J Neurosurg. 1989;70:721-727.
300 Klepper J, Busse M, Strassburg HM, et al. Epilepsy in shunt-treated hydrocephalus. Dev Med Child Neurol. 1998;40:731-736.
301 Noetzel MJ, Blake JN. Prognosis for seizure control and remission in children with myelomeningocele. Dev Med Child Neurol. 1991;33:803-810.
302 Talwar D, Baldwin MA, Horbatt CI. Epilepsy in children with meningomyelocele. Pediatr Neurol. 1995;13:29-32.
303 Clancy CA, McGrath PJ, Oddson BE. Pain in children and adolescents with spina bifida. Dev Med Child Neurol. 2005;47:27-34.
304 Edwards RJ, Witchell C, Pople IK. Chronic headaches in adults with spina bifida and associated hydrocephalus. Eur J Pediatr Surg. 2003;13(Suppl 1):S13-S17.
305 Charney EB, Rosenblum M, Finegold D. Linear growth in a population of children with myelomeningocele. Z Kinderchir. 1981;34:415-419.
306 Rosenblum MF, Finegold DN, Charney EB. Assessment of stature of children with myelomeningocele, and usefulness of arm-span measurement. Dev Med Child Neurol. 1983;25:338-342.
307 Rotenstein D, Adams M, Reigel DH. Adult stature and anthropomorphic measurements of patients with myelomeningocele. Eur J Pediatr. 1995;154:398-402.
308 Satin-Smith MS, Katz LL, Thornton P, et al. Arm span as measurement of response to growth hormone (GH) treatment in a group of children with meningomyelocele and GH deficiency. J Clin Endocrinol Metab. 1996;81:1654-1656.
309 Hayes-Allen MC, Tring FC. Obesity: Another hazard for spina bifida children. Br J Prev Soc Med. 1973;27:192-196.
310 Mita K, Akataki K, Itoh K, et al. Assessment of obesity of children with spina bifida. Dev Med Child Neurol. 1993;35:305-311.
311 Roberts D, Shepherd RW, Shepherd K. Anthropometry and obesity in myelomeningocele. J Paediatr Child Health. 1991;27:83-90.
312 Fernbach SK, McLone DG. Derangement of swallowing in children with myelomeningocele. Pediatr Radiol. 1985;15:311-314.
313 Hesz N, Wolraich M. Vocal-cord paralysis and brainstem dysfunction in children with spina bifida. Dev Med Child Neurol. 1985;27:528-531.
314 Hayes-Allen MC. Obesity and short stature in children with myelomeningocele. Dev Med Child Neurol Suppl. 1972;27:59-64.
315 Grogan CB, Ekvall SM. Body composition of children with myelomeningocele, determined by 40K, urinary creatinine and anthropometric measures. J Am Coll Nutr. 1999;18:316-323.
316 Littlewood RA, Trocki O, Shepherd RW, et al. Resting energy expenditure and body composition in children with myelomeningocele. Pediatr Rehabil. 2003;6:31-37.
317 Shepherd K, Roberts D, Golding S, et al. Body composition in myelomeningocele. Am J Clin Nutr. 1991;53:1-6.
318 van den Berg-Emons HJ, Bussmann JB, Meyerink HJ, et al. Body fat, fitness and level of everyday physical activity in adolescents and young adults with meningomyelocele. J Rehabil Med. 2003;35:271-275.
319 Trollmann R, Strehl E, Wenzel D, et al. Arm span, serum IGF-1 and IGFBP-3 levels as screening parameters for the diagnosis of growth hormone deficiency in patients with myelomeningocele-Preliminary data. Eur J Pediatr. 1998;157:451-455.
320 Hochhaus F, Butenandt O, Ring-Mrozik E. One-year treatment with recombinant human growth hormone of children with meningomyelocele and growth hormone deficiency: A comparison of supine length and arm span. J Pediatr Endocrinol Metab. 1999;12:153-159.
321 Rotenstein D, Breen TJ. Growth hormone treatment of children with myelomeningocele. J Pediatr. 1996;128(5 Pt 2):S28-S31.
322 Rotenstein D, Bass AN. Treatment to near adult stature of patients with myelomeningocele with recombinant human growth hormone. J Pediatr Endocrinol Metab. 2004;17:1195-1200.
323 Trollmann R, Strehl E, Wenzel D, et al. Does growth hormone (GH) enhance growth in GH-deficient children with myelomeningocele? J Clin Endocrinol Metab. 2000;85:2740-2743.
324 Hochhaus F, Butenandt O, Schwarz HP, et al. Auxo-logical and endocrinological evaluation of children with hydrocephalus and/or meningomyelocele. Eur J Pediatr. 1997;156:597-601.
325 Meyer S, Landau H. Precocious puberty in myelomeningocele patients. J Pediatr Orthop. 1984;4:28-31.
326 Perrone L, Del Gaizo D, D’Angelo E, et al. Endocrine studies in children with myelomeningocele. J Pediatr Endocrinol. 1994;7:219-223.
327 Trollmann R, Dorr HG, Strehl E, et al. Growth and pubertal development in patients with meningomyelocele: A retrospective analysis. Acta Paediatr. 1996;85:76-80.
328 Galluzzi F, Bindi G, Poggi G, et al. [Precocious puberty, Gh deficiency and obesity can affect final height in patients with myelomeningocele: Comparison of males and females]. Pediatr Med Chir. 1999;21:73-78.
329 Laurence KM, Beresford A. Continence, friends, marriage and children in 51 adults with spina bifida. Dev Med Child Neurol. 1975;Suppl 35:123-128.
329a Cass AS, Bloom BA, Luxenberg M. Sexual function in adults with myelomeningocele. J Urol. 1986;136:425-426.
329b Sawyer SM, Roberts KV. Sexual and reproductive health in young people with spina bifida. Dev Med Child Neurol. 1999;41:671-675.
329c Decter RM, Furness PD3rd, Nguyen TA, et al. Reproductive understanding, sexual functioning and testosterone levels in men with spina bifida. J Urol. 1997;157:1466-1468.
329d Verhoef M, Barf HA, Vroege JA, et al. Sex education, relationships, and sexuality in young adults with spina bifida. Arch Phys Med Rehabil. 2005;86:979-987.
330 Sandler AD, Worley G, Leroy EC, et al. Sexual function and erection capability among young men with spina bifida. Dev Med Child Neurol. 1996;38:823-829.
331 Palmer JS, Kaplan WE, Firlit CF. Erectile dysfunction in patients with spina bifida is a treatable condition. J Urol. 2000;164(3 Pt 2):958-961.
332 Muller T, Arbeiter K, Aufricht C. Renal function in meningomyelocele: Risk factors, chronic renal failure, renal replacement therapy and transplantation. Curr Opin Urol. 2002;12:479-484.
333 McDonnell GV, McCann JP. Issues of medical management in adults with spina bifida. Childs Nerv Syst. 2000;16:222-227.
334 Milton CA, Sanders P, Steele PM. Late cardiopulmonary complication of ventriculoatrial shunt. Lancet. 2001;358:1608.
335 Vernet O, Rilliet B. Late complications of ventriculoatrial or ventriculoperitoneal shunts. Lancet. 2001;358:1569-1570.
336 Beeker TW, Scheers MM, Faber JA, et al. Prediction of independence and intelligence at birth in meningomyelocele. Childs Nerv Syst. 2006;22:33-37.
337 Fletcher JM, Copeland K, Frederick JA, et al. Spinal lesion level in spina bifida: A source of neural and cognitive heterogeneity. J Neurosurg. 2005;102(3 Suppl):268-279.
338 Brookshire BL, Fletcher JM, Bohan TP, et al. Verbal and nonverbal skill discrepancies in children with hydrocephalus: A five-year longitudinal followup. J Pediatr Psychol. 1995;20:785-800.
339 Fletcher JM, Bohan TP, Brandt ME, et al. Morpho-metric evaluation of the hydrocephalic brain: relationships with cognitive development. Childs Nerv Syst. 1996;12:192-199.
340 Rendeli C, Salvaggio E, Sciascia Cannizzaro G, et al. Does locomotion improve the cognitive profile of children with meningomyelocele? Childs Nerv Syst. 2002;18:231-234.
341 Schoenmakers MA, Uiterwaal CS, Gulmans VA, et al. Determinants of functional independence and quality of life in children with spina bifida. Clin Rehabil. 2005;19:677-685.
342 Davidovitch M, Manning-Courtney P, Hartmann LA, et al. The prevalence of attentional problems and the effect of methylphenidate in children with myelo-menigocele. Pediatr Rehabil. 1999;3:29-35.
343 Kawamura T, Nishio S, Morioka T, et al. Callosal anomalies in patients with spinal dysraphism: Correlation of clinical and neuroimaging features with hemispheric abnormalities. Neurol Res. 2002;24:463-467.
344 Gaston H. Ophthalmic complications of spina bifida and hydrocephalus. Eye. 1991;5(Pt 3):279-290.
345 Pinello L, Bortolin C, Drigo P. [Visual motor and visual defects in spina bifida]. Pediatr Med Chir. 2003;25:437-441.
346 Lennerstrand G, Gallo JE, Samuelsson L. Neuro-ophthalmological findings in relation to CNS lesions in patients with myelomeningocele. Dev Med Child Neurol. 1990;32:423-431.
347 Tubbs RS, Soleau S, Custis J, et al. Degree of tectal beaking correlates to the presence of nystagmus in children with Chiari II malformation. Childs Nerv Syst. 2004;20:459-461.
348 Merguerian PA, Klein RB, Graven MA, et al. Intra-operative anaphylactic reaction due to latex hypersen-sitivity. Urology. 1991;38:301-303.
349 Moneret-Vautrin DA, Mata E, Gueant JL, et al. High risk of anaphylactic shock during surgery for spina bifida. Lancet. 1990;335:865-866.
350 Niggemann B, Buck D, Michael T, et al. Latex provocation tests in patients with spina bifida: Who is at risk of becoming symptomatic? J Allergy Clin Immunol. 1998;102(4 Pt 1):665-670.
351 Obojski A, Chodorski J, Barg W, et al. Latex allergy and sensitization in children with spina bifida. Pediatr Neurosurg. 2002;37:262-266.
352 Shah S, Cawley M, Gleeson R, et al. Latex allergy and latex sensitization in children and adolescents with meningomyelocele. J Allergy Clin Immunol. 1998;101(6 Pt 1):741-746.
353 Buck D, Michael T, Wahn U, et al. Ventricular shunts and the prevalence of sensitization and clinically relevant allergy to latex in patients with spina bifida. Pediatr Allergy Immunol. 2000;11:111-115.
354 Mazon A, Nieto A, Pamies R, et al. Influence of the type of operations on the development of latex sensitization in children with myelomeningocele. J Pediatr Surg. 2005;40:688-692.
355 Pires G, Morais-Almeida M, Gaspar A, et al. Risk factors for latex sensitization in children with spina bifida. Allergol Immunopathol (Madr). 2002;30:5-13.
356 Machado M, Sant’anna C, Aires V, et al. [Latex and banana allergies in children with myelomeningocele in the city of Rio de Janeiro]. Rev Assoc Med Bras. 2004;50:83-86.
357 Cremer R, Kleine-Diepenbruck U, Hering F, et al. Reduction of latex sensitisation in spina bifida patients by a primary prophylaxis programme (five years experience). Eur J Pediatr Surg. 2002;12(Suppl 1):S19-S21.
358 Folio MR, Fewell R. Peabody Developmental Motor Scales and Activity Cards (PDMS). Itasca, IL: River Side, 1983.
359 Russell DJ, Rosenbaum PL, Avery LM, et al. Gross Motor Function Measure (GMFM-66 and GMFM-88) User’s Manual (Clinics in Developmental Medicine). London: Mac Keith Press, 2002.
360 Fedman AB, Haley SM, Coryell J. Concurrent and constructive validity of the pediatric evaluation of disability inventory. Phys Ther. 1990;70:602-610.
361 Haley SM, Coster WJ, Ludlow LH, et al. Pediatric Evaluation of Disability Inventory (PEDI) Version 1.0: Developmental, Standardization, and Administration Manual. Boston: New England Medical Center Hospitals, 1992.
362 Hamilton BB, Granger CV. WeeFIM. Buffalo: Research Foundation of the State University of New York, 1991.
363 Ottenbacher KJ, Msall ME, Lyon NR, et al. Interrater agreement and stability of the Functional Independence Measure for Children (WeeFIM): Use in children with developmental disabilities. Arch Phys Med Rehabil. 1997;78:1309-1315.
364 Damiano DL, Gilgannon MD, Abel MF. Responsiveness and uniqueness of the pediatric outcomes data collection instrument compared to the gross motor function measure for measuring orthopaedic and neurosurgical outcomes in cerebral palsy. J Pediatr Orthop. 2005;25:641-645.
365 Bayley N. Bayley Scales of Infant Development, 2nd ed. San Antonio, TX: The Psychological Corporation, 1993.
366 Roid G. Stanford-Binet Intelligence Scales, 5th ed. Itasca, IL: Riverside, 2003.
367 . Record Form. Weschsler Preschool and Primary Scale of Intelligence. 3rd ed. San Antonio, TX: The Psychological Corporation; 2002.
368 Wechsler Intelligence Scale for Children, 4th ed, Response Booklet 1. San Antonio, TX: The Psychological Corporation, 2003.
369 Achenbach T. Manual for the Child Behavior Checklist and Revised Child Behavior Profile. Burlington: University of Vermont, 1991.
370 Achenbach T. Manual for the Child Behavior Checklist and Youth Self-Report. Burlington: University of Vermont, 1991.
371 Achenbach T. Manual for the Teacher Report Form and the Child Behavior Profile. Burlington: University of Vermont, 1991.
372 Bartonek A, Saraste H. Factors influencing ambulation in myelomeningocele: A cross-sectional study. Dev Med Child Neurol. 2001;43:253-260.
373 Bartonek A, Saraste H, Knutson LM. Comparison of different systems to classify the neurological level of lesion in patients with myelomeningocele. Dev Med Child Neurol. 1999;41:796-805.





