[level-membership-for-neurosurgery-category]
Chapter 117 Molecular Therapies for Movement Disorders
Movement disorders, such as Parkinson disease (PD), are currently treated pharmacologically by systemic administration of drugs that replace, mimic, or potentiate lost neurotransmitters, and more recently with neurosurgical procedures such as deep brain stimulation of the subthalamic nucleus (STN) or internal segment of the globus pallidus (GPi)1,2 to replace older, more destructive procedures such as lesioning of the basal ganglia. However, advances in understanding of the pathophysiology underlying movement disorders coupled with technical improvements in and increasing experience with stereotactic surgery for these diseases, have paved the way for application of molecular therapies to treat human movement disorders. Molecular therapies delivered with stereotactic surgical techniques offer the potential to combine the benefits of focal therapies that would not adversely influence other brain regions with the specificity of intervening in biological processes that may more effectively and efficiently alter or improve abnormal neuronal functions in movement disorders.
Drug/Chemical Infusions
Drug infusion therapy directly into the central nervous system (CNS) is an attractive treatment modality in movement disorder patients with advanced disease. In PD, for example, the effectiveness of oral L-dopa therapy is limited due to its relatively short half-life, and the pulsatile stimulation of dopaminergic neurons can cause significant motor fluctuations and dyskinesias.3 Focal infusion of therapy to the area of dysfunction, however, could help achieve more constant striatal dopaminergic stimulation, and more closely mimic the body’s normal physiologic state.
The first demonstration of the effectiveness of focal infusion of medications within the brain parenchyma was conducted in PD patients, who were tested prior to stereotactic lesioning or electrode placement. These patients were infused with small-volume, low-dose muscimol, a GABA-A agonist, into the globus pallidus just prior to pallidotomy,4 and they displayed a rapid onset of improvement in finger motor speed, reduced extremity tone, and decreased muscle fatigue, which all returned to baseline slowly after approximately 30 minutes. Following subsequent pallidotomy, the patients displayed a similar improvement in motor symptoms. In addition, studies in MPTP-treated nonhuman primates have demonstrated that local inactivation of neurons in the subthalamic nucleus results in an improvement in parkinsonian symptoms.5–7 These observations prompted intraoperative exploration of microinjections of either lidocaine, an anesthetic that selectively blocks axonal sodium channels, or muscimol into the STN of six PD patients to study the effects of local STN inactivation. This resulted in transient improvement in akinesia, rigidity, and limb tremor in patients with PD. Electrophysiologic recordings were also performed, which confirmed inhibition of electrical activity in the nucleus following drug infusion as it spread from the injection site to more distant tissue.
Although muscimol microinjection and electrical stimulation by STN deep brain stimulation (DBS) have the same effects on PD motor symptoms, their mechanisms of action are quite different. The precise mechanism of action of electrical stimulation on symptomatic improvement in movement disorders is still unclear, there is support for one or more functions, including excitation of neighboring axons, activation of recurrent inhibitory circuits, and direct inhibition of neuronal firing.8 As opposed to these more indirect mechanisms, muscimol acts by specifically activating chloride-dependent GABA-A receptors and thereby inhibiting neurons presynaptically. It is still unknown whether continuous infusion of muscimol is superior to stimulation, since the specificity of action provides opportunities for more direct conclusions regarding the relationship between neurotransmitter receptor action and symptomatic improvements, but there may as yet be unknown benefits of some more nonspecific or broader actions of electrical stimulation. In addition, a crucial question that must still be answered is whether patients will develop tolerance to the drug with time. In animals, one unpublished study has demonstrated that parkinsonian rats with unilateral 6OHDA lesions show sustained effects when muscimol is administered by an implanted osmotic pump for 2 weeks,9 but longer-term consequences remain to be seen. However, it is clear to every practitioner of DBS that some patients can have adverse effects due to spread of electrical stimulation outside the STN or to nonmotor areas of the STN despite proper electrode location, and this could be less problematic with a biologically specific therapy such as muscimol infusion. The development of appropriate technology for chronic infusions into brain parenchyma, which have yet to be fully developed, would help facilitate development and ultimate determination of the value of continuous muscimol infusion into basal ganglia targets as a therapy.
In addition, patients with essential tremor (ET) have also been studied through a local drug infusion modality, and one study demonstrated the cessation of tremor in patients microinjected with muscimol into the ventralis intermedius thalamus (Vim).10 In this study, six ET patients undergoing unilateral stereotactic thalamic procedures for relief of tremor refractory to medications were given either saline and/or muscimol intracerebral microinjections prior to thalamotomy or thalamic DBS electrode placement. The patients displayed suppression of tremor for an average of 9 minutes, following a mean latency of 9 minutes to allow for sufficient drug diffusion to influence enough neurons for a therapeutic effect. This not only provides further support for the belief that inhibition of Vim and STN activity can improve symptoms of ET and PD, respectively, but this presents another opportunity for application of direct intraparenchymal drug infusion for a second movement disorder, should technology permit further development of this therapy.
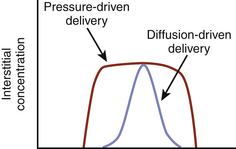
In addition to the therapeutic benefit of local microinjection of drugs such as lidocaine or muscimol, selective infusion of dopamine or dopaminergic medications into the basal ganglia has also been considered, in order to bypass the systemic side effects seen with conventional oral intake of these medications. Chronic intraventricular or intrastriatal dopamine infusions was investigated in 6-OHDA lesioned rats, which demonstrated improvement in motor symptoms following 7 days of continuous dopamine infusion compared to controls.11 However, local examination of the striatum adjacent to the point of intrastriatal dopamine infusion revealed a barrier, which limited diffusion of medication to only 1 to 2 mm from the catheter. In an effort to increase the size of the perfused region, convection techniques have been attempted that move both fluid and drug into the extracellular space by bulk flow driven by a pressure gradient (Fig. 117-1).12–14 The data are still rather limited, and ideal flow rates, catheter diameter, and infusion volume have not yet been determined.15
Other techniques that may allow for efficient delivery and monitoring of drug infusions locally within the brain include microdialysis and voltammetry (Table 117-1). Cerebral microdialysis is a modality that allows measurement of the concentration of extracellular neurochemicals within cerebral structures, locally and in vivo. Initially conducted in 1966 by using a dialysis membrane filled with dextran solution into canine cortex, with measurement of local amino acid concentrations,16 this method has also been applied to PD patients.17 Currently, cerebral microdialysis consists of a double-lumen probe containing an inlet and an outlet port, surrounded by a semipermeable membrane, and sealed above and below a point at which the two tubes are inserted into the membrane lumen. This dialysis probe can be inserted through a burr hole to a preset depth through the brain parenchyma, allowing for perfusion of solutions through the inlet port as well as analysis of the returning solution for extracellular molecule concentrations with techniques such as high-performance liquid chromatography with electrochemical detection (HPLC EC). HPLC EC provides high sensitivity and specificity for the measurement of molecules, such as biogenic amine, including noradrenaline, dopamine, and serotonin, as well as for amino acids, including glutamate, aspartate, and GABA.18–20
| Focal infusion |
| Muscimol (GABA-A agonist). Lidocaine (Na+-channel antagonist). Dopamine (both intrastriatal and intraventricular infusions). |
| Microdialysis |
| Extended monitoring levels of a particular neurotransmitter in probe region; may allow for pump that would administer a drug or chemical in response to a certain level, allowing for achievement of steady-state levels of that neurotransmitter. |
| Voltammetry |
| Detects changes in the concentration of certain neurotransmitters (transmitter oxidation), and allows for excellent temporal and spatial resolution of their release in real time. |
Some limitations of microdialysis, however, include invasiveness, reports of complications in the literature, dependence of accuracy on factors such as flow rate and probe size, and a local fibrotic reaction to the probe tip seen during chronic microdialysis experiments.19,21 In addition, the neurochemicals sampled during microdialysis are not only those released synaptically, but also include neurochemicals of local metabolism, capillary delivery, and neuronal release and uptake from groups of cells.22,23 Despite these limitations, microdialysis has been used successfully in human patients, ranging from traumatic brain injury and subarachnoid hemorrhage to movement disorders and epilepsy.18,22 In movement disorders, particularly PD, there have been published studies such as one demonstrating the changes in concentrations of GABA, cGMP, and glutamate before, during, and after 1 hour in the internal globus pallidus, anteroventral thalamus, and putamen of six patients with refractory PD undergoing DBS in the STN.24,25 In addition, an in vivo microdialysis method was developed permitting the continuous, chronic sampling of local neurotransmitter concentrations in the STN and substantia nigra pars reticulata (SNr) of patients for several days after placement of STN DBS electrodes.26 All six patients in this study had no complications, and the microdialysis probes were successfully removed without disrupting the DBS electrode location. In theory, microdialysis use may be extended to monitoring levels of a particular neurotransmitter in the region where the probe is implanted, which could then be attached to a pump that would administer a drug or chemical in response to a certain level, allowing for achievement of steady-state levels of that neurotransmitter. Microdialysis has in fact been used to monitor systemic drug levels and to infuse agents into the brain in epilepsy patients, and this could easily be extended to movement disorders if improved technology permitted chronic use of these devices.27–29
In addition to microdialysis, voltammetry is another method that can detect changes in the concentration of certain neurotransmitters, and allows for excellent temporal and spatial resolution of their release in real-time.30,31 Voltammetry is based on the principle that oxidation of certain chemicals leads to the release of electrons, and the resultant current can be measured with an electrode.32 Furthermore, a dialysis electrode was developed that combined microdialysis with voltammetry to measure glutamate,33 which specifically contained a dialysis probe with an electrochemical detection system utilizing the enzyme glutamate oxidase to detect glutamate levels. Other systems have been designed that measure the concentrations of molecules such as glucose and oxygen.34,35 Most applications have currently been in animal models, including the construction of a micromanipulator in rats that allowed for both infusion of drugs and voltammetry in freely moving rats36 and mostly studied in tumors, cerebral ischemia, and traumatic brain injury. However, the application of voltammetry has also been extended to the measurement of dopamine in both rats and nonhuman primates. Voltammetry has been used to measure extracellular dopamine concentration changes in rats, such as during certain behaviors or in response to cocaine administration in the nucleus accumbens.37 In regard to movement disorders, there was a recent study using carbon fiber voltammetry to measure the release of dopamine in MPTP-induced dopaminergic lesions in nonhuman primates.38 In addition to its utilization in probing drug effects on dopamine release and uptake, there may also future applications of voltammetry to measuring dopamine release in patients, with tailored therapeutic release of medication by external or internal means in response to certain changes in levels. Although microdialysis can sample any neurotransmitter which can diffuse across the dialysis membrane, voltammetry is currently limited to monitoring a select population of neurotransmitters due to technical constraints of the voltammetry methodology to measure transmitter oxidation. However, because it does not involve a membrane or a tube, physical blockade of fluid diffusion due to luminal obstruction is not a concern and oxidation could be more easily measured by an implanted electrical device compared with chemical analysis of dialysate fluids. Therefore, voltammetry could represent an attractive prospect for long-term monitoring of brain neurotransmission as a component of a dynamic feedback mechanism to tailor intraparenchymal drug delivery to specific patient needs.
Growth Factors/Recombinant Proteins
As an alternative to infusion of drugs into the CNS for the treatment of movement disorders, another potential strategy employs the infusion of growth factors or recombinant proteins. Since the central feature of patients with PD is the loss of dopaminergic neurons in the substantia nigra, researcher proposed the idea of administrating neurotrophic factors to degenerating neurons in order to promote their survival. Specifically, the leading candidate neurotrophic factor was glial cell–line derived neurotrophic factor (GDNF), discovered in 199339 and shown to enhance the survival of dopaminergic neurons in vitro.40 In animal models, the goal of GDNF was to prevent further neuronal loss while encouraging reinnervation of the striatum from any surviving, intact terminal within the striatum. Since GDNF is a 134–amino acid peptide, it does not efficiently cross the blood brain barrier, and thus it must be delivery locally into the CNS either by administration into the ventricular system or directly into the brain parenchyma. In preclinical studies with both 6-OHDA- and MPTP-lesioned animal models, GDNF was delivered using both approaches into the CNS, and it indeed did show some benefit.41–43
Eventually, infusion of a recombinant form of GDNF was administered in human clinical trials (Table 117-2). Initially, 50 patients with moderately advanced PD were treated with intraventricular GDNF; however, the infusion trial was stopped early due to excessive complications from toxicity to the periventricular structures.44 Furthermore, there was a lack of therapeutic efficacy, potentially due to failure of GDNF diffusion across the greater distance between the ventricular system and substantia nigra when compared to nonhuman animal models. Subsequently, this led to direct intraparenchymal infusion of GDNF into the striatum (postcommissural putamen) in an open-label phase 1 study that demonstrated both encouraging safety and efficacy results.45 Five PD patients were treated, with a resulting significant decrease in total Unified Parkinson’s Disease Rating Scale (UPDRS) score in the “off” state at 3 months, as well as mean reduction in UPDRS motors score at 12 months of 48%. There were also correlational 18F-dopa PET scans showing an increased uptake in the area of the GDNF infusion. Since 18F-dopa PET measures dopamine synthetic capacity and by extension dopaminergic innervation of the striatum, these results did support potential enhancement of dopaminergic function in the remaining viable nigrostriatal neurons, and a postmortem case from the study later did demonstrate sprouting of dopaminergic fibers around the GDNF infusion site.46
Table 117-2 Infusion of Recombinant GDNF into CNS of Parkinson Disease Patients
| Central tenet of Parkinson’s Disease |
| Loss of dopaminergic neurons in substantia nigra. |
| Goals |
| Promote survival of degenerating dopaminergic neurons by neurotrophic factors. Encourage reinnervation of the striatum by remaining, intact intrastriatal terminals. |
| Outcomes |
| Intraventricular infusion of GDNF |
| Clinical trials stopped early due to: Toxicity to periventricular structures Question of diffusion limitations of GDNF from ventricular system to substantia nigra |
| Intraparenchymal infusion of GDNF |
| Open-label phase 1 study showed encouraging safety and efficacy results. Improvement in UPDRS motor scores. 18F-dopa PET scans showing an increased uptake in the area of the GDNF infusion. Postmortem sprouting of dopaminergic fibers around the GDNF infusion site. Phase 2 double-blind, placebo-controlled trial results: no significant effect of GDNF was reported in either primary or secondary outcome measures. |
These initial promising results did not translate into success in a phase II double-blind, placebo controlled trial that subsequently followed.47 In this study, 34 patients with moderately advanced PD were bilaterally implanted with a single port catheter to infuse either drug or placebo continuously into the putamen, and no significant effect of GDNF was reported in either primary or secondary outcome measures (except for 18F-dopa PET scan data), leading to considerable debate regarding the reasons for the conflicting results between the open-label and double-blind, placebo-controlled studies. There has long been concern about potential placebo effects confounding trials of new therapies for PD, but the relative absence of substantial benefit in either the treatment or control groups suggests that placebo was not a cause of the different results observed in the two trials. It is possible that the change in the type of infusion catheter and mechanism of infusion (differences in diameter and total amount of GDNF delivered) may have negatively affected the therapy and led to a false-negative result. The study may also not have been adequately powered to demonstrate an effect. This highlights the difficulties that can limit successful translation of promising and potentially efficacious therapies. Given the expense and limited likelihood of success for any new therapy, it can be difficult to develop optimal technology for phase 1 studies. However, when results justify later-phase trials, an incentive is created to improve technology so that the methodology used in the more advanced trial can be more readily extrapolated for general use. Furthermore, patient recruitment and financial considerations often limit the size of randomized studies. These understandable and often unavoidable issues may nonetheless compromise later phase trials and can create difficulties resolving discrepancies with early phase data when subsequent results do not justify continued development. This is not unique to growth factor infusion, and has plagued other promising surgical therapies, including cell transplantation and gene therapy. But these issues must be considered when evaluating results of surgical trials for new molecular therapies, in order to improve future studies and to not automatically reject treatments that may have genuine promise due to disappointing results from a single study.
Gene Therapy
Basic Science of Gene Therapy
Gene therapy strategies can generally be subdivided into two different categories: ex vivo and in vivo (Table 117-3). Ex vivo gene therapy involves transplantation of target cells previously genetically altered in cell culture by plasmid or retroviral vectors before transplantation into the target organ in the patient. These cells can be thought of as genetically engineered inside in situ factories or “minipumps”48 that are able to produce the therapeutic protein or factor of interest within host tissue. One advantage of ex vivo gene therapy is the ability to screen for adequate gene expression prior to introduction into the patient. This method can also be used to alter the characteristics of cells that are themselves intended to be therapeutic, such as modifying stem cells in order to increase survival or improve interaction with host brain. Potential difficulties with ex vivo gene therapy include potential oncogenic properties and unfavorable host immune response against foreign transplanted cells that can lead to rejection of transplanted cells or inflammation in target tissue.49,50
| Ex vivo delivery: Genetically modified cells transplanted into target tissue. |
| Advantages |
| Allows for prior screening for adequate gene expression. Can alter cells to increase survival or improve interaction with host brain. |
| Disadvantages |
| Potential oncogenic properties. Unfavorable host immune. Rejection of transplanted cells or inflammation in target tissue. |
| In vivo delivery: Direct transfer of genetic material into the host cells within the body in situ |
| Advantages |
| Cells already established within neural networks. Bypasses potential adverse host immune response to foreign cells. |
| Disadvantages |
| Limited efficacy of direct transfer of naked or liposome-mediated genetic material. Genetically modified viruses typically used as vehicles, can lead to toxicity due to viral replication or host inflammatory reaction to viral proteins. |
Viral Vectors
Historically, the first viral vectors utilized in human CNS gene therapy were designed to treat malignant brain tumors, with the intention of selectively transferring antineoplastic cytotoxic agents to tumors cells,51 administering oncolytic virus therapy,52 and transferring tumor suppressor genes or immunomodulating genes.53,54 There have been a multitude of different viral vectors employed in gene therapy for PD, including both RNA-based retroviral vectors and DNA viral vectors (Table 117-4). Initially, retroviruses were used, which are RNA viruses that require synthesis of a copy of their own RNA genome following entrance into the host cell, and thus they carry the reverse transcriptase enzyme along with their genome to accomplish this task.55 Furthermore, these agents also require integration into the host cell’s genome for full functionality, thus most retroviral vectors are limited by the necessity of active cell division within the target cell type, and retroviral vector applications in the CNS have mostly been used for tumor therapy.56 Retroviral vectors can be used as part of ex vivo gene therapy to modify cells in culture prior to transplantation into target tissue.57,58 This has been used in preclinical PD studies, but human applications in the brain have mostly been limited to Alzheimer’s disease.59–61 More recently, lentiviruses, which are a unique class of RNA-based retroviral vectors, have emerged as novel agents for both ex vivo and in vivo gene therapy with the CNS. In contrast to other types of retroviruses, lentiviruses are advantageous in theory due to their unique biology of gene transfer in both dividing and nondividing cells.62,63 Included in this family of viruses is human immunodeficiency virus (HIV), which inherently raises some concerns for patient safety. However, most lentiviral vectors used in the CNS for preclinical studies are nonhuman and/or non-HIV lentiviruses, and also further modified to prevent both proliferation and recombination with potentially more dangerous viruses.64,65 Both the safety and efficacy profiles appear promising in animals, and the first human clinical trial using lentiviral vectors to delivery dopamine biosynthetic enzymes to the striatum of human PD patients was recently initiated.66
Table 117-4 Overview of Four Types of Viral Vectors Used in Gene Therapy for Parkinson Disease
| Herpes simplex virus |
| Preclinical data for long-term behavioral recovery in Parkinsonian rats by a herpes simplex virus vector expressing tyrosine hydroxylase. Largely abandoned in Parkinson disease due to cytotoxicity in the central nervous system, low transfection efficiency, and inadequate long-term gene expression. |
| Adenovirus |
| Largely abandoned due to concerns of inflammatory host responses, decreasing transgene expression, and toxicity. |
| Lentivirus |
| Phase 1/2 study of intraputamen lenti-TH-AADC-GCH1 (three dopamine biosynthetic enzymes). Based on preclinical data: MPTP-induced Parkinsonian macaque, demonstrated marked improvement in tremor and bradykinesia, stable for 1 year. |
| Adeno-associated virus |
| Phase 1 study of AAV-GAD gene (glutamic acid decarboxylase) into the subthalamic nucleus of Parkinson disease patients |
| Significant improvement in off- and on-medication UPDRS scores at 3 months, with continuation to the end of study at 1 year. Significant correlation between clinical motor scores and increased brain metabolism in supplementary motor area of the treated side. |
| Phase 1 study of bilateral intrastriatal AAV-neurturin (glial cell-line–derived neurotrophic factor [GDNF] family protein) |
| Significant improvement in off-medication UPDRS score, as well as increase in “on” time without dyskinesia. No change in 18F-levodopa uptake PET. |
| Phase 2 study of AAV-neurturin |
| Failure to meet primary endpoint of improvement in the UPDRS motor off score at 12 months of follow-up |
| Phase 1 study of bilateral intrastriatal AAV-AADC |
| Improvement in UPDRS scores in both the “off” and “on” states. Increased intraputamenal FMT uptake in both cohorts, dose-responsive manner. Increased risk of intracranial hemorrhage seen. |
AADC, aromatic acid decarboxylase; AAV, adeno-associated virus; UPDRS, Unified Parkinson’s Disease Rating Scale.
The more widely used gene transfer agents in clinical trials other than oncology trials are DNA viral vectors, and the most commonly utilized DNA viral vectors are herpes simplex virus (HSV), adenovirus, and adeno-associated virus (AAV). In contrast to retroviruses, DNA viruses use DNA as their genome, and thus they do not require active cell division since DNA enters into the nucleus of the host cell and expression of the desired therapeutic gene occurs without further DNA synthesis. Historically, the first DNA virus used as a gene transfer system was the herpes simplex virus type 1 (HSV1), which naturally targets the nervous system by initial infection of peripheral nerves, with eventual spread into the CNS.67 This innate tropism of HSV1 vectors for the nervous system initially warranted marked enthusiasm, and the first viral vectors were altered to eliminate certain gene required for replication, spread, and/or pathogenesis. However, the HSV vectors presently used in gene therapy, whether recombinant or defective, had multiple limitations including cytotoxicity within the CNS, low transfection efficiency, and inadequate long-term gene expression.68 However, recent technological advances have led to significant improvements that allow for the clinical application of HSV vectors, albeit mostly for disease of peripheral nerves, including diabetic neuropathic and neuropathic pain.69,70 The use of “conditionally replicating” vectors, which proliferate only in dividing cells, has been mostly limited to brain tumor therapy.71 In addition, a variation of the HSV vector, termed an “amplicon,” which contains only HSV replication and packaging signals without HSV genes, requiring another source to provide the HSV gene products and allow packaging of the amplicon into an HSV coat, has displayed efficient gene transfer to neurons in cell culture and living animal brain.72,73 However, production of this system remains cumbersome, and it has not yet reached consideration in human clinical trials.74
Vectors derived from adenovirus have also been used, albeit mainly in clinical trials involving brain tumors. Adenovirus is significantly smaller than HSV, and as a result it contains fewer genes that require modification to provide a safe, efficient transfer system in biological systems. Recent systems involving packaging similar to HSV amplicons have been developed and conditionally-replicating adenovirus vectors have also been created,75 which selectively multiply in dividing cells only. Limitations, however, are abundant, including safety and longevity of gene expression.76,77 Adenoviral-mediated gene transfer currently remains a difficult system to use for gene transfer from a technological standpoint, but advancement in the field may lead to its application in clinical use in the future.
The most widely used vector system for genetic transfer in brain for non-neoplastic diseases is the AAV. Some advantages of AAV include a small size in comparison to the other DNA viruses used as vectors and a lack of association with human disease.78,79 AAV vectors lack the AAV genome, and thus the gene of interest can selectively be packaged into an AAV coat and transferred to host cells.80 Furthermore, continued development of AAV-gene transfer technology has led to the creation of virus in sufficient concentration to allow application in human clinical trials. Of the various serotypes of AAV, only AAV2 has been used in human clinical trials for non-neoplastic neurologic disease. This stems from data demonstrating the ability of AAV2 to become incorporated into neurons with long-term stability and safety, and also functional improvement in animal models of PD.81 AAV2 has also been demonstrated to be safe, stable, and effective in the primate CNS,82 and as demonstrated below and in studies for diseases other than PD, the safety profile of AAV2 has been excellent. In recent years, new subtypes and variants of AAV have emerged, allowing clinicians an even wider array of tools for human gene therapy in the future.83
Gene Therapy in Parkinson Disease
The majority of gene therapy in PD focuses either on replacing enzymes involved in dopamine biosynthesis or neuroprotection with targeted cellular production of neurotrophic factors. With the exception of cancer, gene therapy has been tested more in PD than in any other disorder, and there are currently three main clinical approaches to gene therapy for PD.84–87 In PD, there is substantial loss of dopaminergic cells of the substantia nigra pars compacta, and subsequent diminution of striatal dopaminergic transmission and dysregulation of the circuitry within the basal ganglia modulating cortical control of movement.88 Although the etiology of this pathologic cell loss is still unknown, the anatomic considerations and functional implications are better understood than in most other neurologic diseases, and thus facilitates development of biologically and anatomically targeted technologies such as gene therapy.
The first in vivo gene therapy to be tested in a human neurodegenerative disorder was infusion of AAV carrying the glutamic acid decarboxylase (GAD) gene into the STN.89 GAD is the rate-limiting enzyme in the production of GABA, the main inhibitory neurotransmitter in the brain, and the loss of dopamine in patients with PD leads to a decrease in GABAergic transmission in both the STN and its targets, such as the GPi and SNr.90–93 This leads to reduced thalamocortical activity and symptoms of akinesia and rigidity. Since hyperactivity of the STN in PD is believed to play a central role in the motor dysfunction characteristic of PD, gene therapy with AAV-GAD should allow transduced STN neurons to produce GABA and subsequently release this neurotransmitter both locally and to downstream targets. Thus, AAV-GAD should functionally reestablish the GABAergic tone necessary to the distal areas of the basal ganglia circuitry that are pathologically hyperactive. In rodent models, this theory was confirmed in a study using microdialysis; an increase was measured in evoked GABA release into the SNr following STN AAV-GAD gene therapy.89 Since GAD converts glutamate to GABA, this should reduce glutamate production within the hyperactive STN, which could further normalize basal ganglia circuit activity in PD. Given the fact that preclinical data are often difficult to translate into successful therapies in humans, this approach attempts to capitalize on successful STN surgery in human PD, including DBS and lesioning.93–95 In addition, although STN AAV-GAD gene therapy may initially seem a strictly symptomatic therapy, there is some evidence that it may also exert a neuroprotective function, as preclinical studies in animal models with PD suggest.89 Excessive glutaminergic input to the substantia nigra in PD may result in glutamate-mediated excitotoxicity, and reducing this may block the excitotoxic effect of excessive glutamate on dopaminergic cells seen in this movement disorder.
Following the encouraging preclinical data, a phase 1 study of AAV-GAD gene therapy for PD was conducted,84 involving 12 patients with advanced PD who normally would have fulfilled the criteria necessary to receive DBS. As the first attempt to use in vivo gene therapy in a neurologic disorder, only unilateral therapy was approved to protect against a possible unanticipated, severe adverse reaction with potentially devastating implications if bilateral structures were affected. All 12 patients demonstrated bilateral disease, and the more severe hemisphere was chosen to receive the gene therapy, while the untreated symptomatic side was available for clinical and functional comparison. Thus, although the study was not blinded or placebo-controlled, it differed from most phase 1 trials in the sense that a control hemisphere was available for comparison within each treated patient. This study demonstrated that infusion of AAV-GAD gene therapy in the STN of patients with advanced PD was safe at the doses tested, the primary endpoint of the study,84 and there were no significant adverse events associated with the therapy for the 1 -year duration of the formal study. There was also significant improvement in function as assessed by part III of the standard and widely used USPRS, with total body score significantly improved starting at 3 months after treatment, and continuing for the length of the 1-year follow-up, including both “on” and “off” medication. When analyzed by body side, the effect was restricted to the hemibody opposite the treated hemisphere in both conditions over the time period. Furthermore, there was no significant benefit during the first month, consistent with the natural biology of AAV, since AAV-mediated gene expression normally requires several weeks to reach maximal levels. This is also not consistent with a microlesioning effect, which usually occurs immediately and rarely leads to stable improvement even following insertion of a DBS electrode which with a roughly 10-fold larger diameter and 100-fold larger volume than the infusion catheter used to deliver 50 μl of AAV solution in this study. Functional imaging with flourodeoxyglucose PET (FDG-PET) scan yielded some additional encouraging data, with an improvement in neuronal metabolism within pathologic motor circuits, including in the thalamus and cerebral cortex, only in the hemisphere treated with AAV-GAD gene therapy.84 This was similar to the FDG-PET pattern seen following successful treatment with existing therapies, including medication and DBS. In order to further address and confirm the mechanism of action, a subsequent study compared the FDG-PET from the AAV-GAD gene therapy group with patients who were treated with lesioning of the STN.96 This study further suggested that the outcomes following gene therapy were not due to lesioning, as the effect on the GPi (which intervenes between the STN and thalamus) was completely opposite between the two groups, whereas the effect on downstream circuitry, such as in the thalamus, was similar. As a result, these studies provided encouraging support for both safety and potential efficacy of gene therapy for treatment of PD patients. This also highlights the value of using functional imaging to both help provide more objective biological support for clinical improvements in symptoms and to suggest possible mechanisms that may help answer outcomes questions. Recently, a phase 2 trial was initiated to more definitively assess the potential of this therapy. This study treated patients bilaterally, and used a matched blinded, control group of patients who received partial-thickness burr holes but no gene therapy, to address the possibility of a placebo effect, and the results will hopefully be encouraging.
A second gene therapy approach to human PD used a gene for a growth factor in the GDNF family, neurturin, in an attempt to slow progression of disease and promote dopaminergic neuronal plasticity within the striatum. Neurturin can effectively activate GDNF receptors, and it is postulated to have a trophic role in neurons. There were encouraging preclinical data from neurturin gene therapy, again using AAV vectors, in rodent and primate models (Fig. 117-2), which demonstrated efficacy without any notable adverse effects.97–99 In a phase 1 study, patients with advanced PD were divided into two groups, with each group receiving bilateral striatal infusions of either a high or low concentrations of AAV-neurturin, at a total of six straital injection site through three surgical tracts.87 The procedure was well tolerated with no significant adverse events related to the gene therapy. There were also significant improvements in clinical ratings in treated patients, including “off” medication UPDRS and also improvement in total “on” time. Due to this safety and promising efficacy profile, the study quickly progressed to a phase 2 study, being the first gene therapy approach for PD to enter phase 2. The phase 2 study included 58 patients with advanced PD, with a study design of 2:1, with one treated patient for each control, and controls receiving partial-thickness burr holes. Initial results were disappointing, as the corporate sponsor has reported that they did not meet their primary endpoint of improvement in the UPDRS motor off score at 12 months of follow-up and a few secondary endpoints suggesting very modest benefit. However, they have recently indicated that some patients have shown benefit at 18 months. Although these data have not yet been published, it appears likely that further studies will attempt to resolve potential confounding issues in order to better understand the potential of this gene therapy for human PD.
The final gene therapy approach to patients with PD in human clinical trials involves the expression of genes controlling dopamine biosynthesis locally within the striatum. In the first study of this type to reach human trials, an AAV vector was used to transfer the gene for aromatic acid decarboxylase (AADC), the enzyme that converts L-dopa to dopamine, into the human striatum, with the intention of transducing nondegenerating striatal neurons at the site of dopamine action. Since the AADC gene converts L-dopa to dopamine, F-dopa PET was used as an imaging modality to assess AADC activity and dopamine production.100 Although tyrosine hydroxylase (TH), and not AADC, is the rate-limited enzyme in the biosynthesis of dopamine, initial preclinical studies have been particularly successful, especially in primates, demonstrating improvement in both the magnitude and longevity of response to L-dopa drug therapy.101–104 One advantage of using AAV-AADC in contrast to the other gene therapy approaches is that gene expression is controlled and regulated by the levels of L-dopa, and thus an adverse event due to dopamine overproduction could be modified by lowering the dosage of L-dopa. Similar to the AAV-neurturin trials, this study administered virus by bilateral striatal injections but to a single injection site. This has also demonstrated safety without any significant long-term adverse events related to AAV-AADC.86 The study used PET and the AADC tracer fluoro-L-m-tyrosine (FMT) as an in vivo measure of AADC gene expression. Therefore, the availability of a radiolabelled substrate for AADC provided the unique opportunity to directly image the activity of the enzyme expressed from the gene therapy agent rather than the response of relevant neurons to therapy. A significant increase in intraputamenal FMT uptake was noted in the five treated patients in the study, and with modest improvements in their UPDRS motor score. However, as the study was open label, nonblinded, and with a small sample size of five, a follow-up study employed two cohorts of five patients each (low-dose and high-dose bilateral intraputamenal AAV-AADC injections) studied at 6 months. The surgical procedure was unusually associated with an increased risk of intracranial hemorrhage not seen in other studies, as there was one symptomatic and two asymptomatic intracranial hemorrhages following the operative procedure.105 Given the small sample size, this could simply be a chance event, but it is possible that some unique aspect of this particular methodology did increase hemorrhage risk. Meanwhile, both cohorts of patients had improvements in their UPDRS scores in both the off and on states, and there was an increase in intraputamenal FMT uptake in both cohorts (75% high dose vs. 30% low dose), consistent with increased expression of AADC in the high-dose AAV-AADC cohort. However, there was no significant reduction in the levels of dopaminergic medications as previously described in parkinsonian nonhuman primates.106 Further studies using a placebo group may help clarify the potential benefit due to AAV-AADC in a fashion similar to other gene therapy approaches to date.
As an alternative to single-gene transfer by AAV, the most recent human PD gene therapy trial is using a lentiviral vector to transduce the putamen with all three genes necessary for dopamine biosynthesis, including tyrosine hydroxylase, AADC, and guanosine 5’-triphosphate cyclohydrolase (GCH1). Results using MPTP-induced parkinsonian macaques demonstrated marked improvement in PD symptoms, including both tremor and bradykinesia, stable for 1 year, and with one animal even displaying marked improvements 4 years later.107 In addition, this study attempted to address the relevant medication dosages, timing, and adverse effects associated with chronic L-dopa treatment, most notably dyskinesias, and they found that dopaminergic medication failed to induce dyskinesias in lenti-TH-AADC-GCH1-treated animals, possibly due to continuous production of dopamine, rather than the peaks and troughs typically seen with periodic administration of levodopa therapy. However, the safety profile of lentivirus and its long-term consequences still remain to be demonstrated,108 and the effect of lenti-TH-AADC-GCH1 in human patients with PD in unknown. Therefore, following this encouraging preclinical data, a phase 1–2 clinical trial of intraputaminal lenti-TH-AADC-GCH1 for PD has recently been initiated.
Future
Future advances in molecular therapies for the treatment of movement disorders are expected with the development of nanotechnology for delivery of a variety of molecular agents.106 These include the use of improved liposomes, polymeric or lipidic micro- and nano-particles, polymeric micelles, and most recently dendrimers, all of which may facilitate delivery of drugs across the blood–brain barrier and with high specificity and low toxicity or immunogenicity. Combination of peptide or growth factor infusions with other therapies, such as cell therapies, is likely to help advance both fields and lead to more successful restorative therapies. Finally, the rapid advance of gene therapy is likely to continue, regardless of the outcome of current studies. While the primary focus has been almost entirely on PD, there is great promise in preclinical studies of gene therapies for other movement disorders, such as dystonia and Huntington’s disease. Since these are largely genetic diseases, the attraction of a gene-based therapy to reverse the genetic defect or prevent the consequences of the mutant gene is clear. The widespread and rapid advance of a variety of molecular therapies, along with ongoing technological advances to improve safety and efficacy, are likely to provide a range of new therapeutic options for functional neurosurgeons in the near future. Given the important surgical issues that must be addressed with each new therapy, and the lessons learned from the many prior studies outlined above, it is critical that neurosurgeons not only participate in clinical trials of these new therapies but continue to lead the design of these trials and help drive the scientific advances as well.
Benabid A.L., Chabardes S., Mitrofanis J., Pollak P. Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson’s disease. Lancet Neurol. 2009;8(1):67-81.
Clarke C.E., Worth P., Grosset D., Stewart D. Systematic review of apomorphine infusion, levodopa infusion and deep brain stimulation in advanced Parkinson’s disease. Parkinsonism Relat Disord. 2009;15:728-741.
Eberling J.L., Jagust W.J., Christine C.W., et al. Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology. 2008;70:1980-1983.
Christine C.W., Starr P.A., Larson P.S., et al. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology. 2009;73(20):1662-1669.
Feigin A., Kaplitt M.G., Tang C., et al. Modulation of metabolic brain networks after subthalamic gene therapy for Parkinson’s disease. Proc Natl Acad Sci USA. 2007;104(49):19559-19564.
Galati S., Mazzone P., Fedele E., et al. Biochemical and electrophysiological changes of substantia nigra pars reticulata driven by subthalamic stimulation in patients with Parkinson’s disease. Eur J Neurosci. 2006;23:2923-2928.
Gasmi M., Brandon E.P., Herzog C.D., et al. AAV2-mediated delivery of human neurturin to the rat nigrostriatal system: long-term efficacy and tolerability of CERE-120 for Parkinson’s disease. Neurobiol Dis. 2007;27:67-76.
Gill S.S., Patel N.K., Hotton G.R., et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med. 2003;9(5):589-595.
Kaplitt M.G. Parkinson disease: another player in gene therapy for Parkinson disease. Nat Rev Neurol. 2010;6(1):7-8.
Kaplitt M.G., Feigin A., Tang C., et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet. 2007;369(9579):2097-2105.
Kordower J.H., Herzog C.D., Dass B., et al. Delivery of neurturin by AAV2 (CERE-120)–mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Ann Neurol. 2006;60:706-715.
Lang A.E., Gill S., Patel N.K., et al. Randomised controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson’s disease. Ann Neurol. 2006;59:459-466.
Lou J., Kaplitt M.G., Fitzsimons H.L., et al. Subthalamic GAD gene therapy in a Parkinson’s disease rat model. Science. 2002;298(5592):425-429.
Marks W.J.Jr., Ostrem J.L., Verhagen L., et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: an open-label, phase I trial. Lancet Neurol. 2008;7(5):400-408.
Mattson R.H., Scheyer R.D., Petroff O.A., et al. Novel methods for studying new antiepileptic drug pharmacology. Adv Neurol. 1998;76:105-112.
National Institutes of Health, ClinicalTrials.gov [Internet]. Henri Mondor Hospital (Prof. Stephane Palfi), Paris (France), identifier (NCT00627588). A Phase I/II Study of the Safety, Efficacy and Dose Evaluation of ProSavin®, Administered Using Stereotactic Injection to the Striatum of Patients with Bilateral, Idiopathic Parkinson’s Disease, January 8, 2008, Available at http://clinicaltrials.gov/ct2/show/NCT00627588
Nutt J.F., Burchiel K.J., Comella C.L., et al. Randomized, double-blind trial of glial-derived neurotrophic factor (GDNF) in PD. Neurology. 2003;60(1):69-73.
Pahapill P.A., Levy R., Dostrovsky J.O., et al. Tremor Arrest with thalamic microinjections of muscimol in patients with essential tremor. Ann Neurol. 1999;46(2):249-252.
Perez X.A., Parameswaran N., Huang L.Z., et al. Pre-synaptic dopaminergic compensation after moderate nigrostriatal damage in non-human primates. J Neurochem. 2008;105:1861-1872.
Raghavan R., Brady M.L., Rodríguez-Ponce M.I., et al. Convection-enhanced delivery of therapeutics for brain disease, and its optimization. Neurosurg Focus. 2006;20(4):E12.
Tuszynski M.H., Thal L., Pay M., et al. A phase I clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005;11(5):551-555.
Tisdall M.M., Smith M. Cerebral microdialysis: research technique or clinical tool. Br J Anaesth. 2006;97:18-25.
Troyer K.P., Heien M.L., Venton B.J., Wightman R.M. Neurochemistry and electroanalytical probes. Curr Opin Chem. 2002;6:696-703.
Wichmann T., Bergman H., DeLong M.R. The primate subthalamic nucleus. III. Changes in motor behavior and neuronal activity in the internal pallidum induce by subthalamic inactivation in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:521-530.
1. Benabid A.L., Chabardes S., Mitrofanis J., Pollak P. Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson’s disease. Lancet Neurol. 2009;8(1):67-81.
2. Anderson V.C., Burchiel K.J., Hogarth P., et al. Pallidal vs subthalamic nucleus deep brain stimulation in Parkinson disease. Arch Neurol. 2005;62(4):554-560.
3. Clarke C.E., Worth P., Grosset D., Stewart D. Systematic review of apomorphine infusion, levodopa infusion and deep brain stimulation in advanced Parkinson’s disease. Parkinsonism Relat Disord. 2009;15:728-741.
4. Penn R.D., Kroin J.S., Reinkensmeyer A., Corcos D.M. Injection of GABA-agonist into globus pallidus in patient with Parkinson’s disease. Lancet. 1998;351(9099):340-341.
5. Bergman H., Wichmann T., DeLong M.R. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436-1438.
6. Wichmann T., Bergman H., DeLong M.R. The primate subthalamic nucleus. III. Changes in motor behavior and neuronal activity in the internal pallidum induce by subthalamic inactivation in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:521-530.
7. Guridi J., Herrero M.T., Luquin M.R., et al. Subthalamotomy in parkinsonian monkey. Behavioural and biochemical analysis. Brain. 1996;199:1717-1727.
8. Dostrovsky J.O., Levy R., Wu J.P., et al. Microstimualtion-induced inhibition of neuronal firing in human globus pallidus. J Neurophysiol. 2000;84(1):570-574.
10. Pahapill P.A., Levy R., Dostrovsky J.O., et al. Tremor Arrest with thalamic microinjections of muscimol in patients with essential tremor. Ann Neurol. 1999;46(2):249-252.
11. Kroin J.S., Kao L.C., Zhang T.J., et al. Dopamine distribution and behavioral alteration resulting from dopamine infusion into the brain of the lesioned rat. J Neurosurg. 1991;74(10):105-111.
12. Bobo R.H., Laske D.W., Akbasak A., et al. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci USA. 1994;91(6):2076-2080.
13. Nguyen T.T., Pannu Y.S., Sung C., et al. Convective distribution of macromolecules in the primate brain demonstrated using computerized tomography and magnetic resonance imaging. J Neurosurg. 2003;98(3):584-590.
14. Raghavan R., Brady M.L., Rodríguez-Ponce M.I., et al. Convection-enhanced delivery of therapeutics for brain disease, and its optimization. Neurosurg Focus. 2006;20(4):E12.
15. Gash D.M., Zhang Z., Ai Y., et al. Trophic factor distribution predicts functional recovery in parkinsonian monkeys. Ann Neurol. 2005;58(20):224-233.
16. Bito L., Davson H., Levin E., et al. The concentrations of free amino acids and other electrolytes in cerebrospinal fluid, in vivo dialysate of brain, and blood plasma of the dog. J Neurochem. 1966;13:1057-1067.
17. Meyerson B.A., Linderoth B., Karlsson H., Ungerstedt U. Microdialysis in the human brain: extracellular measurement in the thalamus of parkinsonian patients. Life Sci. 1990;46:301-308.
18. Tisdall M.M., Smith M. Cerebral microdialysis: research technique or clinical tool. Br J Anaesth. 2006;97:18-25.
19. Stamford J.A. Monitoring Neuronal Activity. Oxford: Oxford University Press; 1992.
20. Fried I., Wilson C.L., Maidment N.T., et al. Cerebral microdialysis combined with single-neuron and electroencephalographic recording in neurosurgical patients. Technical notes. J Neurosurg. 1999;91:697-705.
21. Bungay P.M., Morrison P.F., Dedrick R.L. Steady-state theory for quantitative microdialysis of solutes and water in vivo and in vitro. Life Sci. 1990;46:105-119.
22. Hillered L., Vespa P.M., Hovda D.A. Translational neurochemical research in acute human brain injury: the current status and potential future for cerebral microdialysis. J Neurotrauma. 2005;22:3-41.
23. Amara S.G., Methods in Enzymology: Neurotransmitter Transporters, vol 296;San Diego, CA, Academic Press, 1998.
24. Galati S., Mazzone P., Fedele E., et al. Biochemical and electrophysiological changes of substantia nigra pars reticulata driven by subthalamic stimulation in patients with Parkinson’s disease. Eur J Neurosci. 2006;23:2923-2928.
25. Stefani A., Fedele E., Galati S., et al. Subthalamic stimulation activates internal pallidus: evidence from cGMP microdialysis in PD patients. Ann Neurol. 2005;57:448-452.
26. Kaplitt MG, et al. Chronic in vivo microdialysis analysis of neurochemical responses to subthalamic nucleus deep brain stimulation in patients with Parkinson’s disease. In: Abstracts Presented at the Society for Neuroscience, San Diego, CA, November 15, 2001.
27. Mattson R.H., Scheyer R.D., Petroff O.A., et al. Novel methods for studying new antiepileptic drug pharmacology. Adv Neurol. 1998;76:105-112.
28. Scheyer R.D., During M.J., Hochholzer J.M., et al. Phenytoin concentrations in the human brain: an in vivo microdialysis study. Epilepsy Res. 1994;18:227-232.
29. Scheyer R.D., During M.J., Spencer D.D., et al. Measurement of carbamazepine and carbamazepine epoxide in the human brain using in vivo microdialysis. Neurology. 1994;44:1469-1472.
30. Wightman R.M., Zimmerman J.B. Control of dopamine extracellular concentration in rat striatum by impulse flow and uptake. Brain Res Brain Res Rev. 1990;15:135-144.
31. Troyer K.P., Heien M.L., Venton B.J., Wightman R.M. Neurochemistry and electroanalytical probes. Curr Opin Chem. 2002;6:696-703.
32. Marsden C.A., et al. An introduction to in vivo electrochemistry. In: Marsden C.A., editor. Measurement of Neurotransmitter Release In Vivo. Chichester: Wiley; 1984:127-151.
33. Albery W.J., Boutelle M.G., Galley P.T. The dialysis electrode—a new method for in vivo monitoring. J Chem Soc Chem Commun. 1987:900-901.
34. Albery W.J., Bartlett P.N., Cass A.E. Amperometric enzyme electrodes. Phil Trans R Soc B Biol Sci. 1987;316:107-119.
35. Katayama Y, et al. Real time-monitoring of extracellular glutamate concentration with glutamate oxidase-coupled dialysis electrode: intraoperative measurement in patients with gliomas. Proceedings of Second International Meeting on Clinical Aspects of Microdialysis (Uppsala).
36. Revec G.V., Langley P.E., Pierce R.C., et al. A simple micromanipulator for multiple uses in freely moving rats: electrophysiology, voltammetry, and simultaneous intracerebral infusions. J Neurosci Methods. 1993;47:53-59.
37. Robinson D.L., Venton B.J., Heien M.L., Wightman R.M. Detecting subsecond dopamine release with fast-scan cyclic voltammetry in vivo. Clin Chem. 2003;49(10):1763-1773.
38. Perez X.A., Parameswaran N., Huang L.Z., et al. Pre-synaptic dopaminergic compensation after moderate nigrostriatal damage in non-human primates. J Neurochem. 2008;105:1861-1872.
39. Lin L.-F., Doherty D.H., Lile J.D., et al. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130-1132.
40. Yuen E.C., Mobly W.C. Therapeutic potential of neurotrophic factors for neurological disorders. Ann Neurol. 1996;40:346-354.
41. Siegel G.J., Chauhan N.B. Neurotrophic factors in Alzheimer’s and Parkinson’s disease brain. Brain Res Rev. 2000;22:199-227.
42. Ebadi M., Bashir R.M., Heidrick M.L., et al. Neurotrophins and their receptors in nerve injury and repair. Neurochem Int. 1997;30:465-474.
43. Schapira A.H. Treatment options in the modern management of Parkinson’s disease. Arch Neurol. 2007;64:1083-1088.
44. Nutt J.F., Burchiel K.J., Comella C.L., et al. Randomized, double-blind trial of glial-derived neurotrophic factor (GDNF) in PD. Neurology. 2003;60(1):69-73.
45. Gill S.S., Patel N.K., Hotton G.R., et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med. 2003;9(5):589-595.
46. Love S., Plaha P., Patel N.K., et al. Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat Med. 2005;11:703-704.
47. Lang A.E., Gill S., Patel N.K., et al. Randomised controlled trial of intraputamenal glial cell line–derived neurotrophic factor infusion in Parkinson’s disease. Ann Neurol. 2006;59:459-466.
48. Suhr S.T., Gage F.H. Gene therapy for neurologic disease. Arch Neurol. 1993;50(11):1252-1268.
49. Freed W.J., Patel-Vaidya U., Geller H.M. Properties of PC12 pheochromocytoma cells transplanted to the adult rat brain. Exp Brain Res. 1986;82:23-32.
50. Horellou P., Marlier L., Privat A., et al. Exogeneous expression of L-dopa and dopamine in various cell lines following transfer of rat and human tyrosine hydroxylase cDNA: grafting in an animal model of Parkinson’s disase. Prog Brain Res. 1990;82:23-32.
51. Culver K.W., Van Gilder J., Link C.J., et al. Gene therapy for the treatment of malignant brain tumors with in vivo tumor transduction with the herpes simplex thymidine kinase gene/ganciclovir system. Hum Gene Ther. 1994;5(3):343-379.
52. Vaha-Koskela M.J., Heikkilä J.E., Hinkkanen A.E. Oncolytic viruses in cancer therapy. Cancer Lett. 2007;254(2):178-216.
53. Lang F., Bruner J.M., Fuller G.N., et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol. 2003;21(13):2508-2518.
54. Colombo F., Barzon L., Franchin E., et al. Combined HSV-TK/IL-2 gene therapy in patients with recurrent glioblastoma multiforme: biological and clinical results. Cancer Gene Ther. 2005;12(10):835-848.
55. Varmus H. Retroviruses. Science. 1988;240(4858):1427-1435.
56. Barzon L., Zanusso M., Colombo F., Palù G. Clinical trials of gene therapy, virotherapy, and immunotherapy for malignant gliomas. Cancer Gene Ther. 2006;13(6):539-554.
57. Behrstock S., Svendsen C.N. Combining growth factors, stem cells, and gene therapy for the aging brain. Ann N Y Acad Sci. 2004;1019:5-14.
58. Blech A., Tuszynski M. Ex vivo gene therapy for Alzheimer’s disease and spinal cord injury. Clin Neurosci. 1995;3(5):268-274.
59. Tuszynski M.H., Roberts J., Senut M.C., et al. Gene therapy in the adult primate brain: intraparenchymal grafts of cells genetically modified to produce nerve growth factor prevent cholinergic neuronal degeneration. Gene Ther. 1996;3(4):304-314.
60. Tuszynski M.H., Thal L., Pay M., et al. A phase I clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005;11(5):551-555.
61. Tuszynski M.H. Nerve growth factor gene delivery: animal models to clinical trials. Dev Neurobiol. 2007;67(9):1204-1215.
62. Nadini L. Lentiviruses as gene transfer agents for delivery to non-dividing cells. Curr Opin Biotechnol. 1998;9(5):457-463.
63. Naldini L., Kafri T. In vivo gene delivery by lentivirus vectors. Mol Biotechnol. 2007;36(3):184-204.
64. Cockrell A.S., Kafri T. Gene delivery by lentivirus vectors. Mol Biotechnol. 2007;36(3):184-204.
65. Jakobsson J., Lundberg C. Lentiviral vectors for use in the central nervous system. Mol Ther. 2006;13(3):326-336.
66. National Institutes of Health, ClinialTrials.gov [Internet]. Henri Mondor Hospital (Prof. Stephane Palfi), Paris (France), identifier (NCT00627588). A Phase I/II Study of the Safety, Efficacy and Dose Evaluation of ProSavin®, Administered Using Stereotactic Injection to the Striatum of Patients with Bilateral, Idiopathic Parkinson’s Disease, January 8, 2008. http://clinicaltrials.gov/ct2/show/NCT00627588. Available at
67. Glorioso J.C., Fink D.J. Herpes vector-mediated gene transfer in treatment of diseases of the nervous system. Annu Rev Microbiol.. 2004;58:253-271.
68. During M.J., Naegele J.R., O’Malley K.L., Geller A.I. Long-term behavioral recovery in parkinsonian rats by an HSV vector expressing tyrosine hydroxylase. Science. 1994;266(5189):1399-1403.
69. Chattopadhyay M., Krisky D., Wolfe D., et al. HSV-mediated gene transfer of vascular endothelial growth factor to dorsal root ganglia prevents diabetic neuropathy. Gene Ther. 2005;12(18):1377-1384.
70. Hao S., Mata M., Glorioso J.C., Fink D.J. Gene transfer to interfere with TNFalpha signaling in neuropathic pain. Gene Ther. 2007;14(13):1010-1016.
71. Martuza R.L., Rabkin S.D., Yazaki T., et al. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med. 1995;1(9):938-943.
72. Frenkel N. The history of the HSV amplicon: from naturally occurring defective genomes to engineered amplicon vectors. Curr Gene Ther. 2006;6(3):277-301.
73. Kaplitt M.G., Pfaus J.G., Kleopoulos S.P., et al. Expression of a functional foreign gene in adult mammalian brain following in vivo gene transfer via a herpes simplex virus type 1 defective viral vector. Mol Cell Neurosci. 1991;2:320-330.
74. Neve R.L., Neve K.A., Nestler E.J., Carlezon W.A.Jr. Use of herpes virus amplicon vectors to study brain disorders. Biotechniques. 2005;39(3):281-291.
75. Jager L., Ehrhardi A. Emerging adenoviral vectors for stable correction of genetic disorders. Curr Gene Ther. 2007;7(4):272-283.
76. Akli S., Caillaud C., Vigne E., et al. Transfer of a foreign gene into the brain using adenovirus vectors. Nat Genet. 1993;3(3):224-228.
77. Bajocchi G., Feldman S.H., Crystal R.G., Mastrangeli A. Direct in vivo gene transfer to ependymal cells in the central nervous system using recombinant adenovirus vectors. Nat Genet. 1993;3(3):229-234.
78. Hermonat P.L., Muzyczka N. Use of adeno-associated virus as a mammalian DNA cloning vector: transduction of neomycin resistance into mammalian tissue culture cells. Proc Natl Acad Sci USA. 1984;81(20):6466-6470.
79. Tratschin J.D., West M.H., Sandbank T., Carter B.J. A human parvovirus, adeno-associated virus, as a eukaryotic vector: transient expression and encapsidation of the prokaryotic gene for chloramphenicol acetyltransferase. Mol Cell Biol. 1984;4(10):2072-2081.
80. Flotte T.R., Afione S.A., Conrad C., et al. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc Natl Acad Sci USA. 1993;90(220):10613-10617.
81. Kaplitt M.G., Leone P., Samulski R.J., et al. Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet. 1994;8(2):148-154.
82. Bankiewicz K.S., Forsayeth J., Eberling J.L., et al. Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol Ther. 2006;14(4):564-570.
83. Wu Z., Asokan A., Samulski R.J. Adeno-associated virus serotypes: vector toolkit for human gene therapy. Mol Ther. 2006;14(3):316-327.
84. Kaplitt M.G., Feigin A., Tang C., et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: an open label, phase I trial. Lancet. 2007;369(9579):2097-2105.
85. Svendsen C. The first steps towards gene therapy for Parkinson’s disease. Lancet Neurol. 2007;6:754.
86. Eberling J.L., Jagust W.J., Christine C.W., et al. Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology. 2008;70(21):1980-1983.
87. Marks W.J.Jr., Ostrem J.L., Verhagen L., et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: an open-label, phase I trial. Lancet Neurol. 2008;7(5):400-408.
88. Samii A., Nutt J.G., Ransom B.R. Parkinson’s disease. Lancet. 2004;363(9423):1783-1793.
89. Lou J., Kaplitt M.G., Fitzsimons H.L., et al. Subthalamic GAD gene therapy in a Parkinson’s disease rat model. Science. 2002;298(5592):425-429.
90. Obeso J.A., Rodriguez-Oroz M.C., Rodriguez M., et al. Pathophysiologic basis of surgery for Parkinson’s disease. Neurology. 2000;55(12 Suppl 6):S7-S12.
91. Parent A., Cote P.Y., Lavoie B. Chemical anatomy of primate basal ganglia. Prog Neurobiol. 1995;46(2-3):131-197.
92. Smith Y., Charara A., Paquet M., et al. Ionotropic and metabotropic GABA and glutamate receptors in primate basal ganglia. J Chem Neuroanat. 2001;22(1-2):13-42.
93. Wichmann T., DeLong M.R. Pathophysiology of Parkinson’s disease: the MPTP primate model of the human disorder. Ann N Y Acad Sci. 2003;991:199-213.
94. Carbon M., Eidelberg D. Modulation of regional brain function by deep brain stimulation: studies with positron emission tomography. Curr Opin Neurol. 2002;15(4):451-455.
95. Fukuda M., Edwards C., Eidelberg D. Functional brain networks in Parkinson’s disease. Parkinsonism Relat Disord. 2001;8(2):91-94.
96. Feigin A., Kaplitt M.G., Tang C., et al. Modulation of metabolic brain networks after subthalamic gene therapy for Parkinson’s disease. Proc Natl Acad Sci USA. 2007;104(49):19559-19564.
97. Fjord-Larsen L., Johansen J.L., Kusk P., et al. Efficient in vivo protection of nigral dopaminergic neurons by lentiviral gene transfer of a modified neurturin construct. Exp Neurol. 2005;195:49-60.
98. Gasmi M., Brandon E.P., Herzog C.D., et al. AAV2-mediated delivery of human neurturin to the rat nigrostriatal system: long-term efficacy and tolerability of CERE-120 for Parkinson’s disease. Neurobiol Dis. 2007;27:67-76.
99. Kordower J.H., Herzog C.D., Dass B., et al. Delivery of neurturin by AAV2 (CERE-120)-mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Ann Neurol. 2006;60:706-715.
100. Sánchez-Pernaute R., Harvey-White J., Cunningham J., Bankiewicz K.S. Functional effect of adeno-associated virus mediated gene transfer of aromatic L-amino acid decarboxylase into the striatum of 6-OHDA-lesioned rats. Mol Ther. 2001;4:324-330.
101. Bankiewicz K.S., Forsayeth J., Eberling J.L., et al. Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol Ther. 2006;14(3):316-327.
102. Forsayeth J.R., Eberling J.L., Sanftner L.M., et al. A dose-ranging study of AAV-hAADC therapy in Parkinsonian monkeys. Mol Ther. 2006;14(4):571-577.
103. Sanftner L.M., Sommer J.M., Suzuki B.M., et al. AAV2-mediated gene delivery to monkey putamen: evaluation of an infusion device and delivery parameters. Exp Neurol. 2005;194(2):476-483.
104. Bankiewicz K.S., Eberling J.L., Kohutnicka M., et al. Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp Neurol. 2000;164(1):2-14.
105. Christine C.W., Starr P.A., Larson P.S., et al. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology. 2009;73(20):1662-1669.
106. Jarraya B., Boulet S., Ralph G.S., et al. Dopamine gene therapy for Parkinson’s Disease in a nonhuman primate without associated dyskinesia. Sci Trans Med. 2009;1:1-10.
107. Kaplitt M.G. Parkinson disease: another player in gene therapy for Parkinson disease. Nat Rev Neurol. 2010;6(1):7-8.
108. Di Stefano A., Sozio P., Iannitelli A., Cerasa L.S. New drug delivery strategies for improved Parkinson’s disease therapy. Expert Opin Drug Deliv. 2009;6(4):389-404.
[/level-membership-for-neurosurgery-category][not-level-membership-for-neurosurgery-category]
Chapter 117 Molecular Therapies for Movement Disorders
Movement disorders, such as Parkinson disease (PD), are currently treated pharmacologically by systemic administration of drugs that replace, mimic, or potentiate lost neurotransmitters, and more recently with neurosurgical procedures such as deep brain stimulation of the subthalamic nucleus (STN) or internal segment of the globus pallidus (GPi)1,2 to replace older, more destructive procedures such as lesioning of the basal ganglia. However, advances in understanding of the pathophysiology underlying movement disorders coupled with technical improvements in and increasing experience with stereotactic surgery for these diseases, have paved the way for application of molecular therapies to treat human movement disorders. Molecular therapies delivered with stereotactic surgical techniques offer the potential to combine the benefits of focal therapies that would not adversely influence other brain regions with the specificity of intervening in biological processes that may more effectively and efficiently alter or improve abnormal neuronal functions in movement disorders.
Drug/Chemical Infusions
Drug infusion therapy directly into the central nervous system (CNS) is an attractive treatment modality in movement disorder patients with advanced disease. In PD, for example, the effectiveness of oral L-dopa therapy is limited due to its relatively short half-life, and the pulsatile stimulation of dopaminergic neurons can cause significant motor fluctuations and dyskinesias.3 Focal infusion of therapy to the area of dysfunction, however, could help achieve more constant striatal dopaminergic stimulation, and more closely mimic the body’s normal physiologic state.
The first demonstration of the effectiveness of focal infusion of medications within the brain parenchyma was conducted in PD patients, who were tested prior to stereotactic lesioning or electrode placement. These patients were infused with small-volume, low-dose muscimol, a GABA-A agonist, into the globus pallidus just prior to pallidotomy,4 and they displayed a rapid onset of improvement in finger motor speed, reduced extremity tone, and decreased muscle fatigue, which all returned to baseline slowly after approximately 30 minutes. Following subsequent pallidotomy, the patients displayed a similar improvement in motor symptoms. In addition, studies in MPTP-treated nonhuman primates have demonstrated that local inactivation of neurons in the subthalamic nucleus results in an improvement in parkinsonian symptoms.5–7 These observations prompted intraoperative exploration of microinjections of either lidocaine, an anesthetic that selectively blocks axonal sodium channels, or muscimol into the STN of six PD patients to study the effects of local STN inactivation. This resulted in transient improvement in akinesia, rigidity, and limb tremor in patients with PD. Electrophysiologic recordings were also performed, which confirmed inhibition of electrical activity in the nucleus following drug infusion as it spread from the injection site to more distant tissue.
Although muscimol microinjection and electrical stimulation by STN deep brain stimulation (DBS) have the same effects on PD motor symptoms, their mechanisms of action are quite different. The precise mechanism of action of electrical stimulation on symptomatic improvement in movement disorders is still unclear, there is support for one or more functions, including excitation of neighboring axons, activation of recurrent inhibitory circuits, and direct inhibition of neuronal firing.8 As opposed to these more indirect mechanisms, muscimol acts by specifically activating chloride-dependent GABA-A receptors and thereby inhibiting neurons presynaptically. It is still unknown whether continuous infusion of muscimol is superior to stimulation, since the specificity of action provides opportunities for more direct conclusions regarding the relationship between neurotransmitter receptor action and symptomatic improvements, but there may as yet be unknown benefits of some more nonspecific or broader actions of electrical stimulation. In addition, a crucial question that must still be answered is whether patients will develop tolerance to the drug with time. In animals, one unpublished study has demonstrated that parkinsonian rats with unilateral 6OHDA lesions show sustained effects when muscimol is administered by an implanted osmotic pump for 2 weeks,9 but longer-term consequences remain to be seen. However, it is clear to every practitioner of DBS that some patients can have adverse effects due to spread of electrical stimulation outside the STN or to nonmotor areas of the STN despite proper electrode location, and this could be less problematic with a biologically specific therapy such as muscimol infusion. The development of appropriate technology for chronic infusions into brain parenchyma, which have yet to be fully developed, would help facilitate development and ultimate determination of the value of continuous muscimol infusion into basal ganglia targets as a therapy.
In addition, patients with essential tremor (ET) have also been studied through a local drug infusion modality, and one study demonstrated the cessation of tremor in patients microinjected with muscimol into the ventralis intermedius thalamus (Vim).10 In this study, six ET patients undergoing unilateral stereotactic thalamic procedures for relief of tremor refractory to medications were given either saline and/or muscimol intracerebral microinjections prior to thalamotomy or thalamic DBS electrode placement. The patients displayed suppression of tremor for an average of 9 minutes, following a mean latency of 9 minutes to allow for sufficient drug diffusion to influence enough neurons for a therapeutic effect. This not only provides further support for the belief that inhibition of Vim and STN activity can improve symptoms of ET and PD, respectively, but this presents another opportunity for application of direct intraparenchymal drug infusion for a second movement disorder, should technology permit further development of this therapy.
In addition to the therapeutic benefit of local microinjection of drugs such as lidocaine or muscimol, selective infusion of dopamine or dopaminergic medications into the basal ganglia has also been considered, in order to bypass the systemic side effects seen with conventional oral intake of these medications. Chronic intraventricular or intrastriatal dopamine infusions was investigated in 6-OHDA lesioned rats, which demonstrated improvement in motor symptoms following 7 days of continuous dopamine infusion compared to controls.11 However, local examination of the striatum adjacent to the point of intrastriatal dopamine infusion revealed a barrier, which limited diffusion of medication to only 1 to 2 mm from the catheter. In an effort to increase the size of the perfused region, convection techniques have been attempted that move both fluid and drug into the extracellular space by bulk flow driven by a pressure gradient (Fig. 117-1).12–14 The data are still rather limited, and ideal flow rates, catheter diameter, and infusion volume have not yet been determined.15
Other techniques that may allow for efficient delivery and monitoring of drug infusions locally within the brain include microdialysis and voltammetry (Table 117-1). Cerebral microdialysis is a modality that allows measurement of the concentration of extracellular neurochemicals within cerebral structures, locally and in vivo. Initially conducted in 1966 by using a dialysis membrane filled with dextran solution into canine cortex, with measurement of local amino acid concentrations,16 this method has also been applied to PD patients.17 Currently, cerebral microdialysis consists of a double-lumen probe containing an inlet and an outlet port, surrounded by a semipermeable membrane, and sealed above and below a point at which the two tubes are inserted into the membrane lumen. This dialysis probe can be inserted through a burr hole to a preset depth through the brain parenchyma, allowing for perfusion of solutions through the inlet port as well as analysis of the returning solution for extracellular molecule concentrations with techniques such as high-performance liquid chromatography with electrochemical detection (HPLC EC). HPLC EC provides high sensitivity and specificity for the measurement of molecules, such as biogenic amine, including noradrenaline, dopamine, and serotonin, as well as for amino acids, including glutamate, aspartate, and GABA.18–20
| Focal infusion |
| Muscimol (GABA-A agonist). Lidocaine (Na+-channel antagonist). Dopamine (both intrastriatal and intraventricular infusions). |
| Microdialysis |
| Extended monitoring levels of a particular neurotransmitter in probe region; may allow for pump that would administer a drug or chemical in response to a certain level, allowing for achievement of steady-state levels of that neurotransmitter. |
| Voltammetry |
| Detects changes in the concentration of certain neurotransmitters (transmitter oxidation), and allows for excellent temporal and spatial resolution of their release in real time. |
Some limitations of microdialysis, however, include invasiveness, reports of complications in the literature, dependence of accuracy on factors such as flow rate and probe size, and a local fibrotic reaction to the probe tip seen during chronic microdialysis experiments.19,21 In addition, the neurochemicals sampled during microdialysis are not only those released synaptically, but also include neurochemicals of local metabolism, capillary delivery, and neuronal release and uptake from groups of cells.22,23 Despite these limitations, microdialysis has been used successfully in human patients, ranging from traumatic brain injury and subarachnoid hemorrhage to movement disorders and epilepsy.18,22 In movement disorders, particularly PD, there have been published studies such as one demonstrating the changes in concentrations of GABA, cGMP, and glutamate before, during, and after 1 hour in the internal globus pallidus, anteroventral thalamus, and putamen of six patients with refractory PD undergoing DBS in the STN.24,25 In addition, an in vivo microdialysis method was developed permitting the continuous, chronic sampling of local neurotransmitter concentrations in the STN and substantia nigra pars reticulata (SNr) of patients for several days after placement of STN DBS electrodes.26 All six patients in this study had no complications, and the microdialysis probes were successfully removed without disrupting the DBS electrode location. In theory, microdialysis use may be extended to monitoring levels of a particular neurotransmitter in the region where the probe is implanted, which could then be attached to a pump that would administer a drug or chemical in response to a certain level, allowing for achievement of steady-state levels of that neurotransmitter. Microdialysis has in fact been used to monitor systemic drug levels and to infuse agents into the brain in epilepsy patients, and this could easily be extended to movement disorders if improved technology permitted chronic use of these devices.27–29
In addition to microdialysis, voltammetry is another method that can detect changes in the concentration of certain neurotransmitters, and allows for excellent temporal and spatial resolution of their release in real-time.30,31 Voltammetry is based on the principle that oxidation of certain chemicals leads to the release of electrons, and the resultant current can be measured with an electrode.32 Furthermore, a dialysis electrode was developed that combined microdialysis with voltammetry to measure glutamate,33 which specifically contained a dialysis probe with an electrochemical detection system utilizing the enzyme glutamate oxidase to detect glutamate levels. Other systems have been designed that measure the concentrations of molecules such as glucose and oxygen.34,35 Most applications have currently been in animal models, including the construction of a micromanipulator in rats that allowed for both infusion of drugs and voltammetry in freely moving rats36 and mostly studied in tumors, cerebral ischemia, and traumatic brain injury. However, the application of voltammetry has also been extended to the measurement of dopamine in both rats and nonhuman primates. Voltammetry has been used to measure extracellular dopamine concentration changes in rats, such as during certain behaviors or in response to cocaine administration in the nucleus accumbens.37 In regard to movement disorders, there was a recent study using carbon fiber voltammetry to measure the release of dopamine in MPTP-induced dopaminergic lesions in nonhuman primates.38 In addition to its utilization in probing drug effects on dopamine release and uptake, there may also future applications of voltammetry to measuring dopamine release in patients, with tailored therapeutic release of medication by external or internal means in response to certain changes in levels. Although microdialysis can sample any neurotransmitter which can diffuse across the dialysis membrane, voltammetry is currently limited to monitoring a select population of neurotransmitters due to technical constraints of the voltammetry methodology to measure transmitter oxidation. However, because it does not involve a membrane or a tube, physical blockade of fluid diffusion due to luminal obstruction is not a concern and oxidation could be more easily measured by an implanted electrical device compared with chemical analysis of dialysate fluids. Therefore, voltammetry could represent an attractive prospect for long-term monitoring of brain neurotransmission as a component of a dynamic feedback mechanism to tailor intraparenchymal drug delivery to specific patient needs.
Growth Factors/Recombinant Proteins
As an alternative to infusion of drugs into the CNS for the treatment of movement disorders, another potential strategy employs the infusion of growth factors or recombinant proteins. Since the central feature of patients with PD is the loss of dopaminergic neurons in the substantia nigra, researcher proposed the idea of administrating neurotrophic factors to degenerating neurons in order to promote their survival. Specifically, the leading candidate neurotrophic factor was glial cell–line derived neurotrophic factor (GDNF), discovered in 199339 and shown to enhance the survival of dopaminergic neurons in vitro.40 In animal models, the goal of GDNF was to prevent further neuronal loss while encouraging reinnervation of the striatum from any surviving, intact terminal within the striatum. Since GDNF is a 134–amino acid peptide, it does not efficiently cross the blood brain barrier, and thus it must be delivery locally into the CNS either by administration into the ventricular system or directly into the brain parenchyma. In preclinical studies with both 6-OHDA- and MPTP-lesioned animal models, GDNF was delivered using both approaches into the CNS, and it indeed did show some benefit.41–43
Eventually, infusion of a recombinant form of GDNF was administered in human clinical trials (Table 117-2). Initially, 50 patients with moderately advanced PD were treated with intraventricular GDNF; however, the infusion trial was stopped early due to excessive complications from toxicity to the periventricular structures.44 Furthermore, there was a lack of therapeutic efficacy, potentially due to failure of GDNF diffusion across the greater distance between the ventricular system and substantia nigra when compared to nonhuman animal models. Subsequently, this led to direct intraparenchymal infusion of GDNF into the striatum (postcommissural putamen) in an open-label phase 1 study that demonstrated both encouraging safety and efficacy results.45 Five PD patients were treated, with a resulting significant decrease in total Unified Parkinson’s Disease Rating Scale (UPDRS) score in the “off” state at 3 months, as well as mean reduction in UPDRS motors score at 12 months of 48%. There were also correlational 18F-dopa PET scans showing an increased uptake in the area of the GDNF infusion. Since 18F-dopa PET measures dopamine synthetic capacity and by extension dopaminergic innervation of the striatum, these results did support potential enhancement of dopaminergic function in the remaining viable nigrostriatal neurons, and a postmortem case from the study later did demonstrate sprouting of dopaminergic fibers around the GDNF infusion site.46
Table 117-2 Infusion of Recombinant GDNF into CNS of Parkinson Disease Patients
| Central tenet of Parkinson’s Disease |
| Loss of dopaminergic neurons in substantia nigra. |
| Goals |
| Promote survival of degenerating dopaminergic neurons by neurotrophic factors. Encourage reinnervation of the striatum by remaining, intact intrastriatal terminals. |
| Outcomes |
| Intraventricular infusion of GDNF |
| Clinical trials stopped early due to: Toxicity to periventricular structures Question of diffusion limitations of GDNF from ventricular system to substantia nigra |
| Intraparenchymal infusion of GDNF |
| Open-label phase 1 study showed encouraging safety and efficacy results. Improvement in UPDRS motor scores. 18F-dopa PET scans showing an increased uptake in the area of the GDNF infusion. Postmortem sprouting of dopaminergic fibers around the GDNF infusion site. Phase 2 double-blind, placebo-controlled trial results: no significant effect of GDNF was reported in either primary or secondary outcome measures. |
These initial promising results did not translate into success in a phase II double-blind, placebo controlled trial that subsequently followed.47 In this study, 34 patients with moderately advanced PD were bilaterally implanted with a single port catheter to infuse either drug or placebo continuously into the putamen, and no significant effect of GDNF was reported in either primary or secondary outcome measures (except for 18F-dopa PET scan data), leading to considerable debate regarding the reasons for the conflicting results between the open-label and double-blind, placebo-controlled studies. There has long been concern about potential placebo effects confounding trials of new therapies for PD, but the relative absence of substantial benefit in either the treatment or control groups suggests that placebo was not a cause of the different results observed in the two trials. It is possible that the change in the type of infusion catheter and mechanism of infusion (differences in diameter and total amount of GDNF delivered) may have negatively affected the therapy and led to a false-negative result. The study may also not have been adequately powered to demonstrate an effect. This highlights the difficulties that can limit successful translation of promising and potentially efficacious therapies. Given the expense and limited likelihood of success for any new therapy, it can be difficult to develop optimal technology for phase 1 studies. However, when results justify later-phase trials, an incentive is created to improve technology so that the methodology used in the more advanced trial can be more readily extrapolated for general use. Furthermore, patient recruitment and financial considerations often limit the size of randomized studies. These understandable and often unavoidable issues may nonetheless compromise later phase trials and can create difficulties resolving discrepancies with early phase data when subsequent results do not justify continued development. This is not unique to growth factor infusion, and has plagued other promising surgical therapies, including cell transplantation and gene therapy. But these issues must be considered when evaluating results of surgical trials for new molecular therapies, in order to improve future studies and to not automatically reject treatments that may have genuine promise due to disappointing results from a single study.
Gene Therapy
Basic Science of Gene Therapy
Gene therapy strategies can generally be subdivided into two different categories: ex vivo and in vivo (Table 117-3). Ex vivo gene therapy involves transplantation of target cells previously genetically altered in cell culture by plasmid or retroviral vectors before transplantation into the target organ in the patient. These cells can be thought of as genetically engineered inside in situ factories or “minipumps”48 that are able to produce the therapeutic protein or factor of interest within host tissue. One advantage of ex vivo gene therapy is the ability to screen for adequate gene expression prior to introduction into the patient. This method can also be used to alter the characteristics of cells that are themselves intended to be therapeutic, such as modifying stem cells in order to increase survival or improve interaction with host brain. Potential difficulties with ex vivo gene therapy include potential oncogenic properties and unfavorable host immune response against foreign transplanted cells that can lead to rejection of transplanted cells or inflammation in target tissue.49,50
| Ex vivo delivery: Genetically modified cells transplanted into target tissue. |
| Advantages |
| Allows for prior screening for adequate gene expression. Can alter cells to increase survival or improve interaction with host brain. |
| Disadvantages |
| Potential oncogenic properties. Unfavorable host immune. Rejection of transplanted cells or inflammation in target tissue. |
| In vivo delivery: Direct transfer of genetic material into the host cells within the body in situ |
| Advantages |
| Cells already established within neural networks. Bypasses potential adverse host immune response to foreign cells. |
| Disadvantages |
| Limited efficacy of direct transfer of naked or liposome-mediated genetic material. Genetically modified viruses typically used as vehicles, can lead to toxicity due to viral replication or host inflammatory reaction to viral proteins. |
[/not-level-membership-for-neurosurgery-category]
