Molecular Regulation of Cardiac Inward Rectifier Potassium Channels by Pharmacologic Agents
Background
The description in 1949 by Katz1 of the inward rectifier current that he considered “an interesting phenomenon, but difficult to explain” in muscle can be regarded as the scientific birth of inward rectifier currents. Inward rectifier potassium currents (KIR) are characterized by their key feature of allowing more inward than outward potassium flow at equivalent membrane potentials negative and positive of the potassium equilibrium potential. Inward rectifier channels are formed by tetramerization of the underlying pore-forming KIR channel proteins. Ion channel regulation takes place mainly via the intracellularly located amino- and carboxyl-terminal regions of the proteins. Functionally, inward rectifiers define, for a large part, the membrane potential of nonexcitable cells and the resting membrane potential of excitable cells.
The eukaryotic inward rectifier family is encoded by the KCNJ genes and includes seven subfamilies (KIR1-7) (Figure 13-1). Each protein consists of two transmembrane domains, a short pore loop that harbors the potassium selectivity filter (GYG) and intracellularly located N- and C-termini (Figure 13-2). Between subfamilies, sequence homology is as high as 40%, whereas sequence identity rises to approximately 60% within subfamilies. Each of the subfamily gene products display their own characteristics, some having strong and others weak rectifying potential and some responding to metabolic stimuli or neurotransmitters directly. Individual family members are involved in many physiologic processes such as cardiac and neural excitability, insulin release, vascular tone, and potassium homeostasis.2 Members of three different subfamilies are main contributors to cardiac electrophysiology (Table 13-1). The classical inward rectifier current (IK1) channel is formed by KIR2.x proteins and stands at the basis of the resting membrane potential in working cardiomyocytes; the acetylcholine (ACh) responsive rectifiers are built of KIR3.x proteins and play an eminent role in heart rate modulation, whereas the adenosine triphosphate (ATP)-sensitive channels are formed by octamers of KIR6.x and the regulatory sulfonylurea receptor (SUR) proteins and are responsible for coupling the metabolic state of the vulnerable cardiomyocyte to its electrical activity.3,4
Table 13-1
Human Cardiac Inward Rectifier Genes, Expression, and Associated Cardiac Disease
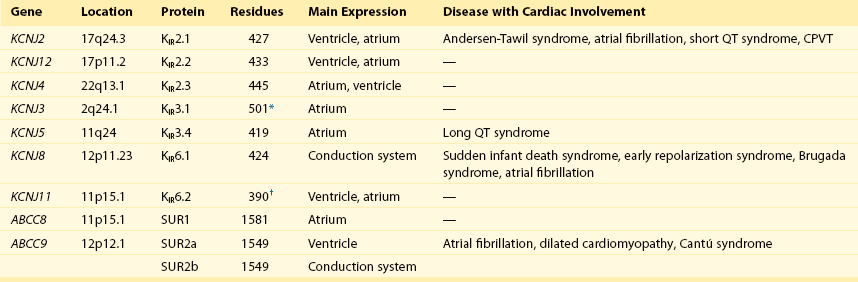
CPVT, Catecholaminergic polymorphic ventricular tachycardia.
*Longest isoform. Shorter isoforms of 235, 253, and 308 amino acids have been reported.
†Longest isoform. Shorter isoform of 303 amino acids has been reported.
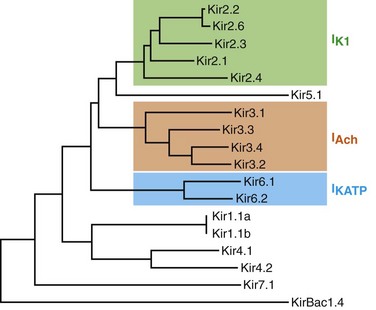
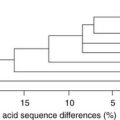
Figure 13-1 Cladogram of the human KCNJ gene family. Amino acid sequences were aligned by a Clustal-W algorithm, phylogeny was determined by the neighbor-joining method. Bacterial KIRBac1.4 was used as outgroup. Subfamilies relevant for cardiac electrophysiology are color-coded: classical inward rectifier genes (green), acetylcholine-regulated channel genes (orange), and ATP-sensitive inward rectifier genes (blue).
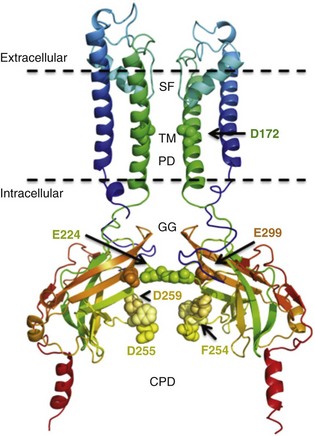
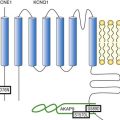
Figure 13-2 Structure model of KIR2.1 ion channel. Negatively charged amino acid residues (D172, E224, F254, D255, and D259) involved in polyamide-mediated rectification face the transmembrane and cytoplasmic pore regions, respectively. CPD, cytoplasmic pore domain; GG, G-gate; SF, selectivity filter; TMPD, transmembrane pore domain. (Used with permission from Van der Heyden MA, Sánchez-Chapula JA: Toward specific cardiac IK1 modulators for in vivo application: old drugs point the way. Heart Rhythm 8:1076–1080, 2011.)
Inward Rectifier Channel Proteins are Encoded by KCNJ Genes
KIR2.x Proteins Constitute the Classical Inward Rectifier Current
Each monomer comprises a transmembrane (TM) and a cytoplasmic part. The transmembrane domain (TMD) consists of the two TM helices, the pore helix positioned between these, and a slide helix located in front of TM1. The cytoplasmic pore domain (CPD) is compiled by interaction of the amino- and carboxyl-termini. In the resulting channel, the four TM2 (inner) helices of each monomer face the central water-filled pore in the TMD, with the potassium selectivity filter (SF) positioned in close proximity to the cellular exterior (see Figure 13-2). It is believed that ion conduction may be regulated by two gates in series: one formed by the inner helices (TM2) of the TMD at the helix-bundle crossing and the other by the G loop at the apex of the CPD.2
Tetramerization of CPDs creates an extended pore region into the cytoplasm, separated from the TM pore region by the G-loops. Polyamine block of inwardly rectifying potassium (KIR) channels underlies their key functional property of preferential conduction of inward K+ currents (inward rectification). Kinetic models describe polyamine block as a multistep process, incorporating sequentially linked “shallow” and “deep” binding steps of polyamines in the KIR pore. Structurally, these shallow and deep binding steps are conceptualized as an initial weakly voltage-dependent binding in the cytoplasmic domain of the channel, followed by a steeply voltage-dependent step in which spermine migrates to a stable binding site in the TM pore (inner cavity). Five negatively charged residues within the cytoplasmic (E224, D259, E299, F254, and D255) and one in the TM pore regions (D172) are involved in interacting with the positively charged polyamines.5
Cardiac Acetylcholine Responsive Inward Rectifiers are Formed by KIR3.1 and KIR3.4 Heteromers
Functional cardiac ACh regulated inward rectifier current (IKACh) channels are formed by heterotetramers of KIR3.1 and KIR3.4 (also known as GIRK1 and GIRK4), encoded by the KCNJ3 and KCNJ5 genes, respectively. Expression is confined mainly to the atrium, and functional expression in the sinus and atrioventricular nodes is an important feature for heart rate and atrioventricular conduction regulation. Homotetramers of KIR3.1 or KIR3.4 produce less or no current when overexpressed in Xenopus oocytes.6 It was established that a stochiometry of 1 : 1 and an alternating position of KIR3.1 and KIR3.4 subunits within the channel produced most robust IKAch channel activity.2
The structural organization of the IKAch channel is similar to the classical IK1 channel. The TM2 domains face the water-filled pore and, confined by the G-gate, an extended pore region protrudes into the cytoplasm. A leucine residue within the C-terminus of KIR3.1 (L333) and KIR3.4 (L339) appears crucial for Gβγ-dependent activation. Interestingly, several domains involved in Gβγ-dependent activation are formed at the cytoplasmic-facing interfaces of the KIR3.1 and KIR3.4 interacting domains.2
IKATP Channels Are Octaheteromers of Four KIR6.x and Four SURX Channel Proteins
Compared with IK1 and IKAch channels, an additional level of complexity is found in the ATP-sensitive rectifier current (IKATP) channels, because a tetramer of SUR proteins embraces the KIR-formed channel.7 Cardiac SUR protein–expressing genes ABCC8 and ABCC9 are members of the large ATP-binding cassette (ABC) superfamily that consists of 48 genes divided into seven subfamilies.8 SUR1 (ABCC8) and SUR2 (ABCC9) contain the typical ABC “core” that consists of two membrane domains (TMD1 and TMD2) of six TM helices, each separated by two cytoplasmic nucleotide–binding domains (NBD1 and NBD2). Furthermore, the proteins contain an additional N-terminal–located membrane domain consisting of five TM helices (TMD0) that are linked to the “core” domain by the L0 linker. Interaction with the KIR6.x-formed pore is established mainly by the TMD0 and L0 domains. Nucleotide binding drives dimerization of NBD1 and NBD2, which results in subsequent rearrangements of TMD1 and TMD2 and alterations in IKATP channel gating. The pore-forming KIR6.x complex resembles the structures of KIR2.x and KIR3.x channels. Cardiac IKATP channels, depending on their location in the heart, consist of octaheteromers of four KIR6.2 (KCNJ11) and four SUR2a subunits (ventricle), KIR6.2 and SUR1 (atrium), or KIR6.1 (KCNJ8)/KIR6.2 and SUR2b (conduction system).
Cardiac Role of Inward Rectifier Channels
KIR2.x Channels Set the Resting Membrane Potential and Contribute to Repolarization
In the working myocardium, KIR2.x-mediated IK1 sets the resting membrane potential close to the potassium reversal potential and contributes outward potassium current during repolarization. With respect to the heart, neonatal KIR2.1 knockout mice were bradycardic and displayed electrocardiogram (ECG) abnormalities such as lengthening of the PR (~50%) and QT (~50%) intervals. Isolated cardiomyocytes from KIR2.1 knockout animals displayed (1) no IK1; (2) action potential prolongation; and (3) spontaneous beating activity.9 In contrast, KIR2.2 knockout mice were viable and lived through adulthood without ECG abnormalities. However, their cardiomyocytes displayed a 50% reduction in IK1 densities. In humans, dominant negative KCNJ2 mutations are associated with Andersen-Tawil syndrome 1, characterized by periodic muscle paralysis, biventricular tachycardia and sometimes long QT times, and developmental bone abnormalities (see Table 13-1).10 Gain-of-function mutations in KCNJ2 have been associated with short QT syndrome, catecholaminergic polymorphic ventricular tachycardia, and congenital atrial fibrillation.11–13 Currently, no disease-causing mutations in KCNJ12 and KCNJ4 have been described.
IKACh Channels and Heart Rate Regulation
IKACh slows heart rate and atrioventricular conduction in response to muscarinic M2 receptor activation by ACh released from the vagus nerve. In this process, Gβγ directly interacts with and activates the channel that thereby antagonizes diastolic depolarization of the nodal cardiomyocytes. KCNJ3–/– mice were viable but showed a lack of carbachol-induced IKACh in atrial cardiomyocytes. Moreover, animals showed blunted responses to pharmacologic interventions directed at vagal-induced bradycardia.14 KCNJ5–/– mice15 displayed loss of cardiac IKAch. Mice were viable; in most studies their resting heart rate increased and animals showed a reduced negative chronotropic response after vagal stimulation. Furthermore, mice were unable to undergo rapid changes in heart frequency. In human heart function, KCNJ5 mutations have been associated with long QT syndrome, syncope, atrial fibrillation, and sudden cardiac death (see Table 13-1).16 No diseases related to mutations in KCNJ3 have been reported.
IKATP Channels Confer Metabolic Status to Electrical Properties
KIR6.1 knockout mice die suddenly after the occurrence of ST elevation and subsequent third-degree atrioventricular block and cessation of heart rhythm, which may result from myocardial ischemia as a result of blunted coronary vasoregulation.17 The presence of KIR6.1 in the cardiac conduction system may provide another or additional mechanism of cardiac pathology and conduction disturbances in these animals. Furthermore, KIR6.1 knockout mice are more susceptible to endotoxin-mediated septic shock, resulting in ST elevation and cardiac death. In the hearts of KIR6.2 knockout mice, a number of stressors induce calcium overload, followed by short-term abnormalities in cardiac rhythm (early afterdepolarization, premature ventricular contractions, and sinus node regulation) and hemodynamics (increased left ventricular end-diastolic pressure).18,19 In the long term, cardiac maladaptive remodeling (exaggerated fibrosis and hypertrophy) finally results in impaired ejection fraction and congestive heart failure.
SUR1 knockout mice are fertile and phenotypically normal but lack atrial IKATP.20 Furthermore, ABCC8 null mutation increases resistance to cardiac, but not neuronal, ischemia-reperfusion injury.21 Knock out of SUR2 resulted in loss of IKATP from all muscles, coronary vasospasms, increased blood pressure, and premature death from week 5 onward. Animals displayed slight cardiac hypertrophy and increased resistance to cardiac ischemia-reperfusion damage.22
In humans, a gain-of-function mutation in KCNJ8 has been associated with early repolarization and Brugada syndrome, both characterized by specific (J-wave) ECG abnormalities and atrial fibrillation.23–26 KCNJ8 loss of function has been associated with sudden infant death syndrome.27 ABCC9 mutations have been linked to atrial fibrillation, dilated cardiomyopathy, and Cantú syndrome, which causes cardiomegaly and pericardial effusion, among other effects (see Table 13-1).28–30 Thus far, KCNJ11 and ABCC8 mutations have not been associated with cardiac phenotypes.
Pharmacology of KIR Channels
Effects of Drugs on KIR Channels
The most commonly used blockers to examine the physiologic roles of KIR channels in native cells and tissues are Ba2+ and Cs+. At micromolar concentration, Ba2+ blocks KIR channels with relative specificity. Externally applied Ba2+ suppresses KIR currents in a voltage-dependent manner and Ba2+ inhibits KIR channels more strongly as the membrane is hyperpolarized.2 However, experiments have shown that BaCl2 infusion in vivo produces strong deleterious effects—mainly skeletal, cardiac, and smooth muscle dysfunction.5
In the intact heart, IK1 blockers produce membrane depolarization (an effect that slows conduction velocity because of a voltage-dependent inactivation of Na+ channels), cause prolongation of the QT and QRS intervals on the surface ECG, and at higher concentrations cause ventricular ectopy and lethal ventricular arrhythmias. Conversely, it has been demonstrated that increasing inward rectifier currents stabilize reentrant arrhythmias. Recently, it has been shown that pharmacologic inhibition of inward rectifier currents by chloroquine induces antifibrillatory effects.31 Different antiarrhythmic (e.g., quinidine, amiodarone, propafenone, disopyramide) and noncardiac (e.g., terfenadine, astemizole) drugs at micromolar concentrations have been found to inhibit the classical cardiac IK1. However, little is known about their mechanisms of inhibition.32
Gambogic acid (GA) is a potent inducer of apoptosis that has been considered for anticancer therapy. At 10 µM, GA inhibited KIR2.1 current differentially during acute applications that produced 30% of inhibition or during prolonged exposures (up to 3 hours), which produced 70% of inhibition. The IC50 of KIR2.1 inhibition by GA during prolonged exposures was 27 nM. GA–induced inhibition is not caused by changes in biogenesis or trafficking to the plasma membrane. It is a lipophilic molecule (LogP ~6.77) that can insert itself into the lipid bilayer and thereby might perturb association of KIR2.1 with regulatory lipids and proteins that specifically facilitate the function of KIR2.1 channels. It has been found that pretreatment of HEK293 cells with GA shifted KIR2.1 and Kv2.1 channels from the Triton X-100–insoluble to the Triton X-100–soluble fraction. GA decreasing KIR2.1 activity is consistent with KIR2.1 being physiologically active in the context of Triton X-100–insoluble microdomains, but not of Triton X-100–soluble microdomains.33 A different drug, the cell-permeable dienone phenol triterpene celastrol, a neuroprotective compound that is also a slow-acting compound, decreased the rate of ion channel transport and caused a reduction of channel density on the cell surface.
Muscarinic ACh receptors in the peripheral nervous system are found primarily on autonomic effector cells innervated by postganglionic parasympathetic nerves. In the heart, the muscarinic receptor M2 is the predominant subtype, which is coupled to the Gi/o protein family. IKACh channels can be activated by muscarinic receptor agonists like cholinomimetic choline esters (ACh, methacholine, carbachol, and bethanechol). The muscarinic receptor antagonist alkaloids such as atropine and synthetic and semisynthetic compounds prevented the effects of ACh by blocking its binding to muscarinic receptors.34
Some antiarrhythmic drugs are known to antagonize at micromolar concentration actions of ACh on IKACh. Evidence from different authors suggests that the mechanisms underlying the anti-ACh effect of antiarrhythmic drugs at micromolar levels are different among such drugs—that is, quinidine, pilsicainide, disopyramide, d,l-sotalol, propafenone, cibenzoline, and amiodarone mainly block the muscarinic ACh receptors—whereas flecainide, verapamil, and propafenone inhibit the K+ channel itself as open channel blockers. Some relatively novel antiarrhythmic drugs have been found to inhibit IKACh at nanomolar concentrations. Dronedarone inhibited IKACh in single cells isolated from the sinoatrial node with an IC50 of 63 nM.35 The benzopyrene derivative NIP-151 potently inhibited the heterologously expressed (in HEK293 cells) channel KIR3.1/3.4, with an IC50 of 1.6 nM. In both cases, it was suggested that dronedarone and NIP-151 inhibited IKACh channels by a direct inhibitory interaction with the channel protein or by disrupting the G-protein–mediated activation.36
There are two major classes of therapeutic compounds that target the IKATP channels: sulfonylureas and IKATP channel openers (KCOs).2 Sulfonylureas on pancreatic β cells bind to SUR1 channel subunits and block IKATP channels, causing membrane depolarization and an influx of Ca2+ channels, stimulating insulin release and hypoglycemia.2 KCOs activate IKATP channels via their action on the SUR subunit of the channel. KCOs that activate cardiac IKATP channels include cromakalim, pinacidil, nicorandil, and diazoxide. KCOs act by opening IKATP channels by decreasing the threshold concentrations of intracellular nucleotides for opening of KIR6.2/SUR2a channels, and they also increase the maximum amplitude.2
Pore-Blocking Drugs
Several compounds that partially mimic polyamine binding to IK1 channels have been identified over the last several years. Block of small molecules such as chloroquine, chloroethylclonidine, pentamidine, and quinacrine have been shown to inhibit IK1 and heterologously expressed KIR2.x channels in a voltage-dependent fashion.37
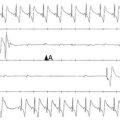
In cardiac myocytes, in contrast to the voltage-independent mode of IK1 inhibition induced by most of the tested drugs, the antimalarial drug chloroquine was found to inhibit IK1 and IKACh in a voltage-dependent manner, with less pronounced blockade at negative test potentials. In addition, unblock was achieved by hyperpolarizing pulses to potentials negative to the current reversal potential (Figure 13-3). This profile of voltage dependence is consistent with a positively charged molecule blocking the channel from the intracellular side and entering the pore to such an extent as to be subjected to the transmembrane electrical field. The blocking effects of chloroquine on IK1 caused an increase in the duration of the ventricular action potential, depolarization of the resting membrane potential, and increase in Purkinje fibers’ automaticity.38
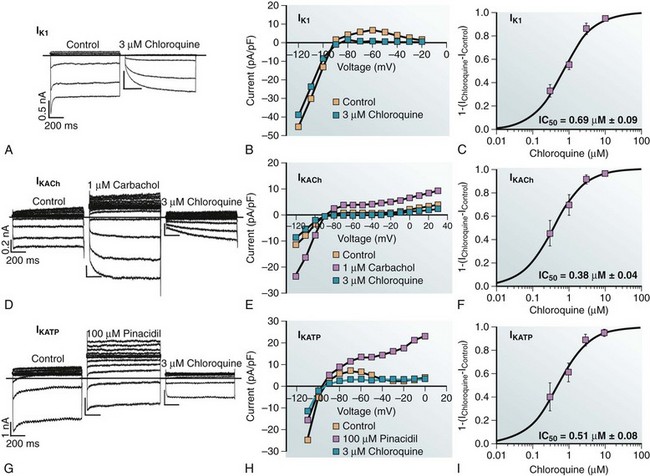
Figure 13-3 Blocking efficacy of chloroquine in different inward rectifier channels. Voltage protocol: steps from −120 mV to +20 mV. A, IK1 current traces in ventricular myocytes. B, I/V relationship from 5 cells. C, Dose-response curve of blocked IK1 at −60 mV. D, Current traces in atrial myocytes in the presence of 1 µM carbachol to activate IKACh. E, I/V relationship from five cells. F, Dose-response curve of blocked IKACh at −60 mV. G, Current traces in ventricular myocytes in the presence of 100 µM pinacidil to activate IKATP. H, I/V relationship from five cells. I, Dose-response curve of blocked IKATP at −60 mV. (Used with permission from Noujaim SF, Stuckey JA, Ponce-Balbuena D, et al: Specific residues of the cytoplasmic domains of cardiac inward rectifier potassium channels are effective antifibrillatory targets. FASEB J 24:4302–4312, 2012.)
Recent studies with small molecules such as chloroquine and pentamidine revealed the molecular basis by which these drugs block KIR2.1 in a voltage-dependent manner. Through site-directed mutagenesis and molecular modeling studies, chloroquine and pentamidine were found to block the KIR2.1 channel by plugging the center of the cytoplasmic conduction pathway via interference with negatively charged residues (E224, D259, and E299) and for chloroquine with an additional aromatic residue (F254), which have previously been shown to be involved in polyamine block (see Figure 13-2).31,38–40 Comparative molecular modeling and ligand docking of the three-dimensional structures of quinidine and chloroquine in the intracellular domain of KIR2.1 predicted that chloroquine effectively blocks potassium flow by binding at the center of the ion permeation vestibule of KIR2.1. In contrast, quinidine binds the vestibular side, only partially blocking ion movement.41 An interesting finding was that neutralization of the D172 residue, which faces the transmembrane pore and is key in the polyamine block of KIR2.1 (termed rectifying controller), does not affect chloroquine KIR2.1-blocking potency. A gain-of-function mutation in KCNJ2 was recently reported to cause one form of short QT syndrome (SQT3). A missense mutation (D172N) located within the transmembrane ion conduction pathway of KIR2.1 was found in a father and daughter with short QT interval.11 The biophysical consequences of D172N mutation include a reduction in the degree of current rectification at depolarized potentials. Interestingly, chloroquine caused a dose- and voltage-dependent reduction in wild type (WT), D172N, and WT-D172N heteromeric KIR2.1 current. As expected, the potency and kinetics of chloroquine block of D172N and WTD172N KIR2.1 current were similar to WT. In silico modeling of the heterozygous WT-D172N KIR2.1 condition predicted that 3 µM chloroquine would normalize inward rectifier K+ current magnitude, action potential duration, and effective refractory period. This suggested that therapeutic concentrations of chloroquine might lengthen cardiac repolarization in SQT3.42,43
Chloroquine also blocked other cardiac inward rectifier channels. In patch-clamp experiments, chloroquine blocked IK1 (IC50 = 0.69 µM), IKACh (IC50 = 0.38 µM), and IKATP (IC50 = 0.51 µM) (see Figure 13-3). Comparative molecular modeling and ligand docking of chloroquine in the intracellular domains (cytoplasmic pore) of KIR2.1, KIR3.1, and KIR6.2 suggested that chloroquine blocks or reduces potassium flow by interacting with negatively charged amino acids facing the ion permeation vestibule of the channel in question. However, these detailed analyses revealed important differences. The molecular modeling indicated that chloroquine binds more effectively to the cytoplasmic domains of KIR2.1 and KIR3.1 than to that of KIR6.2.31 Chloroquine also seemed to interact with acidic residues in the cytoplasmic domain of KIR3.1 and KIR3.4; however, the negative electrostatic potential in the channel pore was lower than in KIR2.1, resulting in partial channel block. In KIR6.2, the molecular modeling and ligand docking of chloroquine in the intracellular domains (cytoplasmic pore) of 6.2 suggested that the drug binds to the side of a large cavity near the membrane side of the protein, leaving a pathway of 10 Å in diameter, which most likely will not impede the passage of potassium ions.
The honey bee venom–isolated toxin tertiapin and the oxidation-resistant tertiapin-Q derivative are potent inhibitors of KIR3.1/3.4 and KIR1.1 channels with nanomolar affinity. In isolated cardiac myocytes, tertiapin Q blocks IKACh without affecting IKATP, IK1, and cardiac potassium voltage–dependent currents like Ito, IKr, or IKs and has shown to have antiarrhythmic efficacy in dogs. Tertiapin binds to the outer vestibule of the conduction pore formed by the linker between the first and second transmembrane (M1–M2) segments. An α-helical structure within the toxin interacts with a short sequence of aromatic residues located in the N-terminal part of the linker that confers high affinity for tertiapin.35
Drugs That Interfere in PIP2–Channel Interaction
Several polyvalent cations, such as polyamines, trivalent metals, neomycin, and polylysine, inhibited KIR channels by screening the negatively charged head groups of phosphatidylinositol 4,5-bisphosphate (PIP2). PIP2 comprises approximately 1% of plasma membrane phospholipids and is required in many plasma membrane processes, including the function of ion channels and transporters.44,45 Activation by PIP2 is a common feature of all KIR channels; after excision of an inside-out membrane patch from a cell expressing KIR channels, current amplitude diminishes over time. Such a rundown of the current results from PIP2 depletion from the cell membrane. The K+ currents can be restored partially by exposing the cytoplasmic face of the patch to PIP2. Each PIP2 molecule consists of an inositol head group and fatty-acid side chains inserted into the membrane. The negatively charged PIP2 head groups are anchored just below the plane of the membrane by the lipid tails of the molecule. It has been proposed that the channel opens when the negative-head charges of PIP2 interact with positively charged residues in the cytoplasmic domain, near the plasma membrane.46,47
The x-ray crystal structure of a chicken KIR2.2 channel in a complex with a short-chain (dioctanoyl) derivative of PIP2 has been resolved recently.48 A simple allosteric mechanism of gating control by PIP2 appears to be responsible for opening the gate at the TM2 helix-bundle crossing. One PIP2 molecule binds to each of the four channel subunits at an interface between the TMD and the CPD. On PIP2 binding, the entire CTD translates 6 Å toward the TMD and becomes tethered to the TMD, and the inner helix gate starts to open. The PIP2-binding site comprises amino acids from the TMD and the CPD, the acyl chains, glycerol backbone, and 1′ (phosphodiester) phosphate of PIP2 interact with the TMD, whereas the inositol head group makes interactions with the CPD. The acyl chains insert into the membrane layer and interact with hydrophobic amino acids on both the inner and outer helices, whereas the 1′ phosphate makes interactions with amino acids forming the sequence arginine-tryptophan-arginine (amino acids 78-80 in KIR2.2). The inositol ring of the head group is oriented toward the CPD, where the 4′ and 5′ phosphates are positioned to interact directly with K183, R186, K188, and K189 of the CPD. Other amino acids on the tether helix, including R190, participate in the formation of a hydrogen-bonding network that seems to strengthen the interaction between the tether helix and other regions of the CPD. Comparing the KIR2.2 amino acids involved in binding to PIP2 by sequence alignment with the other members of the KIR channel family shows that the amino acids are highly conserved among the large family.
A variety of signaling partners influence KIR channel activity by modulating the interaction of KIR channels with PIP2. Some of the results strongly suggest that the apparent affinity of channel-PIP2 interactions is a key element in determining modulation of KIR channels, whereby the strength of channel-PIP2 interactions controls the sensitivity of KIR channels to regulation by modulating factors. Strong interaction between PIP2 and KIR2.1 prevented interference from other modulatory factors like pH, protein kinase C, and membrane receptors such as M1 (type-1 muscarinic) and epidermal growth factor receptors, whereas the weaker interaction between PIP2 and KIR2.3 was interfered further by those modulatory factors such that the function of KIR2.3 or KIR3.4* was reduced.46,47
The cationic amphiphilic drugs (CADs) represent one of the most abundant chemical groups used in pharmacotherapy.49 In contrast to the diverse pharmacologic actions and therapeutic applications, these drugs share several common physicochemical properties. CADs typically contain a hydrophilic domain consisting of one or more primary or substituted nitrogen groups positively charged at physiologic pH and a hydrophobic domain consisting of an aromatic and/or aliphatic ring structure, which may be substituted with one or more halogen moieties. This results in polar hydrophilic cationic side-chain and apolar ring systems within one molecule. CADs bind to the negatively charged phosphoinositides. The cationic group of CADs is normally placed between the polar head groups of phospholipids, and the hydrophobic portion is directed toward the hydrophobic interior of the membrane; thus, the drug molecule intercalates between lipid molecules.
Several widely used CADs have been associated with inward rectifier current disturbances, and recent evidence points to interference of the channel–PIP2 interaction as the underlying mechanism of action.50–55 The inhibitory potency of CADs on the different subfamilies of KIR channels correlates with the sensitivity of the channel to rundown arising from PIP2 depletion. Consistent with this, channels relatively resistant to rundown mediated by PIP2 depletion, such as KIR1.1, 2.1, and 2.2 (“high PIP2 affinity”), are less sensitive to CADs, in contrast to KIR2.3, 3.x, and 6.2, which are more sensitive.
Experimentally, interference of the drug with the interaction of KIR2.x, KIR3.1/3.4, and KIR6.2/SUR2a channels and PIP2 is suggested from four sources of evidence: (1) slow onset of current block when CADs are applied from either the inside or outside of the channel; (2) mutation of KIR2.3 (I213L) and KIR6.2 (C166S) increases their affinity for PIP2 and lowers their sensitivity for CADs; (3) mutations of KIR2.1 (L222I and K182Q), which decrease its affinity for PIP2, increase its sensitivity for CADs (Figure 13-4); and (4) coapplication of CADs with PIP2 lowers CAD-mediated current inhibition.50–55
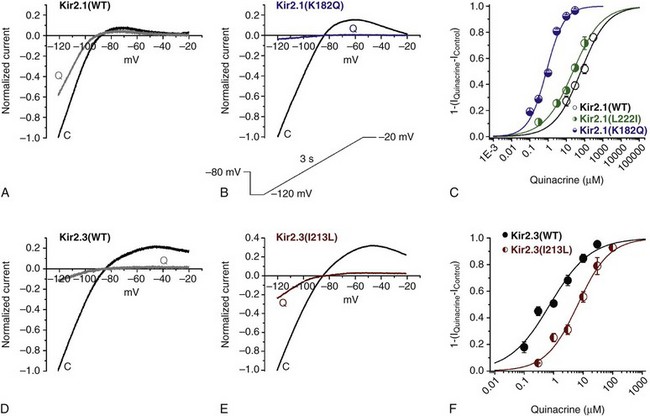
Figure 13-4 Inhibitory effects of quinacrine on KIR2.1 (wild type) (A), KIR2.1 (K182Q) (B), KIR2.3 (wild type) (D), and KIR2.3 (I213L) (E) channels expressed in HEK 293 cells. Each panel shows typical currents recorded under control conditions (trace C) and in the presence of 30 µM quinacrine (trace Q). Experiments were recorded using the whole-cell configuration, elicited by the protocol shown in the inset. Current was normalized to the amplitude recorded at −120 mV under control conditions. Panels C and F show concentration-response curves constructed from current measured at −120 mV. The IC50 of quinacrine on KIR2.1 (wild type) was 65 ± 7 µM, on KIR2.1 (K182Q) was 0.80 ± 0.07 µM, on KIR2.1 (L222I) was 24 ± 4 µM, on KIR2.3 (wild type) was 0.74 ± 0.15 µM, and on KIR2.3 (I213L) was 6.5 ± 0.9 µM (n = 5). Data shown represent mean ± SEM. (Modified from López-Izquierdo A, Aréchiga-Figueroa IA, Moreno-Galindo EG, et al: Mechanisms for Kir channel inhibition by quinacrine: acute pore block of Kir2.x channels and interference in PIP2 interaction with Kir2.x and Kir6.2 channels. Pflugers Arch 462:505–517, 2011.)
Bupivacaine was the first CAD in which the mechanism of inhibition of the inward rectifier channel KIR3.2 by interference with the PIP2–channel interaction was proposed. Bupivacaine is a local anesthetic drug with a long duration of action that produces excellent sensory anesthesia. Bupivacaine produced inhibitory effects on G protein–gated KIR3.x channels expressed in Xenopus laevis oocytes but did not affect KIR1.1 or KIR2.1 channels. The effects of bupivacaine on KIR3.x channels was independent of the method of channel activation, by activation of the muscarinic receptor or directly via coexpressed G protein Gβγ subunits or ethanol, which suggested that bupivacaine’s site of action was at the channel protein itself.50 On the basis that KIR1.1 and 2.1 are high-affinity PIP2 binders and the KIR3x subfamily channel shows a high sensitivity to PIP2 depletion, it was proposed that bupivacaine antagonized the interaction of PIP2 with KIR3.x channels, thereby decreasing the current. Consistent with this hypothesis, it was found that KIR3.2(I234L) and 3.1(M223L)/KIR3.2(I234L) mutant channels, which have increased affinity for PIP2, were less sensitive to bupivacaine.50
The first-generation antihistamines, mepyramine and diphenhydramine, are H1 competitive receptor antagonists. Both drugs reduced the KIR2.3 current amplitude by 25% and 17% at 100-µM concentrations, whereas KIR2.1 current was insensitive to both antihistamines.56 Furthermore, KIR3.4 current was inhibited by mepyramine similarly to KIR2.3. The inhibitory effects of both of agents were voltage independent. Moreover, mepyramine-induced KIR2.3 current inhibition was significantly reduced by a single-point mutation of KIR2.3 (I213L), which enhanced its interaction with membrane PIP2. These results suggested that membrane PIP2 may likely be involved in the KIR current inhibition by the first-generation antihistamines.
Quinacrine is an antimalarial agent that has been used for a number of additional indications, such as other parasitic infections, as an antifibrillatory agent and for treatment of autoimmune disorders. Like other CADs, quinacrine has a high lipophilicity (log P = 5.5) and interacts directly with membrane phospholipids to induce lipidosis. The side chain of the aminoacridine group of quinacrine (pKa = 10.3) is identical to the side chain of the aminoquinoline drug, chloroquine. Quinacrine differentially inhibited KIR channels in the following order of potency: KIR6.2 > KIR2.3 > KIR2.1. In addition, it was found in cardiac myocytes that quinacrine inhibited IKATP > IK1 (see Figure 13-4). It was presented evidence that quinacrine displayed a double action toward KIR2.1 and 2.3 channels, that is, interfering with PIP2 interaction and direct pore block (see Figure 13-4). On KIR6.2/SUR2a channels, the inhibitory effects of quinacrine seemed to be only by interference in PIP2-channel interaction.55
Tamoxifen is a drug used in the treatment of hormone-responsive breast cancer and prevention of breast cancer in women at high risk. At clinically relevant concentrations, tamoxifen inhibited the inward rectifier potassium currents IK1, IKATP and IKACh in feline atrial and ventricular myocytes in the following order of sensitivity: IKACh > IKATP > atrial IK1 > ventricular IK1. Tamoxifen also inhibited in that same order KIR6.2/SUR2A and KIR3.1/3.4 > KIR2.3 > KIR2.1 = KIR2.2 in transfected HEK-293 cells.51,52 The order of sensitivity of the different KIR channels, the slow inhibition time course, the independence of internal or external drug administration, and the voltage independence of the inhibition induced by tamoxifen suggested that tamoxifen probably inserts into the lipid membrane and might interfere with PIP2–channel interactions. Evidence from three other sources—KIR2.3(1213 L), KIR6.2(C166S), and exogenous PIP2—further suggested the interference of tamoxifen with the KIR-PIP2 interaction.
Mefloquine is an antimalarial drug that is used mainly as a prophylactic for malaria and in cases of chloroquine-resistant malaria. Mefloquine produced inhibition of IKATP channels of pancreatic β cells. The effect is mediated by interaction of the drug with the KIR6.2 subunit, rather than the regulatory SUR subunit. Mefloquine inhibited three different types of cardiac KIR channels in the following order of potency, KIR6.2/SUR2A > KIR2.3 > KIR2.1, as demonstrated by determination of the IC50 for mefloquine inhibition of the respective KIR channels.53 Mefloquine also inhibited IKATP and IK1 in cardiac myocytes. Interestingly, the characteristics of the mefloquine inhibition of the KIR channels were similar to those found with tamoxifen.51,52 The effect on KIR channels was concentration dependent but voltage independent and showed a slow time course inhibition. In addition, the effects were similar whether mefloquine was applied externally or internally, suggesting that the inhibitory effect was membrane delimited. In accordance, the KIR2.3 (I213L) mutant was significantly less sensitive and in inside-out patches, continuous application of exogenous PIP2 strikingly prevented the mefloquine inhibition.
Carvedilol, a β- and α-adrenoceptor blocker, is used to treat congestive heart failure, mild to moderate hypertension, and myocardial infarction. Carvedilol is a CAD with a high lipophilicity (log P = 3.8) and an α-hydroxyl secondary amine functional group (pKa 7.9). Ferrer et al. demonstrated that carvedilol inhibits KIR2.3-carried current with at least 100-fold higher potency (IC50 = 0.49 µM) than KIR2.1 (IC50 = 50 µM).54 KIR2.3 channels showed similar inhibition characteristics like tamoxifen and mefloquine, results that suggested that carvedilol, as other cationic amphiphilic drugs, inhibited KIR2.3 channels by interfering with the PIP2–channel interaction. Again, the effect was concentration dependent and voltage-independent, whereas the KIR2.3 (I213L) mutation decreased the potency of block more than 20-fold (IC50 = 11.1 µM), and in the presence of exogenous PIP2, inhibition by carvedilol was strongly reduced (80 vs. 2% current inhibition).54
The acidic compound thiopental is a short-acting barbiturate used intravenously for anesthesia induction. Recently, it has been shown that thiopental inhibited KIR2.1, KIR2.2, KIR2.3, KIR1.1, and KIR6.2/SUR2A currents with similar potency; in whole-cell experiments, 30 µM thiopental decreased KIR2.1, KIR2.2, KIR2.3, and KIR1.1 currents to 55% ± 6%, 39% ± 8%, 42% ± 5%, and 49% ± 5%, respectively.57 Evidence was presented showing that the thiopental effect on KIR channels likely involves disruption of interactions between KIR6.2/SUR2a and PIP2. Thiopental inhibited all KIR channels in a concentration-dependent and voltage-independent manner. In addition, the time course of thiopental inhibition was slow (T1/2-4 minutes) and independent of external or internal drug application, suggesting that the inhibitory effect was membrane delimited. In addition, in the presence of exogenous PIP2, inhibition by thiopental on KIR channels was significantly decreased, suggesting an interference of PIP2-KIR channel interaction. It was proposed that the anionic drug thiopental may have a different mechanism of action on KIR channels compared with polycations like neomycin or CADs. It was suggested that thiopental interacts with the various KIR channels directly at a common site and it interfered with the KIR channel–PIP2 interaction. A candidate residue for thiopental binding may be R218 in KIR2.1, which is conserved among all KIR channels.57 However, that hypothesis requires validation, and more work needs to be done to determine whether the inhibition of thiopental depends specifically on interference with KIR channel–PIP2 interactions.
Drugs That Induce IK1 Activation
The multichannel-blocker antiarrhythmic drug flecainide exhibits effective control of ventricular arrhythmias associated with mutations in KCNJ2 associated with Andersen-Tawil syndrome. Recently, it was found that flecainide acutely increased human IKIR2.1, mainly by reducing the polyamine blockade of the channel. In addition, incubation with flecainide increased functional KIR2.1 channel density. It was proposed that flecainide reduces the inward rectification of the current at potentials positive to the potassium reversal potential. Flecainide interacts with C311 at the G loop of the CPD of the channel. It was also shown that incubation with flecainide increased expression of functional KIR2.1 channels on the plasma membrane, an effect also determined by C311. Interestingly, flecainide pharmacologically rescues R67W, but not R218W, channel mutations found in patients with Andersen-Tawil syndrome.58
The neurosteroid pregnenolone sulfate has been found to preferentially activate KIR2.3 with a half-maximal concentration of 16 µM and a maximal current potentiation of approximately 80%. Pregnenolone appeared to be selective in that it had no effects on KIR1.1, KIR2.1, KIR2.2, or KIR3.1 currents. Preliminary data suggested that pregnenolone sulfate acts directly on KIR2.3 at an extracellular site, although the precise binding site location and mechanism of action have not yet been defined.59
References
1. Katz, B. Les constants electriques de la membrane du muscle. Arch Sci Physiol. 1949; 3:285–299.
2. Hibino, H, Inanobe, A, Furutani, K, et al. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol Rev. 2010; 90:291–366.
3. Anumonwo, JM, Lopatin, AN. Cardiac strong inward rectifier potassium channels. J Mol Cell Cardiol. 2010; 48:45–54.
4. Flagg, TP, Enkvetchakul, D, Koster, JC, et al. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev. 2010; 90:799–829.
5. De Boer, TP, Houtman, MJC, Compier, M, et al. The mammalian KIR2. x inward rectifier ion channel family: expression patterns and pathophysiology. Acta Physiol. 2010; 199:243–255.
6. Krapivinsky, G, Gordon, EA, Wickman, K, et al. The G-protein-gated atrial K+ channel IKAch is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature. 1995; 374:135–141.
7. Inagaki, N, Gonoi, T, Clement, JP, 4th., et al. Reconstitution of IKATP: An inward rectifier subunit pulus the sulfonylurea receptor. Science. 1995; 270:1166–1170.
8. Dean, M, Annilo, T. Evolution of the ATP-Binding Cassette (ABC) transporter superfamily in vertebrates. Annu Rev Genom Hum Genet. 2005; 6:123–142.
9. Zaritsky, JJ, Redell, JB, Tempel, BL, et al. The consequences of disrupting cardiac inwardly rectifying K(+) current (IK1) as revealed by the targeted deletion of the murine KIR2. 1 and KIR2. 2 genes. J Physiol. 2001; 533:697–710.
10. Plaster, NM, Tawil, R, Tristani-Firouzi, M, et al. Mutations in KIR2. 1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell. 2001; 105:511–519.
11. Priori, SG, Pandit, SV, Rivolta, I, et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ Res. 2005; 96:800–807.
12. Vega, AL, Tester, DJ, Ackerman, MJ, et al. Protein kinase A-dependent biophysical phenotype for V227F-KCNJ2 mutation in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2009; 2:540–547.
13. Hattori, T, Makiyama, T, Akao, M, et al. A novel gain-of-function KCNJ2 mutation associated with short-QT syndrome impairs inward rectification of KIR2. 1 currents. Cardiovasc Res. 2012; 93:666–673.
14. Bettahi, I, Marker, CL, Roman, MI, et al. Contribution of the KIR3. 1 subunit to the muscarinic-gated atrial potassium channel IKACh. J Biol Chem. 2002; 277:48282–48288.
15. Wickman, K, Nemec, J, Gendler, SJ, et al. Abnormal heart rate regulation in GIRK4 knockout mice. Neuron. 1998; 20:103–114.
16. Yang, Y, Yang, Y, Liang, B, et al. Identification of a KIR3. 4 mutation in congenital long QT syndrome. Am J Hum Genet. 2010; 86:872–880.
17. Miki, T, Suzuki, M, Shibasaki, T, et al. Mouse model of Prinzmetal angina by disruption of the inward rectifier KIR6. 1. Nat Med. 2002; 8:466–472.
18. Saito, T, Sato, T, Miki, T, et al. Role of ATP-sensitive K+ channels in electrophysiological alterations during myocardial ischemia: A study using KIR6. 2-null mice. Am J Physiol Heart Circ Physiol. 2005; 288:352–357.
19. Liu, XK, Yamada, S, Kane, GC, et al. Genetic disruption of KIR6. 2, the pore-forming subunit of ATP-sensitive K+ channel, predisposes to catecholamine-induced ventricular dysrhythmia. Diabetes. 2004; 53:S165–S168.
20. Flagg, TP, Kurata, HT, Masia, R, et al. Differential structure of atrial and ventricular KATP: Atrial KATP channels require SUR1. Circ Res. 2008; 103:1458–1465.
21. Elrod, JW, Harrell, M, Flagg, TP, et al. Role of sulfonylurea receptor type 1 subunits of ATP-sensitive potassium channels in myocardial ischemia/reperfusion injury. Circulation. 2008; 117:1405–1413.
22. Stoller, D, Kakkar, R, Smelley, M, et al. Mice lacking sulfonylurea receptor 2 (SUR2) ATP-sensitive potassium channels are resistant to acute cardiovascular stress. J Mol Cell Cardiol. 2007; 43:445–454.
23. Haïssaguerre, M, Chatel, S, Sacher, F, et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. J Cardiovasc Electrophysiol. 2009; 20:93–98.
24. Medeiros-Domingo, A, Tan, BH, Crotti, L, et al. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac KATP channel KIR6. 1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm. 2010; 7:1466–1471.
25. Barajas-Martínez, H, Hu, D, Ferrer, T, et al. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm. 2012; 9:548–555.
26. Delaney, JT, Muhammad, R, Blair, MA, et al. A KCNJ8 mutation associated with early repolarization and atrial fibrillation. Europace. 2012.
27. Tester, DJ, Tan, BH, Medeiros-Domingo, A, et al. Loss-of-function mutations in the KCNJ8-encoded KIR6. 1 KATP channel and sudden infant death syndrome. Circ Cardiovasc Genet. 2011; 4:510–515.
28. Bienengraeber, M, Olson, TM, Selivanov, VA, et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004; 36:382–387.
29. Olson, TM, Alekseev, AE, Moreau, C, et al. KATP channel mutation confers risk for vein of Marshall adrenergic atrial fibrillation. Nat Clin Pract Cardiovasc Med. 2007; 4:110–116.
30. Harakalova, M, van Harssel, JJ, Terhal, PA, et al. Dominant missense mutations in ABCC9 cause Cantú syndrome. Nat Genet. 2012; 44:793–796.
31. Noujaim, SF, Stuckey, JA, Ponce-Balbuena, D, et al. Specific residues of the cytoplasmic domains of cardiac inward rectifier potassium channels are effective antifibrillatory targets. FASEB J. 2012; 4:4302–4312.
32. Tamargo, J, Caballero, R, Gómez, R, et al. Pharmacology of cardiac potassium channels. Cardiovasc Res. 2004; 62:9–33.
33. Zaks-Makhina, E, Li, H, Grishin, A, et al. Specific and slow inhibition of the KIR2. 1 K+ channel by gambogic acid. J Biol Chem. 2009; 284:15432–15438.
34. Brown, JH, Laiken, N. Muscarinic receptors agonists and antagonists. In Brunton L, Chabner B, Knollman B, eds. : Goodman and Gilman’s The Pharmacological Basis of Therapeutics, ed 12, New York, McGraw-Hill, 2011.
35. Dobrev, D, Carlsson, L, Nattel, S. Novel molecular targets for atrial fibrillation therapy. Nat Rev Drug Discov. 2012; 11:275–291.
36. McBride, BF. The emerging role of antiarrhythmic compounds with atrial selectivity in the management of atrial fibrillation. J Clin Pharmacol. 2009; 49:258–267.
37. van der Heyden, MAF, Sánchez-Chapula, JA. Towards specific cardiac IK1 modulators for in vivo application; old drugs point the way. Hearth Rhythm. 2011; 8:1076–1080.
38. Sanchez-Chapula, JA, Salinas-Stefanon, E, Torres-Jacome, J, et al. Blockade of currents by the antimalarial drug chloroquine in feline ventricular myocytes. J Pharm Exp Ther. 2001; 297:437–445.
39. Rodríguez-Menchaca, AA, Navarro-Polanco, RA, Ferrer-Villada, T, et al. The structural molecular basis of chloroquine block of the inward rectifier KIR2. 1 channel. Proc Natl Acad Sci U S A. 2008; 105:1364–1368.
40. de Boer, TP, Nalos, L, Stary, A, et al. The anti-protozoal drug pentamidine blocks KIR2. x-mediated inward rectifier current by entering the cytoplasmic pore region of the channel. Br J Pharmacol. 2010; 159:1532–1541.
41. Noujaim, SF, Stuckey, JA, Ponce-Balbuena, D, et al. Structural bases for the different antifibrillatory effects of chloroquine and quinidine. Cardiovasc Res. 2011; 89:1–8.
42. El Harchi, A, McPate, MJ, Zhang, YH, et al. Action potential clamp and chloroquine sensitivity of mutant KIR2. 1 channels responsible for variant 3 short qt syndrome. J Mol Cel Cardiol. 2009; 47:743–747.
43. Lopez-Izquierdo, A, Ponce-Balbuena, D, Ferrer, T, et al. Chloroquine blocks a mutant KIR2. 1 channel responsible for short QT syndrome and normalizes repolarization properties in silico. Cell Physiol Biochem. 2009; 24:153–160.
44. Fan, Z, Makielski, JC. Anionic phospholipids activate ATP-sensitive potassium channels. J Biol Chem. 1997; 272:5388–5395.
45. Hilgemann, DW, Feng, S, Nasuhoglu, C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE. 2001; RE19.
46. Du, X, Zhang, H, Lopes, C, et al. Characteristic interactions with phosphatidylinositol 4,5–bisphosphate determine regulation of KIR channels by diverse modulators. J Biol Chem. 2004; 279:37271–37281.
47. Logothetis, DE, Jin, T, Lupyan, D, et al. Phosphoinositide-mediated gating of inwardly rectifying K+ channels. Pflugers Arch. 2007; 455:83–95.
48. Hansen, SB, Tao, X, MacKinnon, R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel KIR2. 2. Nature. 2011; 477:495–498.
49. Lundbæk, JA. Lipid bilayer–mediated regulation of ion channel function by amphiphilic drugs. J Gen Physiol. 2008; 131:421–429.
50. Zhou, W, Arrabit, C, Choe, S, et al. Mechanism underlying bupivacaine inhibition of G protein-gated inwardly rectifying K+ channels. Proc Natl Acad Sci U S A. 2001; 98:6482–6487.
51. Ponce-Balbuena, D, López-Izquierdo, A, Ferrer, T, et al. Tamoxifen inhibits KIR2. x family of inward rectifier channels by interfering with PIP2-channel intaractions. J Pharm Exp Ther. 2009; 331:563–573.
52. Ponce-Balbuena, D, Moreno-Galindo, EG, López-Izquierdo, A, et al. Tamoxifen inhibits cardiac KATP and KACh currents in part by interfering with PIP2–channel interaction. J Pharm Sci. 2010; 113:66–75.
53. López–Izquierdo, A, Ponce-Balbuena D. Moreno-Galindo, EG, et al. The antimalarial drug mefloquine inhibits cardiac inward rectifier K+ channels: Evidence for inference in PIP2 –channel interaction. J Cardiovasc Pharmacol. 2011; 57:407–415.
54. Ferrer, T, Ponce-Balbuena, D, López-Izquierdo, A, et al. Carvedilol inhibits KIR2. 3 channels by interference with PIP2-channel interaction. Eur J Pharmacol. 2011; 668:72–77.
55. Lopez-Izquierdo, A, Aréchiga-Figueroa, IA, Moreno-Galindo, EG, et al. Mechanisms for KIR channel inhibition by quinacrine: Acute pore block of KIR2. x channels and interference in PIP2 interaction with KIR2. x and KIR6. 2 channels. Pflügers Arch. 2011; 462:505–517.
56. Liu, B, Jia, Z, Geng, X, et al. Selective inhibition of KIR currents by antihistamines. Eur J Pharmacol. 2007; 558:21–26.
57. López-Izquierdo, A, Ponce-Balbuena, D, Ferrer, T, et al. Thiopental inhibits function of different inward rectifying potassium channel isoforms by a similar mechanism. Eur J Pharmacol. 2010; 638:33–41.
58. Caballero, R, Dolz-Gaitón, P, Gómez, R, et al. Flecainide Increases KIR2. 1 currents by interacting with Cysteine 311 decreasing the polyamine-induced rectification. Proc Nat Acad Sci U S A. 2010; 107:15631–15636.
59. Kobayashi, T, Washiyama, K, Ikeda, K. Pregnenolone sulfate potentiates the inwardly rectifying K channel KIR2. 3. PLoS One. 2009; 4:e6311.